


Abstract
Adenine paired with 8-hydroxyguanine (oh8G), a major component of oxidative DNA damage, is excised by MYH base excision repair protein in human cells. Since repair activity of MYH protein on an A:G mismatch has also been reported, we compared the repair activity of His6-tagged MYH proteins, expressed in Spodoptera frugiperda Sf21 cells, on A:oh8G and A:G mismatches by DNA cleavage assay and gel mobility shift assay. We also compared the repair ability of type 1 mitochondrial protein with type 2 nuclear protein, as well as of polymorphic type 1-Q324 and 2-Q310 proteins with type 1-H324 and 2-H310 proteins by DNA cleavage assay and complementation assay of an Escherichia coli mutM mutY strain. In a reaction buffer with a low salt (0–50 mM) concentration, adenine DNA glycosylase activity of type 2 protein was detected on both A:oh8G and A:G substrates. However, in a reaction buffer with a 150 mM salt concentration, similar to physiological conditions, the glycosylase activity on A:G, but not on A:oh8G, was extremely reduced and the binding activity of type 2 protein for A:G, but not for A:oh8G, was proportionally reduced. The glycosylase activity on A:oh8G and the ability to suppress spontaneous mutagenesis were greater for type 2 than type 1 enzyme. There was apparently no difference in the repair activities between the two types of polymorphic MYH proteins. These results indicate that human MYH protein specifically catalyzes the glycosylase reaction on A:oh8G under physiological salt concentrations.
INTRODUCTION
8-Hydroxyguanine (oh8G) is a modified base of oxidative DNA damage and the formation of oh8G in DNA is involved in mutagenesis (1–3). The OGG1 and MYH proteins (4–16), which are functional homologs of the Escherichia coli MutM and MutY proteins, respectively (17,18), have been identified as enzymes which repair oh8G in DNA in human cells. OGG1 possesses the ability to excise oh8G paired with cytosine (4–12), while MYH possesses the ability to excise adenine paired with oh8G (14–16). Since repair activity of MYH on an A:G mismatch had also been reported (15,16), we investigated the differences in the catalytic activities of human MYH protein towards A:oh8G and A:G substrates, in comparison with those of Schizosaccharomyces pombe MYH protein (19).
Multiple forms of human MYH proteins are expressed in human cells due to multiple transcription initiation sites and alternative splicing of MYH mRNA transcripts (14,16). Type 1 protein is localized in the mitochondria, while type 2 protein, which lacks the N-terminal 14 amino acids of type 1 protein, is localized in the nucleus (14,16). Thus, we also compared the repair activities of type 1 and 2 proteins in vitro and in vivo in this study. In a report of the first cloning and sequencing of the MYH gene, a G/C nucleotide variation in exon 12 among cDNA clones was described (13). This nucleotide variation is associated with an amino acid substitution (Gln/His) and was found to be a common single nucleotide polymorphism (SNP) in our studies (Shinmura et al., unpublished data). Thus, four major types of MYH protein, type 1-Q324, type 1-H324, type 2-Q310 and type 2-H310, are expressed in human cells. In the human OGG1 gene we previously identified a Ser326Cys SNP as being associated with distinct oh8G repair activity in the complementation assay of an E.coli mutM mutY strain (20,21). In addition, it was shown that the catalytic efficiency (kcat/Km) of these two polymorphic proteins differed significantly by 2-fold for excision of oh8G from γ-irradiated DNA (22,23). With reference to this example, we also compared the repair activities of these polymorphic MYH proteins in vitro and in vivo.
MATERIALS AND METHODS
Preparation of DNA substrates
The sequences of oligonucleotides used as substrates or as a size marker are listed in Table 1. Three sets of double-stranded oligonucleotides, A:G (Sequences 1 and 2), A:T (Sequences 1 and 3) and A:oh8G (Sequences 1 and 4), were prepared and 3′-end-labeled with Exo– Klenow (Stratagene) and [α-32P]dCTP (Amersham), as previously described (11).
Table 1. Oligonucleotides used in this study.
| Name | Sequence |
|---|---|
| Sequence 1 | 5′-TTGGGGAATGAGTCAGGCCAC-3′ |
| Sequence 2 | 5′-GGTGGCCTGACGCATTCCCCAA-3′ |
| Sequence 3 | 5′-GGTGGCCTGACTCATTCCCCAA-3′ |
| Sequence 4 | 5′-GGTGGCCTGACG*CATTCCCCAA-3′ |
| Sequence 5 | 5′-GTCAGGCCACC-3́ |
G*, 8-hydroxydeoxyguanosine.
Preparation of human and S.pombe MYH expression vectors
PCR products with the coding sequences for types 1-Q324 and 1-H324 and types 2-Q310 and 2-H310 human MYH proteins were inserted into the EcoRI site of the pGEX1λT (Pharmacia) and pFASTBAC HTa (Gibco BRL) vectors. DNA prepared from an S.pombe cDNA library was used as a template in a PCR amplification of the coding sequence for the S.pombe MYH gene. The PCR product was inserted into the BamHI site of the pGEX1λT and pFASTBAC HTb (Gibco BRL) vectors. pGEX1λT-human OGG1 type 1a-Ser326 was prepared as previously (11).
Preparation of MYH proteins
Type 1-Q324/1-H324 and type 2-Q310/2-H310 human MYH proteins and S.pombe MYH proteins, tagged with His6 at their N-terminal ends, were prepared using the BAC-TO-BAC Baculovirus Expression System (Gibco BRL) according to the manufacturer’s protocol. Briefly, plasmid vectors pFASTBAC HTa/human MYH and pFASTBAC HTb/S.pombe MYH were transfected into DH10BAC competent cells which contain the bacmid with a mini-attTn7 target site and the helper plasmid. After selection of colonies containing recombinant bacmids, recombinant bacmid DNAs were isolated and transfected into Spodoptera frugiperda Sf21 cells. Recombinant MYH proteins were expressed in Sf21 cells by infection of the viral stocks and purified with TALON metal affinity resins (Clontech). The proteins were then dialyzed against buffer containing 10 mM Tris–HCl pH 7.6, 0.5 mM dithiothreitol, 0.5 µg/ml leupeptin, 0.5 µg/ml pepstatin, 0.5 µg/ml chymostatin, 0.5 µg/ml antipain, 1 mM phenylmethylsulfonyl fluoride and 10% glycerol. MYH protein concentrations were determined by western blot analysis using an anti-Tetra His antibody (Qiagen) with recombinant His6-tagged chloramphenicol acetyltransferase protein as a quantitative control. The concentration of chloramphenicol acetyltransferase was measured using the method of Bradford (24). Western blot analyses were performed as described previously (25).
DNA cleavage assay
The reaction mixture contained 12 nM MYH protein, 10 mM Tris–HCl pH 7.6, 0.5 mM dithiothreitol, 0.5 mM EDTA, 1.45% glycerol, 50 µg/ml bovine serum albumin, 2 nM labeled DNA and various concentrations of NaCl. The standard reaction was performed with 20 µl of the mixture at 37°C for 60 min. After the reaction, two separate 9 µl aliquots were taken and one aliquot was exposed to 0.1 M NaOH and heated to 95°C for 4 min. Then, 10 µl of denaturing formamide dye was added to both aliquots. The aliquots were then heat denatured and subjected to 7 M urea–10% polyacrylamide gel electrophoresis. Sequence 5 in Table 1 was 5′-end-labeled, as described previously (11), and used as the size marker of cleaved products. The radioactivities of intact and cleaved oligonucleotides were quantified with a bioimaging analyzer (BAS2000; Fuji Photo Film, Tokyo). For a kinetic study of DNA cleavage, MYH proteins were reacted with various amounts (0.5, 1, 2, 4 and 8 nM) of the A:oh8G substrate in a buffer containing 150 mM NaCl at 37°C for 15 min. Lineweaver–Burk plots representing the reciprocal of initial rates of adenine excision versus the reciprocal of substrate concentrations were utilized to determine the Michaelis constant (Km) and the catalytic constant (kcat).
Gel mobility shift assay
Reactions were performed under the same conditions as for the DNA cleavage assay, except for the incubation time of 30 min. Protein–DNA complexes were analyzed as described previously (11).
Complementation assay
pGEX1λT-human MYH, pGEX1λT-S.pombe MYH and pGEX1λT-human OGG1 were used for the complementation assay. The detailed protocol has been described previously (5). Briefly, overnight cultures of YG5132 cells transformed with MYH- or OGG1-overproducing plasmids and of control cells were analyzed for rifampicin-resistant (RifR) mutation events. In each group, four to six plates were prepared and the average numbers of RifR colonies/108 cells were recorded.
RESULTS
Preparation of human and S.pombe MYH proteins
For functional analysis of human MYH proteins we first tried to express GST–MYH fusion proteins in E.coli. However, we could not obtain any bacterial clones with expression of the fusion proteins. Therefore, we next constructed baculovirus expression vectors for human and S.pombe MYH proteins. Vectors for polymorphic MYH proteins of both mitochondrial type (1-Q324 and 1-H324) and nuclear type (2-Q310 and 2-H310), as well as S.pombe MYH protein, were prepared and expressed in Sf21 insect cells. Since the pFASTBAC HT vector with a His6 tag was used for expression of these proteins, human and S.pombe proteins were N-terminally fused with polypeptides with a predicted molecular size of ∼3.7 and 3.3 kDa, respectively. The levels of MYH protein expression in Sf21 cells were low, as in the case of MYH protein expression in bacteria (14–16), however, purified MYH proteins were detected by western blot analysis using an anti-Tetra His antibody. Human MYH type 1 and type 2 and S.pombe MYH proteins with molecular sizes of 64, 62 and 57 kDa, respectively, were detected (Fig. 1). The sizes determined by western blot analysis corresponded to those calculated from the His6–MYH cDNA sequences. Therefore, we used these proteins for further analyses.
Figure 1.
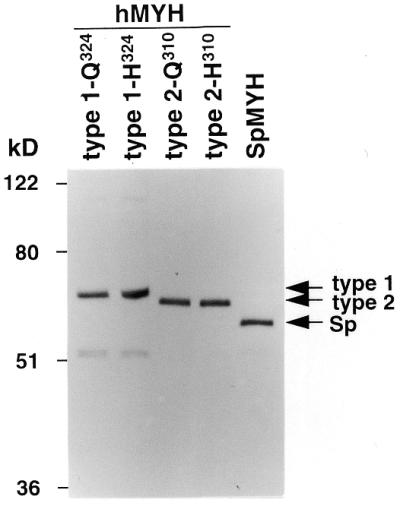
Detection of MYH proteins by western blot analysis. Human MYH (hMYH) types 1-Q324, 1-H324, 2-Q310 and 2-H310 and S.pombe MYH (SpMYH) proteins were expressed in Sf21 cells and purified with TALON metal affinity resins. Twenty nanograms of the purified MYH proteins were applied to each lane. MYH proteins N-terminally fused with polypeptides containing a His6 tag were detected by western blot analysis using an anti-Tetra His antibody. Arrows indicate the positions of the proteins.
Adenine DNA glycosylase and apurinic/apyrimidinic (AP) lyase activities of MYH protein
Type 2-Q310 protein was first prepared as a representative nuclear form of MYH proteins and used for analysis of adenine DNA glycosylase activity and AP lyase activity on the A:oh8G mismatch. These activities were evaluated initially with NaCl-free reaction buffer, which was previously used for analysis of S.pombe MYH protein (19). Since NaOH treatment is known to cleave any AP sites generated by DNA glycosylases (26,27), this treatment was adapted to distinguish adenine DNA glycosylase activity from AP lyase activity. In other words, AP lyase activity was assessed without NaOH treatment of DNA substrates after reaction with MYH proteins, whereas DNA glycosylase activity was assessed after NaOH treatment. Oligonucleotides containing A:T, A:oh8G or A:G reacted with type 2-Q310 protein were treated or not with NaOH before loading onto the gel and the percentage of cleaved products per total oligonucleotides was calculated (Fig. 2). The protein had an activity cleaving oligonucleotides with an A:oh8G or A:G mismatch but did not have an activity cleaving oligonucleotides with A:T (Fig. 2A). Such an activity was more prominent with NaOH treatment than without NaOH treatment. This result indicated that human MYH protein possesses adenine DNA glycosylase activity and AP lyase activity against both A:oh8G and A:G mismatches. AP lyase activity was also shown by production of a stable covalent MYH protein–DNA complex with DNA containing an A:oh8G or A:G mismatch in the presence of borohydride (data not shown).
Figure 2.
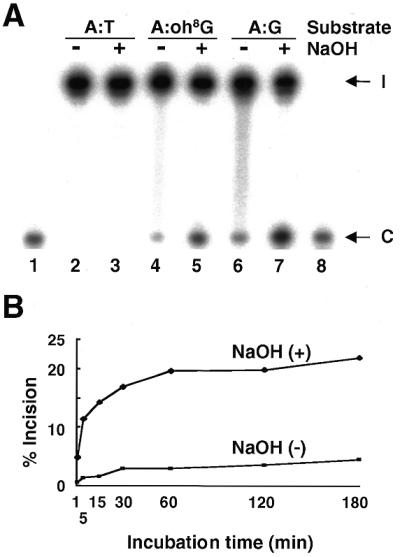
Detection of the glycosylase and AP lyase activities of MYH type 2-Q310 protein. (A) Substrate specificity of the DNA cleavage reaction. The reactivity of the 2-Q310 protein on 3′-end-labeled 22mer oligonucleotides containing either an A:T (lanes 2 and 3), A:oh8G (lanes 4 and 5) or A:G (lanes 6 and 7) base pair at position 12 from the labeled end were examined with an incubation time of 60 min. Samples with (+) or without (–) NaOH treatment were loaded onto the denaturing polyacrylamide gel. The positions of intact (I) and cleaved (C) oligonucleotides are indicated by arrows (lanes 2–7). A 5′-labeled 11mer marker was co-electrophoresed for confirmation of the size of cleaved products (lanes 1 and 8). (B) Time dependency of the DNA cleavage reaction. 3′-End-labeled oligonucleotides containing an A:oh8G mismatch were reacted with 2-Q310 protein for the period indicated. Samples with (+) or without (–) NaOH treatment were loaded onto the gel and the radioactivities of the intact and cleaved oligonucleotides were quantified. The percentages of cleaved products per total oligonucleotides were calculated.
The time-course assay on A:oh8G repair activity of 2-Q310 protein indicated that the amount of cleaved products plateaued within 60 min and was steady in the period of 60–180 min (Fig. 2B). The percentage of AP lyase activity relative to DNA glycosylase activity was ∼15% throughout the course of the experiment. Thus, AP lyase activity of MYH protein almost correlates with adenine DNA glycosylase activity of MYH protein and the reaction time of 60 min was considered to be enough to evaluate these activities. Based on these results, DNA glycosylase activity with NaOH treatment was assessed with a reaction time of 60 min in further experiments.
Salt requirement of the adenine DNA glycosylase activity
We next investigated the effect of NaCl concentration on the DNA glycosylase activity of MYH protein. Type 2-Q310 protein was reacted with either an A:oh8G or A:G substrate in a buffer containing 0–200 mM NaCl (Fig. 3A and B). At low NaCl concentrations the activity against the A:G substrate was higher than that against the A:oh8G substrate. However, the activity of the protein on the A:G substrate was extremely reduced in buffers with >100 mM NaCl, whereas that on the A:oh8G substrate was retained in buffers containing 0–150 mM NaCl. In a report on the repair activity of S.pombe MYH protein, no activity on the A:G substrate at 80 mM NaCl was detected and no experiment on A:oh8G was performed under other than NaCl-free conditions (19). Thus, the activity of human MYH protein on A:G was similar to that of S.pombe MYH protein. On the other hand, it was revealed in this study that the glycosylase activity of the human protein on A:oh8G was retained up to 150 mM NaCl.
Figure 3.

Effect of NaCl concentration on the DNA repair properties of MYH proteins. (A) Effect of NaCl concentration on the glycosylase activity of MYH type 2-Q310 protein was examined by DNA cleavage assay. Oligonucleotides containing an A:G (G) or A:oh8G (8) mismatch were reacted with the protein for 60 min under various NaCl concentrations (mM) and subjected to gel electrophoresis after NaOH treatment. The positions of intact (I) and cleaved (C) oligonucleotides are indicated by arrows. The 5′-labeled 11mer marker was co-electrophoresed on the left for confirmation of the size of the cleaved product. (B) Percentages of oligonucleotides cleaved by MYH protein in the experiment in (A) were calculated and are shown as bars. (C) The effect of NaCl concentration on the binding activity of type 2-Q310 protein to oligonucleotides with an A:oh8G (upper), A:G (middle) or A:T (lower) mismatch was examined by gel mobility shift assay. Reaction mixtures were electrophoresed on a 6% non-denaturing polyacrylamide gel. The specific protein–DNA binding complex under the condition of NaCl concentrations corresponding to experiment (A) is shown.
To clarify whether the effect of NaCl concentration on glycosylase activity is associated with binding activity of the protein to the substrates, the effect of salt concentration on the affinity of MYH protein for both substrates was examined by gel mobility shift assay. The specific binding complexes of 2-Q310 protein and oligonucleotides containing either an A:oh8G or A:G mismatch were compared to each other under various NaCl concentrations (Fig. 3C). Oligonucleotides containing the normal A:T base pair instead of a mismatch base pair were used as a control. The binding activity of type 2-Q310 protein on the A:oh8G substrate was similar at 0–150 mM NaCl, while that on the A:G substrate was extremely reduced at >100 mM NaCl. Thus, binding activity correlated well with glycosylase activity of this protein. These results indicate that MYH glycosylase activity is dependent on NaCl concentration and is regulated by the affinity of the protein for the mismatched DNA.
Next, to elucidate whether glycosylase activity is also affected by KCl, instead of NaCl, we examined activity in a buffer with KCl concentrations of 0–170 mM (Fig. 4A). KCl had the same effect as NaCl on glycosylase activity of the protein. Physiologically, intracellular concentrations of Na+ and K+ ions are maintained at 5–15 and 140 mM, respectively (28), and the nuclear envelop is non-selective for Na+ and K+ ions (29). With reference to these reports, glycosylase activity of the type 2 nuclear protein was further examined under conditions of 10 mM NaCl with various concentrations of KCl (Fig. 4B). The activity on the A:G substrate almost disappeared at 10 mM NaCl + 90 mM KCl, whereas activity on the A:oh8G substrate was retained at up to 10 mM NaCl + 160 mM KCl. Thus it was concluded that glycosylase activity of MYH protein was different in its salt (NaCl and/or KCl) requirements between A:oh8G and A:G substrates. It was also concluded that an A:G mismatch was poorly repaired by MYH protein at a 150 mM salt concentration, which is similar to physiological conditions. Glycosylase activity of S.pombe MYH protein was also examined under various NaCl concentrations (Fig. 4C). The trend of activity of the S.pombe protein against the two substrates was similar to that of the human protein. Thus, the distinct salt requirement of glycosylase activity against A:oh8G and A:G is conserved between S.pombe and human proteins.
Figure 4.

Effect of salt concentrations on the glycosylase activity of MYH proteins. Effect of KCl (A) or NaCl + KCl (B) concentration on the glycosylase activity of type 2-Q310 protein examined by DNA cleavage assay. (C) Effect of NaCl concentration on the glycosylase activity of S.pombe MYH protein examined by DNA cleavage assay. Oligonucleotides containing an A:G or A:oh8G mismatch were reacted under various salt concentrations (mM) and subjected to gel electrophoresis after NaOH treatment. The percentages of oligonucleotides cleaved by MYH proteins were calculated and are shown as bars.
Comparison of the glycosylase activity of the type 1 and type 2 proteins
Both mitochondrial type 1 and nuclear type 2 proteins are expressed in human cells. Thus, glycosylase activity on an A:oh8G mismatch at a 150 mM salt concentration was compared between the 2-Q310 and 1-Q324 proteins (Fig. 5). The activity of type 2-Q310 was 2.7 times higher than that of type 1-Q324 after 60 min reaction. To further confirm the difference in the activities of the type 1 and type 2 proteins, the Michaelis constant (Km) and the catalytic constant (kcat) of the glycosylase reaction of these proteins for an A:oh8G mismatch were determined in a reaction buffer with a 150 mM salt concentration (Table 2). The kcat/Km value, representing catalytic efficiency, of type 2-Q310 protein was 2 times higher than that of type 1-Q324 protein. Thus, it was indicated that glycosylase activity of the type 2 protein is greater than that of the type 1 protein.
Figure 5.
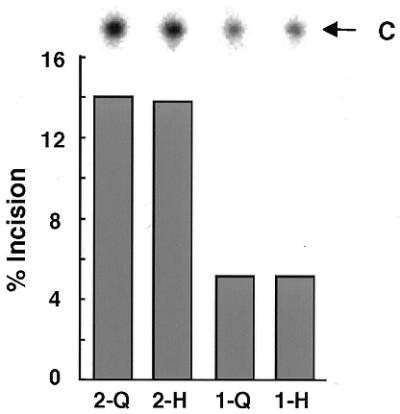
Comparison of glycosylase activity between type 2 and type 1 as well as between polymorphic MYH proteins. Oligonucleotides containing an A:oh8G mismatch were reacted with 2-Q310 (2-Q), 2-H310 (2-H), 1-Q324 (1-Q) or 1-H324 (1-H) protein in the 150 mM NaCl reaction buffer for 60 min. Products treated with NaOH were subjected to gel electrophoresis after heating. The percentages of oligonucleotides cleaved by each protein are presented as bars. A photograph of each cleaved product (C) is shown at the top.
Table 2. Kinetic constants of the glycosylase reaction of MYH proteins on an A:oh8G substrate.
| Type of protein | Km (nM) | kcat (min–1) | kcat/Km (min–1·µM–1) |
|---|---|---|---|
| MYH type 1-Q324 | 1.0 | 5.7 × 10–4 | 0.55 |
| MYH type 1-H324 | 1.2 | 5.8 × 10–4 | 0.50 |
| MYH type 2-Q310 | 2.1 | 2.6 × 10–3 | 1.22 |
| MYH type 2-H310 | 2.2 | 2.1 × 10–3 | 0.99 |
Comparison of the glycosylase activity among polymorphic proteins
A G/C SNP in exon 12 in the MYH gene results in production of type 1-Q324/1-H324 and type 2-Q310/2-H310 proteins. To elucidate whether or not there are interindividual variations in MYH repair ability due to this SNP, glycosylase activity on the A:oh8G mismatch at 150 mM NaCl was compared among these polymorphic proteins. The percentage incision value (Fig. 5) and the kcat/Km value (Table 2) were similar between the type 1-Q324 and 1-H324 proteins as well as between the type 2-Q310 and 2-H310 proteins. Thus, it was indicated that both of the polymorphic MYH proteins efficiently catalyze the glycosylase reaction at similar levels.
Comparison of the ability to suppress spontaneous mutagenesis between type 1 and type 2 proteins as well as polymorphic proteins
The ability to suppress spontaneous mutagenesis was also compared between the type 1 and type 2 as well as polymorphic proteins. Since strain YG5132 is a mutM mutY mutant of E.coli strain CC104, the numbers of RifR mutants in YG5132 transformed with plasmids containing each of the GST–MYH cDNA fragments were compared (Table 3). Plasmids expressing GST–S.pombe MYH and GST–human OGG1 proteins were used as positive controls. The number of RifR YG5132 colonies was significantly higher than that of RifR CC104 colonies. All of the GST–human MYH fusion proteins had activities suppressing spontaneous mutagenesis of the YG5132 strain, as in the cases of the GST–S.pombe MYH and GST–human OGG1 fusion proteins. The activity of type 2 was greater than that of type 1 protein, correlating with the difference in glycosylase activity between them. The activity was similar between polymorphic proteins, also correlating with the similarity in glycosylase activity between them. Based on the analyses of glycosylase activity in vitro and suppression activity in vivo, we concluded that type 2 protein possesses greater repair ability than type 1 protein and that polymorphic proteins of the Gln and His types catalyze the DNA repair reaction at similar levels.
Table 3. Complementation of an E.coli mutM mutY mutant with MYH-overproducing plasmids.
| E.coli strain | Plasmid | No. of RifR cells/108 cellsa |
|---|---|---|
| CC104 | – | 1.7 ± 0.91 |
| YG5132b | – | 516.1 ± 94.6 |
| YG5132 | pGEX-1λT | 402.1 ± 87.1 |
| YG5132 | GST–human MYH type 1-Q324 | 111.1 ± 27.6 |
| YG5132 | GST–human MYH type 1-H324 | 90.9 ± 35.1 |
| YG5132 | GST–human MYH type 2-Q310 | 14.4 ± 6.3 |
| YG5132 | GST–human MYH type 2-H310 | 11.3 ± 2.8 |
| YG5132 | GST–S.pombe MYH | 1.4 ± 0.76 |
| YG5132 | GST–human OGG1 type 1a-Ser326 | 4.6 ± 1.67 |
aMean ± SE is shown.
bStrain YG5132 is a mutM mutY double mutant of E.coli CC104.
DISCUSSION
In the analysis of the DNA repair reaction by DNA cleavage assay and gel mobility shift assay type 2 nuclear protein possessed glycosylase activity as well as binding activity to both A:oh8G and A:G mismatches in a reaction buffer with a low salt concentration. However, in a reaction buffer with a 150 mM salt concentration, which is similar to the physiological condition in the cells, glycosylase activity and affinity of the type 2 protein for A:G, but not A:oh8G, were extremely reduced. Thus, it was strongly indicated that an adenine paired with oh8G is physiologically more preferable as a substrate of MYH protein than an A:G mismatch. The glycosylase activity of MYH protein under the physiological salt concentration is likely to reflect the repair activity in vivo.
The salt concentration-dependent differences in glycosylase activity of the MYH proteins between each mismatch was regulated, at least in part, by the affinity of the MYH proteins for these substrates. Thus, the factor influencing affinity between the protein and DNA substrates may affect the differences in glycosylase activity. In relation to DNA substrates, the presence or absence of a C8-keto group in guanine causes the distinct nature of hydrogen bonding between A:oh8G and A:G (30,31). This difference may be related to the distinct level of dissociation of the protein from substrate DNA when the salt concentration is elevated, although the detailed mechanism is unclear at present. In general, in vitro studies on the efficiency of DNA repair activity are not always done under physiological salt concentrations. However, for instance, G:T mismatch repair activity in vitro was reported to be remarkably different between the physiological and other salt concentrations (32). A difference in DNA cleavage activity of APEX protein was also detected between these two conditions (33). Thus, the experiment at physiological salt concentration should be important in understanding DNA repair activity in vivo.
In both the DNA cleavage and complementation assays the activity of type 2 protein was greater than that of type 1 protein. This result may imply a higher repair ability of the nuclear MYH form compared with the mitochondrial MYH form. Such a difference may be associated with the greater number of nucleotides in chromosomal DNA, compared with those in mitochondrial DNA. However, the type 1 protein is suggested to be processed during mitochondrial transport in cells and the mature form of the protein is unclear (14). Therefore, repair activity of the full-length mitochondrial form may not always reflect the activity in vivo.
Both the type 1-Q324 and 1-H324, as well as the type 2-Q310 and 2-H310, polymorphic proteins showed glycosylase activity and an ability to suppress spontaneous mutagenesis at similar levels. Thus, this SNP is likely to be neutral with respect to repair activity. However, it is possible that addition of GST or His6 polypeptides to native proteins has made it difficult to detect a difference in repair capacities of these polymorphic proteins. Since this SNP exists outside the catalytic domain for DNA glycosylase activity, it is also possible that other properties may differ between these polymorphic proteins.
In this study it was indicated that human MYH protein possesses both adenine DNA glycosylase activity and AP lyase activity. The percentage of AP lyase activity relative to DNA glycosylase activity of MYH protein was ∼15% in the time-course experiment (Fig. 2B). Such a low AP lyase activity of MYH protein in comparison with its DNA glycosylase activity has also been reported by others (15,16,34). In contrast, there is controversy whether E.coli MutY protein possesses AP lyase activity in addition to DNA glycosylase activity (35). Low AP lyase activity relative to DNA glycosylase activity of MutY protein was reported, while a similar level of AP lyase activity to glycosylase activity of the protein was also reported (35). It has been thought that formation of a covalent DNA–protein complex by borohydride reduction of the Schiff base intermediate is a characteristic of DNA glycosylases with AP lyase activity. However, Williams and David demonstrated that MutY protein possesses only glycosylase activity and not AP lyase activity, in spite of forming a covalent complex (35). Thus, although the covalent complex of human MYH protein and substrate DNA was detected in our preliminary experiment, such a finding would not be enough to support the presence of AP lyase activity in MYH protein. Since the AP lyase activity of MYH protein was much lower than its glycosylase activity so far reported, as well as examined in this study, further studies will be needed to elucidate the catalytic mechanism of eukaryotic and prokaryotic MYH proteins.
In summary, it is indicated here that MYH protein specifically catalyzes the glycosylase reaction on A:oh8G under physiological salt concentrations. This approach will provide new insights into the DNA repair machinery surrounding MYH on oxidatively damaged and mismatched DNA in vivo.
Acknowledgments
ACKNOWLEDGEMENTS
We acknowledge the generous gift of a S.pombe cDNA library by Dr Hiroto Okayama (University of Tokyo, Japan). We thank Hiroshi Kasai and Katsuyoshi Fujikawa of the University of Occupational and Environmental Health for help in the determination of the kinetic constants. This work was supported in part by Grants-in-Aid from the Ministry of Health and Welfare for the 2nd term Comprehensive 10-Year Strategy for Cancer Control and from the Ministry of Education, Science, Sports and Culture of Japan.
REFERENCES
- 1.Kasai H. and Nishimura,S. (1991) Formation of 8-hydroxyguanosine in DNA by oxygen radicals and its biological significance. In Sies,H. (ed.), Oxidative Stress: Oxidants and Antioxidants. Academic Press, London, UK, pp. 99–116.
- 2.Cheng K.C., Cahill,D.S., Kasai,H., Nishimura,S. and Loeb,L.A. (1992) 8-Hydroxyguanine, an abundant form of oxidative DNA damage, causes G→T and A→C substitutions. J. Biol. Chem., 267, 166–172. [PubMed] [Google Scholar]
- 3.Shibutani S., Takeshita,M. and Grollman,A.P. (1991) Insertion of specific bases during DNA synthesis past the oxidation-damaged base 8-oxodG. Nature, 349, 431–434. [DOI] [PubMed] [Google Scholar]
- 4.Aburatani H., Hippo,Y., Ishida,T., Takashima,R., Matsuba,C., Kodama,T., Takao,M., Yasui,A., Yamamoto,K., Asano,M., Fukasawa,K., Yoshinari,T., Inoue,H., Ohtsuka,E. and Nishimura,S. (1997) Cloning and characterization of mammalian 8-hydroxyguanine-specific DNA glycosylase/apurinic, apyrimidinic lyase, a functional mutM homologue. Cancer Res., 57, 2151–2156. [PubMed] [Google Scholar]
- 5.Arai K., Morishita,K., Shinmura,K., Kohno,T., Kim,S.-R., Nohmi,T., Taniwaki,S., Ohwada,S. and Yokota,J. (1997) Cloning of a human homolog of the yeast OGG1 gene that is involved in the repair of oxidative DNA damage. Oncogene, 14, 2857–2861. [DOI] [PubMed] [Google Scholar]
- 6.Lu R., Nash,H.M. and Verdine,G.L. (1997) A mammalian DNA repair enzyme that excises oxidatively damaged guanines maps to a locus frequently lost in lung cancer. Curr. Biol., 7, 397–407. [DOI] [PubMed] [Google Scholar]
- 7.Radicella J.P., Dherin,C., Desmaze,C., Fox,M.S. and Boiteux,S. (1997) Cloning and characterization of hOGG1, a human homolog of the OGG1 gene of Saccharomyces cerevisiae. Proc. Natl Acad. Sci. USA, 94, 8010–8015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rosenquist T.A., Zharkov,D.O. and Grollman,A.P. (1997) Cloning and characterization of a mammalian 8-oxoguanine DNA glycosylase. Proc. Natl Acad. Sci. USA, 94, 7429–7434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bjoras M., Luna,L., Johnsen,B., Hoff,E., Haug,T., Rognes,T. and Seeberg,E. (1997) Opposite base-dependent reactions of a human base excision repair enzyme on DNA containing 7,8-dihydro-8-oxoguanine and abasic sites. EMBO J., 16, 6314–6322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Roldan-Arjona T., Wei,Y., Carter,K.C., Klungland,A., Anselmino,C., Wang,R., Augustus,M. and Lindahl,T. (1997) Molecular cloning and functional expression of a human cDNA encoding the antimutator enzyme 8-hydroxyguanine-DNA glycosylase. Proc. Natl Acad. Sci. USA, 94, 8016–8020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shinmura K., Kasai,H., Sasaki,A., Sugimura,H. and Yokota,J. (1997) 8-Hydroxyguanine (7,8-dihydro-8-oxoguanine) DNA glycosylase and AP lyase activities of hOGG1 protein and their substrate specificity. Mutat. Res., 385, 75–82. [DOI] [PubMed] [Google Scholar]
- 12.Tani M., Shinmura,K., Kohno,T., Shiroishi,T., Wakana,S., Kim,S.-R., Nohmi,T., Kasai,H., Takenoshita,S., Nagamachi,Y. and Yokota,J. (1998) Genomic structure and chromosomal localization of the mouse Ogg1 gene that is involved in the repair of 8-hydroxyguanine in DNA damage. Mamm. Genome, 9, 32–37. [DOI] [PubMed] [Google Scholar]
- 13.Slupska M.M., Baikalov,C., Luther,W.M., Chiang,J.H., Wei,Y.F. and Miller,J.H. (1996) Cloning and sequencing a human homolog (hMYH) of the Escherichia coli mutY gene whose function is required for the repair of oxidative DNA damage. J. Bacteriol., 178, 3885–3892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Takao M., Zhang,Q.M., Yonei,S. and Yasui,A. (1999) Differential subcellular localization of human MutY homolog (hMYH) and the functional activity of adenine:8-oxoguanine DNA glycosylase. Nucleic Acids Res., 27, 3638–3644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Slupska M.M., Luther,W.M., Chiang,J.H., Yang,H. and Miller,J.H. (1999) Functional expression of hMYH, a human homolog of the Escherichia coli MutY protein. J. Bacteriol., 181, 6210–6213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ohtsubo T., Nishioka,K., Imaiso,Y., Iwai,S., Shimokawa,H., Oda,H., Fujiwara,T. and Nakabeppu,Y. (2000) Identification of human MutY homolog (hMYH) as a repair enzyme for 2-hydroxyadenine in DNA and detection of multiple forms of hMYH located in nuclei and mitochondria. Nucleic Acids Res., 28, 1355–1364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Boiteux S., O’Connor,T.R. and Laval,J. (1987) Formamidopyrimidine-DNA glycosylase of Escherichia coli: cloning and sequencing of the fpg structural gene and overproduction of the protein. EMBO J., 6, 3177–3183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Au K.G., Cabrera,M., Miller,J.H. and Modrich,P. (1988) Escherichia coli mutY gene product is required for specific A-G→C·G mismatch correction. Proc. Natl Acad. Sci. USA, 85, 9163–9166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lu A.L. and Fawcett,W.P. (1998) Characterization of the recombinant MutY homolog, an adenine DNA glycosylase, from yeast Schizosaccharomyces pombe. J. Biol. Chem., 273, 25098–25105. [DOI] [PubMed] [Google Scholar]
- 20.Kohno T., Shinmura,K., Tosaka,M., Tani,M., Kim,S.-R., Sugimura,H., Nohmi,T., Kasai,H. and Yokota,J. (1998) Genetic polymorphisms and alternative splicing of the hOGG1 gene, that is involved in the repair of 8-hydroxyguanine in damaged DNA. Oncogene, 16, 3219–3225. [DOI] [PubMed] [Google Scholar]
- 21.Shinmura K., Kohno,T., Kasai,H., Koda,K., Sugimura,H. and Yokota,J. (1998) Infrequent mutations of the hOGG1 gene, that is involved in the excision of 8-hydroxyguanine in damaged DNA, in human gastric cancer. Jpn. J. Cancer Res., 89, 825–828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dherin C., Radicella,J.P., Dizdaroglu,M. and Boiteux,S. (1999) Excision of oxidatively damaged DNA bases by the human alpha-hOgg1 protein and the polymorphic alpha-hOgg1(Ser326Cys) protein which is frequently found in human populations. Nucleic Acids Res., 27, 4001–4007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Audebert M., Radicella,J.P. and Dizdaroglu,M. (2000) Effect of single mutations in the OGG1 gene found in human tumors on the substrate specificity of the Ogg1 protein. Nucleic Acids Res., 28, 2672–2678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bradford M.M. (1976) A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein–dye binding. Anal. Biochem., 72, 248–254. [DOI] [PubMed] [Google Scholar]
- 25.Shinmura K., Kohno,T., Takeuchi-Sasaki,M., Maeda,M., Segawa,T., Kamo,T., Sugimura,H. and Yokota,J. (2000) Expression of the OGG1-type 1a (nuclear form) protein in cancerous and non-cancerous human cells. Int. J. Oncol., 16, 701–707. [DOI] [PubMed] [Google Scholar]
- 26.Porello S.L., Leyes,A.E. and David,S.S. (1998) Single-turnover and pre-steady-state kinetics of the reaction of the adenine glycosylase MutY with mismatch-containing DNA substrates. Biochemistry, 37, 14756–14764. [DOI] [PubMed] [Google Scholar]
- 27.Michaels M.L., Tchou,J., Grollman,A.P. and Miller,J.H. (1992) A repair system for 8-oxo-7,8-dihydrodeoxyguanine. Biochemistry, 31, 10964–10968. [DOI] [PubMed] [Google Scholar]
- 28.Alberts B., Bray,D., Lewis,J., Raff,M., Roberts,K. and Watson,J.D. (1994) Principles of membrane transport. In Molecular Biology of the Cell, 3rd Edn. Garland Publishing, New York, NY, pp. 508–512.
- 29.Oberleithner H., Schuricht,B., Wunsch,S., Schneider,S. and Puschel,B. (1993) Role of H+ ions in volume and voltage of epithelial cell nuclei. Pflugers Arch., 423, 88–96. [DOI] [PubMed] [Google Scholar]
- 30.McAuley-Hecht K.E., Leonard,G.A., Gibson,N.J., Thomson,J.B., Watson,W.P., Hunter,W.N. and Brown,T. (1994) Crystal structure of a DNA duplex containing 8-hydroxydeoxyguanine-adenine base pairs. Biochemistry, 33, 10266–10270. [DOI] [PubMed] [Google Scholar]
- 31.Gao X.L. and Patel,D.J. (1988) G(syn)·A(anti) mismatch formation in DNA dodecamers at acidic pH: pH-dependent conformational transition of G·A mispairs detected by proton NMR. J. Am. Chem. Soc., 110, 5178–5182. [Google Scholar]
- 32.Blackwell L.J., Bjornson,K.P. and Modrich,P. (1998) DNA-dependent activation of the hMutSalpha ATPase. J. Biol. Chem., 273, 32049–32054. [DOI] [PubMed] [Google Scholar]
- 33.Carey D.C. and Strauss,P.R. (1999) Human apurinic/apyrimidinic endonuclease is processive. Biochemistry, 38, 16553–16560. [DOI] [PubMed] [Google Scholar]
- 34.Tsai-Wu J.J., Su,H.T., Wu,Y.L., Hsu,S.M. and Wu,C.H. (2000) Nuclear localization of the human mutY homologue hMYH. J. Cell. Biochem., 77, 666–677. [PubMed] [Google Scholar]
- 35.Williams S.D. and David,S.S. (1998) Evidence that MutY is a monofunctional glycosylase capable of forming a covalent Schiff base intermediate with substrate DNA. Nucleic Acids Res., 26, 5123–5133. [DOI] [PMC free article] [PubMed] [Google Scholar]