

Abstract
Japanese encephalitis virus (JEV) is a neurotropic and neuroinvasive flavivirus causing viral encephalitis, which seriously threatens the development of animal husbandry and human health. DNA methylation is a major epigenetic modification involved in viral pathogenesis, yet how DNA methylation affects JEV infection remains unknown. Here, we show genome-wide DNA methylation profiles in the brains of JEV-infected mice compared to mock-infected mice. JEV can significantly increase the overall DNA methylation levels in JEV-infected mouse brains. A total of 14,781 differentially methylated regions associated genes (DMGs) have been identified. Subsequently, KEGG pathway analysis suggested that DNA methylation modulates the tight junction signaling pathway, which can potentially impact the permeability of the blood-brain barrier (BBB). We demonstrate that hypermethylation of the tight junction gene Afdn promoter inhibited AFDN expression and increased monolayer permeability of mouse brain microvascular endothelial (bEnd.3) cells in an in vitro transwell assay. Collectively, this study reveals that DNA methylation is increased in a murine Japanese encephalitis model and that modulation of Afdn expression promotes BBB permeability.
Supplementary Information
The online version contains supplementary material available at 10.1186/s12974-024-03266-6.
Keywords: Japanese encephalitis virus, DNA methylation, WGBS, Blood-brain barrier, Afdn
Background
Japanese encephalitis virus (JEV), a mosquito-borne flavivirus, is one of the leading neurotropic pathogens and a significant cause of mortality, mainly in Southeast Asia and Western Pacific regions [1–4]. In those endemic regions, JEV outbreaks have been largely controlled by vaccination [5, 6]. However, mass vaccinations have not yet taken place outside these regions [7, 8]. Due to climate change, globalization, and other changes to ecosystems, the vertebrate hosts and mosquito vectors associated with the spread of JEV may be able to invade areas that are currently unaffected, such as Europe and America [9, 10]. In 2022, different states of Australia reported outbreaks of JEV (genotype IV) in pig farms, which resulted inseven human deaths, 47 persons with encephalitis and serious economic losses of livestock [11, 12]. Thus, not only in endemic areas but also in some adjacent non-endemic areas, JEV infections have become a concern of public and animal health.
Although JEV infection usually starts in peripheral tissues after mosquito bites, subsequent brain infection is critical to JEV-induced mortality [13]. JEV brain infection leads to encephalitis characterized by high viral loads, robust inflammatory responses and disruption of the Blood-brain barrier (BBB) [3, 14]. Once JEV has established an infection in the brain, vaccination can no long prevent the neurological demage [15]. At this stage, therapeutical medicines are urgently needed [16], but effective treatments for JEV brain infections remain unavailable. Insights into the neurological pathogenesis of JEV infections would facilitate the identification of potential therapeutic targets. Thus, investigation of the underlying mechanisms of JEV infection is important not only to address JEV pathogenesis but also to develop innovative strategies for the treatment of JEV brain infections.
DNA methylation is a major epigenetic modification that plays an important role in tissue homeostasis by modulation of gene expression [17–20]. Alteration of DNA methylation patterns is involved in the pathogenesis of various diseases, including cancers, autoimmune diseases, and neurological disorders [21–23]. For example, DNA methylation in Alzheimer’s disease (AD) has been extensively studied by different technological approaches, such as whole-genome bisulfite sequencing (WGBS), which was instrumental in gaining more insight into the role of DNA methylation in AD development [24, 25]. Furthermore, emerging evidence demonstrates associations between host cell DNA methylation and virus infections [19, 26]. For example, influenza virus NS1 promotes ubiquitination and degradation of DNA methyltransferases 3B (DNMT3B), resulting in demethylation of the promoter of JAK/STAT signaling suppressor genes. As a consequence, the expression of the suppressors is quickly upregulated, thereby inhibiting type I interferon (IFN) signaling [19]. In addition, miRNA29-mediated inhibition of DNMT3A and DNMT3B expression resulted in the demethylation of the COX2 promoter, resulting in an IFN-λ response against influenza virus infection [26]. Other reports suggested that DNA methylation is also involved in the pathogenesis of flavivirus infections [27–29]. ZIKV infection results in the methylation of a number of genes (e.g., WWTR1, RASSF1A, RABGAP1L) that regulate brain size during development, likely linked to ZIKV-induced microcephaly [27, 28]. Another study showed that demethylation of the TNF-α promoter in DENV-infected patients resulted in high expression of this cytokine, which is associated with the development of hemorrhagic fever [30]. Thus, the accumulating knowledge of the effect of DNA (de-)methylation on the pathogenesis of viral diseases might assist in developing potential treatments for those being affected.
In this study, we used WGBS to characterize the alteration of DNA methylation patterns occurring during JEV-induced encephalitis in a murine model. KEGG pathway analysis was used to identify pathways potentially regulated by DNA methylation. And we explored the mechanism by which DNA methylation participates in BBB permeability. Our study paves the way to understanding the role of alteration of DNA methylation in the pathogenesis of JEV-induced encephalitis.
Materials and methods
Cell culture, virus and reagents
bEnd.3 cells were purchased from the National Collection of Authenticated Cell Cultures and were cultured in Phenol-free red DMEM medium (Gibco:21063029, Thermo Fisher Scientific) supplemented with 10% fetal bovine serum (FBS) at 37 °C in 5% CO2 incubator. The JEV NJ2008 strain (GenBank accession No. GQ918133) was stored at our laboratory (Shanghai, China). The Afdn (AF-6/afadin/MLLT4) antibody (Catalog: A18865) was obtained from the ABclonal company. Trichloroacetic acid (TCA) (Catalog: T9159) and sodium fluorescein dye (NaF) (Catalog: F6377-100G) were purchased from Sigma-Aldrich. Transwell inserts (12 mm, 0.4 μm polyester membrane, tissue culture treated, REF:3460) was purchased from Corning. Rat tail collagen (Catalog: C7661-5MG) and FITC-Dextran-10,000 (10kD, FD10S-100MG), and 5-Aza-2′-deoxycytidine (a DNA methylation inhibitor, named 5-AZA, Catalog: A3656-5MG) were also purchased from Sigma-Aldrich.
JEV stocks was prepared by using BHK-21 cells. The information in details as follows: BHK-21 cells were seeded in T75 cell culture flasks. When the cell density reaches about 70%, the cell culture medium was discarded and replaced with 10mL DMEM. Then 200ul stock JEV solution (about 0.2MOI JEV) was added into T75 flasks. After incubation for 2 h, the virus-containing medium was removed. The cells were then washed twice by PBS and maintained in DMEM medium supplemented with 2% FBS. When the cells show cytopathic effect changes (usually about 72 h post-infection), the supernatant was collected and stored in a -80 ℃ refrigerator.
Virus tistration
Virus titers were determined by using the TCID50 assay. Briefly, BHK-21 cells were cultured in the 96-well plate until reaching around 70% confluency. The medium was removed, and cells were inoculated with the certain diluted viuses (100 µL/well) ranging from 10− 1 to 10–11 with eight replicateds per dilution. After incubation, cells were incubated with DMEM medium containing 2% FBS at 37 ℃. When it appeared the clearly cytopathic effect (plaques), the number of wells with or without plaques was recorded at each dilution. The TCID50 of the virus was calculated using the Reed-Muench algorithm [31].
Mouse model of JEV infection
Female C57BL/6 mice (three weeks old) were ordered from Shanghai Lingchang Company (Shanghai, China). After acclimatizing for one week under standard laboratory conditions, the mice (n = 20) were then randomly divided into two groups: a mock-infected control group (n = 10) and a JEV-infected group (n = 10). In the JEV group, ten mice were intraperitoneally injected with 5 × 10^4 PFU JEV. The clinical signs and mortality were monitored daily, and clinical symptoms were scored according to a publishing standard score system [32]. When the mice show severe neurological symptoms (e.g., body stiffening, hind limb paralysis, tremor) with clinical score approaching 5 were euthanized with carbon dioxide (CO2). Then, the mouse’s heart was promptly perfused with PBS and brain tissues were collected quickly. Ethical approval for all animal experiments was obtained from the Institutional Animal Care and Use Committee of Shanghai Veterinary Research Institute, and the experiments were conducted in strict accordance with the Guidelines on the Humane Treatment of Laboratory Animals.
Blood-brain barrier permeability measurement in mouse brain
After JEV infection, mice developed severe neurological symptoms and were intraperitoneally injected with 100 µL (10 mg) sodium fluorescein dye (NaF). After 10 min, the blood samples were collected by eye enucleation, and serum was separated. A 50 µL of the 15% trichloroacetic acid (TCA) was added to the serum, followed by centrifugation at 10,000 rpm for 10 min. The supernatant (45 µL) was mixed with 30 µLof 5 mol/L NaOH and 7.5% TCA. Mice were then perfused with PBS, and the brain tissues were then extracted, weighed, and homogenized with 7.5% TCA. After centrifugation at 10,000 rpm for 10 min, the supernatant was treated with 30 µL of 5 mol/L NaOH. The fluorescence intensity in serum and brain supernatant was measured respectively (excitation wavelength: 485 nm; Emission wavelength: 530 nm), and NaF concentrations were calculated using a standard curve. The permeability index (mL/g) was calculated using the formula:
Permeability index (mL/g) = (NaF content in brain tissue/brain tissue weight)/(NaF content in serum/serum amount).
DNA extraction and quality control
Three mock-infected mouse brain samples and three JEV-infected mouse brain DNA samples were collected. Genomic DNA was extracted using a QIAGEN DNeasy ® Blood & Tissue kit. DNA quality and quantity were determined by an Ultraviolet spectrophotometer.
Whole-genome bisulfite sequencing (WGBS)
For Whole-Genome Bisulfite Sequencing (WGBS), a total of six samples (three mock-infected and three JEV-infected mouse brain DNA samples) were used for WGBS at BGI Genomics (Wuhan, China). After a quality control test for the library preparation, these genomic DNAs were broken into fragments of an average size of approximately 250 bp by Bioruptor (Diagenode, Belgium), followed by the blunt-ending, dA addition to 3’-end, and finally, adaptor ligation (in this case of methylated adaptors to protect from bisulfite conversion). Ligated DNA was bisulfite converted using the EZ DNA Methylation-Gold kit (ZYMO), followed by PCR amplification and purification with the QIAquick Gel Extraction Kit (Qiagen). Finally, sequencing was performed on an appropriate platform, and the raw reads were first filtered. Low-quality adaptor automatically, and duplicated reads were removed to get the clean reads using BSMAP v2.90. The sequencing data for each sample is summarized in Table 1. Then, clean data were mapped to the reference genome, and only high-quality, uniquely mapped data were used for methylation analysis. The bioinformatics analysis included methylation pattern identification and pathway enrichment analysis using OmicStudio tools (https://www.omicstudio.cn/tool). Visualizations tevhniques suach as violin plots, circular plots (circo), heatmaps, and pathway analysis were generated by utilizing the OmicStudio tools.
Table 1.
Sequencing data quality control chart by whole genome bisulfite sequencing (WGBS)
| Sample ID | Clean Reads Q20 Rate (%) |
Clean Data Size (bp) |
Mapping Rate (%) | Bisulfite Conversion Rate (%) | Duplication Rate (%) | Average Depth (X) |
Coverage (%) |
|---|---|---|---|---|---|---|---|
| mock1 | 93.62;94.65 | 96,122,431,400 | 91.60 | 99.59 | 12.36 | 23.68 | 87.322 |
| mock2 | 93.85;94.83 | 105,886,943,400 | 89.97 | 99.61 | 6.80 | 27.65 | 87.318 |
| mock3 | 94.42;94.56 | 96,252,253,600 | 87.89 | 99.52 | 7.56 | 24.58 | 87.222 |
| JEV1 | 94.62;95.18 | 111,946,471,000 | 90.73 | 99.53 | 7.75 | 29.54 | 87.329 |
| JEV2 | 95.20;95.45 | 116,417,847,000 | 90.82 | 99.56 | 5.92 | 31.19 | 87.363 |
| JEV3 | 94.70;93.48 | 98,233,161,800 | 91.95 | 99.50 | 7.64 | 26.34 | 87.276 |
Mapping rate: the proportion of reads that can be mapped to reference genome in total reads
Coverage (%): sequencing coverage of the whole genome
Methylation level calculation
The methylation level was determined by dividing the number of reads covering each mC by the total reads covering that cytosine, which was also equal to the mC/C ratio at each reference cytosine [33]. The formula is shown as follows:
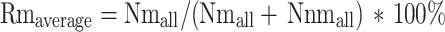 |
Nm represents the reads number of mC, while Nnm represents the number of non-methylation reads.
Differentially methylated regions (DMRs) and pathway analysis
DMRs were identified by comparison of the mock samples and JEV samples methylomes using windows that contained at least five CpG (or CHG, CHH) sites with a minimum two-fold change in methylation level and Fisher’s test p-value < = 0.05. Two nearby DMRs would be considered interdependent and joined into one continuous DMR if the genomic region from the start of an upstream DMR to the end of a downstream DMR also had two-fold methylation level differences between sample1 and sample2 with a p-value < = 0.05. Otherwise, the two DMRs were viewed as independent. After iteratively merging interdependent DMRs, the final dataset of DMRs was made up of those independent of each other. To compare the methylation level of DMR in different models by CIRCOS, the degree of difference of methyl-cytosine (mCG, mCHG, mCHH) was calculated using the following formula:
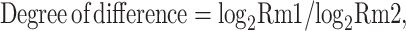 |
Where Rm1 and Rm2 represent the Methylation level of methyl-cytosine for sample1 and sample2, respectively. 0.001 will replace Rm1(or Rm2) while it is 0.
Pathway-based analysis helps to further understand genes’ biological functions. KEGG is used to perform pathway enrichment analysis of DMR-related genes. This analysis identifies significantly enriched metabolic pathways or signal transduction pathways in DMR-related genes compared with the whole genome background.
Bisulfite sequencing PCR (BSP)
Genomic DNA was subjected to bisulfite treatment using EZ DNA Methylation-Gold™ Kit (ZymoResearch, Los Angeles, USA). Bisulfite sequencing primers were designed using the online software MethPrimer tool. The DNA samples treated with bisulfite were amplified by PCR using NEB’s EpiMark heat-activated Taq DNA polymerase. About 20 positive clones were randomly selected for sequencing with the pMD19-T universal sequencing primer. The sequencing data were analyzed using QUMA online software (http://quma.cdb.riken.jp/top/index.html) to assess the non-CpG C-T conversion rate. The methylation status of the target sequence shows the percentage of methylated CpGs in the total number of CpGs. Fisher’s Exact T-test is used for statistical difference analysis.
Quantitative real-time PCR (RT-qPCR)
Total cell RNA was extracted from the TRIzol reagent, followed by reverse transcription using the PrimeScriptTM RT regent kit. Subsequently, RT-qPCR was performed with SYBR Premix Ex TaqTM II (Takara). The relative abundance of transcripts for each gene in each sample was normalized to the GAPDH gene and calculated by the 2−ΔCT method. All qPCR samples were subjected to three replicates. The primer sequences used for RT-qPCR are listed in the Table 2.
Table 2.
Primers used for qPCR in this study
| Primer Name | Primer sequence (5’to 3’) |
|---|---|
| Afdn-Forward | TGAAGAGGAGACCACCGGACT |
| Afdn-Reverse | GAGCGGAGCCGTAGCCA |
| Cldn19-Forward | GGCCCTGGACGGTCATATC |
| Cldn19-Reverse | TTACTGTCTCCAACCCGAGTG |
| Cldn6-Forward | TTCATCGGCAACAGCATCGT |
| Cldn6-Reverse | CAGCGCCAACAGTGAGTCATA |
| Cldn3-Forward | GCGGCTCTGCTCACCTTAGT |
| Cldn3-Reverse | CAGCCCAGCCCACGTACAACC |
| Crb3-Forward | CCCCCAGGTGATGAGTCCAAC |
| Crb3-Reverse | TGCTGGGCCGGTAGGT |
| Cldn14-Forward | GCATGGTGGCCGTGTC |
| Cldn14-Reverse | GAGATGAAGCCCAGGTACAGG |
| GAPDH-Forward | TATGTCGTGGAGTCTACTGGT |
| GAPDH-Reverse | GAGTTGTCATATTTCTCGT |
RNA interference
The Mus-Afdn RNA interference was designed and chemically synthesized (GenePharma, Shanghai, China). And bEnd.3 cell were respectively transfected with 20pmol of Afdn siRNA-1, siRNA-2, or siRNA-3 using Lipofectamine RNAi MAX (Thermo Fisher Scientific), Six hours post-transfection, the cell medium was replaced with DMEM containing 2% FBS. After 72 h of RNA interference, the RNA samples would be collected to detect the expression of Afdn.
The sequences for Afdn siRNA were as follows:
forward of siRNA-1 (5’-GCACCUCGCAUGUGUUUAATT-3’),
forward of siRNA-2 (5’-CCAGGUACCUGGACAAUAUTT-3’).
forward of siRNA-3 (5’-GAGGUGCUAAGGAAAUAAUTT-3’).
Transwell assay for cell permeability
We chose Corning Nested (REF:3460) 12 mm (12-well plate) Transwell (0.4 μm polyester membrane tissue culture treatment) and bEnd.3 cell line for in vitro monolayer cellular BBB simulation experiments. Firstly, rat tail collagen was coated in the transwell chambers for 1 h. The excess collagen solution was discarded, and the membrane surface was gently washed with PBS. Next, pre-equilibrate the chamber with serum-free DMEM (phenol red-free) for at least 1 h. Then seed bEnd.3 cell with an inoculation of 3 × 105 cell density. Add 1.5 mL of DMEM medium with 10% FBS Phenol-free red to the wells of a 12-well plate (bottom chamber), and then put the pre-equilibrated nest into the wells and add 500 µL of cell suspension into the nest (upper chamber). After 48 h, a monolayer was formed and its integrity was assessed by 4 h leakage assay. Subsequently, we followed the above procedures to set up experiments for permeability analysis. Based on the different objectives, bEnd.3 cells were differentially treated by JEV-infected mouse brain supernatant or RNA interference by targeting Afdn gene for the experiment setup: (1) to determine role of JEV supernatant treatment in cell permeability, bEnd.3 was treated by JEV supernatant and mock supernatant, respectively; (2) to determine role of Afdn in cell permeability, bEnd.3 was treated by the Afdn specific siRNA and the negative control siRNA, respectively. The treated cells were returned to 5% CO2 and incubated at 37 °C for 6 h or 72 h. 500 µL of FITC-Dextran-10,000 (10 kD, 100 µg/mL) was then added to the upper chamber of the Transwell and incubated at 37 °C in a 5% CO2 incubator. After 60–120 min of incubation, 100 µL of medium was removed from the upper and bottom chambers. The upper and bottom chambers are then measured for fluorescence intensity (excitation light: 492 nm; emission light: 520 nm). We make a standard curve using standards with different dilution concentrations and then calculate the concentrations of fluorescein sodium after different treatments. The apparent permeability coefficients (Pe) were calculated as described previously. We used the following equation to measure the permeability coefficients: Permeability coefficients (Pe) = C*V/(C0*T*S), [V = sample volume in the basal chamber, C = concentration in the basal chamber, C0 = the initial concentration in the donor compartment T = time variations (hour), S = exposed surface area (cellular monolayer in cm2)].
Statistical analysis
Statistical analysis was conducted using GraphPad Prism version 8.0 (GraphPad Software, La Jolla, CA, USA). Data are reported as mean values ± standard deviations (SD) of three independent experiments with three biological replicates. The significance of differences between groups was evaluated using the Student’s t-test, Log-rank (Mantel-Cox) test and WilcoxonTest method of non-parametric tests. Statistical significance was established at *p < 0.05, **p < 0.01, and ***p < 0.001.
Results
JEV infection significantly increases the permeability of the mouse blood-brain barrier in vivo
Since DNA methylation has an important role in other neurological diseases and viral infections, the aim was to profile whole genome DNA methylation of mouse brains with JEV-induced encephalitis. We first established a mouse model to investigate alterations of genome-wide DNA methylation profiles in the brain during JEV infection. C57BL/6 mice were intraperitoneally injected with 5 × 10^4 PFU JEV and monitored for 14 days post-infection (dpi) with and without symptoms. According to the reported criteria to score the animals for the progression of symptoms [32], sick mice were euthanized with a clinical score of 5, which included signs such as body tremors, hind limb paralysis and other near-death conditions appearing (Fig. 1A). By calculating the survival rate, the results showed that 80% of the mice in this model succumbed to JEV infection within 14 days (Fig. 1B). High viral loads were observed in the JEV-infected brains (Fig. 1C), consistent with the severity of the infection. A known characteristic of JEV-induced encephalitis is increased permeability of the BBB. To detect changes in BBB permeability, sodium fluorescein dye (NaF fluorescence) was intraperitoneally injected into mice, and then values of the NaF fluorescence were measured in JEV-infected and mock-infected mice (Fig. 1D). In comparison to mock-infected mice, JEV-infected mice showed exhibited increased yellow fluorescence in the brain tissue, suggesting the BBB is disrupted (Fig. 1E). Next, the index of BBB permeability in the brain was calculated according to the previously reported formula [34], we found JEV significantly increased permeability ratio by detecting fluorescence values in brains (Fig. 1F), confirming that JEV increases the permeability of the BBB in this model.
Fig. 1.

JEV infection significantly increases the permeability of the mouse blood-brain barrier in vivo. (A) JEV-infected mice clinical score. Mice were intraperitoneally injected with 5 × 10^4 PFU JEV. For clinical symptom score standards, see Methods details. (B) JEV-infected mice survival curve (mock group: n = 10; JEV group: n = 10). Survival curves were analyzed by Log-rank (Mantel-Cox) test. (C) TCID50 detection of virus titer in JEV-infected mouse brain. TCID50 was analysed by the WilcoxonTest method of non-parametric tests. (D) Schematic diagram of establishing a model for detecting BBB permeability in JEV-infected mice. (E) Picture showing the content of sodium fluorescein in mouse brain. (F) BBB permeability ratio was detected and calculated by measuring sodium fluorescein dye using a microplate reader (n = 3). The data presented represent the mean values ± standard deviations (SD) of three independent experiments with three biological replicates using Student’s t-test. Statistical significance was established at **p < 0.01.The schematic diagram was made with the free version of BioRender
Elevated genome-wide methylation levels in the JEV-infected mouse brain
Next, brain DNA was extracted from three JEV-infected mice and three mock mice, respectively. WGBS was performed to investigate changes in the whole-genome DNA methylation profile as a result of JEV infection (Fig. 2A). After filtering out low-quality data, on average, 104.143 Gb clean reads were generated for each sample. These reads were compared to the reference mouse genome using BSMAP software, and the statistical results of the comparison are shown in Table 1. The mapping rates for subsequent analysis ranged from 87.89 to 91.95%, and the bisulfite conversion rate was as high as 99%.
Fig. 2.

Elevated genome-wide methylation levels in the JEV-infected mouse brain. (A) Schematic diagram of DNA samples taken from mouse brains for WGBS sequencing. The schematic diagram was made with BioRender. (B) Violin plot for the overall distribution of methylation levels for different methylation types. mCG, mCHG, and mCHH. (H = A, C or T). (C) The number and proportion of each mC (mCG, mCHG, and mCHH) type in the whole genome in different samples, and the sum of the proportion of three mC types is 100%. A histogram is used to display a quantitative comparison of the pie chart above. The data presented represent the mean values ± standard deviations (SD) of three biological replicates using Student’s t-test. Statistical significance was established at *p < 0.05, **p < 0.01, and ***p < 0.001
To characterize the methylation profile of the whole genome, the average methylation level of the entire genome was analyzed. In mammals, there are three types of methyl-cytosine: mCG, mCHG, and mCHH, with mCG being the primary form. Violin plots were plotted to show the mean methylation levels in the two groups of samples (Fig. 2B). The average methylation level of mCG accounted for around 74–76% in both sample groups, whereas mCHH and mCHG exhibited average methylation levels of less than 2%. The overall methylation level of mCG was significantly higher in the JEV-infected group (75.6%) compared to the mock group (74.7%) (p < 0.01) (Fig. 2B). Conversely, JEV infection led to a noticeable decrease in the overall methylation levels of mCHG (from 1.2 to 1.1%, p < 0.001) and mCHH (from 1.4 to 1.3%, p < 0.001) (Fig. 2B). The proportions of each mC type were further analyzed with the sum of the three types (mCG, mCHG, and mCHH) totaling 100%. Consistently, JEV infections significantly increased the proportion of mCG methylation (average = 75.3%, p < 0.05) compared to the mock group (average = 71.0%) (Fig. 2C). In contrast, the percentages of mCHH (average = 20.9%, p < 0.01) and mCHG (average = 3.7% p < 0.05).
decreased in the JEV group compared with the mock group (mCHH and mCHG, average = 24.6% and 4.4%, respectively) (Fig. 2C).
To further analyze the DNA methylation levels in various functional genomic elements between the mock and JEV group, the whole genome was divided into seven different transcriptional element regions and compared the average methylation levels (mCG, mCHG, and mCHH types) in these regions were compared (Additional file 1: Figure S1). Our analysis revealed that DNA methylation levels were the lowest near the transcription start site (TSS) for the three mC-types. The same patterns were observed for regions, peaking at 76%. Moreover, the methylation levels of the JEV group displayed a slight elevation compared to the mock group in the first exon, internal intron and last exon (Additional file 1: Figure S1A). For mCHG, the overall methylation level remained consistently below 2.4% throughout the genome (Additional file 1: Figure S1B). As for mCHH, the methylation level ranged from approximately 0.5–1.2% (Additional file 1: Figure S1C).
In short, our results demonstrate a significant increase in the overall methylation level of mCG and a decrease in the methylation levels of mCHG and mCHH in the JEV group compared to the mock group.
Methylome map of JEV infection
Next, we identified a total of 184,986 differentially methylated regions (DMRs) in the type of mCG across the genome. To visualize these findings, a DNA methylome map using Circos images was generated, which allowed analysis of the differential methylation levels on each chromosome. Since there are more than 184,000 DMRs, it isn’t easy to distinguish all hypermethylated and hypomethylated regions with different colors in the circle diagram. To better visualize our DMRs, we further filtered and displayed the DMRs with a circle chart where the absolute value of the difference in methylation levels between the two groups was greater than 0.25. The differential methylation levels between the JEV group and the mock group are illustrated in the third circle (from outside to inside) in the Circos images (Fig. 3A). The redder the color, the higher the methylation level after JEV infection, the greener the color, the lower the methylation level after JEV infection. The findings also indicated that JEV infection indeed increased methylation levels of DMRs at the genome-wide scale and on all chromosomes (Fig. 3A).
Fig. 3.

Methylome map of JEV infection. (A) Circos image showed the differential methylation levels between the JEV group and mock group at the genome-wide scale in the type of mCG. Each circle from the outside to the inside depicts: (1) shows a diagram of the mouse genome subdivided by chromosomes; (2) the methylation level of the mock group; (3) represents the absolute value of the differential methylation level value of the JEV group minus the mock group greater than 0.25. Red color represents hypermethylated areas, green color represents hypomethylated areas, the redder the color, the higher the methylation level after JEV infection, the greener the color, the lower the methylation level after JEV infection; (4) the methylation level of the JEV group. (B) The graphs show the numbers of hyper and hypomethylated DMRs distribution in different genomic elements in the mCG types
In addition to the genome-wide analysis, a comprehensive DMRs statistical analysis was performed in mCG by dividing the genome into distinct regions, including upstream 2k, CG island, 5’UTR, First exon, First intron, exon, intron, CDS, 3’UTR, and downstream2k regions. Our analysis showed an intriguing pattern: in all regions, the number of hypermethylated DMRs outnumbered hypomethylated DMRs (Fig. 3B). For example, a total of 6086 hyper-DMRs and 3428 hypo-DMRs were located in upstream2k region. These findings provide compelling evidence that JEV infection induces a substantial increase in DNA methylation across the genome, predominantly characterized by hypermethylation in various genomic regions.
Similarly, we conducted circo plot and DMRs statistical analysis on the other two methylated forms, mCHG and mCHH. Our results indicated that the methylation levels in the JEV group were significantly lower than those in the mock group across all chromosomes and throughout the genome in the type of mCHG (Additional file 2: Figure S2A). Specifically, the number of hypomethylated DMRs in the mCHG type was significantly higher than the number of hypermethylated DMRs in all genetic regions (Additional file 2: Figure S2B). Similarly, for mCHH, the number of hypermethylated DMRs was significantly lower than that of hypomethylated DMRs, and the methylation level of the JEV group on was significantly lower than those in the mock group across chromosome (Additional file 2: Figure S2C and D). These findings contrast with the patterns observed for mCG types.
Identification of DMR-related genes and validation by bisulfite sequencing PCR
By integrating the DMRs with gene annotations, 14,781 DMR-related genes (DMGs) were identified that demonstrated significant changes in methylation status in response to JEV infection. These DMGs represent potential candidates underlying the regulatory effects of methylation on gene expression. To further understand the distribution of DMGs across the genome, we conducted a statistical analysis of DMGs in different genomic regions. Consistent with the patterns observed in the DMR analysis (Fig. 3B), there are far more hypermethylated DMGs than hypomethylated DMGs in all functional genetic elements (Fig. 4A). This observation highlights the prevalent hypermethylation induced by JEV infection across various genomic regions. Given the crucial role of promoter methylation in gene expression regulation [35], we next focused exclusively on DMGs located in upstream 2k regions. As a result, a total of 9514 DMRs mapping to 7990 DMGs were identified. To explore the methylation patterns of these DMGs, a heat map clustering analysis was conducted with 7990 DMGs in the upstream 2k region. Heap cluster analysis found 66% (5277 /7990) of DMGs showed hypermethylation and 34% (2713/7990) of DMGs demonstrated hypomethylation in the upstream 2k, illustrating that JEV increases the DMG methylation levels in the upstream 2k region (Fig. 4B).
Fig. 4.

Identification of DMR-related genes and validation by bisulfite sequencing PCR. (A) The graphs show the numbers of hyper and hypomethylated DMGs distribution in different genomic elements in the mCG types. (B) The heatmap shows the methylation levels of DMGs in the mock and JEV groups in the upstream 2k region. The differential methylation values (JEV group minus mock group methylation levels) were sorted from high to low. The heat map uses a gradient from blue to yellow to orange-red. The more orange-red the color, the higher the methylation level. The different colors represent the different methylation levels value in the upstream 2k region. (C) and (D) show validation of the WGBS DMGs in DNA levels by BSP. Two hypermethylated DMGs (C) and two hypomethylated DMGs (D) were selected and BSP confirmed their methylation levels. Then, sequencing results were analyzed using QUMA online software. Each horizontal line and the circle above represent a clone, which contains several methylation sites. Each circle represents a CG site, with the white circles representing unmethylation and the black circles representing methylation. Data are represented as the mean values ± standard deviations (SD) of at least three biological replicates using Student’s t-test (*p < 0.05, **p < 0.01, and ***p < 0.001) between the indicated groups
Next, to verify the reliability of the WGBS data, we selected four DMGs (two hypermethylated genes and two hypomethylated genes) that potentially affect the pathogenesis of JEV to assess the DNA methylation level through bisulfite sequencing PCR (BSP). Igsf21 and Nfam1, the hypermethylated genes, are implicated in synaptic inhibition, inhibitory presynaptic differentiation, and cytokine production amplification, respectively, suggesting their significant roles in JEV neuropathogenesis and immune responses [36, 37]. BSP analysis illustrated that Igsf21 exhibited a notable increase in methylation levels from 13.6% in the mock group to 26.4% in the JEV group. Similarly, Nfam1 methylation levels significantly increase post-JEV infection from 21 to 50.8% (Fig. 4C). On the other hand, the hypomethylated gene Irf2, which encodes a negative regulator of type I IFN production [38–40], demonstrated a prominent reduction in the methylation level from 8.8% in the mock group to 0% in the JEV group, potentially influencing the antiviral immune responses during JEV infection. In addition, Dll4, which encodes an essential component of the Notch signaling pathway [41], exhibited a dramatic decrease in methylation levels with 6% in the JEV group, compared with 14.3% in the mock group, suggesting its involvement in JEV-mediated dysregulation of cellular signaling. (Fig. 4D). Overall, these BSP results confirmed the differential methylation patterns observed in the WGBS data, indicating that the WGBS data were reliable and suitable for further study.
KEGG analysis reveals that JEV infection modulates tight junction signaling by hypermethylation of the Afdn promoter
To determine the biological functions of the DMGs in the upstream 2k region, we performed a KEGG pathway enrichment analysis, comparing the DMGs between mock and JEV groups. The KEGG pathway analysis showed that these DMGs were predominantly associated with key signaling pathways involved in the tight junction, cytokine-cytokine receptor interaction, MAPK, Ras and AMPK signaling pathways. These signaling pathways are known to play critical roles in processes such as BBB integrity, inflammation and apoptosis (Fig. 5A). Particularly, the tight junction signaling pathway is a crucial mechanism that has been found to affect BBB integrity during JEV infection, which mainly increases the permeability of the BBB by reducing tight junction protein expression [34, 42]. However, it is not clear whether DNA methylation regulates the tight junction signaling pathway and influences the changes in BBB permeability. Hence, we proceeded to analyze the tight junction signaling pathway in greater detail. Firstly, clustered heat maps were conducted to analyze the methylation levels of DMGs in the tight junction signaling pathway and results illustrated the vast majority of these DMGs were hypermethylated in the JEV-infected group; in contrast, hypomethylated DMGs account for a smaller proportion (Fig. 5B).
Fig. 5.

KEGG analysis reveals that JEV infection modulates tight junction signaling by hypermethylation of the Afdn promoter. (A) KEGG pathway analysis of DMGs in upstream2k region. The ordinate represents the enriched pathways, and the abscissa represents the Rich factor of the correspond- ding pathways; the size of the spots represents the number of genes related to DMRs enriched in each pathway. (B) Heatmap analysis of methylation levels of DMGs in the tight junction signaling pathway. (C) Tight junction signaling pathway-related hypermethylated DMGs methylation levels from WGBS-seq. X-axis represents hypermethylated DMGs. The Y-axis represents the fold difference of the methylation level value of the JEV group divided by the methylation level value of the mock group. Among them, those above the dotted line indicate that the multiple of the differential methylation level value of the two groups is greater than 1.5 times. (D) qPCR was performed to verify some hypermethylated DMGs with differential methylation levels greater than 1.5 times. Y axis represents the relative expression of GAPDH (100%). (E) BSP was performed to test Afdn genes’ methylation levels using JEV-infected mouse brain DNA samples. Each circle represents a CG site, with the white circles representing unmethylation and the black circles representing methylation. (F) Western blot measures the expression of AFDN after JEV infection. The histogram below shows a densitometric analysis of the Western blot results. Data are reported as mean values ± standard deviations (SD) of three independent experiments with three biological replicates. The significance of differences between groups was evaluated using the Student’s t-test. Statistical significance was established at *p < 0.05, **p < 0.01, and ***p < 0.001, ns: not significant
In order to determine the effect of DNA methylation of tight junction signaling pathway genes on BBB permeability, the methylation levels of hypermethylated genes in the tight junction signaling pathway in the two groups (JEV/mock) were compared to identify hypermethylated genes with a fold difference greater than 1.5 times (Fig. 5C). Subsequently, DMGs with a noticeable difference were selected for qPCR verification to detect the expression of these gene changes. Our results demonstrated that the expression of the Afdn gene significantly decreased after JEV infection, which was consistent with its hypermethylation status (Fig. 5D). However, the expression of other genes important for the tight junction pathway, Cldn19, Cldn6, Cldn3, Crb3 and Cldn14, did not support the general notion that JEV-induced DNA methylation would reduce gene expression. Considering that Afdn has the highest degree of methylation among all differential genes and can negatively regulate gene expression at the RNA level, the Afdn gene was chosen as a candidate gene for further study.
To quantify the DNA methylation status of the Afdn gene in mouse brains, BSP was conducted on both the mock and JEV-infected groups (Fig. 5E). The BSP results revealed that compared with mock group samples, there was a dramatic increase in the methylation level of the Afdn gene in the JEV group, which was consistent with the hypermethylation observed in the WGBS result. Furthermore, changes in AFDN protein expression were detected using Western blot and our results showed that the protein of AFDN was significantly reduced during JEV infection (Fig. 5F). Overall, our analysis reveals that DNA methylation is involved in modulation of tight junction signaling, which played a crucial role in maintaining the integrity of the BBB during JEV infection. Notably, among the DMGs of tight junction signaling, the Afdn gene promoter is hypermethylated, consisitent with its decreased protein expression.
Modulation of Afdn expression by the hypermethylation of its gene promoter contributes to the increased BBB permeability
Next, we hypothesized that the Afdn gene might also exhibit hypermethylation in vitro in infected cell models and investigated the impact of Afdn expression levels on BBB permeability. In vitro, bEnd.3 cells, representing mouse brain microvascular endothelial cells, serve as a standard cell model for studying BBB permeability. Previous studies have indicated that inflammatory factors in the homogenate supernatant of JEV-infected mouse brain, rather than the JEV infection itself, contribute to increased permeability [34, 43]. In order to further validate the results, we have examined effect of JEV-infected supernatant and UV-inactivated JEV-infected supernant, respectively on expression of AFDN. Our data showed that there was no difference between these two groups. However, compared with mock group, the significant decrease of AFDN expression was observed in both of those JEV-groups ((Additional file 3: Figure S3), consistent that cytokine signalings mainly contribute to BBB disruption. Therefore, to simulate this, homogenates from mock or JEV-infected mouse brains were collected, and the supernatants were obtained after centrifugation. bEnd.3 cells were incubated with either mock or JEV-infected mouse brain supernatant (“mock supernatant” or “JEV supernatant” for short), and DNA and RNA samples were collected for analysis.
Then, BSP was performed to test the DNA methylation changes of the Afdn genes in the bEnd.3 cells treated with different supernatants. It was found that treatment with JEV supernatant significantly elevated DNA methylation levels of this gene in the bEnd.3 cells, with 2.8% in the mock supernatant group compared to 12.5% in the JEV supernatant group (Fig. 6A). Moreover, RT-qPCR results revealed a noticeable downregulation of Afdn gene expression following JEV supernatant treatment (Fig. 6B), consistent with the results observed in the mouse brain (Fig. 5E). Western blot analysis also confirmed a decrease in AFDN protein levels after JEV supernatant treatment (Fig. 6C). Our previous results (Fig. 1E, F) illustrated that JEV contributed to increased BBB permeability in the mouse brain. Therefore, it was investigated whether JEV supernatant treatment could also increase bEnd.3 cell permeability in vitro. Transwell experiments were performed to measure bEnd.3 permeability after mock or JEV supernatant treatments (Fig. 6D). The results illustrated that the JEV supernatant treatment did enhance the bEnd.3 cell permeability, compared with the mock supernatant 120 min after the onset of the treatment (Fig. 6E).
Fig. 6.

Modulation of Afdn expression by hypermethylation of its gene promoter contributes to the increased BBB permeability. (A) Detection of methylation levels of Afdn using JEV supernatant treated bEnd.3 cell by BSP. (B) qPCR was performed to detect the expression of Afdn after JEV-infected supernatant treated in bEnd.3 cell. (C) Western blot was performed to measure Afdn protein expression in bEnd.3 cells treated with JEV-infected supernatant. The right histogram shows a densitometric analysis of the Western blot results. (D) Schematic diagram showing the transwell assay to assess the permeability of bEnd.3 cells after mock or JEV supernatant treatment for 72 h. (E) In vitro, transwell experiment was performed to measure bEnd.3 cells permeability after JEV infected mice brain supernatant treatment. (F) qPCR analysis of Afdn expression changes after Afdn siRNA transfection in the bEnd.3 cell. (G) Western bolt was detected the expression of AFDN after siRNA transfection in bEnd.3 cells. The right histogram shows a densitometric analysis of the Western blot results. (H) Schematic diagram showing the detection of permeability of bEnd.3 cells after transfection with Afdn siRNA. (I) Transwell experiment to measure the permeability of bEnd.3 cell after Afdn siRNA transfection for 72 h. The schematic diagrams were made with the free version of BioRender. Data are represented as the mean values ± standard deviations (SD) of at least three biological replicates using Student’s t-test (*p < 0.05, **p < 0.01, and ***p < 0.001) between the indicated groups
To further investigate the function of the Afdn gene, Afdn gene silencing was performed in bEnd.3 cells through transfection with small interfering (si)RNA. After 72 h post-transfection, the expression level of Afdn was determined by RT-qPCR. The results indicated that all three siRNAs tested effectively silenced the Afdn gene, with the second siRNA (RNAi-2) resulting in the most efficient knockdown of Afdn (Fig. 6F). Western blot analysis further confirmed the effective downregulation of the AFDN protein after RNAi-2 transfection (Fig. 6G). To explore the role of Afdn in influencing permeability in bEnd.3 cells, Afdn silencing was performed for 72 h, followed by a Transwell experiment to assess bEnd.3 cell permeability (Fig. 6H). Our finding suggested that Afdn silencing resulted in increased monolayer cell permeability compared to the negative control (NC) group, as shown by incubation with FITC-Dextran-10,000 for 60 and 120 min, suggesting AFDN as an important protein for maintaining the integrity of the BBB (Fig. 6I). In summary, our study demonstrated that the hypermethylation of the Afdn gene by JEV led to its downregulation at both the mRNA and protein levels. Additionally, experimentally silencing the Afdn gene in bEnd.3 cells resulted in increased cell permeability, indicating a crucial role of Afdn in maintaining BBB integrity. These findings suggest that JEV might affect the integrity of the BBB via hypermethylation and subsequent downregulation of Afdn expression.
DNA methylation inhibitor treatment restores Afdn expression and BBB integrity by counteracting the hypermethylation of Afdn gene promoter induced by JEV supernatant
As far, our study has demonstrated the hypermethylation of the Afdn gene leads to its downregulation at both the mRNA and protein levels, which contributes to the increased cell permeability induced by treatment of the JEV-infected brain supernatant. DNA methylation inhibitors, such as 5-Aza-2′-deoxycytidine (5-AZA), have proven effective in inhibiting DNA methylation levels and treating many diseases, including acute myeloid leukemia (AML) and other cancers [44]. To further gain the logical connection between expression of Afdn, DNA methylation and endothelial cell permeability, we conducted the experiment with and without treatment of the DNA methylation inhibitor 5-AZA. Briefly, bEnd.3 cells were initially treated with JEV supernatant for 48 h and subsequently treated with 5-AZA for 24 h to detect changes in Afdn methylation levels and expression. Our results indicated that the JEV supernatant-AZA group exhibited a significant reduction in Afdn methylation levels (Fig. 7A). Moreover, the expression of Afdn in RNA and protein levels was markedly restored in comparison to the JEV supernatant-DMSO group (Fig. 7B and C), providing further evidence that JEV supernatant induces hypermethylation of Afdn gene. Furthermore, our results show that the permeability of the JEV supernatant -AZA group was significantly reduced compared with the JEV supernatant-DMSO group (Fig. 7D), indicating that Afdn hypermethylation could contribute to the increased BBB permeability.
Fig. 7.

DNA methylation inhibitor treatment restores Afdn expression and BBB integrity by counteracting the hypermethylation of Afdn gene promoter induced by JEV supernatant. bEnd.3 cells were treated with mock or JEV supernatant for 48 h, followed by treatment with either DMSO or AZA for 24 h. (A) BSP method was used to detect changes in Afdn methylation level. (B) The changes in Afdn expression was measured by qPCR. (C) Western bolt was detected the expression of AFDN in protein levels. (D) Transwell experiment was conducted to assess the permeability of bEnd.3 cell. Data are represented as the mean values ± standard deviations (SD) of at least three biological replicates using Student’s t-test (*p < 0.05, **p < 0.01, ***p < 0.001, and ns: no significant) between the indicated groups
Discussion
DNA methylation, a pivotal epigenetic modification, contributes to the pathogenesis of flavivirus infections, as exemplified by the association of changes in DNA methylation in ZIKV-induced microcephaly and DENV hemorrhagic fever [27, 28, 30]. So far, it is not clear whether DNA methylation also plays a critical role in JEV infection. Here, WGBS technology was used to determine the difference in methylation levels between mock and JEV-infected mouse brains. We demonstrate that JEV infection globally increases DNA methylation levels in mouse brains compared with the mock group (Fig. 2). Additionally, our genome-wide profiles of DNA methylation in mouse brains identify more hyper-DMRs than hypo-DMRs across different chromosomes after JEV infection (Fig. 3). Thus, this study reveals that DNA methylation is induced in a murine Japanese encephalitis model.
Japanese encephalitis has been reported to be characterized as disruption of BBB with enhanced permeability [45, 46]. Consistently, we have demonstrated that JEV enhanced BBB permeability in our mouse model (Fig. 1E, F). Of note, the tight junction proteins (such as claudin-5, ZO-1, and occluding protein) are important in controlling the BBB permeability [34, 43, 47]. For example, previous reports have demonstrated that JEV infection reduces claudin-5, ZO-1, and occluding protein expression contributes to the increased BBB permeability. Thus, alteration of the tight junction signaling pathway is important to the JEV-induced disorder of BBB [34, 48, 49]. AFDN (also known as Afadin, AF6, or MLLT4) encodes a multi-domain scaffold protein found in both adherents and tight junctions, which plays structural and signal-modulating roles [50]. AFDN has been reported to regulate tight junctions through direct interaction with tight junctional protein ZO-1, highlighting its role as a peripheral component of tight junctions [51, 52]. In addition, AFDN also plays a crucial role in maintaining barrier function and homeostasis of the intestinal epithelia [53].
In the central nervous system (CNS), tight junction proteins are highly expressed by endothelial cells, which are the primary structural component of the BBB [54, 55]. It has been reported that DNA methylation plays an important role in a leaky BBB by modulation of the tight junction protein expression [56, 57]. For example, ethanol mediates ZO-1 hypermethylation through DNMT3A activation, leading to increased endothelial cell permeability [56]. Consistently, DNA methyltransferase inhibitor treatment can improve the compromised BBB, suggesting that DNA methylation could become a therapeutic target for treatment of diseases with a leaky BBB [56]. Our whole genome sequence data reveals that DNA methylation modulates the tight junction signaling pathway in a murine Japanese encephalitis model (Fig. 5A), which is characterized as a leaky BBB.
Generally, DNA methylation in gene promoter regions tends to downregulate gene expression [58, 59]. Among the DMGs of tight junction signaling, we found the Afdn gene is the highest degree of methylation and we also illustrated that JEV infection decreases AFDN expression by hypermethylation of the Afdn gene promoter (Fig. 5D, E, F) in mouse brains. Interestingly, the qPCR results of several other hypermethylated tight junction genes (Cldn19, Cldn6, Cldn3, Crb3 and Cldn14) we selected did not show a significant decrease in expression, which is not in line with the general notion that DNA methylation would reduce gene expression (Fig. 5D). However, increasing evidence suggests that this association does not always absolute and promoter hypermethylation now also appears to be associated with high transcriptional activity. For example, enhancer interaction with a hypermethylated promoter promotes transcriptional activation, contributing to gene expression [60, 61]. Therefore, our findings are consistent with recent researches, i.e. the actual effects of DNA methylation on gene expression are complex, subtle and highly context-dependent.
Notably, while AFDN as a component of tight junctions has been extensively studied in maintaining the barrier integrity and homeostasis of the intestinal epithelia [50, 53, 62, 63], its effects on BBB permeability are less understood. To address this gap in knowledge, we have determined the role of Afdn in BBB permeability by taking advantage of the in vitro BBB model (bEnd.3), which has been extensively used for the analysis of BBB permeability [34, 64, 65]. In vitro, we found that JEV supernatant could increase bEnd.3 cell permeability (Fig. 6D), which is consistent with the in vivo BBB permeability model (Fig. 1F), indicating that bEnd.3 cells can simulate in vivo permeability results. Significantly, we elucidate the mechanism whereby hypermethylation of the Afdn gene promoter diminishes AFDN expression at both the mRNA and protein levels, subsequently elevating BBB permeability. This mechanistic insight is further reinforced by our observation that Afdn knockdown via siRNAs markedly increases permeability, highlighting Afdn’s regulatory capacity over BBB integrity.
Furthermore, our study introduces a therapeutic perspective by demonstrating that treatment with the DNA methylation inhibitor 5-AZA significantly reduced the hypermethylation-induced suppression of AFDN expression, effectively decreasing BBB permeability. This intervention not only provides a potential therapeutic avenue but also highlights the critical role of DNA methylation in regulating BBB permeability in the context of JEV infection. This study not only fills a critical gap in our understanding of BBB regulation but also identifies hypermethylation of Afdn as a key molecular mechanism affecting BBB integrity during viral infection.
However, it is important to note that, our study primarily focuses on the bEnd.3 cell model, which represents an in vitro approximation of BBB permeability. While very informative, this may not fully capture the complexity of the in vivo brain microenvironment. Moreover, BBB permeability is regulated by a complex network of molecular interactions, and DNA methylation is just one of the many factors involved. Although our current study demonstrates a correlation between Afdn hypermethylation and increased BBB permeability, the precise mechanism(s) by which Afdn hypermethylation contributes to BBB disruption remains unknown. Future studies should be investigated to elucidate the underlying mechanism(s).
By revealing the relationship between methylation of the Afdn gene and BBB permeability, our study sheds light on a novel mechanism of BBB disruption during JEV infection. This finding opens a potential therapeutic avenue using targeted epigenetic interventions to preserve BBB integrity during JEV infection. To the best of our knowledge, our study is the first to establish a link between Afdn DNA methylation and JEV-induced BBB permeability, offering insights that may be applicable to other neurotropic flavivirus infections.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Supplementary Material 1: Figure S1. Analyze differentially methylated levels among the mock and JEV group in different transcriptional elements of the genome. (A), (B), (C), respectively represent the mean methylation level of mCG, mCHG, and mCHH. The Y-axis represents the level of methylation level (values from 0 to 1). The X-axis is divided into the 7 different transcription element regions. The length of each transcription element region is equally divided into an equal number of bin regions (referring to the binning interval containing a certain number of bases). Each colored curve represents the methylation level of each bin area. The yellow dotted line between a and b is TSS (transcription start site) location. Figure S2. Methylome map of JEV infection. (A) and (C) Circos images respectively show the differential methylation levels between the JEV group and mock group at the genome-wide scale in the type of mCHG and mCHH. Each circle from the outside to the inside depicts: (1) a diagram of the mouse genome subdivided by chromosomes; (2) the methylation level of the mock group; (3) the absolute value of the differential methylation level value of the JEV group minus the mock group. Red color represents hypermethylated areas, green color represents hypomethylated areas, the redder the color, the higher the methylation level after JEV infection, the greener the color, the lower the methylation level after JEV infection; (4) the methylation level of the JEV group. (B) and (D) the graphs respectively show the numbers of hyper and hypomethylated DMRs distribution in different genomic elements in the mCHG and mCHH types. Figure S3. Effect on expression of AFDN by treatment of JEV-infected supernatant and UV-inactivated JEV-infected supernatant. bEnd.3 cell was treated by JEV-infected supernatant and UV-inactivated JEV-infected supernantant, respectively. In the meanwhile, treatment by mock brain supernatant was as a control. qPCR was performed to detect the expression of Afdn after each treatment. Data are represented as the mean values ± standard deviations (SD) of at least three biological replicates using Student’s t-test (*p < 0.05, **p < 0.01, and ***p < 0.001) between the indicated groups.
Acknowledgements
We thank Qitong Huang for giving us useful advice and guidance on high-throughput data analysis. We also thank BioRender for providing free drawing elements, which helped visualize our research. Xiao Xiang is enrolled through the graduate school Production Ecology and Resource Conservation (PE&RC) in the 2018 Sino-Dutch joint Ph.D. program between the Chinese Academy of Agricultural Sciences (CAAS) and Wageningen University & Research.
Abbreviations
- JEV
Japanese encephalitis virus
- DMRs
Differentially methylated regions
- BBB
Blood-brain barrier
- WGBS
Whole genome bisulfite sequencing
- AD
Alzheimer’s Disease
- DNMT3B
DNA methyltransferase 3B
- dpi
Day post-infection
- BSP
Bisulfite sequencing PCR
- bEnd3
Brain microvascular endothelial
- 5-AZA
5-Aza-2′-deoxycytidine
Author contributions
X.X. performed most of the experiments, analyzed the WGBS data, and wrote the manuscript. Y.Q. and G.P. supervised the entire project and revised the manuscript. D.Y. contributed to the revision of the manuscript. Z.L. contributed to the mouse infection experiment. M.O. and J.F. contributed to conceptualization of project and the revision of the manuscript. J.W., K.L., Z.L., D.S., B.L., J.K. and Z.M. also contributed to conceptualization and resources for the project. All authors reviewed the manuscript.
Funding
This study was supported by the National Key Research and Development Program of China (No. 2022YFD1800100). This project also was funded by the China Scholarship Council (CSC) (File No.202203250067).
Data availability
No datasets were generated or analysed during the current study.
Declarations
Ethics approval and consent to participate
The animal experiments conducted in this study were granted ethical approval by the Institutional Animal Care and Use Committee of Shanghai Veterinary Research Institute with the approval number (No): SHVRI-SZ-20200110-05 and reference number (IACUC No): Shvri-po-2016060501. Policy No. 2006 398, released by the People’s Republic of China’s Ministry of Science and Technology, details the Guidelines on the Humane Treatment of Laboratory Animals, and all studies were carried out in full compliance with these regulations.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Gorben P. Pijlman, Email: gorben.pijlman@wur.nl
Yafeng Qiu, Email: yafengq@shvri.ac.cn.
References
- 1.Sharma KB, Vrati S, Kalia M. Pathobiology of Japanese encephalitis virus infection. Mol Aspects Med. 2021;81:100994. [DOI] [PubMed] [Google Scholar]
- 2.Kumar S, Verma A, Yadav P, Dubey SK, Azhar EI, Maitra SS, Dwivedi VD. Molecular pathogenesis of Japanese encephalitis and possible therapeutic strategies. Arch Virol. 2022;167:1739–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ghosh D, Basu A. Japanese encephalitis-a pathological and clinical perspective. PLoS Negl Trop Dis. 2009;3:e437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Li-Ta Keng L-YC. Japanese encephalitis. Clin IMAGES. 2018;190(21):E657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Davis EH, Beck AS, Li L, White MM, Greenberg MB, Thompson JK, Widen SG, Barrett ADT, Bourne N. Japanese encephalitis virus live attenuated vaccine strains display altered immunogenicity, virulence and genetic diversity. NPJ Vaccines. 2021;6:112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lee S-IYY-M. Japanese encephalitis:the virus and vaccines. Hum Vaccines Immunotherapeutics. 2014;10(2):263–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mileno MD. Japanese Encephalitis Vaccine. R I Med J (2013) 2020, 103:49–50. [PubMed]
- 8.Chen HL, Chang JK, Tang RB. Current recommendations for the Japanese encephalitis vaccine. J Chin Med Assoc. 2015;78:271–5. [DOI] [PubMed] [Google Scholar]
- 9.Sakamoto R, Tanimoto T, Takahashi K, Hamaki T, Kusumi E, Crump A. Flourishing Japanese Encephalitis, Associated with global warming and urbanisation in Asia, demands widespread Integrated Vaccination Programmes. Annals Global Health 2019, 85. [DOI] [PMC free article] [PubMed]
- 10.Hameed M, Wahaab A, Nawaz M, Khan S, Nazir J, Liu K, Wei JC, Ma ZY. Potential Role of Birds in Japanese Encephalitis Virus Zoonotic Transmission and Genotype Shift. Viruses-Basel 2021, 13. [DOI] [PMC free article] [PubMed]
- 11.Mackenzie JS, Williams DT, van den Hurk AF, Smith DW, Currie BJ. Japanese Encephalitis Virus: the emergence of genotype IV in Australia and its potential endemicity. Viruses 2022, 14. [DOI] [PMC free article] [PubMed]
- 12.Guanlun Xu TG, Zhijie Wang J, Zhang B, Cui X, Shen A, Zhou Y, Zhang J, Zhao. Hong Liu: Re-Emerged Genotype IV of Japanese Encephalitis Virus Is the Youngest Virus in Evolution viruses 2023, 15(3):626. [DOI] [PMC free article] [PubMed]
- 13.Frank JC, Song BH, Lee YM. Mice as an animal model for Japanese Encephalitis Virus Research: mouse susceptibility, infection Route, and Viral Pathogenesis. Pathogens 2023, 12. [DOI] [PMC free article] [PubMed]
- 14.Kumar A, Sharma P, Shukla KK, Misra S, Nyati KK. Japanese encephalitis virus: Associated immune response and recent progress in vaccine development. Microb Pathog 2019, 136. [DOI] [PubMed]
- 15.Filgueira L, Lannes N. Review of emerging Japanese Encephalitis Virus: New aspects and concepts about Entry into the brain and inter-cellular spreading. Pathogens; 2019. p. 8. [DOI] [PMC free article] [PubMed]
- 16.Turtle L, Solomon T. Japanese encephalitis - the prospects for new treatments. Nat Reviews Neurol. 2018;14:298–313. [DOI] [PubMed] [Google Scholar]
- 17.Moore LD, Le T, Fan GP. DNA methylation and its basic function. Neuropsychopharmacology. 2013;38:23–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cui D, Xu XR. DNA methyltransferases, DNA methylation, and Age-Associated cognitive function. Int J Mol Sci 2018, 19. [DOI] [PMC free article] [PubMed]
- 19.Liu S, Liu L, Xu G, Cao ZY, Wang Q, Li S, Peng NF, Yin JC, Yu HS, Li MQ et al. Epigenetic modification is regulated by the Interaction of Influenza A Virus Nonstructural protein 1 with the De Novo DNA methyltransferase DNMT3B and subsequent transport to the cytoplasm for K48-Linked polyubiquitination. J Virol 2019, 93. [DOI] [PMC free article] [PubMed]
- 20.Tirado-Magallanes R, Rebbani K, Lim R, Pradhan S, Benoukraf T. Whole genome DNA methylation: beyond genes silencing. Oncotarget. 2017;8:5629–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mase S, Shinjo K, Totani H, Katsushima K, Arakawa A, Takahashi S, Lai HC, Lin RI, Chan MWY, Sugiura-Ogasawara M, Kondo Y. ZNF671 DNA methylation as a molecular predictor for the early recurrence of serous ovarian cancer. Cancer Sci. 2019;110:1105–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Blanch M, Mosquera JL, Ansoleaga B, Ferrer I, Barrachina M. Altered mitochondrial DNA methylation pattern in Alzheimer Disease-Related Pathology and in Parkinson Disease. Am J Pathol. 2016;186:385–97. [DOI] [PubMed] [Google Scholar]
- 23.Zouali M. DNA methylation signatures of autoimmune diseases in human B lymphocytes. Clin Immunol. 2021;222:108622. [DOI] [PubMed] [Google Scholar]
- 24.De Jager PL, Srivastava G, Lunnon K, Burgess J, Schalkwyk LC, Yu L, Eaton ML, Keenan BT, Ernst J, McCabe C, et al. Alzheimer’s disease: early alterations in brain DNA methylation at ANK1, BIN1, RHBDF2 and other loci. Nat Neurosci. 2014;17:1156–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bennett DA, Yu L, Yang J, Srivastava GP, Aubin C, De Jager PL. Epigenomics of Alzheimer’s disease. Transl Res. 2015;165:200–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fang J, Hao Q, Liu L, Li Y, Wu J, Huo X, Zhu Y. Epigenetic changes mediated by microRNA miR29 activate cyclooxygenase 2 and lambda-1 interferon production during viral infection. J Virol. 2012;86:1010–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Anderson D, Neri JICF, Souza CRM, Valverde JG, De Araujo JMG, Nascimento MDSB, Branco RCC, Arrais NMR, Lassmann T, Blackwell JM, Jeronimo SMB. Zika Virus Changes methylation of genes involved in Immune response and neural development in Brazilian babies born with congenital Microcephaly. J Infect Dis. 2021;223:435–40. [DOI] [PubMed] [Google Scholar]
- 28.Kandilya D, Maskomani S, Shyamasundar S, Tambyah PA, Shiao Yng C, Lee RCH, Hande MP, Mallilankaraman K, Chu JJH, Dheen ST. Zika virus alters DNA methylation status of genes involved in Hippo signaling pathway in human neural progenitor cells. Epigenomics. 2019;11:1143–61. [DOI] [PubMed] [Google Scholar]
- 29.Janssens S, Schotsaert M, Karnik R, Balasubramaniam V, Dejosez M, Meissner A, Garcia-Sastre A, Zwaka TP. Zika Virus Alters DNA Methylation of Neural Genes in an Organoid Model of the Developing Human Brain. mSystems 2018, 3. [DOI] [PMC free article] [PubMed]
- 30.Gomes AV, de Souza Morais SM, Menezes-Filho SL, de Almeida LG, Rocha RP, Ferreira JM, Dos Santos LL, Malaquias LC, Coelho LF. Demethylation profile of the TNF-alpha promoter gene is associated with high expression of this cytokine in Dengue virus patients. J Med Virol. 2016;88:1297–302. [DOI] [PubMed] [Google Scholar]
- 31.Reed LJ. A simple method of estimating fifty per cent endpoints. Cabidigitallibraryorg. 1938;27:493–7. [Google Scholar]
- 32.Sehgal N, Kumawat KL, Basu A, Ravindranath V. Fenofibrate reduces mortality and precludes neurological deficits in survivors in murine model of Japanese encephalitis viral infection. PLoS ONE. 2012;7:e35427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ryan Lister MP, Robert H, Dowen RD, Hawkins G, Hon J, Tonti-Filippini JR, Nery L, Lee Z, Ye Q-M, Ngo. Lee Edsall, Jessica Antosiewicz-Bourget, Ron Stewart, Victor Ruotti, a Harvey Millar, James A Thomson, Bing Ren, Joseph R Ecke: human DNA methylomes at base resolution show widespread epigenomic differences. Nature. 2009;462(7271):315–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang K, Wang HL, Lou WJ, Ma LH, Li YC, Zhang N, Wang C, Li F, Awais M, Cao SB et al. IP-10 promotes blood-brain barrier damage by Inducing Tumor Necrosis Factor Alpha Production in Japanese encephalitis. Front Immunol 2018, 9. [DOI] [PMC free article] [PubMed]
- 35.Tu Y, Fang P, Zhang L, Sun K. Analysis of the Effect of SNAI Family in breast Cancer and Immune Cell. Front Cell Dev Biol. 2022;10:906885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tanabe Y, Naito Y, Vasuta C, Lee AK, Soumounou Y, Linhoff MW, Takahashi H. IgSF21 promotes differentiation of inhibitory synapses via binding to neurexin2alpha. Nat Commun. 2017;8:408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Juchem KW, Gounder AP, Gao JP, Seccareccia E, Yeddula N, Huffmaster NJ, Cote-Martin A, Fogal SE, Souza D, Wang SS, et al. NFAM1 promotes pro-inflammatory cytokine production in mouse and human monocytes. Front Immunol. 2021;12:773445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nakao M, Miyagaki T, Sugaya M, Sato S. Exacerbated Imiquimod-Induced Psoriasis-Like skin inflammation in IRF5-Deficient mice. Int J Mol Sci 2020, 21. [DOI] [PMC free article] [PubMed]
- 39.Harada H, Fujita T, Miyamoto M, Kimura Y, Maruyama M, Furia A, Miyata T, Taniguchi T. Structurally similar but functionally distinct factors, IRF-1 and IRF-2, bind to the same regulatory elements of IFN and IFN-inducible genes. Cell. 1989;58:729–39. [DOI] [PubMed] [Google Scholar]
- 40.Zhang XJ, Jiang DS, Li H. The interferon regulatory factors as novel potential targets in the treatment of cardiovascular diseases. Br J Pharmacol. 2015;172:5457–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kamath AV, Yip V, Gupta P, Boswell CA, Bumbaca D, Haughney P, Castro J, Tsai SP, Pacheco G, Ross S, et al. Dose dependent pharmacokinetics, tissue distribution, and anti-tumor efficacy of a humanized monoclonal antibody against DLL4 in mice. MAbs. 2014;6:1631–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Al-Obaidi MMJ, Bahadoran A, Wang SM, Manikam R, Raju CHS, Sekaran SD. Disruption of the blood brain barrier is vital property of neurotropic viral infection of the central nervous system. Acta Virol. 2018;62:16–27. [DOI] [PubMed] [Google Scholar]
- 43.Chai Q, He WQ, Zhou M, Lu H, Fu ZF. Enhancement of blood-brain barrier permeability and reduction of tight junction protein expression are modulated by chemokines/cytokines induced by Rabies virus infection. J Virol. 2014;88:4698–710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ohtani H, Orskov AD, Helbo AS, Gillberg L, Liu M, Zhou W, Ungerstedt J, Hellstrom-Lindberg E, Sun W, Liang G, et al. Activation of a subset of evolutionarily young transposable elements and innate immunity are linked to clinical responses to 5-Azacytidine. Cancer Res. 2020;80:2441–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Choi JY, Kim JH, Hossain FMA, Uyangaa E, Park SO, Kim B, Kim K, Eo SK. Indispensable role of CX(3)CR1(+) dendritic cells in regulation of Virus-Induced Neuroinflammation through Rapid Development of Antiviral Immunity in Peripheral lymphoid tissues. Front Immunol. 2019;10:1467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hsieh JT, St John AL. Japanese encephalitis virus and its mechanisms of neuroinvasion. PLoS Pathog. 2020;16:e1008260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Castro-Jorge LA, Pretto CD, Smith AB, Foreman O, Carnahan KE, Spindler KR. A protective role for Interleukin-1 signaling during mouse adenovirus type 1-Induced Encephalitis. J Virol 2017, 91. [DOI] [PMC free article] [PubMed]
- 48.Yan Z, Liu YM, Wu WD, Jiang Y, Zhuo LB. Combined exposure of heat stress and ozone enhanced cognitive impairment via neuroinflammation and blood brain barrier disruption in male rats. Sci Total Environ. 2023;857:159599. [DOI] [PubMed] [Google Scholar]
- 49.Feng SR, Chen ZX, Cen JN, Shen HJ, Wang YY, Yao L. [Critical roles of matrix metalloproteinases secreted by leukemic cells in the pathogenesis of central nervous system leukemia]. Zhonghua Xue Ye Xue Za Zhi. 2016;37:1070–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Huxham J, Tabaries S, Siegel PM. Afadin (AF6) in cancer progression: a multidomain scaffold protein with complex and contradictory roles. BioEssays. 2021;43:e2000221. [DOI] [PubMed] [Google Scholar]
- 51.Yamamoto T, Harada N, Kano K, Taya S, Canaani E, Matsuura Y, Mizoguchi A, Ide C, Kaibuchi K. The ras target AF-6 interacts with ZO-1 and serves as a peripheral component of tight junctions in epithelial cells. J Cell Biol. 1997;139:785–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ooshio T, Kobayash R, Iked W, Miyata M, Fukumoto Y, Matsuzawa N, Ogita H. Involvement of the Interaction of Afadin with ZO-1 in the formation of TJ in MDCK cell. J BIOL CHEM. 2020;285:5003–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Tanaka-Okamoto M, Hori K, Ishizaki H, Itoh Y, Onishi S, Yonemura S, Takai Y, Miyoshi J. Involvement of afadin in barrier function and homeostasis of mouse intestinal epithelia. J Cell Sci. 2011;124:2231–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Liu L, Yang C, Lavayen BP, Tishko RJ, Larochelle J, Candelario-Jalil E. Targeted BRD4 protein degradation by dBET1 ameliorates acute ischemic brain injury and improves functional outcomes associated with reduced neuroinflammation and oxidative stress and preservation of blood-brain barrier integrity. J Neuroinflammation. 2022;19:168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Yao Y, Chen ZL, Norris EH, Strickland S. Astrocytic laminin regulates pericyte differentiation and maintains blood brain barrier integrity. Nat Commun. 2014;5:3413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Behera J, Kelly KE, Tyagi N. Hydrogen sulfide prevents ethanol-induced ZO-1 CpG promoter hypermethylation-dependent vascular permeability via miR-218/DNMT3a axis. J Cell Physiol. 2021;236:6852–67. [DOI] [PubMed] [Google Scholar]
- 57.Ihezie SA, Mathew IE, McBride DW, Dienel A, Blackburn SL. Thankamani pandit PK: epigenetics in blood-brain barrier disruption. Fluids Barriers CNS. 2021;18:17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ge Y, Zadeh M, Mohamadzadeh M. Vitamin B12 regulates the transcriptional, metabolic, and Epigenetic Programing in Human Ileal epithelial cells. Nutrients 2022, 14. [DOI] [PMC free article] [PubMed]
- 59.Xu XF, Hu QY, Liang LF, Wu L, Gu WZ, Tang LL, Fu LC, Du LZ. Epigenetics of hyper-responsiveness to allergen challenge following intrauterine growth retardation rat. Respir Res. 2014;15:137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Smith J, Sen S, Weeks RJ, Eccles MR, Chatterjee A. Promoter DNA hypermethylation and paradoxical gene activation. Trends Cancer. 2020;6:392–406. [DOI] [PubMed] [Google Scholar]
- 61.de Mendoza A, Nguyen TV, Ford E, Poppe D, Buckberry S, Pflueger J, Grimmer MR, Stolzenburg S, Bogdanovic O, Oshlack A, et al. Large-scale manipulation of promoter DNA methylation reveals context-specific transcriptional responses and stability. Genome Biol. 2022;23:163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lin Y, Wozniak JM, Grimsey NJ, Girada S, Patwardhan A, Molinar-Inglis O, Smith TH, Lapek JD, Gonzalez DJ, Trejo J. Phosphoproteomic analysis of protease-activated receptor-1 biased signaling reveals unique modulators of endothelial barrier function. Proc Natl Acad Sci U S A. 2020;117:5039–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Faralla C, Bastounis EE, Ortega FE, Light SH, Rizzuto G, Gao L, Marciano DK, Nocadello S, Anderson WF, Robbins JR, et al. Listeria monocytogenes InlP interacts with afadin and facilitates basement membrane crossing. PLoS Pathog. 2018;14:e1007094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kim KA, Kim D, Kim JH, Shin YJ, Kim ES, Akram M, Kim EH, Majid A, Baek SH, Bae ON. Autophagy-mediated occludin degradation contributes to blood-brain barrier disruption during ischemia in bEnd.3 brain endothelial cells and rat ischemic stroke models. Fluids Barriers CNS. 2020;17:21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Hao W, Cui Y, Fan Y, Chen M, Yang G, Wang Y, Yang M, Li Z, Gong W, Yang Y, Gao C. Hybrid membrane-coated nanosuspensions for multi-modal anti-glioma therapy via drug and antigen delivery. J Nanobiotechnol. 2021;19:378. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Material 1: Figure S1. Analyze differentially methylated levels among the mock and JEV group in different transcriptional elements of the genome. (A), (B), (C), respectively represent the mean methylation level of mCG, mCHG, and mCHH. The Y-axis represents the level of methylation level (values from 0 to 1). The X-axis is divided into the 7 different transcription element regions. The length of each transcription element region is equally divided into an equal number of bin regions (referring to the binning interval containing a certain number of bases). Each colored curve represents the methylation level of each bin area. The yellow dotted line between a and b is TSS (transcription start site) location. Figure S2. Methylome map of JEV infection. (A) and (C) Circos images respectively show the differential methylation levels between the JEV group and mock group at the genome-wide scale in the type of mCHG and mCHH. Each circle from the outside to the inside depicts: (1) a diagram of the mouse genome subdivided by chromosomes; (2) the methylation level of the mock group; (3) the absolute value of the differential methylation level value of the JEV group minus the mock group. Red color represents hypermethylated areas, green color represents hypomethylated areas, the redder the color, the higher the methylation level after JEV infection, the greener the color, the lower the methylation level after JEV infection; (4) the methylation level of the JEV group. (B) and (D) the graphs respectively show the numbers of hyper and hypomethylated DMRs distribution in different genomic elements in the mCHG and mCHH types. Figure S3. Effect on expression of AFDN by treatment of JEV-infected supernatant and UV-inactivated JEV-infected supernatant. bEnd.3 cell was treated by JEV-infected supernatant and UV-inactivated JEV-infected supernantant, respectively. In the meanwhile, treatment by mock brain supernatant was as a control. qPCR was performed to detect the expression of Afdn after each treatment. Data are represented as the mean values ± standard deviations (SD) of at least three biological replicates using Student’s t-test (*p < 0.05, **p < 0.01, and ***p < 0.001) between the indicated groups.
Data Availability Statement
No datasets were generated or analysed during the current study.