

Abstract
Inherited mutations in the mitochondrial (mt)DNA are a major cause of human disease, with approximately 1 in 5000 people affected by one of the hundreds of identified pathogenic mtDNA point mutations or deletions. Due to the severe, and often untreatable, symptoms of many mitochondrial diseases, identifying how these mutations are inherited from one generation to the next has been an area of intense research in recent years. Despite large advances in our understanding of this complex process, many questions remain unanswered, with one of the most hotly debated being whether or not purifying selection acts against pathogenic mutations during germline development.
In this review, we synthesize current knowledge regarding the germline transmission of mtDNA mutations and examine the evidence for and against purifying selection during this process, focusing primarily on data from humans and other mammals, but drawing on additional species where relevant.
Mitochondrial DNA mutation: homoplasmy versus heteroplasmy
The mammalian mitochondrial (mt)DNA genome consists of a circular, double‐stranded loop of DNA varying from 15 000 to 17 000 bp in length depending on the species, with the human mtDNA sequence containing 16 569 bp (Chinnery & Hudson 2013). The mtDNA contains 37 genes, encoding 13 subunits of the electron transport chain (ETC), which contribute to the production of energy in the cell via oxidative phosphorylation (OXPHOS); and 24 RNA, comprising 22 tRNA and two ribosomal RNA (16S RNA and 12S RNA), required for the transcription and translation of mtDNA‐encoded proteins (Chinnery & Hudson 2013). Unlike the nuclear genome, mammalian mtDNA is inherited uniparentally, solely via the maternal line, and does not undergo recombination (Hagstrom et al. 2014). mtDNA is present in multiple copies within each cell, and copy number varies from 100 to 10 000 copies, adapting to the cellular needs in a tissue‐specific manner (Chinnery & Hudson 2013). mtDNA has a much higher mutation rate than the nuclear genome, possibly due to the close proximity of mtDNA to mutagenic reactive oxygen species (ROS) (Lagouge & Larsson 2013), continuous replication of mtDNA in post‐mitotic cells, with an error rate several orders of magnitude higher than in the nucleus (Johnson & Johnson 2001), or a less extensive array of DNA repair mechanisms (Kazak et al. 2012; Scheibye‐Knudsen et al. 2015) compared with the nucleus. Novel mtDNA mutations invariably lead to a heterogenic state termed “heteroplasmy”, where wild‐type molecules coexist with mutated mtDNA molecules in the same cell, in contrast to the normal state of “homoplasmy”, where all copies of mtDNA present in the cell share the same sequence (Fig. 1A).
Figure 1.
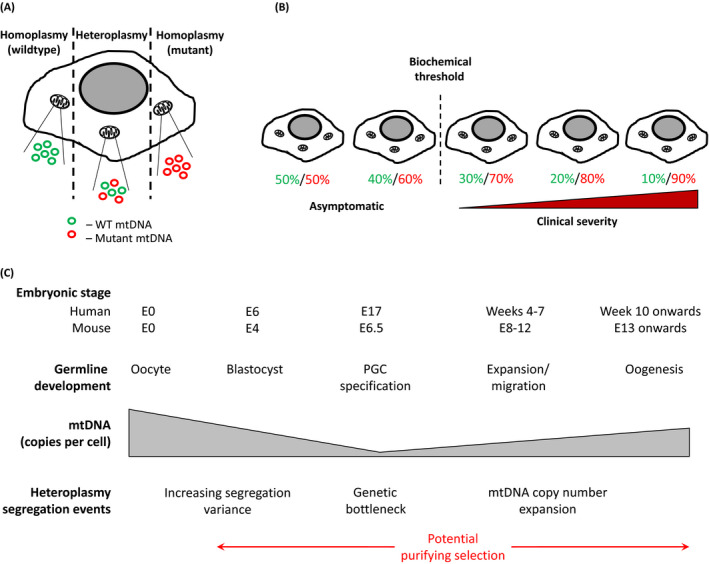
mtDNA heteroplasmy and its transmission through the female germline. (A) Each cell contains multiple mtDNA molecules, a mutation in the mtDNA (red circles) is termed homoplasmic if all copies carry the mutation, or heteroplasmic if only a proportion carry it. (B) In heteroplasmic cells, as the burden of a pathogenic mutation (red figures) increases compared to wildtype mtDNA (green figures), a biochemical threshold is reached. Beyond this point the cell can no longer compensate for the mutation and a respiratory chain defect develops. The severity of this defect tends to worsen with further increase in the levels of mutant mtDNA. (C) Schematic representation of female germline development during embryogenesis, and associated events determining the differential segregation of heteroplasmic mtDNA variants. Whilst the contribution of purifying selection to this process is controversial, such mechanisms could be active from the initial decrease in cellular mtDNA copy number leading up to the genetic bottleneck, during PGC specification and development and through oogenesis into adult life.
mtDNA mutations and purifying selection throughout evolution
In 1964, Muller postulated that asexual inheritance of DNA without recombination should lead to the accumulation and fixation of deleterious mutations. In the absence of purifying selection to remove these mutations, this process will ultimately result in mutational meltdown of the genome, a hypothesis known as “Muller's ratchet” (Muller 1964). Evidence of this predicted build‐up of mutations in mtDNA can be seen in phylogenetic data from human lineages. In theory, all human mtDNA can be traced back to a single woman, the so‐called “Mitochondrial Eve”, who lived in Africa approximately 200 000 years ago (Cann et al. 1987). Since that time the mtDNA genome has been constantly acquiring point mutations, resulting in the development of a number of discrete haplogroups, each defined by a specific subset of variants (Torroni et al. 1997; Wallace et al. 1999). It is important to note that each of these variants must first have existed as a heteroplasmic mutation, before becoming fixed homoplasmic polymorphisms, and some mitochondrial diseases, such as Leber hereditary optic neuropathy, are primarily caused by homoplasmic mutations (Yu‐Wai‐Man et al. 2002). Despite the presence of fixed variants in the mitochondrial genome, which in rare cases can be pathogenic, many mtDNA‐related mitochondrial diseases are caused by heteroplasmic point mutations in the mtDNA coding sequence, which disrupt ETC activity and lead to mitochondrial dysfunction (Stewart & Chinnery 2015). However, it is important to note that heteroplasmic mutations can either be inherited or acquired through de novo somatic mutation during embryogenesis or postnatal development. Although homoplasmic variants may be selected against at the population level (Stewart et al. 2008a), heteroplasmic mutations can additionally be subject to purifying selection within individuals and during transmission from generation to generation (Li et al. 2016), allowing them to be removed from the population before becoming fixed in the mtDNA sequence. The fact that relatively few highly deleterious variants have become fixed in the mtDNA genome, in spite of its elevated mutation rate, suggests that purifying selection against such mutations must be active, preventing the rapid advancement of Muller's ratchet (Rand & Kann 1996, 1998; Elson et al. 2004; Rand 2008).
The efficiency of purifying selection cannot be 100%, because deleterious heteroplasmies are found relatively commonly in the general population (Elliott et al. 2008; Payne et al. 2013), and novel heteroplasmies have been repeatedly allowed to fix in the mtDNA genome throughout evolution, thus “escaping” selection altogether. Additionally, the mutations that “escape” selection are not merely neutral variants, as many of the haplogroup polymorphisms found in human mtDNA are non‐synonymous and potentially affect mitochondrial function (Kazuno et al. 2006; Pello et al. 2008; Gomez‐Duran et al. 2010). It has been suggested that positive selection of advantageous mtDNA variants may explain the presence of some haplogroup polymorphisms. The founding mutations of several haplogroups appear to be linked to major migratory events during human colonization of the globe, and might have allowed survival in colder climates as our ancestors moved out of Africa (Coskun et al. 2003; Mishmar et al. 2003; Ruiz‐Pesini et al. 2004); however, this theory is controversial and not universally accepted (Elson et al. 2004; Kivisild et al. 2006). Whilst it is tempting to envisage a simple model where advantageous heteroplasmies are retained and become fixed, and deleterious variants are purged, this is clearly not the case. Some haplogroup‐specific variants have been shown to predispose to certain diseases (Hudson et al. 2013, 2014; Jimenez‐Sousa et al. 2015), with some affecting individuals during reproductive life. Therefore, a balance clearly exists between the appearance of novel heteroplasmic mtDNA mutations and subsequent selection against the most pathogenic of these variants.
Potential modes of purifying selection
There are a number of levels at which this process of purifying selection could occur (Rand 2001). In addition to the aforementioned selection at the population level, mtDNA mutations may be selected against at the level of individual organisms, via classical Darwinian selection, with higher levels of mutation resulting in reduced fitness and less chance of passing the mutation to the subsequent generation. At the cellular level, variations in mutational load between cells will result in selection against those cells that are least fit (i.e. carry the highest levels of mutation) (Rajasimha et al. 2008). This variation may originate either when cells divide, or during replication of mtDNA genomes in post‐mitotic cells (Chinnery & Samuels 1999). Because mtDNA replication is an ongoing process, even in non‐dividing cells, segregation is active in all tissues throughout life (Burgstaller et al. 2014), and purifying selection may therefore play an important role at all stages. Finally, selection may also occur at the subcellular level, potentially through preferential replication of a subset of mtDNA within the cell prior to division (Blok et al. 1997).
When considering the transmission of heteroplasmic mtDNA mutations, the development of the maternal germline is a unique and intriguing step, as it represents the point at which the single cell and the whole organism converge. Therefore, any selective mechanisms active on the mtDNA at this stage have huge potential to influence the integrity of the mtDNA genome in subsequent generations. This is one of the reasons that germline inheritance of heteroplasmy has been such an intense area of research in recent years, and perhaps one of the most important advances in this field has been the development of the germline genetic bottleneck theory of mtDNA inheritance.
Maternal inheritance of mtDNA heteroplasmy: the genetic bottleneck theory
In patients carrying potentially deleterious heteroplasmic mtDNA mutations, the severity of disease symptoms tends to correlate with the level of heteroplasmy, and if a certain biochemical threshold is reached the individual will develop pathogenic phenotypes (Fig. 1B) (Chinnery et al. 1997; DiMauro & Schon 2001). Very low level mtDNA heteroplasmy appears to be universal in the human population (Payne et al. 2013), and more than 1:200 healthy live births carry a point mutation present at a more than 1% heteroplasmy level (Elliott et al. 2008). Because mtDNA is strictly maternally inherited (Hutchison et al. 1974; Case & Wallace 1981; Pyle et al. 2015) and the heteroplasmy level transmitted from a mother to her offspring varies significantly (Larsson et al. 1992; Blok et al. 1997), providing prognostic advise to healthy women carrying a pathogenic mtDNA mutation is currently very challenging (Poulton et al. 2010; Chinnery et al. 2014).
Shifts in heteroplasmy between a mother and her offspring were first observed in Holstein cows harboring two mitochondrial genotypes, distinguished by a single point mutation. In a single maternal lineage, significant heteroplasmy shifts were observed within a few generations (Hauswirth & Laipis 1982). Hauswirth and Laipis suggested that such variation could be due to a dramatic reduction of the mtDNA copy number during oogenesis, resulting in an increased likelihood of heteroplasmy segregation by genetic drift: the so‐called “genetic bottleneck” theory. This change in copy number was later evaluated in mice and shown to decrease from 100 000 copies in the fertilized oocyte to just 200 copies in the PGC, with an estimated 40 mitochondria per cell containing five mtDNA molecules each at this stage (Fig. 1C) (Nass 1966; Nogawa et al. 1988; Jenuth et al. 1996). This mtDNA bottleneck effect in the PGC has since been corroborated by computational simulation and quantitative PCR measurements at the single cell level (Cree et al. 2008; Wai et al. 2008). Nonetheless, this theory remains controversial because additional work utilizing the same animal model failed to show such a drastic decrease in the mtDNA copy number, with the lowest mtDNA content estimated to be approximately 1500 copies per cell (Cao et al. 2007, 2009).
The mtDNA germline genetic bottleneck seems to be present in other vertebrate species. In zebrafish, Otten et al. measured the mtDNA copy number during embryogenesis, from fertilized oocyte to PGC. They observed a marked decrease from 2.0 × 107 copies per cell in the fertilized oocyte to 170 copies per cell in the PGC (Otten et al. 2016), suggesting a strong bottleneck effect, similar to that observed in mice. Measurements of mtDNA copy number have also been performed at later stages during oogenesis in mammalian species. In sheep, Cotterill et al. compared the mtDNA content at the primordial follicle stage to the metaphase II oocyte stage. They observed an increase from 605 copies per cell in the primordial follicle to 7.5 × 105 copies per cell in mature oocytes, suggesting a bottleneck event following the primordial follicle stage (Cotterill et al. 2013). It is interesting to note that, even though the primordial follicle stage occurs later than the PGC specification during the embryogenesis, the mtDNA copy number observed in this study remains lower than the measurements performed by Cao et al. (2007).
Thus, numerous independent studies point toward the genetic sampling event during oogenesis that was first proposed by Hauswirth and Laipis (1982). The reduction of mtDNA copy number is predicted to lead to a shift in heteroplasmy that can happen within a few generations (Hauswirth & Laipis 1982; Jenuth et al. 1996; Cree et al. 2008). However, over 30 years since this hypothesis was first put forward, the exact timing of the mtDNA copy number decrease during oogenesis and its impact on heteroplasmy shifts between generations remains elusive. Wai et al. (2008) did not observe significant differences between the level of heteroplasmy of female mice and the PGC of their progeny, as had previously been suggested (Hauswirth & Laipis 1982; Jenuth et al. 1996). Instead, they found that shifts in heteroplasmy appeared to occur during postnatal folliculogenesis. The authors suggested that this “late” genetic bottleneck was due to the replication of a subpopulation of mtDNA molecules during the maturation phase of the follicles (Wai et al. 2008). The observation by Cao et al. that mouse PGC contained higher mtDNA copy number than previously predicted (1500 copies, compared with the 200 copies reported by Cree et al.) led to a third bottleneck theory, involving unequal segregation of the mtDNA molecules during cell division, rather than a drastic reduction in copy number (Cao et al. 2007; Cree et al. 2008). However, direct evidence supporting this mechanism is lacking. More recently, Freyer et al. (2012) investigated the timing of heteroplasmy changes of novel mtDNA mutations by backcrossing a female mouse carrying a PolgA exo − mutation (also called the mtDNA mutator mouse (Trifunovic et al. 2004)) with wild‐type males. Using this method, they obtained a maternal lineage carrying an m.3875delC in the tRNAMet gene. Measurement of heteroplasmy levels in maternal cells, embryonic PGC and oocytes/soma of the offspring suggested a shift of heteroplasmy occurring during the early stages of PGC development, lending further support to the “early” bottleneck hypothesis (Freyer et al. 2012).
These contrasting observations highlight the current ambiguity concerning the timing and mechanism of the mitochondrial germline genetic bottleneck. The differences may, in part, reflect true biological differences between different strains and species. However, an alternative explanation is that technical differences, due to the measurement of low quantities of mtDNA in single cells, or the precise timing of the observations during development, could contribute to the different results reported. However, whatever the precise underlying process may be, the unpredictable shifts in heteroplasmy transmitted from mothers carrying pathogenic mutations to their children continue to make this area of research highly relevant. One key question that remains unanswered is whether or not the germline genetic bottleneck plays a role in selection against deleterious mtDNA heteroplasmies during transmission of the mtDNA from mother to offspring (Fig. 1C).
Evidence for and against purifying selection in mouse pedigrees
Much work on germline transmission of mtDNA has utilized invertebrate model organisms, including Drosophila (Hill et al. 2014; Ma et al. 2014) and Caenorhabditis elegans (Wernick et al. 2016). However, whilst these species provide tractable models for such studies, they are far removed from humans in evolutionary terms, and so here we focus on data from a range of studies in mammals that have attempted to address the role (if any) of purifying selection in inheritance of mtDNA heteroplasmies.
Transmission of mtDNA has been studied in a number of mammalian species, including cows, sheep and zebrafish, but many of the studies to date have been conducted in mice. The inability to genetically manipulate mammalian mtDNA has made the study of pathogenic heteroplasmies in mouse models challenging. Early studies relied on fusing cytoplasts from two different mouse strains to generate conplastic heteroplasmic animals carrying two separate mtDNA genotypes. Jenuth et al. (1996) studied segregation of heteroplasmy in conplastic NZB/BALBc mice, concluding that the variance of this non‐pathogenic heteroplasmy in germline cells was due to random genetic drift. Using a similar approach, Meirelles and Smith (1997) generated a conplastic NZB/C57Bl6 mouse strain and saw evidence of stable heteroplasmy across generations, contrasting the variance seen by Jenuth et al. (1997), and inconsistent with the existing bottleneck theories. Interestingly, Sharpley et al. (2012) found that mixing of mtDNA from the NZB and 129S6 strains resulted in animals that developed pathogenic phenotypes, a phenomenon also seen by Acton et al. (2007) in NZB/BALBc conplastic mice. Sharpley et al. (2012) showed that heteroplasmy in NZB/129S6 mice segregated rapidly towards 129S6 homoplasmy over successive generations, suggesting active selection against transmission of NZB mtDNA. These contrasting results highlight the complex nature of mtDNA inheritance, and suggest that pathogenic and non‐pathogenic heteroplasmies may segregate differently, and may be influenced by the nuclear genetic background.
More recently, several groups have been able to generate mice harboring deleterious heteroplasmic mtDNA mutations, allowing more detailed investigation of how pathogenic variants are transmitted through the germline. Fan et al. (2008) used a complex approach involving cytoplast fusion of mouse ES cells, and subsequent injection into C57Bl/6 blastocysts, to introduce two heteroplasmic mutations into the mtDNA of a single female mouse. One mutation was a highly deleterious frame‐shift in the ND6 gene, and the second a less severe missense mutation in the cytochrome c oxidase I (COI) gene. Analysis of heteroplasmy levels in subsequent generations revealed rapid elimination of the severe ND6 mutant within four generations, whilst the milder COI mutant persisted, despite causing myopathy and cardiomyopathy in the mice that carried it (Fan et al. 2008). These results suggest that purifying selection may act rapidly against severe pathogenic mutations, whilst allowing less severe variants to persist in the population. In a separate study, Stewart et al. (2008b) utilized the PolgA exo − mtDNA mutator mouse model to introduce random mutations into the maternal mitochondrial genome and studied their transmission. Using this approach, they also found evidence that non‐synonymous (i.e. possibly pathogenic) mutations in protein coding sequences are rapidly purged over just a few generations, lending further support to the hypothesis that deleterious variants are subject to purifying selection (Stewart et al. 2008b).
The point at which purifying selection occurs in the mouse is currently not well defined. In the aforementioned study by Freyer et al. (2012), segregation of the tRNAMet m.3875delC mutation was seen early in PGC development, consistent with the presence of a germline genetic bottleneck. However, evidence of purifying selection against high levels of mutation was only seen in postnatal tissues, and not during germline development (Freyer et al. 2012). A similar study in mice carrying a point mutation in the tRNAAla gene also identified selection against high levels of heteroplasmy in subsequent generations, but there was no concurrent increase in embryonic death, again suggesting that the selection takes place at the cellular/organellar level after birth (Kauppila et al. 2016). Despite these findings, there is currently very little data available on the transmission of pathogenic mtDNA mutations through the mouse germline, and further work will be required to fully understand the dynamics of purifying selection in this context.
Evidence for and against purifying selection in human pedigrees
Understanding heteroplasmy transmission in the human germline presents an even greater challenge than that faced in the mouse. This is partly due to the difficulty of obtaining embryonic tissues for analysis and the ethical constraints that must be considered when dealing with such sensitive material, but also because of the inherent problem of ascertainment bias when obtaining pedigree data from an affected proband (Wilson et al. 2016). Consequently, the number of studies in this area is few, although a number of groups have managed to make some progress in this challenging field. Monnot et al. (2011), who analyzed embryonic tissues from nine heteroplasmic females carrying the common m.3243A>G mutation, responsible for mitochondrial encephalomyopathy, lactic acidosis and stroke‐like episodes syndrome, found that segregation appears to be governed by random genetic drift during early embryonic development. Similar results were seen in oocytes and embryos for both the m.3243A>G mutation (Brown et al. 2001) and the m.8993T>G neuropathy, ataxia and retinitis pigmentosa mutation in (Blok et al. 1997; Steffann et al. 2006, 2007), suggesting that these variants are not subject to purifying selection. However, it must be noted that drawing firm conclusions from small‐scale studies such as these is difficult because reliable statistics require many independent observations (Wonnapinij et al. 2010). More recently, an analysis of human pedigrees transmitting a number of common pathogenic heteroplasmies found that, although the rate of segregation appears to vary between different mutations, there was no evidence of selection from mother to offspring (Wilson et al. 2016).
Although the above studies all seem to argue against purifying selection acting on mtDNA variants during germline development, these findings contrast with other similar studies; Rebolledo‐Jaramillo et al. (2014) sequenced mtDNA from 39 healthy mother–child pairs, and found that most carried one or more low‐level heteroplasmies, some of which were disease associated. Analysis of these point mutations showed reduced transmission of non‐synonymous compared with synonymous mutations, suggesting that potentially pathogenic variants are selected against. Similarly, Li et al. (2016) also identified selection against novel deleterious mtDNA heteroplasmies in a large dataset obtained from the Genomes of the Netherlands project. Furthermore, in vivo data from recent analysis of oocytes from nine healthy women found evidence of selection against potentially pathogenic mtDNA variants during oogenesis, occurring between the expulsion of the first and second polar bodies (De Fanti et al. 2017). Finally, very recent data from Floros et al. (2018), obtained from early‐gestation human embryos (Carnegie stages 12–21), suggests that non‐synonymous mtDNA mutations are indeed subject to purifying selection during PGC development. These conflicting results highlight the fact that our understanding of the complex mechanisms underpinning mtDNA transmission is far from complete, and much work remains to be done to fully elucidate this key process.
Proposed mechanisms by which purifying selection may occur
The mechanism(s) controlling purifying selection in the mammalian germline are not clear, and there is currently much debate over whether this occurs purely by random genetic drift or by active selection. Whilst this issue remains unresolved, it is quite possible that the underlying processes vary depending on the specific mutation and the level of heteroplasmy present, with the additional complications raised by the different nuclear genetic backgrounds.
Despite the evolutionary differences between mammals and invertebrates alluded to previously, two recent studies in model organisms have shed light on potential mechanisms involved in the transmission of heteroplasmic mtDNA mutations and warrant mention here as potential pathways of interest to investigate further in mammals. Hill et al. (2014) generated a mutant Drosophila line carrying a temperature‐sensitive mtDNA heteroplasmy in the COI gene. During oogenesis, they found that wild‐type mtDNA from “healthy” mitochondria was preferentially amplified over those containing high levels of the mutant version, suggesting that selection against heteroplasmy is dependent upon “mitochondrial fitness”. In contrast, Lin et al. (2016) engineered a C. elegans strain harboring an mtDNA deletion heteroplasmy, and identified that the mitochondrial unfolded protein response (UPRmt) plays an important role in maintenance of this deleterious mutation. It remains to be seen whether either of these proposed mechanisms plays any role in mammalian cells, although recent data from Pezet and colleagues has failed to identify UPRmt activation in human cybrids carrying a heteroplasmic mtDNA deletion, suggesting that this mechanism is not involved in maintenance of heteroplasmy in human cell lines (Mikael Pezet, unpub. data, 2017).
In mice, Battersby and Shoubridge (2001) attempted to identify the mechanism underlying preferential segregation of NZB mtDNA in liver tissue of NZB/BALBc conplastic animals (Jenuth et al. 1997). They concluded that selection against BALBc mtDNA was not due to growth defects or altered OXPHOS capacity, but instead likely depended upon factors involved in mtDNA maintenance (Battersby & Shoubridge 2001). Subsequently, Moreno‐Loshuertos et al. (2006) reported that NZB/BALBc heteroplasmy does result in altered OXPHOS efficiency due to an SNP in the tRNAArg gene, the effects of which are masked by compensatory mechanisms triggered by upregulation of ROS production. However, these findings were refuted by Battersby and Shoubridge (2007), and Freyer et al. (2012) have since published further evidence that selection against heteroplasmy is not dependent upon OXPHOS function. Thus, a definitive mechanism for this selection remains elusive in this context. In the same NZB/BALBc model, Jokinen et al. (2015) studied the preferential selection for BALBc mtDNA in hematopoietic cells and found that the tail‐anchored, ER‐resident GTPases Gimap3 and Gimap5 play a critical role in mtDNA segregation, suggesting that coordination of organelle interactions may be involved in modulating mtDNA segregation and selection.
Studies investigating these selective mechanisms in humans are similarly sparse. Blok et al. (1997) suggested preferential amplification of a subset of mitochondrial genomes, possibly similar to that seen in Drosophila (Hill et al. 2014), to explain skewed segregation of the m.8993T>G mutation in human oocytes, but there is no empirical human data to support this theory. More recently, Ling et al. (2016) found that fibroblasts carrying the m.3243A>G mutation showed increased segregation towards homoplasmy following treatment with ROS, and suggested that this was due to formation of mtDNA concatemers that allow amplification of multiple identical mtDNA copies as a single unit. Whether this mechanism has any role in mtDNA segregation during germline development is currently not clear.
Current tools for studying purifying selection in the germline
Although we currently understand very little about the selective mechanisms active in the female germline, recent advances in both murine and human reproductive biology have provided important new tools that are likely to aid the further investigation of this important subject. Here, we discuss the current in vitro and in vivo technologies that exist to enable study of female germline development and mtDNA transmission.
In vitro models to investigate germline mtDNA transmission
It is only recently that induced pluripotent stem (iPS) cells derived from patients carrying heteroplasmic mtDNA variants have been used to understand the tissue specificity of mitochondrial diseases and to study the impact of the heteroplasmy upon cell fate (Cherry et al. 2013; Folmes et al. 2013; Hamalainen et al. 2013). These iPS‐derived cell models represent a potential tool to investigate the underlying disease mechanisms, but also to screen for new therapeutic drugs in a tissue‐specific manner (Hatakeyama & Goto 2016). This is possibly due to our comprehensive understanding of the molecular pathways involved in the differentiation of the iPS cells into different cell lineages. Such mechanisms, although already well established in iPS cells, have also been intensively studied in the context of PGC specification in recent years.
The PGC are specified from the proximal epiblast by BMP signaling from the extra‐embryonic tissues. This was first identified in homozygous BMP4 knockout mice, which do not develop and PGC, and a similar, but less drastic, phenotype was also observed in BMP8b null mice (Lawson et al. 1999; Ying et al. 2000). BMP signaling activates Blimp‐1, a key transcriptional regulator that is first expressed at E6.25 in mouse embryos. When Blimp1 expression is disrupted, the number of founder PGC drops from 40 to 20 and they no longer migrate to the genital ridge, where the future gonads will be formed (Ohinata et al. 2005). BMP signaling also activates expression of Prdm14, followed by Tcfap2c, which encodes the transcription factor AP2γ. Together Blimp1, Prdm14 and AP2γ control PGC specification and are able to rescue this process when expressed in the absence of BMP signaling (Magnusdottir et al. 2013). Further factors involved in PGC development were identified by Saitou et al. (2002) in a screen for PGC‐specific genes. They identified FGF‐8, a gene that is expressed in the early stages of PGC specification, and Stella, whose expression is germ cell‐specific at E7.25 and continues to be expressed in migrating PGC. This detailed understanding of PGC specification has enabled the in vitro induction of mouse and human ES cells, using growth factors including BMP4, BMP8 and bFGF, to produce primordial germ cell‐like cells (PGCLC), which recapitulate the gene expression profile of in vivo PGC (Hayashi & Saitou 2013; Sugawa et al. 2015). As proof of the robustness of this in vitro model, Hayashi et al. aggregated PGCLC with gonadal cells, which were then into mice depleted of endogenous PGC. These mice were subsequently bred and produced healthy offspring (Hayashi et al. 2012). Similarly, Hikabe et al. have reconstituted in vitro the entire cycle of mouse female germline. Using mouse ES cells, they were able to generate fully mature oocytes that could also give rise to healthy offspring when fertilized and transferred to surrogate mothers (Hikabe et al. 2016).
In the same way that iPS cells have expanded our understanding of the tissue specificity of mitochondrial diseases, in vitro modeling using PGCLC or mature oocytes derived from mouse and human ES cells are likely to represent a malleable tool to study the precise timing and mechanisms underpinning the transmission of mtDNA variants during oogenesis.
Mouse models for in vivo study of mtDNA transmission
In addition to the use of in vitro PGCLC, there are also a number mouse models that allow in vivo examination of mtDNA inheritance, and several of these have already been discussed. A number of groups have generated artificial heteroplasmies by mixing mtDNA from different mouse strains. This approach has a number of drawbacks: first, unlike pathogenic variants in human mtDNA caused by single point mutations, the two mtDNA sequences in conplastic mice often contain a large number of nucleotide differences (e.g. 91 polymorphisms exist between the NZB and 129S6 strains, including 15 non‐synonymous variants [Sharpley et al. 2012]). This makes dissection of any selective mechanisms difficult, as multiple different factors may contribute to the dynamics of segregation in these animals. Also, in many cases, these heteroplasmies are not reported to be pathogenic (Jenuth et al. 1996; Meirelles & Smith 1997), and therefore may not be subject to purifying selection at all. Finally, introduction of a “foreign” mtDNA invariably leads to a nuclear/mtDNA mismatch within the cells, a situation that is exacerbated if two mtDNA sequences are backcrossed onto the nuclear background of a third strain (Sharpley et al. 2012). Because nuclear/mtDNA mismatching in conplastic mice is known to impact upon OXPHOS function and influence health and longevity (Latorre‐Pellicer et al. 2016), caution should be taken when interpreting results from these studies, as there is likely to be overlap between the effects of heteroplasmy and the mismatch with the nuclear genome.
The development of the PolgA exo − mtDNA mutator mouse, which acquires de novo heteroplasmic mtDNA mutations due to an inactivating mutation in the proof‐reading subunit of the mtDNA polymerase gamma (Trifunovic et al. 2004), has resulted in a much more tractable method for generating mice with genuine heteroplasmies. However, the rapid and random introduction of mutations into the mtDNA has made studying the inheritance of individual mtDNA mutations using this model challenging. By backcrossing PolgA exo − females with wild‐type males, Freyer et al. (2012) were able to generate a strain carrying just two mtDNA mutations: a homoplasmic substitution, m.5245T>C, in the tRNACys gene and a heteroplasmic deletion, m.3875delC, in the tRNAMet gene. Although these mice appeared phenotypically normal, a compensatory transcriptional response was seen in tissues carrying high levels of the tRNAMet mutant, suggesting that this heteroplasmy was impacting mitochondrial function (Freyer et al. 2012). More recently, Kauppila et al. (2016) used a similar approach to obtain a strain carrying a single heteroplasmic mutation at m.5024C>T in the tRNAAla gene. Crucially, these animals develop cardiomyopathy and the mutation appears to be selected against in mitotic tissues, making this perhaps the best model currently available for studying inheritance of pathogenic mtDNA mutations in the mouse germline.
A number of tools exist to aid study of the mouse germline, allowing effective identification and isolation of PGC from their induction in the proximal epiblast at embryonic day (E)6.25 (Ohinata et al. 2005) through to oogenesis in late‐stage embryos. Staining of embryonic cells for PGC markers, such as tissue non‐specific alkaline phosphatase (Ginsburg et al. 1990), Stella (Dppa3) (Saitou et al. 2002) and Blimp1 (Prdm1) (Ohinata et al. 2005) allows efficient identification of PGC during early embryonic development; however, use of such stains and antibodies requires fixed tissues, limiting this approach to non‐living material. To enable more versatile studies in live cells, Payer et al. (2006) generated transgenic mice expressing a GFP‐tagged version of the PGC‐specific marker Stella, which allows non‐invasive identification of PGC from E7.5 onwards. Ohinata et al. (2008) subsequently built upon this model, creating a double‐transgenic reporter mouse expressing enhanced cyan fluorescent protein (ECFP)‐tagged Stella and mVenus‐tagged Blimp1. In this strain, Blimp1‐mVenus expression is seen from E7.5, with Stella‐ECFP detectable from E8.5, allowing for robust identification of double‐positive PGC. Crossing transgenic PGC reporter mice with females carrying mtDNA mutations, such as the m.5024C>T tRNAAla mutant, should enable detailed investigation of the dynamics involved in germline transmission of pathogenic mtDNA heteroplasmies and has the potential to dramatically expand our knowledge of this pivotal process.
Studying the human germline in vivo
In vivo study of human germline development is incredibly challenging and fraught with ethical and technical difficulties. Whilst some data is available from oocytes and early embryos of patients carrying pathogenic heteroplasmies (Blok et al. 1997; Brown et al. 2001; Monnot et al. 2011), successful isolation of primary PGC from human embryos has not been reported. However, recently developed flow cytometry‐based protocols have been developed allowing the isolation of human PGC from Carnegie stage 12 onwards (Tang et al. 2015; Floros et al. 2018), representing a significant advance in our ability to investigate development of the human germline.
Conclusions and perspectives
In recent years, huge advances have been made in the field of inherited mitochondrial disease, with a number of therapeutic approaches, such as pre‐implantation genetic screening (Smeets et al. 2015) and mitochondrial replacement therapy (Wolf et al. 2015), offering hope to families affected by these devastating conditions. However, despite the large number of studies aimed at understanding how heteroplasmic mtDNA mutations are transmitted through the germline, many of the mechanisms involved remain elusive, hampering efforts to develop more effective treatment and prevention strategies. The recent development of novel model systems and in vivo techniques for detailed investigation of germline development, both in humans and other mammals, promises to begin shedding light on some of the key unanswered questions that remain regarding mtDNA transmission, and will hopefully lead to tangible progress in the ongoing fight against mitochondrial disease.
Acknowledgments
P. F. C. is a Wellcome Trust Senior Fellow in Clinical Science (101876/Z/13/Z), and a UK NIHR Senior Investigator, who receives support from the Medical Research Council Mitochondrial Biology Unit (MC_UP_1501/2), the Medical Research Council (UK) Centre for Translational Muscle Disease (G0601943), and the National Institute for Health Research (NIHR) Biomedical Research Centre based at Cambridge University Hospitals NHS Foundation Trust and the University of Cambridge. The views expressed are those of the author(s) and not necessarily those of the NHS, the NIHR or the Department of Health.
References
- Acton, B. M. , Lai, I. , Shang, X. , Jurisicova, A. & Casper, R. F. 2007. Neutral mitochondrial heteroplasmy alters physiological function in mice. Biol. Reprod. 77, 569–576. [DOI] [PubMed] [Google Scholar]
- Battersby, B. J. & Shoubridge, E. A. 2001. Selection of a mtDNA sequence variant in hepatocytes of heteroplasmic mice is not due to differences in respiratory chain function or efficiency of replication. Hum. Mol. Genet. 10, 2469–2479. [DOI] [PubMed] [Google Scholar]
- Battersby, B. J. & Shoubridge, E. A. 2007. Reactive oxygen species and the segregation of mtDNA sequence variants. Nat. Genet. 39, 571–572; author reply 572. [DOI] [PubMed] [Google Scholar]
- Blok, R. B. , Gook, D. A. , Thorburn, D. R. & Dahl, H. H. 1997. Skewed segregation of the mtDNA nt 8993 (T–>G) mutation in human oocytes. Am. J. Hum. Genet. 60, 1495–1501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown, D. T. , Samuels, D. C. , Michael, E. M. , Turnbull, D. M. & Chinnery, P. F. 2001. Random genetic drift determines the level of mutant mtDNA in human primary oocytes. Am. J. Hum. Genet. 68, 533–536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgstaller, J. P. , Johnston, I. G. , Jones, N. S. , Albrechtova, J. , Kolbe, T. , Vogl, C. , Futschik, A. , Mayrhofer, C. , Klein, D. , Sabitzer, S. , Blattner, M. , Gully, C. , Poulton, J. , Rulicke, T. , Pialek, J. , Steinborn, R. & Brem, G. 2014. MtDNA segregation in heteroplasmic tissues is common in vivo and modulated by haplotype differences and developmental stage. Cell Rep. 7, 2031–2041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cann, R. L. , Stoneking, M. & Wilson, A. C. 1987. Mitochondrial DNA and human evolution. Nature 325, 31–36. [DOI] [PubMed] [Google Scholar]
- Cao, L. , Shitara, H. , Horii, T. , Nagao, Y. , Imai, H. , Abe, K. , Hara, T. , Hayashi, J. & Yonekawa, H. 2007. The mitochondrial bottleneck occurs without reduction of mtDNA content in female mouse germ cells. Nat. Genet. 39, 386–390. [DOI] [PubMed] [Google Scholar]
- Cao, L. , Shitara, H. , Sugimoto, M. , Hayashi, J. , Abe, K. & Yonekawa, H. 2009. New evidence confirms that the mitochondrial bottleneck is generated without reduction of mitochondrial DNA content in early primordial germ cells of mice. PLoS Genet. 5, e1000756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Case, J. T. & Wallace, D. C. 1981. Maternal inheritance of mitochondrial DNA polymorphisms in cultured human fibroblasts. Somatic Cell Genet. 7, 103–108. [DOI] [PubMed] [Google Scholar]
- Cherry, A. B. , Gagne, K. E. , McLoughlin, E. M. , Baccei, A. , Gorman, B. , Hartung, O. , Miller, J. D. , Zhang, J. , Zon, R. L. , Ince, T. A. , Neufeld, E. J. , Lerou, P. H. , Fleming, M. D. , Daley, G. Q. & Agarwal, S. 2013. Induced pluripotent stem cells with a mitochondrial DNA deletion. Stem Cells 31, 1287–1297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chinnery, P. F. & Hudson, G. 2013. Mitochondrial genetics. Br. Med. Bull. 106, 135–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chinnery, P. F. & Samuels, D. C. 1999. Relaxed replication of mtDNA: a model with implications for the expression of disease. Am. J. Hum. Genet. 64, 1158–1165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chinnery, P. F. , Howell, N. , Lightowlers, R. N. & Turnbull, D. M. 1997. Molecular pathology of MELAS and MERRF. The relationship between mutation load and clinical phenotypes. Brain 120(Pt 10), 1713–1721. [DOI] [PubMed] [Google Scholar]
- Chinnery, P. F. , Craven, L. , Mitalipov, S. , Stewart, J. B. , Herbert, M. & Turnbull, D. M. 2014. The challenges of mitochondrial replacement. PLoS Genet. 10, e1004315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coskun, P. E. , Ruiz‐Pesini, E. & Wallace, D. C. 2003. Control region mtDNA variants: longevity, climatic adaptation, and a forensic conundrum. Proc. Natl Acad. Sci. USA 100, 2174–2176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cotterill, M. , Harris, S. E. , Collado Fernandez, E. , Lu, J. , Huntriss, J. D. , Campbell, B. K. & Picton, H. M. 2013. The activity and copy number of mitochondrial DNA in ovine oocytes throughout oogenesis in vivo and during oocyte maturation in vitro. Mol. Hum. Reprod. 19, 444–450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cree, L. M. , Samuels, D. C. , De Sousa Lopes, S. C. , Rajasimha, H. K. , Wonnapinij, P. , Mann, J. R. , Dahl, H. H. & Chinnery, P. F. 2008. A reduction of mitochondrial DNA molecules during embryogenesis explains the rapid segregation of genotypes. Nat. Genet. 40, 249–254. [DOI] [PubMed] [Google Scholar]
- De Fanti, S. , Vicario, S. , Lang, M. , Simone, D. , Magli, C. , Luiselli, D. , Gianaroli, L. & Romeo, G. 2017. Intra‐individual purifying selection on mitochondrial DNA variants during human oogenesis. Hum. Reprod. 32, 1100–1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiMauro, S. & Schon, E. A. 2001. Mitochondrial DNA mutations in human disease. Am. J. Med. Genet. 106, 18–26. [DOI] [PubMed] [Google Scholar]
- Elliott, H. R. , Samuels, D. C. , Eden, J. A. , Relton, C. L. & Chinnery, P. F. 2008. Pathogenic mitochondrial DNA mutations are common in the general population. Am. J. Hum. Genet. 83, 254–260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elson, J. L. , Turnbull, D. M. & Howell, N. 2004. Comparative genomics and the evolution of human mitochondrial DNA: assessing the effects of selection. Am. J. Hum. Genet. 74, 229–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan, W. , Waymire, K. G. , Narula, N. , Li, P. , Rocher, C. , Coskun, P. E. , Vannan, M. A. , Narula, J. , Macgregor, G. R. & Wallace, D. C. 2008. A mouse model of mitochondrial disease reveals germline selection against severe mtDNA mutations. Science 319, 958–962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Floros, V. I. , Pyle, A. , Dietmann, S. , Wei, W. , Tang, W. W. C. , Irie, N. , Payne, B. A. I. , Capalbo, A. , Noli, L. , Coxhead, J. , Hudson, G. , Crosier, M. , Strahl, H. , Khalaf, Y. , Saitou, M. , Ilic, D. , Surani, M. A. & Chinnery, P. F. 2018. Segregation of mitochondrial DNA heteroplasmy through a development genetic bottleneck in human embryos. Nat. Cell. Biol. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Folmes, C. D. , Martinez‐Fernandez, A. , Perales‐Clemente, E. , Li, X. , McDonald, A. , Oglesbee, D. , Hrstka, S. C. , Perez‐Terzic, C. , Terzic, A. & Nelson, T. J. 2013. Disease‐causing mitochondrial heteroplasmy segregated within induced pluripotent stem cell clones derived from a patient with MELAS. Stem Cells 31, 1298–1308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freyer, C. , Cree, L. M. , Mourier, A. , Stewart, J. B. , Koolmeister, C. , Milenkovic, D. , Wai, T. , Floros, V. I. , Hagstrom, E. , Chatzidaki, E. E. , Wiesner, R. J. , Samuels, D. C. , Larsson, N. G. & Chinnery, P. F. 2012. Variation in germline mtDNA heteroplasmy is determined prenatally but modified during subsequent transmission. Nat. Genet. 44, 1282–1285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ginsburg, M. , Snow, M. H. & McLaren, A. 1990. Primordial germ cells in the mouse embryo during gastrulation. Development 110, 521–528. [DOI] [PubMed] [Google Scholar]
- Gomez‐Duran, A. , Pacheu‐Grau, D. , Lopez‐Gallardo, E. , Diez‐Sanchez, C. , Montoya, J. , Lopez‐Perez, M. J. & Ruiz‐Pesini, E. 2010. Unmasking the causes of multifactorial disorders: OXPHOS differences between mitochondrial haplogroups. Hum. Mol. Genet. 19, 3343–3353. [DOI] [PubMed] [Google Scholar]
- Hagstrom, E. , Freyer, C. , Battersby, B. J. , Stewart, J. B. & Larsson, N. G. 2014. No recombination of mtDNA after heteroplasmy for 50 generations in the mouse maternal germline. Nucleic Acids Res. 42, 1111–1116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamalainen, R. H. , Manninen, T. , Koivumaki, H. , Kislin, M. , Otonkoski, T. & Suomalainen, A. 2013. Tissue‐ and cell‐type‐specific manifestations of heteroplasmic mtDNA 3243A>G mutation in human induced pluripotent stem cell‐derived disease model. Proc. Natl Acad. Sci. USA 110, E3622–E3630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatakeyama, H. & Goto, Y. 2016. Concise Review: Heteroplasmic mitochondrial DNA mutations and mitochondrial diseases: toward iPSC‐based disease modeling, drug discovery, and regenerative therapeutics. Stem Cells 34, 801–808. [DOI] [PubMed] [Google Scholar]
- Hauswirth, W. W. & Laipis, P. J. 1982. Mitochondrial DNA polymorphism in a maternal lineage of Holstein cows. Proc. Natl Acad. Sci. USA 79, 4686–4690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi, K. & Saitou, M. 2013. Generation of eggs from mouse embryonic stem cells and induced pluripotent stem cells. Nat. Protoc. 8, 1513–1524. [DOI] [PubMed] [Google Scholar]
- Hayashi, K. , Ogushi, S. , Kurimoto, K. , Shimamoto, S. , Ohta, H. & Saitou, M. 2012. Offspring from oocytes derived from in vitro primordial germ cell‐like cells in mice. Science 338, 971–975. [DOI] [PubMed] [Google Scholar]
- Hikabe, O. , Hamazaki, N. , Nagamatsu, G. , Obata, Y. , Hirao, Y. , Hamada, N. , Shimamoto, S. , Imamura, T. , Nakashima, K. , Saitou, M. & Hayashi, K. 2016. Reconstitution in vitro of the entire cycle of the mouse female germ line. Nature 539, 299–303. [DOI] [PubMed] [Google Scholar]
- Hill, J. H. , Chen, Z. & Xu, H. 2014. Selective propagation of functional mitochondrial DNA during oogenesis restricts the transmission of a deleterious mitochondrial variant. Nat. Genet. 46, 389–392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudson, G. , Nalls, M. , Evans, J. R. , Breen, D. P. , Winder‐Rhodes, S. , Morrison, K. E. , Morris, H. R. , Williams‐Gray, C. H. , Barker, R. A. , Singleton, A. B. , Hardy, J. , Wood, N. E. , Burn, D. J. & Chinnery, P. F. 2013. Two‐stage association study and meta‐analysis of mitochondrial DNA variants in Parkinson disease. Neurology 80, 2042–2048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudson, G. , Gomez‐Duran, A. , Wilson, I. J. & Chinnery, P. F. 2014. Recent mitochondrial DNA mutations increase the risk of developing common late‐onset human diseases. PLoS Genet. 10, e1004369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutchison, C. A. 3rd , Newbold, J. E. , Potter, S. S. & Edgell, M. H. 1974. Maternal inheritance of mammalian mitochondrial DNA. Nature 251, 536–538. [DOI] [PubMed] [Google Scholar]
- Jenuth, J. P. , Peterson, A. C. , Fu, K. & Shoubridge, E. A. 1996. Random genetic drift in the female germline explains the rapid segregation of mammalian mitochondrial DNA. Nat. Genet. 14, 146–151. [DOI] [PubMed] [Google Scholar]
- Jenuth, J. P. , Peterson, A. C. & Shoubridge, E. A. 1997. Tissue‐specific selection for different mtDNA genotypes in heteroplasmic mice. Nat. Genet. 16, 93–95. [DOI] [PubMed] [Google Scholar]
- Jimenez‐Sousa, M. A. , Tamayo, E. , Guzman‐Fulgencio, M. , Heredia, M. , Fernandez‐Rodriguez, A. , Gomez, E. , Almansa, R. , Gomez‐Herreras, J. I. , Garcia‐Alvarez, M. , Gutierrez‐Junco, S. , Bermejo‐Martin, J. F. , Resino, S. & Spanish Sepsis, G. 2015. Mitochondrial DNA haplogroups are associated with severe sepsis and mortality in patients who underwent major surgery. J. Infect. 70, 20–29. [DOI] [PubMed] [Google Scholar]
- Johnson, A. A. & Johnson, K. A. 2001. Exonuclease proofreading by human mitochondrial DNA polymerase. J. Biol. Chem. 276, 38097–38107. [DOI] [PubMed] [Google Scholar]
- Jokinen, R. , Lahtinen, T. , Marttinen, P. , Myohanen, M. , Ruotsalainen, P. , Yeung, N. , Shvetsova, A. , Kastaniotis, A. J. , Hiltunen, J. K. , Ohman, T. , Nyman, T. A. , Weiler, H. & Battersby, B. J. 2015. Quantitative changes in Gimap3 and Gimap5 expression modify mitochondrial DNA segregation in mice. Genetics 200, 221–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kauppila, J. H. K. , Baines, H. L. , Bratic, A. , Simard, M. L. , Freyer, C. , Mourier, A. , Stamp, C. , Filograna, R. , Larsson, N. G. , Greaves, L. C. & Stewart, J. B. 2016. A phenotype‐driven approach to generate mouse models with pathogenic mtDNA mutations causing mitochondrial disease. Cell Rep. 16, 2980–2990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kazak, L. , Reyes, A. & Holt, I. J. 2012. Minimizing the damage: repair pathways keep mitochondrial DNA intact. Nat. Rev. Mol. Cell Biol. 13, 659–671. [DOI] [PubMed] [Google Scholar]
- Kazuno, A. A. , Munakata, K. , Nagai, T. , Shimozono, S. , Tanaka, M. , Yoneda, M. , Kato, N. , Miyawaki, A. & Kato, T. 2006. Identification of mitochondrial DNA polymorphisms that alter mitochondrial matrix pH and intracellular calcium dynamics. PLoS Genet. 2, e128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kivisild, T. , Shen, P. , Wall, D. P. , Do, B. , Sung, R. , Davis, K. , Passarino, G. , Underhill, P. A. , Scharfe, C. , Torroni, A. , Scozzari, R. , Modiano, D. , Coppa, A. , De Knijff, P. , Feldman, M. , Cavalli‐Sforza, L. L. & Oefner, P. J. 2006. The role of selection in the evolution of human mitochondrial genomes. Genetics 172, 373–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lagouge, M. & Larsson, N. G. 2013. The role of mitochondrial DNA mutations and free radicals in disease and ageing. J. Intern. Med. 273, 529–543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsson, N. G. , Tulinius, M. H. , Holme, E. , Oldfors, A. , Andersen, O. , Wahlstrom, J. & Aasly, J. 1992. Segregation and manifestations of the mtDNA tRNA(Lys) A–>G(8344) mutation of myoclonus epilepsy and ragged‐red fibers (MERRF) syndrome. Am. J. Hum. Genet. 51, 1201–1212. [PMC free article] [PubMed] [Google Scholar]
- Latorre‐Pellicer, A. , Moreno‐Loshuertos, R. , Lechuga‐Vieco, A. V. , Sanchez‐Cabo, F. , Torroja, C. , Acin‐Perez, R. , Calvo, E. , Aix, E. , Gonzalez‐Guerra, A. , Logan, A. , Bernad‐Miana, M. L. , Romanos, E. , Cruz, R. , Cogliati, S. , Sobrino, B. , Carracedo, A. , Perez‐Martos, A. , Fernandez‐Silva, P. , Ruiz‐Cabello, J. , Murphy, M. P. , Flores, I. , Vazquez, J. & Enriquez, J. A. 2016. Mitochondrial and nuclear DNA matching shapes metabolism and healthy ageing. Nature 535, 561–565. [DOI] [PubMed] [Google Scholar]
- Lawson, K. A. , Dunn, N. R. , Roelen, B. A. , Zeinstra, L. M. , Davis, A. M. , Wright, C. V. , Korving, J. P. & Hogan, B. L. 1999. Bmp4 is required for the generation of primordial germ cells in the mouse embryo. Genes Dev. 13, 424–436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, M. , Rothwell, R. , Vermaat, M. , Wachsmuth, M. , Schroder, R. , Laros, J. F. , Van Oven, M. , De Bakker, P. I. , Bovenberg, J. A. , Van Duijn, C. M. , Van Ommen, G. J. , Slagboom, P. E. , Swertz, M. A. , Wijmenga, C. , Genome of Netherlands, C. , Kayser, M. , Boomsma, D. I. , Zollner, S. , De Knijff, P. & Stoneking, M. 2016. Transmission of human mtDNA heteroplasmy in the Genome of the Netherlands families: support for a variable‐size bottleneck. Genome Res. 26, 417–426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin, Y. F. , Schulz, A. M. , Pellegrino, M. W. , Lu, Y. , Shaham, S. & Haynes, C. M. 2016. Maintenance and propagation of a deleterious mitochondrial genome by the mitochondrial unfolded protein response. Nature 533, 416–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ling, F. , Niu, R. , Hatakeyama, H. , Goto, Y. , Shibata, T. & Yoshida, M. 2016. Reactive oxygen species stimulate mitochondrial allele segregation toward homoplasmy in human cells. Mol. Biol. Cell 27, 1684–1693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma, H. , Xu, H. & O'Farrell, P. H. 2014. Transmission of mitochondrial mutations and action of purifying selection in Drosophila melanogaster . Nat. Genet. 46, 393–397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magnusdottir, E. , Dietmann, S. , Murakami, K. , Gunesdogan, U. , Tang, F. , Bao, S. , Diamanti, E. , Lao, K. , Gottgens, B. & Azim Surani, M. 2013. A tripartite transcription factor network regulates primordial germ cell specification in mice. Nat. Cell Biol. 15, 905–915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meirelles, F. V. & Smith, L. C. 1997. Mitochondrial genotype segregation in a mouse heteroplasmic lineage produced by embryonic karyoplast transplantation. Genetics 145, 445–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mishmar, D. , Ruiz‐Pesini, E. , Golik, P. , Macaulay, V. , Clark, A. G. , Hosseini, S. , Brandon, M. , Easley, K. , Chen, E. , Brown, M. D. , Sukernik, R. I. , Olckers, A. & Wallace, D. C. 2003. Natural selection shaped regional mtDNA variation in humans. Proc. Natl Acad. Sci. USA 100, 171–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monnot, S. , Gigarel, N. , Samuels, D. C. , Burlet, P. , Hesters, L. , Frydman, N. , Frydman, R. , Kerbrat, V. , Funalot, B. , Martinovic, J. , Benachi, A. , Feingold, J. , Munnich, A. , Bonnefont, J. P. & Steffann, J. 2011. Segregation of mtDNA throughout human embryofetal development: m.3243A>G as a model system. Hum. Mutat. 32, 116–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreno‐Loshuertos, R. , Acin‐Perez, R. , Fernandez‐Silva, P. , Movilla, N. , Perez‐Martos, A. , Rodriguez De Cordoba, S. , Gallardo, M. E. & Enriquez, J. A. 2006. Differences in reactive oxygen species production explain the phenotypes associated with common mouse mitochondrial DNA variants. Nat. Genet. 38, 1261–1268. [DOI] [PubMed] [Google Scholar]
- Muller, H. J. 1964. The relation of recombination to mutational advance. Mutat. Res. 106, 2–9. [DOI] [PubMed] [Google Scholar]
- Nass, M. M. 1966. The circularity of mitochondrial DNA. Proc. Natl Acad. Sci. USA 56, 1215–1222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nogawa, T. , Sung, W. K. , Jagiello, G. M. & Bowne, W. 1988. A quantitative analysis of mitochondria during fetal mouse oogenesis. J. Morphol. 195, 225–234. [DOI] [PubMed] [Google Scholar]
- Ohinata, Y. , Payer, B. , O'Carroll, D. , Ancelin, K. , Ono, Y. , Sano, M. , Barton, S. C. , Obukhanych, T. , Nussenzweig, M. , Tarakhovsky, A. , Saitou, M. & Surani, M. A. 2005. Blimp1 is a critical determinant of the germ cell lineage in mice. Nature 436, 207–213. [DOI] [PubMed] [Google Scholar]
- Ohinata, Y. , Sano, M. , Shigeta, M. , Yamanaka, K. & Saitou, M. 2008. A comprehensive, non‐invasive visualization of primordial germ cell development in mice by the Prdm1‐mVenus and Dppa3‐ECFP double transgenic reporter. Reproduction 136, 503–514. [DOI] [PubMed] [Google Scholar]
- Otten, A. B. , Stassen, A. P. , Adriaens, M. , Gerards, M. , Dohmen, R. G. , Timmer, A. J. , Vanherle, S. J. , Kamps, R. , Boesten, I. B. , Vanoevelen, J. M. , Muller, M. & Smeets, H. J. 2016. Replication errors made during oogenesis lead to detectable de novo mtDNA mutations in zebrafish oocytes with a low mtDNA copy number. Genetics 204, 1423–1431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Payer, B. , Chuva De Sousa Lopes, S. M. , Barton, S. C. , Lee, C. , Saitou, M. & Surani, M. A. 2006. Generation of stella‐GFP transgenic mice: a novel tool to study germ cell development. Genesis 44, 75–83. [DOI] [PubMed] [Google Scholar]
- Payne, B. A. , Wilson, I. J. , Yu‐Wai‐Man, P. , Coxhead, J. , Deehan, D. , Horvath, R. , Taylor, R. W. , Samuels, D. C. , Santibanez‐Koref, M. & Chinnery, P. F. 2013. Universal heteroplasmy of human mitochondrial DNA. Hum. Mol. Genet. 22, 384–390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pello, R. , Martin, M. A. , Carelli, V. , Nijtmans, L. G. , Achilli, A. , Pala, M. , Torroni, A. , Gomez‐Duran, A. , Ruiz‐Pesini, E. , Martinuzzi, A. , Smeitink, J. A. , Arenas, J. & Ugalde, C. 2008. Mitochondrial DNA background modulates the assembly kinetics of OXPHOS complexes in a cellular model of mitochondrial disease. Hum. Mol. Genet. 17, 4001–4011. [DOI] [PubMed] [Google Scholar]
- Poulton, J. , Chiaratti, M. R. , Meirelles, F. V. , Kennedy, S. , Wells, D. & Holt, I. J. 2010. Transmission of mitochondrial DNA diseases and ways to prevent them. PLoS Genet. 6, e1001066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pyle, A. , Hudson, G. , Wilson, I. J. , Coxhead, J. , Smertenko, T. , Herbert, M. , Santibanez‐Koref, M. & Chinnery, P. F. 2015. Extreme‐depth re‐sequencing of mitochondrial DNA finds no evidence of paternal transmission in humans. PLoS Genet. 11, e1005040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajasimha, H. K. , Chinnery, P. F. & Samuels, D. C. 2008. Selection against pathogenic mtDNA mutations in a stem cell population leads to the loss of the 3243A–>G mutation in blood. Am. J. Hum. Genet. 82, 333–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rand, D. M. 2001. The units of selection on mitochondrial DNA. Annu. Rev. Ecol. Evol. Syst. 32, 415–448. [Google Scholar]
- Rand, D. M. 2008. Mitigating mutational meltdown in mammalian mitochondria. PLoS Biol. 6, e35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rand, D. M. & Kann, L. M. 1996. Excess amino acid polymorphism in mitochondrial DNA: contrasts among genes from Drosophila, mice, and humans. Mol. Biol. Evol. 13, 735–748. [DOI] [PubMed] [Google Scholar]
- Rand, D. M. & Kann, L. M. 1998. Mutation and selection at silent and replacement sites in the evolution of animal mitochondrial DNA. Genetica 102–103, 393–407. [PubMed] [Google Scholar]
- Rebolledo‐Jaramillo, B. , Su, M. S. , Stoler, N. , McElhoe, J. A. , Dickins, B. , Blankenberg, D. , Korneliussen, T. S. , Chiaromonte, F. , Nielsen, R. , Holland, M. M. , Paul, I. M. , Nekrutenko, A. & Makova, K. D. 2014. Maternal age effect and severe germ‐line bottleneck in the inheritance of human mitochondrial DNA. Proc. Natl Acad. Sci. USA 111, 15474–15479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruiz‐Pesini, E. , Mishmar, D. , Brandon, M. , Procaccio, V. & Wallace, D. C. 2004. Effects of purifying and adaptive selection on regional variation in human mtDNA. Science 303, 223–226. [DOI] [PubMed] [Google Scholar]
- Saitou, M. , Barton, S. C. & Surani, M. A. 2002. A molecular programme for the specification of germ cell fate in mice. Nature 418, 293–300. [DOI] [PubMed] [Google Scholar]
- Scheibye‐Knudsen, M. , Fang, E. F. , Croteau, D. L. , Wilson, D. M. 3rd & Bohr V. A. 2015. Protecting the mitochondrial powerhouse. Trends Cell Biol. 25, 158–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharpley, M. S. , Marciniak, C. , Eckel‐Mahan, K. , McManus, M. , Crimi, M. , Waymire, K. , Lin, C. S. , Masubuchi, S. , Friend, N. , Koike, M. , Chalkia, D. , Macgregor, G. , Sassone‐Corsi, P. & Wallace, D. C. 2012. Heteroplasmy of mouse mtDNA is genetically unstable and results in altered behavior and cognition. Cell 151, 333–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smeets, H. J. , Sallevelt, S. C. , Dreesen, J. C. , De Die‐Smulders, C. E. & De Coo, I. F. 2015. Preventing the transmission of mitochondrial DNA disorders using prenatal or preimplantation genetic diagnosis. Ann. N. Y. Acad. Sci. 1350, 29–36. [DOI] [PubMed] [Google Scholar]
- Steffann, J. , Frydman, N. , Gigarel, N. , Burlet, P. , Ray, P. F. , Fanchin, R. , Feyereisen, E. , Kerbrat, V. , Tachdjian, G. , Bonnefont, J. P. , Frydman, R. & Munnich, A. 2006. Analysis of mtDNA variant segregation during early human embryonic development: a tool for successful NARP preimplantation diagnosis. J. Med. Genet. 43, 244–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steffann, J. , Gigarel, N. , Corcos, J. , Bonniere, M. , Encha‐Razavi, F. , Sinico, M. , Prevot, S. , Dumez, Y. , Yamgnane, A. , Frydman, R. , Munnich, A. & Bonnefont, J. P. 2007. Stability of the m.8993T‐>G mtDNA mutation load during human embryofetal development has implications for the feasibility of prenatal diagnosis in NARP syndrome. J. Med. Genet. 44, 664–669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart, J. B. & Chinnery, P. F. 2015. The dynamics of mitochondrial DNA heteroplasmy: implications for human health and disease. Nat. Rev. Genet. 16, 530–542. [DOI] [PubMed] [Google Scholar]
- Stewart, J. B. , Freyer, C. , Elson, J. L. & Larsson, N. G. 2008a. Purifying selection of mtDNA and its implications for understanding evolution and mitochondrial disease. Nat. Rev. Genet. 9, 657–662. [DOI] [PubMed] [Google Scholar]
- Stewart, J. B. , Freyer, C. , Elson, J. L. , Wredenberg, A. , Cansu, Z. , Trifunovic, A. & Larsson, N. G. 2008b. Strong purifying selection in transmission of mammalian mitochondrial DNA. PLoS Biol. 6, e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugawa, F. , Arauzo‐Bravo, M. J. , Yoon, J. , Kim, K. P. , Aramaki, S. , Wu, G. , Stehling, M. , Psathaki, O. E. , Hubner, K. & Scholer, H. R. 2015. Human primordial germ cell commitment in vitro associates with a unique PRDM14 expression profile. EMBO J. 34, 1009–1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang, W. W. , Dietmann, S. , Irie, N. , Leitch, H. G. , Floros, V. I. , Bradshaw, C. R. , Hackett, J. A. , Chinnery, P. F. & Surani, M. A. 2015. A unique gene regulatory network resets the human germline epigenome for development. Cell 161, 1453–1467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torroni, A. , Petrozzi, M. , D'urbano, L. , Sellitto, D. , Zeviani, M. , Carrara, F. , Carducci, C. , Leuzzi, V. , Carelli, V. , Barboni, P. , De Negri, A. & Scozzari, R. 1997. Haplotype and phylogenetic analyses suggest that one European‐specific mtDNA background plays a role in the expression of Leber hereditary optic neuropathy by increasing the penetrance of the primary mutations 11778 and 14484. Am. J. Hum. Genet. 60, 1107–1121. [PMC free article] [PubMed] [Google Scholar]
- Trifunovic, A. , Wredenberg, A. , Falkenberg, M. , Spelbrink, J. N. , Rovio, A. T. , Bruder, C. E. , Bohlooly, Y. M. , Gidlof, S. , Oldfors, A. , Wibom, R. , Tornell, J. , Jacobs, H. T. & Larsson, N. G. 2004. Premature ageing in mice expressing defective mitochondrial DNA polymerase. Nature 429, 417–423. [DOI] [PubMed] [Google Scholar]
- Wai, T. , Teoli, D. & Shoubridge, E. A. 2008. The mitochondrial DNA genetic bottleneck results from replication of a subpopulation of genomes. Nat. Genet. 40, 1484–1488. [DOI] [PubMed] [Google Scholar]
- Wallace, D. C. , Brown, M. D. & Lott, M. T. 1999. Mitochondrial DNA variation in human evolution and disease. Gene 238, 211–230. [DOI] [PubMed] [Google Scholar]
- Wernick, R. I. , Estes, S. , Howe, D. K. & Denver, D. R. 2016. Paths of heritable mitochondrial DNA mutation and heteroplasmy in reference and gas‐1 strains of Caenorhabditis elegans . Front Genet. 7, 51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson, I. J. , Carling, P. J. , Alston, C. L. , Floros, V. I. , Pyle, A. , Hudson, G. , Sallevelt, S. C. , Lamperti, C. , Carelli, V. , Bindoff, L. A. , Samuels, D. C. , Wonnapinij, P. , Zeviani, M. , Taylor, R. W. , Smeets, H. J. , Horvath, R. & Chinnery, P. F. 2016. Mitochondrial DNA sequence characteristics modulate the size of the genetic bottleneck. Hum. Mol. Genet. 25, 1031–1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf, D. P. , Mitalipov, N. & Mitalipov, S. 2015. Mitochondrial replacement therapy in reproductive medicine. Trends Mol. Med. 21, 68–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wonnapinij, P. , Chinnery, P. F. & Samuels, D. C. 2010. Previous estimates of mitochondrial DNA mutation level variance did not account for sampling error: comparing the mtDNA genetic bottleneck in mice and humans. Am. J. Hum. Genet. 86, 540–550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ying, Y. , Liu, X. M. , Marble, A. , Lawson, K. A. & Zhao, G. Q. 2000. Requirement of Bmp8b for the generation of primordial germ cells in the mouse. Mol. Endocrinol. 14, 1053–1063. [DOI] [PubMed] [Google Scholar]
- Yu‐Wai‐Man, P. , Turnbull, D. M. & Chinnery, P. F. 2002. Leber hereditary optic neuropathy. J. Med. Genet. 39, 162–169. [DOI] [PMC free article] [PubMed] [Google Scholar]