

Summary
Mesothelioma is a lethal cancer. Despite promising outcomes associated with immunotherapy, durable responses remain restricted to a minority of patients, highlighting the need for improved strategies that better predict outcome. Here, we described the development of a mesothelioma-specific gene signature that accurately predicts survival. Comprehensive gene expression analysis of asbestos exposed MexTAg Collaborative Cross mouse tumors revealed distinct tumor clusters characterized by epithelial mesenchymal transition/extracellular matrix, or immune infiltrate related gene expression profiles. Weighted gene co-expression network analysis (WGCNA) identified 20 hub genes that drove differential gene expression. Human homologues of these 20 hub genes were refined through univariate Cox regression and least absolute shrinkage and selection operator (LASSO) regression analyses to identify a six-gene mesothelioma-specific prognostic signature that accurately predicted patient survival across four independent human mesothelioma datasets. Furthermore, this six-gene signature demonstrated the potential to predict treatment response, thus advancing the management of this challenging malignancy.
Subject areas: Biological sciences, Genetics, Cancer
Graphical abstract
Highlights
-
•
Asbestos-induced CC-MexTAg mouse tumors are defined by distinct gene expression profiles
-
•
20 hub genes refined to a six-gene, mesothelioma-specific prognostic signature
-
•
The six-gene signature accurately predicted survival across four human mesothelioma datasets
-
•
The six-gene signature demonstrated potential to predict treatment response
Biological sciences; Genetics; Cancer
Introduction
Mesothelioma is an aggressive and universally fatal cancer with a median survival of 14–18 months after diagnosis.1,2 Disease management is mostly palliative, and conventional therapeutic approaches such as surgery, radiotherapy, or chemotherapy provide only marginal improvements in patient survival.3,4 More recently, the addition of the immune checkpoint inhibitors, ipilimumab and nivolumab, to chemotherapy regimens has demonstrated durable anti-tumor immunity, leading to a modest increase in overall survival compared to chemotherapy alone2. However, this response was restricted to 20–30% of mesothelioma patients, most of whom were diagnosed with sarcomatoid mesothelioma. While these results are encouraging, the benefit of combination immunotherapy is restricted to a subset of patients; therefore, there is a clear need to deepen our understanding of the underlying mechanisms of resistance to existing therapies and develop novel approaches that predict treatment response in patients with mesothelioma.
Recent advances in high-throughput RNA sequencing technologies and associated analytical methods have enabled deep profiling of the cancer transcriptome, which has facilitated the discovery of novel biomarkers linked to cancer prognosis and treatment response.5,6 However, the robustness and validity of most transcriptomic analyses in human mesothelioma studies have been impeded by the scarcity of available mesothelioma samples and the paucity of well-defined clinical information pertaining to asbestos exposure history and treatment regimens administered following diagnosis.7,8,9 To overcome these limitations, we used a unique combination of two distinct mouse models. The Collaborative Cross (CC) is a powerful genetic resource developed for the rapid identification of genes that mediate complex traits.10,11,12,13,14,15 The CC consists of a collection of hundreds of recombinant inbred mouse strains developed from eight founder strains selected to maximize genetic diversity and archives over 90% of the common allelic diversity of the entire mouse species.16,17,18 By utilizing the CC, we maximized the genetic polymorphisms, overcoming the constraints imposed by conventional mouse models. In the well-characterized MexTAg model of mesothelioma, expression of the oncogenic SV40 large T antigen (TAg) is restricted to mesothelial cells through the utilization of a cell-type specific mesothelin promoter.19,20 Expression of SV40 TAg in these mice phenocopies p16 loss,21 consistently and reliably recapitulates key features of human mesothelioma, and tumors only develop after asbestos exposure.19,20,21 For this study we crossbred the CC mice with the C57BL/6 MexTAg mouse model to generate the MexTAg-Collaborative Cross mouse model (CCMT) that enabled a high incidence of asbestos-induced mesothelioma development within the context of maximum genetic diversity.22
The CC mouse resource has been successfully utilized to identify genes associated with the development of polygenic diseases including melanoma and prostate cancer.12,17,23,24,25 We recently employed the CC to investigate the role of host genetics in asbestos-related disease (ARD) development, specifically within the context of mesothelioma.22,26 ARD development in asbestos-exposed CCMT mice was monitored for up to 18 months, or until ARD reached a clearly defined welfare endpoint. Across the cohort of 2562 mice, encompassing 72 genetically-distinct CCMT strains, there was greater than a 3-fold variation in overall survival. This was attributed to differences in disease latency (the time between asbestos exposure and the emergence of the first signs of disease, FSD) rather than disease progression (time from FSD to cull), demonstrating that host genetics cannot slow disease progression once the tumor has been established.22
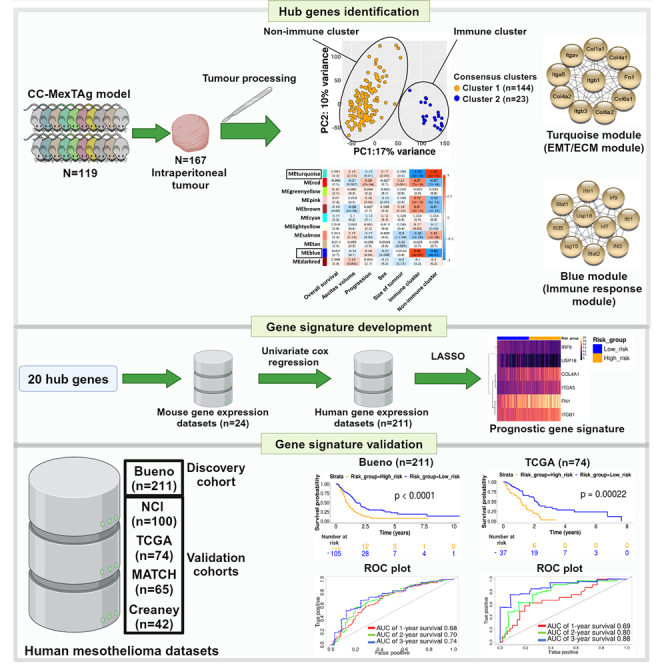
In this study, we performed comprehensive transcriptomic and immunohistochemical analysis of 167 distinct mesothelioma tumors harvested from asbestos-exposed CCMT mice22 and identified a set of 20 optimal hub genes that were responsible for the differential gene expression profiles between two distinct CCMT tumor clusters. Using the human orthologs of these hub genes, we interrogated a large human mesothelioma dataset to develop a six-gene mesothelioma-specific prognostic signature that was validated in three additional independent human mesothelioma datasets (graphical abstract).
Results
Genetically distinct CCMT mice displayed limited variation in tumor gene expression profiles
Of the 72 total CCMT strains, we selected 35 strains that exhibited the greatest variation in overall median survival22 and performed bulk RNA-Seq on 167 distinct asbestos-induced tumors from 119 mice (Figures 1A and 1B). Using unsupervised consensus clustering, we determined the optimal number of clusters (Figure S1) to better differentiate the samples based on gene expression data. The accuracy of this prediction was confirmed by the presence of two distinct clusters (dark blue) in the hierarchical clustering heatmap (Figure 1C, Cluster 1, n = 144; Cluster 2, n = 23) and illustrated using principal component analysis (PCA) (Figure 1D).
Figure 1.

Limited variation in gene expression observed between tumors derived from genetically distinct CC-MexTAg mice
(A) Experimental schema. Asbestos-exposed CCMT were monitored for up to 548 days (18 months) to assess asbestos related disease development (ARD). Phenotypic traits such as disease latency (time from first exposure to first signs of disease, FSD), disease progression (time from FSD to cull), ascites volume and overall survival were quantified. Bulk RNA-seq was performed on tumors harvested from individual asbestos-exposed CCMT mice.
(B) Violin plots displaying the ranked median overall survival (as frequency distribution of the data; median, min and max) of all 72 CCMT strains (2562 total mice). Tumors were selected from individual mice (dots) belonging to strains with the most variation in median survival (35 of the 72 total CCMT strains: highest and lowest ∼40% median survival). The dot color represents the number of tumors per mouse. The orange line corresponds to the median survival of the entire 72 strain CCMT cohort (386 days).
(C) Hierarchical consensus clustering heatmap generated using the ConsensusClusterPlus R package for K = 2, based on the minimum overlap between the two clusters using the 5000 most highly variable genes from the CCMT tumor RNA-seq data.
(D) PCA plot of CCMT tumor RNA-seq data. Orange and blue dots represent tumors characterized as Cluster 1 and Cluster 2, respectively.
(E) PCA plot of 167 tumors based on their gene expression data. Red and yellow dots depict tumors from mice above and below the CCMT cohort median survival (386 days), respectively.
(F) Volcano plot illustrating 237 differentially expressed genes (red dots) in the below CCMT cohort median group relative to the above median survival group. Adjusted p value <0.01 and log2foldchange >1 was applied as a cut off (dashed lines).
(G) Heatmap depicting the expression of all differentially expressed genes identified in 167 tumors derived from mice with overall survival below versus above the CCMT cohort median survival.
PCA also indicated a moderate variation (27% total; 17% PC1 and 10% PC2) within the CCMT tumor gene expression dataset separating the two clusters (Figure 1D). Interestingly, this variation was not associated with median survival or any other traits, including disease latency, ascites volume, disease progression, sex or tumor size (Figures 1E and S2). Differential gene expression analysis revealed 237 differentially expressed genes (adjusted p value <0.01, log2foldchange >1). However, there were no notable differences in differentially expressed genes between tumors from mice with overall survival below, versus above the median survival (386 days) of the entire CCMT cohort (Figures 1F and 1G). A parallel analysis performed on a single tumor from each mouse (n = 119) corroborated this finding, revealing 204 differentially expressed genes (adjusted p < 0.01, log2foldchange >1; Figure S3D), confirming that the inclusion of multiple tumors from a single animal did not have an undue influence on the outcomes of our analyses (Figure S3E). Cluster 2 tumors were primarily derived from two distinct CCMT strains: CC044 and CC053 (Figure S4).
Gene set enrichment analysis identified a subset of tumors characterized by high immune cell infiltration
Gene set enrichment analysis (GSEA), using the Kyoto Encyclopedia of Genes and Genomes (KEGG) and the Molecular Signatures Database (MSigDB), demonstrated an enrichment of adaptive immune response related gene sets in tumors from Cluster 2 (Figure 2A). The ‘Spermatogenesis’ gene set, containing genes related to DNA replication and cell duplication, was the only non-immune related gene set from the MSigDB database that was significantly enriched in Cluster 2 tumors.
Figure 2.

CCMT tumors with an immune phenotype are characterized by increased infiltration of CD4+ T cells and CD19+ B cells
(A) Gene set enrichment analysis (GSEA) depicting the top four significant gene sets enriched in tumors from PCA Cluster 1 (left) and Cluster 2 (right) using MSigDB and KEGG databases. The size and color of the circles represents the number of genes and the q score (false discovery rate) value per gene set, respectively.
(B) Stacked bar plot of CIBERSORT analysis results based on transcriptomes of CCMT tumors, after removing samples with low deconvolution confidence (p-value > = 0.05). The estimated proportion of immune cell subtypes is shown in different colors: B cells (pink), Macrophages (green), CD8+ T cells (red), regulatory T cells (Treg, yellow), and CD4+ T cells (dark blue).
(C) Representative images of tumors from Cluster 1 (EMT/ECM) and Cluster 2 (immune) illustrating the expression of the immune cell markers, including CD4, CD8, CD19 (B cells), IBA1 (Macrophage) and FoxP3 (Treg), and the immune checkpoint receptor, PD-L1. Scale bars = 50 μm.
In addition, CIBERSORT deconvolution analysis indicated that Cluster 2 tumors had a higher proportion of infiltrating CD4+ and CD8+ T cells and B cells, but a lower proportion of macrophages, dendritic cells, mast cells and natural killer (NK) cells than most Cluster 1 tumors (Figures 2B, S5A, and S5B). This finding was further validated through multiplex immunofluorescence, in which the density of key immune cell markers and expression of the immune co-inhibitory receptor programmed cell death ligand 1 (PD-L1) were assessed (Table S1). The results confirmed the significantly higher abundance of CD4+ T and CD19+ B cells in Cluster 2 tumors than in Cluster 1 tumors (Figures 2C and S6).
Additionally, GSEA using the BioCarta database revealed a significantly higher enrichment of cytokine related signaling pathways in Cluster 2 (Figure S7A). Analysis of cytokine expression associated with anti-tumor immune response revealed that Cluster 2 tumors had higher expression of adaptive immune related genes including Tnf, Ifnγ, Tgfb1, Il1b, Il18, Il21 and Il10 in comparison to Cluster 1 tumors (Figure S7B).
These observations collectively suggested that Cluster 2 tumors exhibited an ‘immunologically active’ phenotype, whereas Cluster 1 tumors were predominately characterized by high enrichment scores for gene sets associated with epithelial-to-mesenchymal transition (EMT), transforming growth factor β (TGF-β) signaling and extracellular matrix (ECM) receptor interaction (Figure 2A). Subsequent immunofluorescence screening for well-known EMT and ECM markers, including E-cadherin, Vimentin, Collagen type 1 alpha 1 (COL1A1) and Podoplanin (PDPN), demonstrated a significantly higher level of COL1A1 only in Cluster 1 tumors (Figure S8).
To validate the reliability of examining multiple tumors in the same mouse, we performed similar analyses using randomly selected single tumors from each mouse. The analysis confirmed the presence of two distinct clusters characterized by non-adaptive immune (EMT/ECM) (Cluster 1) and adaptive immune reactive (Cluster 2) phenotypes (Figures S9A–S9D). Additionally, gene expression profiling of multiple tumors within the same mouse demonstrated similar gene expression profiles between tumors from the same animal (Figure S10).
Genes associated with response to immune checkpoint blockade and chemotherapy are highly expressed in cluster 2 tumors
We hypothesized that CCMT tumors with increased adaptive immune cell infiltration (Cluster 2) would have a more favorable response to immune checkpoint immunotherapy. However, as CCMT mice were no longer available to empirically test this hypothesis, we utilized gene sets associated with known positive responses to chemotherapy and/or immune checkpoint blockade (ICB). These gene sets were derived from publicly available mouse and human gene expression studies27,28,29,30,31,32,33,34 and were used to compare gene expression profiles between Cluster 1 and Cluster 2 CCMT tumors.
GSEA revealed significant enrichment of gene sets associated with a positive response to ICB and chemotherapy within the Cluster 2 tumors with increased immune cell infiltration compared to the Cluster 1/non-immune (EMT/ECM) phenotype tumors (Figures 3A and 3B). To further investigate the differential expression of immune checkpoint genes between non-immune (EMT/ECM) and immune-infiltrated tumors, we assessed the expression of 20 immune checkpoint genes, known for their roles in the regulation of T cell immune responses (Table S2). All immune checkpoint genes, except Havcr2 (Tim-3) and Vsir (Vista), were expressed at significantly higher levels in immune cluster tumors than in non-immune (EMT/ECM) cluster tumors (Figure S11). Taken together, these data suggest that tumors derived from CC044 and CC053 CCMT mouse strains (viz., Cluster 2/immune phenotype) are more likely to respond to ICB and/or chemotherapy.
Figure 3.

Tumors with the immune phenotype (Cluster 2) are enriched with gene sets linked to positive response to cancer therapy
(A) GSEA of tumors from the CCMT immune cluster (Cluster 2) was conducted in reference to publicly available mouse gene sets, while (B) shows the same analysis in relation to human gene sets associated with response to ICB/chemotherapy. Circle size represents the number of genes per gene set, and circle color indicates the q score (false discovery rate) value for each gene set signature.
(C) Heatmap of correlation between the first principal component (eigengene) of modules and phenotypic traits, as well as EMT/ECM and immune clusters. Each cell contains the corresponding p value and correlation coefficient for trait-module relationships.
(D) Protein-protein interaction (PPI) network analysis of the top 10 hub genes from each of the Blue (Immune) and Turquoise (EMT/ECM) modules.
(E) Univariate Cox regression analysis of all 20 hub genes, ranked by their hazard ratios. The 95% confidence interval and p values are shown on the right end column of the table. Itgb3 is highlighted by red square and was the only gene that did not significantly associate with disease outcome.
Hub genes identified from co-expressed genetic networks strongly correlate with variation in tumor phenotype
To differentiate the molecular networks and hub genes driving variations in the CCMT tumor phenotypes, we employed Weighted Gene Co-expression Network Analysis (WGCNA). This method allowed us to construct network modules and pinpoint hub genes that correlated with the contrasting gene expression profiles between non-immune (EMT/ECM) and immune tumor clusters (Figures S12A–S12C). Eleven co-expressed modules were constructed and correlated with asbestos-exposed CCMT traits, including overall survival, disease latency or progression, ascites volume, sex, and tumor size (Figures 3C and S12D). Although no correlation was found between modules and CCMT traits (r < 0.3), some modules displayed a significant correlation with the distinct immune and non-immune (EMT/ECM) clusters identified by PCA (p < 0.05, r > 0.5).
The ‘Blue’ and ‘Turquoise’ modules showed the strongest correlation with PCA clusters as well as module membership correlation scores (MMC r > 0.8, p value < 1200), indicating that the genes within each module were strongly co-expressed and likely participated in similar biological pathways (Figures S12E and S12F). Using the gene ontology biological process (GO-BP) database, we found that the ‘Blue’ module was enriched in genes associated with immune responses, while the ‘Turquoise’ module was abundant in genes linked to morphogenesis and cell migration (Figures S12G and S12H). Moreover, intramodular connectivity analysis highlighted the 20 most highly connected hub genes (10 each for Blue and Turquoise modules) that were subsequently utilized to construct a protein-protein interaction network (PPI) for each module using the STRING database (Figure 3D).
The top 20 hits from our hub genes analysis identified key genes implicated in immune responses and included: Irf7, lrf9, lfit3, lfi35, lfih1, Stat1, Stat2, Isg15, Usp15, and lfit1 for the Blue module and for the Turquoise module the ECM and EMT related genes: Col1a1, Col4a1, Col6a2, Col6a1, Col4a2, Itgav, Fn1, Itgb1, Itga5, and Itgb3. To minimize potential bias from including mice with multiple tumors in our analysis, we repeated the analysis on single tumors from each mouse (n = 119) and observed similar sets of hub genes, with the exception that Col1a2 replaced Itgb3 in the EMT/ECM (Turquoise) module (Figures S9C and S9D).
To further probe the prognostic potential of hub genes associated with immune and non-immune (EMT/ECM) tumor clusters from asbestos-exposed CCMT mice, we leveraged tumor RNA sequencing data from BALB/c mice bearing subcutaneous AB1 mesothelioma treated with combination α-PD-L1+α-CTLA4 ICB therapy. Univariate Cox regression analysis of all 20 hub genes demonstrated a significant association between the expression of 19 of the 20 hub genes identified in CCMT tumors with ICB treated subcutaneous AB1 mesotheliomas bearing BALB/c mice35 (n = 24, Figure 3E).
Construction and validation of a mesothelioma-specific prognostic gene signature
Following the identification of similarities between the gene expression profile of Cluster 2 tumors and previously published gene sets strongly associated with positive responses to immunotherapy and chemotherapy in both mouse and human cancers, we sought to assess the prognostic potential of the identified CCMT hub genes to predict a positive outcome in human mesothelioma. The remaining 19 hub genes identified in the ICB treated mouse model were employed to develop a prognostic gene signature using well-defined, publicly available, human mesothelioma datasets. We used the largest human mesothelioma dataset (Bueno, n = 211)27 as the training set, whereas four additional human mesothelioma datasets, NCI (n = 100),36 TCGA-MESO (n = 74),8 MATCH (n = 67)37 and Creaney (n = 42),38 served as validation datasets. Univariate Cox regression analysis identified 10 hub genes that were significantly associated with improved survival post-surgery in the Bueno training dataset (Figure 4A). Next, we conducted least absolute shrinkage and selection operator (LASSO) Cox regression analysis; a variable selection method designed to identify the most refined sets of genes for long-term prediction. This narrowed the candidates to six genes that formed the prognostic model to calculate each patient’s time of death (risk score, Figure S13).
Figure 4.

Development and validation of a six-gene prognostic signature specific to mesothelioma derived from CCMT hub genes
(A) Univariate cox regression analysis of all 20 Hub genes from the Bueno human mesothelioma dataset (n = 211), ranked by their hazard ratio. 95% confidence interval and p values are shown in the right-hand column of the table. Genes significantly associated with survival are highlighted by red squares.
(B) Heatmap illustrating the six-gene prognostic signature expression, survival data and risk score distribution across all 211 tumor samples in the Bueno training dataset. The median risk score, indicated by a red Line, is used to stratify patients into high (orange) and low (blue) risk groups.
(C and D) Kaplan Meier curves showing the overall survival of patients in high (orange) and low (blue) risk groups. ROC curves verified the prognostic performance of the estimated risk score within the Bueno training dataset.
(E and F) Forrest plot of multivariate Cox regression analysis of the gene signature within Bueno and TCGA datasets. The multivariate analysis was adjusted for sex, histological subtype and patient age. The 95% confidence interval and p values are shown on the right end column of the table.
(G) Stacked bar chart of AUC for the six-gene signature model and other published gene signature models across five independent mesothelioma datasets. Area under the curve (AUC) percentages of ROC analysis on gene signature model predictions and CCNB1 in five independent mesothelioma datasets (Bueno, NCI, TCGA, MATCH and Creaney). Colors in each bar correspond to overall survival year in the cohort for the specified gene signature model. The 44-gene signature and CCNB1 signature are from Nair et al., 2023.36 AUC values of every survival year are compared within each datasets using the iid-representation of the AUC estimator from TimeROC package. ∗p < 0.05, ∗∗p < 0.01.
Risk scores for each patient were calculated based on the expression of the six genes identified by LASSO analysis: interferon regulatory factor 9 (IRF9), ubiquitin specific peptidase 18 (USP18), collagen type IV alpha 1 (COL4A1), integrin alpha 5 (ITGA5), fibronectin 1 (FN1) and integrin subunit beta 1 (ITGB1). Risk scores were then used to stratify patients into high or low-risk groups, with the lower-risk group demonstrating better overall survival (Figure 4B). IRF9 and USP18 are known to be involved in interferon (IFN) signaling and their expression, based on the risk score formula (Figure S13), demonstrated a negative impact (i.e., a lower risk score and better survival). Conversely, COL4A1, ITGA5, FN1 and ITGB1, are associated with ECM processes and positively influenced the risk score (i.e., higher risk and lower survival).
We assessed the predictive accuracy of the survival model in the Bueno training dataset27 using receiver operating characteristic (ROC) curve analysis. This yielded area under the curve (AUC) probabilities of 0.68, 0.70 and 0.74 for 1-, 2- and 3-year survival after surgery, respectively (Figure 4C). The prognostic potential of the six-gene signature was validated in the TCGA8 and Creaney38 datasets (Figures 4D, S14B, and S14D), but the gene signature was not significantly associated with prognosis in the MATCH37 mesothelioma dataset (Figures S14A and S14C). Conversely, multivariate Cox regression analysis identified the six-gene signature as an independent prognostic factor for mesothelioma in the Bueno, TCGA and Creaney cohorts, after adjusting for sex, age, and histological subtypes (Figures 4E, 4F, and S14D).
To assess whether the six-gene prognostic signature was specific for mesothelioma, we investigated additional non-mesothelioma TCGA cancer datasets. The six-gene signature failed to significantly stratify patients into high- or low-risk in several cancers including cutaneous melanoma (SKCM), thymoma (THYM), prostate adenocarcinoma (PRAD) and ovarian cancer (OV), the latter having a similar mutational profile to mesothelioma.27 Conversely, the six-gene signature significantly stratified bladder cancer (BLCA) and lung adenocarcinoma (LUAD) patients, although with lower predicative performance compared to mesothelioma-specific datasets (Figure S15). Finally, the predictive performance of the six-gene signature model was benchmarked against other published mesothelioma gene signatures7,33,36,39,40,41 using Time-dependent AUC comparison analysis (timeROC) and demonstrated similar predictive performance in five independent mesothelioma datasets8,27,36,37,38 further validating the capability of the six-gene signature to accurately predict mesothelioma patient survival (Figure 4G and Table S3).
Six-gene mesothelioma-specific gene signature accurately predicted survival and response to ICB therapy in the NCI mesothelioma cohort
To further validate the prognostic potential of the six-gene mesothelioma-specific signature, we used the recently published NCI mesothelioma dataset.36 This dataset is unique in that it contains treatment outcome data and includes an equal number of patients with pleural and peritoneal mesothelioma. While the six-gene signature was not significantly correlated with patient prognosis for the entire NCI dataset (n = 100), it accurately predicted prognosis for most NCI mesothelioma patients (n = 94), excluding a small group (n = 6) with survival over 10 years (Figures 5A and 5B). The predictive accuracy of the six-gene signature in the NCI dataset was further corroborated by the high AUC probabilities (0.84, 0.70, and 0.70) from ROC curves for 1-, 2- and 3-year overall survival post-diagnosis respectively (Figure 5C).
Figure 5.

Six-gene signature successfully predicts overall survival of patients in NCI mesothelioma dataset with less than 10 years survival
(A) Kaplan Meier survival curves for all NCI mesothelioma patients (n = 100).
(B) Kaplan-Meier survival curves for patients with less than 10 years survival, stratified by the six-gene signature into low (orange) and high (blue) risk groups (32).
(C) ROC curves with AUC values for 1, 2, and 3-year survival of mesothelioma patients.
(D) Kaplan Meier survival curves for NCI patients with less than 10 years survival post-biopsy (n = 38) who received different treatment modalities, stratified into low (blue) and high (orange) risk groups.
(E and F) Comparative analysis of the area under the curve (AUC) of ROC analysis of the six-gene and other mesothelioma gene signature models to predict treatment response in the NCI dataset after controlling the effect of age, gender and disease site (pleural or peritoneal). (E) Immune checkpoint blockade (ICB) therapy and (F) chemotherapy (CTX). Patients with complete response (n = 1 for ICB, n = 1 for CTX) or partial response (n = 2 for ICB and n = 23 for CTX) are considered responders and patients with progressive disease (n = 7 for ICB and n = 6 for CTX) or stable disease (n = 6 for ICB and n = 0 for CTX) are considered non-responders. 44-gene, CCNB1 and SELECT gene signatures from Nair et al., 2023.36 AUC of six-gene signature model was compared with other gene signature models using ‘DeLong’ and ‘Venkatraman’ method from the pROC package. ∗p < 0.05 ∗∗p < 0.01.
Interestingly, the prognostic potential of the signature was absent when patients were stratified based on the anatomical location of disease (viz., pleural vs. peritoneal), either in all patients or in patients surviving <10 years (Figure S16). However, the signature accurately stratified patients into low- and high-risk categories, based on the post-biopsy survival of the 38 patients who received treatment (Figure 5D). Comparative analysis using the 'Venkatraman' and ‘DeLong’ method from pROC, of the predictive performance of all gene signatures to predict response to immune checkpoint blockade indicated that the SELECT, CCNB136 and Bai7 signatures had the highest AUC values, although no significant difference was observed between any mesothelioma signatures AUC values after controlling the effect of disease site (pleural or peritoneal), age, and gender (Figure 5E and Table S4). All gene signatures were equally poor at predicating response to chemotherapy (Figure 5F) in the NCI dataset.
Discussion
This study utilized a biobank of solid tumors from asbestos-exposed, genetically diverse CCMT mice.22 These samples provided an optimal platform to model asbestos-related disease development, devoid of numerous confounding factors such as differences in carcinogen exposure, lifestyle/diet impacts, and various cancer treatment modalities encountered in human studies.7,8,9 Interestingly, extensive transcriptomic analysis of 167 tumors from 35 genetically distinct CCMT mouse strains that exhibited the maximum difference in overall survival revealed minimal variation in the gene expression profile of asbestos-induced mesotheliomas and no significant impact on the survival of the collective CCMT cohort. This outcome is consistent with our recent study22 that demonstrated host genetics had little influence on ARD outcomes after disease was established. This observation contrasts with a recent analysis of human RNA sequencing data combining the TCGA-MESO8 and Bueno27 mesothelioma datasets (n = 284), which corroborated a continuum model to explain the variation in patient survival based on tumor gene expression profiles.42 However, it should be noted that patients received treatment (CTX or surgery), whereas CCMT mice were treatment naive.
Our analysis of CCMT tumor gene expression profiles revealed two distinct clusters. Cluster 1 was characterized by higher expression of EMT/ECM-related genes and lower adaptive immune cell infiltration, whereas Cluster 2 displayed an adaptive immune-related phenotype, exhibiting significantly higher infiltration of CD4+ T cells and CD19+ B cells. This aligned with GSEA results of human mesothelioma datasets, where immune pathway expression emerged as a major source of variation.37,42 Furthermore, the high enrichment of EMT/ECM gene sets in Cluster 1 tumors, in conjunction with lower adaptive immune cell infiltration, is consistent with several studies that have demonstrated an inverse correlation between COL1A1 expression and ECM receptor interaction pathway proteins with higher immune infiltration and response to immunotherapy.43,44,45,46 This is also supported by studies suggesting that ECM-related genes expression functions as a barrier to the anti-tumor immune response in the tumor microenvironment.47,48,49,50,51 We also note the significantly higher level of tumor associated macrophages (TAMs) in Cluster 1 tumors compared to Cluster 2 tumors may play a role in limiting adaptive immune cell infiltration and the influence of TAMs in dampening anti-tumor immunity requires further investigation.
The association between immune cell infiltration and low ECM density with anti-tumor immune response in Cluster 2 tumors compared to Cluster 1 tumors led us to hypothesize that Cluster 2 tumors would be more likely to respond to cancer immunotherapy. This was supported by the significant association between the enrichment of both mouse and human gene sets associated with positive responses to cancer ICB/chemotherapy27,28,29,30,31,32,34,52 and the gene expression profile of Cluster 2 tumors. These results prompted further exploration to determine whether the distinction between the gene expression profiles of the two CCMT tumor clusters could be used to develop a prognostic gene signature.
To this end, WGCNA identified 20 hub genes that were strongly correlated with each distinct CCMT cluster, and the human homologues of these 20 hub genes were further refined to an optimal six-gene signature model that accurately predicted survival in four independent human mesothelioma datasets.8,27,36,38 Importantly, the six-gene signature demonstrated the potential to predict positive therapeutic responses with similar AUC values relative to other mesothelioma gene signatures in the NCI mesothelioma dataset, which is currently the only dataset containing data from mesothelioma patients who had received chemo-immunotherapy. These data reaffirm that mesothelioma patients can be stratified into an immunotherapeutic responsive/non-responsive cohort, and that therapeutic response can be defined based on a parsimonious gene signature.
However, there are differences between our data and other studies. Direct comparison of the six-gene (or any other gene) signature with other methods to predict potential therapeutic response (such as the SELECT53 method employed by Nair et al.36) demonstrated that while no significant difference was observed between mesothelioma gene signatures, both SELECT and the CCNB1 gene signatures had higher AUC values for predicting response to ICB. Interestingly, all gene signatures performed poorly at predicting response to chemotherapy only. Additionally, it is important to acknowledge that the relatively small number of treated patients (n = 38 total, 3 of 16 responders to ICB) in the NCI dataset limits the robustness of the predictive capacity of any mesothelioma-specific gene signature. Therefore, the evaluation of mesothelioma-specific prognostic gene signatures in larger (future) mesothelioma patient cohorts with well-defined clinical parameters is essential.
The goal of developing a mesothelioma-specific prognostic gene signature is not new. Although many studies have published mesothelioma-related gene signatures, the considerable variations in cohort composition, statistical methodologies, and analysis workflows may explain why there is little, if any commonality between published mesothelioma gene signatures.7,33,36,39,40,41 Indeed, the six-gene signature described here also demonstrated limited commonality with other published mesothelioma signatures, including the recent NCI cohort-based 48-gene signature.36 In contrast to the Nair study, CCNB1 was not identified in any of our analyses as having an influence on survival. Indeed, when we assessed CCNB1 expression in CCMT tumors we observed higher CCNB1 expression in Cluster 2 tumors that were characterised by the presence of a significant immune infiltrate, compared to non-immune (EMT/ECM) Cluster 1 tumors. However, given that we did not identify CCNB1 as one of the 237 DEGs identified from the CCMT cohort, nor was CCNB1 expression associated with survival outcome, we could not confirm the predictive role of CCNB1 in this study.
Overall, while the development of mesothelioma-specific prognostic gene signatures is encouraging, all proposed prognostic gene signatures developed from mesothelioma studies to date (including the six-gene signature described here) and their corresponding biological pathways require further assessment to clarify possible causal relationships with tumor development. Such advancements would be aided by incorporating known caveats of prognostic gene signature development into the design of future mesothelioma clinical trials. Ideally, the trial design would be adequately powered, and link well-defined clinical information associated with patient survival and treatment response with patient transcriptomic data. This would advance the development of prognostic gene signature(s) that accurately predict both patient survival and therapeutic response and significantly advance the clinical care and management of mesothelioma patients. Furthermore, given the established significance of non-coding transcriptomes in cancer progression, prognosis, and response to therapy,54,55,56 expanding transcriptomic analyses to encompass all transcriptomic forms (i.e., long non-coding RNA/circular RNA) in future mesothelioma clinical trials may have the potential to improve the precision and reliability of disease-associated gene signatures to predict clinical outcomes.
This study utilized a unique cross-species approach that not only demonstrated the translational potential of a mesothelioma-specific gene signature, but also validated the predictive value of well-defined, pre-clinical models that accurately represent the disease endotype for prognostic and therapeutic purposes in human studies. This is particularly useful in the context of rare cancers, where the restricted number of patient samples may limit the power of conventional genetic studies. Indeed, the commonality of the significant genes and pathways identified across species underscores the relevance of our findings to the pathophysiology of human mesothelioma. This cross-species validation bolsters confidence in our prognostic gene signature, highlighting the potential utility of pre-clinical derived data in clinical decision-making for patients with mesothelioma.
In conclusion, while our six-gene signature accurately predicted survival in multiple human mesothelioma cohorts, the limited availability of large multi-omics datasets and comprehensive clinical information restricts its application in predicting response to cancer therapies. This highlights the importance of designing future clinical trials using a standardized set of clinical data to assess the predictive potential of transcriptomic data. Such an approach could significantly improve the stratification of patients and enhance clinical care by identifying mesothelioma patients who are more likely to respond to specific cancer therapies.
Limitations of the study
Out study used RNA-seq of whole tumor tissue derived from asbestos exposed CCMT mice and used CIBRESORT to impute cell type composition. To differentiate tumor cells from stromal cells we would perform single-cell RNA-seq or analyze flow cytometry sorted tumor cells. MexTAg-Collaborative Cross mice were breed specifically for this project and do not exist as a commercial colony. To repeat these experiments each specific CC-MexTAg cross would have to be rebred. Both parental mouse types (i.e., male Collaborative Cross mice and female MexTAg mice) are available.
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Scott A Fisher (scott.fisher@uwa.edu.au).
Material availability
All unique reagents generated in this study are available from the lead contact with a completed materials transfer agreement.
Data code and availability
-
•
The generated RNA-seq datasets used in this manuscript have been deposited at Gene Expression Omnibus (GEO: https://www.ncbi.nlm.nih.gov/geo/) and are publicly available as of 15 February 2024, using accession number GSE232512. The Bueno mesothelioma cohort27 was imported from the European Genome-Phenome Archive (EGA, accession code: EGAS00001001563) in the raw FASTQ file format. TCGA-MESO8 RNA-seq datasets were retrieved in the STAR aligned raw gene count format from the Genomic Data Commons (GDC) portal using ‘TCGAbiolinks’. We sourced FASTQ sequence data files of the NCI cohort from the database of genotypes and phenotypes (dbGaP, accession number: phs002207),36 and those from the Creaney datasets38 from the European Genome-Phenome Archive (EGA accession number EGAS00001005196). The raw gene count table and corresponding clinical data from the MATCH37 datasets were retrieved from Zenodo (accession number 6532036, Zendo Data: https://zenodo.org/record/6532036). DOI’s are listed in the key resources table. Microscopy data reported in this paper will be shared by the lead author upon reasonable request.
-
•
All original code has been deposited in Mendeley Databank and is publicly available as of the date of publication. DOI’s are listed in the key resources table.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
Acknowledgments
This study was funded by the National Health and Medical Research Council of Australia (NHMRC) grant APP1163861. K.B. was supported by an Australian Government Research Training Program (RTP) grant and University Postgraduate Award (UPA). We acknowledge the Genomics WA Laboratory in Perth, Australia for library preparation and sequencing. Genomics WA is supported by Bioplatforms Australia, the State Government of Western Australia, the Harry Perkins Institute of Medical Research, the Telethon Kids Institute, and the University of Western Australia. We gratefully acknowledge the Australian Cancer Research Foundation and the Centre for Advanced Cancer Genomics for providing Illumina Sequencers to Genomics WA.
Author contributions
Each author contributed to the study design, interpretation of the investigations, data analysis, and manuscript review. Conceptualization: S.A.F., G.M., and R.A.L.; Methodology: S.A.F., G.M., R.A.L., S.E.M., K.B., K.P., W.L.C., F.P., and I.N.; Resources: S.A.F., G.M., R.A.L., and F.P.; Investigation: S.A.F., K.P., K.B., F.P., and I.N.; Formal Analysis: S.A.F., K.B., K.P., W.L.C., F.P., I.N., G.M., R.A.L., and S.E.M.; Writing – Original Draft: K.B. and S.A.F.; Writing – Review and Editing: S.A.F., K.B., S.E.M., K.P., W.L.C., F.P., I.N., G.M., and R.A.L.; Funding Acquisition: S.A.F., G.M., and R.A.L.; Supervision: S.A.F., S.E.M., and R.A.L.; Project Administration: S.A.F.
Declaration of interests
The authors declare no competing interests.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| CD4 (D7D2Z) Rabbit mAb | Cell signaling | Cat# 25229S; RRID:AB_2798898 |
| CD8a (D4W2Z) Rabbit mAb | Cell signaling | Cat# 98941; RRID:AB_2756376 |
| FoxP3 (D6O8R) Rabbit mAb | Cell signaling | Cat# 12653; RRID:AB_2797979 |
| PD-L1 (EPR20529) Rabbit mAb | Abcam | Cat# ab213480; RRID:AB_2773715 |
| CD19 (D4V4B) Rabbit mAb | Cell signaling | Cat# 90176; RRID:AB_2800152 |
| IBA1 (Polyclonal) Rabbit | FUJIFILM Wako Pure Chemical Corporation | Cat# 019-19741; RRID:AB_839504 |
| COL1A1 (E8F4L) Rabbit mAb | Cell signaling | Cat# 72026T; RRID:AB_2904565 |
| Vimentin (D21H3) Rabbit mAb | Cell signaling | Cat# 5741T; RRID:AB_10695459 |
| PDPN (Polyclonal) Rabbit | Abcam | Cat# ab109059; RRID:AB_2848181 |
| E-cadherin (24E10) Rabbit mAb | Cell signaling | Cat# 3195S; RRID:AB_2291471 |
| Rabbit IgG, monoclonal (EPR25A) | Abcam | Cat# ab172730; RRID:AB_2687931 |
| DAPI solution (1mg) | BD Bioscience, USA | Cat# 564907; RRID:AB_2869624 |
| Biological samples | ||
| CCMT strain | Mouse ID (Tumour ID) | |
| 266-BOM_GB (Gene Mine.BOM_GB x C57BL/6J.MexTAg 266 HOM | Fisher, S.A et al., 2024.22 | L1 (Tu2) |
| 266-BOON_HF (Gene Mine.BOON_HF x C57BL/6J.MexTAg 266 HOM) | Fisher, S.A et al., 2024.22 | D1 (Tu 1, Tu 2) H2 (Tu1, Tu2, Tu3) I2 (Tu1) I4 (Tu2) |
| 266-CC001/Unc (CC001/Unc x C57BL/6J.MexTAg 266 HOM) | Fisher, S.A et al., 2024.22 | D1 (Tu1, Tu2) H2 (Tu1, Tu2) H3 (Tu1) |
| 266-CC017/Unc (CC017/Unc x C57BL/6J.MexTAg 266 HOM) | Fisher, S.A et al., 2024.22 | H2 (Tu2) I4 (Tu1) |
| 266-CC021/Unc (CC021/Unc x C57BL/6J.MexTAg 266 HOM) | Fisher, S.A et al., 2024.22 | C1 (Tu1) C2 (Tu2) C4 (Tu1) D3 (Tu1) |
| 266-CC035/Unc (CC035/Unc x C57BL/6J.MexTAg 266 HOM) | Fisher, S.A et al., 2024.22 | H1 (Tu1, Tu2, Tu3) H2 (Tu1) |
| 266CC036/Unc (CC036/Unc x C57BL/6J.MexTAg 266 HOM) | Fisher, S.A et al., 2024.22 | C1 (Tu1) C3 (Tu1) F1(Tu1, Tu2) |
| 266-CC040/TauUnc (CC040/Unc x C57BL/6J.MexTAg 266 HOM) | Fisher, S.A et al., 2024.22 | D3 (Tu1) E1(Tu2) E4 (Tu1) H1 (Tu1) |
| 266-CC044/Unc (CC044/Unc x C57BL/6J.MexTAg 266 HOM) | Fisher, S.A et al., 2024.22 | D3 (Tu1, Tu2) F2 (Tu1) F4 (Tu1, Tu2, Tu3) H2 (Tu1) I2 (Tu1) J3 (Tu1, Tu2, Tu3) K1 (Tu1, Tu2, Tu3) |
| 266-CC046/Unc (CC046/Unc x C57BL/6J.MexTAg 266 HOM) | Fisher, S.A et al., 2024.22 | H2 (Tu1) J5 (Tu2) L3 (Tu1) L3 (Tu2) |
| 266-CC053/Unc (CC053/Unc x C57BL/6J.MexTAg 266 HOM) | Fisher, S.A et al., 2024.22 | A1 (Tu1, Tu2) A4 (Tu1) B1 (Tu1) F1 (Tu1, Tu2, Tu4) G3 (Tu1, Tu2) H1 (Tu1) |
| 266-CC058/Unc (CC058/Unc x C57BL/6J.MexTAg 266 HOM) | Fisher, S.A et al., 2024.22 | B2 (Tu2) H4 (TU1) I4 (Tu1, Tu2) |
| 266-CC059/TauUnc (CC059/TauUnc x C57BL/6J.MexTAg 266 HOM) | Fisher, S.A et al., 2024.22 | A2 (Tu1) B1 (Tu1, Tu2) B5 (Tu1) E2 (Tu1, Tu2, Tu3) F2 (Tu1, Tu2) |
| 266-CC060/Unc (CC060/Unc x C57BL/6J.MexTAg 266 HOM) | Fisher, S.A et al., 2024.22 | B1 (Tu1) G2 (Tu1) H2 (Tu2) |
| 266-CC074/Unc (CC074/Unc x C57BL/6J.MexTAg 266 HOM) | Fisher, S.A et al., 2024.22 | B3 (Tu1, Tu2, Tu3, Tu6, Tu8, Tu9) G1 (Tu1) |
| 266-CC081/Unc (OR3269/Unc x C57BL/6J.MexTAg 266 HOM) | Fisher, S.A et al., 2024.22 | A1 (Tu1) C4 (Tu1) C5 (Tu1) |
| 266-CIS_AD (Gene Mine.CIS_AD x C57BL/6J.MexTAg 266 HOM) | Fisher, S.A et al., 2024.22 | E2(Tu1, Tu2) G3 (Tu1, Tu2) G4 (Tu1) |
| 266-FUF_HE (Gene Mine.FUF_HE x C57BL/6J.MexTAg 266 HOM) | Fisher, S.A et al., 2024.22 | H2 (Tu1, Tu2) |
| 266-GIT_GC (Gene Mine.GIT_GC x C57BL/6J.MexTAg 266 HOM) | Fisher, S.A et al., 2024.22 | G2 (Tu1) H3 (Tu1) K1 (Tu1, Tu2) K2 (Tu1) |
| 266-KAV_AF (Gene Mine.KAV_AF x C57BL/6J.MexTAg 266 HOM) | Fisher, S.A et al., 2024.22 | A3 (Tu1, Tu2) D4 (Tu1) G2 (Tu2) |
| 266-LAT_HD (Gene Mine.LAT_HD x C57BL/6J.MexTAg 266 HOM) | Fisher, S.A et al., 2024.22 | A2 (Tu1) D2 (Tu1) D3 (Tu1, Tu2, Tu3, Tu4) E2 (Tu1) F5 (Tu1, Tu2) |
| 266-LAX_FC (Gene Mine.LAX_FC x C57BL/6J.MexTAg 266 HOM) | Fisher, S.A et al., 2024.22 | C2 (Tu1) E2 (Tu1) F2 (Tu1) G3 (Tu1) H1 (Tu1) K2 (Tu1) |
| 266-LOX_GF (Gene Mine.LOX_GF x C57BL/6J.MexTAg 266 HOM) | Fisher, S.A et al., 2024.22 | A3 (Tu1) B4 (Tu1, Tu3) C1 (Tu1) D1 (Tu1, Tu2) E4 (Tu1) F2 (Tu1) F4 (Tu1) |
| 266-MEE_AG (Gene Mine.MEE_AG x C57BL/6J.MexTAg 266 HOM) | Fisher, S.A et al., 2024.22 | A3 (Tu1, Tu2) B1 (Tu1, Tu2) E2 (Tu1, Tu2) I2 (Tu1) |
| 266-PEF2_EC (Gene Mine.PEF2_EC x C57BL/6J.MexTAg 266 HOM) | Fisher, S.A et al., 2024.22 | A2 (Tu1) G2 (Tu1, Tu2) |
| 266-PIPING_BD (Gene Mine.PIPING_BD x C57BL/6J.MexTAg 266 HOM) | Fisher, S.A et al., 2024.22 | I3 (Tu1) J1 (Tu1) N1 (Tu1) |
| 266-PUB_CD (Gene Mine.PUB_CD x C57BL/6J.MexTAg 266 HOM) | Fisher, S.A et al., 2024.22 | I1 (Tu1) |
| 266-ROGAN_CF (Gene Mine.ROGAN_CF x C57BL/6J.MexTAg 266 HOM) | Fisher, S.A et al., 2024.22 | C3 (Tu1, Tu2) E3 (Tu1) E5 (Tu1) J2 (Tu1, Tu2) |
| 266-TAS_FE (Gene Mine.TAS_FE x C57BL/6J.MexTAg 266 HOM) | Fisher, S.A et al., 2024.22 | D3 (Tu1) E3 (Tu1) H2 (Tu1) H5 (Tu1) I1 (Tu1) J2 (Tu1) |
| 266-TOFU_FB (Gene Mine.TOFU_FB x C57BL/6J.MexTAg 266 HOM) | Fisher, S.A et al., 2024.22 | C3 (Tu1) K2 (Tu1) |
| 266-VIT_ED (Gene Mine.VIT_ED x C57BL/6J.MexTAg 266 HOM) | Fisher, S.A et al., 2024.22 | A2 (Tu1) K2 (Tu1) |
| 266-VUX2_HF (Gene Mine.VUX2_HF x C57BL/6J.MexTAg 266 HOM) | Fisher, S.A et al., 2024.22 | B1 (Tu1) D1 (Tu1) E2 (Tu1) G1 (Tu1) |
| 266-WAB2_DH (Gene Mine.WAB2_DH x C57BL/6J.MexTAg 266 HOM) | Fisher, S.A et al., 2024.22 | H2 (Tu1) |
| 266-WOB2_BA (Gene Mine.WOB2_BA x C57BL/6J.MexTAg 266 HOM) | Fisher, S.A et al., 2024.22 | H3 (Tu1) N3 (Tu1) |
| 266-YID_FH (Gene Mine.YID_FH x C57BL/6J.MexTAg 266 HOM) | Fisher, S.A et al., 2024.22 | C1 (Tu1) D1 (Tu1, Tu2) E1 (Tu2) G3 (Tu1) |
| Critical commercial assays | ||
| RNAProtect | Qiagen GmbH | Cat# 76104 |
| RNeasy Mini Kit | Qiagen GmbH | Cat# 74104 |
| TSA FLUORESCEIN REAGENT PACK 100-300 | Akoya Biosciences Inc, MA, USA | Cat# SAT701001EA |
| AF 594 TYRAMIDE 150 SLIDES | ThermoFisher, USA | SKU #B40957 |
| TSA CYANINE 3 SYSTEM 50-150 SLIDES | Akoya Biosciences Inc, MA, USA | Cat# SAT704A001EA |
| TSA CYANINE 5 SYSTEM 50-150 SLIDES | Akoya Biosciences Inc, MA, USA | Cat# SAT705A001EA |
| Background SNIPER | Biocare Medical | Cat# BS966 |
| Peroxidazed 1 | Biocare Medical | Cat# BIC-PX968H |
| Goat anti-Rabbit IgG (H+L) Secondary Antibody, HRP | ThermoFisher, USA | Cat# 31460 |
| D1000 screentapes & reagents | Agilent, USA (Integrated Sciences, Aust) | Cat# 5067-5582; 5067-5583 |
| Qubit HS DNA kit | ThermoFisher, USA | Cat # Q32851 |
| NovaSeq 6000 SP Reagent Kit | Illumina, USA | Cat# 20028402 |
| SureSelect strand specific mRNA library prep kit | Agilent Technologies Australia, VIC, Australia | Cat# G9691B |
| Deposited data | ||
| CCMT RNAseq | This study (Gene Expression Omnibus) | GEO: GSE232512 |
| RNAseq analysis code | Mendeley data | https://doi.org/10.17632/84kjtptmd8.1 |
| MSigDB hallmark gene set collection | Liberzon, A et al., 2015.62 | https://doi.org/10.1016/j.cels.2015.12.004 |
| KEGG | Kanehisa et al., 2022.64 | https://doi.org/10.1093/nar/gkac963 |
| Biocarta | Biocarta | https://maayanlab.cloud/Harmonizome/resource/Biocarta |
| ImmunoPhenoscore (IPS) | Charoentong et al., 2017.27 | https://doi.org/10.1016/j.celrep.2016.12.019 |
| ImmuneCells.sig | Xiong et al. 2020.29 | https://doi.org/10.1038/s41467-020-18546-x |
| Stem.Sig | Zhang et al., 2022.30 | https://doi.org/10.1186/s13073-022-01050-w |
| Inflammatory signature | Thompson et al., 2020.33 | https://doi.org/10.1016/j.lungcan.2019.10.012 |
| T cell infiltration score | Senbabaoglu et al. 2016.28 | https://doi.org/10.1186/s13059-016-1092-z |
| Bueno mesothelioma dataset (Bueno, R et al., 2016)27 | European Genome-Phenome Archive | EGA: EGAS00001001563 |
| TCGA RNAseq datasets (TCGA-MESO, -LUAD, -SKMC, -BLCA, -THYM, -PRCA and -OV) | Genomic Data Commons (GDC) | TCGA datalinks search portal |
| NCI mesothelioma dataset (Nair, N.U et al., 2023)36 | Database of Genotypes and Phenotypes | dbGaP: phs002207 |
| Creaney mesothelioma dataset (Creaney, J et al., 2022)38 | European Genome-Phenome Archive | EGA: EGAS00001005196 |
| MATCH mesothelioma dataset (Mannarino, L et al., 2022)37 | Zenodo | https://doi.org/10.5281/zenodo.6532036 |
| Bai mesothelioma signature | Bai et al., 2020.7 | https://doi.org/10.3389/fgene.2020.00899 |
| Zhou mesothelioma signature | Zhou et al., 2019.41 | https://doi.org/10.3389/fonc.2019.00078 |
| Zhang mesothelioma signature | Zhang et al., 2022.40 | https://doi.org/10.21037/atm-22-527 |
| Shi mesothelioma signature | Shi, J et al., 2023.38 | https://doi.org/10.1007/s00432-023-05128-9 |
| Xiao mesothelioma signature | Xiao, Y et al., 2022.39 | https://doi.org/10.3389/fendo.2022.1056152 |
| C57Bl/6J mouse genome (GRCm38/mm10) | NCBI GenBank | GCF_000001635.27 |
| Human Genome assembly GRCh38 | NCBI GenBank | GCF_000001405.40 |
| Software and algorithms | ||
| Kallisto | Bray et al., 2016.58 | https://doi.org/10.1038/nbt0816-888d |
| Tximport | Soneson et al., 2015.59 | https://doi.org/10.12688/f1000research.7563.2 |
| DESeq2 | Love et al., 2014.60 | https://doi.org/10.1186/s13059-014-0550-8 |
| ConsensusClusterPlus | Wilkerson, A et al., 2010.61 | https://doi.org/10.1093/bioinformatics/btq170 |
| GSEA (v4.2.3) | Broad Institute, Subramanian et al., 200766 and Subramanian et al., 2005.67 |
https://doi.org/10.1093/bioinformatics/btm369 https://doi.org/10.1073/pnas.0506580102 |
| Weighted gene co-expression network analysis (WGCNA) | Langfelder et al., 2008.69 | https://doi.org/10.1186/1471-2105-9-559 |
| CIBERSORTx | Newman et al., 2019.68 | https://doi.org/10.1038/s41587-019-0114-2 |
| Survival | Therneau et al. 2015.76 | Therneau et al. 2015.76 |
| glmnet | Freidman et. Al 2010.78 | https://doi.org/10.18637/jss.v033.i01 |
| survminer | CRAN | https://cran.r-project.org/web/packages/survminer/index.html |
| timeROC | Blanche P et al. 2013.79 | https://doi.org/10.1002/sim.5958 |
| STAR (v2.7.3a) | Dobin et al. 2013.74 | https://doi.org/10.1093/bioinformatics/bts635 |
| featureCounts | Liao et al. 2014.75 | https://doi.org/10.1093/bioinformatics/btt656 |
| Cytohubba plugin | Chin et al., 2014.72 | https://doi.org/10.1186/1752-0509-8-S4-S11 |
| QuantCenter software (v2.5.0.143918). | 3DHistech ltd. | https://www.3dhistech.com/research/quantcenter/ |
| CytoScape (v3.8) | Shannon et al., 2003.73 | https://doi.org/10.1101/gr.1239303 |
| Rstudio (R version 4.1.3) | R studio | https://posit.co/products/open-source/rstudio/ |
| Other | ||
| Qubit™ 4 fluorometer | ThermoFisher, USA | Cat # Q33238 |
| Tapestation 4200 | Agilent, USA (Integrated Sciences, Aust) | Part no# G2991BA |
| iSeq100 sequencer | Illumina, USA | https://sapac.illumina.com/systems/sequencing-platforms/iseq.html |
| NovaSeq 6000 sequencer | Illumina, USA | https://sapac.illumina.com/systems/sequencing-platforms/novaseq/specifications.html |
| 3DHISTECH Pannoramic Flash 250 III Slide Scanner | 3DHISTECH ltd | https://www.3dhistech.com/research/pannoramic-digital-slide-scanners/pannoramic-250-flash-iii/ |
Experimental model and study participant details
Mice
This study did not involve the direct use of animals. However, it did use tumour samples harvested from asbestos exposed CC-MexTAg progeny from our previous study.22 All animal experiments were approved by the University of Western Australia Animal Ethics Committee (RA/3/300/106; RA/3/300/107; RA/3/300/131; RA/3/300/133; RA/3/100/1408; RA/3/100/1730), and were performed in accordance with the National Health and Medical Research Council Australian Code of Practice for the Care and Use of Animals for Scientific Purposes.57 The CC12,15 and MexTAg19,20,21 mouse strains and the generation of CCMT progeny22 have been previously described. Briefly, CC mice were generously provided by Geniad Pty Ltd from its colonies at the Animal Resource Centre (Perth, Western Australia). Additional CC strains (CCXXX format) were obtained from the University of North Carolina CC colony. MexTAg mice were developed in house and colonies maintained at the University of Western Australia Biomedical Resource Facility (Perth, Western Australia). All mice were house at the UWA Animal Care Services M-Block animal facility (Nedlands, Western Australia) and maintained under pathogen-free conditions at 21°C– to 22°C with a 12/12 h light cycle, in single sex cages containing a maximum of 5 mice per cage (a mix of Thoren or Tecniplast individually ventilated cages) on aspen chips bedding (TAPVEI). Mice were fed Rat and Mouse cubes (Specialty Feeds, Perth Western Australia) and had ad libitum access to filtered water. Male mice from different CC strains were mated with female MexTAg mice, and the CCMT progeny (male and female) were administered two intraperitoneal injections of 3 mg Wittenoom crocidolite (blue) asbestos one month apart. Asbestos-exposed CCMT mice were monitored continuously for up to 548 days (18 months/78 weeks) or until signs of asbestos-related disease (ARD, predominantly ascites formation or loss of condition) developed and progressed to a pre-defined animal welfare endpoint, at which time the mice were sacrificed, and tumor and tissues collected for genomic analysis (Figure 1A).
Method details
Sample collection and RNA isolation
Tumors from asbestos exposed CCMT mice were harvested immediately after sacrifice, collected in RNAprotect™ as per manufacturer’s instructions (76104, Qiagen GmbH, Venlo, Netherlands) and stored at -80°C. Large tumors (> 50 mm2) were halved, with half preserved in 4% formaldehyde for at least 24h in preparation for immunohistochemical analysis. Tumors and tissues were homogenised using a handheld rotor-stator homogeniser (Tissue rupture II, Cat No./ID: 9002756, Qiagen). Total RNA was extracted using the RNeasy Mini Kit (74104, Qiagen) as per manufacturer’s instructions.
RNAseq library preparation and sequencing
Stranded PolyA RNAseq libraries were prepared and sequenced by Genomics WA (Perth, Western Australia). A Sureselect strand specific mRNA library prep kit (G9691B, Agilent Technologies Australia, VIC, Australia) was used for cDNA library preparation. The quality and concentrations of the final cDNA libraries were verified using either D1000 screentapes on a Tapestation 4200 or Qubit HS DNA kit (Thermo Fisher Scientific, MA, USA) and a Qubit™ 4 fluorometer. For quality control, initial sequencing was performed using an iSeq100 (Illumina, CA, United States). Thereafter, 300 pM of the cDNA libraries was loaded into SP flow cells for deep sequencing using a NovaSeq 6000 (Illumina) at 2 x 50 cycles, yielding 20 million raw paired-end reads per sample.
RNA sequencing data analysis
Quality control assessment of FASQ sequence reads was performed using FastQC (v0.11.9), and their measures were summarized by MultiQC software (v1.14) prior to RNAseq data analysis. Raw FASQ sequence files were aligned to the murine GRCm38/mm10 reference genome using Kallisto,58 with the number of bootstraps set to 100. The Tximport package59 was used to convert transcript abundance to gene-level estimates. DESeq260 was used to normalize gene expression data and to identify differentially expressed genes in tumors derived from mice based on overall survival (below CCMT cohort median group relative to the above median survival group). The variance-stabilising transformation (Vst) function of DESeq2 was used to transform the gene count data for subsequent principal component analysis (PCA). We used ConsensusClusterPlus61 to perform consensus clustering on 10,000 highly variable genes using 5,000 iterations on 80% of the samples. This facilitated determination of the optimal K value, which denotes the number of clusters assigned to RNAseq data based on fewer color changes and flatter cumulative distribution function (CDF) plots (Figure S1).
GSEA was performed using MSigDB,62,63 KEGG64 databases, and BioCarta pathway database with 1000 permutations executed using the Broad Institute software (GSEA v4.2.3).65,66,67 Gene sets with nominal p values < 0.05 and FDR < 0.25 were considered significant. To estimate the abundance of immune cell types from the gene expression data, we employed the CIBERSORTx deconvolution algorithm68 on transcript per million (TPM) transformed RNAseq data.34
Multiplex immunofluorescence
Multiplex immunofluorescence staining of sequential formalin fixed paraffin embedded (FFPE) tumor sections (5 μm) utilized Tyramide-signal amplification (TSA) reagents. Tissue sections were deparaffinized and rehydrated using graded alcohol and xylene, and antigen retrieval was performed in Tris-EDTA buffer (pH 9.0) using a pressure cooker (Cuisinart, CPC600C). For each successive round, tumor sections were incubated with different primary rabbit anti-mouse antibodies, followed by SuperBoost™ goat anti-rabbit poly HRP (ThermoFisher, Vic, Australia). The antibody specifications, final concentrations and a comprehensive outline of the staining protocol are provided in the key resources table and Table S1. Immunostaining was performed using fluorophore-tagged TSA Reagent kits including TSA-Cyanine 5, TSA-Cyanine 3, TSA-Fluoresceine (Akoya Biosciences Inc, MA, USA) and TSA-AF594 (ThermoFisher). Sequential rounds of antigen retrieval were performed to strip the bound antibodies prior to subsequent staining. Sections were counterstained with DAPI (564907, BD Bioscience, USA) and mounted with ProLong® Diamond Antifade mounting medium (ThermoFisher). Rabbit IgG Isotype control antibody (ab172730) was used as the negative control. The 3DHISTECH Pannoramic Flash 250 III Slide Scanner (3D Histech, Budapest, Hungary) was used to scan whole tumor sections at 25x magnification and images were analyzed using QuantCenter software (3D Histech, v2.5.0.143918).
Treatment response gene set selection
Gene sets associated with response to therapy in mouse mesothelioma were obtained from publicly available studies. These gene sets consisted of genes that were significantly upregulated in subcutaneous tumors from mice that responded to ICB therapy targeting cytotoxic T-lymphocyte–associated antigen 4 (CTLA-4) plus PD-L1 and cyclophosphamide plus 5-flurouracil chemotherapy.29,34 Additionally, published pan-cancer human gene sets, consisting of more than 15 genes, and showing positive correlations with response to cancer therapies, were selected to create human treatment response gene sets. These gene sets included ImmunoPhenoscore (Ips signature),31 Inflammatory signature,30 T cell infiltration score,27 ImmuneCells.Sig,32 and Stem.Sig28 and were then used to perform GSEA on the gene expression profiles identified in this study.
Weighted gene co-expression network analysis (WGCNA)
WGCNA was performed on gene expression data using the ‘WGCNA’ R package69,70 to identify networks of co-expressed genes associated with both immune and non-immune (EMT/ECM) tumor phenotypes. Prior to the analysis, gene expression data were pre-processed by eliminating genes with low expression (< 10 counts) and those lacking formal Mouse Genome Informatics (MGI) symbols. This resulted in a total of 11,016 genes for downstream analysis. RNA sequencing data were transformed into an adjacency matrix using the pickSoftThreshold function with the soft threshold set to b=7 (R2=0.9), ensuring a scale-free network required for the WGCNA. To identify highly connected groups of genes, a Topological Overlap Matrix (TOM) was derived from the adjacency matrix. Finally, we employed the Dynamic Treecut algorithm with a minimum module size of 30 genes to construct highly interconnected clusters of co-expressed genes (modules). A CutHeight of 0.3 was used to merge similar modules (Figures S12A–S12D). The resulting modules from WGCNA, along with a correlation heatmap depicting the relationship between each module, mouse phenotypic traits, and tumor phenotypes, are illustrated in Figure 3C.
Hub gene identification
To identify significant modules within the WGCNA data and their associated hub genes, we performed a correlation analysis between all identified WGCNA modules and CCMT phenotypic traits, along with immune and non-immune (EMT/ECM) tumor phenotypes. Modules demonstrating a significant correlation (p < 0.05, r > 0.8) and strong module membership (MM) (r > 0.8) were selected for further investigation using Gene ontology (GO) enrichment analysis via the clusterProfiler package.71 Genes within these significant modules were then used to construct protein-protein interaction (PPI) networks and identify hub genes. We input a maximum of 2,000 genes from the two most significant modules, representing either the non-immune (EMT/ECM) (Turquoise) or immune (Blue) tumor phenotype, into the STRING online database (http://string-db.org/). We aimed to construct PPI networks with an interaction score of 0.4. To identify the hub genes for each network, we employed the CytoHubba72 plugin in the Cytoscape software (version 3.8)73 to detect the top 10 genes with the highest maximal clique centrality scores.
Mesothelioma transcriptomic datasets
Several human mesothelioma datasets were used to develop and validate the prognostic gene signature derived from CCMT hub genes. All RNA-seq data from the gene signature discovery and validation cohorts were aligned against the same version of the human reference genome (GRCh38). All the aligned RNAseq data files, or raw gene count tables, from all cohorts were subjected to the same processing and normalization workflow as outlined in the RNA sequencing data analysis section. The training cohort, used to develop the six-gene prognostic signature, comprised RNA-seq data from the Bueno cohort.27 This was imported from the European genome-phenome archive (EGA, accession code: EGAS00001001563) in the raw FASTQ file format and aligned with Kallisto (v0.46.1).
For the validation cohorts, TCGA mesothelioma RNA-seq datasets were retrieved in the STAR aligned raw gene count format from the Genomic Data Commons (GDC) portal using ‘TCGAbiolinks’. We sourced FASTQ sequence data files of the NCI cohort from the database of genotypes and phenotypes (dbGaP, accession number: phs002207) and those from the Creaney datasets38 from the EGA (https://ega-archive.org/studies/EGAS00001005196). All sequence data were aligned using STAR (version 2.7.3a)74 and transformed into a raw gene count table using ‘featureCounts’.75 The raw gene count table and corresponding clinical data from the MATCH37 dataset were retrieved from Zenodo (https://zenodo.org/record/6532036).
Gene signature construction and validation using human cancer datasets
We developed a multi-gene prognostic model by screening hub genes using univariate Cox regression analysis on datasets from ICB treated mice (n=24).34 This analysis was performed using the R ‘Survival’ package76,77 and involved screening the 20 CCMT hub genes associated with tumor phenotypes. Subsequently, we used the human orthologs of the remaining 19 CCMT hub genes to perform univariate Cox regression analysis on the Bueno human mesothelioma cohort (training dataset)27 to identify significant genes associated with survival post-surgery. For further analysis, we used the ‘glmnet’ package78 to perform LASSO Cox regression analysis on the 10 most significant genes, incorporating 100 times cross-validation. This analysis selected the optimal set of variables (genes) and generated a refined Cox model to predict patient risk and prognosis. Subsequently, a risk score for each patient was calculated using ∑(Coef×EXPmRNA), where EXPmRNA refers to Vst transformed gene expression, and ‘Coef’ corresponds to coefficients estimated by the LASSO Cox regression analysis. Risk scores were used to divide patients into high and low-risk groups, and the prognostic significance of the model was evaluated through the Kaplan Meier78 curve and ROC plots using ‘survminer’ and ‘timeROC’79 packages. Finally, we evaluated the prognostic potential of the six-gene signature in four independent human mesothelioma cohorts: TCGA-MESO,8 NCI,36 MATCH37 and Creaney.38 We also evaluated gene signature specificity by retrieving transcriptomic data from other non-mesothelioma cancer datasets from the TCGA GDC portal. We processed and analyzed all data using the same workflow outlined in the RNA sequencing data analysis.
Quantification and statistical analysis
All statistical analyses were performed using R studio (R version 4.1.3) and the application noted in reference to the respective results sections or in the figure legends where appropriate. Definitions of respective measures (i.e. median survival, hazard ratio and confidence limits), samples sizes or probabilities (p values) are defined in figure legends. Unless otherwise stated, Benjamini-Hochberg (B-H) correction was employed in differential expression analysis to adjust p values for multiple testing, considering p < 0.01 as significant. Survival analysis was conducted using the Kaplan-Meier method, with P values calculated using the log-rank test (Mantel-Cox). We performed Student’s t-test to compare the expression of EMT/ECM markers and the density of immune cells measured by immunofluorescence image analysis, considering P < 0.05 as significant. Time-dependent AUC comparison analysis (timeROC) was used to compare differences in mesothelioma gene signatures for survival prediction. The ‘Venkatraman/Delong’ methods from the pROC R package80 were used for AUC comparison of the six-gene signature model with other gene signature models.
Published: September 23, 2024
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2024.111011.
Supplemental information
References
- 1.Baas P., Scherpereel A., Nowak A.K., Fujimoto N., Peters S., Tsao A.S., Mansfield A.S., Popat S., Jahan T. First-line nivolumab plus ipilimumab in unresectable malignant pleural mesothelioma (CheckMate 743): a multicentre, randomised, open-label, phase 3 trial. Lancet. 2021;397:375–386. doi: 10.1016/S0140-6736(20)32714-8. [DOI] [PubMed] [Google Scholar]
- 2.Zalcman G., Mazieres J., Margery J., Greillier L., Audigier-Valette C., Moro-Sibilot D., Molinier O., Corre R., Monnet I., Gounant V., et al. Bevacizumab for newly diagnosed pleural mesothelioma in the Mesothelioma Avastin Cisplatin Pemetrexed Study (MAPS): a randomised, controlled, open-label, phase 3 trial. Lancet. 2016;387:1405–1414. doi: 10.1016/S0140-6736(15)01238-6. [DOI] [PubMed] [Google Scholar]
- 3.Janes S.M., Alrifai D., Fennell D.A. Perspectives on the Treatment of Malignant Pleural Mesothelioma. N. Engl. J. Med. 2021;385:1207–1218. doi: 10.1056/NEJMra1912719. [DOI] [PubMed] [Google Scholar]
- 4.Woolhouse I., Bishop L., Darlison L., De Fonseka D., Edey A., Edwards J., Faivre-Finn C., Fennell D.A., Holmes S., Kerr K.M., et al. British Thoracic Society Guideline for the investigation and management of malignant pleural mesothelioma. Thorax. 2018;73:i1–i30. doi: 10.1136/thoraxjnl-2017-211321. [DOI] [PubMed] [Google Scholar]
- 5.Alay A., Cordero D., Hijazo-Pechero S., Aliagas E., Lopez-Doriga A., Marín R., Palmero R., Llatjós R., Escobar I., Ramos R., et al. Integrative transcriptome analysis of malignant pleural mesothelioma reveals a clinically relevant immune-based classification. J. Immunother. Cancer. 2021;9 doi: 10.1136/jitc-2020-001601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kukurba K.R., Montgomery S.B. RNA Sequencing and Analysis. Cold Spring Harb. Protoc. 2015;2015:951–969. doi: 10.1101/pdb.top084970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bai Y., Wang X., Hou J., Geng L., Liang X., Ruan Z., Guo H., Nan K., Jiang L. Identification of a Five-Gene Signature for Predicting Survival in Malignant Pleural Mesothelioma Patients. Front. Genet. 2020;11:899. doi: 10.3389/fgene.2020.00899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hmeljak J., Sanchez-Vega F., Hoadley K.A., Shih J., Stewart C., Heiman D., Tarpey P., Danilova L., Drill E., Gibb E.A., et al. Integrative Molecular Characterization of Malignant Pleural Mesothelioma. Cancer Discov. 2018;8:1548–1565. doi: 10.1158/2159-8290.CD-18-0804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sage A.P., Martinez V.D., Minatel B.C., Pewarchuk M.E., Marshall E.A., MacAulay G.M., Hubaux R., Pearson D.D., Goodarzi A.A., Dellaire G., Lam W.L. Genomics and Epigenetics of Malignant Mesothelioma. High. Throughput. 2018;7 doi: 10.3390/ht7030020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chesler E.J., Miller D.R., Branstetter L.R., Galloway L.D., Jackson B.L., Philip V.M., Voy B.H., Culiat C.T., Threadgill D.W., Williams R.W., et al. The Collaborative Cross at Oak Ridge National Laboratory: developing a powerful resource for systems genetics. Mamm. Genome. 2008;19:382–389. doi: 10.1007/s00335-008-9135-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Churchill G.A., Airey D.C., Allayee H., Angel J.M., Attie A.D., Beatty J., Beavis W.D., Belknap J.K., Bennett B., Berrettini W., et al. The Collaborative Cross, a community resource for the genetic analysis of complex traits. Nat. Genet. 2004;36:1133–1137. doi: 10.1038/ng1104-1133. [DOI] [PubMed] [Google Scholar]
- 12.Collaborative Cross Consortium The genome architecture of the Collaborative Cross mouse genetic reference population. Genetics. 2012;190:389–401. doi: 10.1534/genetics.111.132639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Iraqi F.A., Churchill G., Mott R. The Collaborative Cross, developing a resource for mammalian systems genetics: a status report of the Wellcome Trust cohort. Mamm. Genome. 2008;19:379–381. doi: 10.1007/s00335-008-9113-1. [DOI] [PubMed] [Google Scholar]
- 14.Morahan G., Balmer L., Monley D. Establishment of "The Gene Mine": a resource for rapid identification of complex trait genes. Mamm. Genome. 2008;19:390–393. doi: 10.1007/s00335-008-9134-9. [DOI] [PubMed] [Google Scholar]
- 15.Ram R., Mehta M., Balmer L., Gatti D.M., Morahan G. Rapid identification of major-effect genes using the collaborative cross. Genetics. 2014;198:75–86. doi: 10.1534/genetics.114.163014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Patel S.J., Molinolo A.A., Gutkind S., Crawford N.P.S. Germline genetic variation modulates tumor progression and metastasis in a mouse model of neuroendocrine prostate carcinoma. PLoS One. 2013;8 doi: 10.1371/journal.pone.0061848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ram R., Morahan G. Complex Trait Analyses of the Collaborative Cross: Tools and Databases. Methods Mol. Biol. 2017;1488:121–129. doi: 10.1007/978-1-4939-6427-7_5. [DOI] [PubMed] [Google Scholar]
- 18.Roberts A., Pardo-Manuel de Villena F., Wang W., McMillan L., Threadgill D.W. The polymorphism architecture of mouse genetic resources elucidated using genome-wide resequencing data: implications for QTL discovery and systems genetics. Mamm. Genome. 2007;18:473–481. doi: 10.1007/s00335-007-9045-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Robinson C., van Bruggen I., Segal A., Dunham M., Sherwood A., Koentgen F., Robinson B.W.S., Lake R.A. A novel SV40 TAg transgenic model of asbestos-induced mesothelioma: malignant transformation is dose dependent. Cancer Res. 2006;66:10786–10794. doi: 10.1158/0008-5472.CAN-05-4668. [DOI] [PubMed] [Google Scholar]
- 20.Robinson C., Walsh A., Larma I., O'Halloran S., Nowak A.K., Lake R.A. MexTAg mice exposed to asbestos develop cancer that faithfully replicates key features of the pathogenesis of human mesothelioma. Eur. J. Cancer. 2011;47:151–161. doi: 10.1016/j.ejca.2010.08.015. [DOI] [PubMed] [Google Scholar]
- 21.Robinson C., Dick I.M., Wise M.J., Holloway A., Diyagama D., Robinson B.W.S., Creaney J., Lake R.A. Consistent gene expression profiles in MexTAg transgenic mouse and wild type mouse asbestos-induced mesothelioma. BMC Cancer. 2015;15:983. doi: 10.1186/s12885-015-1953-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fisher S.A., Patrick K., Hoang T., Marcq E., Behrouzfar K., Young S., Miller T.J., Robinson B.W.S., Bueno R., Nowak A.K., et al. The MexTAg collaborative cross: host genetics affects asbestos related disease latency, but has little influence once tumours develop. Front. Toxicol. 2024;6 doi: 10.3389/ftox.2024.1373003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ferguson B., Ram R., Handoko H.Y., Mukhopadhyay P., Muller H.K., Soyer H.P., Morahan G., Walker G.J. Melanoma susceptibility as a complex trait: genetic variation controls all stages of tumor progression. Oncogene. 2015;34:2879–2886. doi: 10.1038/onc.2014.227. [DOI] [PubMed] [Google Scholar]
- 24.Yang C.H., Mangiafico S.P., Waibel M., Loudovaris T., Loh K., Thomas H.E., Morahan G., Andrikopoulos S. E2f8 and Dlg2 genes have independent effects on impaired insulin secretion associated with hyperglycaemia. Diabetologia. 2020;63:1333–1348. doi: 10.1007/s00125-020-05137-0. [DOI] [PubMed] [Google Scholar]
- 25.Yuan J., Tickner J., Mullin B.H., Zhao J., Zeng Z., Morahan G., Xu J. Advanced Genetic Approaches in Discovery and Characterization of Genes Involved With Osteoporosis in Mouse and Human. Front. Genet. 2019;10:288. doi: 10.3389/fgene.2019.00288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Behrouzfar K., Burton K., Mutsaers S.E., Morahan G., Lake R.A., Fisher S.A. How to Better Understand the Influence of Host Genetics on Developing an Effective Immune Response to Thoracic Cancers. Front. Oncol. 2021;11 doi: 10.3389/fonc.2021.679609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bueno R., Stawiski E.W., Goldstein L.D., Durinck S., De Rienzo A., Modrusan Z., Gnad F., Nguyen T.T., Jaiswal B.S., Chirieac L.R., et al. Comprehensive genomic analysis of malignant pleural mesothelioma identifies recurrent mutations, gene fusions and splicing alterations. Nat. Genet. 2016;48:407–416. doi: 10.1038/ng.3520. [DOI] [PubMed] [Google Scholar]
- 28.Charoentong P., Finotello F., Angelova M., Mayer C., Efremova M., Rieder D., Hackl H., Trajanoski Z. Pan-cancer Immunogenomic Analyses Reveal Genotype-Immunophenotype Relationships and Predictors of Response to Checkpoint Blockade. Cell Rep. 2017;18:248–262. doi: 10.1016/j.celrep.2016.12.019. [DOI] [PubMed] [Google Scholar]
- 29.Danaher P., Warren S., Lu R., Samayoa J., Sullivan A., Pekker I., Wallden B., Marincola F.M., Cesano A. Pan-cancer adaptive immune resistance as defined by the Tumor Inflammation Signature (TIS): results from The Cancer Genome Atlas (TCGA) J. Immunother. Cancer. 2018;6:63. doi: 10.1186/s40425-018-0367-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Liberzon A., Birger C., Thorvaldsdóttir H., Ghandi M., Mesirov J.P., Tamayo P. The Molecular Signatures Database (MSigDB) hallmark gene set collection. Cell Syst. 2015;1:417–425. doi: 10.1016/j.cels.2015.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Şenbabaoğlu Y., Gejman R.S., Winer A.G., Liu M., Van Allen E.M., de Velasco G., Miao D., Ostrovnaya I., Drill E., Luna A., et al. Tumor immune microenvironment characterization in clear cell renal cell carcinoma identifies prognostic and immunotherapeutically relevant messenger RNA signatures. Genome Biol. 2016;17:231. doi: 10.1186/s13059-016-1092-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Thompson J.C., Hwang W.T., Davis C., Deshpande C., Jeffries S., Rajpurohit Y., Krishna V., Smirnov D., Verona R., Lorenzi M.V., et al. Gene signatures of tumor inflammation and epithelial-to-mesenchymal transition (EMT) predict responses to immune checkpoint blockade in lung cancer with high accuracy. Lung Cancer. 2020;139:1–8. doi: 10.1016/j.lungcan.2019.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Xiao Y., Huang W., Zhang L., Wang H. Identification of glycolysis genes signature for predicting prognosis in malignant pleural mesothelioma by bioinformatics and machine learning. Front. Endocrinol. 2022;13 doi: 10.3389/fendo.2022.1056152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhang Z., Wang Z.X., Chen Y.X., Wu H.X., Yin L., Zhao Q., Luo H.Y., Zeng Z.L., Qiu M.Z., Xu R.H. Integrated analysis of single-cell and bulk RNA sequencing data reveals a pan-cancer stemness signature predicting immunotherapy response. Genome Med. 2022;14:45. doi: 10.1186/s13073-022-01050-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tilsed C.M., Principe N., Kidman J., Chin W.L., Orozco Morales M.L., Zemek R.M., Chee J., Islam R., Fear V.S., Forbes C., et al. CD4+ T cells drive an inflammatory, TNF-alpha/IFN-rich tumor microenvironment responsive to chemotherapy. Cell Rep. 2022;41 doi: 10.1016/j.celrep.2022.111874. [DOI] [PubMed] [Google Scholar]
- 36.Nair N.U., Jiang Q., Wei J.S., Misra V.A., Morrow B., Kesserwan C., Hermida L.C., Lee J.S., Mian I., Zhang J., et al. Genomic and transcriptomic analyses identify a prognostic gene signature and predict response to therapy in pleural and peritoneal mesothelioma. Cell Rep. Med. 2023;4 doi: 10.1016/j.xcrm.2023.100938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mannarino L., Paracchini L., Pezzuto F., Olteanu G.E., Moracci L., Vedovelli L., De Simone I., Bosetti C., Lupi M., Amodeo R., et al. Epithelioid Pleural Mesothelioma Is Characterized by Tertiary Lymphoid Structures in Long Survivors: Results from the MATCH Study. Int. J. Mol. Sci. 2022;23 doi: 10.3390/ijms23105786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Creaney J., Patch A.M., Addala V., Sneddon S.A., Nones K., Dick I.M., Lee Y.C.G., Newell F., Rouse E.J., Naeini M.M., et al. Comprehensive genomic and tumour immune profiling reveals potential therapeutic targets in malignant pleural mesothelioma. Genome Med. 2022;14:58. doi: 10.1186/s13073-022-01060-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Shi J., Peng B., Zhou X., Wang C., Xu R., Lu T., Chang X., Shen Z., Wang K., Xu C., Zhang L. An anoikis-based gene signature for predicting prognosis in malignant pleural mesothelioma and revealing immune infiltration. J. Cancer Res. Clin. Oncol. 2023;149:12089–12102. doi: 10.1007/s00432-023-05128-9. [DOI] [PubMed] [Google Scholar]
- 40.Zhang X., Huang X., Wang Z., Zhang K. Establishment and validation of a novel immune-related prognostic signature in malignant pleural mesothelioma. Ann. Transl. Med. 2022;10:200. doi: 10.21037/atm-22-527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhou J.G., Zhong H., Zhang J., Jin S.H., Roudi R., Ma H. Development and Validation of a Prognostic Signature for Malignant Pleural Mesothelioma. Front. Oncol. 2019;9:78. doi: 10.3389/fonc.2019.00078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Alcala N., Mangiante L., Le-Stang N., Gustafson C.E., Boyault S., Damiola F., Alcala K., Brevet M., Thivolet-Bejui F., Blanc-Fournier C., et al. Redefining malignant pleural mesothelioma types as a continuum uncovers immune-vascular interactions. EBioMedicine. 2019;48:191–202. doi: 10.1016/j.ebiom.2019.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Geng Q., Shen Z., Li L., Zhao J. COL1A1 is a prognostic biomarker and correlated with immune infiltrates in lung cancer. PeerJ. 2021;9 doi: 10.7717/peerj.11145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pires A., Burnell S., Gallimore A. Exploiting ECM remodelling to promote immune-mediated tumour destruction. Curr. Opin. Immunol. 2022;74:32–38. doi: 10.1016/j.coi.2021.09.006. [DOI] [PubMed] [Google Scholar]
- 45.Ren J., Da J., Hu N. Identification of COL1A1 associated with immune infiltration in brain lower grade glioma. PLoS One. 2022;17 doi: 10.1371/journal.pone.0269533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wang Y., Zheng K., Chen X., Chen R., Zou Y. Bioinformatics analysis identifies COL1A1, THBS2 and SPP1 as potential predictors of patient prognosis and immunotherapy response in gastric cancer. Biosci. Rep. 2021;41 doi: 10.1042/BSR20202564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Abayasiriwardana K.S., Wood M.K., Prêle C.M., Birnie K.A., Robinson B.W., Laurent G.J., McAnulty R.J., Mutsaers S.E. Inhibition of collagen production delays malignant mesothelioma tumor growth in a murine model. Biochem. Biophys. Res. Commun. 2019;510:198–204. doi: 10.1016/j.bbrc.2019.01.057. [DOI] [PubMed] [Google Scholar]
- 48.Blum Y., Meiller C., Quetel L., Elarouci N., Ayadi M., Tashtanbaeva D., Armenoult L., Montagne F., Tranchant R., Renier A., et al. Dissecting heterogeneity in malignant pleural mesothelioma through histo-molecular gradients for clinical applications. Nat. Commun. 2019;10:1333. doi: 10.1038/s41467-019-09307-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gueugnon F., Leclercq S., Blanquart C., Sagan C., Cellerin L., Padieu M., Perigaud C., Scherpereel A., Gregoire M. Identification of novel markers for the diagnosis of malignant pleural mesothelioma. Am. J. Pathol. 2011;178:1033–1042. doi: 10.1016/j.ajpath.2010.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Patil N.S., Righi L., Koeppen H., Zou W., Izzo S., Grosso F., Libener R., Loiacono M., Monica V., Buttigliero C., et al. Molecular and Histopathological Characterization of the Tumor Immune Microenvironment in Advanced Stage of Malignant Pleural Mesothelioma. J. Thorac. Oncol. 2018;13:124–133. doi: 10.1016/j.jtho.2017.09.1968. [DOI] [PubMed] [Google Scholar]
- 51.Wu L., Amjad S., Yun H., Mani S., de Perrot M. A panel of emerging EMT genes identified in malignant mesothelioma. Sci. Rep. 2022;12:1007. doi: 10.1038/s41598-022-04973-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Xiong D., Wang Y., You M. A gene expression signature of TREM2(hi) macrophages and gammadelta T cells predicts immunotherapy response Nat Commun. 2020; 11 (1): 5084. Theor. Biosci. 2020;11:5084. doi: 10.1038/s41467-020-18546-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lee J.S., Nair N.U., Dinstag G., Chapman L., Chung Y., Wang K., Sinha S., Cha H., Kim D., Schperberg A.V., et al. Synthetic lethality-mediated precision oncology via the tumor transcriptome. Cell. 2021;184:2487–2502.e13. doi: 10.1016/j.cell.2021.03.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Solovyov A., Vabret N., Arora K.S., Snyder A., Funt S.A., Bajorin D.F., Rosenberg J.E., Bhardwaj N., Ting D.T., Greenbaum B.D. Global Cancer Transcriptome Quantifies Repeat Element Polarization between Immunotherapy Responsive and T Cell Suppressive Classes. Cell Rep. 2018;23:512–521. doi: 10.1016/j.celrep.2018.03.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Walters K., Sarsenov R., Too W.S., Hare R.K., Paterson I.C., Lambert D.W., Brown S., Bradford J.R. Comprehensive functional profiling of long non-coding RNAs through a novel pan-cancer integration approach and modular analysis of their protein-coding gene association networks. BMC Genom. 2019;20:454. doi: 10.1186/s12864-019-5850-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ye B., Shi J., Kang H., Oyebamiji O., Hill D., Yu H., Ness S., Ye F., Ping J., He J., et al. Advancing Pan-cancer Gene Expression Survial Analysis by Inclusion of Non-coding RNA. RNA Biol. 2020;17:1666–1673. doi: 10.1080/15476286.2019.1679585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Council N.H.a.M.R. 8th ed. National Health and Medical Research Council; 2013. Australian Code for the Care and Use of Animals for Scientific Purposes. [Google Scholar]
- 58.Bray N.L., Pimentel H., Melsted P., Pachter L. Erratum: Near-optimal probabilistic RNA-seq quantification. Nat. Biotechnol. 2016;34:888. doi: 10.1038/nbt0816-888d. [DOI] [PubMed] [Google Scholar]
- 59.Soneson C., Love M.I., Robinson M.D. Differential analyses for RNA-seq: transcript-level estimates improve gene-level inferences. F1000Res. 2015;4:1521. doi: 10.12688/f1000research.7563.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Love M.I., Huber W., Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wilkerson M.D., Hayes D.N. ConsensusClusterPlus: a class discovery tool with confidence assessments and item tracking. Bioinformatics. 2010;26:1572–1573. doi: 10.1093/bioinformatics/btq170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Liberzon A., Subramanian A., Pinchback R., Thorvaldsdóttir H., Tamayo P., Mesirov J.P. Molecular signatures database (MSigDB) 3.0. Bioinformatics. 2011;27:1739–1740. doi: 10.1093/bioinformatics/btr260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Zemek R.M., De Jong E., Chin W.L., Schuster I.S., Fear V.S., Casey T.H., Forbes C., Dart S.J., Leslie C., Zaitouny A., et al. Sensitization to immune checkpoint blockade through activation of a STAT1/NK axis in the tumor microenvironment. Sci. Transl. Med. 2019;11 doi: 10.1126/scitranslmed.aav7816. [DOI] [PubMed] [Google Scholar]
- 64.Kanehisa M., Furumichi M., Sato Y., Kawashima M., Ishiguro-Watanabe M. KEGG for taxonomy-based analysis of pathways and genomes. Nucleic Acids Res. 2023;51:D587–D592. doi: 10.1093/nar/gkac963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Mootha V.K., Lindgren C.M., Eriksson K.F., Subramanian A., Sihag S., Lehar J., Puigserver P., Carlsson E., Ridderstråle M., Laurila E., et al. PGC-1alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat. Genet. 2003;34:267–273. doi: 10.1038/ng1180. [DOI] [PubMed] [Google Scholar]
- 66.Subramanian A., Kuehn H., Gould J., Tamayo P., Mesirov J.P. GSEA-P: a desktop application for Gene Set Enrichment Analysis. Bioinformatics. 2007;23:3251–3253. doi: 10.1093/bioinformatics/btm369. [DOI] [PubMed] [Google Scholar]
- 67.Subramanian A., Tamayo P., Mootha V.K., Mukherjee S., Ebert B.L., Gillette M.A., Paulovich A., Pomeroy S.L., Golub T.R., Lander E.S., Mesirov J.P. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Newman A.M., Steen C.B., Liu C.L., Gentles A.J., Chaudhuri A.A., Scherer F., Khodadoust M.S., Esfahani M.S., Luca B.A., Steiner D., et al. Determining cell type abundance and expression from bulk tissues with digital cytometry. Nat. Biotechnol. 2019;37:773–782. doi: 10.1038/s41587-019-0114-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Langfelder P., Horvath S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinf. 2008;9:559. doi: 10.1186/1471-2105-9-559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Langfelder P., Horvath S. Fast R Functions for Robust Correlations and Hierarchical Clustering. J. Stat. Software. 2012;46 [PMC free article] [PubMed] [Google Scholar]
- 71.Yu G., Wang L.G., Han Y., He Q.Y. clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS. 2012;16:284–287. doi: 10.1089/omi.2011.0118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Chin C.H., Chen S.H., Wu H.H., Ho C.W., Ko M.T., Lin C.Y. cytoHubba: identifying hub objects and sub-networks from complex interactome. BMC Syst. Biol. 2014;8:S11. doi: 10.1186/1752-0509-8-S4-S11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Shannon P., Markiel A., Ozier O., Baliga N.S., Wang J.T., Ramage D., Amin N., Schwikowski B., Ideker T. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13:2498–2504. doi: 10.1101/gr.1239303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Dobin A., Davis C.A., Schlesinger F., Drenkow J., Zaleski C., Jha S., Batut P., Chaisson M., Gingeras T.R. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Liao Y., Smyth G.K., Shi W. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics. 2014;30:923–930. doi: 10.1093/bioinformatics/btt656. [DOI] [PubMed] [Google Scholar]
- 76.Therneau T. A package for survival analysis in S. R package version. 2015;2 [Google Scholar]
- 77.Therneau T.M., Grambsch P.M. Modeling Survival Data: Extending the Cox Model. Springer; 2000. The Cox Model; pp. 39–77. [DOI] [Google Scholar]
- 78.Friedman J., Hastie T., Tibshirani R. Regularization Paths for Generalized Linear Models via Coordinate Descent. J. Stat. Software. 2010;33:1–22. [PMC free article] [PubMed] [Google Scholar]
- 79.Blanche P., Dartigues J.F., Jacqmin-Gadda H. Estimating and comparing time-dependent areas under receiver operating characteristic curves for censored event times with competing risks. Stat. Med. 2013;32:5381–5397. doi: 10.1002/sim.5958. [DOI] [PubMed] [Google Scholar]
- 80.Robin X., Turck N., Hainard A., Tiberti N., Lisacek F., Sanchez J.C., Müller M. pROC: an open-source package for R and S+ to analyze and compare ROC curves. BMC Bioinf. 2011;12:77. doi: 10.1186/1471-2105-12-77. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.