

Abstract
At first recurrence, platinum-sensitive ovarian cancer (PSOC) is frequently treated with platinum-based chemotherapy doublets plus bevacizumab, then single-agent bevacizumab. Most patients' disease progresses within a year after chemotherapy, emphasizing the need for novel strategies. Mirvetuximab soravtansine-gynx (MIRV), an antibody–drug conjugate, comprises a folate receptor alpha (FRα)-binding antibody and tubulin-targeting payload (maytansinoid DM4). In FRα-high PSOC, MIRV plus bevacizumab previously showed promising efficacy (objective response rate, 69% [95% CI: 41–89]; median progression-free survival, 13.3 months [95% CI: 8.3–18.3]; median duration of response, 12.9 months [95% CI: 6.5–15.7]) and safety. The Phase III randomized GLORIOSA trial will evaluate MIRV plus bevacizumab vs. bevacizumab alone as maintenance therapy in patients with FRα-high PSOC who did not have disease progression following second-line platinum-based doublet chemotherapy plus bevacizumab.
Clinical Trial Registration: ClinicalTrials.gov ID: NCT05445778; GOG.org ID: GOG-3078; ENGOT.ESGO.org ID: ENGOT-ov76
Keywords: : bevacizumab, folate receptor alpha, GLORIOSA trial, maintenance therapy, mirvetuximab soravtansine, platinum-sensitive ovarian cancer
Plain Language Summary
Most patients with ovarian cancer are initially treated with platinum-based chemotherapy. If the cancer reappears/recurs after more than 6 months following this therapy, it is called platinum-sensitive ovarian cancer (PSOC). Patients with PSOC usually receive additional platinum-based chemotherapy along with bevacizumab, a drug that reduces tumor growth by decreasing its blood supply. If patients improve or are stable on this therapy, they are usually kept on bevacizumab alone for ‘maintenance therapy’. Unfortunately, this maintenance therapy does not work long-term in all patients, so better long-term treatments are needed. The GLORIOSA (NCT05445778) clinical trial will compare maintenance therapy with bevacizumab alone to maintenance therapy with bevacizumab plus a drug called mirvetuximab soravtansine-gynx (MIRV) to determine which therapy leads to better results in patients with PSOC. MIRV is made up of an antibody that binds to a specific protein (folate receptor alpha [FRα]) on cancer cells to directly deliver a cancer-killing drug. MIRV received US FDA approval to be used as a therapy for patients with ovarian cancer who are resistant to platinum-based chemotherapy and express high levels of FRα. The GLORIOSA trial will study maintenance therapy with MIRV plus bevacizumab in patients with PSOC who have not had cancer progression after second-line platinum-based chemotherapy plus bevacizumab, and whose cancer expresses high amounts of FRα. The main purpose of this trial is to determine if MIRV plus bevacizumab leads to better patient survival and decreases cancer growth and spread when compared with bevacizumab alone.
Tweetable Abstract
GLORIOSA is an ongoing randomized, open-label, Phase III #clinicaltrial of mirvetuximab soravtansine + bevacizumab vs. bevacizumab alone as maintenance therapy in FRα-high platinum-sensitive ovarian, fallopian tube or primary peritoneal cancer.
Plain language summary
Article highlights.
Background
Epithelial ovarian cancer is typically diagnosed at a late stage, and up to 80% of patients experience recurrence after initially responding to platinum-based chemotherapy. Disease that progresses >6 months after platinum-based treatment is traditionally referred to as platinum-sensitive ovarian cancer (PSOC).
Patients with PSOC are frequently treated with platinum-based chemotherapy doublets plus bevacizumab followed by single-agent bevacizumab at first recurrence, and many experience disease progression within a year of chemotherapy.
Folate receptor alpha (FRα) is overexpressed in 72–97% of ovarian tumors.
Mirvetuximab soravtansine-gynx (MIRV) is an antibody–drug conjugate comprising an FRα-binding antibody attached via a cleavable linker to the maytansinoid DM4 payload, a potent microtubule inhibitor.
Findings from the confirmatory, randomized, global, Phase III MIRASOL trial (NCT04209855) supported full approval of MIRV by the US Food and Drug Administration (March 2024) for patients with FRα-high platinum-resistant ovarian cancer (PROC) who have received one to three prior lines of therapy.
The combination of MIRV and bevacizumab previously showed high clinical activity, durable responses, and a manageable safety profile in participants with PSOC and PROC across all FRα expression levels, regardless of prior bevacizumab exposure (Phase Ib/2 FORWARD II trial, NCT02606305).
GLORIOSA Trial
GLORIOSA, a randomized, open-label, Phase III trial (NCT05445778), will evaluate MIRV plus bevacizumab vs. bevacizumab alone as maintenance therapy in participants with FRα-high PSOC whose disease has not progressed after second-line (2L) platinum-based chemotherapy doublet plus bevacizumab.
Prior to randomization, patients must have received four to eight cycles of standard 2L platinum-based chemotherapy doublet with ≥3 cycles of bevacizumab (“triplet therapy”).
Patients can be screened and enrolled via two methods: before initiation (participants complete a run-in before enrollment into the randomized portion of the trial) or after completion (participants are enrolled directly into the randomized portion of the trial) of standard 2L triplet therapy. Entry into the randomized portion of the trial will be limited to patients without disease progression following their 2L triplet therapy.
The primary end point is investigator-assessed progression-free survival (also assessed by blinded independent central review).
The key secondary end point is overall survival.
Other secondary end points include evaluation of adverse events, second progression-free survival, objective response rate, duration of response, disease-free survival, cancer antigen 125 response rate and patient-reported outcomes.
Conclusion
The GLORIOSA trial will define the role of MIRV plus bevacizumab as a novel maintenance regimen in patients with FRα-high PSOC whose disease has not progressed after 2L platinum-based chemotherapy doublet plus bevacizumab.
Infographic
Infographic:

A PDF version of this infographic is available as supplemental material.
1. Introduction
1.1. Background & rationale
Epithelial ovarian cancer (EOC; includes primary peritoneal and fallopian tube cancer) accounts for 95% of all ovarian cancers and is typically diagnosed at an advanced stage [1,2]. EOC is the deadliest gynecologic malignancy; in 2020, approximately 313,959 new EOC cases and 207,252 EOC deaths occurred worldwide (EU, 44,053 deaths; US, 13,438 deaths) [3–6]. First-line treatment consists of debulking surgery along with taxane- and platinum-based chemotherapy with or without bevacizumab, followed by maintenance therapy with bevacizumab and/or poly (adenosine diphosphate [ADP]-ribose) polymerase inhibitors (PARPi) [7–10]. Although most patients with ovarian cancer initially respond to platinum-based therapy, up to 80% experience recurrence, and the 5-year relative survival rate is approximately 50% [11,12]. Ovarian cancer that progresses >6 months after completion of platinum-containing chemotherapy is traditionally considered platinum-sensitive [13]. Re-treatment with platinum-based chemotherapy remains an important tool in managing patients with platinum-sensitive ovarian cancer (PSOC) [14]. Most patients with PSOC receive platinum-based doublets (typically carboplatin plus paclitaxel, gemcitabine or pegylated liposomal doxorubicin [PLD]) at first recurrence [15–17].
Recognition of angiogenesis in ovarian carcinoma led to the emergence of vascular endothelial growth factor A (VEGF-A) as a therapeutic target; bevacizumab was the first VEGF-A–targeted agent approved for EOC [18,19]. Initial bevacizumab approval was based on the AURELIA trial, which found that addition of bevacizumab to chemotherapy significantly improved progression-free survival (PFS) in patients with platinum-resistant ovarian cancer (PROC) [8,20]. Based on results from the GOG-0213 and OCEANS trials, bevacizumab plus platinum-doublet chemotherapy (triplet therapy) followed by bevacizumab maintenance therapy was subsequently approved for PSOC [21–23]. Recent Phase III trial results found that bevacizumab plus carboplatin and PLD was also efficacious in PSOC [24]. Real-world evidence demonstrates that first-line bevacizumab provides a median PFS of approximately 17.4 months in patients with newly diagnosed ovarian cancer [25]. Further, in patients with PSOC who received bevacizumab as part of first-line therapy, bevacizumab re-treatment at first recurrence of PSOC improved PFS compared with chemotherapy alone [26–28].
1.2. Mirvetuximab soravtansine-gynx
Great interest in molecularly targeted strategies for ovarian cancer led to the identification of folate receptor alpha (FRα) as a novel therapeutic target. FRα is a cell surface protein that mediates the active transport of folate into cells via receptor-mediated endocytosis [29]. Although FRα expression is limited in normal adult tissue and absent from normal ovarian epithelium [30], it is expressed in approximately 72–97% of ovarian tumors [31,32,33]. Further, its overexpression may be a negative prognostic factor of chemotherapeutic response [32,34]. Given its almost ubiquitous expression on the surface of high-grade serous ovarian carcinomas and its ability to internalize large molecules containing a toxic payload, FRα is suitable for development as an antibody–drug conjugate (ADC) [35]. Mirvetuximab soravtansine-gynx (MIRV) is an ADC comprising an FRα-binding antibody, a cleavable linker and the maytansinoid DM4 payload, a potent tubulin-targeting agent (Figure 1) [36,37]. MIRV has also demonstrated bystander cytotoxic activity (diffusion of payload metabolites from antigen-targeted cells into neighboring cells), enabling cytotoxic activity in tumors with heterogenous FRα expression [36,37].
Figure 1.
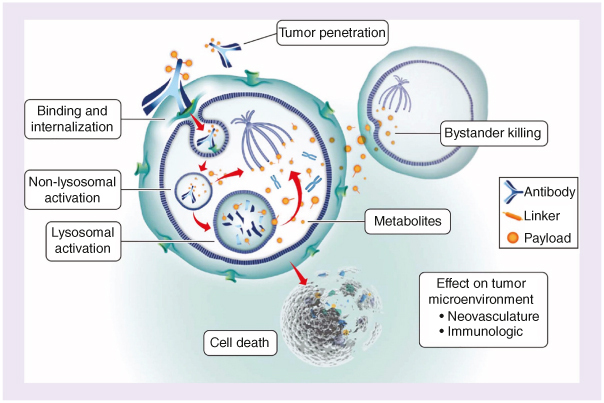
Mirvetuximab soravtansine-gynx mechanism of action. Mirvetuximab soravtansine-gynx (MIRV) binds with high affinity to folate receptor alpha (FRα), which is expressed on the tumor cell surface, prompting receptor-mediated internalization of MIRV via antigen-mediated endocytosis. Lysosomal degradation releases the cytotoxic payload, the maytansinoid DM4, that inhibits tubulin polymerization and microtubule assembly. DM4 metabolites then induce potent antimitotic effects that result in cell cycle arrest and apoptosis. The active metabolites may also diffuse into neighboring cells and induce further cell death (termed “bystander killing”). Reproduced without changes from Moore KN, Vergote I, Oaknin A, et al. FORWARD I: a Phase III study of mirvetuximab soravtansine vs. chemotherapy in platinum-resistant ovarian cancer. Future Oncol. 2018;14(17):1669–1678. https://doi.org/10.2217/fon-2017-0646. © 2018 KN Moore. http://creativecommons.org/licenses/by-nc-nd/4.0/.
The efficacy and safety of MIRV monotherapy have been evaluated in several clinical trials. The first-in-human, Phase I ‘401 trial’ (NCT01609556) of MIRV in patients with EOC and other solid tumors established the recommended Phase II dose as 6 mg/kg adjusted ideal body weight (AIBW) intravenously (IV) once every 3 weeks (Q3W) [37]. The Phase III FORWARD I trial (NCT02631876) then evaluated MIRV monotherapy vs. chemotherapy in participants with FRα-positive PROC (defined as ≥50% of tumor cells expressing FRα using a 10× scoring method) and one to three prior lines of therapy [38,39]. While this trial did not meet its primary end point of PFS, secondary end points consistently favored MIRV, particularly in a prespecified population of participants with FRα-high tumors [38]. Exploratory rescoring of FRα using a method referred to as positive staining 2+ (PS2+) revealed that the 10× scoring method used for screening in FORWARD I had diminished the treatment effect of MIRV by allowing participants to enroll with lower than expected levels of FRα expression [38,40]. These findings supported the use of the PS2+ method for FRα evaluation in subsequent clinical trials of MIRV, including the SORAYA (NCT04296890) and MIRASOL (NCT04209855) trials. The single-arm, Phase II SORAYA trial evaluated MIRV monotherapy in participants with FRα-high PROC and one to three prior therapies; prior bevacizumab was required [11]. The SORAYA trial (N = 105 [efficacy evaluable]) reported an investigator-assessed objective response rate (ORR) of 32.4% (95% CI: 23.6–42.2), including 5 complete responses (CRs) and 29 partial responses (PRs). The median duration of response (DOR) was 6.9 months (95% CI: 5.6–9.7), and MIRV had a distinct and tolerable safety profile with manageable low-grade gastrointestinal, neurosensory and resolvable ocular events [11]. These findings supported accelerated approval of MIRV (US Food and Drug Administration [FDA]; November 2022) for patients with FRα-high PROC who have received one to three prior lines of therapy [41]. The confirmatory, randomized, global, Phase III MIRASOL trial supported full US FDA approval, demonstrating that participants treated with MIRV (n = 227) vs. chemotherapy (n = 226) had a 35% improvement in PFS (hazard ratio [HR], 0.65; 95% CI: 0.52–0.81; p < 0.001), >2× higher ORR (42% with MIRV vs. 16% with chemotherapy; odds ratio, 3.81; 95% CI: 2.44–5.94; p < 0.001) and a 33% improvement in overall survival (OS) (HR: 0.67; 95% CI: 0.50–0.89; p = 0.005) [42,43]. Importantly, there was a consistent benefit seen with MIRV in bevacizumab-naive and bevacizumab-pretreated subgroups [43]. During the MIRASOL trial, MIRV demonstrated a favorable safety and tolerability profile compared with chemotherapy, with fewer grade ≥3 treatment-emergent adverse events (TEAEs) (42 vs. 54%), fewer serious adverse events (SAEs) (24 vs. 33%) and fewer discontinuations due to TEAEs (9 vs. 16%) [43].
The distinct and complementary mechanisms of action and non-overlapping toxicity profiles of MIRV and bevacizumab suggest that they could be combined as a novel therapeutic strategy for EOC. Indeed, MIRV was previously shown to potentiate the activity of bevacizumab in a PROC xenograft model [44]. The Phase Ib/2 FORWARD II trial (NCT02606305) reported dose-finding results and evaluated the MIRV plus bevacizumab combination in two separate cohorts. In the initial cohort of participants with PROC, the MIRV plus bevacizumab combination demonstrated an ORR of 39% (95% CI: 28–52), median PFS of 6.9 months (95% CI: 4.9–8.6) and median DOR of 8.6 months (95% CI: 4.9–14.9) across a range of FRα-expressing tumors [45]. The second cohort included participants who had FRα-high platinum-agnostic ovarian cancer (PROC or PSOC), for whom a non–platinum-based doublet would be appropriate [46]. In this cohort, the combination resulted in a confirmed ORR of 64% (95% CI: 45–80), median PFS of 10.6 months (95% CI: 8.3–13.3) and median DOR of 11.8 months (95% CI: 6.7–13.7). The subset of participants with FRα-high PSOC had a confirmed ORR of 69% (95% CI: 41–89), median PFS of 13.3 months (95% CI: 8.3–18.3) and median DOR of 12.9 months (95% CI: 6.5–15.7) [46]. In the final analysis of combined expansion cohorts, MIRV plus bevacizumab was highly active across all FRα expression levels regardless of prior bevacizumab exposure, and the safety profile consisted of predominantly low-grade adverse events (AEs), as expected based on the safety profiles of each agent [47]. Based on FORWARD II trial results, MIRV plus bevacizumab represents a novel, targeted combination in EOC and warrants further evaluation in the maintenance setting for patients with FRα-expressing PSOC.
1.3. Objectives
Here we present an overview of a trial in progress, the GLORIOSA trial. The primary end point of the GLORIOSA trial will compare investigator-assessed PFS in participants whose disease has not progressed after second-line (2L) platinum-based chemotherapy doublet plus bevacizumab who were randomized to maintenance therapy with MIRV plus bevacizumab vs. bevacizumab alone. PFS will also be assessed via blinded independent central review (BICR) as a sensitivity analysis. The key secondary end point is OS.
1.4. Trial design
GLORIOSA is a randomized, open-label/non-blinded, Phase III trial that will evaluate the safety and efficacy of MIRV plus bevacizumab vs. bevacizumab alone as maintenance therapy in participants without progressive disease (PD) after completion of 2L platinum-based chemotherapy doublet plus bevacizumab for platinum-sensitive, high-grade serous ovarian, fallopian tube or primary peritoneal cancer in first recurrence with high FRα expression (Figure 2).
Figure 2.
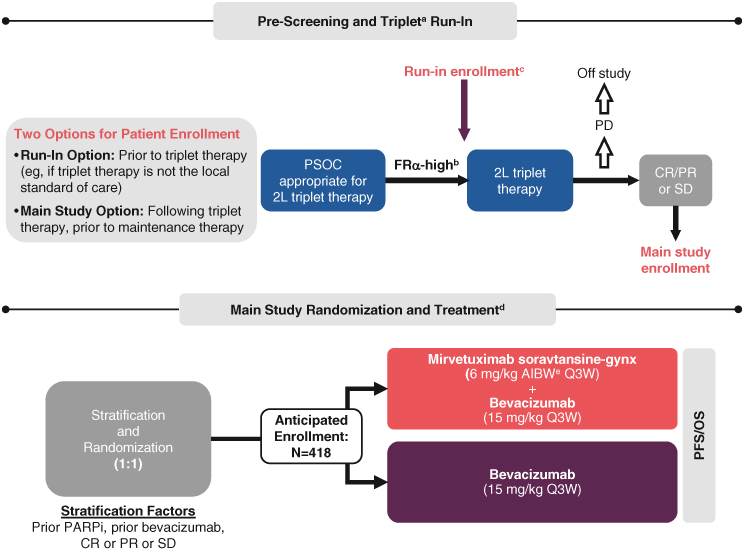
GLORIOSA enrollment and trial design.
aTriplet treatment consists of platinum plus chemotherapy plus bevacizumab for planned six cycles (minimum four and maximum eight cycles), including ≥3 cycles of bevacizumab.
bFRα-high is defined by FRα positivity of ≥75% of tumor membrane staining at ≥2+ intensity (PS2+).
cFRα-high participants who desire to be treated and followed while on their run-in triplet therapy must sign a run-in consent as part of the main consent form if they meet other study eligibility criteria as assessed by the investigator.
dMaintenance treatment must begin ≤12 weeks from last dose of triplet therapy and within 30 days of randomization. Treatment continues until PD, unacceptable toxicity, withdrawal of consent, death or study sponsor termination.
eAIBW, also known as AdjBW, is calculated as IBW (kg) + 0.4 (actual weight – IBW). IBW for females is calculated as 0.9 × height (cm) – 92.
2L: Second-line; AdjBW: Adjusted body weight; AIBW: Adjusted ideal body weight; CR: Complete response; FRα: Folate receptor α; IBW: Ideal body weight; OS: Overall survival; PARPi: Poly (adenosine diphosphate [ADP]-ribose) polymerase inhibitor; PD: Progressive disease; PFS: Progression-free survival; PR: Partial response; PS2+: Positive staining 2+; PSOC: Platinum-sensitive ovarian cancer; Q3W: Three-times per week; SD: Stable disease.
2. Methods
2.1. Study setting
GLORIOSA will be conducted at approximately 250 centers globally, including the US, Europe, Australia, Canada, Israel and The Republic of Korea [48].
2.2. Key eligibility criteria
Table 1 describes key eligibility criteria. Patients are eligible for the GLORIOSA trial if they are aged ≥18 years and have confirmed high-grade serous EOC, primary peritoneal cancer or fallopian tube cancer. Patient tumor samples must demonstrate high FRα expression defined as ≥75% tumor membrane staining at ≥2+ intensity (PS2+) with the VENTANA FOLR1 (FOLR1-2.1) RxDx assay measured centrally; for US-based patients both commercial and local testing with the FDA-approved VENTANA FOLR1 (FOLR1-2.1) RxDx assay is permitted. Patients must have relapsed after first-line platinum-based chemotherapy and have platinum-sensitive disease (defined as progression >6 months from last dose of primary platinum therapy); and must be appropriate for, currently on, or have completed 2L platinum-based chemotherapy doublet plus bevacizumab, including four to eight cycles of platinum-based triplet therapy, with ≥3 cycles of bevacizumab plus platinum-based chemotherapy. Patients must have reached CR, PR or stable disease (SD) after treatment with a platinum-based doublet plus bevacizumab.
Table 1.
Key eligibility criteria.
| Key inclusion criteria |
| ≥18 years of age |
| ECOG performance status of 0 or 1 |
| Confirmed diagnosis of high-grade serous epithelial ovarian, primary peritoneal or fallopian tube cancer |
| FRα-high as detected by IHC with PS2+ staining (≥75% of tumor membrane staining at ≥2+ intensity)a |
| BRCA testing prior to study entry.b Somatic and germ-line BRCA-positive patients must have received prior PARPi therapy as maintenance following 1L therapy |
| Relapsed after 1L platinum-based chemotherapy and platinum-sensitivity (disease progression >6 months after last platinum treatment) |
| Appropriate for, currently on, or completed 2L platinum-based doublet + bevacizumab (‘triplet’) therapy |
| Received four to eight cycles of 2L platinum-based triplet therapy, which included ≥3 cycles of bevacizumab in combination with platinum-based chemotherapyc,d |
| CR, PR or SD after prior treatment with platinum-based triplet therapy in 2L |
| Randomized no later than 8 weeks from the last dose of platinum-based triplet therapy in 2L |
| Stabilized or recovered (grade 1 or baseline) from all prior therapy-related toxicities (except alopecia) and no major surgeries for at least 4 weeks prior to the first dose of maintenance therapy |
| Adequate hematologic, liver and kidney functions |
| Key exclusion criteria |
| Endometroid, clear cell, mucinous, or sarcomatous histology, mixed tumors containing any of the above histologies or low-grade/borderline ovarian tumor |
| ≥1 Line of prior chemotherapye |
| PD while on or following platinum-based triplet therapy |
| Received an intervening dose of bevacizumab after the last dose of triplet therapy before randomization |
| Prior wide-field radiotherapy affecting ≥20% of the bone marrow |
| Grade >1 peripheral neuropathy per CTCAE |
| Active or chronic corneal disorders, history of corneal transplantation or active ocular conditions requiring ongoing treatment/monitoringf |
| History of bowel obstruction related to underlying disease within 6 months before the start of maintenance study treatment |
| Prior treatment with MIRV or other FRα-targeting agents |
FRα staining determined by VENTANA FOLR1 (FOLR1-2.1) RxDx assay.
Local tumor or germline BRCA testing will be acceptable for stratification. If the patient has not been tested, recommend archival tumor samples to be assessed for tissue BRCA. All patients who have received prior 1L PARPi maintenance and/or bevacizumab are eligible.
If the number of cycles received is less than six due to toxicity, this must be documented and toxicity assessed as unlikely related to bevacizumab.
In the case of interval secondary cytoreductive surgery, patients are permitted to receive only two cycles of bevacizumab in combination with the last three cycles of 2L platinum-based chemotherapy doublet plus bevacizumab. In the case of primary cytoreductive surgery before 2L platinum-based chemotherapy doublet plus bevacizumab, patients must have received no fewer than three cycles of bevacizumab in combination with platinum-based chemotherapy after their surgery and before randomization.
Lines of prior anticancer therapy are counted with the following considerations: neoadjuvant ± adjuvant therapies are considered one line of therapy if the neoadjuvant and adjuvant correspond to one fully predefined regimen; otherwise, they are counted as two prior regimens. Maintenance therapy (e.g., bevacizumab, PARPi) will be considered part of the preceding line of therapy (i.e., not counted independently). Change due to toxicity will be considered part of the proceeding line of therapy.
These include uncontrolled glaucoma, wet age-related macular degeneration requiring intravitreal injections, active diabetic retinopathy with macular edema, macular degeneration, presence of papilledema and/or monocular vision.
1L: First-line; 2L: Second-line; CR: Complete response; CTCAE: Common Terminology Criteria for Adverse Events; ECOG: Eastern Cooperative Oncology Group; FRα: Folate receptor alpha; IHC: Immunohistochemistry; MIRV: Mirvetuximab soravtansine-gynx; PARPi: Poly (adenosine diphosphate [ADP]-ribose) polymerase inhibitor; PD: Progressive disease; PR: Partial response; PS2+: Positive staining 2+; SD: Stable disease.
2.3. Interventions & participant timeline
Prior to randomization into the maintenance portion of the trial, patients must have received four to eight cycles of standard 2L platinum-based chemotherapy doublet plus ≥3 cycles of bevacizumab (‘triplet therapy’). Patients can be screened and enrolled via two methods: before initiation (participants complete a run-in before enrollment into the randomized portion of the trial) or after completion (participants are enrolled directly into the randomized portion of the trial) of standard 2L triplet therapy (Figure 2). Pre-screening may be performed prior to chemotherapy completion for participants enrolled directly into the randomized maintenance portion of the trial. Run-in participants will receive their triplet therapy as part of the trial protocol (13 December 2022; version 3.0) as described in Table 2. For both enrollment methods, if previous results from the VENTANA FOLR1 (FOLR1-2.1) RxDx assay are not available, FRα expression will be tested using either an archival or a fresh biopsy tissue sample. Entry into the randomized maintenance portion of the trial will be limited to patients with a CR, PR or SD following their 2L triplet therapy.
Table 2.
Dose and dosing schedule for triplet therapy for run-in participantsa.
| Drug | Dose | Dosing schedule |
|---|---|---|
| Carboplatin plus paclitaxel and bevacizumab | ||
| Carboplatin | AUC5 | Day 1 of a 21-day cycle |
| Paclitaxel | 175 mg/m2 | Day 1 of a 21-day cycle |
| Bevacizumab | 15 mg/kg | Day 1 of a 21-day cycle |
| Carboplatin plus PLD and bevacizumab | ||
| Carboplatin | AUC5 | Day 1 of a 28-day cycle |
| PLD | 30 mg/m2 | Day 1 of a 28-day cycle |
| Bevacizumab | 10 mg/kg | Day 1 and day 15 of a 28-day cycle |
| Carboplatin plus gemcitabine and bevacizumab | ||
| Carboplatin | AUC4 | Day 1 of a 21-day cycle |
| Gemcitabine | 1000 mg/m2 | Day 1 and day 8 of every 21-day carboplatin/bevacizumab cycle |
| Bevacizumab | 15 mg/kg | Day 1 of a 21-day cycle |
In the event of an allergic reaction to carboplatin or a prior severe hypersensitivity reaction, participants may undergo a desensitization protocol per institutional guidelines or may substitute cisplatin at the utilized dose of cisplatin in the three triplets.
AUC: Area under the curve; PLD: Pegylated liposomal doxorubicin.
The dosing schedule for the randomized maintenance portion of the trial (Figure 2) is shown in Table 3. Maintenance therapy must begin ≤12 weeks from the last dose of triplet therapy and within 30 days of randomization. Participants randomized to arm 1 will receive maintenance therapy with MIRV at 6 mg/kg AIBW IV and bevacizumab at 15 mg/kg IV Q3W until disease progression or unacceptable toxicity. Participants randomized to arm 2 will receive maintenance therapy with bevacizumab at 15 mg/kg IV Q3W until disease progression or unacceptable toxicity.
Table 3.
Dose and dosing schedule during randomized maintenance portion of triala.
| Group | Drug | Dose | Dosing schedule |
|---|---|---|---|
| Arm 1 | Mirvetuximab soravtansine-gynx | 6 mg/kg AIBW | Day 1 of a 21-day cycle |
| Bevacizumab | 15 mg/kg | ||
| Arm 2 | Bevacizumab | 15 mg/kg | Day 1 of a 21-day cycle |
Dosing for both arms will continue until disease progression or unacceptable toxicity.
AIBW: Adjusted ideal body weight.
All MIRV-treated participants must receive acetaminophen/paracetamol (orally [PO] or IV), dexamethasone (IV) and diphenhydramine (PO or IV) (equivalent drugs of similar drug classes are also acceptable) before each MIRV infusion. Antiemetic medication (eg, 5-HT3 serotonin receptor antagonist) is recommended before each MIRV dose. Participants receiving MIRV will be required to use lubricating artificial tears ≥four-times daily and as needed; preservative-free lubricating eye drops are strongly recommended. Additionally, participants with slit lamp examination findings of suspected MIRV-related keratopathy (eg, microcystic epithelial change, punctate epithelial keratopathy, and subepithelial inclusion cyst) should start secondary prophylactic corticosteroid eye drops for the remaining cycles of MIRV unless the eye care professional documents that the risks outweigh the benefits of such therapy. If indicated, participants will be directed to self-administer corticosteroid eye drops (1% prednisolone) six-times daily starting the day prior to MIRV infusion until day 4, and then four-times daily on days 5–8 of each subsequent cycle. The Data collection methods: safety evaluations section details further information regarding required ocular examinations. Concomitant non-study anticancer agents during study treatment are prohibited. Concomitant CYP3A-inducers/inhibitors are allowable but require careful monitoring due to potential interaction.
Participants receiving MIRV plus bevacizumab with unacceptable toxicity attributed to one of the treatments may discontinue that treatment and continue participation in the study on the other treatment as monotherapy until a reason for discontinuation arises (toxicity or tumor progression). Participants receiving bevacizumab alone with unacceptable toxicity attributed to bevacizumab should discontinue treatment and proceed to the end of treatment visit and subsequent follow-up, as applicable.
2.4. Outcomes
The primary end point is investigator-assessed PFS per Response Evaluation Criteria in Solid Tumors version 1.1 (RECIST v1.1), defined as the time from date of randomization until investigator-assessed PD or death, whichever occurs first. PFS will also be assessed by BICR as a sensitivity analysis in the same population.
The key secondary end point is OS, defined as the time from randomization to death. Participants alive at the time of analysis will be censored at the last date they were known to be alive. Other secondary end points include safety and tolerability (AEs [except corneal AEs] coded using the latest Medical Dictionary for Regulatory Activities [MedDRA] version, graded using the Common Terminology Criteria for Adverse Events version 5.0 [CTCAE v5.0]); second PFS (PFS2) by the investigator (defined as the time from date of randomization until second disease progression or death, whichever occurs first); investigator- and BICR-assessed ORR in participants with measurable disease per RECIST v1.1 at randomization; investigator- and BICR-assessed DOR in participants who achieved a best overall response of CR or PR upon completion of triplet therapy; investigator- and BICR-assessed disease-free survival in participants without measurable disease per RECIST v1.1 at randomization; cancer antigen 125 (CA-125) response rate per Gynecologic Cancer InterGroup criteria; and patient-reported outcomes/health-related quality of life measures assessed by the National Comprehensive Cancer Network/Functional Assessment of Cancer Therapy Ovarian Cancer Symptom Index – 18 (NFOSI-18).
2.5. Sample size
The GLORIOSA trial will enroll approximately 418 participants into the maintenance portion of the trial (~209 participants in each arm) over a period of roughly 3 years and have 90% power to detect an HR of 0.7. Sample size and power were determined using R-Project package gsDesign version 2 with the following assumptions: median PFS in arm 2 (bevacizumab alone) is 9 months, median PFS in arm 1 (MIRV plus bevacizumab) is 12.9 months, there is exponential distribution for the event process, the overall attrition rate in both arms is approximately 13%, enrollment is uniform over a 36-month period, and duration of follow-up is 24 months.
2.6. Recruitment
Recruitment will occur among a variety of centers in geographically diverse locations to achieve the target sample size of 418 participants [48].
3. Methods: assignment of interventions
3.1. Sequence generation, blinding, implementation & masking
Following CR, PR or SD with 2L triplet therapy, participants will be randomized 1:1 to receive maintenance therapy with either MIRV plus bevacizumab or bevacizumab alone. Randomization will be performed centrally using a block design and stratified by response to 2L triplet therapy (CR vs. PR vs. SD), prior PARPi treatment and prior treatment with bevacizumab as part of first-line therapy. Blinding methods are not applicable, as this is an open-label trial. The treatment randomization schedule will be developed by an interactive response technology vendor. A BICR facility will be used to provide an independent assessment of radiographic tumor assessments.
4. Methods: data collection, management & analysis
4.1. Data collection methods: efficacy evaluations
Efficacy will be evaluated in the intent-to-treat population, which will include all randomized participants. Analysis of PFS will be stratified by the randomization strata. Tumor assessments to evaluate response to triplet therapy (including radiological assessments by computed tomography/magnetic resonance imaging) will be performed in all participants 3–8 weeks after completion of triplet therapy and prior to randomization. Starting on cycle 1 day 1 of maintenance therapy, all subsequent radiological assessments will be conducted every 9 weeks (±2 weeks) for 72 weeks and subsequently every 18 weeks (±3 weeks) until PD, death, start of a new anticancer therapy or withdrawal of consent, whichever occurs first. After maintenance therapy discontinuation, all participants will be followed for survival every 4 months (±1 month) until death, loss to follow-up, withdrawal of consent for survival follow-up or end of trial, whichever comes first. Starting 2 years after maintenance therapy discontinuation, survival assessments will be conducted every 6 months (±1 month). The trial will end 7 years after maintenance treatment on cycle 1 day 1 for the last participant or per discretion of the sponsor.
4.2. Data collection methods: safety evaluations
All participants who receive ≥1 dose of MIRV or ≥1 dose of bevacizumab as maintenance will be included in the safety analyses. Safety will be assessed by evaluating AEs, SAEs and deaths, as well as toxicities determined through evaluations of changes in laboratory parameters and vital signs.
Ocular AEs, primarily blurred vision and keratopathy, have been associated with MIRV and were first recognized in the Phase I ‘401 trial‘ [49,50]. An expansion cohort study investigated the potential benefits of primary prophylactic use of corticosteroid eye drops during MIRV monotherapy and found that implementation was associated with a trend toward ocular symptom improvement and a reduction in ocular AE-related dose modifications [51]. Of note, Hendershot et al. (2023) detail the recommended management and mitigation strategies for ocular AEs associated with MIRV that have been effective during multiple clinical trials of MIRV monotherapy [52].
For the GLORIOSA trial, a complete ophthalmic examination will be performed by an eye care professional (ophthalmologist or optometrist), or other qualified healthcare provider, in all randomized participants at baseline (within 14 days before maintenance cycle 1 day 1 [main screening]), before cycle 7 (between cycles 6 and 7) and at the 30-day safety follow-up visit (±14 days) post-maintenance treatment discontinuation. Complete examination includes, at minimum, manifest refraction, best corrected visual acuity (BCVA), intraocular pressure assessment (IOP), fundoscopy (baseline and 30-day follow-up only) and slit lamp examination to assess the cornea for keratopathy and other corneal abnormalities. Additional ocular examinations (including manifest refraction, BCVA, IOP and slit lamp examination) will be performed prior to cycles 3 and 5 in all MIRV-treated participants who are asymptomatic or have grade ≤1 ocular symptoms. In participants with ocular symptoms (CTCAE v5.0 grade >1), a complete ophthalmic examination will be performed upon emergence of symptoms before the next MIRV dose and at every cycle thereafter until the symptom(s) resolve to baseline or is deemed irreversible in the opinion of the eye care professional. Participants who require an ophthalmic examination due to symptom emergence prior to cycles 2 or 4 will then proceed to every 6-week examinations (i.e., every other cycle [as indicated by findings]) and do not require the routine asymptomatic exams at cycles 3, 5 and 7. For participants with ongoing keratitis/keratopathy, a corneal epithelial defect or ≥3-line loss in BCVA after end of treatment with MIRV, an ophthalmic examination will be done every 30 days until resolution, stabilization or return to baseline.
Assessment of ocular symptoms will be performed by the treating physician or another qualified individual in all participants at baseline (main screening), on day 1 of each maintenance treatment cycle and at the 7-day and the 30-day (±14 days) safety follow-up visits post-maintenance treatment discontinuation. In participants who report CTCAE v5.0 grade ≥1 ocular symptoms, MIRV treatment will be held until the participant is evaluated by an eye care professional for a complete examination. Recommended dose modifications for corneal adverse reactions should be based on both corneal examination findings (with dose modifications according to the grading system for corneal AEs in the trial protocol) and changes in BCVA. Dose modification for noncorneal ocular AEs other than uveitis is at the discretion of the treating physician, in collaboration with an eye care professional (with dose modifications according to the grading system for uveitis in the trial protocol).
4.3. Data management
The clinical database will be developed and maintained by a contract research organization (CRO) or an electronic data capture technology provider as designated by ImmunoGen, Inc. ImmunoGen, Inc. or its designee will be responsible for performing trial data management activities and analyses.
4.4. Statistical methods
The final PFS analysis is planned when ≥330 PFS events have been observed (no interim analysis). If the trial continues until ≥300 OS events are observed, the final analysis for OS will be conducted approximately 4 years after the final PFS analysis. The Kaplan-Meier method will be used for both PFS and OS analyses, and PFS and OS rates will be reported in 3-month intervals. The primary comparison between treatments will use the log-rank test stratified by the randomization strata. In the stratified PFS analysis, the strata will include best overall response to triplet therapy (CR vs. PR vs. SD), prior exposure to PARPi (yes vs. no) and prior exposure to bevacizumab as first-line therapy (yes vs. no). The HR for PFS and OS treatment comparisons will be estimated using a stratified Cox proportional hazards model. For sensitivity analyses, the HR from unstratified Cox proportional hazards models will also be provided for both PFS and OS. Arm 2 (bevacizumab alone) will be used as the reference treatment. A time-to-event HR less than 1.0 would indicate a benefit of MIRV plus bevacizumab (arm 1) over bevacizumab alone (arm 2). For the key secondary end point of OS, the final analysis will be conducted when at least 300 deaths have been observed. Two interim analyses will be conducted for OS: the first will be at the time of final analysis for PFS and the other will be when 240 (80%) of OS events have occurred. The information time will be approximately 60% (180 deaths) at the first interim analysis for OS. A Lan-DeMets α-spending function using an O'Brien-Fleming stopping boundary will be used to control overall Type I error for OS at a 2-sided alpha level of 0.05. Missing data will not be imputed unless otherwise stated.
5. Methods: monitoring
5.1. Data monitoring
An independent data monitoring committee (IDMC) with no involvement in the trial execution will review efficacy data; safety data, including AEs, SAEs and treatment-related mortality; and laboratory data. The IDMC will also review and interpret the interim analysis results and report its findings to the sponsor. Decisions on trial termination, amendments of the protocol or patient recruitment cessation will be made after the sponsor assesses IDMC recommendations. A steering committee will comprise lead investigators from each country enrolling patients and will provide overall guidance regarding safety, efficacy, enrollment and contribution to scientific input for publications.
5.2. Harms
It is the sponsor's responsibility to ensure that each investigator receives a copy of any Suspected Unexpected Serious Adverse Reaction (SUSAR) report regarding the trial drug and that the report is submitted to the appropriate national regulatory agencies.
5.3. Auditing
Monitoring and auditing procedures that comply with current Good Clinical Practice guidelines will be followed.
5.4. Ethics & dissemination
The trial will be performed according to the principles of the Declaration of Helsinki, Good Clinical Practice guidelines of the International Council for Harmonisation and local regulatory requirements. The protocol will be approved by the institutional review board or independent ethics committee at each investigative site. All participants (or legally authorized representatives) will provide written informed consent. Participant names will not be supplied to the sponsor. If a participant name appears on any documents, it will be redacted before a document copy is supplied to the sponsor. Trial findings stored on a computer will be stored in accordance with local data protection laws. Participant blood and tissue samples, and radiologic images sent to outside laboratories and/or CROs, will be identified by trial participant number only to ensure maintenance of confidentiality. The investigator will maintain a personal participant identification list (participant numbers with the corresponding participant names) to enable records to be identified.
6. Conclusion
The Phase III, randomized, open-label GLORIOSA trial will evaluate the potential benefit of MIRV plus bevacizumab maintenance therapy in participants with FRα-high PSOC who have not progressed after 2L platinum-based chemotherapy doublet plus bevacizumab. The primary end point of the trial will evaluate investigator-assessed PFS in participants who receive maintenance therapy with MIRV plus bevacizumab as compared with bevacizumab alone. Overall, results from this trial will help define the role of this combination for improving the management and outcomes of patients with PSOC [53].
Supplementary Material
Acknowledgments
This clinical trial is funded by ImmunoGen, Inc. We thank the patients and their families, investigators co-investigators and study teams at each of the participating centers. Medical writing and editorial support were provided by P Sridhar (freelance) and GM Bravante and A Agazio of Precision AQ (Yardley, PA, USA). All were funded by ImmunoGen, Inc.
Funding Statement
This clinical trial is funded by ImmunoGen, Inc.
Author contributions
The investigators designed the trial. All authors contributed to the development of this manuscript, approved the final version and claim responsibility for the fidelity of the trial and protocol. Medical writing services were funded by the study sponsor.
Financial disclosure
The authors have disclosed any financial involvements with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending, or royalties.
Competing interests disclosure
DM O'Malley: Funding for clinical research from AbbVie Inc, Agenus Inc, Ajinomoto Co, Inc, Amgen Inc, Array BioPharma Inc, AstraZeneca Pharmaceuticals LP, Bristol Myers Squibb Company, Clovis Oncology, Inc, Daré Bioscience, Inc, Eisai Co, Ltd, EMD Serono, Ergomed plc, Genentech, Inc, Genmab A/S, The GOG Foundation Inc, ImmunoGen, Inc, Iovance Biotherapeutics, Inc, Janssen Biotech, Inc, Johnson & Johnson Pharmaceuticals, Ludwig Institute for Cancer Research Ltd, Merck & Co, Inc, Merck Serono, Mersana Therapeutics, Inc, New Mexico Cancer Care Alliance, Novocure GmbH, PRA Health Sciences, Regeneron Pharmaceuticals, Inc, Seagen Inc, Stemcentrx, Inc, Sumitomo Dainippon Pharma Oncology, Inc, Syneos Health, TESARO Inc, TRACON Pharmaceuticals, Inc, VentiRx Pharmaceuticals, Inc and Yale University; personal fees from Agenus Inc, Myriad Genetics, Inc, Rubius Therapeutics, Inc and Tarveda Therapeutics; and fees for consulting or advisory boards from AbbVie Inc, Ambry Genetics Corporation, Amgen Inc, Arquer Diagnostics Ltd, AstraZeneca Pharmaceuticals LP, Celsion Corporation, Clovis Oncology, Inc, Corcept Therapeutics Incorporated, Eisai Co, Ltd, Elevar Therapeutics, Inc, Genentech, Inc, The GOG Foundation, Inc, ImmunoGen, Inc, InxMed Co, Ltd, Iovance Biotherapeutics, Inc, Janssen Biotech, Inc, Johnson & Johnson Pharmaceuticals, Merck & Co, Inc, Mersana Therapeutics, Inc, Novartis, Novocure GmbH, Regeneron Pharmaceuticals, Inc, Roche Diagnostics MSA, Seagen Inc, Sorrento Therapeutics, Inc, Sumitomo Dainippon Pharma Oncology, Inc, Takeda Pharmaceuticals USA, Inc, TESARO Inc and Toray Industries, Inc.
T Myers: Honoraria for lecture to ImmunoGen, Inc. Institutional research support from GSK.
P Wimberger: Research funding from Amgen Inc, AstraZeneca, MSD, GlaxoSmithKline, Novartis, Pfizer Inc, Roche Pharma, Clovis Oncology, Inc and Eli Lilly and Company and honoraria from Amgen Inc, AstraZeneca, MSD, GlaxoSmithKline, Novartis, Pfizer Inc, Roche Pharma, Clovis Oncology, Inc, Teva Pharmaceutical Industries Ltd, Eisai Co, Ltd, Eli Lilly and Company, Gilead Sciences, Inc and Daiichi Sankyo Company, Limited. Participation in advisory boards for Amgen Inc, AstraZeneca, MSD, GlaxoSmithKline, Novartis, Pfizer Inc, Roche Pharma, Clovis Oncology, Inc, Teva Pharmaceutical Industries Ltd, Eisai Co, Ltd, Eli Lilly and Company, Gilead Sciences, Inc and Daiichi Sankyo Company, Limited.
T Van Gorp: Consulting/advising with AstraZeneca, BioNTech SE, Eisai Co, Ltd, GSK, ImmunoGen, Inc, Incyte Corporation, MSD/Merck & Co, Inc, OncXerna Therapeutics, Inc, Seagen Inc, Tubulis GmbH and Zentalis Pharmaceuticals, Inc. Travel, accommodations, and/or expenses from AstraZeneca, GSK, ImmunoGen, Inc, MSD/Merck and PharmaMar. Research funding from Amgen Inc, AstraZeneca and Roche. All payments institutional.
A Redondo: Honoraria, advisory/consultancy services and speakers bureau for MSD, AstraZeneca, GSK and PharmaMar. Travel/accommodations/expenses from AstraZeneca, GSK and PharmaMar.
D Cibula: Advisory board participation with Akeso Bio, AstraZeneca, GSK, MSD, Novocure GmbH, Roche, Seagen Inc and Sotio Biotech. Travel expenses from AstraZeneca.
S Nicum: Institutional research funding from AstraZeneca and GSK. Participation in advisory boards, as a speaker, and/or on a steering committee for AstraZeneca, Clovis Oncology, Inc and GSK.
M Rodrigues: Honoraria: Immunocore Ltd; consulting or advisory role: AstraZeneca and GlaxoSmithKline; research funding: Daiichi Sankyo Company, Limited/AstraZeneca; and travel, accommodations and expenses: AstraZeneca.
FJ Backes: Grant/research/clinical trial support: Clovis Oncology, Inc; Eisai Co, Ltd; ImmunoGen, Inc; Merck; and Natera, Inc. Consultant/advisory boards: Agenus Inc; AstraZeneca; Clovis Oncology, Inc; Eisai Co, Ltd; GlaxoSmithKline; ImmunoGen, Inc; and Merck.
JN Barlin: Consulting or advisory role: AstraZeneca, Clovis Oncology, Inc and OncoC4. Speakers bureau: AstraZeneca, Clovis Oncology, Inc and Merck.
SN Lewin: Research funding from Tesoro, The GOG Foundation Inc, Merck and ImmunoGen, Inc. Speakers bureau for ImmunoGen, Inc, AstraZeneca, GSK, Merck and Myriad Genetic Laboratories Inc.
P Lim: Nothing to disclose.
B Pothuri: Grants, consulting and advisory board fees; institutional PI for industry-sponsored trials from Tesaro Inc/GSK, Duality Biologics, AstraZeneca, Merck, Genentech, Inc/Roche, Celsion Corporation, ImmunoGen, Inc, Karyopharm Therapeutics Inc, Mersana Therapeutics, Inc, Takeda Pharmaceutical Company Limited, Toray, I-Mab Biopharma Co, Ltd, Sutro Biopharma, Inc, Seagen Inc and Clovis Oncology, Inc. Compensated advisory boards include Tesaro Inc/GSK, AstraZeneca, Eli Lilly and Company, Mersana Therapeutics, Inc, Onconova Therapeutics, Inc, Merck, Clovis Oncology, Inc, Eisai Co, Ltd, Toray, Sutro Biopharma, Inc, Deciphera Pharmaceuticals, Inc, I-Mab Biopharma Co, Ltd, Seagen Inc, Imvax, Inc and The GOG Foundation Inc.
E Diver: Employment by ImmunoGen, Inc.
S Banerjee: Institutional research funding from AstraZeneca and GlaxoSmithKline; consulting fees from AstraZeneca, Epsilogen Ltd, GlaxoSmithKline, ImmunoGen, Inc, Merck Sharp & Dohme, Mersana Therapeutics, Inc, Novartis, OncXerna Therapeutics, Inc, Seagen Inc, Shattuck Labs, Inc and Regeneron Pharmaceuticals, Inc; and honoraria from Amgen Inc, AstraZeneca, Clovis Oncology, Inc, GlaxoSmithKline, ImmunoGen, Inc, Merck Sharp & Dohme, Mersana Therapeutics, Inc, Novocure GmbH, Pfizer Inc, Roche and Takeda Pharmaceutical Company Limited.
D Lorusso: Research funding and consulting or advisory fees: PharmaMar, Clovis Oncology, Inc, GSK, MSD, AstraZeneca, Amgen Inc, Seagen Inc/Genmab A/S, ImmunoGen, Inc and Merck Serono. Research funding: Incyte Corporation, Novartis, Roche, Sutro Biopharma, Inc and Corcept Therapeutics Inc.
The authors have no other competing interests or relevant affiliations with any organization or entity with the subject matter or materials discussed in the manuscript apart from those disclosed.
Writing disclosure
Medical writing and editorial support were provided by P Sridhar (freelance) and GM Bravante and A Agazio of Precision AQ (Yardley, PA, USA). All were funded by ImmunoGen, Inc.
Ethical conduct of research
The trial is being performed according to the principles of the Declaration of Helsinki, Good Clinical Practice guidelines of the International Council for Harmonisation and local regulatory requirements. The institutional review board or independent ethics committee at each investigative site approved the protocol. Informed written consent will be obtained from all participants (or legally authorized representatives).
Data availability statement
The study sponsor, ImmunoGen, Inc., is committed to responsible sharing of clinical trial data. Data from this clinical trial, once available, can be requested by any qualified investigator who engages in relevant research. Data requests can be submitted to medicalinformation@immunogen.com.
Previous presentation
Portions of the GLORIOSA clinical trial protocol were presented previously at the American Society of Clinical Oncology (ASCO) Annual Meeting 2023, June 2–6, 2023; Chicago, IL; Abstract TPS5622 [53].
References
Papers of special note have been highlighted as: • of interest; •• of considerable interest
- 1.Moore KN, Martin LP, O'Malley DM, et al. A review of mirvetuximab soravtansine in the treatment of platinum-resistant ovarian cancer. Future Oncol. 2018;14(2):123–136. doi: 10.2217/fon-2017-0379 [DOI] [PubMed] [Google Scholar]; • Review of mechanism of action, pharmacology and clinical evaluation of mirvetuximab soravtansine in ovarian cancer.
- 2.Moore KN, Martin LP, O'Malley DM, et al. Safety and activity of mirvetuximab soravtansine (IMGN853), a folate receptor α-targeting antibody–drug conjugate, in platinum-resistant ovarian, fallopian tube, or primary peritoneal cancer: a Phase I expansion study. J Clin Oncol. 2017;35(10):1112–1118. doi: 10.1200/JCO.2016.69.9538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kuroki L, Guntupalli SR. Treatment of epithelial ovarian cancer. BMJ. 2020;371:m3773. doi: 10.1136/bmj.m3773 [DOI] [PubMed] [Google Scholar]
- 4.World Health Organization, International Agency for Research on Cancer . Ovary. https://gco.iarc.fr/today/data/factsheets/cancers/25-Ovary-fact-sheet.pdf (Accessed December 1, 2023).
- 5.Globocan 2020 Europe . International Agency for Research on Cancer. Lyon, France: World Health Organization; 2020. Fact Sheet 908. [Google Scholar]
- 6.Cancer Statistics at a Glance . Centers for Disease Control and Prevention. November 2023. (Accessed December 6, 2023). https://gis.cdc.gov/Cancer/USCS/#/AtAGlance/
- 7.Poveda AM, Selle F, Hilpert F, et al. Bevacizumab combined with weekly paclitaxel, pegylated liposomal doxorubicin, or topotecan in platinum-resistant recurrent ovarian cancer: analysis by chemotherapy cohort of the randomized Phase III AURELIA trial. J Clin Oncol. 2015;33(32):3836–3838. doi: 10.1200/JCO.2015.63.1408 [DOI] [PubMed] [Google Scholar]
- 8.Pujade-Lauraine E, Hilpert F, Weber B, et al. Bevacizumab combined with chemotherapy for platinum-resistant recurrent ovarian cancer: the AURELIA open-label randomized Phase III trial. J Clin Oncol. 2014;32(13):1302–1308. doi: 10.1200/JCO.2013.51.4489 [DOI] [PubMed] [Google Scholar]; • Phase III AURELIA trial results reporting efficacy and safety results for bevacizumab plus chemotherapy in platinum-resistant ovarian cancer; based on these data, the US Food and Drug Administration approved the use of bevacizumab plus chemotherapy (December 2014) in patients with platinum-resistant ovarian cancer, who have received up to two prior chemotherapy regimens.
- 9.Hamanishi J, Takeshima N, Katsumata N, et al. Nivolumab vs. gemcitabine or pegylated liposomal doxorubicin for patients with platinum-resistant ovarian cancer: open-label, randomized trial in Japan (NINJA). J Clin Oncol. 2021;39(33):367–681. doi: 10.1200/JCO.21.00334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lheureux S, Braunstein M, Oza AM. Epithelial ovarian cancer: evolution of management in the era of precision medicine. CA Cancer J Clin. 2019;69(4):280–304. doi: 10.3322/caac.21559 [DOI] [PubMed] [Google Scholar]
- 11.Matulonis UA, Lorusso D, Oaknin A, et al. Efficacy and safety of mirvetuximab soravtansine in patients with platinum-resistant ovarian cancer with high folate receptor alpha expression: results from the SORAYA study. J Clin Oncol. 2023;41(13):2436–2445. doi: 10.1200/JCO.22.01900 [DOI] [PMC free article] [PubMed] [Google Scholar]; •• Phase II SORAYA trial results reporting efficacy and safety of mirvetuximab soravtansine in platinum-resistant ovarian cancer; based on these data, the US Food and Drug Administration granted accelerated approval to mirvetuximab soravtansine (November 2022) for the treatment of adult patients with FRα-positive, platinum-resistant ovarian cancer who have received one to three prior systemic treatment regimens.
- 12.National Institute of Health National Cancer Institute Surveillance, Epidemiology, and End Results Program . Cancer Stat Facts: Ovarian Cancer. https://seer.cancer.gov/statfacts/html/ovary.html (Accessed September 25, 2023).
- 13.Gupta S, Nag S, Aggarwal S, et al. Maintenance therapy for recurrent epithelial ovarian cancer: current therapies and future perspectives – a review. J Ovarian Res. 2019;12(1):103. doi: 10.1186/s13048-019-0579-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Della Pepa C, Banerjee S. Bevacizumab in combination with chemotherapy in platinum-sensitive ovarian cancer. Onco Targets Ther. 2014;7:1025–1032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Parmar MK, Ledermann JA, Colombo N, et al. Paclitaxel plus platinum-based chemotherapy vs. conventional platinum-based chemotherapy in women with relapsed ovarian cancer: the ICON4/AGO-OVAR-2.2 trial. Lancet. 2003;361(9375):2099–2106. doi: 10.1016/S0140-6736(03)13718-X [DOI] [PubMed] [Google Scholar]
- 16.Pfisterer J, Plante M, Vergote I, et al. Gemcitabine plus carboplatin compared with carboplatin in patients with platinum-sensitive recurrent ovarian cancer: an intergroup trial of the AGO-OVAR, the NCIC CTG, and the EORTC GCG. J Clin Oncol. 2006;24(29):4699–4707. doi: 10.1200/JCO.2006.06.0913 [DOI] [PubMed] [Google Scholar]
- 17.Pujade-Lauraine E, Wagner U, Aavall-Lundqvist E, et al. Pegylated liposomal doxorubicin and carboplatin compared with paclitaxel and carboplatin for patients with platinum-sensitive ovarian cancer in late relapse. J Clin Oncol. 2010;28(20):3323–3329. doi: 10.1200/JCO.2009.25.7519 [DOI] [PubMed] [Google Scholar]
- 18.Rossi L, Verrico M, Zaccarelli E, et al. Bevacizumab in ovarian cancer: a critical review of Phase III studies. Oncotarget. 2017;8(7):12389–12405. doi: 10.18632/oncotarget.13310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Haunschild CE, Tewari KS. Bevacizumab use in the frontline, maintenance and recurrent settings for ovarian cancer. Future Oncol. 2020;16(7):225–246. doi: 10.2217/fon-2019-0042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lippincott Williams & Wilkins, Inc. Philadelphia, PA, USA . FDA approves avastin plus chemotherapy for ovarian cancer. Oncology Times. 2014;36(23):10. doi: 10.1097/01.COT.0000459154.18451.1e [DOI] [Google Scholar]
- 21.Aghajanian C, Blank SV, Goff BA, et al. OCEANS: a randomized, double-blind, placebo-controlled Phase III trial of chemotherapy with or without bevacizumab in patients with platinum-sensitive recurrent epithelial ovarian, primary peritoneal, or fallopian tube cancer. J Clin Oncol. 2012;30(17):2039–2045. doi: 10.1200/JCO.2012.42.0505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Coleman RL, Brady MF, Herzog TJ, et al. Bevacizumab and paclitaxel-carboplatin chemotherapy and secondary cytoreduction in recurrent, platinum-sensitive ovarian cancer (NRG Oncology/Gynecologic Oncology Group study GOG-0213): a multicentre, open-label, randomised, Phase III trial. Lancet Oncol. 2017;18(6):779–791. doi: 10.1016/S1470-2045(17)30279-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.FDA Approves Genentech's Avastin® (Bevacizumab) Plus Chemotherapy for a Specific Type of Advanced Ovarian Cancer. News Release. Genetech; December 6, 2016. (Accessed November 30, 2023). https://www.gene.com/media/press-releases/14647/2016-12-06/fda-approves-genentechs-avastin-bevacizu#:~:text=Genentech%2C%20a%20member%20of%20the,chemotherapy%2C%20followed%20by%20Avastin%20alone%2C [Google Scholar]
- 24.Pfisterer J, Shannon CM, Baumann K, et al. Bevacizumab and platinum-based combinations for recurrent ovarian cancer: a randomised, open-label, Phase III trial. Lancet Oncol. 2020;21(5):699–709. doi: 10.1016/S1470-2045(20)30142-X [DOI] [PubMed] [Google Scholar]
- 25.Berton D, Floquet A, Lescaut W, et al. Real-world experience of bevacizumab as first-line treatment for ovarian cancer: the GINECO ENCOURAGE Cohort of 468 French patients. Front Pharmacol. 2021;12:711813. doi: 10.3389/fphar.2021.711813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pignata S, Lorusso D, Joly F, et al. Carboplatin-based doublet plus bevacizumab beyond progression vs. carboplatin-based doublet alone in patients with platinum-sensitive ovarian cancer: a randomised, Phase III trial. Lancet Oncol. 2021;22(2):267–276. doi: 10.1016/S1470-2045(20)30637-9 [DOI] [PubMed] [Google Scholar]
- 27.Burger RA, Brady MF, Bookman MA, et al. Incorporation of bevacizumab in the primary treatment of ovarian cancer. N Engl J Med. 2011;365(26):2473–2483. doi: 10.1056/NEJMoa1104390 [DOI] [PubMed] [Google Scholar]
- 28.Perren TJ, Swart AM, Pfisterer J, et al. A Phase III trial of bevacizumab in ovarian cancer. N Engl J Med. 2011;365(26):2484–2496. doi: 10.1056/NEJMoa1103799 [DOI] [PubMed] [Google Scholar]
- 29.Birrer MJ, Betella I, Martin LP, et al. Is targeting the folate receptor in ovarian cancer coming of age? Oncologist. 2019;24(4):425–429. doi: 10.1634/theoncologist.2018-0459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kelemen LE. The role of folate receptor alpha in cancer development, progression and treatment: cause, consequence or innocent bystander? Int J Cancer. 2006;119(2):243–250. doi: 10.1002/ijc.21712 [DOI] [PubMed] [Google Scholar]
- 31.Markert S, Lassmann S, Gabriel B, et al. α-folate receptor expression in epithelial ovarian carcinoma and non-neoplastic ovarian tissue. Anticancer Res. 2008;28(6a):3567–3572. [PubMed] [Google Scholar]
- 32.Kalli KR, Oberg AL, Keeney GL, et al. Folate receptor alpha as a tumor target in epithelial ovarian cancer. Gynecol Oncol. 2008;108(3):619–626. doi: 10.1016/j.ygyno.2007.11.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Martin LP, Konner JA, Moore KN, et al. Characterization of folate receptor alpha (FRα) expression in archival tumor and biopsy samples from relapsed epithelial ovarian cancer patients: a Phase I expansion study of the FRα-targeting antibody–drug conjugate mirvetuximab soravtansine. Gynecol Oncol. 2017;147(2):402–407. doi: 10.1016/j.ygyno.2017.08.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Toffoli G, Russo A, Gallo A, et al. Expression of folate binding protein as a prognostic factor for response to platinum-containing chemotherapy and survival in human ovarian cancer. Int J Cancer. 1998;79(2):121–126. doi: 10.1002/(SICI)1097-0215(19980417)79:2 [DOI] [PubMed] [Google Scholar]
- 35.Chelariu-Raicu A, Mahner S, Moore KN, et al. Integrating antibody drug conjugates in the management of gynecologic cancers. Int J Gynecol Cancer. 2023;33(3):420–429. doi: 10.1136/ijgc-2022-003701 [DOI] [PubMed] [Google Scholar]
- 36.Ab O, Whiteman KR, Bartle LM, et al. IMGN853, a folate receptor-alpha (FRα)-targeting antibody–drug conjugate, exhibits potent targeted antitumor activity against FRα-expressing tumors. Mol Cancer Ther. 2015;14(7):1605–1613. doi: 10.1158/1535-7163.MCT-14-1095 [DOI] [PubMed] [Google Scholar]; • The preclinical development and characterization of mirvetuximab soravtansine.
- 37.Moore KN, Borghaei H, O'Malley DM, et al. Phase I dose-escalation study of mirvetuximab soravtansine (IMGN853), a folate receptor α-targeting antibody–drug conjugate, in patients with solid tumors. Cancer. 2017;123(16):3080–3087. doi: 10.1002/cncr.30736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Moore KN, Oza AM, Colombo N, et al. Phase III, randomized trial of mirvetuximab soravtansine vs. chemotherapy in patients with platinum-resistant ovarian cancer: primary analysis of FORWARD I. Ann Oncol. 2021;32(6):757–765. doi: 10.1016/j.annonc.2021.02.017 [DOI] [PubMed] [Google Scholar]
- 39.VENTANA FOLR1 (FOLR1-2.1) RxDx Assay [Interpretation Guide for EOC]. Tucson, AZ: Ventana Medical Systems, Inc. and Roche Diagnostics International, Inc.; 2022. [Google Scholar]
- 40.Moore KN, Oza AM, Columbo N, et al. FORWARD I (GOG 3011): A Phase III study of mirvetuximab soravtansine, a folate receptor alpha (FRα)-targeting antibody–drug conjugate, vs. chemotherapy in patients with platinum-resistant ovarian cancer. Presented at: European Society for Medical Oncology Congress. Barcelona, Spain; September 27, 2019-October 1, 2019. [Google Scholar]
- 41.Dilawari A, Shah M, Ison G, et al. FDA approval summary: mirvetuximab soravtansine-gynx for FRα-positive, platinum-resistant ovarian cancer. Clin Cancer Res. 2023;29(19):3835–3840. doi: 10.1158/1078-0432.CCR-23-0991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Moore KN, Konecny GE, Banerjee S, et al. Phase III MIRASOL (GOG 3045/ENGOT-ov55) Study: Mirvetuximab soravtansine vs. investigator's choice of chemotherapy in platinum-resistant, advanced high-grade epithelial ovarian, primary peritoneal or fallopian tube cancers with high folate receptor-alpha (FRα) expression. Presented at: American Society of Clinical Oncology Annual Meeting. Chicago, IL; June 2, 2023-June 6, 2023. doi: 10.1200/JCO.2023.41.17_suppl.LBA5507 [DOI] [Google Scholar]
- 43.Moore KN, Angelergues A, Konecny GE, et al. Mirvetuximab soravtansine in FRα-positive, platinum-resistant ovarian cancer. N Engl J Med. 2023;389(23):2162–2174. doi: 10.1056/NEJMoa2309169 [DOI] [PubMed] [Google Scholar]; •• Results from the global, confirmatory, randomized Phase III MIRASOL trial showing efficacy and safety of mirvetuximab soravtansine vs. investigator's choice chemotherapy in patients with FRα-high platinum-resistant ovarian cancer. In the MIRASOL trial, mirvetuximab soravtansine was the first novel therapy to demonstrate a statistically significant and clinically meaningful overall survival (OS) advantage over single-agent chemotherapy in a Phase III trial of patients with platinum-resistant ovarian cancer.
- 44.Ponte JF, Ab O, Lanieri L, et al. Mirvetuximab soravtansine (IMGN853), a folate receptor α-targeting antibody–drug conjugate, potentiates the activity of standard of care therapeutics in ovarian cancer models. Neoplasia. 2016;18(12):775–784. doi: 10.1016/j.neo.2016.11.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.O'Malley DM, Matulonis UA, Birrer MJ, et al. Phase Ib study of mirvetuximab soravtansine, a folate receptor alpha (FRα)-targeting antibody–drug conjugate (ADC), in combination with bevacizumab in patients with platinum-resistant ovarian cancer. Gynecol Oncol. 2020;157(2):379–385. doi: 10.1016/j.ygyno.2020.01.037 [DOI] [PubMed] [Google Scholar]; •• Results from Phase Ib/II FORWARD II trial showing efficacy and safety of mirvetuximab soravtansine plus bevacizumab in platinum-resistant ovarian cancer (data cutoff date of 22 April 2019).
- 46.O'Malley DM, Matulonis UA, Mantia-Smaldone GM, et al. Mirvetuximab soravtansine, a folate receptor α-targeting antibody drug conjugate, in combination with bevacizumab in patients with platinum agnostic ovarian cancer: final analysis. Presented at: American Society of Clinical Oncology Annual Meeting. 2021 June 4–8; Fully Online. doi: 10.1200/JCO.2021.39.15_suppl.5504 [DOI] [Google Scholar]; •• Results from the Phase Ib/II FORWARD II trial demonstrating the efficacy and safety of mirvetuximab soravtansine plus bevacizumab in platinum-agnostic ovarian cancer.
- 47.Gilbert L, Oaknin A, Matulonis UA, et al. Safety and efficacy of mirvetuximab soravtansine, a folate receptor alpha (FRα)-targeting antibody–drug conjugate (ADC), in combination with bevacizumab in patients with platinum-resistant ovarian cancer. Gynecol Oncol. 2023;170:241–247. doi: 10.1016/j.ygyno.2023.01.020 [DOI] [PubMed] [Google Scholar]; •• Results from the Phase Ib/II FORWARD II trial showing the efficacy and safety of mirvetuximab soravtansine plus bevacizumab in FRα-expressing platinum-resistant and platinum-sensitive ovarian cancer (final analysis; data cutoff date of 21 June 2021).
- 48.Clinical Trials Identifier: NCT05445778 . Mirvetuximab soravtansine with bevacizumab vs. bevacizumab as maintenance in platinum-sensitive ovarian, fallopian tube, or peritoneal cancer (GLORIOSA). https://www.clinicaltrials.gov/ct2/show/NCT05445778 (Accessed August 30, 2023).
- 49.Moore KN, Borghaei H, O'Malley DM, et al. Phase I dose-escalation study of mirvetuximab soravtansine (IMGN853), a folate receptor α-targeting antibody–drug conjugate, in patients with solid tumors. Cancer. 2017;123(16):3080–3087. doi: 10.1002/cncr.30736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Clinical Trials Identifier: NCT01609556 . First-in-human study to evaluate the safety, tolerability, pharmacokinetics and pharmacodynamics of mirvetuximab soravtansine in adults with ovarian cancer and other folate receptor 1 (FOLR1)-positive solid tumors (IMGN853-0401). https://www.clinicaltrials.gov/study/NCT01609556 (Accessed November 30, 2023).
- 51.Matulonis UA, Birrer MJ, O'Malley DM, et al. Evaluation of prophylactic corticosteroid eye drop use in the management of corneal abnormalities induced by the antibody–drug conjugate mirvetuximab soravtansine. Clin Cancer Res. 2019;25(6):1727–1736. doi: 10.1158/1078-0432.CCR-18-2474 [DOI] [PubMed] [Google Scholar]
- 52.Hendershot A, Slabaugh M, Riaz KM, et al. Strategies for prevention and management of ocular events occurring with mirvetuximab soravtansine. Gynecol Oncol Rep. 2023;47:101155. doi: 10.1016/j.gore.2023.101155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.O'Malley DM, Myers T, Zamagni C, et al. GLORIOSA: a randomized, open-label, Phase III study of mirvetuximab soravtansine with bevacizumab vs. bevacizumab as maintenance in platinum-sensitive ovarian, fallopian tube or primary peritoneal cancer. Presented at: American Society for Clinical Oncology Annual Meeting. Chicago, IL; June 2, 2023-June 6, 2023. doi: 10.1200/JCO.2023.41.16_suppl.TPS5622 [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The study sponsor, ImmunoGen, Inc., is committed to responsible sharing of clinical trial data. Data from this clinical trial, once available, can be requested by any qualified investigator who engages in relevant research. Data requests can be submitted to medicalinformation@immunogen.com.