

Abstract
Aims/hypothesis
All forms of diabetes result from insufficient functional beta cell mass. Due to the relatively limited expression of several antioxidant enzymes, beta cells are highly vulnerable to pathological levels of reactive oxygen species (ROS), which can lead to the reduction of functional beta cell mass. During early postnatal ages, both human and rodent beta cells go through a burst of proliferation that quickly declines with age. The exact mechanisms that account for neonatal beta cell proliferation are understudied but mitochondrial release of moderated ROS levels has been suggested as one of the main drivers. We previously showed that, apart from its conventional role in protecting beta cells from oxidative stress, the nuclear factor erythroid 2-related factor 2 (NRF2) is also essential for beta cell proliferation. We therefore hypothesised that NRF2, which is activated by ROS, plays an essential role in beta cell proliferation at early postnatal ages.
Methods
Beta cell NRF2 levels and beta cell proliferation were measured in pancreatic sections from non-diabetic human cadaveric donors at different postnatal ages, childhood and adulthood. Pancreatic sections from 1-, 7-, 14- and 28-day-old beta cell-specific Nrf2 (also known as Nfe2l2)-knockout mice (βNrf2KO) or control (Nrf2lox/lox) mice were assessed for beta cell NRF2 levels, beta cell proliferation, beta cell oxidative stress, beta cell death, nuclear beta cell pancreatic duodenal homeobox protein 1 (PDX1) levels and beta cell mass. Seven-day-old βNrf2KO and Nrf2lox/lox mice were injected daily with N-acetylcysteine (NAC) or saline (154 mmol/l NaCl) to explore the potential contribution of oxidative stress to the phenotypes seen in βNrf2KO mice at early postnatal ages. RNA-seq was performed on 7-day-old βNrf2KO and Nrf2lox/lox mice to investigate the mechanisms by which NRF2 stimulates beta cell proliferation at early postnatal ages. Mitochondrial biogenesis and function were determined using dispersed islets from 7-day-old βNrf2KO and Nrf2lox/lox mice by measuring MitoTracker intensity, mtDNA/gDNA ratio and ATP/ADP ratio. To study the effect of neonatal beta cell-specific Nrf2 deletion on glucose homeostasis in adulthood, blood glucose, plasma insulin and insulin secretion were determined and a GTT was performed on 3-month-old βNrf2KO and Nrf2lox/lox mice fed on regular diet (RD) or high-fat diet (HFD).
Results
The expression of the master antioxidant regulator NRF2 was increased at early postnatal ages in both human (1 day to 19 months old, 31%) and mouse (7 days old, 57%) beta cells, and gradually declined with age (8% in adult humans, 3.77% in adult mice). A significant correlation (R2=0.568; p=0.001) was found between beta cell proliferation and NRF2 levels in human beta cells. Seven-day-old βNrf2KO mice showed reduced beta cell proliferation (by 65%), beta cell nuclear PDX1 levels (by 23%) and beta cell mass (by 67%), and increased beta cell oxidative stress (threefold) and beta cell death compared with Nrf2lox/lox control mice. NAC injections increased beta cell proliferation in 7-day-old βNrf2KO mice (3.4-fold) compared with saline-injected βNrf2KO mice. Interestingly, RNA-seq of islets isolated from 7-day-old βNrf2KO mice revealed reduced expression of mitochondrial RNA genes and genes involved in the electron transport chain. Islets isolated from 7-day old βNrf2KO mice presented reduced MitoTracker intensity (by 47%), mtDNA/gDNA ratio (by 75%) and ATP/ADP ratio (by 68%) compared with islets from Nrf2lox/lox littermates. Lastly, HFD-fed 3-month-old βNrf2KO male mice displayed a significant reduction in beta cell mass (by 35%), a mild increase in non-fasting blood glucose (1.2-fold), decreased plasma insulin (by 14%), and reduced glucose tolerance (1.3-fold) compared with HFD-fed Nrf2lox/lox mice.
Conclusions/interpretation
Our study highlights NRF2 as an essential transcription factor for maintaining neonatal redox balance, mitochondrial biogenesis and function and beta cell growth, and for preserving functional beta cell mass in adulthood under metabolic stress.
Data availability
Sequencing data are available in the NCBI Gene Expression Omnibus, accession number GSE242718 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE242718).
Keywords: Beta cell death, Beta cell proliferation, Diabetes, Mitochondria, Neonatal development, NRF2, Oxidative stress, Redox balance
Introduction
Since all forms of diabetes are associated with insufficient functional beta cell mass, many efforts are focused on discovering the mechanisms that regulate beta cell expansion and function [1]. Type 2 diabetes is often associated with increased oxidative stress, a condition where supraphysiological levels of reactive oxygen species (ROS) are generated. Prolonged exposure to high ROS concentrations leads to diminished insulin content, defective insulin secretion, decreased beta cell identity and reduced beta cell proliferation and even death, all of which result in the reduction of functional beta cell mass [1, 2]. On the other hand, moderate ROS levels are important for maintaining beta cell function beta cell identity and beta cell proliferation [2–4]. This raises the question of how beta cells maintain proper redox balance.
Nuclear factor erythroid 2-related factor 2 (NRF2, encoded by Nrf2 [also known as Nfe2l2]), plays a key role managing oxidative stress. Upon any increase in ROS levels, the NRF2 pathway is activated, turning on the expression of multiple ROS-detoxifying enzymes to prevent oxidative stress within the beta cells. Once ROS levels are back within the physiological range, the NRF2 inhibitor (Kelch-like ECH-associated protein 1 [KEAP1]) takes control, restoring the normal NRF2 protein levels using the proteasomal machinery [1, 2]. Interestingly, several SNPs in the NRF2 pathway have been linked to diabetes [5–9]. We previously showed that apart from its conventional role in protecting beta cells from oxidative stress, NRF2 is also essential for adaptive beta cell proliferation under situations of metabolic stress, such as high glucose and obesogenic diet [1].
During early postnatal ages, both human and rodent beta cells undergo a burst of proliferation that quickly declines with age [10–12]. Although the mechanisms that regulate neonatal beta cell proliferation are not completely understood, this early burst of proliferation is the major contributor towards reaching an appropriate functional beta cell mass later in adulthood, to supply sufficient insulin under conditions of metabolic stress [13]. Interestingly, mitochondrial release of moderated ROS levels has been suggested as one of the main drivers for neonatal beta cell proliferation [4].
Here, we address the novel role of NRF2 in neonatal beta cell proliferation and its impact on compensatory beta cell mass expansion and glucose homeostasis in adult mice with overnutrition.
Methods
Mouse models
Beta cell-specific Nrf2-knockout (βNrf2KO) mice were generated by crossing male Ins1Cre mice [14] (B6(Cg)-Ins1tm1.1(cre)Thor/J, The Jackson Laboratory, USA; https://www.jax.org/strain/026801, RRID: IMSR_JAX:026801) with female Nrf2lox/lox mice [15] (C57BL/6-Nfe2l2tm1.1Sred/SbisJ, The Jackson Laboratory, USA; https://www.jax.org/strain/025433, RRID: IMSR_JAX:025433). Ins1Cre mice were kept in a heterozygous state. Both male mice and female mice were used in most experiments. Adult 2-month-old mice were randomly placed for 1 month on a lard-based high-fat diet (HFD) (41% energy from fat; no. TD 96001; Harlan Teklad) or a regular diet (RD) (13.1% energy from fat; Purina PicoLab 5053; LabDiet). Newborn mice were given an s.c. injection of 75 mg/kg N-acetylcysteine (NAC) or saline (154 mmol/l NaCl) daily for 7 days as previously described [16]. All studies were performed with the approval of and in accordance with guidelines established by the institutional animal care and use committee of the Icahn School of Medicine at Mount Sinai.
Mouse islet isolation
Islets were isolated from adult mice as previously reported [1]. Islets were isolated from neonatal mice after collagenase P injection into the pancreas, followed by hand picking of islets. See ESM Methods for details.
Immunostaining
Paraffin-embedded mouse pancreatic sections were immunolabelled to assess the levels of beta cell proliferation, beta cell mass, NRF2, DNA and RNA oxidative damage, pancreatic duodenal homeobox protein 1 (PDX1), MAF bZIP transcription factor A (MAFA) and cyclin D1 in insulin-positive cells See electronic supplementary material (ESM) Methods for details. The list of antibodies used in this study is listed in ESM Table 1.
TUNEL assay
TUNEL labelling was performed according to the manufacturer instructions, using the DeadEnd Fluorometric TUNEL System (G3250; Promega). Samples were immunostained with insulin antibody. The percentage of TUNEL-positive/total insulin-positive cells was calculated.
Determination of mitochondrial biogenesis and function
Dispersed islets from mice were pretreated with MitoTracker Orange CM-H2TMRos (M7511; Invitrogen) followed by immunostaining with insulin antibody. The ratio of mitochondrial DNA (mtDNA)/genomic DNA (gDNA) was determined by quantitative real-time PCR (qPCR) using primers previously described in [17] (ESM Table 2). The ATP/ADP ratio was determined using ADP/ATP ratio assay kit (ab65313; Abcam).
qPCR
Total RNA was extracted from islets isolated from mice and qPCR was performed as previously described [1]. See ESM Methods for details. Primers sequences are listed in ESM Table 2.
RNA isolation and RNA-seq
RNA was extracted from 7-day-old Nrf2lox/lox and βNrf2KO mouse islets to generate four pools of five mice each. Samples were then sequenced. See ESM Methods for details.
Glucose homeostasis and insulin content
Blood glucose, IPGTT, ITT, glucose-stimulated insulin secretion (GSIS) and insulin content measurements were performed as previously described [1]. See ESM Methods for details.
Statistical analysis
Data are presented as means ± SEM. The number of biologically independent replicates (n) for each experiment is indicated in the figure legends. Statistical analysis was performed using unpaired two-tailed t test, one-way ANOVA and two-way ANOVA with Tukey multiple comparison test using GraphPad Prism version 8.4.3 (https://www.graphpad.com/). A p value of <0.05 was considered statistically significant. For all experiments, mice were divided into groups based on genotype and the specific mouse chosen per group was randomised. Daily NAC/saline injections in neonates and glucose homeostasis measurements in adult mice were done blinded. Otherwise, blinding was not carried out. Data from mice that died prematurely were excluded.
Results
Beta cell proliferation and NRF2 expression are associated in early postnatal ages in humans
NRF2 is necessary for rat, mouse and human beta cell proliferation under situations of metabolic stress, such as high glucose or HFD feeding [1, 18]. Metabolic adaptation to changes in diet (transitioning from mother’s milk to solid food) occurs during early postnatal ages. We therefore analysed the expression of NRF2 in human beta cells and human beta cell proliferation at different neonatal ages (1 day to 19 months), childhood (age 2–7 years) and adulthood (age 26–56 years). Pancreatic sections from non-diabetic human cadaveric donors (ESM Table 3) were immunolabelled for Ki67 and insulin (Fig. 1a,b) or for NRF2 and insulin (Fig. 1c,d). Beta cell proliferation, which was the highest in the neonatal group (1.2±0.2%), was significantly diminished in the adult group. Similarly, beta cell NRF2 levels in the neonatal group (31±5.9%) were significantly higher than in the adult group (8±1.6%). Moreover, a significant correlation (R2=0.568; p=0.001) was found between Ki67 and NRF2 levels in beta cells (Fig. 1e). Hence, our data suggest that NRF2 expression plays an important role in human postnatal beta cell proliferation.
Fig. 1.
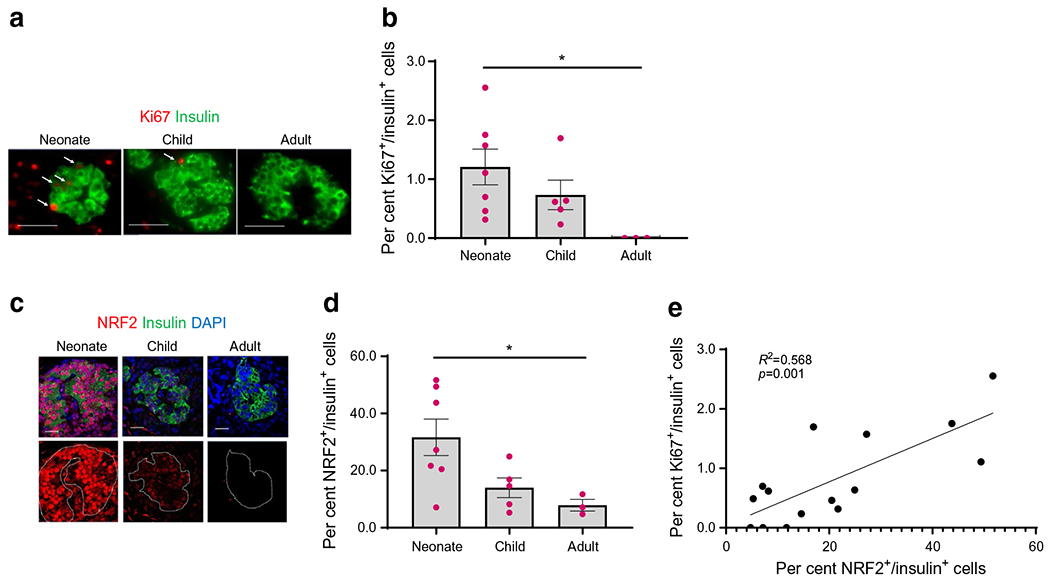
NRF2 levels are upregulated in human beta cells during neonatal ages. (a–d) Pancreatic sections from non-diabetic human juvenile cadaveric donors were embedded and immunolabelled for Ki67 and insulin (scale bar, 50 μm) (a, b), or insulin and NRF2 (scale bar, 20 μm) (c, d). The percentage of Ki67- and insulin-positive and NRF2- and insulin-positive cells was calculated. (e) Simple linear regression was calculated between the per cent Ki67+/insulin+ to per cent NRF2+/insulin+ cells. Representative images were selected from an average of 30 images per individual donor. Data are the means ± SEM for n=3–7, *p<0.05
NRF2 is necessary for neonatal beta cell proliferation, beta cell mass expansion and maintaining redox balance
To explore the link between NRF2 and beta cell proliferation during the neonatal period, we generated a beta cell-specific Nrf2 deletion mouse model (βNrf2KO) by crossing Ins1Cre [14] with Nrf2lox/lox [15] mice (Fig. 2a). Adult βNrf2KO mouse islets displayed a 67±10.9% reduction in Nrf2 mRNA levels compared with control Nrf2lox/lox mouse islets (Fig. 2b). Since Nrf2 is primarily regulated at the protein level [19], we assessed NRF2 levels in beta cells by immunostaining pancreatic sections from 1-, 7-, 14- and 28-day-old male and female Nrf2lox/lox control or βNrf2KO mice (Fig. 2c,d). Like in humans, we observed the highest levels of NRF2 at neonatal ages, with NRF2-positive/insulin-positive cells peaking at 7 days of age (57±6.4%); this proportion gradually declined with age, reaching 3.77±1.7% in adult Nrf2lox/lox mice. Interestingly, most Proliferating cell nuclear antigen (PCNA)-positive beta cells were also positive for NRF2 (72±4.1%) in 7-day-old Nrf2lox/lox mice (ESM Fig. 1a, b). Additionally, Nrf2 deletion in 1- and 7-day-old βNrf2KO mice significantly reduced beta cell proliferation by 100±0.0% and 65±6.3%, respectively (Fig. 2e,f). This suggests that NRF2 is essential for beta cell proliferation at early postnatal ages. No differences in beta cell proliferation were found between male and female mice at these ages.
Fig. 2.
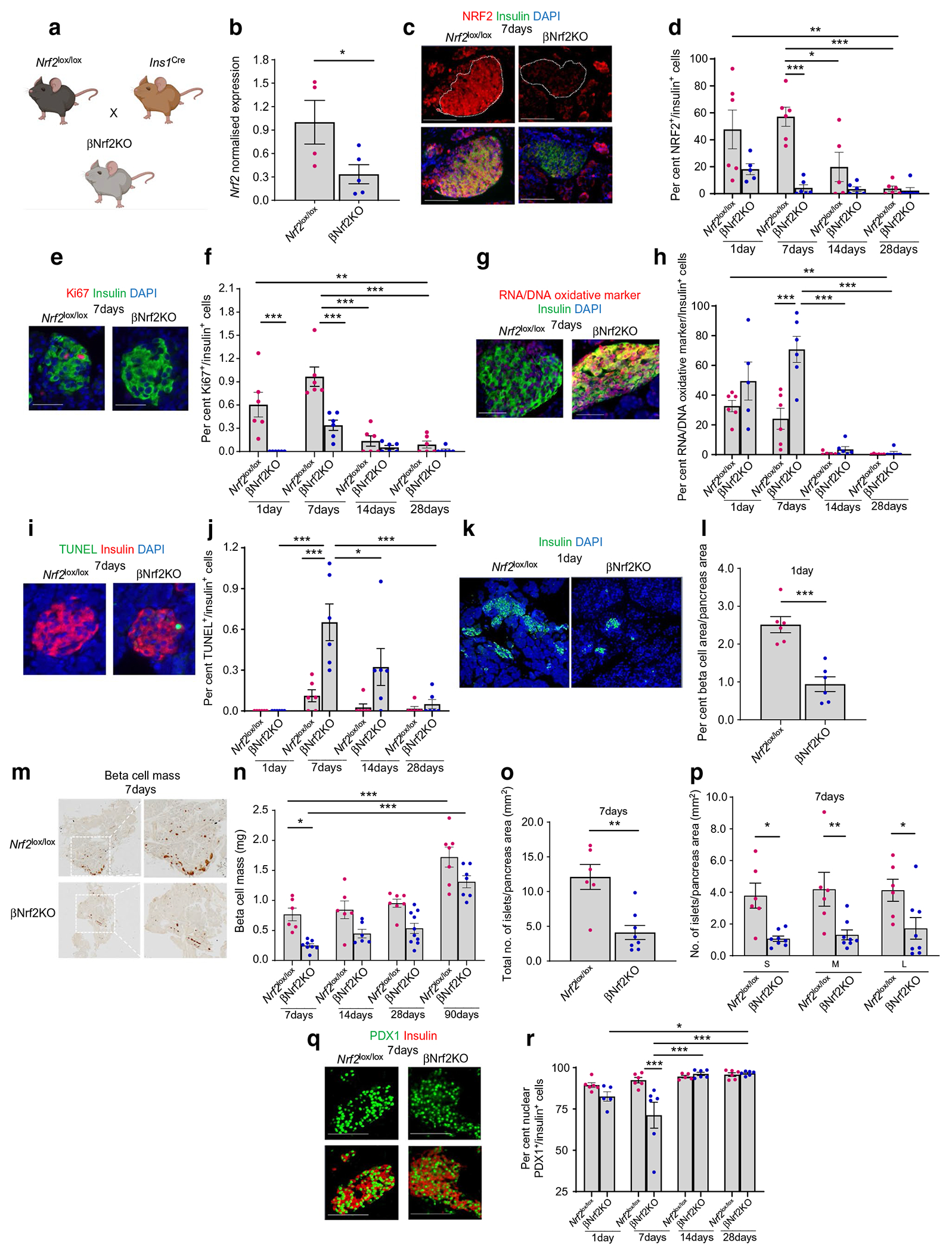
NRF2 is necessary for mouse beta cell proliferation, maintaining proper redox balance, promoting cell survival and increasing nuclear PDX1 localisation at early postnatal ages. (a) A postnatal beta cell-specific Nrf2 deletion mouse model (βNrf2KO) was generated by crossing Ins1Cre with Nrf2lox/lox mice. (b) Islets were isolated from adult βNrf2KO mice, RNA was extracted and expression of Nrf2 was measured. (c–j) Pancreatic sections from 1-, 7-, 14- and 28-day-old male and female Nrf2lox/lox control or βNrf2KO mice were immunolabelled for NRF2 and insulin (c, d), Ki67 and insulin (e, f), oxidative stress marker and insulin (g, h) or TUNEL and insulin (i, j). Scale bar, 50 μm. The percentage of NRF2-positive, Ki67-positive, TUNEL-positive or oxidative-stress-marker-positive and insulin-positive cells was calculated. (k, l) Pancreatic sections from 1-day-old βNrf2KO or Nrf2lox/lox mice were immunolabelled for insulin. Scale bar, 200 μm. Beta cell area/pancreas area was calculated. (m–p) Pancreatic sections from 7-, 14-, 28- and 90-day-old male and female Nrf2lox/lox control or βNrf2KO mice were immunolabelled with insulin. Beta cell mass and islet morphometry were determined. For islet morphometry, islet size was divided into three groups, including small (S) (<1000 μm2), medium (M) (1001–2200 μm2) and large (L) (>2201 μm2), and islet numbers per pancreas area were calculated. (q, r) Pancreatic sections from 1-, 7-, 14- and 28-day-old male and female Nrf2lox/lox control or βNrf2KO mice were immunolabelled for PDX1 and insulin. Scale bar 100 μm. The percentage of PDX1- and insulin-positive cells was calculated. Representative images were selected from an average of 20 images per individual mouse. Data are mean ± SEM for n=4–10. *p<0.05, **p<0.01, ***p<0.001. Fig. 2a was created using BioRender.com
As a redox balance regulator, any increase in oxidative stress promotes NRF2 protein stability and transcriptional activity [2]. To test whether the increase in beta cell Nrf2 expression during early postnatal ages results from increased oxidative stress, pancreatic sections from 1-, 7-, 14- and 28-day-old Nrf2lox/lox control or βNrf2KO mice were immunolabelled for the oxidative stress marker and insulin (Fig. 2g,h). In agreement with previous data published by Sander’s group [4], moderate levels of beta cell oxidative stress were observed in 1-day-old control mice, followed by a gradual decrease with age (99±0.3% reduction in 28-day-old compared with 1-day-old Nrf2lox/lox control mice). Conversely, deletion of Nrf2 resulted in even higher levels of oxidative stress, reaching up to 70±7.9% of beta cells in 7-day-old βNrf2KO mice (3±0.3-fold compared with 7-day-old Nrf2lox/lox mice). Not surprisingly, this beta cell redox unbalance seen in 7-day-old βNrf2KO mice increased beta cell death (Fig. 2i,j), suggesting that NRF2 is necessary for mitigating damage from oxidative stress in beta cells at early postnatal ages.
We then investigated whether the inhibition of beta cell proliferation and survival seen in βNrf2KO mice during early postnatal ages affected the expansion of beta cell mass at those ages and later in life. For this purpose, pancreatic sections from 1-, 7-, 14-, 28- and 90-day-old Nrf2lox/lox control or βNrf2KO mice were immunostained for insulin, chromogranin A and glucagon. One-day-old βNrf2KO mice displayed a significant reduction in per cent beta cell area/pancreas area (63±7.0% reduction) (Fig. 2k,l) and in per cent beta cell area/islet area (37±1.6% reduction) (ESM Fig. 1c, d) compared with Nrf2lox/lox control mice. No difference was found in the percentage of alpha cell area/pancreas area (ESM Fig. 1e), suggesting that Nrf2 deletion in beta cells did not affect alpha cell mass. Similar to the 1-day-old mice, 7-day-old βNrf2KO mice displayed a decrease (67±3.6%) in beta cell mass compared with Nrf2lox/lox littermates (Fig. 2m,n). This reduction in beta cell mass correlated with a decrease in total islet number across all islet sizes (Fig. 2o,p) without affecting pancreas weight, indicating that overall pancreas development was not affected (ESM Fig. 1f). It is thus likely that after maturation, a compensatory increase in beta cell mass takes place in βNrf2KO mice from day 14 onwards since beta cell mass is not significantly different compared with Nrf2lox/lox control mice, although a tendency towards lower beta cell mass remains.
We next analysed whether the levels of PDX1, a key transcription factor required for beta cell identity, function and survival [20], were affected by Nrf2 deletion in beta cells. Supraphysiological oxidative stress inhibits PDX1 nuclear localisation by changes in its phosphorylation status [20, 21]. We previously found that beta cell-specific Nrf2 deletion reduces PDX1 nuclear levels in adult mice with increased oxidative stress [1]. Here we found a significant decrease in nuclear PDX1 levels in 7-day-old βNrf2KO mice (23±7.7% reduction compared with 7-day-old Nrf2lox/lox mice) (Fig. 2q,r). This effect was transient, as PDX1 levels returned to normal levels at 14 and 28 days post-birth. These results indicate that Nrf2 deletion temporarily reduces PDX1 nuclear localisation, suggesting alterations in beta cell maturation at early postnatal ages. No changes were detected in total PDX1 levels (ESM Fig. 2a).
Thus, NRF2, which is upregulated in both human and mouse beta cells during early postnatal ages, is essential for beta cell proliferation, beta cell mass expansion, maintaining proper redox balance, reducing beta cell death and promoting beta cell identity in neonatal mice.
Antioxidant administration partially restores the effects of Nrf2 deletion on beta cells
We then investigated whether the effects observed in early postnatal βNrf2KO mouse beta cells result from changes in redox balance or from Nrf2 deletion. To tackle this question, neonatal 7-day-old βNrf2KO and Nrf2lox/lox mice were injected with the potent oxidative stress inhibitor NAC (75 mg/kg per day), or with saline (Fig. 3a), as previously described [16]. Mouse pancreatic sections were harvested and immunostained for oxidative stress markers and insulin. As expected, saline-injected 7-day-old βNrf2KO mouse beta cells displayed increased oxidative stress levels (fivefold) compared with Nrf2lox/lox control mouse beta cells (Fig. 3b,c). NAC administration completely blunted this increase.
Fig. 3.
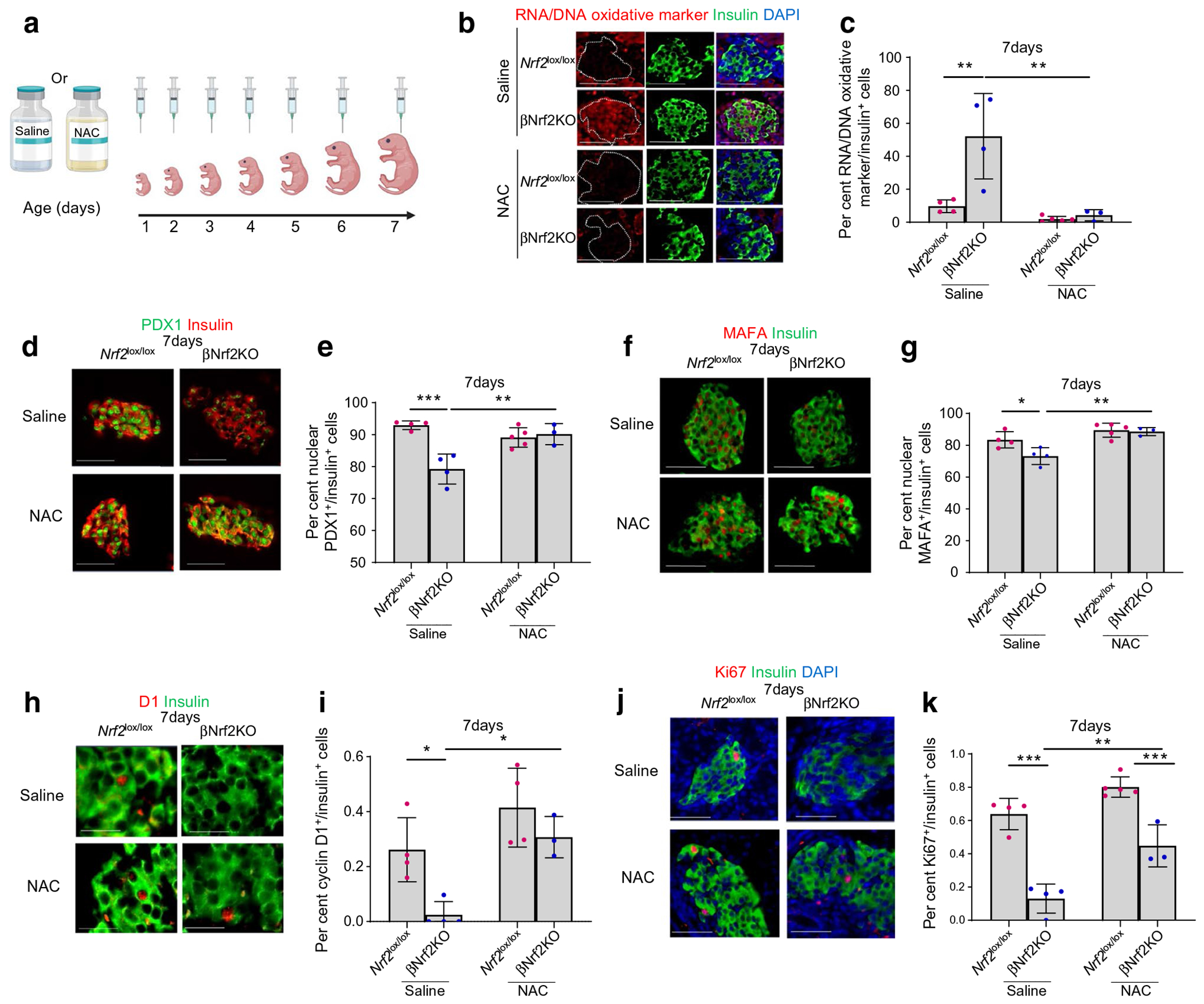
Antioxidant administration partially restores the effects of Nrf2 deletion on beta cells. (a) Neonatal βNrf2KO and Nrf2lox/lox mice were injected daily (day 1 to day 7) with 75 mg/kg NAC, a potent oxidative stress inhibitor, or with saline. At 7 days, pancreases were harvested, embedded and immunolabelled for oxidative stress marker and insulin (scale bar 50 μm) (b, c) PDX1 and insulin (scale bar, 50 μm) (d, e), MAFA and insulin (scale bar, 50 μm) (f, g), cyclin D1 and insulin (scale bar, 20 μm) (h, i), or Ki67 and insulin (scale bar, 50 μm) (j, k). The percentage of Ki67-, oxidative stress marker-, PDX1-, MAFA-, or cyclin D1- and insulin-positive cells were calculated. Representative images were selected from an average of 20 images per individual mouse. Data are mean ± SEM for n=3–5. *p<0.05, **p<0.01, ***p<0.001. Fig. 3a was created using BioRender.com
Next, we studied the effect of NAC on PDX1 levels in βNrf2KO mice. Immunostaining for PDX1 and insulin revealed a 15±2.1% reduction in beta cell nuclear PDX1 levels in saline-injected βNrf2KO mice compared with control mice (Fig. 3d,e). NAC administration fully restored beta cell nuclear PDX1 levels in βNrf2KO mice, reaching levels comparable with those in Nrf2lox/lox control mice. This finding was in line with our in vitro data showing NAC-dependent restoration of beta cell nuclear PDX1 levels in Nrf2lox/lox islets transfected with Cre-expressing adenovirus following incubation with high glucose (a known oxidative stress-associated condition) (ESM Fig. 2b). As with PDX1, MAFA nuclear localisation is also affected by oxidative stress [21, 22]. We therefore performed immunostaining for MAFA and insulin and found that, similarly to PDX1, beta cell nuclear MAFA levels were reduced (by 13±2.7%) in saline-injected βNrf2KO mice compared with Nrf2lox/lox control mice, whereas NAC administration reversed this reduction (Fig. 3f,g).
Cyclin D1, which plays an important role in postnatal beta cell proliferation [23], is the primary cyclin affected by changes in redox balance. Exposure of cells to oxidative stress results in decreased cyclin D1 translation and stabilisation [24, 25]. To test whether Nrf2 deletion affects beta cell cyclin D1 levels at early postnatal ages, we performed immunostaining for cyclin D1 and insulin. Our data clearly show a 90±8.0% reduction of beta cell cyclin D1 levels in saline-injected 7-day-old βNrf2KO mice compared with Nrf2lox/lox mice (Fig. 3h,i). When injected with NAC, the βNrf2KO mouse beta cells displayed increased cyclin D1 levels (12±1.4-fold) compared with cells from saline-injected βNrf2KO mice, reaching levels similar to those seen in saline- and NAC-injected Nrf2lox/lox control mice. This suggests that NRF2 controls cyclin D1 levels via regulation of redox balance. These changes in cyclin D1 prompted us to interrogate whether the reduction in beta cell proliferation seen in 7-day-old βNrf2KO mice resulted from changes in beta cell redox balance. Immunostaining for Ki67 and insulin showed that 7-day-old saline-injected βNrf2KO mice displayed a sharp decrease (80±5.9%) in beta cell proliferation compared with saline-injected Nrf2lox/lox mice (Fig. 3j,k). NAC-injected βNrf2KO mice showed an increase in beta cell proliferation compared with saline-injected βNrf2KO mice (3.4±0.4-fold). Although no significant difference in beta cell proliferation was observed when comparing NAC-injected βNrf2KO mice with saline-injected Nrf2lox/lox mice, NAC-injected βNrf2KO mice had reduced beta cell proliferation compared with NAC-injected Nrf2lox/lox mice (45±7.4%). These results suggest that NRF2 promotes neonatal beta cell proliferation by controlling additional mechanisms beyond mitigating oxidative stress.
Inhibition of NRF2 at early postnatal ages suppresses mitochondrial ATP synthesis
To uncover mechanisms by which NRF2 stimulates beta cell proliferation in neonatal mice, RNA from islets of 7-day-old βNrf2KO or Nrf2lox/lox mice was sequenced. Due to the limited amount of RNA obtained from a single 7-day-old mouse, samples from islets of five mice were pooled (Fig. 4a). A principal component analysis (PCA) plot of βNrf2KO and Nrf2lox/lox islets showed moderate separation of the two groups along the first component (ESM Fig. 3a). Differential gene expression determined some 1591 significantly expressed genes (p<0.05) (ESM Fig. 3b), including genes related to pro-apoptosis, oxidative stress response, endoplasmic reticulum stress response, cell-cycle regulation, beta cell identity, ion channels and mitophagy (ESM Fig. 3c). Of note, we found a significant downregulation of mitochondria-encoded genes, including mitochondrial tRNAs and electron transport chain (ETC) genes, such as mitochondrial complex-I (mitochondrial NADH dehydrogenase-I), cytochrome c and complex V (mitochondrial ATP synthase) (ESM Fig. 3b, c). GO pathway analysis revealed inhibition of the mitochondrial electron transport-coupled ATP synthesis pathway in βNrf2KO mouse islets (Fig. 4b,c).
Fig. 4.
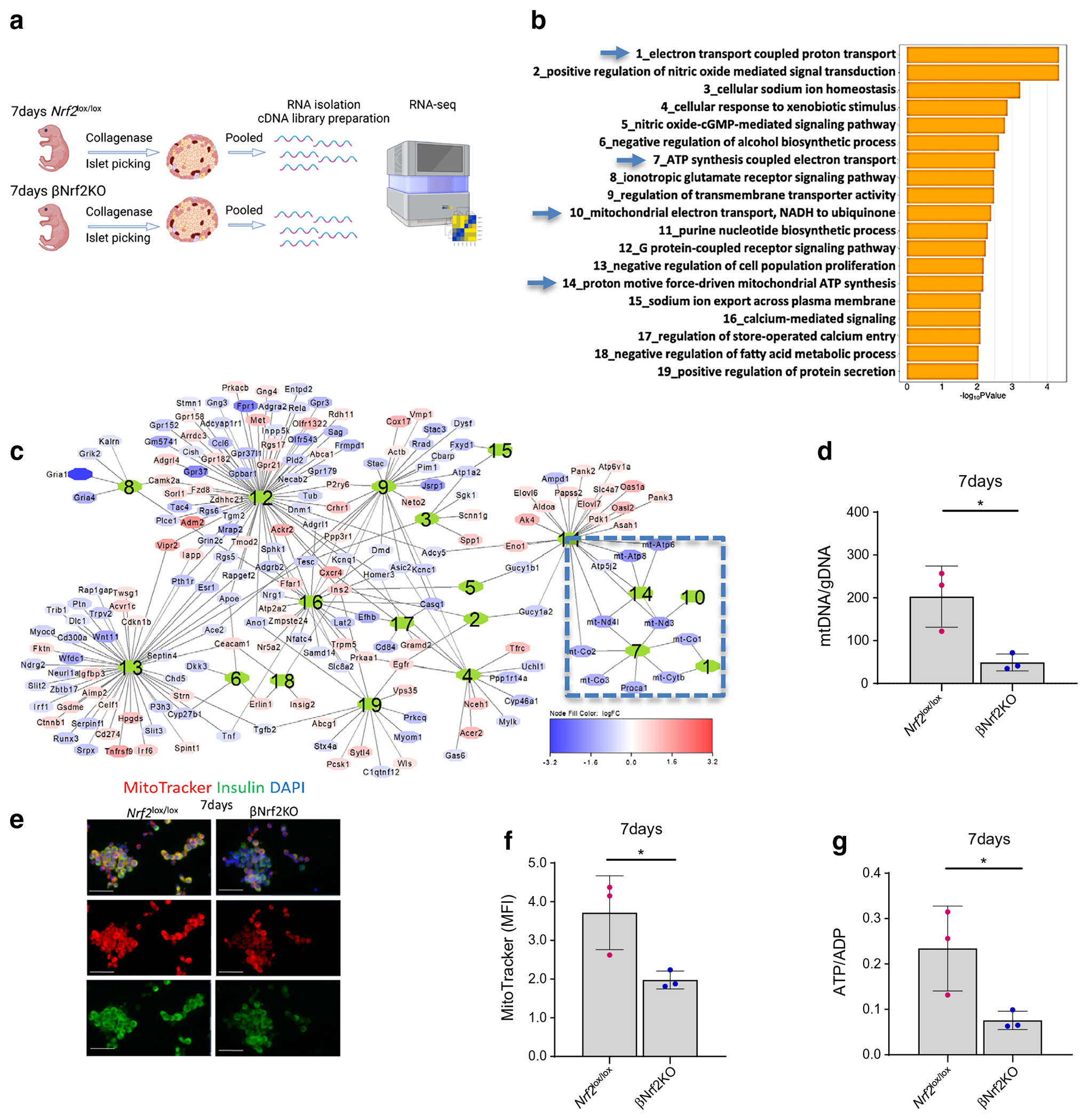
Inhibition of NRF2 at early postnatal ages suppresses mitochondrial ATP synthesis. (a) Islets from 7-day-old βNrf2KO or Nrf2lox/lox mice were isolated, pooled and RNA was isolated. cDNA libraries were then constructed and RNA-seq was performed. (b) GO pathways analysis enriched by differentially expressed genes. Blue arrows indicate shared pathways involved in mitochondrial ETC ATP synthesis. (c) Gene network generated by charting the genes annotated to the pathways shown in (b). Numbers in the centre of each gene subnetwork refer to the numbered pathways shown in (b). Boxed are the subnetworks identified by a blue arrow in (b). (d) The mtDNA/gDNA ratio was measured in 7-day-old βNrf2KO or Nrf2lox/lox mouse islets. (e, f) Islets from 7-day-old βNrf2KO or Nrf2lox/lox mice were dispersed, incubated with MitoTracker, and immunolabelled for insulin. Scale bar 20 μm. The mean fluorescent intensity (MFI) of MitoTracker in insulin-positive cells was calculated. Representative images were selected from an average of 20 images per individual mouse. (g) ATP/ADP ratio was measured in 7-day-old βNrf2KO or Nrf2lox/lox mouse islets. Representative images were selected from three individual experiments. Data for (d–g) are mean ± SEM for n=3. *p<0.05. Fig. 4a was created using BioRender.com
These results suggest that NRF2 might regulate mitochondrial biogenesis (mitobiogenesis) in beta cells via regulation of the expression of genes involved in mtDNA replication and transcription [2, 18, 26]. To test this, and since the mtDNA/gDNA ratio is a good indicator for mitobiogenesis [27], DNA was isolated from 7-day-old Nrf2lox/lox or βNrf2KO mouse islets and mtDNA/gDNA was measured. Our data showed that there was a 75±4.5% reduction in mtDNA/gDNA ratio in βNrf2KO compared with Nrf2lox/lox mouse islets (Fig. 4d). Additionally, immunostaining for insulin and MitoTracker showed a 47±2.9% reduction of active mitochondria in βNrf2KO compared with Nrf2lox/lox mouse islets (Fig. 4e,f). Lastly, since the RNA-seq data showed downregulation of the expression of genes involved in mitochondrial ATP synthesis, we measured the ATP/ADP ratio. Our results showed a 68±4.0% decline in ATP/ADP ratio in βNrf2KO compared with Nrf2lox/lox mouse islets (Fig. 4g). Thus, deletion of Nrf2 at early postnatal ages suppressed mitobiogenesis and mitochondrial ATP synthesis, both of which are required for beta cell proliferation.
Postnatal beta cell Nrf2-deleted mice display compromised glucose homeostasis under metabolic stress
Analysis of glucose homeostasis in both male and female adult βNrf2KO mice showed no significant changes in blood glucose (Fig. 5a and ESM Fig. 4a, e, f), plasma insulin (Fig. 5b), glucose tolerance (Fig. 5c,d and ESM Fig. 4h, i), body weight (ESM Fig. 4b, g), insulin sensitivity (ESM Fig. 4c, d, j, k) or GSIS (Fig. 5e) compared with control mice.
Fig. 5.
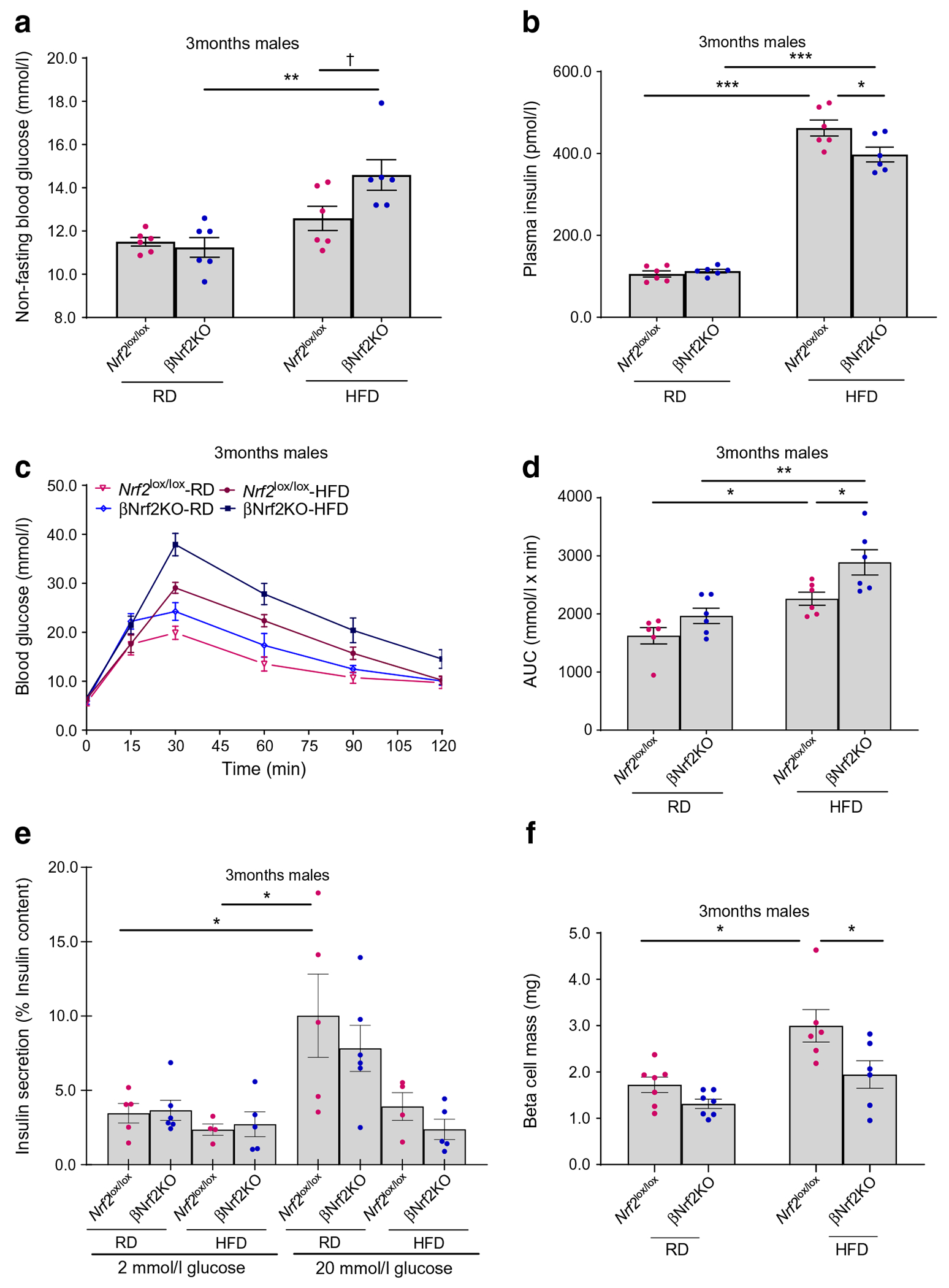
Postnatal βNrf2KO mice display compromised glucose homeostasis under metabolic stress. (a–e) Non-fasting blood glucose (a), plasma insulin (b), IPGTT (c, d) and GSIS (e) were measured in 3-month-old male βNrf2KO or Nrf2lox/lox mice fed on RD or HFD for 1 month. (f) Pancreatic sections from 3-month-old male βNrf2KO or Nrf2lox/lox mice fed on RD or HFD for 1 month were immunolabelled for insulin. Beta cell mass was calculated. Note that data from RD-fed 90-day-old mice in this figure was also used for generation of Fig. 2n. Data are mean ± SEM for n=4–7; 3–5 technical replicates were used for each experiment in GSIS. *p<0.05, **p<0.01, ***p<0.001; †p=0.055
We previously showed that beta cell inducible Nrf2 deletion in adult male mice did not lead to changes in beta cell mass or glucose homeostasis when the mice were fed an RD [1]. On the other hand, when these mice were metabolically stressed by HFD feeding, a condition associated with increased oxidative stress, they displayed attenuated expansion of beta cell mass, reduced plasma insulin, hyperglycaemia and glucose intolerance [1]. To test whether beta cell Nrf2 deletion at embryonic ages affects beta cell mass and glucose homeostasis in metabolically stressed adults, 3-month-old Nrf2lox/lox or βNrf2KO male mice were fed an HFD for 1 month. HFD-fed βNrf2KO male mice displayed a significant reduction (35±9.0%) in beta cell mass (Fig. 5f), a mild non-significant increase (p=0.055) in non-fasting blood glucose (1.2±0.0-fold) (Fig. 5a), decreased (14±3.5%) plasma insulin (Fig. 5b) and reduced glucose tolerance (1.3±0.0-fold) (Fig. 5c,d) compared with HFD-fed Nrf2lox/lox mice. No significant changes were observed in GSIS (Fig. 5e), fasting blood glucose (ESM Fig. 4a), body weight (ESM Fig. 4b) or insulin sensitivity (ESM Fig. 4c, d). Conversely, HFD-fed adult βNrf2KO female mice displayed no change in glucose homeostasis variables (ESM Fig. 4e–j). Thus, adult male mice harbouring beta cell-specific Nrf2 deletion from embryonic ages display compromised glucose homeostasis under metabolic stress but not under basal conditions.
Discussion
During early postnatal ages, a time when beta cell metabolism predominately relies on amino acids and lipids, beta cells produce moderate levels of mitochondrial ROS as a byproduct of increased mitochondrial aerobic respiration [4, 28, 29]. Unlike high levels of ROS, moderate ROS levels are beneficial to beta cell health and function, activating signalling intermediates, enhancing GSIS and stimulating beta cell proliferation [2–4]. Indeed, moderate levels of ROS in the late G1 phase of the cell cycle promote gene transcription and protein synthesis needed for dividing cells. In addition, moderate ROS levels regulate the activation of protein phosphatases, protein kinases, growth factors and transcription factors through oxidation of redox-sensitive cysteine and tyrosine residues [30]. One of the best characterised transcription factors that is activated by ROS is NRF2. Accordingly, NRF2 protein stabilisation and consequent activation takes place when critical cysteine residues on KEAP1, the NRF2 inhibitor, are oxidised, enabling the escape of NRF2 from the proteasomal degradation machinery [1, 2]. Consistent with our findings, the mild increase in oxidative stress at early postnatal ages correlates with an upregulation of NRF2 levels both in human and mouse beta cells. We cannot exclude that other factors that are increased in postnatal ages contribute to NRF2 upregulation. For example, the nutrient sensor target of rapamycin (mTORC1), which is upregulated in beta cells during neonatal ages, phosphorylates p62 during selective autophagy, thus inhibiting KEAP1 from targeting NRF2 for degradation [17, 31]. However, the involvement of mTORC1 in NRF2 upregulation during neonatal beta cell growth has yet to be studied.
During early postnatal ages, beta cells go through a burst of proliferation that will further affect the beta cell mass in adulthood [4, 10–12]. Consistent with our previous work in adult mice fed a diabetogenic diet [1], we show that during early postnatal ages Nrf2 deletion disrupts beta cell redox balance leading to high levels of oxidative stress, which promotes beta cell death and reduced beta cell proliferation. Exceedingly high levels of oxidative stress inhibit cell proliferation, in part, by translational inhibition and proteasomal degradation of cyclin D1. This serves as a check point for the cell to stop dividing due to potential risk for DNA damage [24]. Seven-day-old βNrf2KO mice injected with NAC showed partial restoration of beta cell proliferation. This shows that with appropriate ROS levels, postnatal beta cell proliferation can be sustained in the absence of NRF2 but only up to a certain degree. It is possible that the concentration used for NAC injections was not optimal for increasing beta cell proliferation up to control levels. Alternatively, it might be that redox-independent mechanisms are also involved.
Consistent with our RNA-seq results, our findings show that beta cell-specific Nrf2 deletion attenuates mitochondrial ATP production. Mitochondrial aerobic respiration is the major source of cellular ATP [32]. Since dividing cells require ATP for the synthesis of new RNA and proteins [32], we speculate that NRF2 supports the neonatal burst of beta cell proliferation by maintaining an adequate production of mitochondrial ATP. We previously found that activation of NRF2 via inhibition of KEAP1 increases the expression of genes that are part of the PPP pathway to produce NADPH [1]. Considering that the PPP pathway is used for the synthesis of DNA and that NADPH is used for the synthesis of membrane lipids in dividing cells [1, 32], this might be an additional way by which NRF2 supports neonatal beta cell proliferation. Further research is warranted to decipher the metabolic mechanisms regulated by NRF2 to promote beta cell proliferation.
Our data indicate that Nrf2 deletion affects mitochondrial biogenesis in beta cells. However, since incubation of mouse beta cells with pharmacological NRF2 activators preserves mitochondrial membrane potential [33], it is possible that mitochondrial function is also affected. Indeed, our data show that beta cells from 7-day-old βNrf2KO mice display diminished intensity of MitoTracker, a mitochondrial membrane potential-sensitive fluorochrome [34]. NRF2 also promotes degradation of dysfunctional mitochondria via the mitophagy pathway, thus preventing high ROS levels [35, 36]. However, we did not find significant changes in most mitophagy-related genes in the RNA-seq dataset. Nevertheless, this does not rule out the possibility that altered mitophagy can contribute to dysfunctional mitochondria in the absence of NRF2.
Our results show that 7-day-old βNrf2KO mice display reduced nuclear presence of PDX1 and MAFA in beta cells. Since these transcription factors control insulin biosynthesis [37], it is possible that some beta cells in βNrf2KO mice displayed decreased insulin content and therefore were not detected as beta cells. In this case, the observed reduction of PDX1 and MAFA in βNrf2KO beta cells would be even greater. Yet, this inhibition was reversed upon injection with NAC, suggesting that NRF2 controls their localisation via regulation of redox balance. While NAC antioxidant treatment is known to restore PDX1 and MAFA nuclear levels under oxidative stress conditions [22, 38–40], the role of NRF2 in this phenomenon has never been studied. Interestingly, two NRF2 target genes encoding glutathione peroxidase 1 and superoxide dismutase 1 can regulate PDX1 and MAFA levels [41–43]. However, changes in the expression of these genes were not observed in the RNA-seq data, suggesting that, at least in this setting, the mechanism might be different.
One-day-old βNrf2KO mice displayed reduced beta cell area compared with Nrf2lox/lox littermates, suggesting that the effects of Nrf2 deletion had an impact on beta cell growth during embryogenesis. This aligns with Cre-mediated recombination occurring as early as embryonic day 13.5 under control of the Ins1 promoter [44, 45]. Interestingly, under non-stressful conditions, the reduction in beta cell mass seen at early postnatal ages was restored to normal levels in adulthood. This compensation could have resulted from accelerated proliferation in those beta cells that had evaded the Nrf2 deletion. Alternatively, this compensation may rely on NRF2-independent pathways. Future lineage tracing studies are needed to tackle this question. Nonetheless, reduced beta cell mass and defective glucose homeostasis were observed in adult βNrf2KO mice fed an HFD [1, 2]. This suggests that the protective effects of NRF2 are necessary only under stressful conditions. Accordingly, beta cell Nrf2 deletion in adult mice overexpressing inducible nitric oxide synthase in beta cells leads to reduced beta cell mass and attenuated glucose homeostasis [46]. Interestingly, the extent of beta cell mass reduction seen in HFD-fed adult βNrf2KO mice was lower than that seen in 7-day-day-old βNrf2KO mice vs respective littermate controls. This may result from differences in the amount of ROS generated, differences in beta cell sensitivity to ROS or differences in KEAP1 expression at different developmental ages. We also observed a relatively mild interference with glucose homeostasis in adult mice lacking NRF2 after 1 month of HFD feeding. However, we speculate that continued HFD feeding would aggravate their compromised glucose homeostasis.
To conclude, this study shows that NRF2 is essential for maintaining beta cell redox balance and survival, as well as the burst of beta cell proliferation and expansion at early postnatal ages by preserving mitochondrial biogenesis and function. NRF2 is also required for maintaining functional beta cell mass in adulthood under metabolic stress. Future studies are needed to investigate whether NRF2 uses distinct proliferative mechanisms in beta cells at neonatal ages compared with metabolic stress. Additional studies are also needed to find whether NRF2 plays a role in stimulating beta cell proliferation in other situations of increased insulin demand or growth such as pregnancy and embryonic development.
Supplementary Material
Research in context.
What is already known about this subject?
During early postnatal ages, both human and rodent beta cells go through a burst of proliferation that quickly declines with age
Mitochondrial release of moderated ROS levels has been suggested as one of the main drivers for the neonatal burst of beta cell proliferation
NRF2, which is activated by ROS, plays an essential role in adult beta cell proliferation
What is the key question?
Does NRF2 play a role in beta cell proliferation at early postnatal ages?
What are the new findings?
NRF2 levels are increased during the postnatal burst of beta cell proliferation in both humans and rodents
NRF2 is required for beta cell proliferation, survival, identity and mass expansion at early stages of life
NRF2 regulates neonatal beta cell proliferation by promoting mitochondrial ATP synthesis
How might this impact on clinical practice in the foreseeable future?
Early intervention using NRF2 pharmacological modulators may improve glucose homeostasis during metabolic stress and obesity
Acknowledgements
The authors thank the Icahn School of Medicine at Mount Sinai Microscopy Core, the Biorepository and Pathology core and the Einstein/Sinai Diabetes Research Center Human Islet and Adenovirus Core P30DK020541 (AGO and DKS). The authors thank the Developmental Studies Hybridoma Bank, Department of Biology, The University of Iowa (Iowa City, IA, USA), for providing the human insulin antibody. The human pancreatic sections were generously provided by A. C. Powers and M. Brissova from the Vanderbilt Pancreas Biorepository, which was created with the support of NIH DK106755, DK120456, DK108120 and DK104211, The Leona M. and Harry B. Helmsley Charitable Trust and as part of efforts in the Vanderbilt Diabetes Research and Training Center (DK020593). This study used paediatric pancreas samples from the Human Atlas of the Neonatal Development and Early Life Pancreas and associated Immune Organs, a project supported by The Leona M. and Harry B. Helmsley Charitable Trust.
Funding
This study was supported by National Institutes of Health, National Institute of Diabetes and Digestive and Kidney Diseases, Mentored Research Scientist Development Award K01 DK128387-01A1 (to SBA), the Mindich Child Health and Development Institute Pilot and Feasibility Grant (to SBA), R01DK114338 and R01DK130300 (to DKS).
Abbreviations
- ETC
Electron transport chain
- gDNA
Genomic DNA
- GSIS
Glucose-stimulated insulin secretion
- HFD
High-fat diet
- KEAP1
Kelch-like ECH-associated protein 1
- MAFA
MAF bZIP transcription factor A
- mtDNA
Mitochondrial DNA
- NAC
N-acetylcysteine
- NRF2
Nuclear factor erythroid 2-related factor 2
- PDX1
Pancreatic duodenal homeobox protein 1
- qPCR
Quantitative real-time PCR
- RD
Regular diet
- ROS
Reactive oxygen species
Footnotes
Supplementary Information The online version contains peer-reviewed but unedited supplementary material available at https://doi.org/10.1007/s00125-023-06071-7.
Authors’ relationships and activities The authors declare that there are no relationships or activities that might bias, or be perceived to bias, their work.
Data availability
The mouse lines generated in this study are available from the corresponding author upon reasonable request. The sequencing data included in this publication have been deposited in NCBI’s Gene Expression Omnibus [47] and are accessible through GEO Series accession number GSE242718.
References
- 1.Baumel-Alterzon S, Katz LS, Brill G et al. (2022) Nrf2 regulates beta-cell mass by suppressing beta-cell death and promoting beta-cell proliferation. Diabetes 71(5):989–1011. 10.2337/db21-0581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Baumel-Alterzon S, Katz LS, Brill G, Garcia-Ocana A, Scott DK (2021) Nrf2: the master and captain of beta cell fate. Trends Endocrinol Metab 32(1):7–19. 10.1016/j.tem.2020.11.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ahmed Alfar E, Kirova D, Konantz J, Birke S, Mansfeld J, Ninov N (2017) Distinct levels of reactive oxygen species coordinate metabolic activity with beta-cell mass plasticity. Sci Rep 7(1):3994. 10.1038/s41598-017-03873-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zeng C, Mulas F, Sui Y et al. (2017) Pseudotemporal ordering of single cells reveals metabolic control of postnatal beta cell proliferation. Cell Metab 25(5):1160–1175 e1111. 10.1016/j.cmet.2017.04.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jimenez-Osorio AS, Gonzalez-Reyes S, Garcia-Nino WR et al. (2016) Association of nuclear factor-erythroid 2-related factor 2, thioredoxin interacting protein, and heme oxygenase-1 gene polymorphisms with diabetes and obesity in Mexican patients. Oxid Med Cell Longev 2016:7367641. 10.1155/2016/7367641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Matana A, Ziros PG, Chartoumpekis DV et al. (2020) Rare and common genetic variations in the Keap1/Nrf2 antioxidant response pathway impact thyroglobulin gene expression and circulating levels, respectively. Biochem Pharmacol 173:113605. 10.1016/j.bcp.2019.08.007 [DOI] [PubMed] [Google Scholar]
- 7.Wang X, Chen H, Liu J et al. (2015) Association between the NF-E2 related factor 2 gene polymorphism and oxidative stress, anti-oxidative status, and newly-diagnosed type 2 diabetes mellitus in a chinese population. Int J Mol Sci 16(7):16483–16496. 10.3390/ijms160716483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zazueta C, Jimenez-Uribe AP, Pedraza-Chaverri J, Buelna-Chontal M (2022) Genetic variations on redox control in cardiometabolic diseases: the role of Nrf2. Antioxidants (Basel) 11(3):507. 10.3390/antiox11030507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shin JH, Lee KM, Shin J, Kang KD, Nho CW, Cho YS (2019) Genetic risk score combining six genetic variants associated with the cellular NRF2 expression levels correlates with type 2 diabetes in the human population. Genes Genomics 41(5):537–545. 10.1007/s13258-019-00791-0 [DOI] [PubMed] [Google Scholar]
- 10.Gregg BE, Moore PC, Demozay D et al. (2012) Formation of a human beta-cell population within pancreatic islets is set early in life. J Clin Endocrinol Metab 97(9):3197–3206. 10.1210/jc.2012-1206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Beamish CA, Mehta S, Strutt BJ, Chakrabarti S, Hara M, Hill DJ (2017) Decrease in Ins(+)Glut2(LO) beta-cells with advancing age in mouse and human pancreas. J Endocrinol 233(3):229–241. 10.1530/JOE-16-0475 [DOI] [PubMed] [Google Scholar]
- 12.Bonner-Weir S, Aguayo-Mazzucato C, Weir GC (2016) Dynamic development of the pancreas from birth to adulthood. Ups J Med Sci 121(2):155–158. 10.3109/03009734.2016.1154906 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Spears E, Serafimidis I, Powers AC, Gavalas A (2021) Debates in pancreatic beta cell biology: proliferation versus progenitor differentiation and transdifferentiation in restoring beta cell mass. Front Endocrinol (Lausanne) 12:722250. 10.3389/fendo.2021.722250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Thorens B, Tarussio D, Maestro MA, Rovira M, Heikkila E, Ferrer J (2015) Ins1(Cre) knock-in mice for beta cell-specific gene recombination. Diabetologia 58(3):558–565. 10.1007/s00125-014-3468-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Reddy NM, Potteti HR, Mariani TJ, Biswal S, Reddy SP (2011) Conditional deletion of Nrf2 in airway epithelium exacerbates acute lung injury and impairs the resolution of inflammation. Am J Respir Cell Mol Biol 45(6):1161–1168. 10.1165/rcmb.2011-0144OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Puente BN, Kimura W, Muralidhar SA et al. (2014) The oxygen-rich postnatal environment induces cardiomyocyte cell-cycle arrest through DNA damage response. Cell 157(3):565–579. 10.1016/jxell.2014.03.032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jaafar R, Tran S, Shah AN et al. (2019) mTORC1 to AMPK switching underlies beta-cell metabolic plasticity during maturation and diabetes. J Clin Invest 129(10):4124–4137. 10.1172/JCI127021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kumar A, Katz LS, Schulz AM et al. (2018) Activation of Nrf2 is required for normal and ChREBPalpha-augmented glucose-stimulated beta-cell proliferation. Diabetes 67(8):1561–1575. 10.2337/db17-0943 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bryan HK, Olayanju A, Goldring CE, Park BK (2013) The Nrf2 cell defence pathway: Keap1-dependent and -independent mechanisms of regulation. Biochem Pharmacol 85(6):705–717. 10.1016/j.bcp.2012.11.016 [DOI] [PubMed] [Google Scholar]
- 20.Baumel-Alterzon S, Scott DK (2022) Regulation of Pdx1 by oxidative stress and Nrf2 in pancreatic beta-cells. Front Endocrinol (Lausanne) 13:1011187. 10.3389/fendo.2022.1011187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kaneto H, Matsuoka TA (2012) Involvement of oxidative stress in suppression of insulin biosynthesis under diabetic conditions. Int J Mol Sci 13(10):13680–13690. 10.3390/ijms131013680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Harmon JS, Stein R, Robertson RP (2005) Oxidative stress-mediated, post-translational loss of MafA protein as a contributing mechanism to loss of insulin gene expression in glucotoxic beta cells. J Biol Chem 280(12):11107–11113. 10.1074/jbc.M410345200 [DOI] [PubMed] [Google Scholar]
- 23.Kushner JA, Ciemerych MA, Sicinska E et al. (2005) Cyclins D2 and D1 are essential for postnatal pancreatic beta-cell growth. Mol Cell Biol 25(9):3752–3762. 10.1128/MCB.25.9.3752-3762.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pyo CW, Choi JH, Oh SM, Choi SY (2013) Oxidative stress-induced cyclin D1 depletion and its role in cell cycle processing. Biochim Biophys Acta 1830(11):5316–5325. 10.1016/j.bbagen.2013.07.030 [DOI] [PubMed] [Google Scholar]
- 25.Wang J, Wang H (2017) Oxidative stress in pancreatic beta cell regeneration. Oxid Med Cell Longev 2017:1930261. 10.1155/2017/1930261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lee HC, Wei YH (2005) Mitochondrial biogenesis and mitochondrial DNA maintenance of mammalian cells under oxidative stress. Int J Biochem Cell Biol 37(4):822–834. 10.1016/j.biocel.2004.09.010 [DOI] [PubMed] [Google Scholar]
- 27.Popov LD (2020) Mitochondrial biogenesis: an update. J Cell Mol Med 24(9):4892–4899. 10.1111/jcmm.15194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Helman A, Cangelosi AL, Davis JC et al. (2020) A nutrientsensing transition at birth triggers glucose-responsive insulin secretion. Cell Metab 31(5):1004–1016 e1005. 10.1016/j.cmet.2020.04.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jacovetti C, Regazzi R (2022) Mechanisms underlying the expansion and functional maturation of beta-cells in newborns: impact of the nutritional environment. Int J Mol Sci 23(4):2096. 10.3390/ijms23042096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sies H, Jones DP (2020) Reactive oxygen species (ROS) as pleiotropic physiological signalling agents. Nat Rev Mol Cell Biol 21(7):363–383. 10.1038/s41580-020-0230-3 [DOI] [PubMed] [Google Scholar]
- 31.Ichimura Y, Waguri S, Sou YS et al. (2013) Phosphorylation of p62 activates the Keap1-Nrf2 pathway during selective autophagy. Mol Cell 51(5):618–631. 10.1016/j.molcel.2013.08.003 [DOI] [PubMed] [Google Scholar]
- 32.da Veiga Moreira J, Peres S, Steyaert JM et al. (2015) Cell cycle progression is regulated by intertwined redox oscillators. Theor Biol Med Model 12:10. 10.1186/s12976-015-0005-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Schultheis J, Beckmann D, Mulac D, Muller L, Esselen M, Dufer M (2019) Nrf2 activation protects mouse beta cells from glucolipotoxicity by restoring mitochondrial function and physiological redox balance. Oxid Med Cell Longev 2019:7518510. 10.1155/2019/7518510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sorvina A, Bader CA, Darby JRT et al. (2018) Mitochondrial imaging in live or fixed tissues using a luminescent iridium complex. Sci Rep 8(1):8191. 10.1038/s41598-018-24672-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Marasco MR, Conteh AM, Reissaus CA et al. (2018) Interleukin-6 reduces beta-cell oxidative stress by linking autophagy with the antioxidant response. Diabetes 67(8):1576–1588. 10.2337/db17-1280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.East DA, Fagiani F, Crosby J et al. (2014) PMI: a DeltaPsim independent pharmacological regulator of mitophagy. Chem Biol 21(11):1585–1596. 10.1016/j.chembiol.2014.09.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhu Y, Liu Q, Zhou Z, Ikeda Y (2017) PDX1, Neurogenin-3, and MAFA: critical transcription regulators for beta cell development and regeneration. Stem Cell Res Ther 8(1):240. 10.1186/s13287-017-0694-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kaneto H, Kajimoto Y, Miyagawa J et al. (1999) Beneficial effects of antioxidants in diabetes: possible protection of pancreatic beta-cells against glucose toxicity. Diabetes 48(12):2398–2406. 10.2337/diabetes.48.12.2398 [DOI] [PubMed] [Google Scholar]
- 39.Robertson RP, Harmon J, Tran PO, Tanaka Y, Takahashi H (2003) Glucose toxicity in beta-cells: type 2 diabetes, good radicals gone bad, and the glutathione connection. Diabetes 52(3):581–587. 10.2337/diabetes.52.3.581 [DOI] [PubMed] [Google Scholar]
- 40.Schuurman M, Wallace M, Sahi G et al. (2022) N-acetyl-L-cysteine treatment reduces beta-cell oxidative stress and pancreatic stellate cell activity in a high fat diet-induced diabetic mouse model. Front Endocrinol (Lausanne) 13:938680. 10.3389/fendo.2022.938680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wang XD, Vatamaniuk MZ, Wang SK, Roneker CA, Simmons RA, Lei XG (2008) Molecular mechanisms for hyperinsulinaemia induced by overproduction of selenium-dependent glutathione peroxidase-1 in mice. Diabetologia 51(8):1515–1524. 10.1007/s00125-008-1055-3 [DOI] [PubMed] [Google Scholar]
- 42.Wang X, Vatamaniuk MZ, Roneker CA, Pepper MP, Hu LG, Simmons RA, Lei XG (2011) Knockouts of SOD1 and GPX1 exert different impacts on murine islet function and pancreatic integrity. Antioxid Redox Signal 14(3):391–401. 10.1089/ars.2010.3302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Harmon JS, Bogdani M, Parazzoli SD et al. (2009) Beta-cell-specific overexpression of glutathione peroxidase preserves intranuclear MafA and reverses diabetes in db/db mice. Endocrinology 150(11):4855–4862. 10.1210/en.2009-0708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hasegawa Y, Daitoku Y, Mizuno S et al. (2014) Generation and characterization of Ins1-cre-driver C57BL/6N for exclusive pancreatic beta cell-specific Cre-loxP recombination. Exp Anim 63(2):183–191. 10.1538/expanim.63.183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cheng Y, Su Y, Shan A et al. (2015) Generation and characterization of transgenic mice expressing mouse Ins1 promoter for pancreatic beta-cell-specific gene overexpression and knockout. Endocrinology 156(7):2724–2731. 10.1210/en.2015-1104 [DOI] [PubMed] [Google Scholar]
- 46.Yagishita Y, Fukutomi T, Sugawara A et al. (2014) Nrf2 protects pancreatic beta-cells from oxidative and nitrosative stress in diabetic model mice. Diabetes 63(2):605–618. 10.2337/db13-0909 [DOI] [PubMed] [Google Scholar]
- 47.Edgar R, Domrachev M, Lash AE (2002) Gene expression omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res 30(1):207–210. 10.1093/nar/30.1.207 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Sequencing data are available in the NCBI Gene Expression Omnibus, accession number GSE242718 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE242718).
The mouse lines generated in this study are available from the corresponding author upon reasonable request. The sequencing data included in this publication have been deposited in NCBI’s Gene Expression Omnibus [47] and are accessible through GEO Series accession number GSE242718.