

Abstract
The biotechnological potential of Rieske Oxygenases (ROs) and their cognate reductases remains unmet, in part because these systems can be functionally short-lived. Here, we describe a set of experiments aimed at identifying both functional and structural stability limitations of ROs, using terephthalate (TPA) dioxygenase (from Comamonas strain E6) as a model system. Successful expression and purification of a cofactor-complete, histidine-tagged TPA dioxygenase and reductase protein system requires induction with the E. coli host at stationary phase as well as a chaperone inducing cold-shock and supplementation with additional iron, sulfur, and flavin. The relative stability of the Rieske cluster and mononuclear iron can then be assessed using spectroscopic and functional measurements following dialysis in an iron chelating buffer. These include measurements of the overall lifetime of the system via total turnover number using both UV-Visible absorbance and HPLC analyses, as well specific activity as a function of temperature. Important methods for assessing the stability of these multi-cofactor, multi-protein dependent systems at multiple levels of structure (secondary to quaternary) include differential scanning calorimetry, circular dichroism, and metallospectroscopy. Results can be rationalized in terms of three-dimensional structures and bioinformatics. The experiments described here provide a roadmap to a detailed characterization of the limitations of ROs. With a few notable exceptions, these issues are not widely addressed in current literature.
Keywords: Rieske oxygenase, oxygenase, iron, plastic, lignin, aromatic catabolism, protein stability
1. Introduction
In the golden age of protein discovery, driven by sequencing and bioinformatics, there is a need to have a consistent metric to compare the stability of both structure and function within a class of like proteins. In addition, understanding the detailed limitations of an enzyme of interest allows for engineering efforts of complex multimeric, cofactor-dependent proteins using rational design (Goldenzweig & Fleishman, 2018). This is especially important as these proteins are often not amenable to directed evolution approaches. Here, we will describe a set of five standard protocols that can be used to provide a metric for the functional and structural stability of complex, multimeric metalloproteins. We use terephthalic acid dioxygenase (TPADO), a heteroheximeric Rieske oxygenase (RO) from Comamonas sp E6, as a model system for which we have defined in detail the observed instabilities. The protocols give a multifaceted overview of how and when loss of protein structure leads to functional failure.
1.1. Multimeric Mononuclear Iron Oxygenases and their Engineering: Historical Perspectives
The first Rieske iron sulfur cluster was described in 1964 via its unique EPR signal at g=1.9 in a sample of a reduced co-enzyme Q cytochrome C reductase (Complex III) (John S. Rieske et al., 1964; Rieske et al., 1964). The signal identified in this publication was consistent with an electron transfer mediating, iron-containing complex, but did not match the EPR signals from any existing standard. Four years later in 1968, a benzene dioxygenase was described as the first Rieske cluster containing oxygenase (Gibson et al., 1970; Gibson et al., 1968). Gibson et al. proposed an enzymatic mechanism for O2 incorporation into chlorobenzene at the expense of NADH, as observed in Psuedomonas putida. At the time, known mammalian hydroxylation events catalyzed by cytochrome P450s were described exclusively as a pair of trans monoxygenations; however, Jerina and coworkers. rigorously identified the bacterial product of naphthalene-fed Pseudomonas sp. to be a cis-dihydroxylated, dearomatized 1,2-dihydroxy-1,2-dihydronaphthalene, its production catalyzed by a homologous RO, naphthalene dioxygenase. This observation suggested a new pathway to a novel enzymatic product (Jerina et al., 1971).
ROs have since been identified in the genomes of over 16,000 organisms spanning all kingdoms of life. To date, there are approximately 70,000 identified ROs represented by 20 unique X-ray crystal structures (Capyk et al., 2009; Daughtry et al., 2012; Dong et al., 2005; Dumitru et al., 2009; Friemann et al., 2005; Furusawa et al., 2004; Hou et al., 2021; Inoue et al., 2014; Jakoncic et al., 2007; Kauppi et al., 1997; Kincannon et al., 2022; Kumari et al., 2017; Liu et al., 2022; Lukowski et al., 2020; Mahto et al., 2022; Martins et al., 2005; Quareshy et al.). The novel chemistry of both mono- and dioxygenations ROs has the potential fill critical needs in medical, industrial, and environmental applications including the catabolism of various toxic aromatics. RO activity has been identified on pollutants leeched from waste products include toluene, benzene, biphenyl, and phthalate (Axcell & Geary, 1975; Correll et al., 1992; Furusawa et al., 2004; Gibson et al., 1970; Mahto et al., 2021; Martinez-Martinez et al., 2014; Neidle et al., 1991; Parales et al., 1998). The potential economic and societal impact of these applications provides a driving force to discover, characterize, and improve this class of enzymes.
Structurally, ROs are characterized by the presence of a 2Fe-2S cluster where one iron center is coordinated to the protein by two cysteine residues and the other by two histidine residues. The iron coordinated by cysteines maintains a ferric state while the other passes through both ferrous and ferric states before and after electron transfer, respectively. ROs also contain a mononuclear non-heme iron center which accepts electrons from the cluster and directly catalyzes the insertion of either one or both oxygens from molecular O2 into an organic substrate. ROs can have either homotrimeric α3 or heteroheximeric α3β3 quaternary protein structures. Each comma-shaped α subunit contains both metallo-cofactors though only half of a complete active site. The head-to-tail orientation of α subunits results in a 12 Å gap between the Rieske cluster in the tail of one subunit and the mononuclear iron in the head of the adjacent subunit. The β subunits are not directly involved in catalysis and likely provide structural support to the α subunits and/or a reductase as it docks with and delivers electrons to the RO. The NAD(P)H derived electrons are delivered to the RO by either a single flavin and plant-type 2Fe-2S cluster containing reductase, or from separate 2Fe-2S containing ferredoxin and flavin containing NAD(P)H reductase proteins acting together. The utilization of one or both electron transfer proteins, the RO quaternary structure, and the regiospecificity of oxygenation are used to classify ROs (Figure 1) (Chakraborty et al., 2017).
Figure 1.
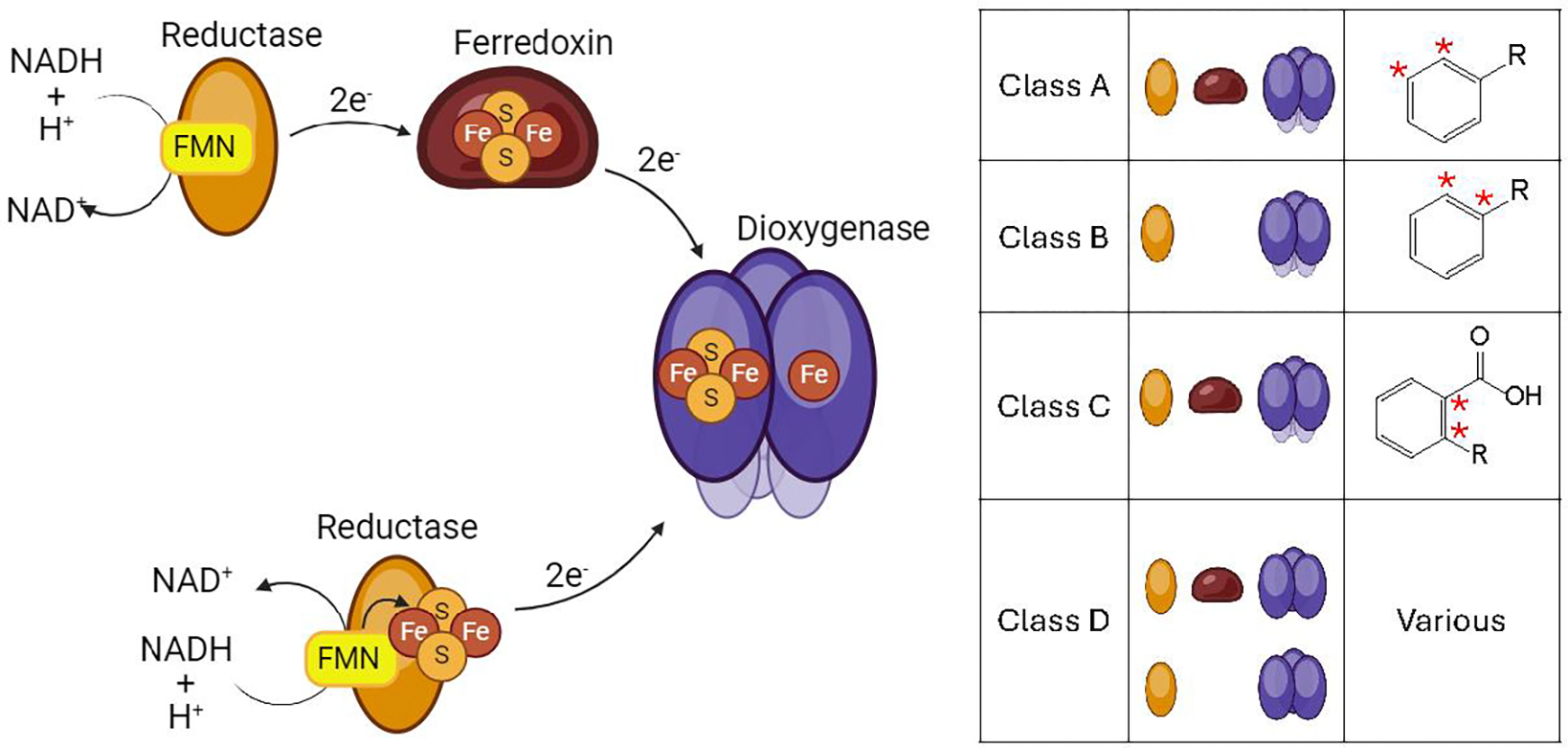
LEFT Pathway for electron flow from NAD(P)H t to the catalytic dioxygenase. Electron transfer proteins mediating the transfer can include a FMN containing reductase, a plant-type or Rieske type iron sulfur cluster ferredoxin, or a combined FMN and iron sulfur cluster containing reductase. RIGHT General classification scheme for the four classes of ROs as defined by Chakraborty and coworkers. Classification is based on dioxygenase structure and composition of electron transport proteins.
1.2. Rieske Iron Oxygenases and their Engineering: Efforts Towards Substrate Expansion and Functional Improvement
The publication of crystal structures from representative ROs such as naphthalene dioxygenase and nitrobenzene dioxygenase led to a variety of site-directed mutagenesis efforts in the late 1990s (Karlsson et al., 2003; Kauppi et al., 1997). The earliest efforts aimed to confirm structure-function hypotheses, including mapping electron transfer pathways and catalytically relevant active site residues (Kauppi et al., 1997; Kumari et al., 2017; Massmig et al., 2020; Parales, 2003; Tarasev et al., 2009; Tiwari et al., 2011; Zhu et al., 2014).
As the community’s understanding of structure-function relationships developed, the desire to alter existing chemistry arose. Xenobiotic aromatic catabolism is environmentally important, and it makes use of the ROs’ unique catalytic capabilities. The regio- and site selectivity and catalytic efficiency of xenobiotic-catabolizing ROs became early targets for engineering, typically through targeted site-directed mutagenesis (Ang et al., 2009; Lessner et al., 2002; Liu et al., 2022; Lukowski et al., 2020; Özgen & Schmidt, 2019; Parales, Lee, et al., 2000; Parales, Resnick, et al., 2000). For example, a single point mutation in a biphenyl dioxygenase exhibited an increased kcat/KM activity on environmentally significant chlorinated biphenyls.41 Another recent study successfully altered p-toluenesulfonate methyl monooxygenase (TsaM) to accept non-native substrate dicamba, a xenobiotic herbicide. Work by this group also expanded the para- specific regioselectivity of TsaM to oxygenate at both meta- and ortho- positions.43 Additionally, engineering efforts at the subunit interfaces have been undertaken with a view toward understanding communication between cofactor-containing sites involved in O2 activation and/or electron transfer (Brimberry et al., 2023; Tsai et al., 2022).
With notable exceptions, such as documenting the lability of the mononuclear iron cofactor, (Bernhardt & Meisch, 1980; Correll et al., 1992; Suen & Gibson, 1993; Wolfe et al., 2002; Wolfe & Lipscomb, 2003) the overall quaternary stability of ROs remains a largely undiscussed theme. The stabilities of multimeric proteins and multiprotein systems are challenging to quantify stability is an emergent property that depends on all levels of structure concurrently. An approach to oligomers began with a modified Gibbs free energy equation including a new coefficient, Keff, to describe the equilibrium constant for denaturation, expressed in monomer units (Park & Marqusee, 2004) and accounting for the equilibrium of folded and multiple potential unfolded states. Despite the large number of unfolded states, multimeric proteins are often more stable than monomeric proteins due to the increased rigidity of their globular protein structure (Balcão & Vila, 2015; Carvalho et al., 2012; Chakraborty et al., 2017; Das & Gerstein, 2000; Flores & Ellington, 2002; Gakhar et al., 2005; Goldenzweig & Fleishman, 2018; Jaenicke & Böhm, 1998; Jaenicke et al., 1996; Lisi et al., 2014; Park & Marqusee, 2004). Immobilization of the multimer to a solid surface can further increase stability (Arana-Pena et al., 2020; Balcão et al., 2001; Hobisch et al., 2021; Kumar, 2010; Thompson et al., 2018).
1.3. Multimeric Rieske Iron Oxygenases and their Engineering: Outline of Unmet Potential
Studies characterizing ROs frequently report catalytic efficiency (kcat/KM) or degrees of uncoupling of O2 reduction from substrate oxygenation (Ang et al., 2009; Kincannon et al., 2022). However, overall lifetime, turnover number, and structural stability measurements are remarkably absent. The potential use of ROs in bioremediation is a beacon application for this class, and further applications in organic synthesis have been envisioned (Capyk et al., 2009; Friemann et al., 2005; Kumar et al., 2011; Liu et al., 2022; Lukowski et al., 2020; Zhu et al., 2014). One group lead by Hudlicky has begun the insertion of ROs into biosynthesis pathways for morphine derivatives and antifungals (Hudlicky et al., 1992; Hudlicky et al., 1999; Hudlicky & Reed, 2009; Özgen & Schmidt, 2019), but the utilization of these enzymes is far below their projected potential. Practical application of any enzyme system requires some understanding of its lifetime and efficiency, which may need to be expanded by engineering efforts. Here, we outline an experimental roadmap which can be used to identify several relevant measures of stability for ROs and other multimeric metalloenzymes. While loss of structure can be induced with either heat or chemicals, we have focused on heat as it can be administered progressively and in many experimental contexts. The results from these experiments can be used to develop stability models for the class as a whole and as a foundation on which future stability engineering efforts can be built.
In addition to these methods, we have recently published a paper adding two more structural studies, small-angle X-ray scattering (SAXS) and circular dichroism (CD). SAXS allows users to observe the solution-state globular structure of the protein. When recorded as a function of increasing temperature, SAXS can identify the physical melting curve of a protein and can potentially predict the mechanism through which the protein dissociation. This method, however useful, is a specialized technique not readily available to most users and is consequently left out of this chapter. CD measures the relative content of secondary protein structures in a sample. As protein structure is lost to temperature, the change in of α-helices and β-sheets can be used to predict where unfolding occurs, further detailing the mechanism of dissociation. CD is commonly used in studies characterizing protein stability, so we have chosen not to elaborate on that method in this chapter.
1.4. Overview of Methods and Key Parameters Derived from Each
| Method | Sample Type | Sample Parameters | Information Content |
|---|---|---|---|
| Atomic Absorption Spectroscopy | Purified dioxygenase, purified reductase | Iron content 1–20 μM | Total iron content, correlated to protein concentration to estimate upper limit on complete active site content |
| UV-Visible Rieske Cluster Analysis | Purified dioxygenase | Protein concentration 3–10 μM | Qualitative Rieske cluster content reported as a ratio of Rieske peak absorbance intensity at 450 and 550 nm to protein absorbance at 280 nm |
| Steady State Activity | Purified dioxygenase, purified reductase | 2 μM dioxygenase, 6 μM reductase | Activity following protein incubation at elevated temperatures relative to specific activity at temperature optimum |
| Lifetime | Purified dioxygenase, purified reductase | 2 μM dioxygenase, 6 μM reductase | Total number of catalytic cycles each enzyme can undergo prior to loss of activity, typically irreversible; Here we outline real-time monitoring of NADH absorbance and discontinuous substrate quantification by HPLC. |
| DSC | Purified dioxygenase or reductase | 20 μM protein | Heat capacity variation due to structural transition event as samples exposed to heat. The result of this method is a quantitative ΔH and mapping of the thermal dissociation pathway for the protein. |
2. Expression and Purification of an RO System
2.1. Equipment
Shaker (Innova 44, New Brunswick Scientific)
2.8 L Fernbach Flask
UV-Vis spectrophotometer (Cary 60, Agilent)
Centrifuge (Beckman)
Sonicator (Branson)
FPLC (Akta)
Electrophoresis system (EPS 3500, Pharmacia)
Amicon centrifugal protein concentration devices (Sigma)
Desalting column (PD-10, Cytiva)
Buffers, Strains, and Reagents
Lemo (DE3) competent E. coli cells (NEB) transformed with pETDuet plasmid (Genscript) containing RO α and β (TPADO) (Kincannon et al., 2022) (see note 1)
Lemo (DE3) competent cells (NEB) transformed with pET 45b(+) plasmid (Genscript) containing RO reductase (TPARED) (Kincannon et al., 2022)
TB media
Ampicillin 50 mg mL−1 in water
Chloramphenicol 34 mg mL−1 in ethanol
Rhamnose 1 M in water
IPTG 1 M in water
FeCl3 1 M in water
L-cystine 10 mg mL−1 in water
Protease inhibitor phenylmethylsufonyl fluoride
Chicken egg white lysozyme
Buffer A (20 mM Tris pH 8.0, 150 mM NaCl, 10% v:v glycerol)
Buffer B (20 mM Tris pH 8.0, 150 mM NaCl, 10% v:v glycerol, 250 mM imidazole)
Buffer C(20 mM Tris pH 8.0, 300 mM NaCl, 15% v:v glycerol)
Buffer D (20 mM Tris pH 8.0, 300 mM NaCl, 15% v:v glycerol, 250 mM imidazole)
Ni-NTA agarose resin (McLAB) in FPLC compatible glass column (XK 16/20, Cytiva)
2.2. Expression and Purification of TPADO
E. coli glycerol stocks containing the plasmids with the cloned genes are taken from −80°C freezer and kept in a Cryo-Safe box.
Use a flame-sterilized steel loop to gently scrape the surface of a glycerol stock. Inoculate an LB agar plate containing 50 g L−1 ampicillin and 34 mg L−1 chloramphenicol. Incubate for 8–16 hours at 37°C.
The following day, transfer a single colony from the plate to a flask containing 50 mL TB supplemented with 50 g L-1 ampicillin and 34 mg L-1 chloramphenicol. Incubate overnight (16–18 hours) at 37°C with constant shaking at 200 RPM.
The next morning, inoculate a 2.8 L Fernbach flask containing 500 mL sterile TB media supplemented with 50 mg L−1 ampicillin,34 mg L−1 chloramphenicol, and 2 mM rhamnose.
Grow the cultures at 37°C, 225 RPM until the OD600nm reaches ~2.5 (5–6 hours) (see note 2).
At OD600nm ~2.5, rest cultures in ice bath for 45 minutes. Meanwhile, supplement culture with 0.1 mM ferric chloride, 10 g L-1 L-cystine, and 1 mM IPTG (see note 3).
Return culture flasks to incubator and grow at 25°C, 200 RPM overnight (16–18 hours) (see note 4)
The next morning, harvest cells by centrifugation at 12,000 × g for 15 minutes, discarding supernatant (see note 5).
Resuspend cell pellet in Buffer A, ~10 mL buffer per gram cell paste in a sonication safe beaker. Add 0.5 mg mL−1 (each) chicken egg white lysozyme and phenylmethylsufonyl fluoride.
Place in a bath of ice and water.. Sonicate for 20 minutes at 45% amplitude, pulsing 10 seconds on, 20 seconds off, while gently stirring.
Centrifuge the resulting lysate for 30 minutes at 40,000 × g at 4°C. Discard solid cell debris.
Attach the glass column containing Ni-NTA resin to the FPLC and wash with 5–10 column volumes (cv)milli-Q water.
Equilibrate the column with 10 cv Buffer A.
Using a superloop if necessary, load the clarified supernatant to the column.
Wash with 20 cv Buffer A, or until A280 returns to baseline.
To remove non-target proteins from the resin, rinse with 10 cv at 30% Buffer B. Discard eluent (see note 6).
Elute target protein with an isocratic wash at 100% Buffer B. collecting 2–5 mL fractions.
Monitor fractions by SDS-PAGE. (Figure 2)
Concentrate pooled pure protein fractions to 5 mL using an Amicon centrifugal filtration device with appropriate molecular weight cutoff.
Remove imidazole from the protein mixture using a PD-10 desalting column (Cytiva) under gravity flow conditions, following manufacture protocol. Here, PD-10 columns were equilibrated using 25 mL Buffer A. Protein was introduced to the equilibrated column in 2.5 mL increments, then was eluted from the column with 3.5 mL Buffer A.
Following desalting, protein is concentrated once more using an Amicon centrifugal filtration device until proteins are approximately 10 mg mL−1. Purified protein is then aliquoted (100 μL), flash frozen in liquid N2, and stored at −80°C.
Figure 2.
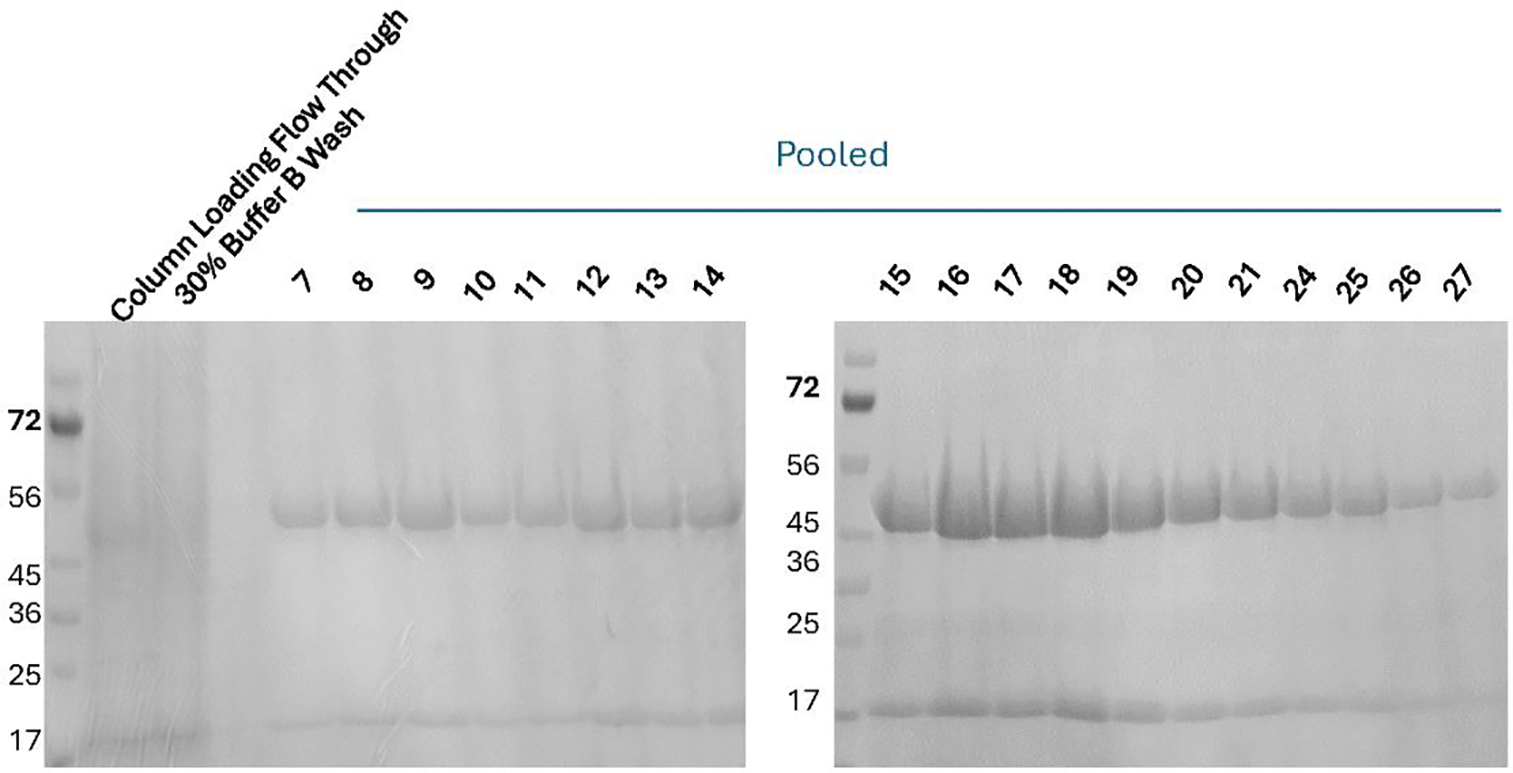
SDS page analysis of fractions collected during the purification of 6x Histidine tagged TPADO seen as bands at 48 and 17 kDa. Fractions (5 mL) were collected during the elution at 100% Buffer B. Fractions 8 through 27 were determined to be of sufficient purity (>95%), pooled together, desalted, concentrated, and stored.
2.3. Expression and purification of TPARED
Starter cultures are prepared as for TPADO and inoculated into 2.8 L Fernbach flasks. E. coli cells containing the TPARED expression plasmid are cultivated and protein expression is induced in the same manner as for TPADO, with the addition of 0.5 mg mL−1 riboflavin to the cultures before induction. For purification:
Attach the glass column containing Ni-NTA resin to the FPLC and wash with 5–10 cv mili-Q water.
Equilibrate the column with 10 cv Buffer C.
Using a superloop if necessary, load the clarified supernatant to the column.
Wash with 20 cv Buffer C, or as much is necessary before A280 returns to baseline. Discard eluent.
Nontarget protein and TPARED are separated by a linear gradient elution from 0 to 50% Buffer D over 8 cv.
Determine purity of fractions through SDS-PAGE and pool only uncontaminated fractions. (Figure 3)
Pooled protein fractions are then concentrated to 5 mL final volume using an Amicon centrifugal filtration device with appropriate molecular weight cutoff.
Imidazole is removed from the protein mixture using a PD-10 column (Cytiva) under gravity flow conditions, following manufacturer’s protocol.
Following desalting, protein is concentrated once more using an Amicon centrifugal filtration device until proteins are approximately 15 mg mL−1. Purified protein is then aliquoted to 100 μL aliquots, flash frozen in liquid N2, and stored at −80°C.
Figure 3.
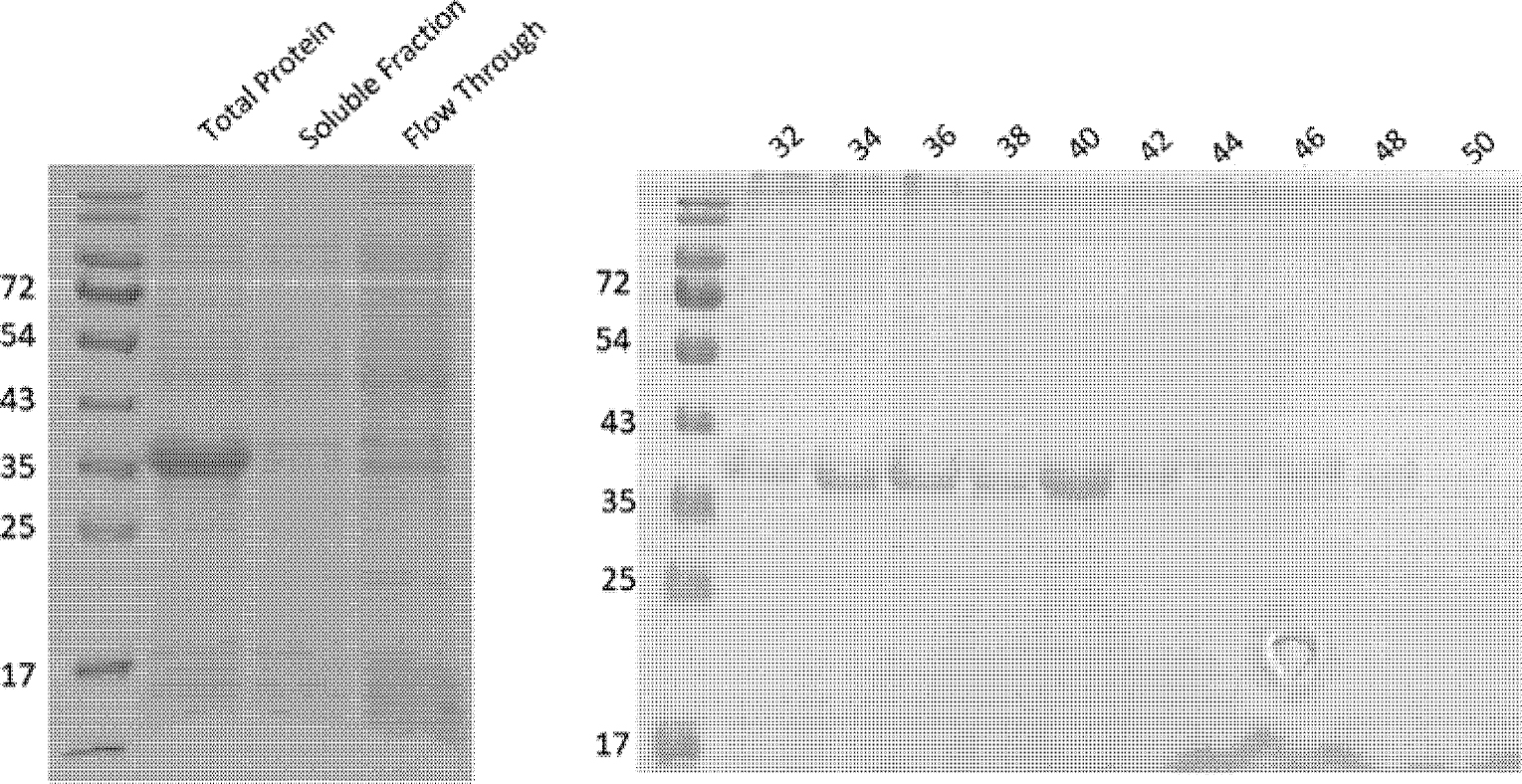
SDS page analysis of fractions collected during the purification of 6x Histidine tagged TPARED seen as a single band at 37 kDa. Fractions (5 mL) were collected during the linear elution 0–50% Buffer D over 8 cv. Fractions 32 through 48 were determined to be of sufficient purity (>95%), pooled together, desalted, concentrated, and stored.
2.4. Notes
For TPADO, a TEV-cleavable C-terminal His6 tag was appended to the α-subunit (TphA2, BAE47077), while the β-subunit (TphA3, BAE47078)) was untagged. For TPARED (TphA1, BAE47080) a C-terminal His6 tag was appended (Kincannon et al., 2022).
Traditional induction of protein expression occurs at 0.3–0.6 OD600nm; however, induction at a stationary-phase OD600nm results in a higher yield of soluble, cofactor complete protein.
The process of exposing growth cultures to an ice bath encourages expression of cold-shock chaperones which has been shown to increase the yield of properly folded protein.
The Lemo(DE3) system uses rhamnose to encourage the production of a T7 polymerase inhibitor that can be used to adjust levels of target protein expression. A small-scale preliminary expression test while varying the concentration of rhamnose can be used to determine optimum expression and solubility conditions for your protein.
Cell mass can be flash-frozen in liquid N2 and stored at −80°C for future use. Under these conditions, TPADO protein typically yields between 20 and 25 g wet cell mass per L culture.
The expression system used here includes a 6-His tag available to interact with the affinity column at each of the three α subunits found in TPADO. Other expression systems may require alternative concentrations of Buffer B to remove impurities while allowing the target protein to remain bound to the column.
3. Iron Cofactor Lability
Metalloproteins rely not only on the preservation of peptide structure, but also on the integrity of the cofactors. Here, we evaluate the relative stability of the coordination of both the Rieske FeS cluster and the mononuclear non-heme iron of TPADO. By using dialysis against a chelating agent (EDTA), weakly bound iron ions are stripped from the protein. UV-Vis, AA, and kinetic measurements are used to quantify how much iron was lost, of which type, and to what degree function was compromised.
3.1. Equipment
UV-Visible Spectrophotometer (Cary 60, Agilent)
Atomic Absorption spectrophotometer (SpectrAA 220 Fast Sequential Atomic Absorption Spectrometer, Varian)
Slide-a-lyzer (ThermoScientific)
Buffers and Reagents
Iron chelating buffer (20 mM Tris, pH 8.0, 150 mM NaCl, 10% v:v glycerol, 5 mM EDTA)
TPADO storage buffer (20 mM Tris, pH 8.0, 150 mM NaCl, 10% v:v glycerol)
2x Reaction master mix (40 mM MOPS, pH 7.2, 200 μM NaCl, 20% v:v DMSO, 400 μM NADH)
Terephthalic acid (TPA) (500 μM, in DMSO )
3.2. Procedure
To determine iron content for a newly purified TPADO sample, first atomic absorption (AA) is used. A series of TPADO samples at 0.5, 1, and 2 μM TPADO (approximately 4.5, 9, and 18 μM iron, respectively, assuming each α subunit contains a single 2Fe-2S cluster and single mononuclear non-heme iron) are prepared. Samples are acidified with 20% nitric acid, incubated at >90°C for 10 minutes, then filtered through a 0.22 μm filter. Absorption measurements for each sample are compared to a standard curve to determine total iron-per-protein concentration.
The relative concentrations of FeS clusters are observed with UV-Visible spectroscopy. Rieske clusters have a characteristic spectrum with two peaks, one at 450 nm and one at 550 nm. TPADO is diluted to 3, 6, and 12 μM into storage buffer (20 mM Tris, pH 8.0, 150 mM NaCl, 10% v:v glycerol) in a quartz cuvette and spectra (280 to 700 nm) are measured (see note 1, Figure 4A).
Finally, TPADO activity is measured. To a cuvette containing 150 μM 2x master mix, 50 μL TPA is added. A series of measurements monitoring NADH concentration at 340 nm is measured over time to determine background NADH oxidation. Finally, 50 μL TPADO (final concentration 2 μM protein) and TPARED (final concentration 6 μM protein) are added. Absorbance changes at 340 nm were measured every 10 seconds for 10 minutes total.
To remove highly labile iron, 300 μL TPADO (10 mg mL-1) are added to a slide-a-lyzer dialysis cassette. The cassette was placed in a 2L beaker containing iron chelation buffer and gently stirred at 4°C for 16 hours.
EDTA is removed by a second dialysis into TPADO storage buffer.
A Bradford assay of the EDTA-treated protein is used to confirm the new concentration (Noble, 2014).
Atomic Absorption, UV-Vis, and activity assays are conducted following identical protocols to pre-EDTA treatment.
Figure 4.
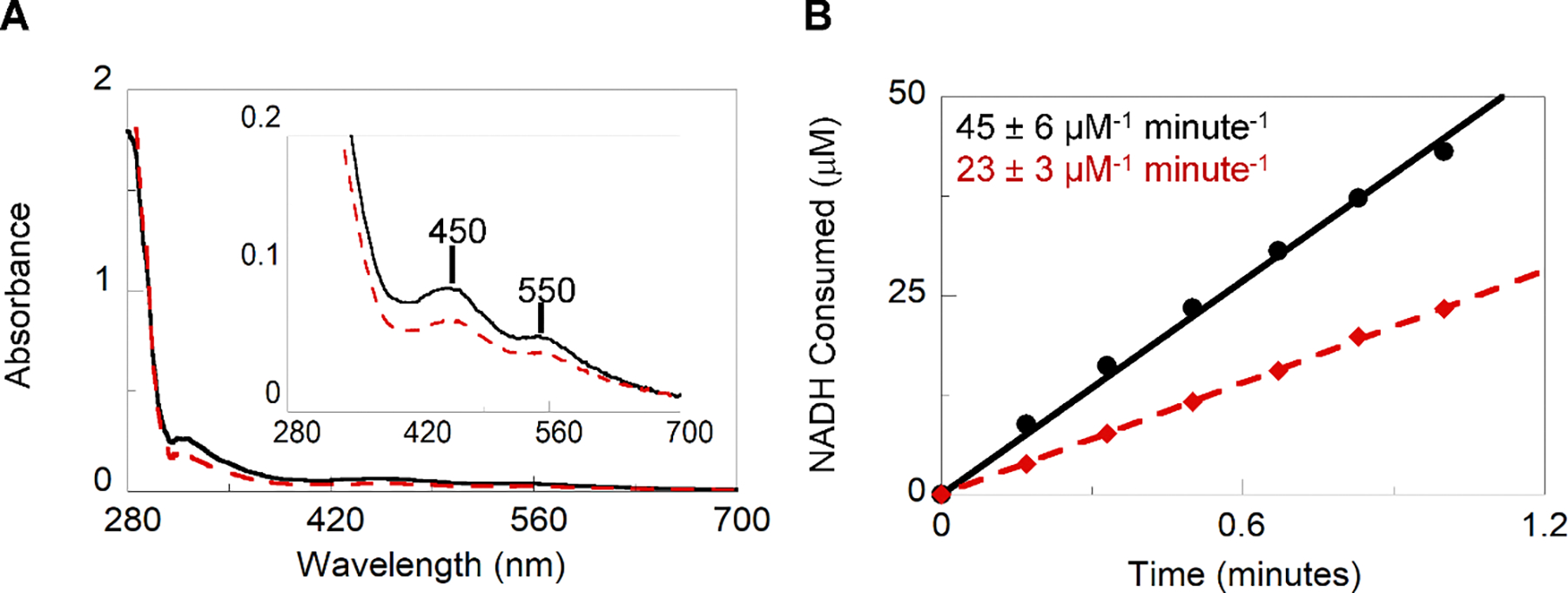
EDTA removal of labile iron from TPADO. A. Qualitative UV-Visible absorbance spectra of the Rieske cluster consistent with that published in previous literature2, 67, 68 indicate two distinct peaks with λmax at 450 and 550 nm (inset). Relative concentrations for the as-isolated (solid black line) and EDTA dialyzed (red dashed line) samples can be referenced to the protein absorbance at 280 nm B. Specific activity for as-isolated and post-dialysis proteins.
3.3. Data Analysis
UV-Vis measurements are a qualitative method for comparing relative concentrations of 2Fe-2S clusters. After spectra have been obtained for both the as-isolated and post-dialysis samples, A450/A280 and A550/A280 values can be calculated as an indication of concentration cofactor per concentration protein. A percentage of signal remaining after dialysis can be reported by dividing the post-dialysis ratios by the as-isolated ratios (Figure 4A).
These measurements can be used with the quantitative AA analysis to report if any of the iron lost due to EDTA treatment belongs to the FeS cluster. Ideally, the FeS clusters, which are more strongly bound than the mono-Fe, should remain intact while the mononuclear Fe is chelated out of the protein (Figure 4B).
-
Activity measurements assess whether fully intact active sites (including the mono-Fe) remain post-dialysis, since both the mononuclear Fe and adjacent Rieske cluster are necessary for catalysis. Beer’s law can be used to convert absorbance at 340 nm to concentration of NADH. The change in NADH concentration measured continuously or discontinuously over a short, linear time interval at the initial portion of the reaction gives the activity, which can be referenced to [protein]:
Eq. 1 Where concentration is calculated by dividing the absorbance at 340nm by the multiplication product of the pathlength of the cuvette in centimeters and the extinction coefficent of NADH at 340 nm in units of μm−1 cm−1 (0.00622 μm−1 cm−1).
These concentrations can then be plotted as a function of time to give initial velocities. Additionally, the total NADH consumed during the 10 minute duration can be calculated using Equation 2.
| Eq. 2 |
Comparing these calculations for the pre- and post-EDTA treatment can provide a measure to the overall lability of both metallo-cofactors.
3.4. Notes
Use of glycerol in a quartz cuvette can cause wetting of the cuvette surface that is not removed by a standard water and ethanol rinse. An additional soak with a commercially available cuvette specific detergent or 0.1 M NaOH followed by rigorous washes with deionized water can remove the film left on the cuvette surface.
4. Temperature Dependent Kinetics
Function can be compromised by loss of structure, cofactor, or the dioxygenase-reductase interface as a result of thermally induced destabilization of the dioxygenase/reductase system. Measurements of initial reaction rates are conducted at increasing temperatures and referenced to a normalized temperature that is optimal for the enzyme and/or the host organism from which it derives.
4.1. Equipment
Plate reader (VarioSkan, Thermo Scienfic; or H1M1, Biotek) (see note 1)
96 well plates (Thermo Scientific)
Thermocycler (Mastercycler EPGradient S, Eppendorf)
Buffers and Reagents
4.2. Procedure
Divide enzymes into 200 μL aliquots, one for each temperature being evaluated.
Place a single aliquot of protein in the thermocycler. Incubate at room temperature for 20 minutes. (see note 4)
Meanwhile, add 125 μL of the 2X Master Mix to three wells of the 96 well plate.
Add 125 μL each 2X Master Mix blank and 20 mM MOPS to a fourth well.
After the protein has incubated, add 50 μL each reductase and RO to each reaction well.
Initiate the reaction by addition of 25 μL TPA. Immediately begin a kinetic absorbance reading at 340 nm, scanning every 10 seconds for 10 minutes total (Figure 5B).
Repeat all steps, adjusting the incubation to 10 minutes at elevated temperatures (ex. 30, 35, 40, 45°C) followed by a 10 minute re-equilibration to room temperature. (see note 5, 6). Figure 5A shows general schematic for the experiment.
Figure 5.
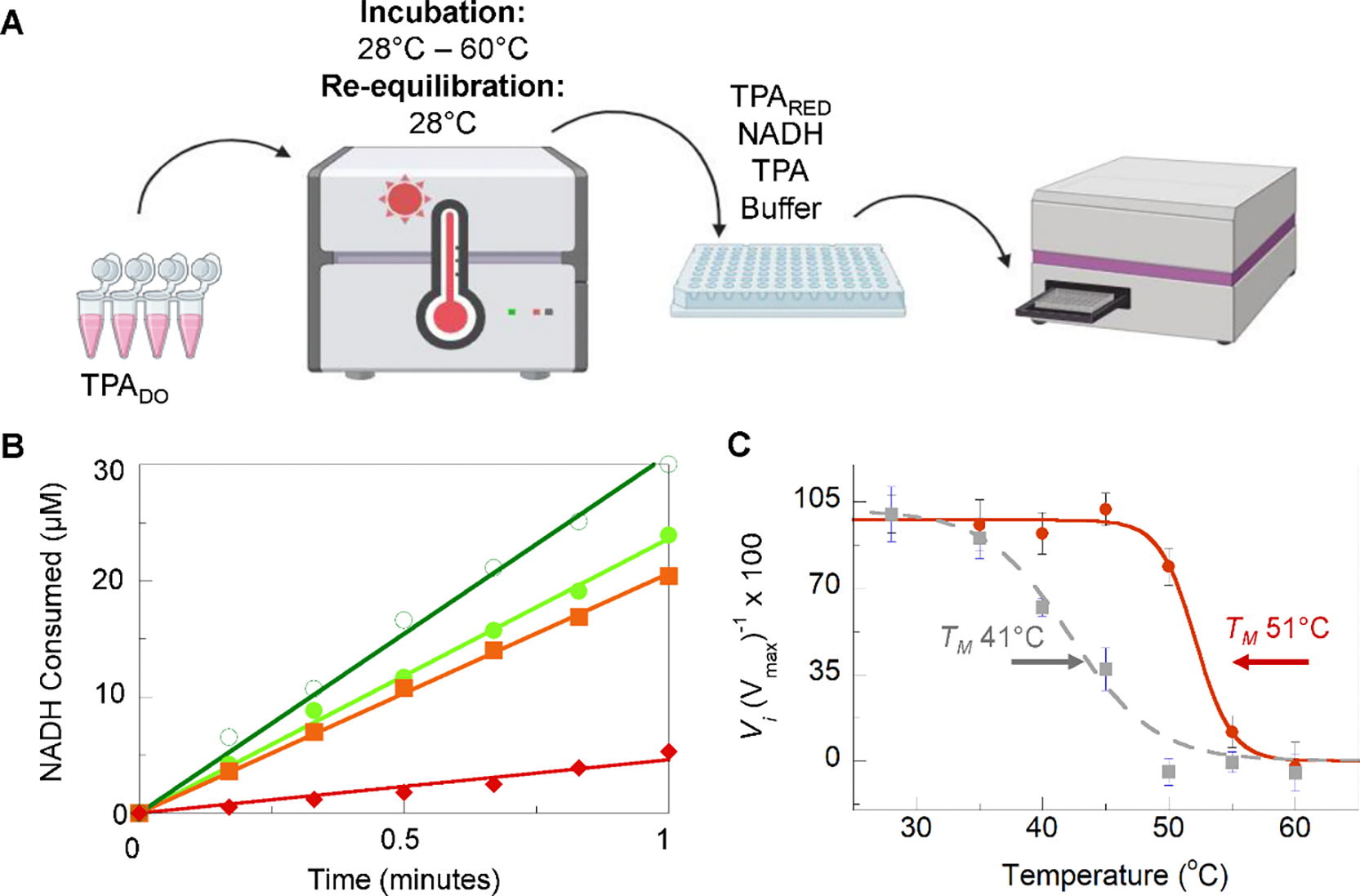
Determination of functional thermotolerance. A. Scheme for measuring temperature-dependent function. TPADO was diluted into a working condition into MOPS buffer then incubated in a thermocycler for 10 minutes at a series of elevated temperatures. The temperature in the thermocycler was then dropped back down to the reaction temperature (28°C) for an additional 10 minutes. The incubated protein was added to reaction wells containing TPARED, substrates NADH and TPA, and a buffer. Reactions were monitored in real-time by change in NADH λmax = 340 nm. This process is repeated for each temperature and can be repeated incubating both TPADO and TPARED. B. Specific activity plots showing the molar consumption of NADH over time for the first minute of reaction for 28°C (dark green, hollow circle), 40°C (light green, filled circle), 50°C (orange, square), and 55°C (red, diamond). C. Relative initial velocity (Vi) for samples when TPADO is inculbated (red, solid line) and when both TPADO and TPARED are incubated (gray, dashed line) where the functional melting temperature (TM) is indicated by arrows.
4.3. Data Analysis
Convert absorbance to concentration using the Beer Lambert law shown in Equation 1 the extinction coefficient 0.00622 μM−1 min−1 for NADH at 340 nm, and a pathlength of 0.779 cm for a ThermoScientific F-bottom 96 well plate filled with 250 μL solution.
- The background NADH hydrolysis (uncoupled to TPA dioxygenation) can be subtracted from the measured time course using the value recorded for the blank well. Initial velocity (vi) is defined as the concentration of NADH consumed over time within the initial, linear portion of the reaction (see note 7). A value for vi is obtained from a line fitted to the points of the [NADH] versus time plot. Since reaction rates are defined as positive, a negative sign is inserted into Equation 3 (see note 8):
Eq. 3 - The initial velocity at a reference temperature (25°C) is associated with 100% activity. Relative activities measured at higher temperatures can be calculated using Equation 4 where is the initial velocity at a given temperature.
Eq. 4 - The resulting reverse sigmoidal curve can be modeled using Equation 5.
Eq. 5
4.4. Notes
For room temperature experiments and those up to 45°C, the VarioSkan model plate reader or those with similar features will be sufficient. At the time of writing, the BioTek Synergy H1 is the only available plate reader with temperature control up to 70°C.
These concentrations have been optimized through a specific activity screen of buffer type, pH, salt concentration, and DMSO concentration. For a 250 μL reaction, 200 μM final concentration NADH nears the UV-Vis saturation limit.
A final reaction concentration of 200 μM for TPA since this is approximately saturating and higher concentrations lead to substrate inhibition.
One or both of the RO system proteins (oxygenase/reductase) can be thermally incubated (Figure 5C).
TPADO has a temperature optimum of 28°C. Thermal incubation for 10 minutes at elevated temperatures was followed by re-equilibration to 28°C prior to activity measurement at 28°C.
The above protocol can additionally be used to identify the temperature optimum of an RO system by performing the kinetic measurements at various temperatures. It is noteworthy that Arrhenius effects will affect the rate of kinetics at elevated temperatures. Additional controls can be used to account for increased background hydrolysis of NADH in the absence of enzymes as well as background hydrolysis due to enzyme activity in the absence of TPA.
With the reported conditions, the linear portion of the reaction for TPADO/TPARED typically consists of the first 60 seconds. A plot of [NADH] over the 10 minute duration of the measurement will assist in identifying appropriate values to use for specific activity.
Specific activity can be derived from this value by referencing the concentration of enzyme active site. Alternatively, values of vi can be compared at a series of temperatures using an equivalent concentration of each enzyme.
5. Lifetime
One of the most useful metrics for applications is enzyme lifetime. Here we describe two methods for determining total turnover number (TTON) for an RO system. First, NADH consumption is monitored by UV-Visible absorbance of NADH at 340 nm. This is limited by the saturation limit of the spectrophotometer, so TPA and NADH must be added periodically until RO activity is no longer observed. In the second method, high substrate loading is used to circumvent the need for repeated substrate addition. Here, residual substrate is quantified by HPLC.
5.1. Equipment
UV-Vis spectrophotometer (Cary 60, Agilent)
2 mL cuvettes
HPLC (LC-300, Shimadzu)
Buffers, Strains, and Reagents
5.2. Procedure
UV-Visible analysis (see note 1)
To a 2 mL cuvette, add 1.7 mL master mix and 100 μL each enzyme.
Record a full spectrum for background subtraction.
Add 50 μL each NADH and TPA. Begin measuring spectra every 5 minutes. To maintain oxygenation of the reaction, gently agitate by pipetting (see note 2).
When the absorbance at 340 nm reaches baseline, add an additional 50 μL NADH and TPA before the next absorbance reading (see note 3, Figure 6).
Periodically add fresh aliquots of NADH and TPA until absorbance does not change after new addition of NADH and TPA.
Figure 6.
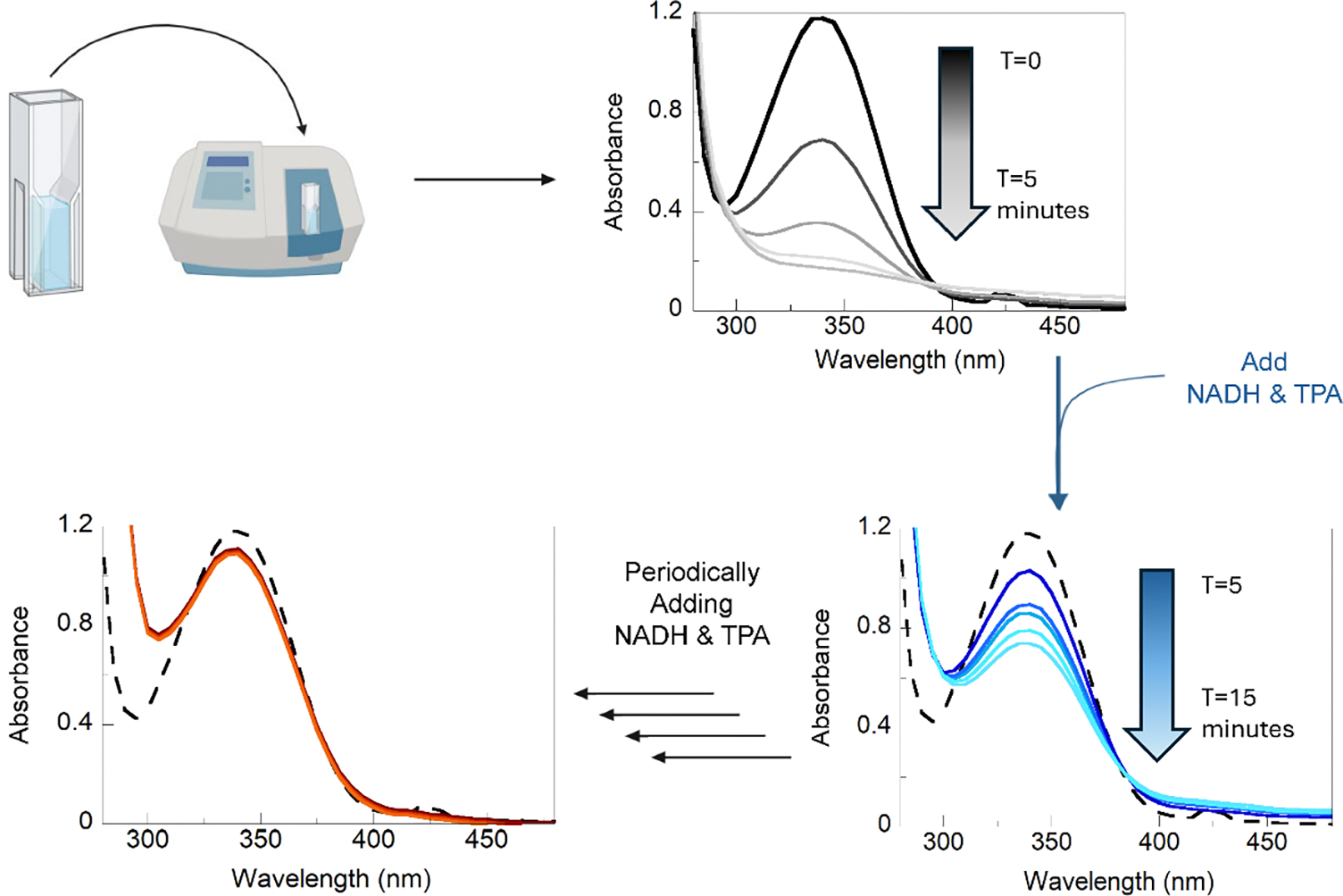
UV-Visible analysis of enzyme lifetime. Function of the RO system is monitored by decrease of signal at the λmax = 340 nm. The reaction containing TPADO, TPARED, and buffer was initiated by addition of 100 μM NADH and TPA. After 5 minutes of continuous measurements, decreased absorbance at 340 nm indicated that the NADH had been consumed, as shown by the black absorbance spectrum. NADH and TPA were then added again in 100 μM increments, as indicated by the blue absorbance spectrum, until no further consumption of NADH was monitored after regeneration (red spectrum).
HPLC analysis (see note 4)
To a 2 mL cuvette, add 1.3 mL master mix and 100 μL of each enzyme.
Add 250 μL each of NADH and TPA. Quickly mix and remove 20 μL from the reaction for a time = 0 HPLC sample. Quench sample in 20 μL ice cold methanol and store at −20°C.
After 5 minutes has elapsed, remove 20 μL from the reaction for a 5 minute HPLC sample. Quench sample in 20 μL ice cold methanol and store at −20°C.
Continue this process removing and quenching HPLC samples every 10 minutes.
5.3. Data Analysis
UV-Visible Analysis
Beer’s law (Equation 1) was used to convert Abs340nm to NADH concentration in μM.
The total amount of NADH consumed was calculated by summing the added volumes and multiplying the total by the concentration of the NADH stock.
TTON was calculated by dividing the moles of NADH consumed by and the moles of TPADO active site.
HPLC analysis
A Shimadzu instrument with a diode array detector and Thermo Scientific™ Hypersil GOLD™ (4.6 mm × 250 mm, 5 μm particle size) column were used. The column was pre-equilibrated in 95% buffer A and 0% buffer B. Buffer A consisted of 0.1% (v/v) TFA (Fisher) in water. Buffer B consisted of 0.1% (v/v) TFA (Fisher) in HPLC grade acetonitrile (Fisher). The reaction components were eluted at a rate of 1 mL min-1 with the following program: 5% B from 0.0 to 10.0 minutes, 0 to 15% B from 10.0 to 18.0 minutes, 15 to 75% B from 18.0 to 24.0 minutes, 75 to 100% B from 24.0 to 24.1 minutes, 100% B from 24.1 to 30.1 minutes, 100% to 0%B from 30.1 to 30.2 minutes, and 0% B from 30.2 to 38.0 minutes. NADH (Αlfa Aesar) and TPA (Sigma-Aldrich), were used to generate standard curves from 1 mM to 50 μM. Openlab PostRun software was used to generate standard curves comparing area under the peak to known substrate concentrations.
Substrate analyses for each sampled timepoint was reported via integration of peaks monitored at 240 nm and retention times of 16.25 and 23.1 min for NADH and TPA, respectively (Figure 6). Sample peak areas were then plotted against the standard curve to report molar concentration.
As with the UV-Visible measurements, TTON was calculated by converting concentration at each segment to moles and dividing by the total moles of the TPADO enzyme active site.
5.4. Notes
Due to substrate inhibition, this experiment is designed to run under low substrate concentration, augmenting substrate periodically as it is consumed. For spectroscopic analysis, NADH concentration is limited to the saturation limit of the spectrophotometer.
Agitation with a stir bar is also acceptable.
This procedure assumes close coupling of the consumption of NADH and TPA; consequently. when NADH is depleted, TPA will be as well. By adding NADH and TPA directly prior to the absorbance measurement, we can confirm the new mixture’s NADH concentration using Beer’s Law.
HPLC and UV-Vis measurements can be performed in tandem; however, it is recommended that UV-Vis is performed first to determine approximate rate of substrate consumption and total reaction time.
6. DSC Differential Scanning Calorimetry
While there are several methods for monitoring loss of protein structure, DSC provides detailed quantification of the dissociation landscape. DSC has long been used to characterize the thermostability of proteins and quantify their folding integrity by reporting variations in heat capacity of a protein sample. Transitions in protein complex dissociation and unfolding are indicated by peaks in heat capacity. The number of peaks, their relative resolution from other peaks, and their areas communicate information about the thermal dissociation profile for a protein sample. Each peak provides a thermal transition midpoint (Tm), and can be used to calculate the apparent enthalpy for the transition. This method can be modified in temperature range as well as the speed at which heat is introduced to the protein so that maximum resolution can be obtained for unique samples.
6.1. Equipment
Differential scanning calorimeter (VP-capillary DSC, MicroCal)
Slide-a-lyzer
Buffers and Reagents
6.2. Procedure
Target protein must be first dialyzed into DSC appropriate buffer (see note 2). This can be done using centrifugation buffer exchange or in a small volume dialysis cassette such as a slide-a-lyzer. Following buffer exchange, protein was concentrated to a final concentration 0.5 mg mL−1.
Prior to experimentation, both buffer and protein samples must be degassed with stirring under vacuum pressure.
Define the parameters for the DSC experiment. Here, we set the starting temperature to 25°C and final temperature to 100°C with a pre-scan incubation of 5 minutes. The scans performed here were taken at a rate of 90°C hr−1 (See note 3).
Triplicate measurements for the protein sample as well as the DSC buffer were taken. Background thermal changes observed in the buffer sample were subtracted from the protein samples as a reference.
6.3. Data Analysis
Subtract background (buffer) data from experimental data.
Normalize the data for concentration by dividing by the number of moles of the sample in the cell.
To create a baseline, a software such as Origin lets you adjust the left and right linear segments and allows you to connect the segments.
In Origin, the data is fit by choosing a model, either two state or non-two state. The models use the Levenberg/Marquardt (LM) non-linear least-square method.
Some programs provide fit models to auto-integrate DSC data to report melting temperature (TM), heat capacity ΔCp and enthalpy (ΔH). (See note 4)
The raw data exported from the cap-DSC included temperature (°C), sample weight (mole), and heat flow (kcal s−1). To determine enthalpy, we generated a plot of [Heat flow per weight] on the y-axis and time on the x-axis. The transition state peaks of this plot can then be integrated to report enthalpy (ΔH) in units of kcal mole−1.
Heat capacity for the system was calculated by dividing the enthalpy by temperature. Plotting heat capacity as a function of temperature can be used to find specific heat capacity for each transition state at the TM.
6.4. Notes
The VP-Capillary DSC with its autosampling, higher scan rates, and higher temperature capabilities is ideal for protein and therefore we limit our discussions in this section to this instrument, although many considerations apply to the conventional DSC as well.
Some buffers, such as Tris, have a low heat of ionization and a high temperature dependent pKa and are consequently inappropriate choices for temperature dependent experiments. Additionally, the chosen buffer must be suitable for long-term stability of the protein. Here, we conducted an evaluation of activity following buffer exchange. A steady-state assay as described in section 4 was measured for 10 minutes every hour for five hours following buffer exchange. Here, we evaluated HEPES, and various concentrations of potassium phosphate (20–100 mM), using MOPS as a control. For TPADO, 20 mM potassium phosphate was determined to have the most maintained activity for the pH-stable buffer options.
The scan-rate parameter will affect the sensitivity and resolution of the DSC data. Higher scan rates (the VP-Capillary DSC can go up to 200°C/h, the conventional equivalent has a maximum rate of 90°C/h) offer higher sensitivity and lower noise, but can lead to lost resolution or loss of equilibrium as compared to rates at a slow scan rate (<60°C/h).
The DSC chromatograms for TPADO had significant overlap in the two-phase change peaks. The modeling software was used to deconvolute this overlap and give approximate peak areas for each peak.
Figure 7.
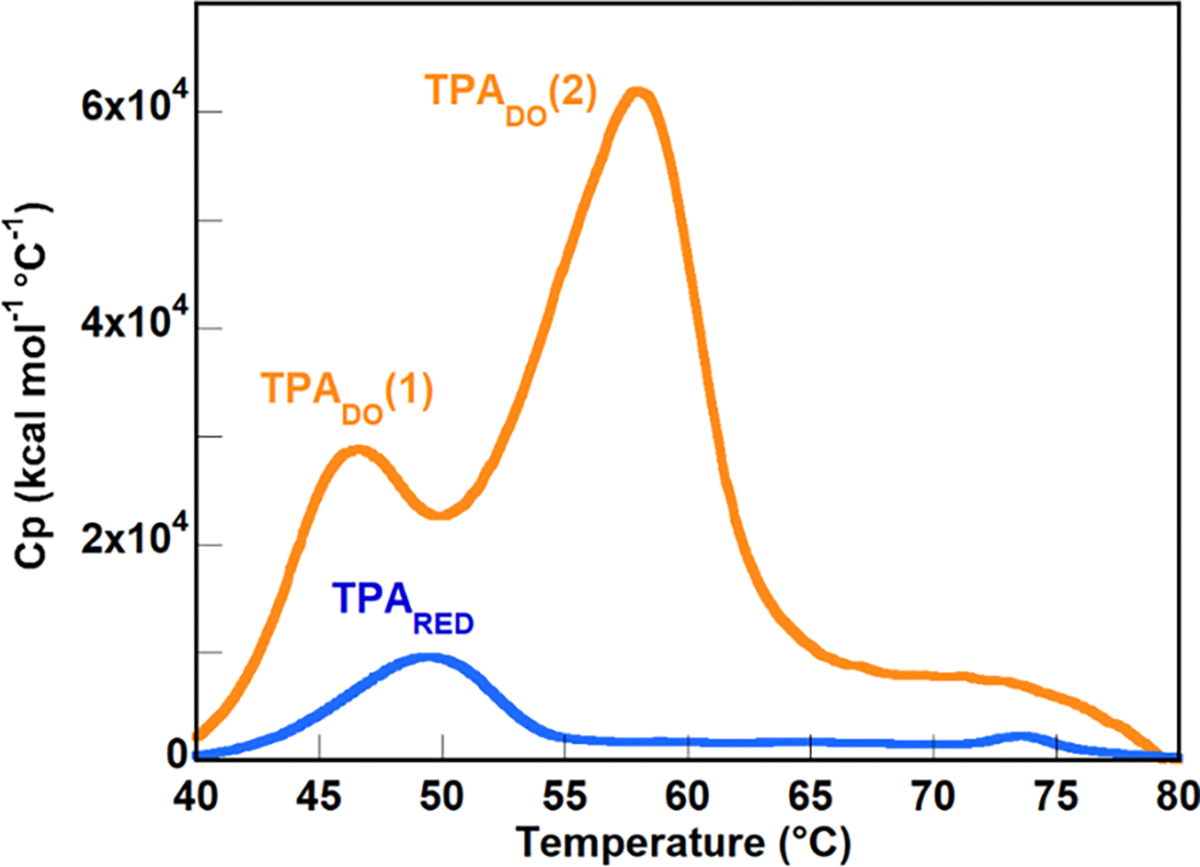
The DSC plot for the thermal dissociation of TPADO contains two distinct but overlapping peaks indicating a non-concerted melting pattern. The first, smaller transition peaks at 48°C while the second, much larger transition has a peak at 58°C. Origin software is used to deconvolute the overlapping area for the two peaks to report TM, ΔCp, and ΔH for each peak.
References
- Ang EL, Obbard JP, & Zhao H (2009). Directed evolution of aniline dioxygenase for enhanced bioremediation of aromatic amines. Applied Microbiol Biotechnology, 81(6), 1063–1070. 10.1007/s00253-008-1710-0 [DOI] [PubMed] [Google Scholar]
- Arana-Pena S, Carballares D, Morellon-Sterlling R, Berenguer-Murcia A, Alcantara AR, Rodrigues RC, & Fernandez-Lafuente R (2020). Enzyme co-immobilization: Always the biocatalyst designers’ choice...or not? Biotechnology Advances, 107584. 10.1016/j.biotechadv.2020.107584 [DOI] [PubMed] [Google Scholar]
- Axcell BC, & Geary PJ (1975). Purification and Some Properties of a Soluble Benzene-Oxidizing System from a Strain of Pseudomonas. Biochemistry Journal, 146, 173–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balcão VM, Mateo C, Fernández-Lafuente R, Malcata FX, & Guisán JM (2001). Structural and Functional Stabilization of L-Asparaginase via Multisubunit Immobilization onto Highly Activated Supports. Biotechnology Progress, 17(3), 537–542. 10.1021/bp000163r [DOI] [PubMed] [Google Scholar]
- Balcão VM, & Vila MM (2015). Structural and functional stabilization of protein entities: state-of-the-art. Advances in Drug Delivery Reviews, 93, 25–41. 10.1016/j.addr.2014.10.005 [DOI] [PubMed] [Google Scholar]
- Bernhardt F-H, & Meisch H-U (1980). Reactivation studies on putidamonooxin — The monooxygenase of a 4-methoxybenzoate o-demethylase from Pseudomonas putida. Biochemical and Biophysical Research Communications, 93(4), 1247–1253. 10.1016/0006-291X(80)90623-3 [DOI] [PubMed] [Google Scholar]
- Brimberry M, Garcia AA, Liu J, Tian J, & Bridwell-Rabb J (2023). Engineering Rieske oxygenase activity one piece at a time. Current Opinions in Chemical Biology, 72, 102227. 10.1016/j.cbpa.2022.102227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capyk JK, D’Angelo I, Strynadka NC, & Eltis LD (2009). Characterization of 3-Ketosteroid 9α-Hydroxylase, a Rieske Oxygenase in the Cholesterol Degradation Pathway of Mycobacterium tuberculosis. Journal of Biological Chemistry, 284(15), 9937–9946. 10.1074/jbc.M900719200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carvalho JW, Santiago PS, Batista T, Salmon CE, Barbosa LR, Itri R, & Tabak M (2012). On the temperature stability of extracellular hemoglobin of Glossoscolex paulistus, at different oxidation states: SAXS and DLS studies. Biophysical Chemistry, 163–164, 44–55. 10.1016/j.bpc.2012.02.004 [DOI] [PubMed] [Google Scholar]
- Chakraborty J, Suzuki-Minakuchi C, Okada K, & Nojiri H (2017). Thermophilic bacteria are potential sources of novel Rieske non-heme iron oxygenases. AMB Express, 7(1), 17. 10.1186/s13568-016-0318-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Correll CC, Batiet CJ, Ballou DP, & Ludwig ML (1992). Phthalate Dioxygenase Reductase: A Modular Structure for Electron Transfer from Pyridine Nucleotides to [2Fe-2S]. Science, 258, 1604–1609. [DOI] [PubMed] [Google Scholar]
- Das R, & Gerstein M (2000). The stability of thermophilic proteins: a study based on comprehensive genome comparison. Functional Integrated Genomics, 1(1), 76–88. 10.1007/s101420000003 [DOI] [PubMed] [Google Scholar]
- Daughtry KD, Xiao Y, Stoner-Ma D, Cho E, Orville AM, Liu P, & Allen KN (2012). Quaternary Ammonium Oxidative Demethylation: X-ray Crystallographic, Resonance Raman, and UV–Visible Spectroscopic Analysis of a Rieske-Type Demethylase. Journal of the American Chemical Society, 134(5), 2823–2834. 10.1021/ja2111898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong X, Fushinobu S, Fukuda E, Terada T, Nakamura S, Shimizu K, Nojiri H, Omori T, Shoun H, & Wakagi T (2005). Crystal structure of the terminal oxygenase component of cumene dioxygenase from Pseudomonas fluorescens IP01. Journal of Bacteriology, 187(7), 2483–2490. 10.1128/JB.187.7.2483-2490.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dumitru R, Jiang WZ, Weeks DP, & Wilson MA (2009). Crystal Structure of Dicamba Monooxygenase: A Rieske Nonheme Oxygenase that Catalyzes Oxidative Demethylation. Journal of Molecular Biology, 392(2), 498–510. 10.1016/j.jmb.2009.07.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fee JA, Findling KL, Yoshida T, Hille R, Tarr GE, Hearshen DO, Dunham WR, Day EP, Kent TA, & Münck E (1984). Purification and characterization of the Rieske iron-sulfur protein from Thermus thermophilus. Evidence for a [2Fe-2S] cluster having non-cysteine ligands. Journal of Biological Chemistry, 259(1), 124–133. 10.1016/s0021-9258(17)43630-1 [DOI] [PubMed] [Google Scholar]
- Flores H, & Ellington AD (2002). Increasing the thermal stability of an oligomeric protein, beta-glucuronidase. Journal of Molecular Biology, 315(3), 325–337. 10.1006/jmbi.2001.5223 [DOI] [PubMed] [Google Scholar]
- Friemann R, Ivkovic-Jensen MM, Lessner DJ, Yu CL, Gibson DT, Parales RE, Eklund H, & Ramaswamy S (2005). Structural insight into the dioxygenation of nitroarene compounds: the crystal structure of nitrobenzene dioxygenase. Journal of Molecular Biology, 348(5), 1139–1151. 10.1016/j.jmb.2005.03.052 [DOI] [PubMed] [Google Scholar]
- Furusawa Y, Nagarajan V, Tanokura M, Masai E, Fukuda M, & Senda T (2004). Crystal structure of the terminal oxygenase component of biphenyl dioxygenase derived from Rhodococcus sp. strain RHA1. Journal of Molecular Biololgy, 342(3), 1041–1052. 10.1016/j.jmb.2004.07.062 [DOI] [PubMed] [Google Scholar]
- Gakhar L, Malik ZA, Allen CC, Lipscomb DA, Larkin MJ, & Ramaswamy S (2005). Structure and increased thermostability of Rhodococcus sp. naphthalene 1,2-dioxygenase. Journal of Bacteriology, 187(21), 7222–7231. 10.1128/JB.187.21.7222-7231.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibson DT, Cardini GE, Máseles FC, & Kallio RE (1970). Incorporation of Oxygen-18 into Benzene by Pseudomonas putida. Biochemistry, 9, 1631–1635. [DOI] [PubMed] [Google Scholar]
- Gibson DT, Koch JR, Schuld CL, & Kallio RE (1968). Oxidative Degradation of Aromatic Hydrocarbons by Microorganisms. II. Metabolism of Halogenated Aromatic Hydrocarbons. Biochemistry, 7, 3795–3802. [DOI] [PubMed] [Google Scholar]
- Goldenzweig A, & Fleishman SJ (2018). Principles of Protein Stability and Their Application in Computational Design. Annual Reviews in Biochemistry, 87, 105–129. 10.1146/annurev-biochem-062917-012102 [DOI] [PubMed] [Google Scholar]
- Hobisch M, Holtmann D, Gomez de Santos P, Alcalde M, Hollmann F, & Kara S (2021). Recent developments in the use of peroxygenases - Exploring their high potential in selective oxyfunctionalisations. Biotechnology Advances, 51, 107615. 10.1016/j.biotechadv.2020.107615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou Y-J, Guo Y, Li D-F, & Zhou N-Y (2021). Structural and Biochemical Analysis Reveals a Distinct Catalytic Site of Salicylate 5-Monooxygenase NagGH from Rieske Dioxygenases. Applied and Environmental Microbiology, 87(6), e01629–01620. 10.1128/AEM.01629-20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudlicky T, Fan R, Luna H, Olivo H, & Price J (1992). Enzymatic hydroxylation of arene and symmetry considerations in efficient synthetic design of oxygenated natural products. Pure and Applied Chemistry, 64(8), 1109–1113. 10.1351/pac199264081109 (Pure and Applied Chemistry) [DOI] [Google Scholar]
- Hudlicky T, Gonzalez D, & Gibson D (1999). Enzymatic dihydroxylation of aromatics in enantioselective synthesis: Expanding asymmetric methodology. Aldrichimica acta, 32, 35–62. [Google Scholar]
- Hudlicky T, & Reed JW (2009). Applications of biotransformations and biocatalysis to complexity generation in organic synthesis [ 10.1039/B901172M]. Chemical Society Reviews, 38(11), 3117–3132. 10.1039/B901172M [DOI] [PubMed] [Google Scholar]
- Inoue K, Usami Y, Ashikawa Y, Noguchi H, Umeda T, Yamagami-Ashikawa A, Horisaki T, Uchimura H, Terada T, Nakamura S, Shimizu K, Habe H, Yamane H, Fujimoto Z, & Nojiri H (2014). Structural Basis of the Divergent Oxygenation Reactions Catalyzed by the Rieske Nonheme Iron Oxygenase Carbazole 1,9a-Dioxygenase. Applied and Environmental Microbiology, 80(9), 2821–2832. 10.1128/AEM.04000-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaenicke R, & Böhm G (1998). The stability of proteins in extreme environments. Current Opinions in Structural Biology, 8(6), 738–748. 10.1016/S0959-440X(98)80094-8 [DOI] [PubMed] [Google Scholar]
- Jaenicke R, Schurig H, Beaucamp N, & Ostendorp R (1996). Structure and Stability of Hyperstable Proteins: Glycolytic Enzymes From Hyperthermophilic Bacterium Thermotoga Maritima. In Richards FM, Eisenberg DS, & Kim PS (Eds.), Advances in Protein Chemistry (Vol. 48, pp. 181–269). Academic Press. 10.1016/S0065-3233(08)60363-0 [DOI] [PubMed] [Google Scholar]
- Jakoncic J, Jouanneau Y, Meyer C, & Stojanoff V (2007). The catalytic pocket of the ring-hydroxylating dioxygenase from Sphingomonas CHY-1. Biochemical and Biophysical Research Communications, 352(4), 861–866. 10.1016/j.bbrc.2006.11.117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jerina DM, Daly JW, Jeffrey AM, & Gibson DT (1971). Cis-1,2-dihydroxy-1,2-dihydronaphthalene: A bacterial metabolite from naphthalene. Archives of Biochemistry and Biophysics, 142(1), 394–396. [DOI] [PubMed] [Google Scholar]
- Rieske John S., MacIennan DH., & Colema R. (1964). Isolation and properties of an iron-protein from the (reduced coenzyme Q)-cytochrom C reductase complex of the respiration chain. Biochemical and Biophysical Research Communications, 15, 338–344. [Google Scholar]
- Karlsson A, Parales JV, Parales RE, Gibson DT, Eklund H, & Ramaswamy S (2003). Crystal Structure of Naphthalene Dioxygenase: Side-on Binding of Dioxygen to Iron. Science, 299. [DOI] [PubMed] [Google Scholar]
- Kauppi B, Lee K, Carredano E, Parales RE, Gibson DT, Eklund H, & Ramaswamy S (1997). Structure of an aromatic-ring-hydroxylating dioxygenase— naphthalene 1,2-dioxygenase. Structure, 6, 571–586. [DOI] [PubMed] [Google Scholar]
- Kincannon WM, Zahn M, Clare R, Lusty Beech J, Romberg A, Larson J, Bothner B, Beckham GT, McGeehan JE, & DuBois JL (2022). Biochemical and structural characterization of an aromatic ring-hydroxylating dioxygenase for terephthalic acid catabolism. Proceedings of the National Academy of Sciences of the United States of America, 119(13), e2121426119. 10.1073/pnas.2121426119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar P, Mohammadi M, Viger JF, Barriault D, Gomez-Gil L, Eltis LD, Bolin JT, & Sylvestre M (2011). Structural insight into the expanded PCB-degrading abilities of a biphenyl dioxygenase obtained by directed evolution. Journal of Molecular Biology, 405(2), 531–547. 10.1016/j.jmb.2010.11.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar S (2010). Engineering cytochrome P450 biocatalysts for biotechnology, medicine and bioremediation. Expert Opinions on Drug Metabolism and Toxicology, 6(2), 115–131. 10.1517/17425250903431040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumari A, Singh D, Ramaswamy S, & Ramanathan G (2017). Structural and functional studies of ferredoxin and oxygenase components of 3-nitrotoluene dioxygenase from Diaphorobacter sp strain DS2. PLoS One, 12(4), Article e0176398. 10.1371/journal.pone.0176398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lessner DJ, Johnson GR, Parales RE, Spain JC, & Gibson DT (2002). Molecular characterization and substrate specificity of nitrobenzene dioxygenase from Comamonas sp. strain JS765. Applied Environmental Microbiology, 68(2), 634–641. 10.1128/AEM.68.2.634-641.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lisi GP, Png CYM, & Wilcox DE (2014). Thermodynamic Contributions to the Stability of the Insulin Hexamer. Biochemistry, 53(22), 3576–3584. 10.1021/bi401678n [DOI] [PubMed] [Google Scholar]
- Liu J, Tian J, Perry C, Lukowski AL, Doukov TI, Narayan ARH, & Bridwell-Rabb J (2022). Design principles for site-selective hydroxylation by a Rieske oxygenase. Nature Communications, 13(1), 255. 10.1038/s41467-021-27822-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukowski AL, Liu J, Bridwell-Rabb J, & Narayan ARH (2020). Structural basis for divergent C-H hydroxylation selectivity in two Rieske oxygenases. Nature Communications, 11(1), 2991. 10.1038/s41467-020-16729-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahto JK, Neetu N, Sharma M, Dubey M, Vellank BP, & Kumar P (2022). Structural Insights into Dihydroxylation of Terephthalate, a Product of Polyethylene Terephthalate Degradation. Journal of Bacteriology, 204(3). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahto JK, Neetu N, Waghmode B, Kuatsjah E, Sharma M, Sircar D, Sharma AK, Tomar S, Eltis LD, & Kumar P (2021). Molecular insights into substrate recognition and catalysis by phthalate dioxygenase from Comamonas testosteroni. Journal of Biological Chemistry, 297(6), 101416. 10.1016/j.jbc.2021.101416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez-Martinez M, Lores I, Pena-Garcia C, Bargiela R, Reyes-Duarte D, Guazzaroni ME, Pelaez AI, Sanchez J, & Ferrer M (2014). Biochemical studies on a versatile esterase that is most catalytically active with polyaromatic esters. Microbial Biotechnology, 7(2), 184–191. 10.1111/1751-7915.12107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martins BM, Svetlitchnaia T, & Dobbek H (2005). 2-Oxoquinoline 8-Monooxygenase Oxygenase Component: Active Site Modulation by Rieske-[2Fe-2S] Center Oxidation/Reduction. Structure, 13(5), 817–824. 10.1016/j.str.2005.03.008 [DOI] [PubMed] [Google Scholar]
- Massmig M, Reijerse E, Krausze J, Laurich C, Lubitz W, Jahn D, & Moser J (2020). Carnitine metabolism in the human gut: characterization of the two-component carnitine monooxygenase CntAB fromAcinetobacter baumannii. Journal of Biological Chemistry, 295(37), 13065–13078. 10.1074/jbc.RA120.014266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neidle EL, Hartnett C, Ornston LN, Bairoch A, Rekik M, & Harayama S (1991). Nucleotide sequences of the Acinetobacter calcoaceticus benABC genes for benzoate 1,2 diozygenase reveal evolutionary relationships among multicomponent oxygenases Journal of Bacteriology, 173(17), 5385–5395. 10.1128/jb.173.17.5385-5395.1991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noble JE (2014). Quantification of protein concentration using UV absorbance and Coomassie dyes. Methods in Enzymology, 536, 17–26. 10.1016/B978-0-12-420070-8.00002-7 [DOI] [PubMed] [Google Scholar]
- Özgen FF, & Schmidt S (2019). Rieske Non-Heme Iron Dioxygenases: Applications and Future Perspectives. In Biocatalysis (pp. 57–82). 10.1007/978-3-030-25023-2_4 [DOI] [Google Scholar]
- Parales JV, Parales RE, Resnik SM, & Gibson DT (1998). Enzyme Specificity of 2-Nitrotoluene 2,3-Dioxygenase from Pseudomonas sp. Strain JS42 Is Determined by the C-Terminal Region of the α Subunit of the Oxygenase Component. Journal of Bacteriology, 180, 1194–1199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parales RE (2003). The role of active-site residues in naphthalene dioxygenase. Journal of Industrial Microbiology and Biotechnology, 30(5), 271–278. 10.1007/s10295-003-0043-3 [DOI] [PubMed] [Google Scholar]
- Parales RE, Lee K, Resnick SM, Jiang H, Lessner DJ, & Gibson DT (2000). Substrate Specificity of Naphthalene Dioxygenase: Effect of Specific Amino Acids at the Active Site of the Enzyme. Journal of Bacteriology 82, 1641–1649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parales RE, Resnick SM, Sharma ND, Yu C-L, Boyd DR, & Gibson DT (2000). Regioselectivity and Enantioselectivity of Naphthalene Dioxygenase during Arene cis-Dihydroxylation: Control by Phenylalanine 352 in the alpht Subunit. Journal of Bacteriology 182, 5495–5504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park C, & Marqusee S (2004). Analysis of the stability of multimeric proteins by effective DeltaG and effective m-values. Protein Sci, 13(9), 2553–2558. 10.1110/ps.04811004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quareshy M, Shanmugam M, Townsend E, Jameson E, Bugg TDH, Cameron AD, & Chen Y Structural basis of carnitine monooxygenase CntA substrate specificity, inhibition, and intersubunit electron transfer. Journal of Biological Chemistry 296, 100038. 10.1074/jbc.RA120.016019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rieske JS, Hansen RE, & Zaugg WS (1964). Studies on the Electron Transfer System. Journal of Biological Chemistry, 239(9), 3017–3022. 10.1016/s0021-9258(18)93846-9 [DOI] [PubMed] [Google Scholar]
- Suen W-C, & Gibson DT (1993). Isolation and Preliminary Characterization of the Subunits of the Terminal Component of Naphthalene Dioxygenase from Pseudomonas putida NCIB 9816–4. Journal of Bacteriology 175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarasev M, Pullela S, & Ballou DP (2009). Distal end of 105–125 loop - A putative reductase binding domain of phthalate dioxygenase. Archives of Biochemistry and Biophysics, 487(1), 10–18. 10.1016/j.abb.2009.05.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson MP, Peñafiel I, Cosgrove SC, & Turner NJ (2018). Biocatalysis Using Immobilized Enzymes in Continuous Flow for the Synthesis of Fine Chemicals. Organic Process Research & Development, 23(1), 9–18. 10.1021/acs.oprd.8b00305 [DOI] [Google Scholar]
- Tiwari MK, Lee J-K, Moon H-J, & Zhao H (2011). Further biochemical studies on aminopyrrolnitrin oxygenase (PrnD). Bioorganic & Medicinal Chemistry Letters, 21(10), 2873–2876. 10.1016/j.bmcl.2011.03.087 [DOI] [PubMed] [Google Scholar]
- Tsai P-C, Chakraborty J, Suzuki-Minakuchi C, Terada T, Kotake T, Matsuzawa J, Okada K, & Nojiri H (2022). The a- and b-Subunit Boundary at the Stem of the MushroomLike a3 b3-Type Oxygenase Component of Rieske Non-Heme Iron Oxygenases Is the Rieske-Type Ferredoxin-Binding Site. Applied and Environmental Microbiology, 88(15). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsang HT, Batie CJ, Ballou DP, & Penner-Hahn JE (1989). X-ray absorption spectroscopy of the [2-iron-2-sulfur] Rieske cluster in Pseudomonas cepacia phthalate dioxygenase. Determination of core dimensions and iron ligation. Biochemistry, 28(18), 7233–7240. 10.1021/bi00444a015 [DOI] [PubMed] [Google Scholar]
- Wolfe MD, Altier DJ, Stubna A, Popescu CV, Münck, E, & Lipscomb JD (2002). Benzoate 1,2-Dioxygenase from Pseudomonas putida: Single Turnover Kinetics and Regulation of a Two-Component Rieske Dioxygenase†. Biochemistry, 41, 9611–9626. [DOI] [PubMed] [Google Scholar]
- Wolfe MD, & Lipscomb JD (2003). Hydrogen peroxide-coupled cis-diol formation catalyzed by naphthalene 1,2-dioxygenase. Journal of Biological Chemistry, 278(2), 829–835. 10.1074/jbc.M209604200 [DOI] [PubMed] [Google Scholar]
- Zhu Y, Jameson E, Crosatti M, Schafer H, Rajakumar K, Bugg TD, & Chen Y (2014). Carnitine metabolism to trimethylamine by an unusual Rieske-type oxygenase from human microbiota. Proceedings of the National Academy of Science of the United States of America, 111(11), 4268–4273. 10.1073/pnas.1316569111 [DOI] [PMC free article] [PubMed] [Google Scholar]