

Abstract
Corynebacterium efficiens is a gram-positive nonpathogenic bacterium which can grow and produce glutamate at 40°C or above. By using the cumulative GC profile method, we have identified four genomic islands which have many unifying genomic island-specific features in the C. efficiens genome. The presence of the gene encoding an aspartate kinase in a genomic island helps explain the unexpected low thermal stability of this enzyme; i.e., the adaptive mutations have not occurred extensively due to the recent horizontal gene transfer.
Corynebacterium efficiens is a gram-positive nonpathogenic bacterium, and it is a close relative of Corynebacterium glutamicum, which is widely used for the fermentative production of amino acids, e.g., glutamate, on an industrial scale (2). At 40°C or above, C. efficiens can still grow and produce glutamate, whereas C. glutamicum cannot. The remarkable thermostability of C. efficiens is a useful trait from an industrial viewpoint, as it reduces the considerable cost of the cooling needed to dissipate the heat generated during glutamate fermentation (9). The complete genome sequence of C. efficiens, which became available recently, provides an opportunity to investigate the mechanisms for the high thermostability of C. efficiens at the sequence level. Indeed, the GC content of C. efficiens is 10% higher than that of C. glutamicum. In addition, some biased amino acid substitutions that increase protein stability were found (9).
Genomic islands contain clusters of genes that are horizontally transferred. By transferring genes across species boundaries, horizontal gene transfer (HGT) alters the genotype of a bacterium, which leads to increased genetic diversity and even new species. Now it is becoming increasingly clear that HGT has critical roles throughout bacterial evolution (1, 3-6, 8, 11).
Although the complete genome sequence of C. efficiens is available, no horizontally transferred genomic islands have been identified in the genome. Among the methods for detecting genomic islands, assessing the changes in GC content remains an established way. In the paper describing the genome, Nishio et al. used a window-based method, i.e., 20-kb sliding windows with a 1-kb step, to display the GC content distribution (see Fig. 1 in reference 9). The window-based method has a low resolution in detecting the GC content change. We recently proposed a windowless method for the GC content computation, the cumulative GC profile, which is much more sensitive to detecting GC content change than the traditional window-based method (12, 14, 15). In the present study, we used the cumulative GC profile to identify genomic islands in the C. efficiens genome. Consequently, four genomic islands which have much lower GC contents than those of the rest of the genome were found. In addition, these four genomic islands have many conserved genomic island-specific features, such as biased codon usage, the presence of mobile genes, and the presence of direct repeats and a tRNA locus at junctions.
FIG. 1.

(A) The cumulative GC profile for the C. efficiens genome and (B) the distribution of the codon usage bias along the genome as determined by use of 22-kb sliding windows. In the cumulative GC profiles, an increase means a decrease in GC content, and any sharp minimum (or maximum) point indicates a turning point, in which the GC content undergoes a relatively abrupt decrease (or increase). If the cumulative GC profile is approximately described by a straight line, the corresponding region is approximately constant in GC content. Therefore, some regions of the C. efficiens genome have an abrupt decrease in GC content, and these regions are fairly homogeneous in GC content. A quantitative index, h, was used to measure the homogeneity of genomic islands. Many of such low-GC-content regions correspond to peaks in the distribution curve of codon usage bias, indicating that genes in these regions have more-biased codon usages. The peak around kb 550 corresponds to a cluster of 18 ribosomal protein genes, whereas the peaks at about kb 266, kb 405, kb 875, and kb 1275 correspond to regions that have many conserved features of genomic islands. See the text for details.
The thermal stabilities of 13 orthologous enzymes of C. efficiens and C. glutamicum were compared by Nishio et al. (9). Most of the tested enzymes from C. efficiens were more thermostable than their C. glutamicum orthologs; unexpectedly, however, an aspartate kinase in C. efficiens was found to be comparatively less stable than that in C. glutamicum (9). We found that the gene encoding this enzyme from C. efficiens is located in one of the identified genomic islands. Therefore, one explanation is that due to the recent horizontal transfer of this gene, the adaptive mutations of the C. efficiens genome have not occurred extensively enough to increase the thermal stability of this aspartate kinase, and indeed, it lacks the biased amino acid substitutions that increase protein stability.
MATERIALS AND METHODS
The complete genome sequence of C. efficiens YS-314 (NC_004369) was downloaded from GenBank (http://www.ncbi.nlm.nih.gov/).
The methods of the cumulative GC profile, the computation of codon usage bias, and the definition of homogeneity of the genomic islands have been detailed previously (15). Here we briefly summarize the methods.
Use of the cumulative GC profile to calculate GC content.
We define
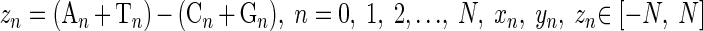 |
(1) |
where An, Cn, Gn, and Tn are the cumulative numbers of the bases A, C, G, and T, respectively, occurring in the subsequence from the first base to the nth base in the inspected DNA sequence with length N. zn is one of the components of the Z curve, which is a three-dimensional curve that uniquely represents a DNA sequence (13, 16). To amplify the deviations of zn, the curve of zn ∼ n is fitted by a straight line by the least-squares technique,
 |
(2) |
where (z, n) is the coordinate of a point on the straight line fitted and k is its slope. Instead of using the curve of zn ∼ n, we will use the z′ curve, or cumulative GC profile, hereafter, where
 |
(3) |
With GC denoting the average GC content within a region Δn in a sequence, we find from equations 1, 2, and 3 that
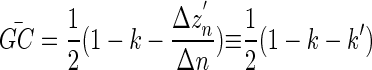 |
(4) |
where k′ = Δzn/Δn represents the average slope of the z′ curve within the region Δn. The region Δn is usually chosen to be a fragment of a natural DNA sequence, e.g., a genomic island. Equation 4 describes the windowless technique for the GC content computation (12).
An index to measure codon usage bias.
The occurrence frequencies of codons (the stop codons are excluded) in a protein-coding gene may be deemed a 61-dimension codon usage vector. The mean codon usage vector determined for all genes in a genome is denoted by c̄. Suppose that the codon usage vector for the ith gene in the genome under study is denoted by ci. Then, the codon usage bias of this gene with respect to the average vector can be calculated by using the index of codon usage bias, cubi,
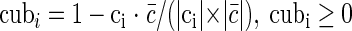 |
(5) |
, where |ci| and |c̄| are the modules of the vectors ci and c̄, respectively. The larger the cubi, the more the codon usage bias of this gene.
An index to measure the homogeneity of the GC content of genomic islands.
We noticed that genomic islands have fairly homogeneous GC contents. The fact that a genomic island has a fairly homogeneous GC content implies that zn is ∼0. The variation of zn may be described by the deviation dgi defined by
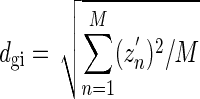 |
(6) |
where zn ∼ n is the cumulative GC profile defined in equation 3 for a genomic island (gi) and M is its length. Similarly, the deviation of the GC content from a constant for a whole genome may be described by dgenome, defined by
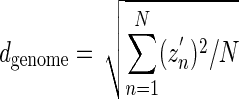 |
(7) |
where zn ∼ n is the cumulative GC profile defined in equation 3 for a whole genome and N is its length. A homogeneity index hgi is defined by the following equation.
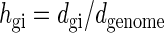 |
(8) |
RESULTS AND DISCUSSION
Figure 1 shows the cumulative GC profile of the C. efficiens genome. An increase in the cumulative GC profile means a decrease in GC content. Any sharp minimum (maximum) point indicates a turning point in which the GC content undergoes an abrupt change from a GC-rich (-poor) region to a GC-poor (-rich) region. If the cumulative GC profile is approximately described by a straight line, the corresponding region is approximately constant in GC content. Therefore, the cumulative GC profile of the C. efficiens genome shows some regions that have an abrupt decrease in GC content. Due to the high resolution, the boundaries of the low-GC-content regions can be more precisely determined (compare to Fig. 1 in reference 9). The cumulative GC profiles were used to study genomic islands in several genomes, and some conserved features have been found, e.g., the genomic island is usually associated with a relatively abrupt jump in the cumulative GC profile, which corresponds to an abrupt change and a fairly homogeneous GC content (15). Based on the behaviors of the cumulative GC profiles of the C. efficiens genome, the regions that have low GC contents are candidates for horizontally transferred genomic islands.
Some low-GC-content regions correspond to peaks in the distribution curve of codon usage bias along the genome (Fig. 1B), indicating that DNA sequences located at these regions have a much more biased codon usage than that of the rest of the genome. It is known that ribosomal protein genes have much more codon usage bias than those of other genes in genomes. Therefore, the regions corresponding to ribosomal protein genes should be excluded. The peak around kb 550 corresponds to a cluster of 18 ribosomal protein genes, which are located from kb 529 to kb 570. There is a small peak at about kb 2980, which corresponds to six rRNA genes and two ribosomal protein genes located at a region from kb 2977 to kb 3000.
In the cumulative GC profile, four low-GC-content regions that correspond to four peaks in the distribution curve of codon usage bias, i.e., the peaks at about kb 266, kb 405, kb 875, and kb 1275, do not have ribosomal genes; instead, they have many genomic island-specific features. Therefore, these four regions are likely to be horizontally transferred genomic islands, which are designated CEGI-1, CEGI-2, CEGI-3, and CEGI-4, respectively.
Genomic islands are usually different in many characteristics from other regions of the core genome. For instance, genomic islands are different in GC content and codon usage from the rest of the genome. In addition, genomic islands have many unifying features. For instance, genomic islands are usually flanked by direct repeat elements, and an integrase gene is frequently located at the 5′ junction. Furthermore, tRNA loci, which are usually located in the junctions, presumably are utilized as the integration sites. Finally, genomic islands often possess genes, such as integrase and transposase genes, that code for genetic mobility.
The GC contents of CEGI-1, CEGI-2, CEGI-3, and CEGI-4 are 0.595, 0.555, 0.595, and 0.566, respectively, much lower than that of the rest of the genome, 0.635. The codon usage biases of CEGI-1, CEGI-2, CEGI-3, and CEGI-4 are 0.181, 0.205, 0.186, and 0.197, respectively, values which are statistically larger than that of the rest of the genome, 0.146 (P of <0.001 for all four genomic islands) (Table 1).
TABLE 1.
Features of the four genomic islands in the C. glutamicum genome
| Segment | Start | End | Length (bp) | No. of genes | Codon usage bias ± SD | GC content | Junction feature(s) | h valuea | Other features |
|---|---|---|---|---|---|---|---|---|---|
| Genomeb | 1 | 3147090 | 2,949,299 | 2,755 | 0.146 ± 0.080 | 0.635 | 0.903 | ||
| CEGI-1 | 225152c | 295658d | 70,507 | 60 | 0.181 ± 0.179e | 0.595 | A recombination protein gene at the 5′ junction; a tRNAPro gene at the 3′ junction | 0.175 | 6 transposase genes |
| CEGI-2 | 371109f | 425727g | 54,619 | 51 | 0.205 ± 0.101e | 0.555 | Unknown | 0.256 | 5 transposase genes |
| CEGI-3 | 873060h | 913441i | 40,382 | 50 | 0.186 ± 0.078e | 0.595 | Direct repeats at both ends | 0.093 | Except for an integrase and a phage-associated protein, all ORFs encode hypothetical proteins |
| CEGI-4 | 1270647j | 1302930k | 32,284 | 34 | 0.197 ± 0.069e | 0.566 | Insertion element at the 3′ junction | 0.061 | 11 transposase genes |
h value, homogeneity index.
Genomic islands are not included.
Start of the ORF CE0211, which codes for a recombination protein.
End of a tRNAPro gene.
P < 0.001 compared to value for genome.
Start of the ORF CE0340, which codes for a transposase.
End of the ORF CE0390, which codes for a hypothetical protein.
Start of the repeat 1.
End of the repeat 2.
Start of the ORF CE1212, which codes for a hypothetical protein.
End of the ORF CE1245, which is in an insertion element.
CEGI-1 is about 70 kb in length and possesses 60 genes. A gene coding for a recombination protein is located at the 5′ junction of CEGI-1. A tRNA locus is located at the 3′ junction, suggesting that this tRNA locus is the integration site. CEGI-1 contains some open reading frames (ORFs) that encode various enzymes, such as aspartate kinase, aspartate-semialdehyde dehydrogenase, and glycerate kinase. CEGI-2 is 55 kb in length and possesses 51 genes. The 51 genes also include some enzymes, such as tyrosine phosphatase and GDP-d-mannose dehydratase. CEGI-3 has a length of about 40 kb and possesses 50 genes. CEGI-3 has a very conserved structure of genomic islands. CEGI-3 is flanked by two 26-bp direct-repeat elements. An integrase gene is located at the 5′ junction (Fig. 2). In addition, it is striking that among the 50 genes, except for an integrase gene and a phage-related gene, all ORFs encode hypothetical proteins (96% hypothetical proteins). Indeed, it was previously found that genomic islands often have a high percentage of genes with unknown functions, e.g., the genomic island of the C. glutamicum genome (15). Therefore, the high percentage of hypothetical protein genes is also an indication that this segment of the genome has a foreign source. CEGI-4 is 32 kb in length and possesses 34 genes. Bacterial insertion sequences are segments of DNA that are capable of inserting at multiple sites in genomes. An insertion sequence is located at the 3′ junction of CEGI-4. In addition, CEGI-1, CEGI-2, and CEGI-4 have 6, 5, and 11 transposase genes, respectively.
FIG. 2.

(A) CEGI-3 has a conserved structure of genomic islands. CEGI-3 is flanked by two direct repeats, and an integrase gene is located at the 5′ junction. The figure is not drawn to scale. (B) Alignment of the two direct repeats.
We previously found that genomic islands are fairly homogeneous in GC content (15). Indeed, compared with the other regions of the genome, the four identified genomic islands are much more homogeneous in GC content, as reflected by the fact that the cumulative GC profiles associated with genomic islands are almost straight lines. To quantify the homogeneity, we defined a homogeneity index, h, to assess the homogeneity of genomic islands. The h values for CEGI-1, CEGI-2, CEGI-3, and CEGI-4 are 0.175, 0.256, 0.093, and 0.061, respectively, much smaller than 0.903, the h value for the rest of the genome. In a previous analysis of the homogeneity of some genomic islands, h values were all less than 0.1. Therefore, it seems that the homogeneity index h should not be used as a threshold to define genomic islands; instead, it should be used to compare the homogeneity of genomic islands with that of the rest of the genome.
The features, such as low GC content, biased codon usage, the presence of repeat elements, an integrase gene, and a tRNA locus at the junctions, the presence of transposase genes, and the homogeneity in terms of GC content, strongly suggest that the four regions, i.e., CEGI-1, CEGI-2, CEGI-3, and CEGI-4, are horizontally transferred genomic islands.
Among the species belonging to the genus Corynebacterium, C. efficiens can grow at the highest temperature, and it is the only one able to produce glutamate above 40°C (9). Therefore, the C. efficiens proteins are likely to be more thermostable than those of other members of the genus Corynebacterium, such as C. glutamicum. To test this, the thermal stabilities of 13 pairs, i.e., orthologs of enzymes, on the Glu and Lys biosynthetic pathways of the two species were compared on the basis of the enzymatic activities remaining after heat treatment of crude extracts. Most of the tested enzymes from C. efficiens were more thermostable than their C. glutamicum orthologs; unexpectedly, however, the enzyme aspartate kinase from C. glutamicum was more thermostable than that from C. efficiens (9). This phenomenon is hard to explain. We found that the gene encoding this enzyme, ORF CE0220, which is the only aspartate kinase gene in the C. efficiens genome, is located in CEGI-1. Therefore, one explanation is that the adaptive mutations of the C. efficiens genome have not occurred extensively due to the recent horizontal transfer of this gene. Indeed, a recent comparative study of C. efficiens, C. glutamicum, and Corynebacterium diphtheriae suggested that the evolutionary events of gene loss and HGT must have been responsible for the functional differentiation in amino acid biosynthesis of the three species of corynebacteria (10). The finding that C. efficiens may harbor four genomic islands is consistent with this result, i.e., events of HGT have happened during the evolution of the C. efficiens genome. Among the 13 tested enzymes, a diaminopimelate dehydrogenase from C. glutamicum was also more thermostable than its C. efficiens ortholog (9), but the ORF coding for this enzyme, CE2498, does not seem to be horizontally transferred.
Both C. efficiens and C. glutamicum belong to the genus Corynebacterium (2, 10). If the four genomic islands were integrated before the divergence of C. efficiens and C. glutamicum, it is likely that C. glutamicum would have the genomic islands that C. efficiens has. However, none of the four genomic islands is present in the C. glutamicum genome. Therefore, it is very likely that the four genomic islands were integrated after the divergence of C. efficiens and C. glutamicum. Therefore, the length of time that the four genomic islands have been present in the C. efficiens genome is relatively short compared to the length of time of the whole evolutionary process of C. efficiens from the origin of this species. In addition, based on a comparison of all orthologous ORFs between C. efficiens and C. glutamicum, there is tremendous bias in amino acid substitutions (9). Three substitutions were found to be important for the stability of the C. efficiens proteins: the substitutions of arginine for lysine, alanine for serine, and threonine for serine. A point system was defined previously as the difference between the sum of the three substitutions from C. glutamicum to C. efficiens (of Arg for Lys, Ala for Ser, and Thr for Ser) and the sum of the reverse substitutions. Based on this point system, the point value for the aspartate kinase is −1, suggesting that this protein does not have the three biased amino acid substitutions that lead to increased thermal stability (9). This observation further supports our hypothesis that during evolution, due to the recent HGT, the adaptive mutations have not occurred extensively enough to increase the thermal stability of this aspartate kinase.
C. efficiens is of particular interest from an industrial viewpoint because of its ability to produce amino acids at high temperatures. Aspartate kinase is the first enzyme in the aspartate-derived amino acid biosynthesis pathways for the production of lysine, methionine, threonine, and isoleucine (7). Because the gene coding for this aspartate kinase is the only aspartate kinase gene in the C. efficiens genome, it is possible that the increase in the thermostability of this enzyme can potentially further increase the ability of C. efficiens to produce certain amino acids at high temperatures. Our results may provide some insight in explaining the relatively low thermostability of this aspartate kinase and some clues to the work necessary to increase its thermostability.
Acknowledgments
The present study was supported in part by the National Natural Science Foundation of China (grant 90408028).
REFERENCES
- 1.de la Cruz, F., and J. Davies. 2000. Horizontal gene transfer and the origin of species: lessons from bacteria. Trends Microbiol. 8:128-133. [DOI] [PubMed] [Google Scholar]
- 2.Fudou, R., Y. Jojima, A. Seto, K. Yamada, E. Kimura, T. Nakamatsu, A. Hiraishi, and S. Yamanaka. 2002. Corynebacterium efficiens sp. nov., a glutamic-acid-producing species from soil and vegetables. Int. J. Syst. Evol. Microbiol. 52:1127-1131. [DOI] [PubMed] [Google Scholar]
- 3.Hacker, J., and J. B. Kaper. 2000. Pathogenicity islands and the evolution of microbes. Annu. Rev. Microbiol. 54:641-679. [DOI] [PubMed] [Google Scholar]
- 4.Hentschel, U., and J. Hacker. 2001. Pathogenicity islands: the tip of the iceberg. Microbes Infect. 3:545-548. [DOI] [PubMed] [Google Scholar]
- 5.Jain, R., M. C. Rivera, J. E. Moore, and J. A. Lake. 2003. Horizontal gene transfer accelerates genome innovation and evolution. Mol. Biol. Evol. 20:1598-1602. [DOI] [PubMed] [Google Scholar]
- 6.Koonin, E. V., K. S. Makarova, and L. Aravind. 2001. Horizontal gene transfer in prokaryotes: quantification and classification. Annu. Rev. Microbiol. 55:709-742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Malumbres, M., and J. F. Martin. 1996. Molecular control mechanisms of lysine and threonine biosynthesis in amino acid-producing corynebacteria: redirecting carbon flow. FEMS Microbiol. Lett. 143:103-114. [DOI] [PubMed] [Google Scholar]
- 8.Nakamura, Y., T. Itoh, H. Matsuda, and T. Gojobori. 2004. Biased biological functions of horizontally transferred genes in prokaryotic genomes. Nat. Genet. 36:760-766. [DOI] [PubMed] [Google Scholar]
- 9.Nishio, Y., Y. Nakamura, Y. Kawarabayasi, Y. Usuda, E. Kimura, S. Sugimoto, K. Matsui, A. Yamagishi, H. Kikuchi, K. Ikeo, and T. Gojobori. 2003. Comparative complete genome sequence analysis of the amino acid replacements responsible for the thermostability of Corynebacterium efficiens. Genome Res. 13:1572-1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nishio, Y., Y. Nakamura, Y. Usuda, S. Sugimoto, K. Matsui, Y. Kawarabayasi, H. Kikuchi, T. Gojobori, and K. Ikeo. 2004. Evolutionary process of amino acid biosynthesis in Corynebacterium at the whole genome level. Mol. Biol. Evol. 21:1683-1691. [DOI] [PubMed] [Google Scholar]
- 11.Ochman, H., J. G. Lawrence, and E. A. Groisman. 2000. Lateral gene transfer and the nature of bacterial innovation. Nature 405:299-304. [DOI] [PubMed] [Google Scholar]
- 12.Zhang, C. T., J. Wang, and R. Zhang. 2001. A novel method to calculate the G+C content of genomic DNA sequences. J. Biomol. Struct. Dyn. 19:333-341. [DOI] [PubMed] [Google Scholar]
- 13.Zhang, C. T., and R. Zhang. 1991. Analysis of distribution of bases in the coding sequences by a diagrammatic technique. Nucleic Acids Res. 19:6313-6317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhang, R., and C. T. Zhang. 2003. Identification of genomic islands in the genome of Bacillus cereus by comparative analysis with Bacillus anthracis. Physiol. Genomics 16:19-23. [DOI] [PubMed] [Google Scholar]
- 15.Zhang, R., and C. T. Zhang. 2004. A systematic method to identify genomic islands and its applications in analyzing the genomes of Corynebacterium glutamicum and Vibrio vulnificus CMCP6 chromosome I. Bioinformatics 20:612-622. [DOI] [PubMed] [Google Scholar]
- 16.Zhang, R., and C. T. Zhang. 1994. Z curves, an intuitive tool for visualizing and analyzing the DNA sequences. J. Biomol. Struct. Dyn. 11:767-782. [DOI] [PubMed] [Google Scholar]