

Abstract
We present an ultraviolet (UV) - extreme-ultraviolet (XUV) pump-probe beamline with applications in ultrafast time-resolved photoelectron spectroscopy. The UV pump pulses, tuneable between 255 and 285 nm and with µJ-level energy, are generated by frequency up-conversion between ultrashort visible/infrared pulses and visible narrow-band pulses. Few-femtosecond XUV probe pulses are produced by a high-order harmonic generation source equipped with a state-of-the-art time-delay compensated monochromator. Two-colour UV-XUV sidebands are used for a complete in situ temporal characterization of the pulses, demonstrating a temporal resolution of better than 20 fs. We validate the performances of the beamline through a UV-XUV pump-probe measurement on 1,3-cyclohexadiene, resolving the ultrashort dynamics of the first conical intersection. This instrument opens exciting possibilities for investigating ultrafast UV-induced dynamics of organic molecules in ultrashort time scales.
Subject terms: High-harmonic generation, Ultrafast photonics, Photochemistry
Introduction
Femtosecond and attosecond spectroscopies intend to follow real-time ultrafast nuclear and electron dynamics in atoms, molecules, and solids1. In a time-resolved spectroscopy experiment, the sample is irradiated with an ultrashort pump pulse that drives the system out of its equilibrium state. Subsequently, a second pulse, named probe pulse, is employed to track the ultrafast processes induced by the pump pulse. The choice of the observable (spectral absorption, photoelectrons, photoions…) depends on the information most suitable to unveil the mechanism triggered by the pump pulse. One of the most extended schemes is time-resolved photoemission spectroscopy (TRPES)2–4, in which the kinetic energy of the ejected photoelectrons is mapped as a function of the pump-probe delay. Being sensitive to both vibrational dynamics and electronic configurations, it is well suited for the investigation of ultrafast non-adiabatic processes.
In a traditional TRPES setup, a pulse in the visible or ultraviolet (UV) range is used to resonantly excite the sample, while a probe pulse, typically lying in the deep UV range, ionizes the photo-excited system. This UV-UV scheme has been proven to be a powerful technique for disclosing relaxation pathways and non-adiabatic dynamics on molecules5. Still, most studies have been conducted with a temporal resolution of about 100 fs thus limiting the investigation of the primary steps after photo-excitation. In particular, little is known about the mechanisms governing light-matter interactions on organic molecules on time scales of a few tens of femtoseconds. These primary events determine the response of the system and are associated with electronic transitions that might trigger diverse photochemical processes6–11. The possibility of investigating such mechanisms has indeed aroused great interest in the scientific community, which has dedicated many efforts in the last few years towards the generation of UV pulses with temporal durations below 10 fs12–17 – a challenging task due to the intricate strong spectral dispersion and lack of amplifying materials in this regime.
Furthermore, in the UV-UV TRPES scheme, employing UV probes leads to the generation of low-kinetic energy photoelectrons, which are subject to the influence of inelastic scattering and restrict the observation window. This calls for probe pulses with photon energies in the extreme-ultraviolet (XUV), which can be obtained by high-order harmonic generation (HHG)18,19. In conventional HHG setups, an intense femtosecond pulse is focused onto a gas target. When macroscopic phase matching in the gas is ensured, the highly non-linear response of the medium leads to the coherent production of XUV radiation, resulting in a train of attosecond pulses. For specific applications (including TRPES), selecting a single harmonic from the broad spectrum is essential (e.g. avoiding the excitation of numerous ionization channels)20. This selectivity can be achieved utilizing spectral filters, monochromators, or time-delay compensated monochromators (TDCM)21,22.
The combination of ultrashort pump pulses in the UV range with attosecond or few-femtosecond XUV probe pulses is therefore a promising tool for investigating and resolving the electronic dynamics on the few-femtosecond time scale23 and has become an exciting field of research in the last few years. Suzuki and co-workers developed TRPES beamlines exploiting UV pulses generated by four-wave mixing or third-harmonic generation (THG) and single XUV pulses, which have been successfully employed for measurements of both isolated molecules24–26 and liquid jets27,28. The Artemis facility exploits spectrally selected XUV harmonics combined with THG or optical parametric amplifiers to investigate molecular dissociations29,30. Titov et al. employed near-UV pulses obtained by second-harmonic generation and single XUV probe pulses for investigating aminoazobenzenes31. Finally, Woerner’s group recently used UV pump pulses obtained by a THG setup and an XUV harmonic to explore the stilbene isomerization32. In all these studies the instrumental response function (IRF) ranged from 50 to 180 fs, with the exception of a few recent works reporting IRFs below 30 fs26,33–36.
Here we present the development and application of a UV-XUV time-resolved spectroscopy beamline with an IRF better than 20 fs and full spectral tunability. We generate sub-20 fs UV pulses tuneable between 4.35 and 4.85 eV by a frequency up-conversion scheme. We then combine these UV pulses with sub-10 fs XUV pulses spectrally selected between 20 and 45 eV from an HHG source equipped with a TDCM. To the best of our knowledge, we demonstrate for the first time the extension of IR-XUV temporal characterization methods based on two-colour photo-ionization37,38 to the UV-XUV regime, which allows us to provide a full direct retrieval of the pump and probe pulses and precisely determine the IRF of the apparatus. Finally, we show the application of the beamline to the study of the ring-opening dynamics of 1,3-cyclohexadiene (CHD), resolving the ultrafast dynamics of the first conical intersection passage.
Methods
The scheme of the experimental setup for the UV-XUV beamline is shown in Fig. 1. A fraction of the output energy (1.2 mJ) from a Ti: Sa laser system (Astrella, Coherent), which delivers 35-fs pulses at 800 nm and operates at 1-kHz repetition rate, is coupled into a 1-m-long hollow core fibre (HCF)39,40 with a core diameter of 320 μm and filled with neon gas at a constant pressure of 1.8 bar. The accumulated positive dispersion of the pulse is overcompensated at the output of the HCF with a battery of chirped mirrors, resulting in slightly negatively chirped pulses. The post-compressed IR pulses were temporally characterized using a second-harmonic FROG (SH-FROG)41, obtaining IR pulses with a duration of 9.5 fs (full-width half-maximum (FWHM)) and energy of 0.84 mJ. This IR beam is split into two parts. One part, with 80% of the beam power, is used as input for HHG and focused onto a 1-cm-long gas cell filled with argon; an XUV single harmonic is then spectrally selected by a TDCM. The latter consists of two equal sections equipped with two toroidal mirrors and two 400 gr/mm gratings in the off-plane mount. The first section performs the spectral selection of a single harmonic on the intermediate slit, the second section compensates for the pulse-front tilt due to the diffraction. After selection, the typical energy of the XUV pulses is of the order of a few tens of pJ. The probe photon energy can be tuned from 20 eV (HH13) to 45 eV (HH29) with a total transmission (from the HH source to the sample) higher than 15% in the 30–50 eV range, and in particular, peaked at 18% at 40 eV. For the experiments reported in this paper, we selected the 25th harmonic, with a central energy of ∼39.5 eV and a photon bandwidth of 0.4 eV. A pair of fused silica wedges on the IR beam path is used prior to generation to achieve fine-tuning of the dispersion and optimization of the HHG process.
Fig. 1.

Scheme of the TRPES beamline. The output of a Ti: Sa laser is post-compressed in a hollow core fiber compressor; additional chirped mirrors (CMs) impose a slightly negative chirp. A fraction of the beam, whose energy is controlled by a waveplate (WP) and a linear polarizer (LP), is upconverted in a broadband sum frequency generation (SFG) scheme with a narrowband second harmonic, resulting in sub-20 fs UV pulses. Spherical mirrors (SM) are used for focusing and collimation. The remaining beam is used for HHG. The generated harmonics are spectrally selected by a TDCM, composed of two stages consisting of two toroidal mirrors (TM) and a grating (G). The delay between the UV pump and the XUV probe pulses is controlled by a delay line (DL). Both pulses are focused into the time of flight spectrometer (TOF).
The remaining portion of the IR beam (150 µJ) is employed for UV generation in a frequency up-conversion scheme exploiting the mechanism of indirect phase transfer13. To this end, the IR pulse is mixed with a narrowband beam at 400 nm, obtained by frequency-doubling the output of the Ti: Sa system in a 200-µm thick Type-I BBO crystal. To prevent self-phase modulation effects and avoid residual non-converted IR/visible light, a lossless geometry was chosen. Both beams were focused in a slightly non-collinear configuration using a spherical mirror with a focal length of 400 mm onto a free-standing 25-µm Type-I BBO crystal. Precise control of the energy of each beam was achieved through variable attenuators, consisting of a linear polarizer and a half-wave plate, placed along the respective paths. In the BBO plane, the maximum energies of the IR and second-harmonic (SH) beams reached 40 µJ and 50 µJ, respectively. The sizes (FWHM) of the beams at the interaction point were around 500 μm. The temporal overlap was finely adjusted by a delay stage placed along the SH beam path. To remove any residual IR and SH beams after the BBO crystal for sum-frequency generation (SFG), we spatially isolated the UV signal taking advantage of the noncollinear configuration and employed two dielectric mirrors to filter out the remaining IR and SH beams. By slightly changing the phase matching angle, tuneable pulses with central wavelengths ranging from 255 to 285 nm (4.85 to 4.35 eV, see Fig. 2(a)) and energies of up to 7 µJ were achieved.
Fig. 2.

(a) Tuneable UV spectra. (b) Measured XFROG trace for a central wavelength of 270 nm. (c) Retrieved XFROG trace of (b) using an ePIE algorithm. (d) The retrieved intensity profile of the UV pulse with a FWHM is 18.9 ± 0.6 fs. (e) Spectrum and phase of the retrieved UV pulse (shaded purple area and black lines respectively). The shaded area in the phase represents the standard deviation calculated from 10 reconstructions with different input parameters.
A crucial aspect when dealing with UV pulses is the management of the spectral phase. This scheme exploits the mechanism of indirect phase transfer42,43, in which the phase of the broadband IR/visible pulse is transferred to the UV beam by the SFG process. Fine-tuning can thus be achieved by adding a controlled amount of material dispersion to the negatively chirped IR/visible pulses. The temporal characterization of the UV pulses was performed with an all-in-vacuum cross-correlation FROG (XFROG) between the UV pulse and an IR gate pulse in a difference frequency generation (DFG) scheme. A pick-off mirror, mounted in a motorized stage, intercepted the beams before they entered the TOF spectrometer chamber and sent them to the XFROG setup. The UV and IR pulses, both vertically polarized, were focused onto a 10-µm thick Type II BBO crystal for DFG. To control the delay between the IR and the UV pulses, a piezoelectric stage with a step time of 0.3 fs was employed. The spectrum of the generated DFG signal was recorded as a function of the relative delay between the two pulses. An example of an XFROG trace, for a pulse centred at 270 nm (4.6 eV), is shown in Fig. 2 (b).
Particular attention was devoted to the retrieval algorithm. The reconstruction of the temporal profile of the object pulse (in this case, the UV pulse) from the XFROG traces usually requires a well-characterized gate pulse44. Our approach employs instead a blind FROG method based on an extended ptychographic iterative engine (ePIE) algorithm, allowing the retrieval of both UV and IR pulses without the necessity of a precisely characterized IR reference pulse45–47.
Figure 2(c) shows the retrieved XFROG spectrogram, with an error G = 0.002 (representing the root mean square (RMS) difference between traces). A total of 10 reconstructions were performed changing the initial guesses. Each reconstruction was obtained after 1000 iterations of the ePIE algorithm. Figure 2(e) shows the experimental spectrum (purple shaded area) together with the retrieved spectral phase (black solid line), which presents a residual third-order dispersion. The retrieved UV intensity profile is shown in Fig. 2(d), which presents a temporal FWHM of 18.9 ± 0.6 fs. The shaded areas (in the retrieved pulse and phase) represent the uncertainties obtained with the standard deviation from all the reconstructed traces.
The UV pump and the XUV probe pulses are then non-collinearly focused with a small angle (∼ 1º) into the gas target. The photoelectrons generated from the UV-XUV interaction are collected by a time-of-flight (TOF) spectrometer (Kaesdorf ToF spectrometer ETF10), allowing the acquisition of the photoelectron spectrum as a function of the delay between the two beams. The energy resolution of the TOF spectrometer is  250 meV48, though it is the XUV bandwidth that ultimately sets an upper limit to the spectral resolution of the experiments. The pressure of the target gas jet is carefully regulated, and a chopper is used to switch the UV radiation on and off during measurements, enabling the collection of the XUV-only signal. The XUV photon spectrum is analysed using an XUV spectrometer located at the end of the beamline.
250 meV48, though it is the XUV bandwidth that ultimately sets an upper limit to the spectral resolution of the experiments. The pressure of the target gas jet is carefully regulated, and a chopper is used to switch the UV radiation on and off during measurements, enabling the collection of the XUV-only signal. The XUV photon spectrum is analysed using an XUV spectrometer located at the end of the beamline.
Results
XUV pump – UV probe in argon: in situ temporal characterization
Pump-probe spectroscopy experiments require a precise characterization of the IRF to extract information on the rise and decay times of the different species. In the following, we demonstrate how IR-XUV cross-correlation methods can be extended to the UV domain.
A common procedure for the characterization of XUV femtosecond pulses is to combine them with a portion of the IR driver in a pump-probe fashion. The resulting spectrogram is composed of main bands (direct photoionization signal by the harmonic) and sidebands (cross-correlation signals due to the interaction of the XUV with an absorbed/emitted IR photon). In comparison to attosecond pulses, the narrower bandwidth of a single harmonic and the associated loss of sub-cycle resolution, reduce the level of information redundancy on the trace making it necessary the use of a robust iterative algorithm37. In addition, when dealing with UV pulses, the signal-to-noise ratio dramatically drops as the relative intensity of the sideband with respect to the main band scales as λ448.
Figure 3 (a) shows the experimental spectrogram obtained by a photoionization cross-correlation measurement in argon by mixing the 25th harmonic (∼39.5 eV) with UV pulses at a central wavelength of 270 nm. The lens parameter of the TOF spectrometer was set to guarantee an optimal detection of the weak sideband signal. Due to large signal amplitude difference, this resulted in a distorted main band peak; this energy region (21–25 eV) has been thus set to zero in Fig. 3(a). We start by examining the upper sideband (highlighted by the dotted lines in Fig. 3(a)). The integrated signal along the electron kinetic energy axis (upper panel in Fig. 3(a)) corresponds to the intensity cross-correlation of the two pulses49 and provides the estimated IRF, resulting in 20 fs.
Fig. 3.

(a) Experimental spectrogram obtained by an XUV pump – UV probe photoelectron measurement in argon. The main band has been removed (see text for details). Top: integrated photoelectron signal along the kinetic energy axis for the upper sideband in the region highlighted by the dotted lines. (b) Retrieved spectrogram using STRIPE. Experimental spectrum (shaded area) and retrieved phase for (c) the UV and (d) the XUV pulses and corresponding intensity profiles in the temporal domain (e, f). The grey shaded area represents the standard deviation calculated from 10 reconstructions with different input parameters.
The low signal-to-noise ratio and the lack of information contained in the main band challenge the use of the commonly used retrieval algorithms. We have recently introduced a method called simplified trace reconstruction in the perturbative regime (STRIPE) for the retrieval of ultrashort XUV pulses, which is particularly robust against noise and does not necessitate redundant information50. We applied STRIPE to the experimental spectrogram of Fig. 3(a). The reconstructed trace, resulting from 10 independent retrievals with different starting parameters, is shown in Fig. 3(b). Panels (c) and (d) show the experimental spectra and the retrieved spectral phases for the UV and the XUV pulses, corresponding to temporal durations of 18.5±0.4 fs for the UV pulse (Fig. 3(e)) and 5.0±0.1 fs for the XUV pulse (Fig. 3(f)). The reconstructed UV phase agrees with that retrieved by the XFROG measurement (Fig. 2(e)) within the error margin. The residual XUV phase is attributed to the residual group delay dispersion introduced by the TDCM and is in good agreement with that of an independent IR-XUV measurement37.
Similar IRF values were obtained for different UV central wavelengths. Figure 4 reports the upper sideband and the corresponding integrated signal for UV central wavelengths of 257 nm (panel a) and 287 nm (panel b), entailing an IRF of 20 fs at 257 nm and 16.5 fs at 287 nm. The energy resolution of these measurements was estimated to 0.47 eV.
Fig. 4.
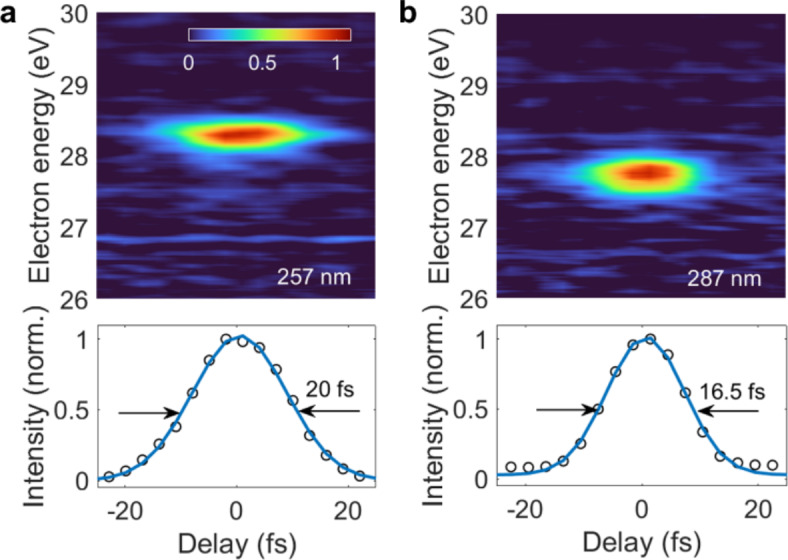
Upper sideband obtained by an XUV pump – UV probe photoelectron measurement in argon (top) and integrated signals along the kinetic energy axis (bottom) for UV central wavelengths of (a) 257 nm and (b) 287 nm.
UV pump – XUV probe in cyclohexadiene: resolving its ultrafast excited-state dynamics
We now demonstrate the application of the developed beamline to a TRPES experiment in 1,3-cyclohexadiene (CHD), a prototype molecule undergoing a ring-opening after UV excitation which has been widely investigated in the literature25,34,51–56. According to the generally accepted description, schematically shown in Fig. 5(a), by exciting CHD in the UV the 1B state is populated. In the diabatic model, the ring-opening reaction is thought to proceed to the dark 2A state (or the 3A state according to most recent works55), which is accessed by a conical intersection. From 2A it subsequently reaches the pericyclic minimum, from which the population can decay to the ground state of either CHD or 1,3,5-hexatriene (HT). It must be noticed that the involvement of an intermediate state and its nature is still an issue of intense debate25,54–56.
Fig. 5.

(a) Schematic representation of the decay pathways describing the electrocyclic ring-opening reaction of CHD as inferred from different works in literature25,53. Inset: chemical structure of CHD. (b) 2D map of photoelectron spectra. (c) Temporal lineouts of the integrated signals (solid lines) labelled as I and II in panel (b) and their least-squares fitting (dotted lines).
In our experiment, the energy of the pump pulses, with a central wavelength of 270 nm (4.6 eV) and a temporal duration of 18.5 fs (corresponding to the measurements shown in Fig. 3(c) and (e)), was adjusted to 450 nJ. The beam spot at the target was 200 μm (diameter at 1/e2), as measured by a CCD camera calibrated for the UV. To probe the dynamics induced, we selected the HH25 (centred at ∼ 39.5 eV and with a temporal duration of 5 fs, as shown in Fig. 3 (d) and (f)). The focal spot was estimated to be ∼ 120 μm at 1/e2. The experiment was conducted in the gas phase at room temperature. The time delay between the two pulses was controlled by the piezoelectric stage, with a delay step of 5 fs. The kinetic energy of the generated photoelectrons was measured by the TOF spectrometer as a function of the delay between both pulses, obtaining the photoelectron spectrograms in Fig. 5. The UV pump pulse was blocked every 5 delay steps to collect the photoelectron spectrum of the XUV-only signal to correct any variation in the harmonic signal along the scan.
Figure 5(b) shows the average of 4 photoelectron spectrograms in terms of binding energy (i.e., the difference between the probe photon energy and the photoelectron kinetic energy measured by the TOF). Two bands are clearly distinguished in the spectrogram: one at binding energies between 3.4 and 5.3 eV (labelled as I in Fig. 5(b)) and another comprising binding energies between 5.4 and 7 eV (feature II). Recent works25,34 have assigned signal I to the photoionization of the excited state to the ground state of the cation. This band exhibits a strong blueshift, which is a spectroscopic fingerprint of the delay-dependent behaviour of the potential energy surface of the excited state. Signal II shows a more complex structure with a first band peaking at about 6.5 eV, and a second delayed signal appearing at slightly lower binding energies. The former has been attributed to photoionization of 11B to the ground state of the cation, whereas the second to the transition from 21A (or 3A) to the first cationic state25,55. Figure 5(c) shows the temporal profiles of signals I and II and the corresponding fits. A decay time of ∼17.5 fs was found for signal I after deconvolution with the IRF. We tentatively assign this time constant to the time it takes the wave packet to reach the first conical intersection. Band II is instead fitted by the sum of two delayed exponentially modified Gaussian functions, showing decay times of 17.5 fs and 35.4 fs. The short time constant coincides with the decay time of band I and we therefore assign it to the arrival time to the first conical intersection. The second time constant can instead be attributed to the time required to access the second conical intersection, and therefore the relaxation of the system to the electronic ground state.
Discussion
We have described a novel beamline combining UV and XUV pulses for applications in time-resolved spectroscopy. Sub-20 fs UV pump pulses, tuneable between 4.35 and 4.85 eV, were generated by a frequency up-conversion scheme while sub-10 fs XUV probe pulses were spectrally selected between 20 and 45 eV from an HHG source equipped with a TDCM. To the best of our knowledge, we have demonstrated for the first time a full in situ temporal characterization of the UV and XUV pulses by extending IR-XUV cross-correlation methods to the UV domain. In particular, we have performed a two-colour photo-ionization experiment in argon and employed the STRIPE approach to retrieve the information on the pulses. Such measurement has allowed us to obtain a precise value of the IRF, demonstrating a temporal resolution below 20 fs at all pump wavelengths.
We have shown the application of the beamline to the study of the ultrafast dynamics of 1,3-cyclohexadiene (CHD). The high temporal resolution of our setup has allowed us to properly resolve spectroscopic signals characterized by two time constants as short as 17.5 and 35.4 fs. We have tentatively assigned them to the passages through the conical intersections. This provides compelling evidence of the capabilities of this setup for UV pump – XUV probe measurements in molecules. Our beamline therefore offers a unique platform for the future investigation of ultrafast excited-state dynamics triggered by UV light.
Acknowledgements
This project has received funding from the European Union’s Horizon 2020 research and innovation programme (grant agreement No 871161, IMPULSE) and European Research Council (ERC): ERC Synergy grant agreement no. 951224, TOMATTO and ERC StG grant no. 848411, AUDACE. We also thank financial support from Fondazione Cariplo (grant n. 2020–4380, DINAMO), Ministero dell’Università e della Ricerca (202239HFZN) and COST (European Cooperation in Science and Technology) through the COST Action CA18222 (AttoChem). A.C. acknowledges support from the Ministerio de Universidades and the European Union NextGenerationEU/PRTR via her Margarita Salas Fellowship through the University of Salamanca.
Author contributions
R.B.-V., M.N., M.R. and M.L. conceived the experiments. A.C., F.M., S.S., L.M., F.F, L.P., M.L. and R.B.-V. developed the experimental setup. A.C., S.S., F.M., L.M. and F.V. performed the time-resolved measurements. A.C., S.S., L.M., M.L. and R.B.-V. analysed the data. All authors contributed to the discussion of the results. A.C. and R.B.V. wrote the first version of the paper to which all authors contributed.
Data availability
The datasets generated during and/or analysed during the current study are available from the corresponding author on reasonable request.
Declarations
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Borrego-Varillas, R., Lucchini, M. & Nisoli, M. Attosecond spectroscopy for the investigation of ultrafast dynamics in atomic, molecular and solid-state physics. Rep. Prog Phys. 85, 006401 (2022). [DOI] [PubMed] [Google Scholar]
- 2.Stolow, A. Femtosecond time-resolved photoelectron spectroscopy of polyatomic molecules. Annu. Rev. Phys. Chem. 54, 89–119 (2003). [DOI] [PubMed] [Google Scholar]
- 3.Stolow, A., Bragg, A. E. & Neumark, D. M. Femtosecond time-resolved photoelectron spectroscopy. Chem. Rev. 104, 1719–1758 (2004). [DOI] [PubMed] [Google Scholar]
- 4.Suzuki, T. Time-resolved photoelectron spectroscopy of non-adiabatic electronic dynamics in gas and liquid phases. Int. Rev. Phys. Chem. 31, 265–318 (2012). [Google Scholar]
- 5.Stolow, A. Time-resolved photoelectron spectroscopy: Non-adiabatic dynamics in polyatomic molecules. Int. Rev. Phys. Chem. 22, 377–405 (2003). [DOI] [PubMed] [Google Scholar]
- 6.Xue, B., Yabushita, A. & Kobayashi, T. Ultrafast dynamics of uracil and thymine studied using a sub-10 fs deep ultraviolet laser. Phys. Chem. Chem. Phys. 18, 17044–17053 (2016). [DOI] [PubMed] [Google Scholar]
- 7.Borrego-Varillas, R., Ganzer, L., Cerullo, G. & Manzoni, C. Ultraviolet transient absorption spectrometer with sub-20-fs time resolution. Appl. Sci. 8, 989 (2018). [Google Scholar]
- 8.Squibb, R. J. et al. Acetylacetone photodynamics at a seeded free-electron laser. Nat. Commun. 9, 63 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Picchiotti, A. et al. Pyrene, a test case for deep-ultraviolet molecular photophysics. J. Phys. Chem. Lett. 10, 3481–3487 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chergui, M. Ultrafast molecular photophysics in the deep-ultraviolet. J. Chem. Phys. 150, 070901 (2019). [DOI] [PubMed] [Google Scholar]
- 11.Borrego-Varillas, R. et al. Tracking excited state decay mechanisms of pyrimidine nucleosides in real time. Nat. Commun. 12, 7285 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Baum, P., Lochbrunner, S. & Riedle, E. Tunable sub-10-fs ultraviolet pulses generated by achromatic frequency doubling. Opt. Lett. 29, 1686–1688 (2004). [DOI] [PubMed] [Google Scholar]
- 13.Borrego-Varillas, R. et al. Microjoule-level, tunable sub-10 fs UV pulses by broadband sum-frequency generation. Opt. Lett. 39, 3849–3852 (2014). [DOI] [PubMed] [Google Scholar]
- 14.Travers, J. C., Grigorova, T. F., Brahms, C. & Belli, F. High-energy pulse self-compression and ultraviolet generation through soliton dynamics in hollow capillary fibres. Nat. Photonics. 13, 547–554 (2019). [Google Scholar]
- 15.Brahms, C., Grigorova, T., Belli, F. & Travers, J. C. High-energy ultraviolet dispersive-wave emission in compact hollow capillary systems. Opt. Lett. 44, 2990–2993 (2019). [DOI] [PubMed] [Google Scholar]
- 16.Galli, M. et al. Generation of deep ultraviolet sub-2-fs pulses. Opt. Lett. 44, 1308–1311 (2019). [DOI] [PubMed] [Google Scholar]
- 17.Reduzzi, M. et al. Direct temporal characterization of sub-3-fs deep UV pulses generated by resonant dispersive wave emission. Opt. Express. 31, 26854–26864 (2023). [DOI] [PubMed] [Google Scholar]
- 18.Krause, J. L., Schafer, K. J. & Kulander, K. C. High-order harmonic generation from atoms and ions in the high intensity regime. Phys. Rev. Lett. 68, 3535 (1992). [DOI] [PubMed] [Google Scholar]
- 19.Corkum, P. B. Plasma perspective on strong-field multiphoton ionization. Phys. Rev. Lett. 71, 1994–1997 (1993). [DOI] [PubMed] [Google Scholar]
- 20.Lucchini, M. et al. Few-femtosecond C2H4 + internal relaxation dynamics accessed by selective excitation. J. Phys. Chem. Lett. 13, 11169–11175 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Poletto, L. et al. Intense femtosecond extreme ultraviolet pulses by using a time-delay-compensated monochromator. Opt. Lett. 32, 2897–2899 (2007). [DOI] [PubMed] [Google Scholar]
- 22.Poletto, L. et al. Time-delay compensated monochromator for the spectral selection of extreme-ultraviolet high-order laser harmonics. Rev. Sci. Instrum. 80, 123109 (2009). [DOI] [PubMed] [Google Scholar]
- 23.Calegari, F. & Martin, F. Open questions in attochemistry. Commun. Chem. 6, 184 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nishitani, J., West, C. W., Higashimura, C. & Suzuki, T. Time-resolved photoelectron spectroscopy of polyatomic molecules using 42-nm vacuum ultraviolet laser based on high harmonics generation. Chem. Phys. Lett. 684, 397–401 (2017). [Google Scholar]
- 25.Karashima, S. et al. Ultrafast ring-opening reaction of 1,3-cyclohexadiene: identification of nonadiabatic pathway via doubly excited state. J. Am. Chem. Soc. 143, 8034–8045 (2021). [DOI] [PubMed] [Google Scholar]
- 26.Uenishi, R., Boyer, A., Karashima, S., Humeniuk, A. & Suzuki, T. Signatures of conical intersections in extreme ultraviolet photoelectron spectra of furan measured with 15 fs time resolution. J. Phys. Chem. Lett. 15, 2222–2227 (2024). [DOI] [PubMed] [Google Scholar]
- 27.Yamamoto, Y. I. & Suzuki, T. Ultrafast geminate recombination facilitated by hydrogen-atom transfer in charge transfer reactions from hydroxide and methoxide ions. J. Phys. Chem. Lett. 14, 10463–10468 (2023). [DOI] [PubMed] [Google Scholar]
- 28.Orimo, N., Yamamoto, Y. I., Karashima, S., Boyer, A. & Suzuki, T. Ultrafast electronic relaxation in 6-methyluracil and 5-fluorouracil in isolated and aqueous conditions: substituent and solvent effects. J. Phys. Chem. Lett. 14, 2758–2763 (2023). [DOI] [PubMed] [Google Scholar]
- 29.Downes-Ward, B. et al. Photodissociation dynamics of methyl iodide across the A-band probed by femtosecond extreme ultraviolet photoelectron spectroscopy. J. Phys. B: Mol. Opt. Phys. 54, 134003 (2021). [DOI] [PubMed] [Google Scholar]
- 30.Warne, E. M. et al. Time resolved detection of the S(1D) product of the UV induced dissociation of CS2. J. Chem. Phys. 154, 034302 (2021). [DOI] [PubMed] [Google Scholar]
- 31.Titov, E., Hummert, J., Ikonnikov, E., Mitrić, R. & Kornilov, O. Electronic relaxation of aqueous aminoazobenzenes studied by time-resolved photoelectron spectroscopy and surface hopping TDDFT dynamics calculations. Faraday Discuss. 228, 226–241 (2021). [DOI] [PubMed] [Google Scholar]
- 32.Wang, C. et al. Different timescales during ultrafast stilbene isomerization in the gas and liquid phases revealed using time-resolved photoelectron spectroscopy. Nat. Chem. 14, 1126–1132 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Latka, T. et al. Femtosecond wave-packet revivals in ozone. Phys. Rev. A. 99, 063405 (2019). [Google Scholar]
- 34.Karashima, S., Chen, C. J. & Suzuki, T. Generation of sub-10-fs deep and extreme ultraviolet pulses for time-resolved photoemission spectroscopy. Opt. Lett. 49, 3777–3780 (2024). [DOI] [PubMed] [Google Scholar]
- 35.Wanie, V. et al. A flexible beamline combining XUV attosecond pulses with few-femtosecond UV and near-infrared pulses for time-resolved experiments. Rev. Sci. Instrum. 95, 083004 (2024). [DOI] [PubMed] [Google Scholar]
- 36.Koga, M. et al. Extreme ultraviolet time-resolved photoelectron spectroscopy of adenine, adenosine and adenosine monophosphate in a liquid flat jet. Phys. Chem. Chem. Phys. 26, 13106–13117 (2024). [DOI] [PubMed] [Google Scholar]
- 37.Lucchini, M. et al. Few-femtosecond extreme-ultraviolet pulses fully reconstructed by a ptychographic technique. Opt. Express. 26, 6771 (2018). [DOI] [PubMed] [Google Scholar]
- 38.Murari, M., Lucarelli, G. D., Lucchini, M. & Nisoli, M. Robustness of the ePIE algorithm for the complete characterization of femtosecond, extreme ultra-violet pulses. Opt. Express. 28, 10210–10224 (2020). [DOI] [PubMed] [Google Scholar]
- 39.Nisoli, M., De Silvestri, S. & Svelto, O. Generation of high energy 10 fs pulses by a new pulse compression technique. Appl. Phys. Lett. 68, 2793–2795 (1996). [Google Scholar]
- 40.Nisoli, M. Hollow fiber compression technique: a historical perspective. IEEE J. Sel. Top. Quantum Electron. 30, 1–14 (2024). [Google Scholar]
- 41.Delong, K. W., Trebino, R., Hunter, J. & White, W. E. Frequency-resolved optical gating with the use of second-harmonic generation. J. Opt. Soc. Am. B. 11, 2206–2215 (1994). [Google Scholar]
- 42.Tan, H. S., Schreiber, E. & Warren, W. S. High-resolution indirect pulse shaping by parametric transfer. Opt. Lett. 27, 439–441 (2002). [DOI] [PubMed] [Google Scholar]
- 43.Shimizu, S., Nabekawa, Y., Obara, M. & Midorikawa, K. Spectral phase transfer for indirect phase control of sub-20-fs deep UV pulses. Opt. Express. 13, 6345–6353 (2005). [DOI] [PubMed] [Google Scholar]
- 44.Linden, S., Kuhl, J. & Giessen, H. Amplitude and phase characterization of weak blue ultrashort pulses by downconversion. Opt. Lett. 24, 569–571 (1999). [DOI] [PubMed] [Google Scholar]
- 45.Maiden, A. M. & Rodenburg, J. M. An improved ptychographical phase retrieval algorithm for diffractive imaging. Ultramicroscopy. 109, 1256–1262 (2009). [DOI] [PubMed] [Google Scholar]
- 46.Lucchini, M. et al. Ptychographic reconstruction of attosecond pulses. Opt. Express. 23, 29502–29513 (2015). [DOI] [PubMed] [Google Scholar]
- 47.Witting, T. et al. Time-domain ptychography of over-octave-spanning laser pulses in the single-cycle regime. Opt. Lett. 41, 4218–4221 (2016). [DOI] [PubMed] [Google Scholar]
- 48.Lucchini, M. et al. Controlling Floquet states on ultrashort time scales. Nat. Commun. 13, 7103 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Moio, B. et al. Time-frequency mapping of two-colour photoemission driven by harmonic radiation. J. Phys. B: Mol. Opt. Phys. 54, 154003 (2021). [Google Scholar]
- 50.Dolso, G. L. et al. Versatile and robust reconstruction of extreme-ultraviolet pulses down to the attosecond regime. APL Photonics. 8, 076101 (2023). [Google Scholar]
- 51.Fuß, W., Schmid, W. E. & Trushin, S. A. Time-resolved dissociative intense-laser field ionization for probing dynamics: Femtosecond photochemical ring opening of 1,3-cyclohexadiene. J. Chem. Phys. 112, 8347–8362 (2000). [Google Scholar]
- 52.Kosma, K., Trushin, S. A., Fuß, W. & Schmid, W. E. Cyclohexadiene ring opening observed with 13 fs resolution: Coherent oscillations confirm the reaction path. Phys. Chem. Chem. Phys. 11, 172–181 (2009). [DOI] [PubMed] [Google Scholar]
- 53.Attar, A. R. et al. Femtosecond x-ray spectroscopy of an electrocyclic ring-opening reaction. Sci. (1979). 356, 54–59 (2017). [DOI] [PubMed] [Google Scholar]
- 54.Polyak, I., Hutton, L., Crespo-Otero, R., Barbatti, M. & Knowles, P. J. Ultrafast photoinduced dynamics of 1,3-cyclohexadiene using XMS-CASPT2 surface hopping. J. Chem. Theory Comput. 15, 3929–3940 (2019). [DOI] [PubMed] [Google Scholar]
- 55.Travnikova, O. et al. Photochemical ring-opening reaction of 1,3-cyclohexadiene: identifying the true reactive state. J. Am. Chem. Soc. 144, 21878–21886 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Salazar, E. X., Menger, M. F. S. J. & Faraji, S. Ultrafast photoinduced dynamics in 1,3-cyclohexadiene: a comparison of trajectory surface hopping schemes†. J. Chem. Theory Comput. 20, 5796–5806 (2024). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The datasets generated during and/or analysed during the current study are available from the corresponding author on reasonable request.