

Abstract
Reverse transcriptase (RT) with its associated RNase H (RH) domain and integrase (IN) are key enzymes encoded by retroviruses and retrotransposons. Several studies have implied a functional role of the interaction between IN and RT during the replication of retroviral and retrotransposon genomes. In this study, IN deletion mutants were used to investigate the role of IN on the RT activity of the yeast Saccharomyces cerevisiae retrotransposon Ty1. We have identified two domains of Ty1 integrase which have effects on RT activity in vivo. The deletion of a domain spanning amino acid residues 233 to 520 of IN increases the exogenous specific activity of RT up to 20-fold, whereas the removal of a region rich in acidic amino acid residues between residues 521 and 607 decreases its activity. The last result complements our observation that an active recombinant RT protein can be obtained if a small acidic tail mimicking the acidic domain of IN is fused to the RT-RH domain. We suggest that interaction between these acidic amino acid residues of IN and a basic region of RT could be critical for the correct folding of RT and for the formation of an active conformation of the enzyme.
Reverse transcriptase (RT) and integrase (IN) play a central role in the replication and integration of retroviral and retrotransposon genomes into the host cell chromosomes. Several studies suggest that functional interactions between these two enzymes occur during replication. For the Saccharomyces cerevisiae retrotransposon Ty3, mutations in the nonconserved amino- and carboxy-terminal domains of IN affect multiple stages of the retrotransposon's life cycle, including RT and IN expression, 3′-end processing of the cDNA, and the amount of DNA associated with virus-like particles (VLPs) (15, 26, 27). These results, coupled with the observation that two forms of Ty3 RT (the mature 55-kDa RT species and a 115-kDa RT-IN fusion protein) were detected by immunoblot analyses (7, 14), imply a functional role of the interaction between IN and RT. Moreover, mutations of Ty3 IN that caused a reduced accumulation of full-length cDNA in VLPs could be rescued only by an RT-IN protein (expressed as a capsid [CA]-IN-RT fusion protein) delivered in trans, but not by a CA-IN protein; this observation supports a model where RT and IN domains function closely or even in cis during Ty3 replication (27). A model where IN and RT are closely associated and might be components of a Ty3 RT/RT-IN heterodimer has been proposed. Precedents of an association between RT and IN proteins exist in retroviral systems (4, 8, 9, 11, 20, 28, 29, 31, 33, 39, 41, 42). In avian leukosis retroviruses, where this association has been well studied, IN is an integral part of an α-β heterodimeric form of RT, where α is the RT protein and β is an RT-IN fusion protein (6, 13, 34, 35). In human T-cell leukemia virus type 1, the RT and RT-IN proteins are likely associated in an oligomeric structure of the α3/β type (32). In human immunodeficiency virus type 1 (HIV-1) and murine retroviruses, RT and IN are fully separated by proteolytic processing during virion maturation. However, an important biological role for IN in the initiation of reverse transcription has been demonstrated for HIV-1. Several mutations of IN displayed an in vivo DNA synthesis defect and a block of viral replication at the level of reverse transcription (18, 39, 42), while other mutations increased virus fitness by augmenting the initiation of reverse transcription (29). Similarly, mutations of Moloney murine leukemia virus IN have been reported to affect cDNA production (17). Wu et al. (39) showed previously that a direct physical interaction between the IN and RT of HIV-1 could explain the role of IN in the increase of HIV-1 DNA synthesis. Pulldown assays with antibodies generated against RT or IN have also demonstrated that the HIV-1 or murine leukemia virus proteins interact physically in vitro (9, 11, 31). Furthermore, biochemical analyses showing that HIV-1 RT and IN inhibit each other suggest a regulatory role for the interaction between these two proteins (28, 31). More recently, Zhu et al. (42) have found that a C-to-S substitution at position 130 in HIV-1 IN conferred an inability to initiate reverse transcription and that IN with the C130S substitution failed to interact with RT, resulting in a defect of reverse transcription. Hehl et al. (8) have mapped the domains of interaction on both protein partners and shown that both monomeric and heterodimeric forms of HIV-1 RT can interact with IN.
In the yeast retrotransposon Ty1, IN and RT are expressed and assembled in the VLPs as part of a large Gag-Pol-p199 precursor protein (Fig. 1) (1, 5, 19, 22, 23, 25, 40). After the assembly of VLPs, the precursor is processed by the pol-encoded protease to liberate the mature Gag-p45, PR-p20, IN-p71, and RT-RNase H (RH)-p63 proteins. In protease-deficient VLPs, Ty1 RT is functionally active as part of the Gag-Pol-p199 precursor (40). By systematically mutagenizing the cleavage sites between the protein domains of the Ty1 Gag-Pol precursor, Merkulov et al. (22) have shown that PR-IN-RT and IN-RT fusion proteins make amounts of Ty1 cDNA similar to the amounts produced by wild-type (WT) proteins. This indicates that Ty1 RT retains its full activity in vivo when it is fused to the IN domain. These results and the observation that an active Ty1 recombinant protein can be obtained only after including amino acid residues encoded by the IN gene (37) suggest that interactions between IN and RT might also be important for the function of Ty1 RT.
FIG. 1.

(A) Schematic representation of the Ty1 genome, mRNA, and proteins. The portions of the Gag-Pol-p199 polyprotein containing the GAG protein, protease (PR), IN, and RT-RH are indicated. LTR, long terminal repeat; PBS, primer binding site. (B) Schematic representation of the WT and deletion mutant proteins. The end points of each deletion are indicated by vertical dotted lines at positions 2173, 2737, 3600, and 3855. These numbers are the Ty1-H3 positions (2) where SacI restriction sites were inserted to allow the construction of the deletion mutants; they correspond to amino acid residues 45, 233, 521, and 605 of the IN protein, which contains 635 amino acids.
In the present study, IN deletion mutants were used to investigate the role of IN on the activity of RT in Ty1 VLPs. Ty1 IN, a 71-kDa protein containing 635 amino acid residues, has two phylogenetically conserved regions. The N-terminal region contains a conserved zinc binding sequence, HHCC, which is likely involved in substrate binding. The central core domain contains the conserved catalytic DDE motif required to bind divalent metal ions and to perform the DNA cleavage and joining reactions during transposition. The C-terminal region of Ty1 IN is not as well conserved as and is larger than the C-terminal region of retroviral RTs, and its function is not well understood. The IN deletion mutants studied here were able to synthesize minus-strand strong-stop DNA (−sssDNA) and showed RNase H activity. However, they failed to perform the first-strand transfer or to synthesize plus-strand DNA. Moreover, the exogenous polymerase activities as measured by a standard oligo(dG)/poly(rC) primer template assay were very different for WT and mutant VLPs. The deletion of half of the C-terminal region (residues 233 to 520) or a larger deletion including the C-terminal end of the zinc binding sequence, the core domain, and half of the C-terminal region (residues 45 and 520) did not impair the polymerase activity of Ty1 RT. On the contrary, these deletions increased the exogenous polymerase activity of Ty1 RT up to 20-fold. In contrast, the deletion of a domain comprising amino acid residues 521 to 605 strongly decreased the specific activity of RT. A striking feature of this domain is its high content of acidic amino acid residues which might be involved in the formation of a complex between IN and RT through some ionic interactions. Notably, in a previous report (37) we showed that an active recombinant RT could be obtained only if the 115-amino-acid contiguous C-terminal portion of the IN spanning this acidic domain was fused to the RT-RH domain. We have now expressed an active recombinant fusion protein bearing a much shorter IN domain (36 amino acid residues), in which a few acidic residues mimicking the acidic domain of IN are added at its N terminus. This suggests that the acidic amino acid residues in the C-terminal region of IN might be involved in the proper folding of the protein and could play a role in the activity of Ty1 RT.
MATERIALS AND METHODS
Ty1 VLP purification.
Ty1 VLPs were isolated from yeast strain AGY9 (MATa leu2Δ1 ura3-52 trp1Δ63 his4-539 lys2-801 spt3-202 GAL+) (21) transformed with WT plasmid pJEF1105 (3), kindly provided by J. D. Boeke, and with deletion mutant versions of this plasmid. The VLPs were purified using a method described previously by Eichinger and Boeke (3). RT activity was assayed in purified VLP preparations with added exogenous oligo(dG)/poly(rC): 4 μl of purified VLPs was mixed with 16 μl of assay mix and incubated for 60 min at room temperature (22°C to 24°C). The standard assay mix (20 μl) contained final concentrations of 50 mM Tris-HCl (pH 7.8), 40 mM KCl, 20 mM MgCl2, 0.05% NP-40, 1.5 10−2 mM dGTP, 8 mM β-mercaptoethanol, 0.01 U oligo(dG)/poly(rC), and 1 μCi [α-32P]dGTP. The incorporation of 32P-radiolabeled dGTP into high-molecular-weight poly(dG) was determined by scintillation counting. Aliquots of the reaction mixture were spotted onto DE81 filters (Whatman). The filters were washed three times in 5% Na2HPO4 to remove unincorporated [α-32P]dGTP, washed once in deionized water, dried after one ethanol wash, and counted.
Expression and purification of recombinant Ty1 IN-RT fusion proteins.
Recombinant hexahistidine-tagged Ty1 IN-RT-RH fusion proteins were expressed from plasmids p6H3601 and p6HEK3838 as described previously (37). Plasmid p6H3601 (construct 2 in the work of Wilhelm et al. [37]) contained the Ty1 RT-RH domain linked to a 115-amino-acid contiguous C-terminal fragment of Ty1 integrase fused to the N terminus of the RT-RH domain. Six histidine residues were fused at the amino terminus of this IN (115)-RT-RH protein. Plasmid p6HEK3838 contained the Ty1 RT-RH domain plus a 36-amino-acid contiguous C-terminal fragment of Ty1 integrase encoded by Ty1-H3 nucleotides (nt) 3838 to 3945 (for nucleotide numbering, see reference 2) fused to the N terminus of the RT-RH domain. An enterokinase (EK) site, DDDDK, and six histidine residues were fused at the amino terminus of the IN (36)-RT-RH protein. The p6HEK3838 plasmid was constructed as described previously by Wilhelm et al. (37). The IN-RT-RH region spanning nt 3838 to 5562 was amplified by PCR and cloned into the p6H vector. The 3′ amplification primer contained a SalI site (underlined below) and was complementary to the 3′-terminal coding sequence (including the stop codon) of the TyB open reading frame (5′-ACGCGTCGACCTAATGAATCCATTTG-3′). The 5′ amplification primer contained a BamHI site, the coding sequence for the EK site (underlined below), and the sequence beginning at nt 3838 of Ty1-H3 that codes for the N-terminal amino acid residues of the fusion protein (5′-CGCGGATCCGACGACGACGACAAGTTAGAAGATAATGAAACTG-3′).
The recombinant proteins were expressed in Escherichia coli strain M15 containing pREP4 (QIAGEN) and purified by Ni2+-nitriloacetic acid-agarose (QIAGEN) affinity chromatography as described previously (37).
RNA and DNA oligonucleotides and RNA labeling.
The RNA and DNA oligonucleotides were purchased from Thermo Electron (Ulm, Germany). The chemically synthesized RNAs were labeled at their 5′ termini with 32P using [γ-32P]ATP and T4 polynucleotide kinase.
Primer template annealing and RNase H assays.
To prepare the RNA/DNA duplexes, a 3 to 5 molar excess of template DNA was hybridized to the 32P-labeled primer RNA in a buffer containing 50 mM Tris-HCl (pH 7.8), 15 mM NaCl, and 8 mM β-mercaptoethanol. The mixture was heated at 90°C for 1 min and then incubated for 10 min at 70°C, 10 min at 42°C, and 10 min at room temperature. After addition of the Ty1 RT at a final concentration of 200 to 300 nM and at a 2:5 RT-to-primer template molar ratio, the samples were incubated at 23°C for 10 min. RNase H cleavage was initiated with MgCl2 at a final concentration of 20 mM. The reactions were stopped by the addition of formamide sample buffer containing 25 mM EDTA. The products of the reaction were separated on denaturing 20% polyacrylamide gels and visualized by autoradiography.
Western blot analysis.
Immunoblot analyses were performed on the fractions of the VLP sucrose gradients. For detection of RT, 10 μl of each gradient fraction was heated at 70°C in sodium dodecyl sulfate-polyacrylamide gel electrophoresis sample buffer and run onto 3% stacking/10% separating sodium dodecyl sulfate polyacrylamide gels. Proteins were transferred onto a 0.45-μm polyvinylidene difluoride membrane (Immobilon-P; Millipore Corporation) in transfer buffer (25 mM Tris, 192 mM glycine, 10% methanol) at 70 V for 2 h. Membranes were washed once with 25 ml phosphate-buffered saline for 5 min at room temperature and incubated for 1 h in 10 ml blocking buffer (phosphate-buffered saline containing 0.1% Tween 20 and 1% bovine serum albumin) and then for 1 h at 4°C with the Ty1 RT antibody (1/20,000 dilution) in 10 ml blocking buffer. The Ty1 RT antibody was a gift from T. Menees (12). Membranes were washed three times for 5 min each time in 10 ml blocking buffer and incubated for 1 h at room temperature with a 1/10,000 dilution of alkaline phosphatase-linked anti-rabbit immunoglobulin G (New England Biolabs) in 10 ml of blocking buffer. Membranes were washed again three times for 5 min each time in 10 ml blocking buffer and visualized with the Phototope-Star Western Blot detection kit (New England Biolabs).
Plasmid construction and mutagenesis.
Deletion mutants were made in two steps. First, new SacI restriction sites were introduced by site-directed mutagenesis at Ty1-H3 positions 2173, 3600, and 3855 in plasmid pJEF1105 (2). A 3,597-bp BamHI-KpnI fragment from pJEF1105 was subcloned into similarly digested pSL1190 (a superlinker phagemid derived from pUC118, purchased from Amersham) and mutagenized. Site-directed mutagenesis was performed as described by Kunkel (16). Following mutagenesis, the 3,597-bp fragment was cloned back into pJEF1105. The presence of the desired mutations was confirmed by sequencing. The Δ2173-3600 and Δ2173-3855 deletion mutants were then constructed by digesting the plasmids containing two new SacI sites with SacI and by self-ligating the largest fragments. The pJEF Δ2173-3600 plus Hrb1 and pJEF Δ2173-3855 plus Hrb1 plasmids were constructed by digesting the Δ2173-3601 and Δ2173-3855 plasmids with SacI, ligating them with a similarly digested PCR-amplified Hrb1 gene, and subsequently identifying the correctly oriented inserts. The oligonucleotides used for generating the new SacI sites are the following, with the new SacI sites underlined: Sac2173, 5′CTAGACCAGAGCTCATCTGATTCG3′; Sac3600, 5′CGATGAATTGAGCTCCGGTGGAG3′; and Sac3855, 5′GTGATACCTTGAGCTCAGTTTCATTATC3′.
The primers used to amplify the Hrb1 gene were as follows, with SacI sites at the 5′ ends of the primers underlined: 5′Hrb, 5′GTATGAGCTCAGTGATGATCATGGTTATG3′, and 3′Hrb, 5′ATACGAGCTCGA GGCGTTTAGCGTACG3′.
Reiterative primer extension.
To detect −sssDNA and plus-strand strong-stop DNA (+sssDNA) replication intermediates, a reiterative primer extension was carried out with 5′-end-labeled strand-specific oligonucleotide primers (38). Nucleic acids extracted from VLPs were incubated with the labeled primer in a 20-μl volume using the following PCR buffer (10 mM Tris-HCl [pH 8.8], 50 mM KCl, 0.1% Triton X-100, 2 mM MgCl2) along with 2 U of Taq polymerase. Primer extension products were generated by 30 cycles of denaturation (30 s at 95°C), annealing (30 s at 42°C), and extension (60 s at 72°C). At the end of the reaction, 10% formamide was added to the reaction mixture. Products were denatured by being heated at 80°C for 2 min prior to being loaded on an 8% polyacrylamide-8 M urea denaturing gel and then analyzed by phosphorimaging. The primers used to detect +sssDNA and −sssDNA, respectively, were 5′CAATCCTTGCGTTTCAGCTTCCAC3′ (complementary to Ty1-H3 positions 5716 to 5697) and 5′GGAGAACTTCTAGTATATTCTGTATACC3′ (complementary to Ty1-H3 positions 243 to 270 or 5827 to 5854).
RESULTS AND DISCUSSION
Acidic amino acid residues in the C-terminal region of IN have a role in Ty1 RT activity.
To examine the role of IN in the activity of Ty1 RT, we generated IN deletion mutants of the galactose-inducible, plasmid-based Ty1-H3 element. Their structures are shown in Fig. 1. Yeast cells were transformed with these constructs, and VLP formation was induced by growing the strains at 22°C for 2 to 3 days on a selective medium containing galactose. Following induction, VLPs were fractionated on a sucrose step gradient. Immunoblot analyses and RT assays were performed on fractions from the density gradients to measure the activities and amounts of RT in WT and mutant versions of VLPs (Fig. 2). In one mutant, the Δ2173-3855 mutant, the VLP-associated RT was not processed efficiently by the Ty1 protease. Plausible explanations for this incomplete processing could be improper folding of the mutant polyprotein or steric hindrance between the PR-IN and IN-RT cleavage sites, which are brought too close together after deletion of the fragment from nt 2173 to 3855. To test the second hypothesis, we separated the two sites by inserting the yeast gene Hrb1 (10) (a putative RNA binding protein of approximately the same size and pKi as the deleted IN fragment) into the Δ2173-3855 and Δ2173-3600 mutants. As shown in Fig. 2 and 3, the insertion of the Hrb1 gene into the deletion mutant restored the correct processing of the Ty1 polyprotein.
FIG. 2.

Characterization of VLPs isolated from WT and IN deletion mutant cells. (A) Sucrose step gradient fractionation of VLPs. VLP-associated RT activity was assayed using an oligo(dG)/poly(rC) primer template. (B) Immunoblots of gradient fractions 3 to 7 with anti-RT antibodies. For clarity, only the major bands corresponding to p63-RT are shown, except with the Δ2173-3855 mutant, in which a precursor band corresponding to unprocessed IN (Δ2173-3855)-RT is also shown. The other precursor bands observed in the WT and the Δ2173-3600 and Δ2173-3855 mutants are shown in Fig. 3.
FIG. 3.

Immunoblots of WT and mutant IN deletion VLPs. The WT, Δ2173-3600, and Δ2173-3855 VLPs shown in lanes 1, 2, and 3, respectively, were not prepared in the same experiment as were the WT, Δ2173-3600 plus Hrb1, and Δ2173-3855 plus Hrb1 VLPs that are shown in lanes 4, 5, and 6, respectively. Thus, the relative levels of RT can be compared only between lanes 1, 2, and 3 or between lanes 4, 5, and 6. In each experiment, the volumes of VLPs that were found to incorporate the same amount of dGTP into high-molecular-mass poly(dG) by the oligo (dG)/poly(rC) primer template assay were loaded onto the gel and probed with Ty1 RT antibodies. The molecular masses (MM) of two markers are indicated in lane M. In lane 3, three bands are apparent; according to their sizes, they correspond to mature RT (MM, 63 kDa), unprocessed IN (Δ 2173-3855)-RT (MM, 71 kDa), and PR-IN (Δ2173-3855)-RT (MM, 88 kDa). Bands corresponding to unprocessed WT PR-IN-RT (MM, 134 kDa) and unprocessed PR-IN (Δ2173-3600)-RT (MM, 97 kDa) are also apparent in lanes 1 and 2, respectively. Δ, deletion.
The exogenous polymerase activities of the VLPs were measured using the standard oligo(dG)/poly(rC) primer template assay. For WT and Δ2173-3600 VLPs, a peak of RT activity was observed when the VLPs were sedimented through the sucrose step gradient (Fig. 2). Some level of activity was also detected in Δ2173-3855 VLPs, but it was low compared to that of WT and Δ2173-3600 VLPs (see below). The peaks of RT activity of the WT VLPs were always higher than those of the mutant VLPs. However, the activities of RT did not correlate with the amounts of RT protein detected by immunoblot analyses (Fig. 3). The amount of RT protein loaded onto the gel was adjusted to normalize for the differences in RT activities, i.e., volumes of VLPs previously determined to incorporate the same amount of dGTP into high-molecular-mass poly(dG) in the exogenous oligo(dG)/poly(rC) assay were loaded on the gel. Most notably, the Δ2173-3600 and Δ2173-3600 plus Hrb1 VLPs contained RT proteins with 6- and 20-fold higher specific activities, respectively. This result can be compared to the previous observation of Youngren et al. (40) that Ty1 VLPs prepared from a strain expressing a Ty1 element with a Δ1702-3301 deletion contained 15-fold higher exogenous RT activities than those of Ty1 VLPs from a strain expressing a WT element, implying that some interactions between IN and RT inhibit the exogenous activity of RT in WT VLPs.
If we assume that the exogenous activity represents an in vivo mechanism, this suggests that IN may play a regulatory role in RT activity. Using two mutants with smaller deletions (Δ2173-2737 and Δ2738-3600), we determined (data not shown) that the specific polymerase activity of RT was increased only in the mutant with deletion Δ2738-3600 (residues 233 to 520 of IN). We next deleted more IN residues (Δ2173-3855 deletion mutant). In this case, the activity of RT did not increase any more. On the contrary, it decreased 5- to 10-fold. Therefore, important amino acid residues in the region encoded by nt 3601 to 3855 (residues 521 to 607) play a role in RT activity.
A striking feature of this domain is its high content of acidic residues (pKi, 4.3) (Fig. 4) with several acidic residues clustered near its N-terminal end. It is noteworthy that the active Ty1 recombinant IN-RT fusion protein p6H3601 (obtained previously and found to bear 115 amino acid residues from the IN C-terminal region fused to the RT-RH domain) included this acidic domain. Our attempts to express shorter active fusion proteins with 79, 48, 36, 30, and 26 residues from the C-terminal region of IN fused to the RT-RH domain, but which did not include this acidic domain, were unsuccessful. In two of the shorter constructs with 30- and 36-amino-acid residue extensions, we introduced an EK cleavage site between the His tag and the N terminus of the fusion protein. Surprisingly, we found that construct p6HEK3838 (bearing 36 amino acid residues of IN encoded by nt 3838 to 3945 fused to the EK cleavage site) had the same polymerase-specific activity as the p6H3601 recombinant RT and had RNase H activity (see Fig. 6). In this construct, the EK cleavage site DDDDK is immediately followed by the cluster of acidic residues located between the two motifs of the bipartite nuclear localization signal characterized by Moore et al. (24). Thus, the C-terminal region of the EK3838 fusion protein forms a very acidic domain, DDDDKLEDNETE, where 8 of 12 residues are acidic. This domain could mimic the acidic region of IN encoded by nt 3600 to 3856 and participate in the complex formation between RT and IN.
FIG. 4.

Sequence of the IN-RT junction in the Gag-Pol-p199 polyprotein. The 85 amino acid residues necessary for full activity of the Ty1 RT in the VLPs are underlined. The negatively charged amino acid residues in the IN domain are indicated by asterisks, and the positively charged amino acid residues near the N terminus of RT are highlighted by open circles. The numbers represent amino acid (AA) residues of IN or RT. The Ty1-H3 nucleotides that code for these amino acids are indicated in parentheses.
FIG. 6.

RNase H cleavage by WT and mutant VLPs of an RNA primer containing the PPT sequence used to initiate +sssDNA synthesis. (A) Sequence of the 61-nt DNA template and the −9/+7 RNA primer used in the experiment shown in panel B. The arrow indicates the cleavage site between nt −1 and nt +1 in the RNA primer. The asterisk at the 5′ end of the RNA primer represents the 32P label. (B) The primer template shown in panel A was incubated with WT, Δ2173-3600, and Δ2173-3855 VLPs for 10 min. A control experiment was done with recombinant p6H3601 Ty1 RT at high and low concentrations (conc). Also shown is the result of RNase H cleavage by recombinant p6HEK3838 RT.
It is possible that some basic amino acid residues are involved in the formation of the interface between the two proteins through ionic interactions. A possible interacting partner is the highly basic (pKi of 9.89) region of Ty1 RT between amino acid residues 1 and 85 which possesses a cluster of five basic amino acid residues, uninterrupted by acidic residues, near its N-terminal end (Fig. 4). To test this hypothesis, we tried to demonstrate the interaction of the IN acidic domain with the RT basic region by two-hybrid and pulldown analyses. We used a GAL4-based two-hybrid system to detect the protein interactions in yeast. The acidic domain of IN encoded by nt 3601 to 3855 was fused to the GAL4 DNA binding domain, and the 85 N-terminal amino acid residues of RT containing the basic domain of RT were fused to the GAL4 activation domain. Unfortunately, this construction did not make the protein interaction phenotypically detectable. We then tried a pulldown analysis. The 115 amino acid residues encoded by nt 3601 to 3945 from the C terminus of IN were fused to a His tag and used as bait to test for binding of the 85 N-terminal residues of RT. Unfortunately, the 85-amino-acid residue peptide of RT was not stably expressed, and it was not possible to perform the pulldown experiment. We then reasoned that if there is an interaction between the C-terminal region of IN and the N-terminal region of RT, a peptide encoded by nt 3601 to 4200 with the 115 C-terminal amino acid residues of IN fused to the 85 N-terminal amino acids of RT (Fig. 4) could be stably expressed, despite the fact that the N-terminal peptide of RT was not expressed alone. We indeed observed a stable expression of this fusion peptide, suggesting that the interaction between the two domains of the fusion peptide enables its expression. To test the role of the acidic residues of IN in the expression of the fusion peptide, we replaced the negatively charged acidic residues (Fig. 4) by alanine residues. We found that the mutant fusion peptide was not stably expressed anymore (data not shown). These observations point to the fact that the acidic residues of IN play a role in the stability and therefore in the structure of the fusion peptide. This could explain why the level of RT activity was decreased in Ty1 VLPs prepared from a strain expressing a Ty1 element with a deletion of the acidic region of IN.
The IN deletion mutants synthesize −sssDNA but do not synthesize +sssDNA.
The homopolymer primer template assay used above allowed us to rapidly test whether the RT present in VLPs had retained exogenous polymerase activity. However, the assay did not tell us anything about the endogenous polymerase activity of the RT. To evaluate the endogenous polymerase activity, we carried out a reiterative primer extension analysis to determine the presence of −sssDNA and +sssDNA replication intermediates in WT and mutant versions of VLPs (38). 5′-end-labeled primers complementary to −sssDNA or +sssDNA were annealed to VLP-derived nucleic acids and repeatedly extended with Taq polymerase. Since the 5′ ends of the −sssDNAs and +sssDNAs are fixed by the 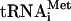 and by the polypurine tract (PPT) used as a primer to initiate +sssDNA synthesis, respectively, discrete labeled extension products should be generated. With the primers used in this study, 94-base and 135-base extension products were expected for −sssDNA and +sssDNA, respectively. Primer extension was carried out with WT and three deletion mutant VLP-derived nucleic acids (the Δ2173-3600, Δ2173-2737, and Δ2738-3600 mutants). As shown in Fig. 5, the highest level of −sssDNA was detected in WT VLPs, but a significant amount of −sssDNA was also detected in mutant VLPs, indicating that the IN deletions do not inhibit −sssDNA synthesis. We next used a 24-nt primer, complementary to U3, to determine the presence of the +sssDNA replication intermediate in VLP-derived nucleic acids. As shown in Fig. 5, a high level of +sssDNA was detected in WT VLPs, whereas the level of +sssDNA was very low or absent in mutant VLPs. There are two possible explanations of the +sssDNA synthesis defect observed in IN deletions: (i) a defect in first-strand transfer or (ii) an inhibition of RNase H activity in the mutant VLPs. To choose between the two alternatives, we tested the RNase H activity of the mutant VLPs.
and by the polypurine tract (PPT) used as a primer to initiate +sssDNA synthesis, respectively, discrete labeled extension products should be generated. With the primers used in this study, 94-base and 135-base extension products were expected for −sssDNA and +sssDNA, respectively. Primer extension was carried out with WT and three deletion mutant VLP-derived nucleic acids (the Δ2173-3600, Δ2173-2737, and Δ2738-3600 mutants). As shown in Fig. 5, the highest level of −sssDNA was detected in WT VLPs, but a significant amount of −sssDNA was also detected in mutant VLPs, indicating that the IN deletions do not inhibit −sssDNA synthesis. We next used a 24-nt primer, complementary to U3, to determine the presence of the +sssDNA replication intermediate in VLP-derived nucleic acids. As shown in Fig. 5, a high level of +sssDNA was detected in WT VLPs, whereas the level of +sssDNA was very low or absent in mutant VLPs. There are two possible explanations of the +sssDNA synthesis defect observed in IN deletions: (i) a defect in first-strand transfer or (ii) an inhibition of RNase H activity in the mutant VLPs. To choose between the two alternatives, we tested the RNase H activity of the mutant VLPs.
FIG. 5.

Reiterative primer extension analysis of Ty1 replication intermediates. 5′-end-labeled primers complementary to −sssDNA or +sssDNA were annealed to VLP-derived nucleic acids and repeatedly extended with Taq polymerase. The asterisk at the 5′ end of the primer represents the 32P label. The sequences at the 5′ ends of the −sssDNA and +sssDNA are indicated. The extension products were loaded on an 8% denaturing polyacrylamide gel along with a sequence which was used as a size marker. The sequence was determined with the primer used in the primer extension assay so that the extension product would migrate like the sequencing fragments bearing the last nucleotide of the strong-stop DNA (indicated by an asterisk in the sequencing gel). In fact, the primer extension product migrated slightly faster, because the primer used in the primer extension assay was phosphorylated at its 5′ end, whereas the primer used in the sequencing reaction was not phosphorylated. PBS, primer binding site.
IN deletion mutants have RNase H activity.
The RNase H activity of RT in the WT and mutant VLPs was examined by monitoring the cleavage of an RNA primer spanning the PPT plus-strand initiation site. A −9/+7 primer containing the −9/−1 PPT sequence UGGGUGGUA and 7 nt (+1/+7) complementary to the 3′ U3 sequence was annealed to a 61-nt minus-strand DNA template spanning the PPT region (Fig. 6). This hybrid substrate was incubated with the VLPs to induce RNase H cleavage, and the digestion products were analyzed on a 20% denaturing polyacrylamide gel. Using the same primer template, we showed previously that the recombinant RT was able to cleave the RNA primer specifically between positions −1 and +1 to generate a product 9 nt in length with the correct PPT primer 3′ terminus at position −1 (36). As shown in Fig. 6, the WT and mutant VLPs were also able to make a specific cleavage between positions −1 and +1. This indicates that the lack of plus-strand synthesis cannot be attributed to the inhibition of RNase H activity in the deletion mutants and could be explained by a defect of strand transfer.
Conclusion.
Increasing evidence suggests that two key catalytic enzymes, IN and RT, encoded by retroviruses and retrotransposons interact with each other and that this interaction has a functional role (see the introduction). In this report, we have identified two domains of Ty1 integrase which have effects on RT activity in vivo. The deletion of a domain spanning amino acid residues 233 to 520 of IN increases the exogenous specific activity of RT up to 20-fold, whereas the removal of a region rich in acidic amino acid residues between residues 521 and 605 decreases its activity. This last result is complementary to the observation that recombinant RT is active if a short fragment of IN extended by a small acidic tail is added at its N terminus and suggests that the negatively charged amino acids are involved in an ionic interaction with some residues of RT. Numerous observations support the conclusion that the folding of a protein is a hierarchical process and that it begins locally (30), i.e., the secondary structure of a protein is determined primarily by interactions among amino acids that are close in sequence. In Ty1 RT, a region rich in basic amino acids is located near the N terminus. During synthesis of the Gag-Pol-p199 precursor protein, this region is close in sequence to the IN region that is rich in the acidic amino acids necessary to confer full activity to the RT. Interaction between these two regions might be critical for the correct folding of RT and for the formation of an active conformation of the enzyme. Our observation that replacement of the acidic residues by alanine residues in a small fusion peptide spanning these two regions affects the peptide's expression and structure corroborates this hypothesis. Further structural studies and X-ray crystallographic analyses will be needed to determine the interface between the IN and RT proteins and to determine if there is a direct interaction between these two regions.
Acknowledgments
We thank T. Menees (University of Missouri, Kansas City) for kindly providing the Ty1 RT antibodies and B. Senger (IBMC, Strasbourg, France) for the plasmid containing the Hrb1 gene. We thank A. Gabriel (Rutgers University, Piscataway, N.J.) for helpful discussions during the early stages of this work and for manuscript review.
REFERENCES
- 1.Adams, S. E., J. Mellor, K. Gull, R. B. Sim, M. F. Tuite, S. M. Kingsman, and A. J. Kingsman. 1987. The functions and relationships of Ty-VLP proteins in yeast reflect those of mammalian retroviral proteins. Cell 49:111-119. [DOI] [PubMed] [Google Scholar]
- 2.Boeke, J. D., D. Eichinger, D. Castrillon, and G. R. Fink. 1988. The Saccharomyces cerevisiae genome contains functional and nonfunctional copies of transposon Ty1. Mol. Cell. Biol. 8:1432-1442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Eichinger, D. J., and J. D. Boeke. 1988. The DNA intermediate in yeast Ty1 element transposition copurifies with virus-like particles: cell-free Ty1 transposition. Cell 54:955-966. [DOI] [PubMed] [Google Scholar]
- 4.Engelman, A., G. Englund, J. M. Orenstein, M. A. Martin, and R. Craigie. 1995. Multiple effects of mutations in human immunodeficiency virus type 1 integrase on viral replication. J. Virol. 69:2729-2736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Garfinkel, D. J., A.-M. Hedge, S. D. Youngren, and T. D. Copeland. 1991. Proteolytic processing of pol-TYB proteins from the yeast retrotransposon Ty1. J. Virol. 65:4573-4581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Grandgenett, D., T. Quinn, P. J. Hippenmeyer, and S. Oroszlan. 1985. Structural characterization of the avian retrovirus reverse transcriptase and endonuclease domains. J. Biol. Chem. 260:8243-8249. [PubMed] [Google Scholar]
- 7.Hansen, L. J., and S. B. Sandmeyer. 1990. Characterization of a transpositionally active Ty3 element and identification of the Ty3 integrase protein. J. Virol. 64:2599-2607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hehl, E. A., P. Joshi, G. V. Kalpana, and V. R. Prasad. 2004. Interaction between human immunodeficiency virus type 1 reverse transcriptase and integrase proteins. J. Virol. 78:5056-5067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hu, S. C., D. L. Court, M. Zweig, and J. G. Levin. 1986. Murine leukemia virus pol gene products: analysis with antisera generated against reverse transcriptase and endonuclease fusion proteins expressed in Escherichia coli. J. Virol. 60:267-274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hurt, E., M.-J. Luo, S. Röther, R. Reed, and K. Strässer. 2004. Cotranscriptional recruitment of the serine-arginine-rich (SR)-like proteins Gbp2 and Hrb1 to nascent mRNA via the TREX complex. Proc. Natl. Acad. Sci. USA 101:1858-1862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ishikawa, T., N. Okui, N. Kobayashi, R. Sakuma, T. Kitamura, and Y. Kitamura. 1999. Monoclonal antibodies against the minimal DNA-binding domain in the carboxyl-terminal region of human immunodeficiency virus type 1 integrase. J. Virol. 73:4475-4480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Karst, S. M., M.-L. Rütz, and T. M. Menees. 2000. The yeast retrotransposons Ty1 and Ty3 require the RNA lariat debranching enzyme, Dbr1p, for efficient accumulation of reverse transcripts. Biochem. Biophys. Res. Commun. 268:112-117. [DOI] [PubMed] [Google Scholar]
- 13.Katz, R. A., and A. M. Skalka. 1994. The retroviral enzymes. Annu. Rev. Biochem. 63:133-173. [DOI] [PubMed] [Google Scholar]
- 14.Kirchner, J., and S. Sandmeyer. 1993. Proteolytic processing of Ty3 proteins is required for transposition. J. Virol. 67:19-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kirchner, J., and S. B. Sandmeyer. 1996. Ty3 integrase mutants defective in reverse transcription or 3′-end processing of extrachromosomal Ty3 DNA. J. Virol. 70:4737-4747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kunkel, T. A. 1985. Rapid and efficient site-specific mutagenesis without phenotypic selection. Proc. Natl. Acad. Sci. USA 82:488-492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lai, L., H. Liu, X. Wu, and J. C. Kappes. 2001. Moloney murine leukemia virus integrase protein augments viral DNA synthesis in infected cells. J. Virol. 75:11365-11372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Leavitt, A. D., G. Robles, N. Alesandro, and H. E. Varmus. 1996. Human immunodeficiency virus type 1 integrase mutants retain in vitro integrase activity yet fail to integrate viral DNA efficiently during infection. J. Virol. 70:721-728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Martin-Rendon, E., D. W. Hurd, G. Marfany, S. M. Kingsman, and A. J. Kingsman. 1996. Identification of proteolytic cleavage sites within the gag-analogue protein of Ty1 virus-like particles. Mol. Microbiol. 22:1035-1043. [DOI] [PubMed] [Google Scholar]
- 20.Masuda, T., V. Planelles, P. Krogstad, and I. S. Y. Chen. 1995. Genetic analysis of human immunodeficiency virus type 1 integrase and the U3 att site: unusual phenotype of mutants in the zinc finger-like domain. J. Virol. 69:6687-6696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mathias, S. L., A. F. Scott, H. J. Kazazian, J. D. Boeke, and A. Gabriel. 1991. Reverse transcriptase encoded by a human transposable element. Science 254:1808-1810. [DOI] [PubMed] [Google Scholar]
- 22.Merkulov, G. V., J. F. Lawler, Jr., Y. Eby, and J. D. Boeke. 2001. Ty1 proteolytic cleavage sites are required for transposition: all sites are not created equal. J. Virol. 75:638-644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Merkulov, G. V., K. M. Swiderek, C. B. Brachmann, and J. D. Boeke. 1996. A critical proteolytic cleavage site near the C terminus of the yeast retrotransposon Ty1 Gag protein. J. Virol. 70:5548-5556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Moore, S. P., L. A. Rinckel, and D. J. Garfinkel. 1998. A Ty1 integrase nuclear localization signal required for retrotransposition. Mol. Cell. Biol. 18:1105-1114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Muller, F., K. H. Bruhl, K. Freidel, K. V. Kowallik, and M. Ciriacy. 1987. Processing of Ty1 proteins and formation of Ty1 virus-like particles in Saccharomyces cerevisiae. Mol. Gen. Genet. 207:421-429. [DOI] [PubMed] [Google Scholar]
- 26.Nymark-McMahon, M. H., and S. B. Sandmeyer. 1999. Mutations in nonconserved domains of Ty3 integrase affect multiple stages of the Ty3 life cycle. J. Virol. 73:453-465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nymark-McMahon, M. H., N. S. Beliakova-Bethell, J.-L. Darlix, S. F. J. Le Grice, and S. B. Sandmeyer. 2002. Ty3 integrase is required for initiation of reverse transcription. J. Virol. 76:2804-2816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Oz, I., O. Avidan, and A. Hizi. 2002. Inhibition of the integrases of human immunodeficiency viruses type 1 and type 2 by reverse transcriptases. Biochem. J. 361:557-566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Padow, M., L. Lai, C. Deivanayagam, L. J. DeLucas, R. B. Weiss, D. M. Dunn, X. Wu, and J. C. Kappes. 2003. Replication of chimeric human immunodeficiency virus type 1 (HIV-1) containing HIV-2 integrase (IN): naturally selected mutations in IN augment DNA synthesis. J. Virol. 77:11050-11059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Roder, H. 2004. Stepwise helix formation and chain compaction during protein folding. Proc. Natl. Acad. Sci. USA 101:1793-1794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tasara, T., G. Maga, M. O. Hottiger, and U. Hübscher. 2001. HIV-1 reverse transcriptase and integrase enzymes physically interact and inhibit each other. FEBS Lett. 507:39-44. [DOI] [PubMed] [Google Scholar]
- 32.Trentin, B., N. Rebeyrotte, and R. Z. Mamoun. 1998. Human T-cell leukemia virus type 1 reverse transcriptase (RT) originates from the pro and pol open reading frames and requires the presence of RT-RNase H (RH) and RT-RH-integrase proteins for its activity. J. Virol. 72:6504-6510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tsurutani, N., M. Kubo, Y. Maeda, T. Ohashi, N. Yamamoto, M. Kannagi, and T. Masuda. 2000. Identification of critical amino acid residues in human immunodeficiency virus type 1 IN required for efficient proviral DNA formation at steps prior to integration in dividing and nondividing cells. J. Virol. 74:4795-4806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Werner, S., P. Hindmarsh, M. Napirei, K. Vogel-Bachmayr, and B. M. Wöhrl. 2002. Subcellular localization and integration activities of Rous sarcoma virus reverse transcriptase. J. Virol. 76:6205-6212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Werner, S., and B. M. Wöhrl. 2000. Asymmetric subunit organization of heterodimeric Rous sarcoma virus reverse transcriptase αβ: localization of the polymerase and RNase H active sites in the α subunit. J. Virol. 74:3245-3252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wilhelm, F.-X., M. Wilhelm, and A. Gabriel. 2003. Extension and cleavage of the polypurine tract plus-strand primer by Ty1 reverse transcriptase. J. Biol. Chem. 278:47678-47684. [DOI] [PubMed] [Google Scholar]
- 37.Wilhelm, M., M. Boutabout, and F.-X. Wilhelm. 2000. Expression of an active form of recombinant Ty1 reverse transcriptase in Escherichia coli: a fusion protein containing the C-terminal region of the Ty1 integrase linked to the reverse transcriptase-RNase H domain exhibits polymerase and RNase H activities. Biochem. J. 348:337-342. [PMC free article] [PubMed] [Google Scholar]
- 38.Wilhelm, M., T. Heyman, M. Boutabout, and F.-X. Wilhelm. 1999. A sequence immediately upstream of the plus-strand primer is essential for plus-strand DNA synthesis of the Saccharomyces cerevisiae Ty1 retrotransposon. Nucleic Acids Res. 27:4547-4552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wu, X., H. Liu, H. Xiao, J. A. Conway, E. Hehl, G. V. Kalpana, V. Prasad, and J. C. Kappes. 1999. Human immunodeficiency virus type 1 integrase protein promotes reverse transcription through specific interactions with the nucleoprotein reverse transcription complex. J. Virol. 73:2126-2135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Youngren, S. D., J. D. Boeke, N. J. Sanders, and D. J. Garfinkel. 1988. Functional organization of the retrotransposon Ty from Saccharomyces cerevisiae: Ty protease is required for transposition. Mol. Cell. Biol. 8:1421-1431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yung, E., M. Sorin, A. Pal, E. Craig, A. Morozov, O. Delattre, J. Kappes, D. Ott, and G. V. Kalpana. 2001. Inhibition of HIV-1 virion production by a transdominant mutant of integrase interactor 1. Nat. Med. 7:920-926. [DOI] [PubMed] [Google Scholar]
- 42.Zhu, K., C. Dobard, and S. A. Chow. 2004. Requirement for integrase during reverse transcription of human immunodeficiency virus type 1 and the effect of cysteine mutations of integrase on its interactions with reverse transcriptase. J. Virol. 78:5045-5055. [DOI] [PMC free article] [PubMed] [Google Scholar]