

Introductory paragraph
Posttranslational modifications (PTMs) on histones are a key source of regulation on chromatin through impacting cellular processes, including gene expression1. These PTMs often arise from metabolites and are thus impacted by metabolism and environmental cues2–7. One class of metabolically regulated PTMs are histone acylations, which include histone acetylation, butyrylation, crotonylation, and propionylation3,8. Since these PTMs can be derived from short chain fatty acids (SCFAs), which are generated by the commensal microbiota in the intestinal lumen9–11, we aimed to define how microbes impact the host intestinal chromatin landscape, mainly in female mice. Here we show that in addition to acetylation, intestinal epithelial cells (IECs) from the cecum and distal mouse intestine also harbor high levels of butyrylation and propionylation on lysines 9 and 27 of histone H3. We demonstrate that these acylations are regulated by the microbiota, and histone butyrylation is additionally regulated by the metabolite tributyrin. Tributyrin-regulated gene programs are correlated with histone butyrylation, which is associated with active gene regulatory elements and levels of gene expression. Together, our study uncovers a regulatory layer of how the microbiota and metabolites influence the intestinal epithelium through chromatin, demonstrating a physiological setting in which histone acylations are dynamically regulated and associated with gene regulation.
Cells are exposed to various stimuli in the environment, which can induce signals that converge onto chromatin and impact cellular processes. Posttranslational modifications (PTMs) on histones, or histone marks, represent one major way in which the chromatin landscape is dynamically regulated. Some PTMs have crucial regulatory roles in processes such as transcription, chromatin organization, and more1. Understanding how PTMs are regulated and their functions are critical for determining molecular mechanisms of fundamental biological processes such as gene regulation.
Recently, different modifications have been identified that greatly expand the diversity of histone PTMs4,8,12. However, the physiological functions of many of these PTMs have not been defined. Here, we focused our studies on histone acylations, which include acetylation, butyrylation, crotonylation, and propionylation3. These are structurally similar PTMs that are generally associated with gene expression and sensitive to cellular metabolism2,3,13–15. Furthermore, histone acetylation has been linked to circadian transcriptional dynamics in the gut16.
The donor molecules for many histone PTMs are small metabolites. Histone PTMs are thus not only regulated by enzymes that deposit or remove modifications, but also by the availability of donor molecules5–7. Short chain fatty acids (SCFAs) are metabolites generated at high concentrations in the intestinal lumen, through the action of microbes that ferment fiber9. Acetate, butyrate, and propionate are estimated to be at millimolar concentrations in the human intestinal lumen10. These metabolites can then act on neighboring intestinal epithelial cells (IECs) or distal tissues via circulation. SCFAs bind to receptors, feed into energy pathways, and act as inhibitors of histone deacetylase (HDAC) enzymes11,17–22. Acetylation can be traced from bacterial fermentation to histone PTMs23. Alterations in the commensal microbiota or SCFAs also regulate select histone acetylation and methylation PTMs, and crotonylation24,25. Together, these studies suggest that metabolites and the microbiota regulate select PTMs on chromatin.
Here, we aimed to study histone acylations in vivo, predominantly focusing on the less characterized PTM of butyrylation. We hypothesized that since the gut harbors high concentrations of SCFAs, here these metabolites may be deposited onto chromatin and have functional roles as histone acylations in intestinal gene regulation.
To investigate different histone acylations in a physiological setting, we decided to first identify where these modifications occur in the body. We hypothesized that tissues may harbor different levels of histone acylations depending on their local environment, and that the gut particularly may be conducive to histone acylations due to high levels of SCFAs (Fig. 1a). We primarily focused on mouse abdominal tissues, from which we purified histones and then probed for different PTMs (Fig. 1b). We used pan-lysine modification antibodies, which recognize PTMs of interest independent of the specific modification sites. We found that the cecum and, to a lesser extent the large intestine, displayed higher levels of histone butyrylation and propionylation compared to other tissues. These findings are consistent with the cecum and colon being major sites of microbial fermentation and production of SCFAs11. Based on this data, we focused our studies on the cecum for subsequent experiments.
Fig. 1: The intestine is an environment that harbors a variety of histone acyl marks.
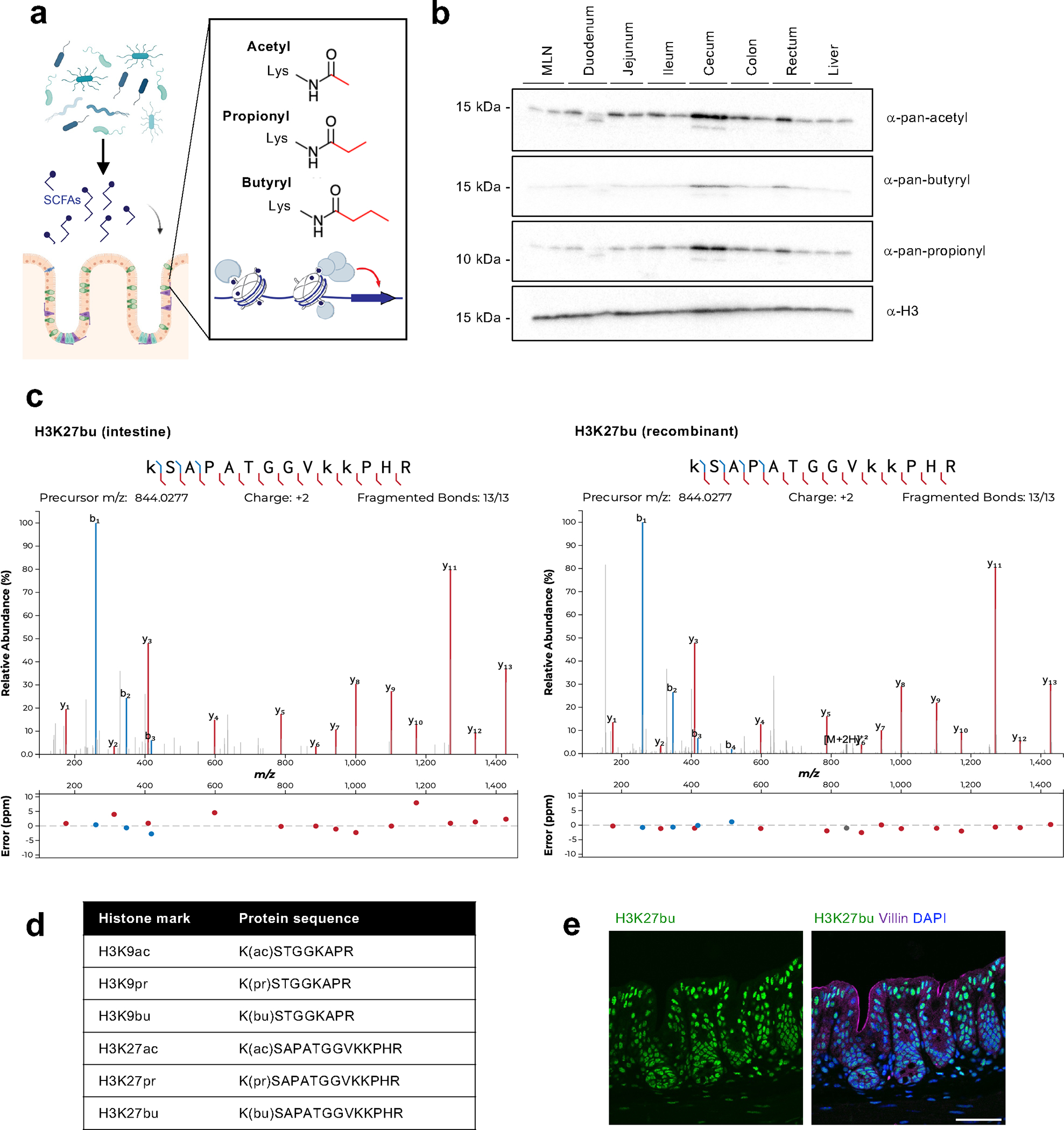
(A) Schematic of overall hypothesis and the structure of select histone acylations. Created with BioRender.com. (B) Tissue distribution of histone acylations. Histones were purified from tissues from female C57BL/6 mice (n = 2 shown) using acid extraction and then subject to immunoblotting with pan-lysine antibodies targeting different acylation modifications. H3 serves as a loading control. (C) Characterization of specific sites of histone acylation. LC-MS/MS analysis of extracted histone from the mouse cecum. Representative fragmentation spectra are shown for endogenous H3K27bu in the intestine (left) and for recombinant H3K27bu on modified nucleosomes (right). (D) Table summarizing the different histone acyl marks detected in the mouse cecum. (E) H3K27bu displays bright staining in the intestinal epithelium. Representative immunofluorescence images of H3K27bu, Villin (marker of epithelium), and DAPI in mouse cecal sections, n = 5 female C57BL/6 mice. Scale bar = 50 μm.
We next aimed to identify specific sites of histone butyrylation and propionylation in the mouse gut by performing mass spectrometry (MS) on extracted histones from the cecum. We identified four specific PTMs with high confidence: butyrylation and propionylation on histone H3 lysine 27 (H3K27br and H3K27pr, respectively) and histone H3 lysine 9 (H3K9bu and H3K9pr) (Fig. 1c,d, Extended Data Fig. 1–2). The acylations on H3K27 displayed stronger signals than those on H3K9, which may be due to differences in abundance and/or ionization properties. We also examined the dose-response behavior of the different histone acyl marks. Upon testing a dilution series of recombinant acylated nucleosomes spiked into unmodified nucleosomes, we observed that H3K27bu produces a lower signal than H3K27ac at similar relative abundances and that the butyryl mark for both H3K9 and H3K27 becomes undetectable at levels where the acetyl mark remains visible (Extended Data Fig. 2b). This suggests that the abundance of histone butyrylation and perhaps other acylation marks may be underestimated unless synthetic standards are utilized for quantification. While previous studies detected crotonylation on H3K18 in the colon25, we did not observe crotonylation on lysine 9, 18, or 27 of histone H3 in the cecum by parallel reaction monitoring MS (Extended Data Fig. 3).
Since acyl modifications are structurally similar, we next tested antibodies targeting these PTMs for selectivity. We utilized recombinant nucleosomes to test butyryl and acetyl antibodies, which displayed selectivity towards their specific modifications. The H3K27bu antibody has some cross-reactivity with H3K9bu and crotonylation, but very minimal cross-reactivity with H3K27ac (Extended Data Fig. 4). While we acknowledge that our efforts may also be detecting H3K9bu with this antibody, we were not concerned about detecting crotonylation based on our MS results. Due to both the importance of histone butyrylation in various aspects of physiology and disease, as well as the strong MS signal intensity of H3K27bu, we decided to largely focus our efforts on butyrylation over propionylation. We then stained intestinal sections from the mouse gut to visualize H3K27bu in the setting of the tissue architecture. We found that H3K27bu is widespread and displays bright staining in the cecal epithelia (Fig. 1e). Together, these data suggest that select non-canonical histone acylations, specifically H3K27bu, are in intestinal epithelial cells in regions that harbor fermenting bacteria.
We next investigated whether these PTMs were regulated by the commensal microbiota. We found that germ-free mice had reduced levels of histone acetylation, butyrylation, and propionylation compared to conventional mice (Fig. 2a,b). We also treated conventional mice with an antibiotic cocktail, which reduced levels of these same PTMs (Fig. 2c,d). This antibiotic cocktail treatment, as well as the single agent ampicillin alone, both resulted in expected increased cecal weights and alterations to the gut microbiome composition (Extended Data Fig. 5). Together, these data suggest these histone acylations are sensitive to changes in the commensal microbiota in vivo.
Fig. 2: Histone acyl marks, including select non-acetyl acyl marks, are dependent on the microbiota.
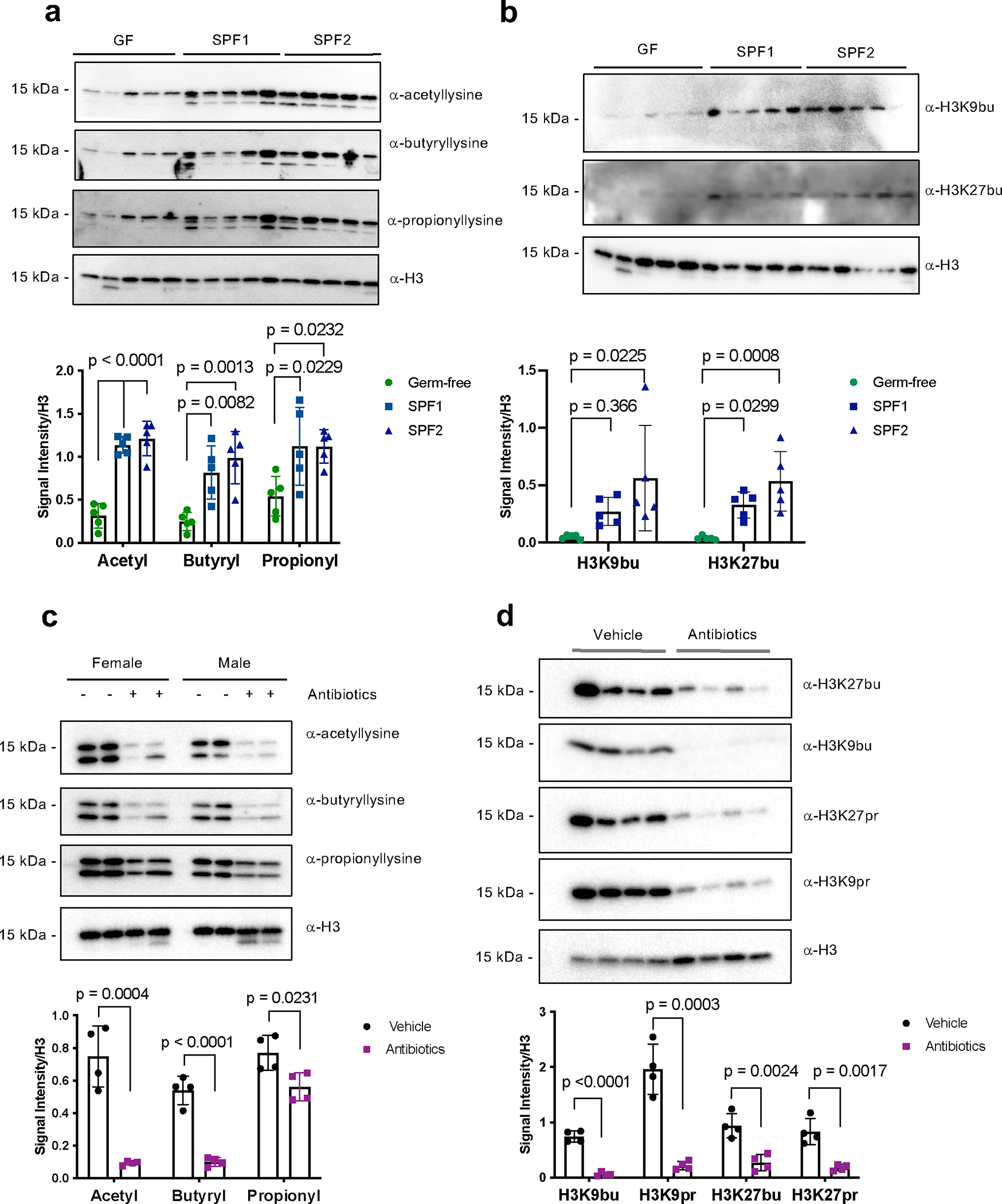
Immunoblotting (top) and quantification of signal intensity relative to histone H3 (bottom). Histones were extracted from the cecum and each lane represents an individual mouse. Quantification was performed in ImageJ and H3 serves as a loading control. All graphs depict mean and standard error. (A) Pan-acylations and (B) specific histone acylations are reduced in germ-free mice. Germ-free (GF) mice (n = 5), conventional mice (SPF = specific pathogen free, SPF1 = C57BL/6NTac, n = 5, SPF2 = C57BL/6J, n =5, all mice in A-B are female). Statistical significance was determined by one-way ANOVA and adjusted for multiple comparisons using Dunnett’s test. (C) Pan-acylations and (D) specific histone acylations are reduced in antibiotic-treated mice. C57BL/6J mice were treated with (n = 4) or without (n = 4) antibiotics (ampicillin, vancomycin, neomycin, and metronidazole) for seven days. Panel C shows results from both male and female mice (indicated), and panel D shows results from only female mice. Statistical significance was determined using the two-tailed Student’s t-test.
We next tested whether the microbial metabolites themselves were sufficient to regulate histone acylations, using a setting where we reduced the microbiota and added an exogenous metabolite to the mouse gut (Fig. 3a). We treated mice with or without ampicillin, which as a sole agent is sufficient to reduce select histone acylations. Then, we orally gavaged mice with vehicle control or an analog of butyrate (tributyrin), which increases the concentration of butyrate from undetectable to approximately 20 mM in the cecal lumen following oral gavage in antibiotic-treated mice26. Indeed, these treatments altered intracellular levels of butyryl-CoA, and tributyrin had minimal effects on other cellular metabolites (Fig. 3b,c). In addition, tributyrin treatment partially rescued histone butyrylation in the cecum (Fig. 3d,e), suggesting that a specific metabolite alone is partially sufficient to regulate these PTMs in the intestine.
Fig. 3: Microbial metabolites regulate select histone acyl marks and gene expression.
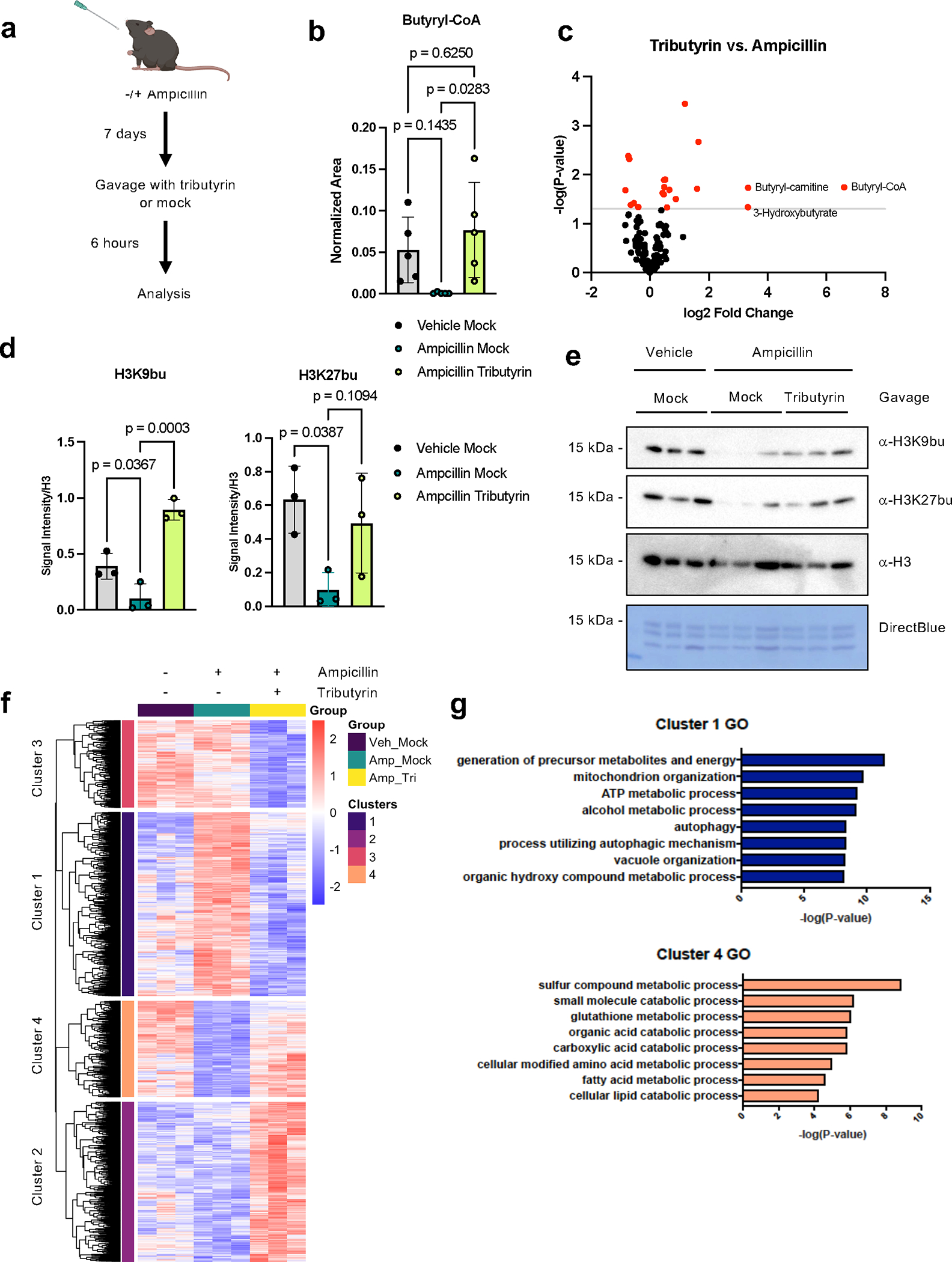
(A) Schematic of experimental system. Mice are treated with or without Ampicillin for seven days, followed by gavage with vehicle or tributyrin. Created with BioRender.com. (B) Intracellular butyryl-CoA is altered upon antibiotic and tributyrin treatment. Cecal tissues from mice were subject to metabolomic analysis by LC-MS, n = 5 female mice per group. Graph displays mean and standard error; statistical significance was determined by one-way ANOVA and adjusted for multiple comparisons. (C) Volcano plots displaying changes in polar metabolites across different conditions. Red dots are metabolites that are significantly changed (p-value < 0.05 by unpaired two-tailed Student’s t-test) and grey line indicates significance threshold. (D-E) Histone butyrylation dynamically changes in cecal tissues with butyrate treatment. Immunoblotting quantification (D) and image (E) showing levels of histone butyrylation in the cecum with different treatments. Representative experiment shown, n = 3 female mice per group. Quantification was performed in ImageJ and H3 serves as a loading control. Graphs depict mean and standard error; statistical significance was determined by one-way ANOVA and adjusted for multiple comparisons. (F) Heatmap of significant genes changing with tributyrin treatment by RNA-seq in cecal IECs. Significant genes changing between mice treated with ampicillin plus tributyrin vs. ampicillin with mock gavage, padj < 0.05 by pairwise test, are shown. The Wald test was used as part of the DESeq2 package for differential gene analysis, along with multiple testing correction using Benjamini-Hochberg false discovery rate to get the padj value. Hierarchical clustering was performed after determining optimal number of clusters using the elbow method. The heatmap intensity corresponds to the rlog counts as shown in the legend. Individual mice serve as biological replicates, n = 3 per group. (G) Gene ontology (GO) of significant differentially expressed genes in clusters 1 and 4. Top eight most significantly enriched categories are shown. Over Representation Analysis was performed using a one-sided version of Fisher’s exact test in the clusterProfiler package.
We then analyzed gene expression changes in intestinal epithelial cells (IECs) when mice are treated with antibiotics and tributyrin, to address potential functional consequences of altering histone butyrylation. Our RNA-seq results indicate that ampicillin and ampicillin plus tributyrin induced robust changes in gene expression in cecal IECs (Fig. 3f, Extended Data Fig. 6). We focused our analysis on genes that changed with tributyrin treatment compared to ampicillin alone. Hierarchical clustering resulted in four clusters with distinct patterns of gene expression (Fig. 3f). While tributyrin impacted expression of some genes that were not affected by ampicillin (clusters 2 and 3), we reasoned that these genes may reflect a different mechanism than the potential action of butyrate on chromatin (i.e., binding of butyrate to cell surface receptors). We therefore focused on genes that displayed a pattern of expression matching the histone acyl marks: genes that were altered in expression with ampicillin treatment, and then rescued back to a vehicle baseline (clusters 1 and 4, Fig. 3g, Extended Data Fig. 4d). In these clusters, tributyrin treatment resulted in significant enrichment in gene ontology (GO) categories related to metabolism, consistent with previously published roles of butyrate in energy metabolism in the colon27. Tributyrin treatment downregulated genes related to mitochondria and oxidative phosphorylation, as well as autophagy, while upregulated genes were related to glutathione metabolism and catabolic processes (Fig. 3g, Extended Data Fig. 6e, Supplementary Table 1). Together, this suggests that tributyrin increases histone butyrylation and results in gene expression changes that are enriched in metabolic programs.
Given the enrichment of differential genes in metabolic GO categories, we performed metabolomics to determine whether our treatments may be broadly altering cellular metabolism in the cecum (Extended Data Fig. 7a, Supplementary Table 1). We observed some changes in cecal metabolism following ampicillin treatment. Of the 136 metabolites measured, the most decreased metabolite following ampicillin treatment was butyryl-CoA, followed by N-acetylornithine and butyryl-carnitine (Extended Data Fig. 7). Tributyrin itself did not cause many additional changes in metabolites, except butyryl-CoA levels were rescued and metabolites related to butyrate metabolism, such as butyryl-carnitine and 3-hydroxyburyrate, were also increased compared to ampicillin treatment alone (Fig. 3c, Extended Data Fig. 7c). Our metabolomics data suggests that the changes in gene expression and histone butyrylation are not simply due to alterations in cellular metabolic pathways.
To investigate the genomic landscape of histone acylations in IECs, we performed ChIP-seq for different histone PTMs. Here, we focused our analysis on the localization of H3K27bu, yet we acknowledge that our efforts may also be detecting H3K9bu (Extended Data Fig. 4) and our observations may be more general to histone H3 butyrylation. We observed that H3K27bu largely overlaps with other active histone modifications, including H3K27ac and H3K4me3 (Fig. 4a, Extended Data Fig. 3e). In contrast, H3K27me3, which is associated with gene repression, is localized to distinct regions. We also analyzed the genomic localization of H3K27bu peaks, which is enriched at active gene regulatory elements (Fig. 4b), as expected from our visualization of ChIP-seq signal. H3K27bu peaks showed pronounced enrichment versus the genome at promoter regions, especially within 1 kb of the transcription start site (TSS). We further analyzed the top 10% of H3K27bu peaks, in which the most significantly enriched GO categories were related to oxidative stress, development, and phosphorylation, while other significantly enriched categories in cellular metabolism are related to phosphorus, carbohydrate, and DNA metabolism (Extended Data Fig. 8, Supplementary Table 1). In particular, categories related to carbohydrate metabolism were highly represented. While histone butyrylation and tributyrin treatment (Fig. 3g) both induce overlapping changes in metabolic gene programs, there are also gene programs unique to each analysis, suggesting both shared and distinct mechanisms of tributyrin treatment and histone butyrylation. We next asked how H3K27bu occupancy is related to levels of gene expression. In analyzing IECs from untreated mice, we binned our RNA-seq data into quartiles based on expression level. H3K27bu signal was highest in the quartile with highest gene expression and lowest in the quartile with lowest expression (Fig. 4c). Thus, we conclude that H3K27bu is associated with active gene regulatory elements and gene expression in IECs.
Fig. 4: Histone butyrylation on H3K27 is associated with active gene regulatory elements and gene expression in intestinal epithelial cells.
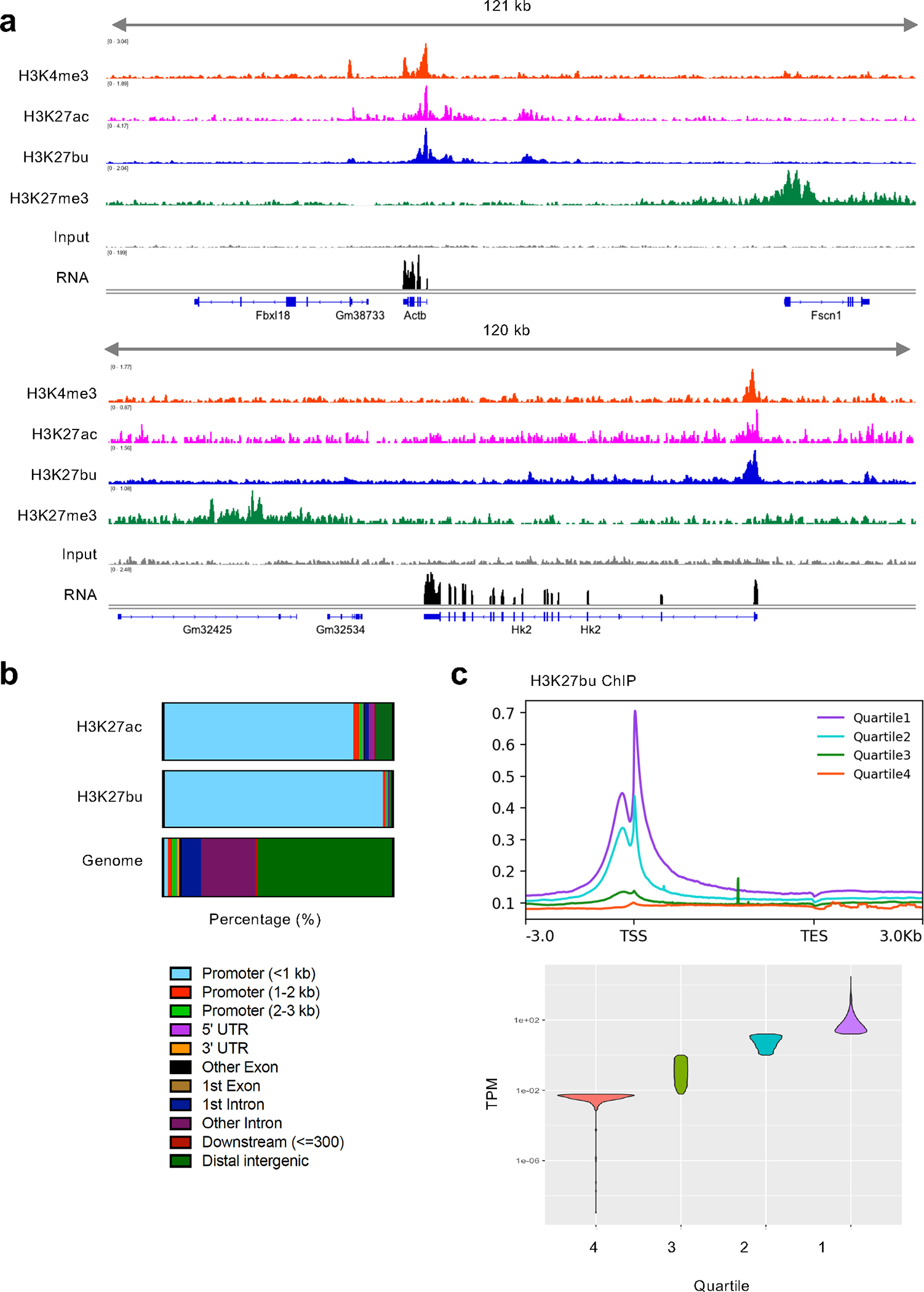
(A) Representative tracks of ChIP-seq data in cecal intestinal epithelial cells from female C57BL/6J mice. At least two biological replicates were performed for each ChIP. A representative RNA-seq track is also shown (n = 3). ChIP and RNA signal on a representative housekeeping gene Actb (top) and tributyrin-regulated gene Hk (bottom). (B) Genomic localization of both H3K27ac and H3K27bu consensus peaks of two replicates each and genomic localizations (Genome). X-axis depicts percentage of sites. (C) H3K27bu localizes to highly expressed genes. Meta plots of a representative H3K27bu ChIP signal shown above violin plot displaying the quartiles of gene expression.
Our studies have identified understudied histone PTMs in the physiological context of the mammalian intestine that are associated with gene expression. Namely, we detected histone butyrylation and propionylation on histone H3 lysines 9 and 27, which are localized in cecal intestinal epithelial cells, along with histone acetylation. We were not able to detect histone crotonylation in the cecum, which differs from previous studies in the colon25. We speculate that this difference could be due to many reasons, including but not limited to potential differences in microbial environments, microbial or host metabolic activity, or chromatin biology. Future studies will be important to tease apart these differences.
The PTMs that we describe here are regulated by the microbiota, and in the case of histone butyrylation, the availability of butyrate. Changes in histone butyrylation may be part of a mechanism of action of butyrate that has so far been overlooked. The landscape of histone PTMs is diverse in the intestine and can be regulated by the availability of donor molecules, and important future studies include in vivo tracing from microbes to deposition on histones via chromatin writer enzymes. The connection between how metabolism regulates histone PTMs and vice versa is an open question in the field. Some studies suggest that histone PTMs can function as a reservoir, where excess metabolites are deposited onto chromatin as histone PTMs for storage, and then recycled by chromatin modifying enzymes28,29. This hypothesis would fit with our study, where SCFAs are at high concentrations in the intestinal lumen and could then accumulate in cells. However, the metabolic reservoir hypothesis is also not mutually exclusive from these PTMs also having functional roles.
A major question from our work is: what is the function of histone butyrylation in gene expression, specifically in the intestinal epithelium? Interestingly, most genes rescued by tributyrin treatment in mice are downregulated (Fig. 3c). This may be surprising, as many studies have found that histone acyl marks are activating in transcription, including butyrate on histone H415. In addition, H3K27bu levels are correlated with gene expression (Fig. 4d). In contrast, histone crotonylation has a repressive role during yeast metabolic cycling30. These paradoxical observations suggest that more studies are needed to determine the functional role of H3K27bu in vivo. In addition, using traditional RNA-seq gives us a snapshot of gene expression in time, yet not direct versus indirect effects of H3K27bu. Investigating transcription factor-mediated responses and reader proteins (which are often effectors for histone PTMs) for these acylations and their potential impact on chromatin structure may be key future directions. For example, different structural domains differ in their abilities to recognize lysine acylations; compared to bromodomains, generally YEATS and double PHD finger domains could accommodate longer acyl side chains like butyrylation3. In addition, the top peaks of H3K27bu ChIP-seq in mouse cecal epithelial cells are enriched in oxidative stress and broadly cellular adaptations (Extended Data Fig. 8). This suggests that histone butyrylation potentially has a functional role in the cellular response to stress in the intestinal epithelium, consistent with butyrate and tributyrin reducing oxidative stress31–34. Future studies will be important to delineate the potential function of histone butyrylation through identifying specific contexts for when this histone mark may impact the cell stress response.
Another important course of future study is to separate the effects of histone butyrylation vs. acetylation. Sodium butyrate is an HDAC inhibitor, and as such has important biological roles, both in intestinal epithelial cells and other cell populations in the gut35,36. The use of additional model systems would be useful to tease apart the effects of these two similar histone PTMs37. Furthermore, our analyses are focused on gene expression in bulk epithelial cells from the intestine, and an important question is how cellular responses to butyrate and other microbial metabolites varies between different cell types in the gut. Finally, another limitation of our study is that our experiments are skewed towards female mice. An important area of future study will be investigating any potential sex differences in intestinal histone acylations.
Methods
Animals
C57BL/6J mice were purchased from Jackson Laboratories (000664). B6NTac murine pathogen free (MPF) or germ-free (GF) mice were purchased from Taconic Biosciences. All animals utilized were between 6–10 weeks old. Female mice were used for experiments unless otherwise noted. Mice were housed in Specific Pathogen Free conditions with controlled temperature, 12-hour light/dark cycles, and sterilized food (LabDiet 5053) and water were provided ad libitum unless otherwise noted. For treatment with antibiotics, single or multiple antibiotics (1 g/L ampicillin, 1 g/L neomycin, 0.5 g/L metronidazole, and/or 0.5 g/L vancomycin) were dissolved in drinking water with 10 g/L Splenda. Mice were acclimated to water with Splenda alone for two days prior to administration of antibiotics. For gavage experiments, gavages were performed using either 200 μl tributyrin (Sigma W222322) or with 200 μl equimolar glycerol solution for the mock gavage groups. Food was provided ad libitum until four hours prior to gavage, at which point it was removed. Mice were randomized into experimental groups according to ARRIVE guidelines using stratified randomization, with controlling for differences in bodyweight due to experimental designs with gavaging. Randomization was not performed for analysis of germ-free mice compared to conventional controls due to mouse availability and housing requirements. A power analysis was performed on a pilot experiment measuring the difference in global levels of histone acylations by immunoblotting following antibiotic treatment, and studies were designed with an alpha of 0.05 and 90% power to detect an effect size with a Cohen’s d of 2. Animal care and use followed NIH guidelines and was approved by the Institutional Animal Care and Use Committee (IACUC) at The Rockefeller University.
Histone extractions from tissues
All tissue samples were collected at time of sacrifice and snap frozen in liquid nitrogen and stored at −80 °C until processing. All tissues of the gastrointestinal tract were first minced in cold PBS with protease inhibitors. The tissue was spun briefly and the pellet brought up in cold PBS with protease inhibitors and 0.1% NP-40. Tissue homogenization was carried out using a Pellet Pestle for approximately 20–30 seconds per sample. Samples were then centrifuged at 200xg for 5 minutes and the supernatant removed. The pellet was then brought up in Modified RIPA Buffer (50 mM Tris-HCL pH 8.0, 150 mM NaCl, 1 mM EDTA, 1% Triton-X, 0.25% Sodium Deoxycholate, 0.1% SDS, Complete Protease Inhibitor, and 1 mM PMSF) and incubated on ice for 20 minutes. Samples were then centrifuged for 10 minutes at 16,000xg. The supernatant was saved as tissue lysate, and the pellet was processed for acid extraction of histones38. Briefly, pellets were vortexed in 0.4 N H2SO4 and incubated overnight, then precipitated with 100% TCA. After precipitation, pellets were washed with acetone and allowed to air dry. Histones were then resuspended in water and protein concentration was assessed with BCA assay (Thermo 23225) and Coomassie staining.
Mass spectrometry of histone PTMs
Histones were prepared for MS analysis using derivatization with propionic anhydride39. Isotopically labeled propionic anhydride (D10, 98%, Cambridge Isotope Laboratories Inc. DLM-3305-PK) was used to differentiate endogenous (light) from artificial (heavy) histone propionylation. Briefly, 1 volume of 25% propionic anhydride in 2-propanol was mixed with 2 volumes of 0.1M ammonium bicarbonate containing 10–20 μg of histone extracts or 2 μg of mono-nucleosome extracts. After allowing the reaction to proceed for 15 mins at 37°C, the histones were dried in a speed-vac. This derivatization was performed once more prior to digestion with trypsin (1 μg per 20 μg histones) overnight at room temperature in 0.1M ammonium bicarbonate. After digestion, two additional rounds of derivatization were performed to propionylate the free N-termini. Peptides were then desalted with C18 stage tips for analysis by LC-MS/MS.
Peptides were resolved by reversed phase liquid chromatography using EASY-nLC 1000, Dionex Ultimate3000, or NeoVanquish (Thermo) systems fitted with 75 μm i.d. columns (either 15–20 cm packed in-house with 2.4 μm C18 material or 25 cm Acclaim PepMap 3 μm from Thermo) positioned in line with a QE or QE-HF mass spectrometer (Thermo). The chromatography gradient typically started at 2% B for 2 mins, increasing to 10% B from 2–4 mins, increasing to 30% B from 4–49 mins, and increasing to 90% B from 49–52 mins. In some cases, another gradient was used increasing from 2 to 45% B from 0–40 mins and then increasing to 95% B from 40–41 mins. Solvents A and B consisted of 0.1% formic acid in water and 80% acetonitrile with 0.1% formic acid, respectively. Parallel reaction monitoring (PRM) was used to analyze peptides of interest, including the unmodified, acetylated, butyrylated, and endogenously propionylated forms of H3K9 and H3K27. Typical parameters of the PRM were as follows: positive polarity; 17,500 or 30,000 resolution; 5e5 or 1e6 AGC; 50 or 100 ms max injection time; 11 or 20 loop count; 1.2 m/z isolation width or 3.0 m/z isolation width with 0.7 m/z offset; 30 NCE; centroid mode. Each cycle also included a full MS scan with typical settings as follows: positive polarity; 60,000 or 70,000 resolution; 1e6 AGC; 50 or 100 ms max injection time; 200–1200 or 250–1000 m/z scan range; profile mode. Data were analyzed with Skyline and fragmentation patterns were visualized with the Interactive Peptide Spectral Annotator (interactivepeptidespectralannotator.com40). Peptide modifications that were considered included artificial D5-propionyl (+61.057598 to K and N-termini), endogenous propionyl (+56.026215 to K), butyryl (+70.041865 to K), and acetyl (+42.010565 to K).
To construct the titration curve of histone acyl marks, 2 μg of unmodified (Epicypher 16–0006) or acylated mono-nucleosomes (H3K9ac, H3K9bu, H3K27ac, H3K27bu; Epicypher 16–0314, 16–0371, 16–0365, 16–0384 respectively) were separately propionylated, digested, and analyzed by LC-MS/MS using 0.2 μg injections. Based on the observed peak areas of selected unmodified peptides from H2A, H2B, H3, and H4, peptide concentrations were equalized again to 110 ng/μl. A 10-fold dilution series was then prepared from a standard mixture of acyl marks, originally containing 3 μl of each acyl mark. 8 μl of each dilution, including the starting mixture, was then combined with 2 μl of peptides from unmodified nucleosomes. In this way, accounting for the unmodified H3K9 and H3K27 present in acylated H3K27 or H3K9 mono-nucleosomes, respectively, in addition to the unmodified H3K9 and H3K27 in unmodified mono-nucleosomes, the ratio of each acyl mark to its corresponding unmodified background ranges from 3.3E-1 to 2E-7. Final peptide concentrations were equalized to 22 ng/μl with solvent and 4.5 μl (100 ng) injections were used for LC-MS/MS analysis.
Immunoblotting
Samples were run on 16% or 4–20% tris glycine SDS-PAGE gels (Invitrogen XP00165BOX) and transferred to PVDF membranes. Blocking and all antibody incubations were done in 5% milk, with TBST washes in between. Immunoblots were imaged using Immobilon ECL (Millipore WBKLS0500) and an Amersham Imager 600 (GE). Antibodies used are as follows: pan-acetyllysine (PTM-Biolabs PTM-105, 1:5,000), pan-butyryllysine (PTM-Biolabs PTM-301, 1:5,000), pan-propionyllysine (PTM-Biolabs PTM-201, 1:5,000), H3 (Abcam, ab1791, 1:100,000), H3K27bu (Millipore ABE2854, 1:2,000), H3K9bu (PTM-Biolabs PTM-305, 1:1,000), H3K27pr (Millipore ABE2853, 1:2,000), H3K9pr (Millipore ABE2852, 1:2,000), H3K27ac (Active Motif 39133, 1:5,000).
Immunofluorescence
Intestinal tissues were fixed with 2% paraformaldehyde in PBS overnight. After fixation, tissues were washed with B1n buffer, pH 7.0 (0.3 M Glycine, 0.1% v/v Triton-X 100) followed by PBS, and then stored in 70% ethanol. Tissues were embedded in parafilm and sectioned to 5 μm sections by HistoWiz, Inc. After rehydrating, sections were boiled in sodium citrate buffer, pH 6.0 (10 mM sodium citrate, 0.05% Tween 20), and then stained. Imaging was performed on a Zeiss Inverted LSM 780 laser scanning confocal microscope using a 40x objective. Stainings and dilutions: α-H3K27bu (Millipore ABE2854, 1:500), α-Villin-647 (SantaCruz sc58897–647 lot D2920, 1:200), Goat anti-Rabbit Alexa Fluor 488 (Invitrogen A-11008, 1:1,000), DAPI (1:1,000).
16S Sequencing
16S sequencing was performed in the Microbiome Core Lab at Weill Cornell Medicine23. Cecal contents were subject to DNA extraction using the Max Prep and the Maxwell RSC 48 Instrument (Promega). Primers targeting the V4 and V5 regions (515F-926R) were used for amplification according to the protocol from the Earth Microbiome Project (https://earthmicrobiome.org/protocols-and-standards/16s/)41 and paired-end sequencing was performed on the Illumina MiSeq with 15% PhiX spike-in control. After demultiplexing, reads were merged using usearch v11.0.66742 with the following options: fastq_maxdiffs 5, -fastq_pctid 90, -fastq_minovlen 17, and -fastq_minmergelen 300. The following commands were used: usearch filter_phix (removal of PhiX reads), usearch-fastq_filter -fastq_maxee 1.0 (quality filtering), usearch -cluster_otus (OUT clustering), usearch -otutab (mapping merged pre-filtered reads to OTUs). Finally, taxonomic prediction was performed with usearch -sintax -strand both -sintax_cutoff 0.8. The RDP 16S training set (v16) was used as a reference database43,44.
Metabolomics
Small sections of mouse cecum (several mm3 and weighing approximately 10 mg) were collected immediately after dissection and flash-frozen in liquid nitrogen. No statistical methods were used to pre-determine sample size, but our sample sizes are similar to those reported in previous publications and consistent with published guidelines45. Samples were grinded in a mortar and pestle in liquid nitrogen and then weighed on an analytical scale. Polar metabolites were extracted in equal weight/volume 80% methanol that included 15N and 13C fully-labeled amino acid standards (MSK-A2–1.2, Cambridge Isotope Laboratories, Inc). Samples were shaken for ten minutes in the cold room and then centrifuged at 16,000xg for ten minutes to remove debris and proteins. Samples were then dried under nitrogen and stored at −80°C until analysis by liquid chromatography mass spectrometry (LC-MS). The samples were resuspended in 60 uL 2:2:1 acetonitrile:methanol:water, vortexed for 30 seconds, then centrifuged at 16,000xg and 4°C for 30 minutes. LC-MS analysis was performed46,47 and a QExactive orbitrap mass spectrometer was coupled to a Vanquish UPLC System (Thermo Fisher Scientific). The mass spectrometer was operated with a spray voltage set to 3.5 kV and heated capillary temperature of 350°C. MS1 data was acquired with the scan ranges of 55–440 and 438–876 m/z. Relative metabolite abundances were quantified using XCalibur QualBrowser 2.2 and Skyline Targeted Mass Spec Environment (MacCoss Lab) using a 5 ppm mass tolerance and a pooled-library of metabolite standards to verify metabolite identity. Relative metabolite levels were calculated by normalizing to total protein levels, as measured by Bicinchoninic Acid Assay (BCA).
Intestinal epithelial cell isolation
Epithelial cell isolation was carried out using established methods for the intestine48,49, which were further optimized for the cecum. All steps were performed on ice or 4 °C unless otherwise noted. After excising the cecum, any visible fat was removed. Tissues were then opened longitudinally and cleaned of feces with sequential washing in PBS. Once all visible feces were removed and tissues appeared clean, tissues were cut into approximately 1 cm sections and placed in Dissociation Reagent 1 (30 mM EDTA, 1.5 mM DTT in PBS) on ice for 20 minutes. Tissues were then transferred to Dissociation Reagent 2 (30 mM EDTA in DMEM + 2% FCS) and incubated at 37 °C for 10 minutes. After incubation, tissues were shaken by hand for 30 seconds until the solution became cloudy. The supernatant was then filtered through a stainless-steel sieve and spun at 600xg for 5 mins. The pellet was then washed with PBS + 10% FBS and centrifuged again. Finally, the pellet was resuspended in 10 mL HBSS with 0.5 U/ml Dispase (Corning 354235) and incubated for 5–10 minutes at 37 °C, shaking every 2 mins. Cells were checked for dissociation starting at 5 minutes, during which samples were placed back on ice. Once mostly single cells were observed, 10% FBS was added to quench the reaction. Cells were sequentially pass cells through 70 μm and 40 μm filters, spun down and washed once with 10% FBS. The final pellet was resuspended in FACS buffer (2.5% FBS in PBS), and cells were counted and visualized using a hemocytometer.
Chromatin Immunoprecipitation
Isolated cecal IECs from pooled mice (n =3) were fixed in PBS with 1% formaldehyde for 10 minutes, followed by quenching with 125 mM glycine for 5 minutes. Crosslinked cells were washed once with PBS and then lysed using the NEXSON protocol50. Briefly, cells were resuspended in FL Buffer (5 mM PIPES, pH 8.0, 85 mM KCl, 0.5% NP-40, protease inhibitor cocktail) and sonicated in the Bioruptor Pico (Diagenode) on low power at 15 seconds on 30 seconds off cycles until at least 70% cell membranes lysed and intact nuclei were visible. Nuclei were then washed in FL Buffer and resuspended in D3 Sonication Buffer (10 mM Tris-HCl, pH 8.0, 0.1% SDS, 1 mM EDTA, protease inhibitor cocktail) for sonication using the Covaris E220 (Covaris). Sonication was optimized in an assay-dependent manner and input DNA was checked prior to proceeding with immunoprecipitations (IPs). For IPs, sonicated chromatin was diluted into ChIP Dilution Buffer (10 mM Tris-HCl, pH 8.0, 1 mM EDTA, 150 mM NaCl, 1% Triton-X, protease inhibitor cocktail) and added to antibody-bound Protein A Dynabeads (Invitrogen 10002D) and incubated overnight in the cold room, rotating. Antibodies used were as follows: H3K27ac (Active Motif 39133), H3K27bu (Millipore ABE2854), H3K4me3 (Active Motif 39159), H3K27me3 (Cell Signaling 9733, clone 19). The next day, beads were washed six times with RIPA buffer (50 mM HEPES-KOH, pH 7.5, 100 mM LiCl, 1 mM EDTA, 0.7% Na-Deoxycholate, 1% NP-40) followed by one wash with TE-NaCl Buffer (10 mM Tris-HCl, pH 8.0, 50 mM NaCl, 1 mM EDTA). DNA was eluted from the beads using Elution Buffer (50 mM Tris-HCl, pH 8.0, 10 mM EDTA, 1% SDS). DNA was reverse crosslinked at 65 °C overnight, followed by RNase and ProteinaseK digestion and purification using Phenol-Chloroform Isoamyl Alcohol mix (Millipore 516726) and PhaseLock tubes (QuantaBio 2302830). DNA was quantified using Qubit 4 Fluorometer (Thermo) prior to proceeding with library preparation.
RNA isolation
Total RNA was isolated from cecal IECs using RNeasy Mini Kit (Qiagen 74104) with on-column DNA digestion. RNA samples were analyzed on the Bioanalyzer RNA 6000 Pico (Agilent) prior to library preparation.
Library preparations & sequencing
RNA libraries were converted to cDNA libraries using NEBNext Poly(A) mRNA Magnetic Isolation Module (NEB E7490L) and NEBNext Ultra II RNA Library Prep Kit for Illumina (NEB E7770L) according to the manufacturer’s instructions. ChIP libraries were prepared using NEBNext Ultra II DNA Library Prep Kit for Illumina (NEB E7645L) according to the manufacturer’s instructions. All libraries were analyzed by TapeStation prior to sequencing. Single-end sequencing was performed on the Illumina NextSeq 500 sequencer.
Bioinformatics analysis
Sequence and transcript coordinates for mouse mm10 UCSC genome and gene models were retrieved from the Bioconductor Bsgenome.Mmusculus.UCSC.mm10 (version 1.4.0) and TxDb.Mmusculus.UCSC.mm10.knownGene (version 3.4.0) Bioconductor libraries respectively.
For the analysis of RNAseq data, transcript expressions were calculated using the Salmon quantification software51(version 0.8.2) and gene expression levels as TPMs and counts retrieved using Tximport52(version 1.8.0). Normalization and rlog transformation of raw read counts in genes, PCA and differential gene expression analysis were performed using DESeq2 52(version 1.20.0). Additional sample to sample variability assessment was made with heat maps of between sample distances using Pheatmap (version 1.0.10). Significant genes (padj < 0.05 ) from a pairwise comparison between mice treated with ampicillin plus tributyrin vs. ampicillin with mock gavage were used for hierarchical clustering. The Z-score of the rlog of gene counts was used as the input for clustering. Clustering was done using Pheatmap after determining optimal number of clusters using the elbow method. Gene set enrichment tests were conducted using clusterProfiler (version 3.18.1)53.
For the analysis of ChIPseq data, reads were mapped using the Rsubread package’s align function (version 1.30.6)54. Peaks of enrichment were determined using the MACS2 peak caller (version 2.1.1)55. Consensus peaks were determined to be peaks that are found in common across replicates. Peaks were annotated and genome distribution was determined using the ChIPseeker package (Version 1.26.2)56. Heatmaps and metaplots were generated with deeptools (version 3.5). Normalized, fragment extended signal bigWigs are created using the rtracklayer package (version 1.40.6), and then visualized and exported from IGV.
Statistics
Details for statistical tests and replicates are described in the figure legends. All measurements shown were taken from distinct samples of individual mice unless otherwise indicated. No animals or data points were excluded from analysis. Data collection and analysis were not performed blind due to the conditions of the experiments. Data distribution was assumed to be normal but this was not formally tested, unless otherwise noted. Prism 7 (GraphPad v9) was used to generate plots and perform statistical tests. Error bars represent the standard error. Unpaired two-tailed Student’s t-test or one-way ANOVA with multiple comparisons was used to assess significance and is indicated in the figure legends. P<0.05 was considered statistically significant.
Extended Data
Extended Data Fig. 1: Chromatograms of histone acylations in the mouse intestine by mass spectrometry.
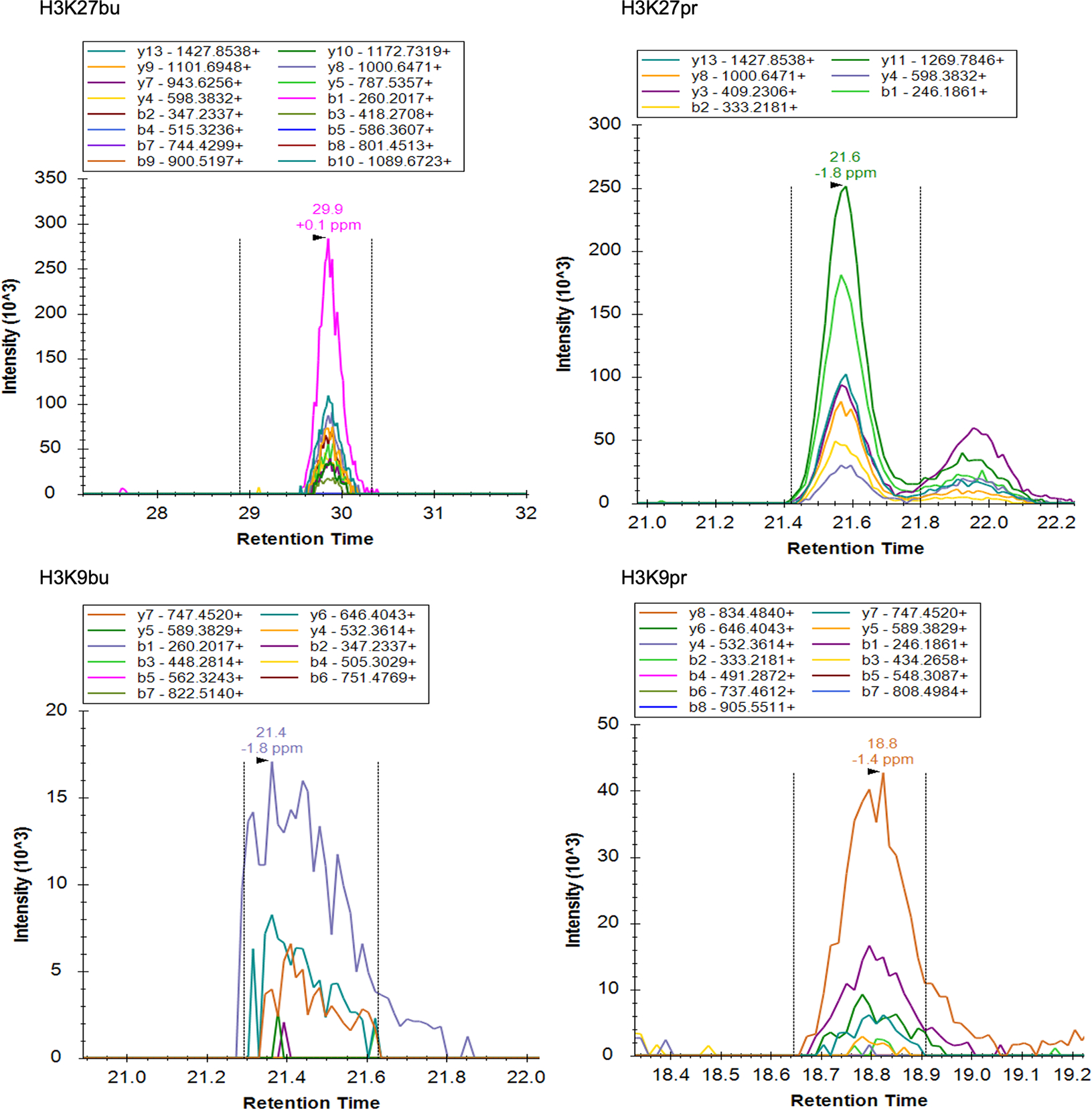
(A) Chromatograms of fragment ions from precursor histone peptides with acyl marks from female C57BL/6J mouse cecum. Representative traces are shown out of n = 3 mice.
Extended Data Fig. 2: Fragmentation spectra and comparison of histone acylations.

(A) Fragmentation spectra of unmodified peptides and peptides with H3K27pr, H3K9bu, and H3K9pr. All representative spectra are from histones extracted from female C57BL/6J mouse cecal samples. Representative traces are shown out of n = 3 mice. (B) Butyrylated peptides are not as readily detected as acetylated peptides at low relative abundances. Concomitant serial dilution of recombinant human nucleosomes with acetylated and butyrylated H3K9 and H3K27 nucleosomes into unmodified nucleosomes were analyzed by mass spectrometry.
Extended Data Fig. 3: Targeted mass spectrometry for crotonylated histones in the mouse cecum.
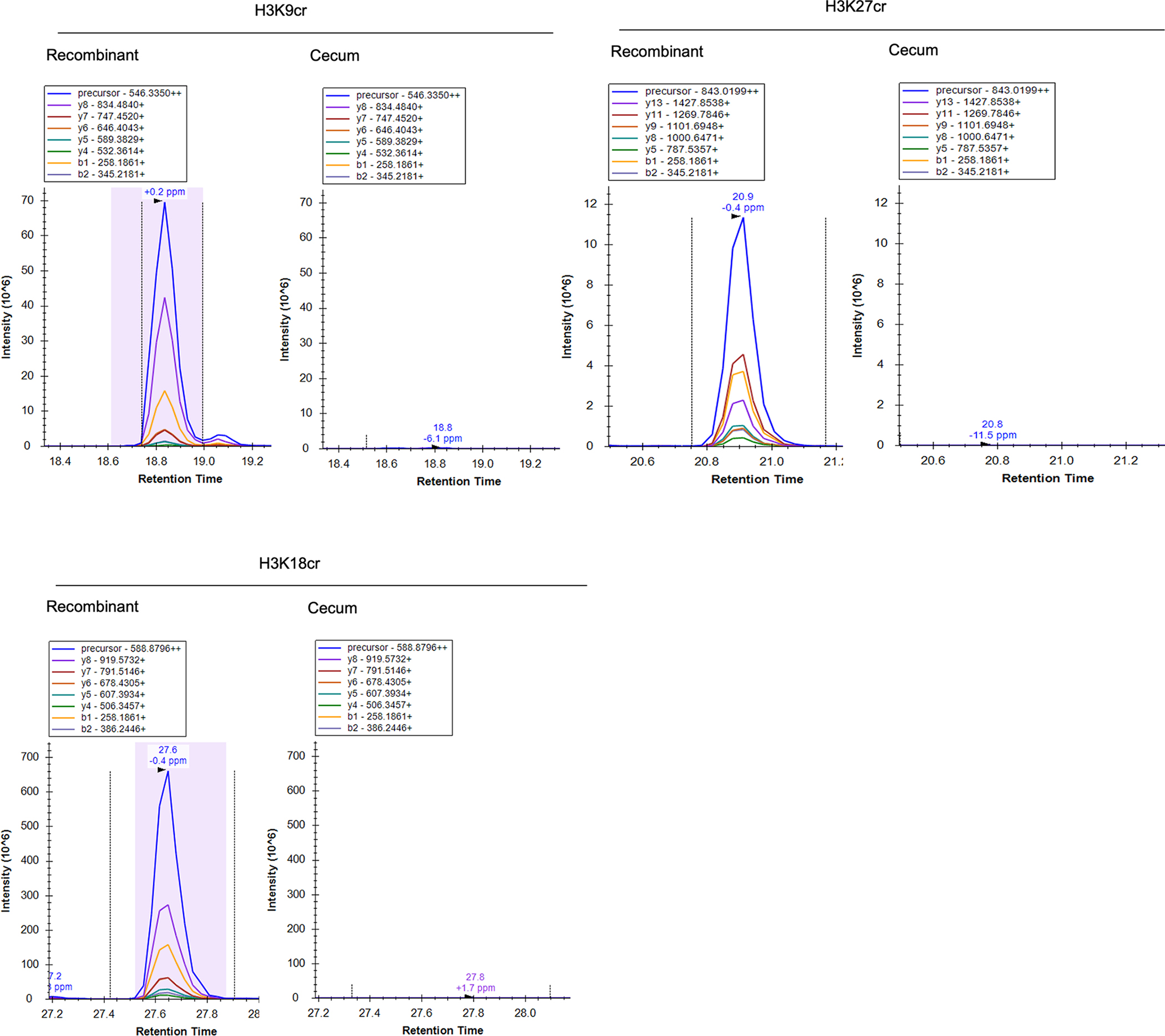
Chromatograms of fragment ions from precursor histone peptides from recombinant standards with crotonylated residues (left) or female C57BL/6J mouse cecum (right). Representative traces are shown out of n = 9 mice.
Extended Data Fig. 4: Characterization of site-specific butyryl antibodies.

Abbreviations are as follows: ac = acetyl, bu = butyryl, and cr = crotonyl. Testing of antibody specificity using recombinant nucleosomes. 30 or 100 ng recombinant human nucleosomes were run on an SDS-PAGE gel and subjected to immunoblotting. A representative blot is shown from two independent experiments.
Extended Data Fig. 5: Gut microbial composition is altered following antibiotic treatments.

C57BL/6J female mice were treated either with vehicle (10 g/l Splenda, n = 4 and n = 5), an antibiotic cocktail of 1 g/l ampicillin, 1 g/l neomycin, 0.5 g/l vancomycin, and 0.5 g/l metronidazole (n = 4) or ampicillin alone (n = 5). (A) Cecal weights are increased upon antibiotic treatment. Mice and intact ceca were weighed and the ratio of cecal weight to bodyweight is displayed. Statistical significance was determined by Student’s t-test. Graphs display mean and standard error. (B) Treatment with antibiotics causes alterations in the gut microbiota. 16S sequencing was performed on DNA extracted from mouse feces and the top ten bacterial families detected are shown.
Extended Data Fig. 6: Supplemental RNA-seq data.
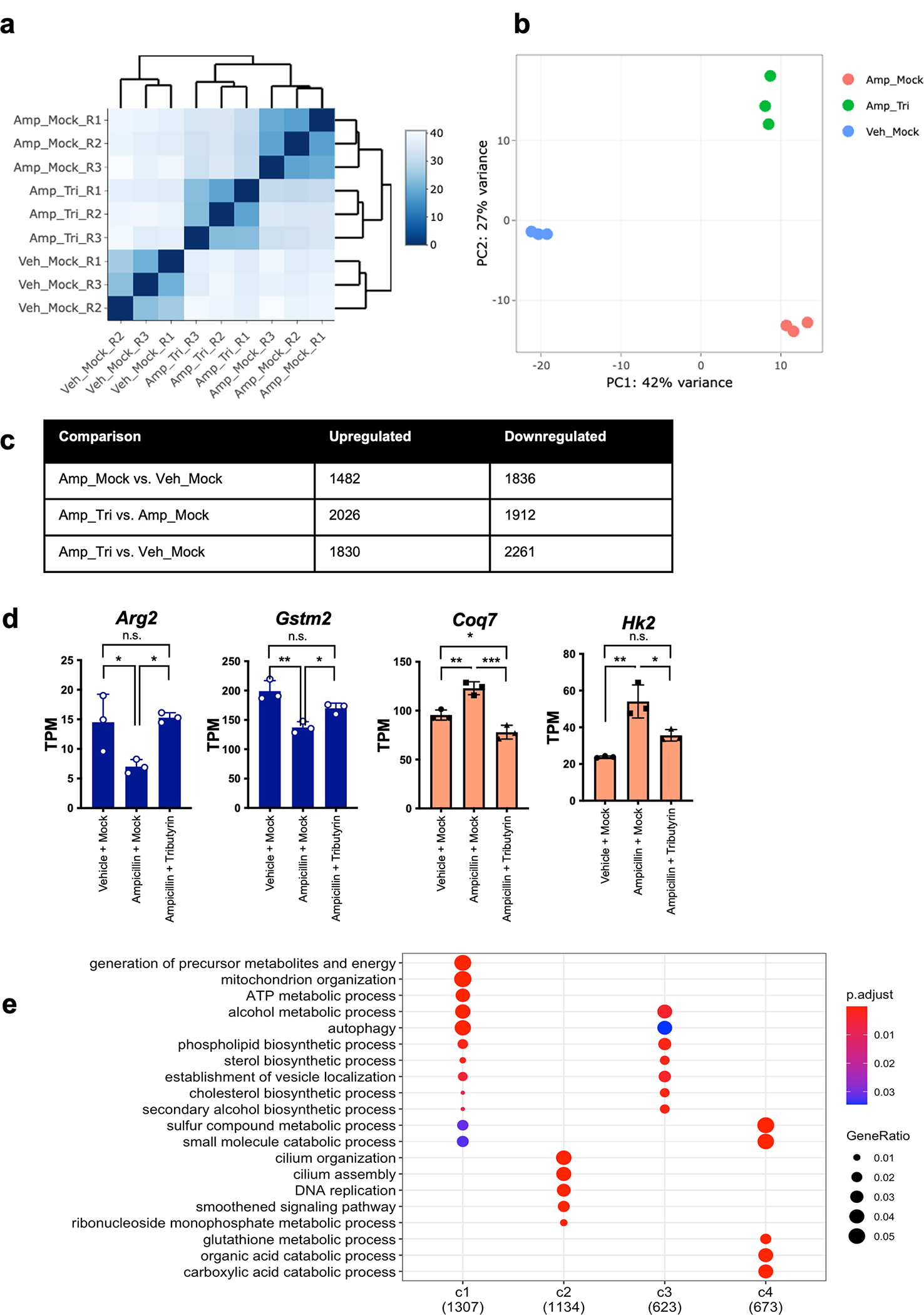
C57BL/6J female mice were divided into three different groups (n = 3 biologically independent animals per group): vehicle treated and mock gavaged (Veh_Mock), Ampicillin treated and mock gavaged (Amp_Mock), and Ampicillin treated with tributyrin gavage (Amp_Tri). Cecal IECs were isolated for RNA sequencing. (A) Heatmap of correlation between samples. Pearson correlation was performed of gene expression measurements across samples using DEseq2. (B) Principal component analysis of RNA-seq samples. (C) Table of significant differential gene expression across groups. Gene expression changes were identified using DEseq2 and p-values less than 0.05 were considered significant. The Wald test was used as part of the DESeq2 package for differential gene analysis, along with multiple testing correction using Benjamini-Hochberg false discovery rate to get the padj value. (D) Visualization of expression of select genes in clusters 1 (blue) and 4 (red). Graphs display mean and standard error. * p-value = < 0.05, ** p-value = < 0.01, *** p-value = < 0.001 by one-way ANOVA and adjusted for multiple comparisons. (E) Gene ontology analysis of all clusters of differential gene expression. Over Representation Analysis was performed using a one-sided version of Fisher’s exact test in the clusterProfiler package.
Extended Data Fig. 7: Metabolic profiling of cecal tissues following treatments.
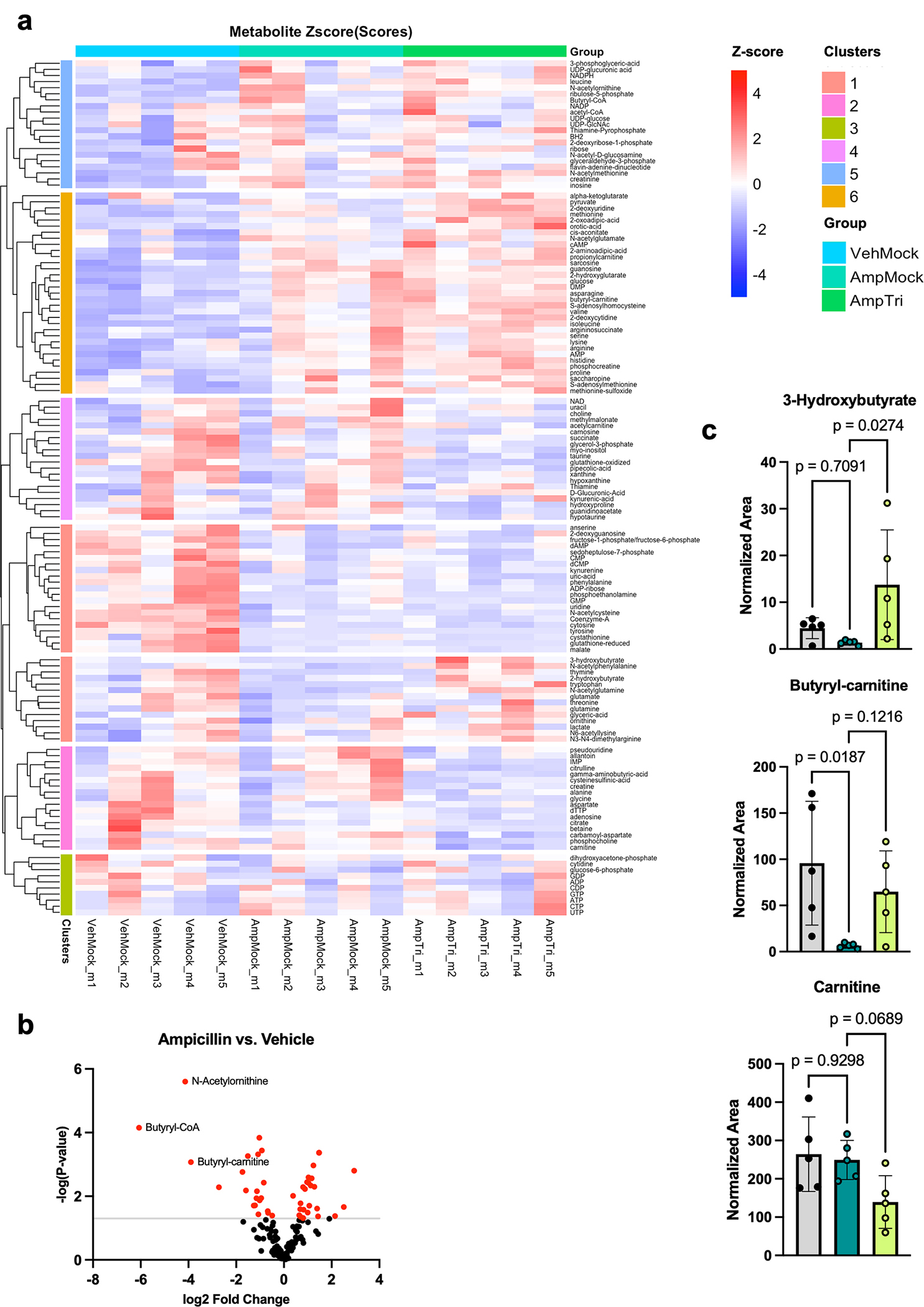
C57BL/6J female mouse cecal tissue was processed for polar metabolomics analysis by mass spectrometry. (A) Metabolomics heatmap of all metabolites detected. Heatmap displays z-scores of normalized area under the curve for each metabolite across different samples (n=5 biologically independent animals per group) and treatments. List of individual metabolites can be found in Supplementary Table 2. (B) Volcano plots displaying changes in metabolites comparing ampicillin treated mice to vehicle control. Red dots are metabolites that are significantly changed (p-value < 0.05 by unpaired two-tailed Student’s t-test) and grey line indicates significance threshold. (C) Metabolites related to butyrate metabolism are changing with ampicillin or tributyrin treatments in mice. Metabolites related to butyrate metabolism (3-hydroxybutyrate and butyryl-carnitine) are altered following mouse treatments, while carnitine serves as an example of a metabolite that is largely unchanging and does not follow the same pattern. Graphs display mean and standard error. Statistical significance was determined by one-way ANOVA and adjusted for multiple comparisons (n=5 biologically independent animals per group).
Extended Data Fig. 8: Gene ontology of top H3K27bu ChIP-seq peaks.
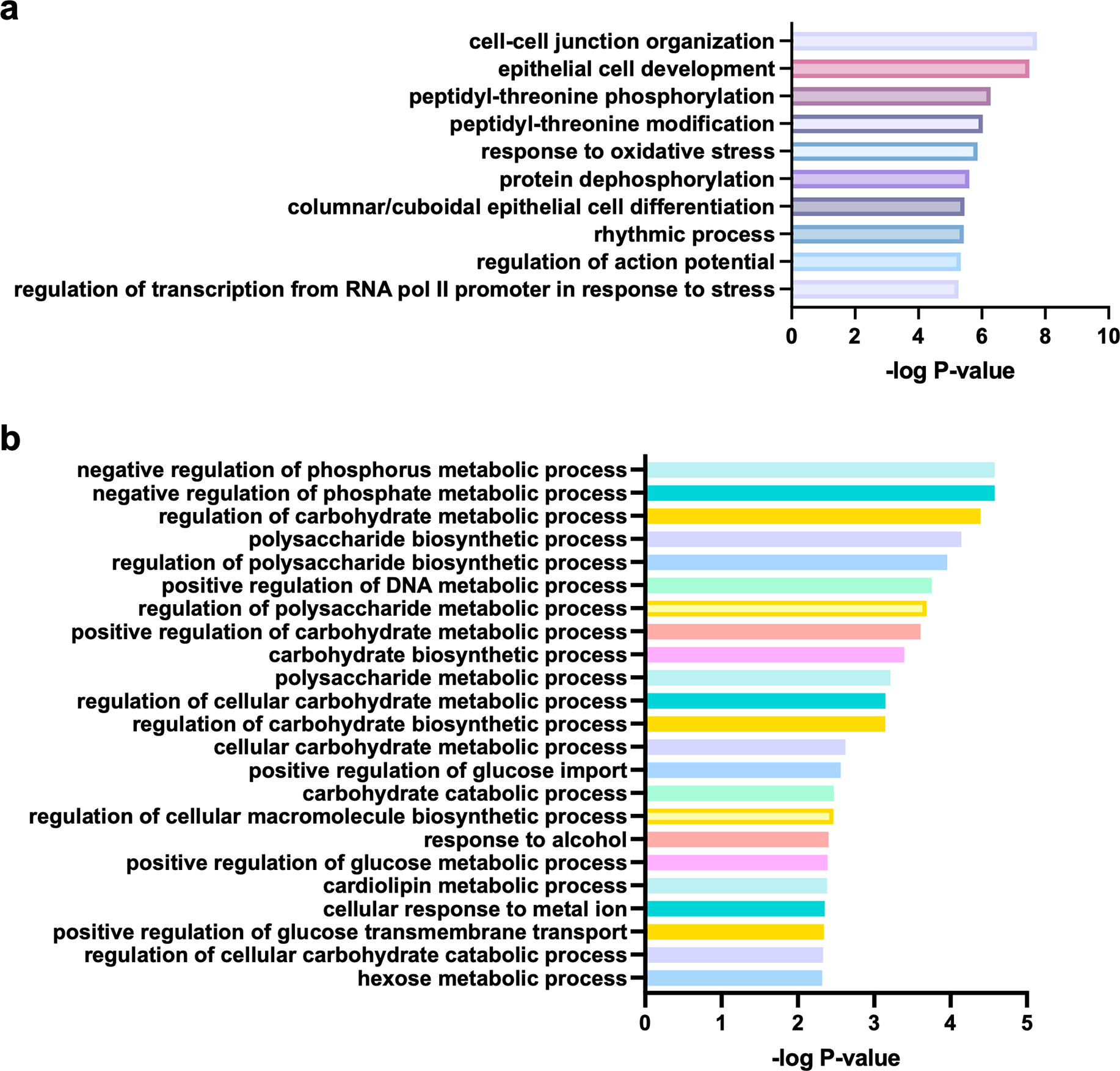
The top 10% of H3K27bu peaks from female C57BL/6J mouse cecal intestinal epithelial cells were analyzed for enrichment of gene ontology categories. The peak counts of each of two replicate ChIP-seq experiments from biologically independent mice were averaged, and then the top 10% used for analysis. Over Representation Analysis was performed using a one-sided version of Fisher’s exact test in the clusterProfiler package. (A) Top 10 most significantly enriched GO categories are displayed. (B) All GO categories related to cellular metabolism, excluding any related to oxidative stress, are displayed.
Supplementary Material
Acknowledgements
We thank all members of the Allis and Mucida laboratories. We thank Steven Josefowicz (Weill Cornell Medical College), Rachel Niec (Memorial Sloan Kettering Cancer Center), Yadira Soto-Feliciano (Massachusetts Institute of Technology), and Alexey Soshnev (University of Texas San Antonio) for helpful discussions and support. C.D.A. wishes to acknowledge the encouragement of the above Tri-Institutional faculty and The Rockefeller University for financial support. Select schematics were generated using BioRender. L.A.G. was supported by the Shapiro-Silverberg Fund for the Advancement of Translational Research at The Rockefeller University, a National Institutes of Health (NIH) Ruth L. Kirschstein NRSA fellowship (F32GM134560), and a NIH MOSAIC Career Award (K99GM143550). P.J.L. was supported by the Crohn’s and Colitis Foundation (RFA 598467 to P.J.L) and the NIH (T32CA009140). B.A.G was supported by the NIH (R01AI118891 and R01HD106051 to B.A.G.). We thank MilliporeSigma for the generation of H3K27bu, H3K27pr, and H3K9pr antibodies. We acknowledge the help and support of the following Resource Centers at Rockefeller University: Bio-Imaging, Genomics, Bioinformatics, Proteomics, and Comparative Bioscience Center, and the Microbiome Core Lab at Weill Cornell Medicine. This article is dedicated to the memory of C. David Allis, who died on January 8, 2023.
Footnotes
Competing Interests Statement
The authors declare no competing interests.
Code Availability
No custom code was used for this study.
Data Availability
The RNA-seq and ChIP-seq data have been deposited to the NCBI GEO accession database under accession number GSE216319. The histone mass spectrometry data has been deposited to the MassIVE database under dataset number MSV000090586. Metabolomics data has been deposited to the Metabolomics Workbench under Study ID ST002969. Source data is available at Figshare (https://doi.org/10.6084/m9.figshare.24749973) and is provided with the manuscript. Any additional data that support the findings of this study or materials are available from the corresponding author upon request.
References
- 1.Allis CD & Jenuwein T The molecular hallmarks of epigenetic control. Nat Rev Genet 17, 487–500 (2016). [DOI] [PubMed] [Google Scholar]
- 2.Sabari BR et al. Intracellular Crotonyl-CoA Stimulates Transcription through p300-Catalyzed Histone Crotonylation. Molecular Cell 58, 203–215 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sabari BR, Zhang D, Allis CD & Zhao Y Metabolic regulation of gene expression through histone acylations. Nature Reviews Molecular Cell Biology 18, 90–101 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhang D et al. Metabolic regulation of gene expression by histone lactylation. Nature 574, 575–580 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wellen KE et al. ATP-Citrate Lyase Links Cellular Metabolism to Histone Acetylation. Science 324, 1076–1080 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Carey BW, Finley LWS, Cross JR, Allis CD & Thompson CB Intracellular α-ketoglutarate maintains the pluripotency of embryonic stem cells. Nature 518, 413–416 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dai Z, Ramesh V & Locasale JW The evolving metabolic landscape of chromatin biology and epigenetics. Nat Rev Genet 21, 737–753 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tan M et al. Identification of 67 Histone Marks and Histone Lysine Crotonylation as a New Type of Histone Modification. Cell 146, 1016–1028 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Koh A, Vadder FD, Kovatcheva-Datchary P & Bäckhed F From Dietary Fiber to Host Physiology: Short-Chain Fatty Acids as Key Bacterial Metabolites. Cell 165, 1332–1345 (2016). [DOI] [PubMed] [Google Scholar]
- 10.Cummings JH, Pomare EW, Branch WJ, Naylor CP & Macfarlane GT Short chain fatty acids in human large intestine, portal, hepatic and venous blood. Gut 28, 1221 (1987). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.den Besten G et al. The role of short-chain fatty acids in the interplay between diet, gut microbiota, and host energy metabolism. J. Lipid Res. 54, 2325–2340 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Farrelly LA et al. Histone serotonylation is a permissive modification that enhances TFIID binding to H3K4me3. Nature 567, 535–539 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Brownell JE et al. Tetrahymena Histone Acetyltransferase A: A Homolog to Yeast Gcn5p Linking Histone Acetylation to Gene Activation. Cell 84, 843–851 (1997). [DOI] [PubMed] [Google Scholar]
- 14.Kebede AF et al. Histone propionylation is a mark of active chromatin. Nature Structural & Molecular Biology 24, 1048–1056 (2017). [DOI] [PubMed] [Google Scholar]
- 15.Goudarzi A et al. Dynamic Competing Histone H4 K5K8 Acetylation and Butyrylation Are Hallmarks of Highly Active Gene Promoters. Molecular Cell 62, 169–180 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tognini P et al. Distinct Circadian Signatures in Liver and Gut Clocks Revealed by Ketogenic Diet. Cell Metabolism 26, 523–538.e5 (2017). [DOI] [PubMed] [Google Scholar]
- 17.Fleming SE, Fitch MD, DeVries S, Liu ML & Kight C Nutrient Utilization by Cells Isolated from Rat Jejunum, Cecum and Colon. The Journal of Nutrition 121, 869–878 (1991). [DOI] [PubMed] [Google Scholar]
- 18.Roediger WEW Utilization of Nutrients by Isolated Epithelial Cells of the Rat Colon. Gastroenterology 83, 424–429 (1982). [PubMed] [Google Scholar]
- 19.Donohoe DR et al. The Warburg Effect Dictates the Mechanism of Butyrate-Mediated Histone Acetylation and Cell Proliferation. Mol Cell 48, 612–626 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Brown AJ et al. The Orphan G Protein-coupled Receptors GPR41 and GPR43 Are Activated by Propionate and Other Short Chain Carboxylic Acids. Journal of Biological Chemistry 278, 11312–11319 (2003). [DOI] [PubMed] [Google Scholar]
- 21.Poul EL et al. Functional Characterization of Human Receptors for Short Chain Fatty Acids and Their Role in Polymorphonuclear Cell Activation. Journal of Biological Chemistry 278, 25481–25489 (2003). [DOI] [PubMed] [Google Scholar]
- 22.Vidali G, Boffa LC, Bradbury EM & Allfrey VG Butyrate suppression of histone deacetylation leads to accumulation of multiacetylated forms of histones H3 and H4 and increased DNase I sensitivity of the associated DNA sequences. Proceedings of the National Academy of Sciences 75, 2239–2243 (1978). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lund PJ et al. Stable Isotope Tracing in vivo Reveals A Metabolic Bridge Linking the Microbiota to Host Histone Acetylation. Biorxiv 2021.07.05.450926 (2021) doi: 10.1101/2021.07.05.450926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Krautkramer KA et al. Diet-Microbiota Interactions Mediate Global Epigenetic Programming in Multiple Host Tissues. Mol Cell 64, 982–992 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fellows R et al. Microbiota derived short chain fatty acids promote histone crotonylation in the colon through histone deacetylases. Nature Communications 1–15 (2018) doi: 10.1038/s41467-017-02651-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kelly CJ et al. Crosstalk between Microbiota-Derived Short-Chain Fatty Acids and Intestinal Epithelial HIF Augments Tissue Barrier Function. Cell Host and Microbe 17, 662–671 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Donohoe DR, Wali A, Brylawski BP & Bultman SJ Microbial Regulation of Glucose Metabolism and Cell-Cycle Progression in Mammalian Colonocytes. PLoS ONE 7, e46589–9 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ye C & Tu BP Sink into the Epigenome: Histones as Repositories That Influence Cellular Metabolism. Trends in Endocrinology and Metabolism 29, 626–637 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mendoza M et al. Enzymatic transfer of acetate on histones from lysine reservoir sites to lysine activating sites. Science Advances 8, eabj5688 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gowans GJ et al. Recognition of Histone Crotonylation by Taf14 Links Metabolic State to Gene Expression. Mol Cell 76, 909–921.e3 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rosignoli P et al. Protective activity of butyrate on hydrogen peroxide-induced DNA damage in isolated human colonocytes and HT29 tumour cells. 22, 1675–1680 (2001). [DOI] [PubMed] [Google Scholar]
- 32.Hamer HM et al. Butyrate modulates oxidative stress in the colonic mucosa of healthy humans. 28, 88–93 (2009). [DOI] [PubMed] [Google Scholar]
- 33.Leonel AJ et al. Antioxidative and immunomodulatory effects of tributyrin supplementation on experimental colitis. British Journal of Nutrition 109, 1396–1407 (2013). [DOI] [PubMed] [Google Scholar]
- 34.Liu P et al. The role of short-chain fatty acids in intestinal barrier function, inflammation, oxidative stress, and colonic carcinogenesis. 165, 105420 (2021). [DOI] [PubMed] [Google Scholar]
- 35.Furusawa Y et al. Commensal microbe-derived butyrate induces the differentiation of colonic regulatory T cells. Nature 504, 446–450 (2013). [DOI] [PubMed] [Google Scholar]
- 36.Wu S et al. Microbiota-derived metabolite promotes HDAC3 activity in the gut. Nature (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Liu X et al. MOF as an evolutionarily conserved histone crotonyltransferase and transcriptional activation by histone acetyltransferase-deficient and crotonyltransferase-competent CBP/p300. Cell Discov 3, 17016 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shechter D, Dormann HL, Allis CD & Hake SB Extraction, purification and analysis of histones. Nat Protoc 2, 1445–1457 (2007). [DOI] [PubMed] [Google Scholar]
- 39.Lund PJ et al. Isotopic Labeling and Quantitative Proteomics of Acetylation on Histones and Beyond. in 43–70 (Springer New York, 2019). doi: 10.1007/978-1-4939-9232-4_5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Brademan DR, Riley NM, Kwiecien NW & Coon JJ Interactive Peptide Spectral Annotator: A Versatile Web-based Tool for Proteomic Applications. Mol Cell Proteomics 18, S193–S201 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ul-Hasan S et al. Community ecology across bacteria, archaea and microbial eukaryotes in the sediment and seawater of coastal Puerto Nuevo, Baja California. PLoS ONE 14, e0212355 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Edgar RC Search and clustering orders of magnitude faster than BLAST. Bioinformatics 26, 2460–2461 (2010). [DOI] [PubMed] [Google Scholar]
- 43.Edgar RC SINTAX: a simple non-Bayesian taxonomy classifier for 16S and ITS sequences. bioRxiv (2016) doi: 10.1101/074161. [DOI] [Google Scholar]
- 44.Cole JR et al. Ribosomal Database Project: data and tools for high throughput rRNA analysis. 42, D633–D642 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Turtoi E et al. Analysis of polar primary metabolites in biological samples using targeted metabolomics and LC-MS. 4, 102400 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Unlu G et al. Metabolic-scale gene activation screens identify SLCO2B1 as a heme transporter that enhances cellular iron availability. Mol Cell 82, 2832–2843 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kenny TC et al. Integrative genetic analysis identifies FLVCR1 as a plasma-membrane choline transporter in mammals. Cell Metab. 35, (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gracz AD, Puthoff BJ & Magness ST Somatic Stem Cells, Methods and Protocols. Methods Mol Biology 879, 89–107 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Reis BS, Rogoz A, Costa-Pinto FA, Taniuchi I & Mucida D Mutual expression of the transcription factors Runx3 and ThPOK regulates intestinal CD4+ T cell immunity. Nature Immunology 14, 271–280 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Arrigoni L et al. Standardizing chromatin research: a simple and universal method for ChIP-seq. Nucleic Acids Res 44, e67–e67 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Patro R, Duggal G, Love MI, Irizarry RA & Kingsford C Salmon provides fast and bias-aware quantification of transcript expression. Nature Methods 14, 417–419 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Love MI, Hogenesch JB & Irizarry RA Modeling of RNA-seq fragment sequence bias reduces systematic errors in transcript abundance estimation. Nature Biotechnology 34, 1287–1291 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Yu G, Wang L-G, Han Y & He Q-Y clusterProfiler: an R Package for Comparing Biological Themes Among Gene Clusters. OMICS: A Journal of Integrative Biology 16, 284–287 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Liao Y, Smyth GK & Shi W The R package Rsubread is easier, faster, cheaper and better for alignment and quantification of RNA sequencing reads. Nucleic Acids Research 47, e47–e47 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zhang Y et al. Model-based Analysis of ChIP-Seq (MACS). Genome Biology 9, R137 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Yu G, Wang L-G & He Q-Y ChIPseeker: an R/Bioconductor package for ChIP peak annotation, comparison and visualization. Bioinformatics 31, 2382–2383 (2015). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The RNA-seq and ChIP-seq data have been deposited to the NCBI GEO accession database under accession number GSE216319. The histone mass spectrometry data has been deposited to the MassIVE database under dataset number MSV000090586. Metabolomics data has been deposited to the Metabolomics Workbench under Study ID ST002969. Source data is available at Figshare (https://doi.org/10.6084/m9.figshare.24749973) and is provided with the manuscript. Any additional data that support the findings of this study or materials are available from the corresponding author upon request.