

Abstract
Mechanisms of DNA oxidation by copper complexes of 3-Clip-Phen and its conjugate with a distamycin analogue, in the presence of a reductant and air, were studied. Characterisation of the production of 5-methylenefuranone (5-MF) and furfural, associated with the release of nucleobases, indicated that these copper complexes oxidised the C1′ and C5′ positions of 2-deoxyribose, respectively, which are accessible from the DNA minor groove. Oxidation at C1′ was the major degradation route. Digestion of DNA oxidation products by P1 nuclease and bacterial alkaline phosphatase allowed characterisation of glycolic acid residues, indicating that these copper complexes also induced C4′ oxidation. However, this pathway was not associated with base propenal release. The ability of the copper complex of the 3-Clip-Phen conjugate with the distamycin analogue to produce sequence-selective DNA cleavage allowed confirmation of these mechanisms of DNA oxidation by PAGE. Comparison of DNA cleavage activity showed that conjugation of 3-Clip-Phen with a DNA minor groove binder, like the distamycin analogue, decreased both its ability to perform C1′ oxidation as well as the initial rate of the reaction, but this conjugate is still active after 5 h at 37°C, making it an efficient DNA cleaver.
INTRODUCTION
Molecules able to irreversibly modify nucleic acids have received considerable interest because of their potential applications as biological tools or as chemotherapeutic agents. Transition metal complexes endowed with redox properties and DNA affinity have been developed as chemical nucleases. Typical examples of these DNA cleavers are iron–bleomycin, Fe(II)–EDTA, Mn(III)–porphyrin, nickel complexes and Co, Rh and Ru complexes of phenanthroline or bipyridine and [Cu(Phen)2]2+ (1–5). All these complexes are able to mediate oxidative damage to nucleobases and/or to the 2-deoxyribose moiety. Knowledge of their DNA oxidation mechanisms is highly useful to explore the potential applications of these molecules as biological tools or therapeutic agents, since their toxic or therapeutic effects could be associated with their ability to produce DNA damage that is difficult to repair (6,7). For example, the alkylation products of malondialdehyde derived from base propenals produced by iron–bleomycin action on DNA are toxic (8); oxidised abasic sites of 2-deoxyribose are relatively resistant to cleavage by many apurinic/apyrimidic endonucleases (9–12); and the toxicity of neocarzinostatin is associated with its ability to produce double-strand oxidation of DNA (13).
1,10-Phenanthroline (Phen) is a versatile ligand able to chelate copper to give mono-Phen or bis-Phen complexes in oxidation state +I or +II (14). The (Phen)2Cu(I) complex in the presence of hydrogen peroxide efficiently cleaves double-stranded DNA by oxidative attacks on C1′ and C4′ of 2-deoxyribose hydrogens by interacting within the minor groove (15,16) while the Phen-Cu(I) complex is less efficient (17). DNA cleavage products include 5′- and 3′-monophosphate ester termini, free bases and 5-methylenefuranone (5-MF) resulting from a major C1′ attack in the floor of the minor groove, but also a small amount of 3′-phosphoglycolate, which is a marker of a minor amount of C4′ oxidation (18–20). Hydrogen peroxide can be generated in close proximity to DNA by (Phen)2Cu(II) in the presence of a reductant and molecular oxygen.
We have recently prepared 2- and 3-Clip-Phen derivatives containing two Phen ligands linked at the 2′ or 3′ position by a serinol bridge in order to favor a 2:1 Phen:Cu ratio (21,22). The oxidative nuclease activity of copper complexes of these new ligands was found to be higher than that of Phen itself, by a factor of 2 for 2-Clip-Phen but by a factor of 60 for 3-Clip-Phen. Conjugates of 2-Clip-Phen with the natural polyamine spermine (a minor groove binder) (23) or intercalators such as acridine derivatives (24) also had enhanced nuclease activity.
The higher nuclease activity of (3-Clip-Phen)CuCl2 allowed us to prepare promising conjugates with potential chemotherapeutic properties. However, the steric constraints resulting from the linking group between the two phenanthrolines or the presence of a DNA binding entity in the conjugate could influence the mechanism of DNA cleavage in comparison with (Phen)2CuCl2, the parent compound. Recently, the DNA cleavage activity of 3-Clip-Phen has been targeted to A·T tracts by conjugation with a distamycin analogue (25) able to bind preferentially five successive A·T base pairs in the DNA minor groove (Fig. 1) (26–28). In the present paper, the ability of this conjugate to produce sequence-specific DNA cleavage allowed us to analyse the DNA oxidation mechanism of copper complexes of 3-Clip-Phen and its conjugates by PAGE. Oxidation of 2-deoxyribose C1′, C4′ and also C5′, confirming an interaction within the DNA minor groove, were observed by HPLC and GC-MS characterisation of oxidation markers, and these results are further supported by PAGE data (Fig. 2).
Figure 1.

Structures of conjugates 1, 2 and 3.
Figure 2.

Proposed mechanisms for DNA damage by C1′, C4′ and C5′ hydrogen abstraction induced by copper complexes of 3-Clip-Phen or 3-Clip-Phen–distamycin conjugates. Marker products of the C1′, C4′ and C5′ mechanisms analysed by HPLC or GC-MS (released nucleic acid bases before or during a heating step, 5-MF, furfural and phosphoglycolate ends giving glycolic acid) are in red boxes. 5′- and 3′-phosphate ends of cleavage fragments and proposed structure for the NaBH4 reactive 5′-end observed by PAGE analysis are in green boxes. βE, β-elimination; Δ, heating step (0.1 M HEPES–NaOH buffer, pH 8.0, 90°C).
MATERIALS AND METHODS
3-Clip-Phen (1) (22), 3-Clip-Phen–distamycin analogue (2) (25) and Boc–distamycin analogue (3) (25) were prepared as previously described.
Metalation
Stock solutions of ligands (2.5 mM) were prepared in water for the 3-Clip-Phen–distamycin (2) and Boc–distamycin (3) conjugates or in DMF for 3-Clip-Phen (1). Metalation of the different ligands used in the DNA cleavage experiments was carried out using a 1 mM CuCl2 concentration and 1 equiv. of ligand in water for 1 h at room temperature. Complexes were diluted to the appropriate concentration with water just before use.
HPLC analysis of released DNA nucleobases and oxidised sugars
Experiments were conducted in 50 µl volumes and all indicated concentrations are final concentrations.
To a solution of calf thymus DNA (700 µM in base pairs) in Tris–HCl (or sodium phosphate) buffer (40 mM, pH 7.2), 50 mM NaCl and 10 mM MgCl2 was added (3-Clip-Phen)CuCl2 or (3-Clip-Phen–distamycin)CuCl2 (35 or 100 µM). After 30 min at room temperature, mercaptopropionic acid (MPA) or ascorbate (4 mM) was added and samples were incubated for 1 or 5 h at 37°C and then reactions were stopped by fast freezing in liquid nitrogen.
Some samples were precipitated with 50 µl of sodium acetate buffer (3 M, pH 5.2) and 500 µl of ethanol for 2 h at –20°C. Pellets were rinsed with 200 µl of ethanol (twice) then lyophilised for 5 min in a Speed Vac concentrator. An aliquot of 50 µl of HEPES–NaOH buffer (0.1 M, pH 8.0) was added and the samples were heated for 15 min at 90°C. Reactions were stopped by freezing the samples in liquid nitrogen.
Samples were analysed at 260 and 280 nm on a Nucleosyl C18 10µ HPLC analytical column (250 × 4.6 mm) coupled to a diode array detector (Waters 994) with triethylammonium acetate buffer (0.1 M, pH 6.5), 5% acetonitrile isocratic elution (1 ml/min). Nucleobases, 5-MF and furfural, released during DNA oxidation or the heating step, were analysed by injection of authentic samples and from their UV-vis profiles. They were quantified by comparison of peak areas versus standard injections of authentic samples at different concentrations. Retention times were as follows: Rt ascorbate = 3.6 min; C = 3.8 min; G = 5.0 min; T = 5.9 min; A = 9.3 min, furfural = 12.6 min; 5-MF = 13.7 min.
GC-MS conditions
GC-MS analyses were performed with a Hewlett-Packard 5973 instrument using electron impact ionisation at 70 eV. The carrier vector was He and a HP-5MS non-polar capillary column (30 m × 0.25 mm × 0.25 µm film thickness, crosslinked 5% PH MC siloxane) was used. The injector temperature was 250°C; the column temperature was 70°C for 1 min and then a linear gradient to 150°C (10°C/min) was used.
GC-MS characterisation of 5-MF and furfural
Calf thymus DNA cleavage experiments with (3-Clip-Phen)CuCl2 (35 µM) in the presence of ascorbate and air were conducted for 1 h at 37°C on the 2.5 ml scale as described for HPLC analyses. After the heating step, 5-MF and furfural were extracted with 1.8 ml of CH2Cl2. The volume was reduced to 10 µl and samples were analysed by GC-MS by comparison with authentic standards. Rt furfural = 2.193 min; 5-MF = 2.588 min.
GC-MS characterisation of glycolate
The method was adaptated from McGall et al. (29).
Calf thymus DNA cleavage experiments with (3-Clip-Phen)CuCl2 (35 µM) in the presence of ascorbate and air were conducted for 1 h at 37°C on the 1 ml scale as described for HPLC analysis. Then samples were precipitated by addition of 800 µl of sodium acetate buffer (3 M, pH 5.2) and 4.4 ml of ethanol for 1 h at –20°C. Pellets were rinsed with 800 µl of ethanol (twice), then lyophilised for 5 min in a Speed Vac concentrator. Samples were dissolved in 400 µl of sodium acetate buffer (50 mM, pH 5.2) and digested with 10 U P1 nuclease (Sigma) for 1 h at 37°C. Samples were then diluted with 400 µl of Tris–HCl buffer (400 mM, pH 8.0) and digested again with 2.2 U bacterial alkaline phosphatase (Sigma) for 1 h at 37°C before being heated for 5 min at 90°C. Solutions were applied to a 4 × 1 cm column of DEAE–Sephadex A25 equilibrated with ammonium formate buffer (1 M, pH 7.5) then water prior to use. The column was washed with 10 ml of water and then glycolic acid was eluted with 10 ml of 1% aqueous solution of formic acid. The acid was neutralised with a few drops of concentrated NH4OH and samples were lyophilised and then dried over P2O5 in a dessicator under vacuum. Samples were heated for 1 h at 70°C in 10 µl of dry CH3CN and 10 µl of N,O-bis(trimethylsilyl)trifluoroacetamide containing 1% trimethylchlorosilane then analysed by GC-MS from comparison of authentic standards. Rt bis-trimethylsilyl derivative of glycolic acid = 4.858 min.
Characterisation of malondialdehyde
The method was adaptated from Murugesan et al. (30). Standard malondialdehyde solutions were prepared by acid hydrolysis of 1,1,3,3-tetramethoxypropane.
Calf thymus DNA cleavage by copper complexes of 1 or 2 were conducted for 1 h at 37°C on the 50 µl scale as described for HPLC analysis, then 500 µl of an aqueous solution of 0.6% 2-thiobarbituric acid (TBA) was added and samples were heated for 20 min at 85°C. In control experiments the reductant was added after TBA but before the heating step. UV-vis spectra (from 190 to 820 nm) of reaction mixtures were then obtained and the quantity of malondialdehyde chromophore (ɛ = 150 000 M–1 cm–1 at 531 nm) was determined from TBA reaction with a standard. Subtraction of malondialdehyde formed during the control experiment (3 µM) from the value obtained under DNA cleavage conditions gave the quantity of malondialdehyde formed during the oxidation step.
Comparison of the cleavage pattern of ODN I–ODN II by copper complexes of 1, 2 and 3
The two complementary ODNs I and II (Fig. 3) were synthesised by the standard solid phase β-cyanoethylphosphoramidite method and purified on a 15% polyacrylamide gel. Concentrations of single-stranded ODNs were determined by UV titration at 260 nm. The ODNs were end-labeled with 32P using standard procedures with T4 polynucleotide kinase (New England BioLabs) and [γ-32P]ATP for the 5′-end or terminal deoxynucleotidyl transferase (Gibco BRL) and [α-32P]ddATP for the 3′-end, before being purified on a MicroSpin G25 column (Pharmacia) (31).
Figure 3.

Sequence of ODN I–ODN II. Preferential binding sites for the distamycin analogue of conjugate 2 (five successive A·T base pairs) are shown. Cleavage sites for conjugate 2 are indicated by arrows; stars show secondary cleavage sites only observed with ascorbate.
The 35mer target (2.5 µM in each ODN) was annealed in 160 µl of Tris–HCl buffer (100 mM, pH 7.2) and 125 mM NaCl by heating at 90°C for 5 min followed by slow cooling to room temperature and, finally, addition of 40 µl of 0.1 M MgCl2.
For cleavage experiments, 2 µl of the desired complex (40 µM) was added to a 4 µl solution of the ODN I–ODN II duplex. After 30 min at room temperature cleavage was initiated by addition of 2 µl of an aqueous solution of MPA or ascorbate (400 µM) and samples were incubated for 1 h at 37°C. An aliquot of 10 µl of sodium acetate buffer (3 M, pH 5.2), 0.1 mM EDTA and 0.2 µg salmon sperm DNA were then added and samples were precipitated with 150 µl of ethanol for 2 h at –20°C. Pellets were rinsed with 100 µl of ethanol (twice) and lyophilised for 5 min in a Speed Vac concentrator.
In order to study the DNA cleavage mechanism, additional treatments were performed on some samples: (i) heating for 30 min at 90°C in 50 µl of HEPES–NaOH buffer (0.1 M, pH 8.0) followed by precipitation with 50 µl of sodium acetate buffer (3 M, pH 5.2) and 500 µl of ethanol; (ii) heating for 30 min at 90°C in 50 µl of aqueous piperidine (1 M) then lyophilisation; (iii) after the heating step in HEPES buffer and precipitation some samples were dissolved in 20 µl of appropriate buffer and 3′-end phosphates were removed by treatment with 10 U T4 polynucleotide kinase (New England Biolabs) for 30 min at 37°C, followed by precipitation of samples with 20 µl of sodium acetate buffer (3 M, pH 5.2) and 300 µl of ethanol; (iv) after the heating step in HEPES buffer (and before precipitation), 5 µl of Tris–HCl buffer (1 M, pH 8.0) were added and 5′-end phosphates were removed by treatment with 150 U bacterial alkaline phosphatase (Gibco BRL) for 1 h at 37°C, then samples were precipitated with 50 µl of sodium acetate buffer (3 M, pH 5.2) and 500 µl of ethanol; (v) in order to reduce reactive DNA ends some samples were dissolved in 20 µl of Tris–HCl buffer (40 mM, pH 8.0) then reacted with 2 µl of HEPES–NaOH buffer (1 M, pH 8.0) and 2 µl of aqueous 1 M NaBH4 for 30 min at room temperature, after which the excess NaBH4 was quenched with 2 µl of acetone and samples were precipitated with 20 µl of sodium acetate buffer (3 M, pH 5.2) and 300 µl of ethanol.
Fragments of DNA were separated by denaturing 20% polyacrylamide gel electrophoresis and identified by comparison with Maxam–Gilbert sequencing ladders (32).
RESULTS AND DISCUSSION
In order to study the mechanisms of DNA degradation by (3-Clip-Phen)CuCl2 (1) and to compare its reactivity with a conjugate with a distamycin analogue (2) (Fig. 1), we first searched for oxidative markers of C1′ and C4′ oxidation of DNA previously observed with the parent compound (Phen)2CuCl2 (Fig. 2) (4). C1′ oxidation could be easily characterised by HPLC analysis of the direct release of nucleobases during the oxidation step associated with formation, after a heating step, of 5-MF. The phosphoglycolate fragments resulting from C4′ oxidation could be characterised by GC-MS analysis of glycolic acid. In a second test we have confirmed the observed mechanisms of oxidation by PAGE analysis of cleavage patterns resulting from oxidation of a 35mer double-stranded target by copper complexes of 1 and 2.
Characterisation of marker products resulting from C1′ and C5′ oxidation by HPLC and GC-MS
Calf thymus DNA was oxidised by copper complexes of 1 and 2 in the presence of a reductant and air. Nucleobases and marker products of the oxidation of 2-deoxyribose, released before or after heating were analysed by HPLC (Table 1). Experimental conditions were chosen in order to have <10% DNA degradation (estimated from nucleobase release), in order to decrease the probability of formation of secondary oxidation products. Different parameters were modified, including time of reaction (1 or 5 h), complex concentration (35 or 100 µM), nature of the reductant (ascorbate or MPA) and buffer (Tris–HCl or sodium phosphate). After DNA oxidation a precipitation step quenched the reaction by removing the reductant or H2O2 generated during the reaction (copper complexes of 1 or 2 were difficult to remove but were inactive without reductant) and also served to remove nucleobases that had been directly released during the oxidation step.
Table 1. Quantification of release of nucleobases, furfural and 5-MF by copper complexes of 1 and 2 (35 or 100 µM).
| Ligand | Buffer | Conca (µM) | Red. | Time (h) | Cb (µM) | G (µM) | T (µM) | A (µM) | 5-MF (µM) | GΔ (µM) | TΔ (µM) | AΔ (µM) | 5-MFΔ (µM) | FurΔ (µM) | G+T+A (µM) | G+T+A+GΔ+TΔ+AΔ (µM) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | Phos | 100 | Asc | 1 | ndc | 12 | 9 | 16 | 0 | 2 | 3 | 3 | 19 | 3 | 37 | 45 |
| 1 | Tris | 100 | Asc | 1 | nd | 12 | 8 | 16 | 0.4 | 1 | 1 | 2 | 11 | 3 | 36 | 40 |
| 1 | Phos | 100 | Asc | 5 | nd | 14 | 12 | 22 | 2 | 1 | 1 | 1 | 23 | 2 | 48 | 51 |
| 1 | Tris | 100 | Asc | 5 | 21 | 15 | 12 | 22 | 3 | 0.6 | 1 | 0.4 | 9 | 2 | 49 | 51 |
| 1 | Phos | 35 | Asc | 1 | nd | 6 | 6 | 11 | 0 | 1 | 2 | 2 | 14 | 2 | 23 | 28 |
| 1 | Tris | 35 | Asc | 1 | nd | 5 | 4 | 11 | 0 | nd | nd | nd | 7 | 0.4 | 20 | nd |
| 1 | Phos | 35 | Asc | 5 | nd | 8 | 10 | 17 | 0 | 1 | 1 | 0.6 | 18 | 1.5 | 35 | 38 |
| 1 | Tris | 35 | Asc | 5 | nd | 9 | 6 | 17 | 1 | nd | nd | nd | 7 | 0.8 | 32 | nd |
| 1 | Phos | 100 | MPA | 1 | 11 | 4 | 4 | 12 | 3 | 1 | 1 | 1 | 9 | 3 | 20 | 23 |
| 1 | Tris | 100 | MPA | 1 | 9 | 5 | 5 | 14 | 2 | 1 | 1 | 1 | 9 | 3 | 24 | 27 |
| 1 | Phos | 100 | MPA | 5 | 11 | 4 | 5 | 12 | 2 | 1 | 2 | 1 | 7 | 2 | 21 | 25 |
| 1 | Tris | 100 | MPA | 5 | 9 | 5 | 6 | 15 | 2 | 1 | 0.6 | 1 | 8 | 2 | 26 | 28 |
| 2 | Phos | 100 | Asc | 1 | nd | 1 | 4 | 7 | 0 | 3 | 2 | 7 | 8 | 3 | 12 | 24 |
| 2 | Phos | 100 | Asc | 5 | nd | 7 | 21 | 12 | 0 | 9 | 7 | 17 | 23 | 4 | 40 | 73 |
The cleavage reactions (1 or 5 h at 37°C) of calf thymus DNA (700 µM in base pairs) were initiated by 4 mM reductant (MPA or ascorbate, indicated in ‘Red.’ column) in 40 mM sodium phosphate or Tris–HCl, pH 7.2 (indicated in ‘Buffer’ column), 50 mM NaCl, 10 mM MgCl2. C, G, T, A and 5-MF, quantification of nucleic acid bases and 5-MF released before the heating step; GΔ, TΔ, AΔ, FurΔ and 5-MFΔ, quantification of nucleobases, furfural and 5-MF released during the heating step (0.1 M HEPES–NaOH buffer, pH 8.0, 15 min at 90°C).
aConc, concentration of the copper complex of 1 or 2.
bC was often unquantifiable because it was contaminated by the large HPLC peak of ascorbate (or buffer).
cnd, not determined.
DNA degradation by (3-Clip-Phen)CuCl2 (1) in the presence of air and a reductant gave nucleobase release before and, to a lesser extent, after the heating step: 5-MF and a small quantity of furfural were mainly observed after the thermal step. Control experiments showed that nucleobases, 5-MF and furfural were not released in the absence of copper complex or reductant (results not shown).
The release of furfural and 5-MF was confirmed by GC-MS analysis for calf thymus DNA oxidation by (3-Clip-Phen)CuCl2 activated by ascorbate. They are markers of C1′ and C5′ DNA oxidation, respectively (Fig. 2). C1′ oxidation gave direct nucleobase release during the oxidation step, whereas two β-elimination steps (occurring during the heating step) are necessary to release nucleobases after C5′ oxidation.
C5′ oxidation has not been detected for DNA oxidation by 1,10-phenanthroline–copper complexes. The pro-S H-atoms at C5′ of 2-deoxyribose, located at the entrance to the minor groove, are more accessible sites for oxidants. The higher reactivity of (3-Clip-Phen)CuCl2 and steric constraints due to the linking group between the two phenanthrolines of 3-Clip-Phen could explain this type of oxidation.
The facility of production of free nucleobases was A ≈ C > G ≈ T, while calf thymus DNA contains 39% GC. With the HPLC conditions used cytosine was only quantifiable in Tris buffer because of HPLC profile contamination by the phosphate buffer peak and, for activation by ascorbate, which co-eluted with cytosine, after 5 h reaction which was necessary for transformation of all the reductant (one part could be used for the synthesis of H2O2 by the cupric complex in the presence of air).
5-MF and furfural are heat sensitive. A previous study concerning the stability of 5-MF produced during DNA oxidation by Mn-TMPyP/KHSO5 has shown that maximum release of 5-MF occurred after 10–15 min heating at 90°C at pH 8.0 (33). Thus, heating steps were conducted for 15 min at 90°C in HEPES–NaOH buffer, pH 8.0. However, the quantities observed were only a part of the total 5-MF or furfural that had actually been produced during DNA oxidation. Another part might not be released or could be degraded during the heating step [this can be estimated as 33% according to Pratviel et al. (33)] or during the relatively long oxidation step (1 or 5 h at 37°C). It can be proposed that the quantity observed was ≤66% of the quantity of precursor formed during the oxidation step.
The yield of nucleobases released was comparable when DNA oxidation was performed in phosphate or Tris buffer, but less 5-MF was observed in Tris. The quantity of 5-MF was also lower in Tris buffer after a 5 h reaction than for 1 h, indicating that 5-MF or its precursor were degraded during the reaction time and were less stable in Tris than in phosphate. A minor quantity of 5-MF was also observed before the heating step, in particular in Tris buffer, that could probably act as a base and catalyse the β-elimination steps leading to release of 5-MF.
When MPA was used as reductant the reaction was complete after 1 h and nucleobase release was only 50% of the yield obtained with ascorbate, in accordance with the lower activity previously observed for DNA oxidation by (2-Clip-Phen)CuCl2 (23).
With ascorbate, when the reaction time was increased from 1 to 5 h the yield of nucleobases released increased to only ∼30% and ∼60% for 100 and 35 µM (3-Clip-Phen)CuCl2, respectively. Analysis of HPLC chromatograms showed that after a 1 h reaction a higher residual quantity of ascorbate was present for 35 µM of complex (∼40%) than for 100 µM (∼10%). All ascorbate was consumed after 5 h reaction, although 70% was recovered after 5 h at 37°C in a control experiment without complex (data not shown). This consumption of ascorbate could explain the poor evolution of the reaction after 1 h for 100 µM complex.
With 35 µM complex the yield of nucleobases released was ∼60% of the yield observed with 100 µM complex. However, based on a linear dependence of nucleobase release on complex concentration, this value must be 35%. It is reasonable to propose that for 35 µM (3-Clip-Phen)CuCl2 (1 complex per 17 bp) a higher quantity of the complex was interacting with DNA than for 100 µM (1 complex per 7 bp), allowing a higher efficiency of oxidative cleavage.
For experiments conducted with 35 µM (3-Clip-Phen)CuCl2, 38 µM adenine, guanine and thymine was released after 5 h reaction. Since the yield of free released cytosine was ∼15 µM, the total of nucleobases released was ∼50 µM, in accordance with a system able to catalyse the oxidative degradation of DNA (nearly two catalytic cycles).
Two major differences were observed when calf thymus DNA was oxidised by (3-Clip-Phen–distamycin)CuCl2 (2) in the presence of ascorbate. (i) The quantity of nucleobases released after the heating step was similar to the quantity of nucleobases released before heating, although this value was ∼30% in the case of calf thymus DNA oxidation by (3-Clip-Phen)CuCl2. Since C1′ oxidation leads only to direct nucleobase release, this type of DNA oxidation seemed to be less important in the case of (3-Clip-Phen–distamycin)CuCl2 than for (3-Clip-Phen)CuCl2. (ii) After a 1 h reaction the efficiency of the copper complex of 1 was higher than that of 2 (release of adenine, guanine and thymine was 45 and 24 µM for 1 and 2, respectively) but less ascorbate was consumed with the copper complex of 2 (40% of the residual ascorbate was present in this case). After a 5 h reaction the yield of free nucleobases increased by 30% for 1 but the increase was 300% for 2. In fact, DNA oxidation mediated by the copper complex of 2 was slower than with (3-Clip-Phen)CuCl2, but more efficient (51 and 73 µM adenine, guanine and thymine released after 5 h reaction with 1 and 2, respectively).
These experimental results suggest that steric constraints resulting from conjugation with the distamycin analogue decreased the accessibility of 2-deoxyribose C1′ in the inside of the minor groove, favouring other oxidation pathways. In contrast, the distamycin entity of the conjugate increased DNA affinity and thus the efficiency of this oxidative nuclease, but probably decreased accessibility to the reductant, which decreased the rate of the cleavage reaction.
Evidence for C4′ oxidation of DNA deoxyriboses by (3-Clip-Phen)CuCl2 and (3-Clip-Phen–distamycin)CuCl2
In order to characterise C4′ oxidation leading to 3′-phosphoglycolate end groups (Fig. 2), calf thymus DNA was oxidised by (3-Clip-Phen)CuCl2 in the presence of ascorbate and then hydrolysed by P1 nuclease and bacterial alkaline phosphatase in order to release glycolic acid, which was isolated on a DEAE–Sephadex A25 ion exchange column. After silylation, formation of this marker of C4′ oxidation in the reaction mixture was confirmed by GC-MS analysis (29).
In the case of DNA oxidation by iron–bleomycin, the formation of 3′-phosphoglycolate termini was associated with release of base propenals, which are easily characterised by HPLC (34). Base propenals can be hydrolysed under acidic conditions and release malondialdehyde, which can then react with TBA to produce a chromophore having a maximum absorption at 531 nm (30). However, in the case of calf thymus DNA oxidation by copper complexes of 1 or 2 in the presence of a reductant and air attempts to characterise base propenals by HPLC analyses failed (standards were obtained by DNA oxidation by ferrous bleomycin and also by ferric bleomycin activated by ascorbate in the buffers used for DNA degradation by copper complexes of 1 and 2, indicating that base propenals are analysable under the conditions used for this study; data not shown).
We also tested TBA reactivity on calf thymus DNA oxidised by copper complexes of 1 and 2 in the presence of a reductant and air. A chromophore having a maximal absorption at 531 nm was observed (7 µM malondialdehyde equiv. were formed in 1 h at 37°C for experiments conducted in phosphate buffer with 100 µM copper complex of 1 or 2 activated by ascorbate). These results are in agreement with the recent work of von Sonntag and colleagues on γ-radiolysis of DNA aqueous solutions (known to produce DNA fragments with 3′-phosphoglycolate ends). No base propenals were detected, although malondialdehyde was observed, indicating that the ‘glycolate pathway’ does not always lead to formation of base propenals (35). However, in the present case of DNA oxidation by (3-Clip-Phen)CuCl2 or by its conjugate with a distamycin analogue, due to the drastic conditions used for the TBA reaction, the known formation of malondialdehyde from other DNA oxidation products than ones resulting from C4′ oxidation cannot be totally discarded.
PAGE analysis of DNA breaks resulting from oxidation of ODN I–ODN II by copper complexes of 1, 2 and 3
The sequence-selective cleavage pattern of an EcoRI–RsaI restriction fragment by the copper complex of conjugate 2 was the origin of the choice of the sequence of the ODN I–ODN II target (25). The base composition contains the major cleavage sites which have been observed in the vicinity of seven successive A·T base pairs which constitute three overlapping major sites of interaction for the distamycin analogue entity of the conjugate (Fig. 3). This sequence was located in the middle of a relatively short 35mer in order to allow the analysis of minor variations in electrophoretic migration resulting from cleavage fragments exhibiting different oxidised termini.
Cleavage patterns of ODN I–ODN II (1 µM) by copper complexes (10 µM) of 1 and 2 were compared. The redox activity of the copper complexes was triggered by addition of 100 µM MPA or ascorbate in the presence of air. Samples were analysed by PAGE and cleavage sites were determined with Maxam–Gilbert sequencing reactions. The Boc–distamycin analogue (3) was also tested as a control in the presence of 1 equiv. CuCl2 under the same experimental conditions (Fig. 1).
Figures 4 and 5 show the results obtained for the duplex labeled on the 5′-end of ODN I and ODN II, respectively. The copper complex of 3-Clip-Phen (1) led to a non-specific cleavage pattern where the target was oxidised at all nucleotides in a random fashion. In contrast, the copper complex of the distamycin conjugate of 3-Clip-Phen (2) produced a sequence-selective cleavage pattern. Cleavage was observed at C12, A13, A26, C27 and, to a lesser extent, at C23 for ODN I and at T11, A12, T25, G26, A27 and, slightly, A15 for ODN II. The target was less cleaved by copper complexes of 1 and 2 when activation was with MPA rather than with ascorbate. Cleavage patterns were roughly the same with the two reductants, but with conjugate 2 scission at C23 on ODN I and at A15 on ODN II was only observed under more reactive conditions (with ascorbate). This suggests that C23 on ODN I and A15 on ODN II are probably secondary cleavage sites when using conjugate 2.
Figure 4.

PAGE analysis of cleavage of the ODN I–ODN II duplex, 5′-end-labeled on ODN I, by conjugates 1, 2 and 3. Conjugates (10 µM) were complexed with 1 equiv. CuCl2. The duplex (1 µM) cleavage reactions (1 h at 37°C) were initiated with 100 µM reductant (MPA or ascorbate) in 40 mM Tris–HCl, pH 7.2, 50 mM NaCl, 10 mM MgCl2. Lanes A + G and G, Maxam–Gilbert A + G and G ladders, respectively; last lanes, control experiments with copper complexes of 2 and 1, respectively, without reductant but after piperidine treatment. Heat treatment (30 min at 90°C) in 100 mM HEPES–NaOH buffer, pH 8.0, or in 1 M piperidine is indicated at the top of the gel.
Figure 5.

PAGE analysis of cleavage of the ODN I–ODN II duplex (1 µM), 5′-end-labeled on ODN II, by copper complexes of 1 and 2 (10 µM). The cleavage reactions (1 h at 37°C) were initiated with 100 µM reductant (MPA or ascorbate) in 40 mM Tris–HCl, pH 7.2, 50 mM NaCl, 10 mM MgCl2. Lanes A + G and G, Maxam–Gilbert A + G and G ladders, respectively. Heat treatment (30 min at 90°C) in 100 mM HEPES–NaOH buffer, pH 8.0, or in 1 M piperidine or T4 polynucleotide kinase digestion steps are indicated at the top of the gel.
These results are in agreement with the peptide entity of either one or two conjugate molecules interacting within the A·T box, in either the 5′→3′ or 3′→5′ direction, directing interaction of the 3-Clip-Phen moiety of the conjugate to nucleotides in the vicinity of the binding site (26,28). The absence of cleavage observed with conjugate 3 (the peptide portion alone) in the presence of CuCl2 (Fig. 4) and the random cleavage pattern produced by (3-Clip-Phen)CuCl2 confirm that the cleavage pattern observed with copper conjugate 2 resulted from a DNA interaction targeted by the distamycin entity of 2 and a nuclease activity resulting from the 3-Clip-Phen entity of the conjugate.
Additionally, analysis of the cleavage of ODN I–ODN II by conjugate 2 showed that scissions on both strands were shifted by 2 bp towards the 3′-ends (Fig. 3). The 3′ shift confirmed that the chemical nuclease oxidised sugar residues within the minor groove (an attack in the major groove would have given a 5′ shift) (4). Modified products resulting from C1′ or C4′ oxidation can be observed by PAGE for a 5′-end-labeled target (Fig. 2).
Since most of the intense cleavage fragments co-migrated with the Maxam–Gilbert lane and were sensitive to T4 polynucleotide kinase (Fig. 5), it can be assumed that these fragments have 3′-phosphate ends. However, some minor cleavage products, with an increased PAGE mobility, were also observed for small cleavage fragments (indicated by arrows in Figs 4 and 5). These products were estimated by phosphorimaging as being <20% of the major cleavage products in the case of cleavage of ODN I–ODN II by (3-Clip-Phen)CuCl2. This percentage was more variable for (3-Clip-Phen–distamycin)CuCl2. The resolution of the gel was insufficient to detect the presence of these minor cleavage products on longer fragments since the difference in electrophoretic migration with fragments having 3′-phosphate ends was reduced when the length of the oligodeoxyribonucleotides increased. These minor cleavage products appeared to be stable to further heat treatment under alkaline conditions (30 min at 90°C in HEPES–NaOH buffer, pH 8.0, or in 1 M aqueous piperidine). They were probably fragments with 3′-phosphoglycolate ends resulting from oxidation at C4′ of 2-deoxyribose.
A different oxidation pathway at C4′ of 2-deoxyribose can generate abasic oligonucleotides with a ketone group at C4′ (Fig. 6) (4,13,34). This product is sensitive to hydrazine and to reduction by NaBH4 and can be cleaved by alkaline treatment. It has been previously characterised on 5′-end-labeled fragments in the case of DNA oxidation by chemical nucleases like iron–bleomycin or neocarzinostatin. In the case of oxidation of ODN I–ODN II by copper complexes of 1 and 2 all attempts to characterise it with hydrazine, NaBH4 or thermal treatment failed (data not shown). Under the experimental conditions used (or perhaps in general) copper complexes of 3-Clip-Phen and its conjugates are probably unable to produce such a ketone at C4′.
Figure 6.

C4′ oxidation mechanism proposed for iron–bleomycin and neocarzinostatin. βE, β-elimination; Δ, heating step; pip, heating step at 90°C in 1 M piperidine.
A smear was also observed after cleavage of ODN I–ODN II by copper complexes of 1 and 2 (more easily observed with 2), resulting from metastable products that decomposed during electrophoresis. A heating step at pH 8.0 was sufficient to remove this smear, but no new cleavage products were detected, thus it is reasonable to propose that the metastable products were transformed to fragments with 3′-phosphate ends. These smears probably resulted from metastable products generated by C1′ oxidation (abasic ribonolactone oligonucleotides or, more probably, fragments with a 3′-ribonolactone end) since a simple heating step at pH 8.0 was able to release fragments with 3′-phosphate ends and 5-MF (the marker product of C1′ oxidation) (33). Major cleavage products with a 3′-phosphate end could also result from C1′ oxidation, as previously observed for DNA oxidation by 1,10-phenanthroline–copper complexes, since Chen and Greenberg have shown that copper complexes of phenanthroline catalyse β-elimination leading to fragments with 3′-phosphate ends (36). The heating step prior to loading samples on the gel could also degrade another portion of these particularly unstable oxidative intermediates (37).
In order to detect eventual nucleobase oxidation, a heating step in 1 M piperidine was performed (5). A comparison did not show clear differences from cleavage patterns obtained after a heating step under lower alkaline conditions at pH 8.0, where the modification observed in comparison analyses without heating could be attributed to 2-deoxyribose oxidation. Since no nucleobase oxidation was detected, copper complexes of 1 and 2 probably preferentially oxidise deoxyribose, as previously observed for Phen–copper complexes (1).
Marker products of C5′ oxidation (Fig. 2) could be characterised by PAGE using 3′-end-labeled oligonucleotides. Thus, cleavage of the ODN I–ODN II target was studied by labeling the 3′-end of ODN I. Figure 7 shows the results obtained with the copper complex of conjugate 2 activated with MPA. In general, cleavage fragments had 5′-phosphate ends, since they co-migrated with the Maxam–Gilbert lanes and were sensitive to bacterial alkaline phosphatase. No clear modification of the cleavage pattern was observed after alkaline treatment or reaction with NaBH4. These results are consistent with the C1′ and C4′ oxidations proposed after analysis of the cleavage patterns of 5′-end-labeled target (Fig. 2).
Figure 7.
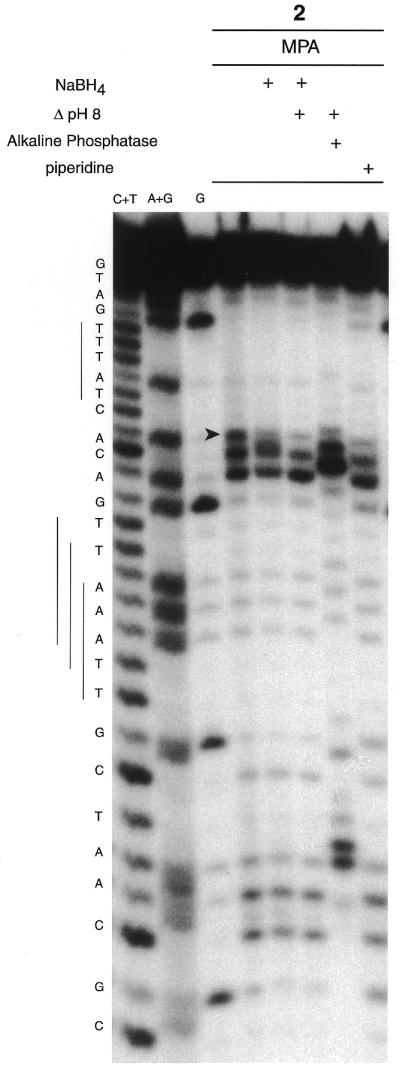
PAGE analysis of cleavage of the ODN I–ODN II duplex (1 µM), 3′-end-labeled on ODN I, by the copper complex of conjugate 2 (10 µM). The cleavage reactions (1 h at 37°C) were initiated with 100 µM MPA in 40 mM Tris–HCl, pH 7.2, 50 mM NaCl, 10 mM MgCl2. Lanes C + T, A + G and G, Maxam–Gilbert C + T, A + G and G ladders, respectively. A NaBH4 (0.1 M) reduction step, heat treatment (30 min at 90°C) in 100 mM HEPES–NaOH buffer, pH 8.0, or in 1 M piperidine or a bacterial alkaline phosphatase digestion step are indicated at the top of the gel.
After the oxidation step by the copper complex three cleavage products can be observed on the 5′-side of the binding site, but one break (indicated by an arrow in Fig. 7) was sensitive to reduction by NaBH4. This fragment was transformed into a new product whose electrophoretic mobility was poorly separated from the adjacent band resulting from oxidation of the deoxyribose of C12. After a heating step at pH 8.0 only two cleavage fragments with 3′-phosphate ends (sensitive to alkaline phosphatase) were observed. This sensitivity of the metastable fragment to reduction and heat under slightly alkaline conditions has been previously observed after C5′ oxidation of DNA by neocarzinostatin or Mn-TMPyP (4) and can probably be attributed to a cleavage fragment with a C5′ aldehyde resulting from oxidation at the C5′ position of 2-deoxyribose by the copper complex of conjugate 2 (Fig. 2).
As previously observed for a 5′-label, target breaks were observed at C12, A13, A26 and C27 of ODN I. Ascorbate activation generates the same cleavage pattern, but target degradation was higher (data not shown).
Only random cleavage on all nucleotides was observed with conjugate 1 (data not shown). All fragments seemed to have 5′-phosphate ends, since they co-migrated with a Maxam–Gilbert lane and were sensitive to bacterial alkaline phosphatase and because no clear modification of the cleavage pattern was observed after alkaline treatment or reduction by NaBH4. However, the presence of minor cleavage products resulting from C5′ oxidation could not be excluded since, in the case of random cleavage of the target, the probability that they co-migrated with other cleavage fragments is not negligible.
Experiments were conducted in Tris–HCl buffer (pH 7.2) in the presence of NaCl and MgCl2. The same cleavage patterns were obtained in sodium phosphate buffer, but residual phosphate decreased the level of digestion of fragments by bacterial alkaline phosphatase and T4 polynucleotide kinase (results not shown).
CONCLUSION
In the presence of a reductant and air the copper complex of 3-Clip-Phen degraded DNA in a rather random fashion by oxidation at C1′, C4′ and C5′ of 2-deoxyribose within the DNA minor groove. C1′ oxidation was the major chemical pathway observed, generating nucleobase release and unstable intermediates able to release 5-MF after a thermal step. Production of DNA fragments with 3′-phosphoglycolate ends resulted from C4′ oxidation, which was not associated with base propenal release but a TBA-sensitive product was detected. C5′ oxidation has not been previously observed during DNA oxidation by the (Phen)2Cu(II) parent complex. This oxidation generated direct DNA cleavage and release of furfural and nucleobases after heating. This reactivity reflects the ability of (3-Clip-Phen)CuCl2 to react not only in the floor of the DNA minor groove (perhaps by intercalation, since phenanthroline is able to do so) leading to C1′ oxidation, but also, as a minor pathway, at the edge of the DNA minor groove to generate C4′ and C5′ oxidation.
The study reported in this paper allows estimation of the different oxidation mechanisms if one considers: (i) that (3-Clip-Phen)CuCl2 cleaves DNA without sequence selectivity; (ii) the relative quantities of nucleobases released before and after heating; (iii) the relative proportions of 5-MF and furfural observed; (iv) the quantity of TBA observed; (v) the phosphorimaging quantification, after a heating step, of 3′-phosphate and 3′-phosphoglycolate termini formed in the case of degradation of the ODN I–ODN II target. Thus it can be proposed that (3-Clip-Phen)CuCl2 activated by a reductant in the presence of air for a reaction time of 1 h produces >50% C1′ oxidation, ∼15 ± 5% C4′ oxidation and ∼15 ± 5% C5′ oxidation of 2-deoxyribose on double-stranded DNA.
When 3-Clip-Phen was conjugated with a DNA minor groove binder such as a distamycin analogue, sequence-selective cleavage was observed. This behavior resulted from targeting of the oxidative activity of the copper complex of 3-Clip-Phen to the vicinity of successive A·T base pairs by the distamycin entity of the conjugate. The conjugate degrades DNA by C1′, C4′ and C5′ oxidation, like (3-Clip-Phen)CuCl2, but conjugation with the DNA binder markedly influences the regioselectivity of oxidation between the three sites. C1′ oxidation is less predominant, suggesting that steric constraints force the large conjugate to preferentially interact with the edge of the DNA minor groove. DNA degradation by this conjugate was slower, but more efficient, than that by (3-Clip-Phen)CuCl2 when considering consumption of the reducing agent as the reaction time increased.
REFERENCES
- 1.Sigman D.S., Mazumder,A. and Perrin,D.M. (1993) Chemical nucleases. Chem. Rev., 93, 2295–2316. [Google Scholar]
- 2.Dervan P.B. (1986) Design of sequence-specific DNA-binding molecules. Science, 232, 464–471. [DOI] [PubMed] [Google Scholar]
- 3.Meunier B. (ed.) (1996) DNA and RNA Cleavers and Chemotherapy of Cancer and Viral Diseases. Kluwer, Dordrecht, The Netherlands.
- 4.Pratviel G., Bernadou,J. and Meunier,B. (1995) Carbon-hydrogen bonds of DNA sugar units as targets for chemical nucleases and drugs. Angew. Chem. Int. Ed. Engl., 34, 746–769. [Google Scholar]
- 5.Burrows C.J. and Muller,J.G. (1998) Oxidative nucleobase modifications leading to strand scission. Chem. Rev., 98, 1109–1151. [DOI] [PubMed] [Google Scholar]
- 6.Sancar A. and Sancar,G.B. (1988) DNA repair enzymes. Annu. Rev. Biochem., 57, 29–67. [DOI] [PubMed] [Google Scholar]
- 7.David S.S. and Williams,S.D. (1998) Chemistry of glycosylases and endonucleases involved in base-excision repair. Chem. Rev., 98, 1221–1261. [DOI] [PubMed] [Google Scholar]
- 8.Dedon P.C., Plastaras,J.P., Rouzer,C.A. and Marnett,L.J. (1998) Indirect mutagenesis by oxidative DNA damage: formation of the pyrimidopurinone adduct of deoxyguanosine by base propenal. Proc. Natl Acad. Sci. USA, 95, 11113–11116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kappen L.S., Chen,C. and Goldberg,I.H. (1988) Atypical abasic sites generated by neocarzinostatin at sequence-specific cytidilate residues in oligodeoxynucleotides. Biochemistry, 27, 4331–4340. [DOI] [PubMed] [Google Scholar]
- 10.Povirk L.F. and Houlgrave,C.W. (1988) Effect of apurinic/apyrimidic endonucleases and polyamines on DNA treated with bleomycin and neocarzinostatin: specific formation and cleavage of closely opposed lesions in complementary strands. Biochemistry, 27, 3850–3857. [DOI] [PubMed] [Google Scholar]
- 11.Häring M., Rüdiger,H., Demple,B., Boiteux,S. and Epe,B. (1994) Recognition of oxidized abasic sites by repair endonucleases. Nucleic Acids Res., 22, 2010–2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Xu Y., Kim,E.Y. and Demple,B. (1998) Excision of C-4′-oxidized deoxyribose lesions from double-stranded DNA by human apurinic/apyrimidic endonuclease (Ape1 protein) and DNA polymerase β*. J. Biol. Chem., 273, 28837–28844. [DOI] [PubMed] [Google Scholar]
- 13.Goldberg I.H. (1991) Mechanism of neocarzinostatin action: role of DNA microstructure in determination of chemistry of bistranded oxidative damage. Acc. Chem. Res., 24, 191–198. [Google Scholar]
- 14.James B.R. and Williams,R.J.P. (1961) The oxidation-reduction potentials of some copper complexes. J. Chem. Soc., 2007–2019. [Google Scholar]
- 15.Marshall L.E., Graham,D.R., Reich,K.A. and Sigman,D.S. (1981) Cleavage of deoxyribonucleic acid by the 1,10-phenanthroline-cuprous complex. Hydrogen peroxide requirement and primary and secondary structure specificity. Biochemistry, 20, 244–250. [DOI] [PubMed] [Google Scholar]
- 16.Veal J.M., Merchant,K. and Rill,R.L. (1991) Noncovalent DNA binding of bis(1,10-phenanthroline)copper(I) and related compounds. Biochemistry, 30, 1132–1140. [DOI] [PubMed] [Google Scholar]
- 17.Veal J.M., Merchant,K. and Rill,R.L. (1991) The influence of reducing agent and 1,10-phenanthroline concentration on DNA cleavage by phenanthroline + copper. Nucleic Acids Res., 19, 3383–3388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kuwabara M., Yoon,C., Goyne,T.E., Thederahn,T. and Sigman,D.S. (1986) Nuclease activity of 1,10-phenanthroline-copper ion: reaction with CGCGAATTCGCG and its complexes with netropsin and EcoRI. Biochemistry, 25, 7401–7408. [DOI] [PubMed] [Google Scholar]
- 19.Goyne T.E. and Sigman,D.S. (1987) Nuclease activity of 1,10-phenanthroline-copper ion. Chemistry of deoxyribose oxidation. J. Am. Chem. Soc., 109, 2846–2848. [Google Scholar]
- 20.Zelenko O., Gallagher,J. and Sigman,D.S. (1997) Scission of DNA with bis(1,10-phenanthroline)copper without intramolecular hydrogen migration. Angew. Chem. Int. Ed. Engl., 36, 2776–2778. [Google Scholar]
- 21.Pitié M., Donnadieu,B. and Meunier,B. (1998) Preparation of the new bis(phenanthroline)ligand “Clip-Phen” and evaluation of the nuclease activity of the corresponding copper complex. Inorg. Chem., 37, 3486–3489. [DOI] [PubMed] [Google Scholar]
- 22.Pitié M., Sudres,B. and Meunier,B. (1998) Dramatic increase of the DNA cleavage activity of Cu(Clip-phen) by fixing the bridging linker on the C3 position of the phenanthroline units. Chem. Commun., 2597–5798. [Google Scholar]
- 23.Pitié M. and Meunier,B. (1998) Preparation of a spermine conjugate of the bis-phenanthroline ligand “Clip-Phen” and evaluation of the corresponding copper complex. Bioconjugate Chem., 9, 604–611. [DOI] [PubMed] [Google Scholar]
- 24.Ross S.A., Pitié,M. and Meunier,B. (1999) Synthesis of two acridine conjugates of the bis(phenanthroline) ligand “Clip-Phen” and evaluation of the nuclease activity of the corresponding copper complexes. Eur. J. Inorg. Chem., 557–563. [DOI] [PubMed] [Google Scholar]
- 25.Pitié M., Van Horn,J.D., Brion,D., Burrows,C.J. and Meunier,B. (2000) Targeting the DNA cleavage activity of copper phenanthroline and Clip-Phen to A.T tracts via linkage to a poly-N-methypyrrole. Bioconjugate Chem., 11, 892–900. [DOI] [PubMed] [Google Scholar]
- 26.Bailly C. and Chaires,C.B. (1998) Sequence specific DNA minor groove binders. Design and synthesis of netropsin and distamycin analogues. Bioconjugate Chem., 9, 513–538. [DOI] [PubMed] [Google Scholar]
- 27.Baird E.E. and Dervan,P.B. (1996) Solid phase synthesis of polyamides containing imidazole and pyrrole amino acids. J. Am. Chem. Soc., 118, 6141–6146. [DOI] [PubMed] [Google Scholar]
- 28.White S., Szewczyk,J.W., Turner,J.M., Baird,E.E. and Dervan,P.B. (1998) Recognition of the four Watson-Crick base pairs in the DNA minor groove by synthetic ligands. Nature, 391, 468–471. [DOI] [PubMed] [Google Scholar]
- 29.McGall G.H., Rabow,L.E., Ashley,G.W., Wu,S.H., Kozarich,J.W. and Stubbe,J. (1992) New insight into the mechanism of base propenal formation during bleomycin-mediated DNA degradation. J. Am. Chem. Soc., 114, 4958–4967. [Google Scholar]
- 30.Murugesan N., Xu,C., Ehrenfeld,G.M., Sugiyama,H., Kilkuskie,R.E., Rodriguez,L.O., Chang,L.-H. and Hecht,S.M. (1985) Analysis of products formed during bleomycin-mediated DNA degradation. Biochemistry, 24, 5735–5744. [DOI] [PubMed] [Google Scholar]
- 31.Sambrook J., Fritsch,E.F. and Maniatis,T. (1989) Molecular Cloning: A Laboratory Manual, 2nd Edn. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- 32.Maxam A.M. and Gilbert,W. (1980) Sequencing end-labeled DNA with base specific chemical cleavages. Methods Enzymol., 65, 499–599. [DOI] [PubMed] [Google Scholar]
- 33.Pratviel G., Pitié,M., Bernadou,B. and Meunier,B. (1991) Mechanism of DNA cleavage by cationic manganese porphyrins: hydroxylations at the 1′-carbon and 5′-carbon atoms of deoxyriboses as initial damages. Nucleic Acids Res., 19, 6283–6288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Burger R.M. (1998) Cleavage of nucleic acids by bleomycin. Chem. Rev., 98, 1153–1169. [DOI] [PubMed] [Google Scholar]
- 35.Rashid R., Langfinger,D., Wagner,R., Schuchmann,H.-P. and von Sonntag,C. (1999) Bleomycin versus OH-radical-induced malonaldehydic-product formation in DNA. Int. J. Radiat. Biol., 75, 101–109. [DOI] [PubMed] [Google Scholar]
- 36.Chen T. and Greenberg,M.M. (1998) Model studies indicate that copper phenanthroline induces direct strand breaks via β-elimination of the 2′-deoxyribonolactone intermediate observed in enediyne mediated DNA damage. J. Am. Chem. Soc., 120, 3815–3816. [Google Scholar]
- 37.Hwang J.T., Tallman,K.A. and Greenberg,M.M. (1999) The reactivity of the 2-deoxyribonolactone lesion in single-stranded DNA and its implication in reaction mechanisms of DNA damage and repair. Nucleic Acids Res., 19, 3805–3810. [DOI] [PMC free article] [PubMed] [Google Scholar]