

Abstract
Evading programmed cell death (PCD) is a hallmark of cancer that allows tumor cells to survive and proliferate unchecked. Endocytosis, the process by which cells internalize extracellular materials, has emerged as a key regulator of cell death pathways in cancer. Many tumor types exhibit dysregulated endocytic dynamics that fuel their metabolic demands, promote resistance to cytotoxic therapies, and facilitate immune evasion. This review examines the roles of endocytosis in apoptotic resistance and immune escape mechanisms utilized by cancer cells. We highlight how inhibiting endocytosis can sensitize malignant cells to therapeutic agents and restore susceptibility to PCD. Strategies to modulate endocytosis for enhanced cancer treatment are discussed, including targeting endocytic regulatory proteins, altering membrane biophysical properties, and inhibiting Rho-associated kinases. While promising, challenges remain regarding the specificity and selectivity of endocytosis-targeting agents. Nonetheless, harnessing endocytic pathways represents an attractive approach to overcome apoptotic resistance and could yield more effective therapies by rendering cancer cells vulnerable to PCD. Understanding the interplay between endocytosis and PCD regulation is crucial for developing novel anticancer strategies that selectively induce tumor cell death.
Introduction
Cancer remains a leading cause of mortality worldwide, with many tumor types exhibiting resistance to standard chemotherapies and radiation treatments that aim to induce cancer cell death [1,2]. Overcoming this therapeutic challenge requires developing novel strategies that not only induce or enhance cancer cell death but also circumvent mechanisms of resistance inherent in conventional treatments [3–5]. Unlike traditional approaches that broadly target cellular proliferation, these new strategies focus on selectively exploiting vulnerabilities within cancer cells to initiate programmed cell death (PCD), while preserving healthy tissues.
Endocytosis, the mechanism by which cells internalize extracellular materials and molecules, has emerged as an attractive target for cancer therapy [6–9]. Cancer cells often exhibit dysregulated endocytic pathways that support their increased metabolic demands and rapid division [6,10]. For example, many cancer cells overexpress receptors like the transferrin receptor or growth factor receptors, which are internalized via clathrin-mediated endocytosis (CME) to fuel tumor growth [7,9,11]. Disrupting endocytosis in these malignant cells can deprive them of essential nutrients and signaling factors, thereby sensitizing them to PCD [12]. Furthermore, inhibiting endocytosis can prevent the internalization and trafficking of therapeutic agents, rendering cancer cells more susceptible to cytotoxic drugs and other anti-cancer modalities [9,13].
In this review, we first define PCD and the mechanisms of how cancer cells evade this process. We will then examine the current understanding of how endocytic pathways are altered in cancer and discuss strategies to target these processes as a means to enhance cancer cell susceptibility to PCD. We highlight recent studies demonstrating the therapeutic potential of modulating endocytosis in various tumor types. Finally, we consider the challenges and future directions in translating approaches targeted at endocytic pathways into effective cancer treatments that selectively trigger cell death in tumors.
PCD in cancer
Cancer is a complex and heterogeneous disease that is characterized by uncontrolled cell proliferation and evasion of cell death mechanisms [14–17]. PCD, composed of apoptosis, autophagy, and programmed necrosis, is essential for maintaining tissue homeostasis and eliminating aberrant cells [5,17]. However, in cancer, the balance between cell survival and death is disrupted, leading to tumor progression and treatment resistance [3,16,17].
Apoptosis, the most extensively studied form of PCD, serves as a critical mechanism for eliminating damaged or unwanted cells. The process of apoptosis is orchestrated by two primary pathways: the extrinsic pathway and the intrinsic pathway [18–20]. In the extrinsic pathway, external death signals activate death receptors on the cell surface, such as Fas (CD95) and tumor necrosis factor receptor 1, leading to the formation of the death-inducing signaling complex (DISC) [21–23]. The DISC recruits and activates procaspase-8, initiating a cascade of caspase activation and ultimately resulting in cell death. Conversely, the intrinsic pathway is initiated by intracellular stress signals, such as DNA damage or metabolic imbalance, leading to mitochondrial outer membrane permeabilization [20,24]. This process releases cytochrome c into the cytosol, activating the apoptosome and triggering caspase activation. Dysregulation of apoptotic pathways in cancer often occurs through genetic mutations, epigenetic alterations, or dysregulated expression of apoptosis-related proteins, allowing cancer cells to evade apoptotic signals and promote tumor survival [16,17].
Autophagy is a conserved catabolic process and plays a dual role in cancer biology. Under physiological conditions, autophagy maintains cellular homeostasis by degrading dysfunctional organelles and proteins [4,17,25]. However, in cancer, autophagy can act as either a pro-survival mechanism or a pro-death pathway, depending on the cellular context and environmental conditions. In nutrient-poor or hypoxic environments, autophagy promotes cancer cell survival by providing essential nutrients and energy substrates. This adaptive response enables cancer cells to withstand metabolic stress and resist apoptosis induced by therapeutic agents [3,17,26]. Alternatively, autophagy can induce a form of non-apoptotic cell death known as autophagic cell death [26]. This process involves excessive or prolonged autophagy leading to cellular self-digestion and eventual cell demise, independent of apoptosis. The dual nature of autophagy in cancer underscores its complexity and context-dependent effects, influencing tumor progression, therapeutic responses, and overall cellular fate modulation [27,28].
Programmed necrosis, once considered a chaotic and unregulated form of cell death [29], has recently emerged as a regulated process with distinct mechanisms, including necroptosis, ferroptosis, and pyroptosis [3,30,31]. Necroptosis, mediated by receptor-interacting protein kinases, occurs when apoptosis is inhibited or compromised, leading to necrotic cell death with inflammatory consequences [32]. Ferroptosis, characterized by iron-dependent lipid peroxidation and membrane damage, represents a novel form of regulated cell death implicated in cancer progression and therapy resistance [33]. Pyroptosis, triggered by inflammasome activation and caspase-1 cleavage, results in inflammatory cell death and immune responses [5,30]. The regulation of programmed necrosis in cancer is complex and context-dependent, involving cross-talk with other cell death pathways and interactions with the tumor microenvironment [30,31,34].
The interplay between different forms of PCD — apoptosis, autophagy, and programmed necrosis — is intricate and multifaceted. Cross-talk between these pathways can either promote or inhibit cell death, depending on the cellular context and environmental conditions. For example, apoptosis and autophagy can synergize to eliminate cancer cells under certain conditions [25], whereas in other scenarios, autophagy may promote cancer cell survival and therapy resistance in the absence of apoptosis [24,27,28]. Additionally, programmed necrosis can serve as a backup mechanism for apoptosis when caspase activation is impaired [32], contributing to the resilience of cancer cells against cell death signals.
Although numerous mechanisms drive the initiation of PCD, cancer cells have evolved a diverse repertoire of strategies to evade these processes. Having established an understanding of the various modalities of cell death, our focus will now shift towards exploring how cancer cells modulate these PCD pathways. Specifically, we will delve into the mechanisms through which cancer cells resist apoptotic signals, with a particular emphasis on extrinsic apoptosis.
Extrinsic mechanisms of apoptotic resistance in cancer
Cancer cells are under constant stress, facing oncogenic stress, genomic instability, cellular hypoxia, and extracellular apoptotic signals [3,5,16]. Typically, cells undergo PCD in response to stress, but cancer cells often evade this response by disabling apoptotic pathways, which is a hallmark of cancer [16]. They achieve this by down-regulating pro-apoptotic factors like caspases or up-regulating apoptosis inhibitors such as inhibitor of apoptosis proteins (IAPs) [25]. Additionally, cancer cells can desensitize themselves to extrinsic apoptotic signals by modulating death receptors [5,23]. In this context, we will explore two critical extrinsic mechanisms of immune-mediated cell death evasion by cancer cells: granule-mediated cytotoxicity and receptor-mediated cytotoxicity.
Granule-mediated cytotoxicity, employed by cytotoxic T lymphocytes (CTLs) and natural killer (NK) cells, releases cytotoxic molecules like perforin and granzyme B toward target cells, inducing intrinsic apoptosis [35–37]. Cancer cells counteract this process by degrading granzyme B or inhibiting cytotoxicity via hypoxia-induced autophagy [38,39]. Interestingly, alterations in mechanical properties on the cell membrane, such as lipid order, has been shown to impact perforin binding and cytotoxicity, with low-order lipids favored for perforin-mediated apoptosis [40,41]. For example, breast cancer cells resistant to lymphocyte cytotoxicity exhibit elevated lipid order, rendering them less susceptible to perforin-induced lysis [40,42]. Other mechanical features such as cell stiffness have been shown to influence susceptibility to perforin, with softer cancer cells evading T cell cytotoxicity [43].
Granule-mediated cytotoxicity serves as the primary mechanism for eliminating target cells in the presence of a large number of T cells. However, this process relies on CTLs to initiate target recognition, leading to the expansion of specific T cell populations capable of identifying target cells through specific peptides presented with the major histocompatibility complex class I (MHC-I) [44]. This presentation of peptides by MHC-I is indispensable for the binding of death receptors to ligands, initiating extrinsic apoptosis.
Cancer cells paradoxically express both the Fas (CD95) death receptor and Fas ligand (FasL), yet often exhibit resistance to Fas-mediated apoptosis [23,45,46]. Fas, a surface receptor from the tumor necrosis factor receptor (TNFR) superfamily, is primarily recognized for initiating cell death upon binding to its ligand, FasL. Despite abundant Fas expression on cancer cells, they often resist this apoptotic pathway, enabling them to evade cell death mechanisms [45,47]. Common evasion strategies involve the down-regulation of key components of the Fas signaling cascade, such as caspase-8 or Fas-associated death domain, which are essential for transmitting apoptotic signals initiated by Fas activation. Additionally, cancer cells often up-regulate cellular FLICE inhibitory protein, a potent inhibitor at the DISC, thereby preventing caspase-8 activation and subsequent apoptosis despite Fas receptor engagement [48]. Deregulation of B-cell lymphoma 2 (Bcl-2) family proteins or inhibitors of apoptotic proteins contributes to the loss of apoptosis signaling through Fas, promoting tumor survival [49]. Cancer cells also use FasL expression to indirectly target immune cells, inducing apoptosis in Fas-expressing CD8+ T cells and evading immune surveillance [50,51]. Remarkably, Fas activation can even boost cancer cell survival by enhancing their motility and invasiveness [45].
Similar to granule-mediated killing, receptor-mediated killing by CTLs or NK cells relies on the mechanical features of the cell membrane. The interaction between T-cell receptors (TCRs) of CD8+ T cells and MHC-I peptides is crucial for initiating apoptosis [44]. Soft membranes prevent effective TCR-MHC-I-peptide interactions, leading to insufficient downstream apoptotic signaling [52,53]. Moreover, MHC-I molecules are localized to higher order lipid regions, and depletion of cholesterol disrupts CTL recognition of MHC-I peptide complexes [36].
These findings suggest that cancer cells evade apoptosis not only through the regulation of biochemical players but also by modulating mechanical features, such as membrane stiffness. In the following section, we will examine the roles of receptor-mediated cytotoxicity, with a particular focus on how endocytosis contributes to cancer cell death. Finally, we will discuss strategies designed to enhance apoptotic sensitivity in these cells through targeting endocytosis.
Roles of endocytosis in apoptotic evasion
Endocytosis plays a crucial role in various cellular processes, including nutrient uptake, receptor internalization, and signal transduction [54,55]. Defined as the process by which cells engulf extracellular molecules and particles by forming vesicles derived from the plasma membrane, endocytosis serves as a fundamental mechanism for maintaining cellular homeostasis and regulating cell signaling pathways [8,56,57].
Endocytosis is tightly regulated by various cellular factors, including membrane composition, cytoskeletal dynamics, and signaling pathways [56,58,59]. Membrane lipid composition, particularly the presence of cholesterol and sphingolipids, influences the formation and stability of endocytic vesicles [60]. Moreover, cytoskeletal elements such as actin filaments and microtubules provide the structural framework necessary for vesicle formation and intracellular trafficking [56,58]. Signaling molecules such as small GTPases, including dynamin and Rab proteins, regulate the budding and fusion of endocytic vesicles with target membranes [54].
There are several types of endocytosis, each serving distinct functions in cellular physiology [61]. CME is the most well-characterized form, involving the formation of clathrin-coated vesicles that transport cargo molecules into the cell [54,58,59]. Caveolae-mediated endocytosis occurs through invaginations of lipid raft domains enriched in caveolin proteins, facilitating the internalization of specific membrane components [62]. Additionally, macropinocytosis involves the nonspecific uptake of extracellular fluid and solutes through large, actin-driven membrane protrusions called macropinosomes [61].
CME, in particular, plays a significant role in oncogenesis and cancer cell proliferation [7,63]. Genetic mutations affecting endocytic proteins have been implicated in leukemia, underscoring the importance of endocytosis in cancer pathogenesis [64]. Moreover, posttranslational ubiquitination of endocytic proteins and receptors serves as a sorting signal in this pathway, influencing cellular processes crucial for cancer progression [64,65]. For example, ubiquitination regulates the internalization and trafficking of receptors such as epidermal growth factor (EGFR), impacting downstream signaling pathways that promote tumor growth and metastasis [66,67]. Additionally, ubiquitination of the E3 ubiquitin ligase Nedd4 can modulate the stability and function of CME machinery, affecting the turnover of membrane proteins involved in cancer cell signaling and survival, including EGFR [65,68]. Active Src kinase has been shown to promote degradation of Cbl, an important regulator of CME, resulting in elevated EGFR expression and signaling in tumors [66,67].
Tumor cells significantly diverge from normal cells in their cell membrane’s structure and composition, resulting in the development of distinct signaling pathways that provide them with a survival edge [9]. Endocytosis plays a pivotal role in this process, as it can selectively engage in the uptake of extracellular molecules, thereby influencing apoptotic pathways and directly affecting cancer cell survival. By internalizing death receptors such as Fas/CD95 and TNFRs, cancer cells can sequester these receptors away from the cell surface, preventing their engagement with extracellular ligands and subsequent initiation of apoptotic signaling cascades [69,70]. Furthermore, endocytosis facilitates the internalization of anti-apoptotic proteins, such as Bcl-2 family members and IAPs, which inhibit pro-apoptotic signaling pathways and promote cell survival [25,71]. Endocytosis also influences cancer immunity by modulating the presentation of tumor-associated antigens. Down-regulation of surface display of MHC-I, facilitated by endocytosis, can impede T-cell-mediated cytotoxicity and promote immune evasion by tumors [72].
Concurrently, cancer cells evade immune surveillance by decreasing ‘eat me’ signals that promote their engulfment, such as exposure of phosphatidylserine on the outer membrane, modifying surface glycosylation patterns and epitopes of intercellular adhesion molecules, while increasing signals that inhibit phagocytosis (such as CD47, PD-L1, and beta-2 microglobulin) [73]. Alternatively, cancer cells release ‘find me’ signals that recruit monocyte or macrophage recruitment toward apoptotic cells, including lipid lysophosphatidylcholine, sphingosine 1-phosphate, fractalkine CX3CL1, and nucleotides ATP and UTP [73]. Although these signals can facilitate the efficient removal of these dying cells before they undergo secondary necrosis, which can trigger inflammation and tissue damage, cancer cells can evade detection and clearance due to their lack of expression of ‘eat me’ signals [73,74].
Dysregulating endocytic pathways has been linked to the altered expression and activity of key oncogenes and tumor suppressor genes, further influencing cancer cell fate [6]. In colon cancer, inhibiting CME has been found to impede tumor growth and enhance the therapeutic efficacy of immune checkpoint blockade, indicating that selective targeting of endocytic pathways could be a viable strategy in cancer treatment [72]. In addition, inhibiting endocytosis, particularly of death-inducing proteins [12], could enhance antitumor efficacy by preventing immune evasion mechanisms employed by cancer cells. Understanding the intricate mechanisms underlying endocytic regulation and its impact on apoptotic signaling pathways is essential for developing targeted therapeutic strategies aimed at overcoming cancer resistance to PCD.
Inhibiting endocytosis sensitizes cancer cells to PCD
Endocytic dynamics are increasingly recognized as a valuable target in anticancer strategies, mainly because of their role in facilitating targeted and efficient drug delivery [13] (Figure 1). Targeted drug delivery systems aim to minimize off-target effects, overcome multidrug resistance, ensure specific distribution to cancerous tissues, and improve the permeability of anticancer agents across cell membranes, ultimately enhancing the vulnerability of cancer cells to treatment [75]. Endocytosis plays a vital role in the uptake of drug-delivery vehicles, allowing therapeutics such as antibody-drug conjugates (ADCs) and radioligands to be efficiently transported into tumor cells [76].
Figure 1. Schematic representation illustrating the potential therapeutic strategy of inhibiting endocytosis in cancer cells.
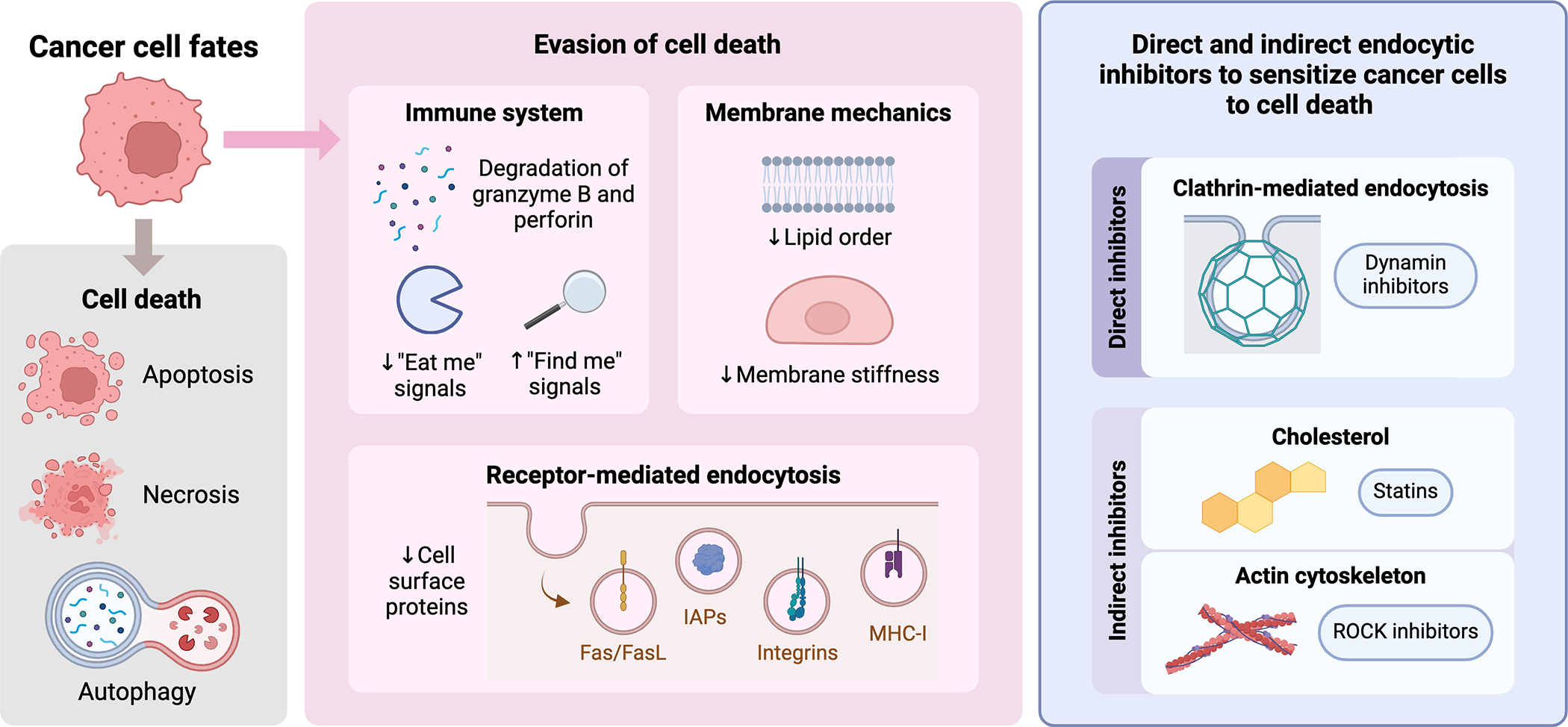
Inhibiting endocytosis can block the internalization of death receptors, restore immune surveillance, and improve the delivery of therapeutics to sensitize cancer cells to programmed cell death. Figure created by Biorender.com.
One promising approach to target endocytosis in cancer is to inhibit the GTPase dynamin, a central regulator of multiple endocytic pathways [77]. In particular, dynamin-1, typically neuron-specific, has been shown to be activated in nonneuronal cells via cancer-relevant signaling pathways, establishing a feedback loop between CME and signaling to enhance cancer cell survival, migration, and proliferation [78,79]. Chemical inhibitors of dynamin, such as Dyngo [80], Dynasore [81], and phenothiazine [82] block the cell’s GTPase activity, disrupting a wide range of dynamin-dependent endocytic processes. This inhibition of endocytosis has shown remarkable efficacy in suppressing proliferation and inducing apoptotic cell death across various cancer cell lines, including leukemia and lymphoma [83,84]. Notably, the disruption of endocytosis through dynamin inhibition has also been observed to overcome chemoresistance in leukemia stem cells, highlighting its potential to sensitize even the most recalcitrant tumor cells to cell death [85].
Beyond dynamin, other endocytic targets have been explored as a means to sensitize cancer cells. Compounds like Pitstop, which interfere with the clathrin-mediated endocytic machinery [86,87], have demonstrated the ability to enhance cancer cell susceptibility to cell death [88]. However, despite mutations in clathrin heavy chain that would theoretically block its supposed binding site, Pitstop 2 inhibits endocytosis indiscriminately [87,89–91]. This non-specific action indicates that Pitstop 2 may not be suitable for clinical use. Although Filipin III, which blocks caveolae/raft-mediated endocytosis [92], has been shown to overcome EGFR inhibitor resistance in lung cancer cells [93], it would not be effective on cells such as PC3 cells that lack cavin-1, a protein essential for caveolae formation [94]. These findings underscore the importance of various modes of endocytic regulation and combination therapies in modulating the response of cancer cells to targeted therapies.
The modulation of endocytosis can also be achieved through indirect approaches that alter the biophysical properties of the cell membrane [95–97]. Cancer cells often exhibit distinct membrane characteristics, such as altered cholesterol content and fluidity [98,99], which can significantly impact endocytic dynamics and the associated signaling cascades crucial for their survival. Agents such as statins, which target membrane cholesterol, have demonstrated anticancer effects in both preclinical and clinical studies. Lovastatin, simvastatin, and rosuvastatin, have shown promise in preclinical studies by temporarily enhancing tumor cell surface receptor density, thereby increasing the accumulation of monoclonal antibodies used in cancer therapies [94]. Lovastatin has also been reported to decrease markers associated with metastasis in breast cancer cells [100]. Furthermore, lovastatin has been shown to enhance apoptosis in brain cancer cells by increasing the activity of doublecortin, a brain-specific gene [101]. In some clinical studies, simvastatin, either used alone or in combination with other chemotherapeutic agents, has been demonstrated to significantly improve treatment outcomes and reduce mortality rates in patients with certain types of cancer [102–104].
Cancer cells are notably softer compared with healthy cells, facilitating rapid membrane remodeling during cancer progression [105–107]. This reduced stiffness is crucial to explain the observed increase in endocytosis in many cancer cells, particularly in a localized manner, which may be attributed to variations in local membrane composition, tension, and cooperative processes like actin remodeling [59]. These attributes play a vital role not only in facilitating cancer cell survival, invasion, and metastasis but also significantly impact the interactions between cancer cells and immune cells. For instance, T cells demonstrate diminished cytoskeletal forces and produce fewer effector cytokines when interacting with softer surfaces [53]. Thus, beyond biochemical immune checkpoints, mechanical checkpoints play a vital role in T cell-mediated cytotoxicity against cancer cells.
There has been growing interest in investigating Rho-associated kinases (ROCK) inhibitors as potential therapies for cancer. ROCK play a pivotal role in regulating the actomyosin cytoskeleton and contractile force generation [108]. This ROCK-driven contractility governs various cellular processes, including cell morphology, migration, invasion, proliferation, immune responses, and apoptosis resistance [109–111]. Inhibiting ROCK leads to increased membrane tension, which subsequently reduces endocytic dynamics [12]. Currently, several ROCK inhibitors such as Fasudil, Netarsudil, Belumosudil, and Ripasudil are approved for clinical use, primarily for treating hypertension [112,113]. While clinical trials using these inhibitors for cancer treatment have not yet been successful, numerous preclinical studies suggest that ROCK inhibition, when combined with chemotherapies, targeted therapies, and immunotherapies, leads to enhanced responses [113,114]. The promise of ROCK inhibitors lies in their ability to modulate the tumor microenvironment, improve drug delivery, and sensitize cancer cells to apoptosis, which preclinical models have shown to be effective in overcoming resistance mechanisms [113]. We have recently demonstrated that ROCK inhibition with Fasudil increases membrane tension in cancer cells and facilitates apoptosis by promoting the retention of Fas receptors on the cell surface [12]. This reduction in endocytosis has been observed to retain Fas receptors across multiple cancer cell lines without altering normal cells, enhancing sensitivity to the soluble Fas ligand and inducing cell death in two-dimensional culture, organoids, and in vivo models [12].
A word of caution is warranted when using endocytosis disruptors, whether genetic or pharmaceutical, to study endocytic regulators in cancer. Many small-molecule inhibitors lack specificity, disrupting multiple endocytic pathways [115]. Common strategies like altering membrane lipid composition or receptor distribution impact all endocytic pathways and essential signaling [94,116]. Inhibiting one pathway may up-regulate alternative routes, such as dynamin-independent endocytosis when dynamin is inhibited [117]. Broad dynamin targeting results in poor selectivity and off-target effects [118]. For instance, Dyngo has been shown to inhibit Trop2 endocytosis in prostate cancer cells, which can potentially reduce the effectiveness of Trop2-targeting ADCs [76,119]. Agents targeting membrane cholesterol like statins, while promising in cancer treatment, may interfere with uptake mechanisms and signaling due to altered fluidity, and affect cytoskeleton organization [120]. Methyl-β-cyclodextrin targeting cholesterol-rich lipid rafts is limited by cytotoxicity [121]. Manipulating intracellular cholesterol trafficking has shown efficacy in slowing melanoma growth, but strategies must carefully balance specificity and safety considerations [122].
Nevertheless, despite these challenges, targeting endocytosis to sensitize cancer cells to PCD remains a worthwhile endeavor in cancer therapy. Combining endocytic inhibitors with therapies such as ADCs and monoclonal antibodies holds promise for enhancing treatment efficacy while reducing off-target toxicity [85,94,123]. Temporary inhibition of CME can prevent the internalization of ADCs, increasing their retention on the cell surface, which in turn enhances antibody-dependent cellular cytotoxicity [90]. When this endocytosis inhibition is lifted, it has been shown that the ADC payload is then delivered to the endosomes in ex vivo tumor samples, enhancing its effectiveness while minimizing adverse effects on normal tissues [124]. By using endocytic inhibitors that offer transient and reversible inhibition, such as the dopamine receptor inhibitor prochloroperazine [82,123], systemic effects can be mitigated, ensuring the inhibition is cell-specific and temporary.
Disrupting dysregulated endocytic pathways that support tumor growth and survival holds promise for improving treatment outcomes across various cancer types. Endocytic inhibitors, whether administered alone, in combination with ADCs or radioligands, represent a critical strategy in cancer therapy. However, the journey to identifying optimal candidates for clinical use will require extensive research into their effects on the entire metastatic process. Addressing concerns of specificity, dosage, timing, safety, and the relevance of in vivo models is paramount to study and treat dysregulated endocytosis within the tumor microenvironment. As our understanding of the intricate relationship between endocytosis and cancer cell biology continues to evolve, the development of more selective and potent agents targeting these pathways holds the potential to significantly improve cancer treatment outcomes in the future.
Perspectives.
Cancer cells evade PCD through various mechanisms, enabling unchecked survival and proliferation. Endocytosis plays a key role in regulating PCD pathways, providing opportunities to target this process and sensitize cancer cells to cytotoxic agents.
Inhibiting endocytosis can prevent internalization of death receptors, restore immune surveillance, and enhance delivery of therapeutic payloads, rendering cancer cells more susceptible to PCD.
Investigating the intricate relationship between endocytic trafficking and apoptotic signaling pathways is crucial for identifying new targets in cancer therapy. There is a growing demand to explore combined approaches that merge endocytic modulation with chemotherapies, targeted therapies, and immunotherapies to improve anticancer effectiveness.
Funding
E.T.C. was supported by the Ohio State University Molecular Biophysics Training Program (NIH T32 GM118291-04) and the Pelotonia Graduate Scholars Program. C.K. was supported by NSF Faculty Early Career Development Program (award number: 1751113) and NIH R01GM127526. Any opinions and conclusions expressed in this material are those of the authors and do not necessarily reflect those of the Molecular Biophysics Training Program, Pelotonia Fellowship Program, or The Ohio State University.
Abbreviations
- ADC
antibody-drug conjugate
- CME
clathrin-mediated endocytosis
- CTL
cytotoxic T lymphocytes
- DISC
death-inducing signaling complex
- EGFR
epidermal growth factor
- FasL
Fas ligand
- IAP
inhibitor of apoptosis protein
- MHC-I
major histocompatibility complex class I
- NK
natural killer cells
- PCD
programmed cell death
- ROCK
Rho-associated kinases
- TCR
T-cell receptors
- TNFR
tumor necrosis factor receptor
Footnotes
Competing Interests
The authors declare that there are no competing interests associated with the manuscript.
References
- 1.Vasan N, Baselga J and Hyman DM (2019) A view on drug resistance in cancer. Nature 575, 299–309 10.1038/s41586-019-1730-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gottesman MM (2002) Mechanisms of cancer drug resistance. Annu. Rev. Med. 53, 615–627 10.1146/annurev.med.53.082901.103929 [DOI] [PubMed] [Google Scholar]
- 3.Ouyang L, Shi Z, Zhao S, Wang F-T, Zhou T-T, Liu B et al. (2012) Programmed cell death pathways in cancer: a review of apoptosis, autophagy and programmed necrosis. Cell Prolif. 45, 487–498 10.1111/j.1365-2184.2012.00845.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Peng F, Liao M, Qin R, Zhu S, Peng C, Fu L et al. (2022) Regulated cell death (RCD) in cancer: key pathways and targeted therapies. Sig. Transduct Target Ther. 7, 286 10.1038/s41392-022-01110-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yuan J and Ofengeim D (2024) A guide to cell death pathways. Nat. Rev. Mol. Cell Biol. 25, 379–395 10.1038/s41580-023-00689-6 [DOI] [PubMed] [Google Scholar]
- 6.Guo H, Zhou C, Zheng M, Zhang J, Wu H, He Q et al. (2024) Insights into the role of derailed endocytic trafficking pathway in cancer: from the perspective of cancer hallmarks. Pharmacol. Res. 201, 107084 10.1016/j.phrs.2024.107084 [DOI] [PubMed] [Google Scholar]
- 7.Khan I and Steeg PS (2021) Endocytosis: a pivotal pathway for regulating metastasis. Br. J. Cancer 124, 66–75 10.1038/s41416-020-01179-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pathak C, Vaidya FU, Waghela BN, Jaiswara PK, Gupta VK, Kumar A et al. (2023) Insights of endocytosis signaling in health and disease. Int. J. Mol. Sci. 24, 2971 10.3390/ijms24032971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mellman I and Yarden Y (2013) Endocytosis and cancer. Cold Spring Harb. Perspect. Biol. 5, a016949 10.1101/cshperspect.a016949 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yarwood R, Hellicar J, Woodman PG and Lowe M (2020) Membrane trafficking in health and disease. Dis. Model. Mech. 13, dmm043448 10.1242/dmm.043448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Witsch E, Sela M and Yarden Y (2010) Roles for growth factors in cancer progression. Physiology 25, 85–101 10.1152/physiol.00045.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kural MH, Djakbarova U, Cakir B, Tanaka Y, Chan ET, Arteaga Muniz VI et al. (2024) Mechano-inhibition of endocytosis sensitizes cancer cells to Fas-induced Apoptosis. Cell Death Dis. 15, 440 10.1038/s41419-024-06822-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bareford LM and Swaan PW (2007) Endocytic mechanisms for targeted drug delivery. Adv. Drug Deliv. Rev. 59, 748–758 10.1016/j.addr.2007.06.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kastan MB and Bartek J (2004) Cell-cycle checkpoints and cancer. Nature 432, 316–323 10.1038/nature03097 [DOI] [PubMed] [Google Scholar]
- 15.Evan GI and Vousden KH (2001) Proliferation, cell cycle and apoptosis in cancer. Nature 411, 342–348 10.1038/35077213 [DOI] [PubMed] [Google Scholar]
- 16.Hanahan D and Weinberg RA (2011) Hallmarks of cancer: the next generation. Cell 144, 646–674 10.1016/j.cell.2011.02.013 [DOI] [PubMed] [Google Scholar]
- 17.Galluzzi L, Vitale I, Aaronson SA, Abrams JM, Adam D, Agostinis P et al. (2018) Molecular mechanisms of cell death: recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 25, 486–541 10.1038/s41418-017-0012-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lossi L (2022) The concept of intrinsic versus extrinsic apoptosis. Biochem. J. 479, 357–384 10.1042/BCJ20210854 [DOI] [PubMed] [Google Scholar]
- 19.Fulda S and Debatin K-M (2006) Extrinsic versus intrinsic apoptosis pathways in anticancer chemotherapy. Oncogene 25, 4798–4811 10.1038/sj.onc.1209608 [DOI] [PubMed] [Google Scholar]
- 20.Tait SWG and Green DR (2010) Mitochondria and cell death: outer membrane permeabilization and beyond. Nat. Rev. Mol. Cell Biol. 11, 621–632 10.1038/nrm2952 [DOI] [PubMed] [Google Scholar]
- 21.Nagata S (1997) Apoptosis by death factor. Cell 88, 355–365 10.1016/S0092-8674(00)81874-7 [DOI] [PubMed] [Google Scholar]
- 22.Lee K, Feig C, Tchikov V, Schickel R, Hallas C, Schütze S et al. (2006) The role of receptor internalization in CD95 signaling. EMBO J. 25, 1009–1023 10.1038/sj.emboj.7601016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Peter ME and Krammer PH (2003) The CD95(APO-1/Fas) DISC and beyond. Cell Death Differ. 10, 26–35 10.1038/sj.cdd.4401186 [DOI] [PubMed] [Google Scholar]
- 24.Li H, Zhu H, Xu C and Yuan J (1998) Cleavage of BID by caspase 8 mediates the mitochondrial damage in the Fas pathway of apoptosis. Cell 94, 491–501 10.1016/S0092-8674(00)81590-1 [DOI] [PubMed] [Google Scholar]
- 25.Eum K-H and Lee M (2011) Crosstalk between autophagy and apoptosis in the regulation of paclitaxel-induced cell death in v-Ha-ras-transformed fibroblasts. Mol. Cell. Biochem. 348, 61–68 10.1007/s11010-010-0638-8 [DOI] [PubMed] [Google Scholar]
- 26.Dalby KN, Tekedereli I, Lopez-Berestein G and Ozpolat B (2010) Targeting the prodeath and prosurvival functions of autophagy as novel therapeutic strategies in cancer. Autophagy 6, 322–329. 10.4161/auto.6.3.11625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li X, He S and Ma B (2020) Autophagy and autophagy-related proteins in cancer. Mol. Cancer 19, 12 10.1186/s12943-020-1138-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Debnath J, Gammoh N and Ryan KM (2023) Autophagy and autophagy-related pathways in cancer. Nat. Rev. Mol. Cell Biol. 24, 560–575 10.1038/s41580-023-00585-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Syntichaki P and Tavernarakis N (2002) Death by necrosis. EMBO Rep. 3, 604–609 10.1093/embo-reports/kvf138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tang R, Xu J, Zhang B, Liu J, Liang C, Hua J et al. (2020) Ferroptosis, necroptosis, and pyroptosis in anticancer immunity. J. Hematol. Oncol. 13, 110 10.1186/s13045-020-00946-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Karsch-Bluman A, Feiglin A, Arbib E, Stern T, Shoval H, Schwob O et al. (2019) Tissue necrosis and its role in cancer progression. Oncogene 38, 1920–1935 10.1038/s41388-018-0555-y [DOI] [PubMed] [Google Scholar]
- 32.Degterev A, Huang Z, Boyce M, Li Y, Jagtap P, Mizushima N et al. (2005) Chemical inhibitor of nonapoptotic cell death with therapeutic potential for ischemic brain injury. Nat. Chem. Biol. 1, 112–119 10.1038/nchembio711 [DOI] [PubMed] [Google Scholar]
- 33.Yao X, Li W, Fang D, Xiao C, Wu X, Li M et al. (2021) Emerging roles of energy metabolism in ferroptosis regulation of tumor cells. Adv. Sci. 8, 2100997 10.1002/advs.202100997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yee PP and Li W (2021) Tumor necrosis: a synergistic consequence of metabolic stress and inflammation. Bioessays 43, e2100029 10.1002/bies.202100029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lieberman J (2003) The ABCs of granule-mediated cytotoxicity: new weapons in the arsenal. Nat. Rev. Immunol. 3, 361–370 10.1038/nri1083 [DOI] [PubMed] [Google Scholar]
- 36.Tuomela K, Ambrose AR and Davis DM (2022) Escaping death: how cancer cells and infected cells resist cell-mediated cytotoxicity. Front. Immunol. 13, 867098 10.3389/fimmu.2022.867098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Prager I and Watzl C (2019) Mechanisms of natural killer cell-mediated cellular cytotoxicity. J. Leukoc. Biol. 105, 1319–1329 10.1002/JLB.MR0718-269R [DOI] [PubMed] [Google Scholar]
- 38.Baginska J, Viry E, Berchem G, Poli A, Noman MZ, van Moer K et al. (2013) Granzyme B degradation by autophagy decreases tumor cell susceptibility to natural killer-mediated lysis under hypoxia. Proc. Natl Acad. Sci. U.S.A. 110, 17450–17455 10.1073/pnas.1304790110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Noman MZ, Janji B, Kaminska B, Van Moer K, Pierson S, Przanowski P et al. (2011) Blocking hypoxia-induced autophagy in tumors restores cytotoxic T-cell activity and promotes regression. Cancer Res. 71, 5976–5986 10.1158/0008-5472.CAN-11-1094 [DOI] [PubMed] [Google Scholar]
- 40.Rudd-Schmidt JA, Hodel AW, Noori T, Lopez JA, Cho H-J, Verschoor S et al. (2019) Lipid order and charge protect killer T cells from accidental death. Nat. Commun. 10, 5396 10.1038/s41467-019-13385-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Antia R, Schlegel RA and Williamson P (1992) Binding of perforin to membranes is sensitive to lipid spacing and not headgroup. Immunol. Lett. 32, 153–157 10.1016/0165-2478(92)90108-Z [DOI] [PubMed] [Google Scholar]
- 42.Li Y and Orange JS (2021) Degranulation enhances presynaptic membrane packing, which protects NK cells from perforin-mediated autolysis. PLoS Biol. 19, e3001328 10.1371/journal.pbio.3001328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Liu Y, Zhang T, Zhang H, Li J, Zhou N, Fiskesund R et al. (2021) Cell softness prevents cytolytic T-cell killing of tumor-repopulating cells. Cancer Res. 81, 476–488 10.1158/0008-5472.CAN-20-2569 [DOI] [PubMed] [Google Scholar]
- 44.Halle S, Halle O and Förster R (2017) Mechanisms and dynamics of T cell-mediated cytotoxicity in vivo. Trends Immunol. 38, 432–443 10.1016/j.it.2017.04.002 [DOI] [PubMed] [Google Scholar]
- 45.Peter ME, Hadji A, Murmann AE, Brockway S, Putzbach W, Pattanayak A et al. (2015) The role of CD95 and CD95 ligand in cancer. Cell Death Differ. 22, 549–559 10.1038/cdd.2015.3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kischkel FC, Hellbardt S, Behrmann I, Germer M, Pawlita M, Krammer PH et al. (1995) Cytotoxicity-dependent APO-1 (Fas/CD95)-associated proteins form a death-inducing signaling complex (DISC) with the receptor. EMBO J. 14, 5579–5588 10.1002/j.1460-2075.1995.tb00245.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Algeciras-Schimnich A, Pietras EM, Barnhart BC, Legembre P, Vijayan S, Holbeck SL et al. (2003) Two CD95 tumor classes with different sensitivities to antitumor drugs. Proc. Natl Acad. Sci. U.S.A. 100, 11445–11450 10.1073/pnas.2034995100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Irmler M, Thome M, Hahne M, Schneider P, Hofmann K, Steiner V et al. (1997) Inhibition of death receptor signals by cellular FLIP. Nature 388, 190–195 10.1038/40657 [DOI] [PubMed] [Google Scholar]
- 49.Igney FH and Krammer PH (2002) Death and anti-death: tumour resistance to apoptosis. Nat. Rev. Cancer 2, 277–288 10.1038/nrc776 [DOI] [PubMed] [Google Scholar]
- 50.Green DR, Droin N and Pinkoski M (2003) Activation-induced cell death in T cells. Immunol. Rev. 193, 70–81 10.1034/j.1600-065X.2003.00051.x [DOI] [PubMed] [Google Scholar]
- 51.Bonfoco E, Stuart PM, Brunner T, Lin T, Griffith TS, Gao Y et al. (1998) Inducible nonlymphoid expression of Fas ligand is responsible for superantigen-induced peripheral deletion of T cells. Immunity 9, 711–720 10.1016/S1074-7613(00)80668-8 [DOI] [PubMed] [Google Scholar]
- 52.Majedi FS, Hasani-Sadrabadi MM, Thauland TJ, Li S, Bouchard L-S and Butte MJ (2020) T-cell activation is modulated by the 3D mechanical microenvironment. Biomaterials 252, 120058 10.1016/j.biomaterials.2020.120058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Saitakis M, Dogniaux S, Goudot C, Bufi N, Asnacios S, Maurin M et al. (2017) Different TCR-induced T lymphocyte responses are potentiated by stiffness with variable sensitivity. eLife 6, e23190 10.7554/eLife.23190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kaksonen M and Roux A (2018) Mechanisms of clathrin-mediated endocytosis. Nat. Rev. Mol. Cell Biol. 19, 313–326 10.1038/nrm.2017.132 [DOI] [PubMed] [Google Scholar]
- 55.Cullen PJ and Steinberg F (2018) To degrade or not to degrade: mechanisms and significance of endocytic recycling. Nat. Rev. Mol. Cell Biol. 19, 679–696 10.1038/s41580-018-0053-7 [DOI] [PubMed] [Google Scholar]
- 56.Di Fiore PP and von Zastrow M (2014) Endocytosis, signaling, and beyond. Cold Spring Harb. Perspect. Biol. 6, a016865 10.1101/cshperspect.a016865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Adler M, Mayo A, Zhou X, Franklin RA, Jacox JB, Medzhitov R et al. (2018) Endocytosis as a stabilizing mechanism for tissue homeostasis. Proc. Natl Acad. Sci. U.S.A. 115, E1926–E1935 10.1073/pnas.1714377115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Boulant S, Kural C, Zeeh J-C, Ubelmann F and Kirchhausen T (2011) Actin dynamics counteract membrane tension during clathrin-mediated endocytosis. Nat. Cell Biol. 13, 1124–1131 10.1038/ncb2307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Djakbarova U, Madraki Y, Chan ET and Kural C (2021) Dynamic interplay between cell membrane tension and clathrin-mediated endocytosis. Biol. Cell 113, 344–373 10.1111/boc.202000110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Casares D, Escribá PV and Rosselló CA (2019) Membrane lipid composition: effect on membrane and organelle structure, function and compartmentalization and therapeutic avenues. Int. J. Mol. Sci. 20, 2167 10.3390/ijms20092167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Joseph JG and Liu AP (2020) Mechanical regulation of endocytosis: new insights and recent advances. Adv. Biosyst. 4, 1900278 10.1002/adbi.201900278 [DOI] [PubMed] [Google Scholar]
- 62.Pelkmans L and Helenius A (2002) Endocytosis via caveolae. Traffic 3, 311–320 10.1034/j.1600-0854.2002.30501.x [DOI] [PubMed] [Google Scholar]
- 63.Floyd S and De Camilli P (1998) Endocytosis proteins and cancer: a potential link? Trends Cell Biol. 8, 299–301 10.1016/S0962-8924(98)01316-6 [DOI] [PubMed] [Google Scholar]
- 64.HuangFu W-C and Fuchs SY (2010) Ubiquitination-dependent regulation of signaling receptors in cancer. Genes Cancer 1, 725–734 10.1177/1947601910382901 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Haglund K and Dikic I (2012) The role of ubiquitylation in receptor endocytosis and endosomal sorting. J. Cell Sci. 125, 265–275 10.1242/jcs.091280 [DOI] [PubMed] [Google Scholar]
- 66.Tice DA, Biscardi JS, Nickles AL and Parsons SJ (1999) Mechanism of biological synergy between cellular Src and epidermal growth factor receptor. Proc. Natl Acad. Sci. U.S.A. 96, 1415–1420. 10.1073/pnas.96.4.1415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Summy JM and Gallick GE (2003) Src family kinases in tumor progression and metastasis. Cancer Metastasis Rev. 22, 337–358 10.1023/a:1023772912750 [DOI] [PubMed] [Google Scholar]
- 68.Katz M, Shtiegman K, Tal-Or P, Yakir L, Mosesson Y, Harari D et al. (2002) Ligand-independent degradation of epidermal growth factor receptor involves receptor ubiquitylation and Hgs, an adaptor whose ubiquitin-interacting motif targets ubiquitylation by Nedd4. Traffic 3, 740–751 10.1034/j.1600-0854.2002.31006.x [DOI] [PubMed] [Google Scholar]
- 69.Sharma S, Carmona A, Skowronek A, Yu F, Collins MO, Naik S et al. (2019) Apoptotic signalling targets the post-endocytic sorting machinery of the death receptor Fas/CD95. Nat. Commun. 10, 3105 10.1038/s41467-019-11025-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Bennett M, Macdonald K, Chan S-W, Luzio JP, Simari R and Weissberg P (1998) Cell surface trafficking of Fas: a rapid mechanism of p53-mediated apoptosis. Science 282, 290–293 10.1126/science.282.5387.290 [DOI] [PubMed] [Google Scholar]
- 71.Popgeorgiev N, Jabbour L and Gillet G (2018) Subcellular localization and dynamics of the Bcl-2 family of proteins. Front. Cell Dev. Biol. 6, 13 10.3389/fcell.2018.00013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Zhang B, Li J, Hua Q, Wang H, Xu G, Chen J et al. (2023) Tumor CEMIP drives immune evasion of colorectal cancer via MHC-I internalization and degradation. J. Immunother. Cancer 11, e005592 10.1136/jitc-2022-005592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Ravichandran KS (2011) Beginnings of a good apoptotic meal: the find-me and eat-me signaling pathways. Immunity 35, 445–455 10.1016/j.immuni.2011.09.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Khalaji A, Yancheshmeh FB, Farham F, Khorram A, Sheshbolouki S, Zokaei M et al. (2023) Don’t eat me/eat me signals as a novel strategy in cancer immunotherapy. Heliyon 9, e20507 10.1016/j.heliyon.2023.e20507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Veselov VV, Nosyrev AE, Jicsinszky L, Alyautdin RN and Cravotto G (2022) Targeted delivery methods for anticancer drugs. Cancers 14, 622 10.3390/cancers14030622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Hammood M, Craig AW and Leyton JV (2021) Impact of endocytosis mechanisms for the receptors targeted by the currently approved antibody-drug conjugates (ADCs)—a necessity for future ADC research and development. Pharmaceuticals 14, 674 10.3390/ph14070674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Trochet D and Bitoun M (2021) A review of Dynamin 2 involvement in cancers highlights a promising therapeutic target. J. Exp. Clin. Cancer Res. 40, 238 10.1186/s13046-021-02045-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Schmid SL (2017) Reciprocal regulation of signaling and endocytosis: implications for the evolving cancer cell. J. Cell Biol. 216, 2623–2632 10.1083/jcb.201705017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Reis CR, Chen P-H, Bendris N and Schmid SL (2017) TRAIL-death receptor endocytosis and apoptosis are selectively regulated by dynamin-1 activation. Proc. Natl Acad. Sci. U.S.A. 114, 504–509 10.1073/pnas.1615072114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.McCluskey A, Daniel JA, Hadzic G, Chau N, Clayton EL, Mariana A et al. (2013) Building a better dynasore: the dyngo compounds potently inhibit dynamin and endocytosis. Traffic 14, 1272–1289 10.1111/tra.12119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Preta G, Cronin JG and Sheldon IM (2015) Dynasore - not just a dynamin inhibitor. Cell Commun. Signal. 13, 24 10.1186/s12964-015-0102-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Daniel JA, Chau N, Abdel-Hamid MK, Hu L, von Kleist L, Whiting A et al. (2015) Phenothiazine-derived antipsychotic drugs inhibit dynamin and clathrin-mediated endocytosis. Traffic 16, 635–654 10.1111/tra.12272 [DOI] [PubMed] [Google Scholar]
- 83.Joshi S, Braithwaite AW, Robinson PJ and Chircop M (2011) Dynamin inhibitors induce caspase-mediated apoptosis following cytokinesis failure in human cancer cells and this is blocked by Bcl-2 overexpression. Mol. Cancer 10, 78 10.1186/1476-4598-10-78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.von Beek C, Alriksson L, Palle J, Gustafson A-M, Grujic M, Melo FR et al. (2021) Dynamin inhibition causes context-dependent cell death of leukemia and lymphoma cells. PLoS One 16, e0256708 10.1371/journal.pone.0256708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Tremblay CS, Chiu SK, Saw J, McCalmont H, Litalien V, Boyle J et al. (2020) Small molecule inhibition of Dynamin-dependent endocytosis targets multiple niche signals and impairs leukemia stem cells. Nat. Commun. 11, 6211 10.1038/s41467-020-20091-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.von Kleist L, Stahlschmidt W, Bulut H, Gromova K, Puchkov D, Robertson MJ et al. (2011) Role of the clathrin terminal domain in regulating coated pit dynamics revealed by small molecule inhibition. Cell 146, 471–484 10.1016/j.cell.2011.06.025 [DOI] [PubMed] [Google Scholar]
- 87.Willox AK, Sahraoui YME and Royle SJ (2014) Non-specificity of Pitstop 2 in clathrin-mediated endocytosis. Biol. Open 3, 326–331 10.1242/bio.20147955 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Smith CM, Haucke V, McCluskey A, Robinson PJ and Chircop M (2013) Inhibition of clathrin by pitstop 2 activates the spindle assembly checkpoint and induces cell death in dividing HeLa cancer cells. Mol. Cancer 12, 4 10.1186/1476-4598-12-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Dutta D, Williamson CD, Cole NB and Donaldson JG (2012) Pitstop 2 is a potent inhibitor of clathrin-independent endocytosis. PLoS ONE 7, e45799 10.1371/journal.pone.0045799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Banushi B, Joseph SR, Lum B, Lee JJ and Simpson F (2023) Endocytosis in cancer and cancer therapy. Nat. Rev. Cancer 23, 450–473 10.1038/s41568-023-00574-6 [DOI] [PubMed] [Google Scholar]
- 91.Liashkovich I, Stefanello ST, Vidyadharan R, Haufe G, Erofeev A, Gorelkin PV et al. (2023) Pitstop-2 and its novel derivative RVD-127 disrupt global cell dynamics and nuclear pores integrity by direct interaction with small GTPases. Bioeng. Transl. Med. 8, e10425 10.1002/btm2.10425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.von Gersdorff K, Sanders NN, Vandenbroucke R, Smedt SCD, Wagner E and Ogris M (2006) The internalization route resulting in successful gene expression depends on both cell line and polyethylenimine polyplex type. Mol. Ther. 14, 745–753 10.1016/j.ymthe.2006.07.006 [DOI] [PubMed] [Google Scholar]
- 93.Kim B, Park YS, Sung JS, Lee JW, Lee SB and Kim YH (2021) Clathrin-mediated EGFR endocytosis as a potential therapeutic strategy for overcoming primary resistance of EGFR TKI in wild-type EGFR non-small cell lung cancer. Cancer Med. 10, 372–385 10.1002/cam4.3635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Pereira PMR, Mandleywala K, Ragupathi A and Lewis JS (2020) Acute statin treatment improves antibody accumulation in EGFR- and PSMA-expressing tumors. Clin. Cancer Res. 26, 6215–6229 10.1158/1078-0432.CCR-20-1960 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Ferguson JP, Huber SD, Willy NM, Aygün E, Goker S, Atabey T et al. (2017) Mechanoregulation of clathrin-mediated endocytosis. J. Cell Sci. 130, 3631–3636 10.1242/jcs.205930 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Willy NM, Ferguson JP, Huber SD, Heidotting SP, Aygün E, Wurm SA et al. (2017) Membrane mechanics govern spatiotemporal heterogeneity of endocytic clathrin coat dynamics. Mol. Biol. Cell 28, 3480–3488 10.1091/mbc.E17-05-0282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Akatay AA, Wu T, Djakbarova U, Thompson C, Cocucci E, Zandi R et al. (2022) Endocytosis at extremes: formation and internalization of giant clathrin-coated pits under elevated membrane tension. Front. Mol. Biosci. 9, 959737 10.3389/fmolb.2022.959737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Alves AC, Ribeiro D, Nunes C and Reis S (2016) Biophysics in cancer: the relevance of drug-membrane interaction studies. Biochim. Biophys. Acta 1858, 2231–2244 10.1016/j.bbamem.2016.06.025 [DOI] [PubMed] [Google Scholar]
- 99.Szlasa W, Zendran I, Zalesińska A, Tarek M and Kulbacka J (2020) Lipid composition of the cancer cell membrane. J. Bioenerg. Biomembr. 52, 321–342 10.1007/s10863-020-09846-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Zheng C, Yan S, Lu L, Yao H, He G, Chen S et al. (2021) Lovastatin inhibits EMT and metastasis of triple-Negative breast cancer stem cells through dysregulation of cytoskeleton-associated proteins. Front. Oncol. 11, 656687 10.3389/fonc.2021.656687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Amadasu E, Kang R, Usmani A and Borlongan CV (2022) Effects of lovastatin on brain cancer cells. Cell Transplant 31, 09636897221102903 10.1177/09636897221102903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Mangelinck A, Habel N, Mohr A, Gaspar N, Stefanovska B and Fromigué O (2021) Synergistic anti-tumor effect of simvastatin combined to chemotherapy in osteosarcoma. Cancers 13, 5869 10.3390/cancers13225869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Beckwitt CH, Brufsky A, Oltvai ZN and Wells A (2018) Statin drugs to reduce breast cancer recurrence and mortality. Breast Cancer Res. 20, 144 10.1186/s13058-018-1066-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Duarte JA, de Barros ALB and Leite EA (2021) The potential use of simvastatin for cancer treatment: a review. Biomed. Pharmacother. 141, 111858 10.1016/j.biopha.2021.111858 [DOI] [PubMed] [Google Scholar]
- 105.Fuhs T, Wetzel F, Fritsch AW, Li X, Stange R, Pawlizak S et al. (2022) Rigid tumours contain soft cancer cells. Nat. Phys. 18, 1510–1519 10.1038/s41567-022-01755-0 [DOI] [Google Scholar]
- 106.Alibert C, Goud B and Manneville J-B (2017) Are cancer cells really softer than normal cells? Biol. Cell 109, 167–189 10.1111/boc.201600078 [DOI] [PubMed] [Google Scholar]
- 107.Händel C, Schmidt BUS, Schiller J, Dietrich U, Möhn T, Kießling TR et al. (2015) Cell membrane softening in human breast and cervical cancer cells. New J. Phys. 17, 083008 10.1088/1367-2630/17/8/083008 [DOI] [Google Scholar]
- 108.Feng J, Ito M, Ichikawa K, Isaka N, Nishikawa M, Hartshorne DJ et al. (1999) Inhibitory phosphorylation site for Rho-associated kinase on smooth muscle myosin phosphatase. J. Biol. Chem. 274, 37385–37390 10.1074/jbc.274.52.37385 [DOI] [PubMed] [Google Scholar]
- 109.Olson MF and Sahai E (2009) The actin cytoskeleton in cancer cell motility. Clin. Exp. Metastasis 26, 273–287 10.1007/s10585-008-9174-2 [DOI] [PubMed] [Google Scholar]
- 110.Ridley AJ (2001) Rho GTPases and cell migration. J. Cell Sci. 114, 2713–2722 10.1242/jcs.114.15.2713 [DOI] [PubMed] [Google Scholar]
- 111.Kümper S, Mardakheh FK, McCarthy A, Yeo M, Stamp GW, Paul A et al. (2016) Rho-associated kinase (ROCK) function is essential for cell cycle progression, senescence and tumorigenesis. eLife 5, e12203 10.7554/eLife.12203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Feng Y, LoGrasso PV, Defert O and Li R (2016) Rho kinase (ROCK) inhibitors and their therapeutic potential. J. Med. Chem. 59, 2269–2300 10.1021/acs.jmedchem.5b00683 [DOI] [PubMed] [Google Scholar]
- 113.Barcelo J, Samain R and Sanz-Moreno V (2023) Preclinical to clinical utility of ROCK inhibitors in cancer. Trends Cancer 9, 250–263 10.1016/j.trecan.2022.12.001 [DOI] [PubMed] [Google Scholar]
- 114.Whatcott CJ, Ng S, Barrett MT, Hostetter G, Hoff DDV and Han H (2017) Inhibition of ROCK1 kinase modulates both tumor cells and stromal fibroblasts in pancreatic cancer. PLoS ONE 12, e0183871 10.1371/journal.pone.0183871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Rennick JJ, Johnston APR and Parton RG (2021) Key principles and methods for studying the endocytosis of biological and nanoparticle therapeutics. Nat. Nanotechnol. 16, 266–276 10.1038/s41565-021-00858-8 [DOI] [PubMed] [Google Scholar]
- 116.Yue H-Y and Xu J (2015) Cholesterol regulates multiple forms of vesicle endocytosis at a mammalian central synapse. J. Neurochem. 134, 247–260 10.1111/jnc.13129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Damke H, Baba T, van der Bliek AM and Schmid SL (1995) Clathrin-independent pinocytosis is induced in cells overexpressing a temperature-sensitive mutant of dynamin. J. Cell Biol. 131, 69–80 10.1083/jcb.131.1.69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Sasso L, Purdie L, Grabowska A, Jones AT and Alexander C (2018) Time and cell-dependent effects of endocytosis inhibitors on the internalization of biomolecule markers and nanomaterials. J. Interdiscip. Nanomed. 3, 67–81 10.1002/jin2.39 [DOI] [Google Scholar]
- 119.Wanger TM, Dewitt S, Collins A, Maitland NJ, Poghosyan Z and Knäuper V (2015) Differential regulation of TROP2 release by PKC isoforms through vesicles and ADAM17. Cell. Signal. 27, 1325–1335 10.1016/j.cellsig.2015.03.017 [DOI] [PubMed] [Google Scholar]
- 120.Tatè R, Zona E, De Cicco R, Trotta V, Urciuoli M, Morelli A et al. (2017) Simvastatin inhibits the expression of stemness-related genes and the metastatic invasion of human cancer cells via destruction of the cytoskeleton. Int. J. Oncol. 51, 1851–1859 10.3892/ijo.2017.4158 [DOI] [PubMed] [Google Scholar]
- 121.Hao M, Mukherjee S, Sun Y and Maxfield FR (2004) Effects of cholesterol depletion and increased lipid unsaturation on the properties of endocytic membranes. J. Biol. Chem. 279, 14171–14178 10.1074/jbc.M309793200 [DOI] [PubMed] [Google Scholar]
- 122.Kuzu OF, Gowda R, Sharma A and Robertson GP (2014) Leelamine mediates cancer cell death through inhibition of intracellular cholesterol transport. Mol. Cancer Ther. 13, 1690–1703 10.1158/1535-7163.MCT-13-0868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Chew HY, De Lima PO, Gonzalez Cruz JL, Banushi B, Echejoh G, Hu L et al. (2020) Endocytosis inhibition in humans to improve responses to ADCC-mediating antibodies. Cell 180, 895–914.e27 10.1016/j.cell.2020.02.019 [DOI] [PubMed] [Google Scholar]
- 124.Joseph SR, Gaffney D, Barry R, Hu L, Banushi B, Wells JW et al. (2019) An ex vivo human tumor assay shows distinct patterns of EGFR trafficking in squamous cell carcinoma correlating to therapeutic outcomes. J. Invest. Dermatol. 139, 213–223 10.1016/j.jid.2018.06.190 [DOI] [PubMed] [Google Scholar]