


Abstract
A nuclear recessive mutant in Saccharomyces cerevisiae, mhr1-1, is defective in mitochondrial genetic recombination at 30°C and shows extensive vegetative petite induction by UV irradiation at 30°C or when cultivated at a higher temperature (37°C). It has been postulated that mitochondrial DNA (mtDNA) is oxidatively damaged by by-products of oxidative respiration. Since genetic recombination plays a critical role in DNA repair in various organisms, we tested the possibility that MHR1 plays a role in the repair of oxidatively damaged mtDNA using an enzyme assay. mtDNA isolated from cells grown under standard (aerobic) conditions contained a much higher level of DNA lesions compared with mtDNA isolated from anaerobically grown cells. Soon after a temperature shift from 30 to 37°C the number of mtDNA lesions increased 2-fold in mhr1-1 mutant cells but not in MHR1 cells. Malonic acid, which decreased the oxidative stress in mitochondria, partially suppressed both petite induction and the temperature-induced increase in the amount of mtDNA damage in mhr1-1 cells at 37°C. Thus, functional mitochondria require active MHR1, which keeps the extent of spontaneous oxidative damage in mtDNA within a tolerable level. These observations are consistent with MHR1 having a possible role in mtDNA repair.
INTRODUCTION
Through the process of oxidative respiration, mitochondria supply cells with most of the energy they need to carry out a diverse range of biological functions. Reactive oxygen species, respiration by-products formed by the oxidation of O2 by reduced components of the electron transport chain, cause oxidative damage to mitochondrial DNA (mtDNA) (1; for a review see 2). The accumulation of oxidative damage in mtDNA could be a major cause of the age-associated DNA aberrations observed in non-dividing cells of mammals, such as nerve and muscle cells (3,4). The accumulation of such aberrations in mtDNA might eventually lead to aging and underlie age-related degenerative diseases (for reviews see 5,6). How mitochondria deal with damaged mtDNA is of considerable biological interest.
It has been suggested that mitochondria have no efficient DNA repair system and that mitochondria, having multiple DNA copies, simply dismiss damaged DNA. More recent studies have shown that many types of DNA lesion are repaired through base excision or damage reversal in mitochondria (for reviews see 7–9). In various organisms, ranging from viruses to man, it is well known that recombinational repair is an efficient pathway for removing DNA lesions and recombination-related genes are important for DNA repair (for reviews see 10–12). However, whether mitochondria have a system for recombinational DNA repair is still unclear. mtDNA recombination was shown to occur in yeast (for a review see 13), but in mammalian cells recombination of mtDNA has not been clearly demonstrated in vivo (cited in 7). Mitochondrial recombination or genes involved in mitochondrial recombination, except yeast PIF1, have not been tested for their roles in mtDNA repair. PIF1 encodes a DNA helicase (14). Yeast cells with a mutant allele of pif1 exhibited normal recombination frequency between mitochondrial alleles in the crossing of ρ+ cells, but prevented the integration of a class of markers of ρ– genomes into the ρ+ genome. In addition, pif1 mutant cells showed a significantly higher level of ρ– induction by UV irradiation, suggesting a role for this gene in mtDNA repair (15).
Some years ago we succeeded in isolating a yeast nuclear recessive mutation (mhr1-1) which rendered mitochondria defective in the mitochondrial recombination observed in crossing of ρ+ cells. MHR1 is a nuclear gene (DDBJ/EMBL/GenBank accession no. AB016430) encoding a unique polypeptide of 226 amino acid residues (26.9 kDa, identical to a hypothetical polypeptide of unknown function, YDR296w) of which no homologs were found in the genome databases. The mhr1-1 mutation is a single base mutation in the open reading frame, resulting in replacement of a single amino acid residue (F.Ling, submitted for publication). mhr1-1 cells show enhanced UV induction of respiration-deficient progeny (vegetative petite) at 30°C and the fraction of mhr1-1 cells having non-functional deleted mtDNA (ρ–) or no mtDNA (ρ0) gradually increases during cultivation at 37°C (but not at 30°C; 16). We observed a temperature-dependent delay in the transmission of mtDNA into daughter cells in mhr1-1 cells (F.Ling, submitted for publication). This explains the production of ρ0 cells during cultivation of mhr1-1 cells at 37°C, but the extensive induction of ρ– mhr1-1 cells at 37°C requires another explanation. Considering the general role of genetic recombination in DNA repair, we assumed that the induction of ρ– cells in mhr1-1 cultures at 37°C was caused by spontaneous oxidative damage to mtDNA, which was efficiently repaired in wild-type cells by the function of the MHR1 gene. In this study this possible role of MHR1 was tested, and is supported by the results obtained.
MATERIALS AND METHODS
Strains and media
The yeast strains IL166-187 (MATα, his1, trp1, can1, ω+ chlr) and FL67 (MATα, mhr1-1, his1, trp1, can1, ω+ chlr) used in this study as MHR1 and mhr1-1 strains, respectively, have been described previously (16).
YPD medium consisted of 1% yeast extract, 2% Bacto peptone and 2% glucose. YPGly medium consisted of 1% yeast extract, 2% peptone, 3% glycerol and 50 mM KH2PO4, pH 6.25. Solid media (YPD and YPGly plates) were prepared by adding 2% Bacto agar (Difco) to the liquid media described above. YPD and YPGly indicate liquid media, unless the word plate is specifically mentioned.
Reagents
Sodium azide (NaN3) was purchased from Merck. Malonic acid (disodium salt monohydrate) was purchased from Nacalai Tesque (Kyoto, Japan). T4 endonuclease V (T4 endoV) was purified as described by Inaoka et al. (17). S1 nuclease was obtained from Takara Shuzo (Kyoto, Japan). Alloxan was purchased from Sigma Chemical Co. (St Louis, MO). All chemicals were analytical grade.
Measurement of vegetative petite induction
Cells of mhr1-1 and MHR1 strains were cultured in YPGly (to select ρ+ cells) at 30°C with aeration through vigorous shaking for 3 days. About 2 × 105 cells from each culture were inoculated into 2 ml of YPD and cultivated at 37 or 30°C with aeration by vigorous shaking for 48 h. The YPD contained various concentrations of malonic acid (up to 50 mM) or NaN3 (up to 100 µM) in some experiments. Then the cells from each culture (∼1 × 108 cells/ml) were spread on YPD plates, after appropriate dilution, so that ∼300 colonies formed on each plate. After incubation at 30°C for 3 days the colonies on each YPD plate were replicated onto both YPGly and YPD plates, followed by incubation at 30°C for 5 days. Respiration-deficient cells can grow on YPD but not on YPGly plates, whereas respiration-proficient cells can grow on both types of plate.
Preparation of whole-cell DNA for DNA damage assay
Yeast cells were inoculated into 300 ml of YPGly at 1 × 105 cells/ml and allowed to grow to 1 × 108 cells/ml at 30°C with aeration for 50 h. The cells were transferred into a fresh 300 ml volume of YPGly and cultured with aeration at 37 or 30°C for the indicated period. The YPGly was supplemented with various amounts of malonic acid or NaN3 in some experiments. Then the cells were collected and whole-cell DNA was prepared according to the method described by Philippsen et al. (18).
Treatment of mitochondria with an oxidizing reagent
To test the validity of a DNA damage assay using T4 endoV we prepared mtDNA with various levels of oxidative damage. It was reported that alloxan acts in vivo mainly as a generator of reactive oxygen species (19,20). To introduce various amounts of oxidative damage into mtDNA we treated isolated mitochondria with alloxan for various periods. First, mitochondria was prepared from anaerobically grown cells to reduce the assay background signal as follows. To examine variations in oxidative damage to mtDNA, IL166-187 (MHR1) cells were inoculated into 1000 ml of YPD overlaid with 250 ml of liquid paraffin in a 5000 ml flask at a concentration of 1 × 105 cells/ml and allowed to grow without shaking at 30°C for 4 days. Cell numbers increased to 1 × 108 cells/ml during cultivation. Mitochondria were isolated from the MHR1 cells as described by Daum et al. (21). The isolated mitochondria were then treated with 5 mM alloxan for various periods. DNA was then purified from the treated mitochondria and subjected to the DNA damage assay.
The S1-assisted T4 endoV DNA damage assay for mtDNA lesions
The S1-assisted T4 endoV DNA damage assay developed in this study included two successive enzyme reactions; nicking (single-stranded breakage) of mtDNA by T4 endoV at the sites of oxidative damage, followed by conversion of the nicks into double-stranded breaks with S1 nuclease. The amounts of small double-stranded DNA fragments produced by the enzyme treatments were measured by quantitative Southern hybridization using a 32P-labeled mtDNA probe after gel electrophoresis. The standard reaction mixture (25 µl) for the T4 endoV treatment consisted of ∼1.0 µg whole-cell DNA to be tested, 10 mM potassium phosphate buffer, pH 7.4, 100 mM KCl and 4.8 µg/ml T4 endoV. The reaction was carried out at 37°C for 1 h. A control reaction was carried out under the same conditions, but T4 endoV was omitted. Aliquots (5 µl) of the T4 endoV reaction mixture (or the control reaction mixture) were then subjected to S1 nuclease treatment. The reaction mixture (10 µl) for the S1 nuclease treatment contained ∼200 ng DNA, 30 mM sodium acetate buffer, pH 4.6, 280 mM NaCl, 1 mM ZnSO4, 3 U/µl S1 nuclease and the components derived from the T4 endoV reaction mixtures (5 µl). The reaction was carried out at 37°C for 30 min. The reactions were terminated by adding 50 mM EDTA mixed with loading buffer consisting of 5% glycerol, 10 mM EDTA, 0.025% bromophenol blue and 0.025% xylene cyanol. The DNA samples were immediately subjected to electrophoresis through a 1% agarose gel using Tris–acetate buffer (22; but without ethidium bromide).
After electrophoresis the DNA in the gel was transferred to a positively charged nylon membrane (Amersham) using Southern blotting (23). To detect mtDNA on the membrane, purified mtDNA was labeled with 32P using a random primer DNA labeling kit (Pharmacia Bioprocess Technology, Sweden) and used as probe (mtDNA probe). After hybridization the radioactivity level of the bound probe was measured directly using a two-dimensional radioactivity analyzer (BAS-2000 Bio Image Analyzer; Fuji Film). The hybridization profile was recorded on X-ray film by exposure to the membrane at –80°C.
The extent of fragmentation due to the enzyme treatments was expressed as the ratio of the signal from fragmented DNA in a sample (radioactivity in region F in each lane of the gels) to the total signal from the sample (radioactivity in regions F + U in the lane). Signals from region U represent those from unfragmented DNA. Before the calculation, background values were subtracted. To improve the reproducibility of the measurements we used gels with fixed dimensions [13.5 (width) × 15 (length) × 0.7 cm (thickness) with two rows of 24 wells of 3 (w) × 1.5 mm (l)] and allowed the bromophenol blue front to run 5 cm from the wells at 80 V (∼2.5 h). We applied fixed sizes of windows for the measurement of radioactivity in regions F and U throughout the experiments. For calculation of the extent of T4 endoV-dependent fragmentation the control ratio of fragmentation by S1 nuclease treatment alone was subtracted from the ratio of fragmentation by T4 endoV treatment followed by S1 nuclease treatment.
RESULTS
The assay for mtDNA damage
For quantitative comparison of damage in mtDNA between anaerobic and aerobic conditions and between MHR1 cells and mhr1-1 cells we used a semi-quantitative assay (S1-assisted T4 endoV DNA damage assay). T4 endoV has long been considered to have an N-glycosylase activity specific to pyrimidine dimers in UV-irradiated DNA (24). In addition to N-glycosylase activity, T4 endoV has nicking activity specific for abasic sites created either by the N-glycosylase activity or by other activities (25–27). Abasic sites also represent a type of oxidative DNA damage or are generated in vivo by damage-specific DNA N-glycosylases, if present.
Although T4 endoV does not recognize 8-oxo-7,8-dihydro-2′-deoxyguanosine, a typical product of oxidatively damaged DNA, in DNA (S.Koizuma and E.Ohtsuka, unpublished observations), it was recently revealed that T4 endoV has an N-glycosylase activity on the oxidative product 4,6-diamino-5-formamidopyrimidine (FapyAde) in DNA (28). FapyAde was shown to be a substrate of repair enzymes such as Fpg protein (29) and endonuclease III (Nth protein; 30) of Escherichia coli and yeast homologs of endonuclease III (the Ngt1 and Ngt2 proteins; 31) and thus can be taken as a representative of repairable oxidative DNA damage. We used T4 endoV as an endonuclease to nick double-stranded DNA at FapyAde and abasic sites, as representatives of oxidative damage, since this enzyme is available in sufficient quantities.
Various damage-specific endonuclease-based assays that have been widely used to detect mtDNA damage in mammalian cells include gel electrophoresis under denaturing conditions. We have had problems with the reliability of this type of assay (see Discussion), therefore, to improve the reproducibility of detecting mtDNA damage, we further treated T4 endoV-treated DNA with S1 nuclease to convert the nicks into double-strand breaks (32,33). The double-stranded DNA fragments produced by the enzyme treatments were separated by agarose gel electrophoresis under native conditions. Mitochondria-specific DNA was detected by Southern hybridization using a 32P-labeled mtDNA probe. The radioactivity in the bound mtDNA probe was measured using a two-dimensional radioactivity analyzer. Figure 1A and the insert in Figure 1C show that signals are linearly dependent on the number of DNA lesions (in linearized plasmid pUC119 DNA UV-irradiated in vitro).
Figure 1.
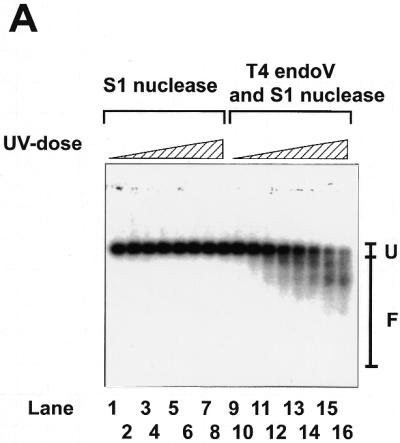
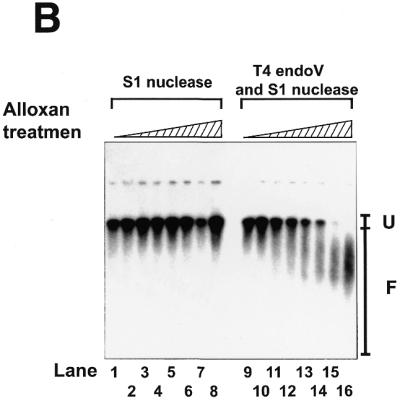

Linear response of the S1-assisted T4 endoV DNA damage assay to the number of DNA lesions. (A and insert in C) Control experiments with damage-free plasmid DNA. pUC119 plasmid DNA purified from E.coli XL1-blue cells using the alkaline SDS method was completely linearized by treatment with restriction endonuclease HindIII and precipitated with ethanol at –80°C. About 20 ng linearized pUC119 DNA suspended in 10 µl of H2O was subjected to UV irradiation for various periods (0 s in lanes 1 and 9; 5 s in lanes 2 and 10; 15 s in lanes 3 and 11; 30 s in lanes 4 and 12; 45 s in lanes 5 and 13; 60 s in lanes 6 and 14; 90 s in lanes 7 and 15; 120 s in lanes 8 and 16; 10 s = 12.3 J/m2). The irradiated DNA (10 ng) was subjected to the DNA damage assay. Signals arising from the fragments of linearized pUC119 were detected using 32P-labeled pUC119 DNA (∼50 ng) as probe. The hybridization profile, recorded on X-ray film by exposure to the membrane, is shown in (B). Lanes 1–8, controls of linearized pUC119 DNA irradiated by UV and then treated with S1 nuclease, but not with T4 endoV; lanes 9–16, linearized pUC119 DNA irradiated by UV and then treated with T4 endoV, followed by treatment with S1 nuclease. F, the region from which the radioactivity of fragmented DNA on the membrane was counted directly with a two-dimensional radioactivity analyzer; U, the region from which the radioactivity of unfragmented DNA was counted. The ratio of fragmentation was calculated by dividing the signal from region F by the total signal (regions F + U) from the sample in each lane. The extent of T4 endoV-dependent fragmentation was calculated and is plotted against UV dosage in the insert in (C). (B and C) The assay for oxidative damage in mtDNA. To decrease the background signal to the minimum level, MHR1 cells were anaerobically grown in YPD medium overlaid with liquid paraffin without shaking at 30°C for 4 days. Cell numbers increased up to ∼1 × 108 cells/ml during cultivation. Mitochondria were isolated and treated with 5 mM alloxan for various times to introduce oxidative damage into mtDNA. Then, mtDNA was extracted and subjected to the DNA damage assay to detect lesions in mtDNA. The hybridization profile is shown in (B). Lane 1, mtDNA from mitochondria without alloxan treatment treated with S1 nuclease alone; lanes 2–9, mtDNA from mitochondria treated with alloxan, then treated with S1 nuclease alone; lane 10, mtDNA isolated from mitochondria without alloxan treatment, then treated with T4 endoV followed by treatment with S1 nuclease; lanes 11–18, mtDNA from mitochondria with alloxan treatment, then treated with T4 endoV followed by treatment with S1 nuclease. The periods of alloxan treatment of mitochondria were 0 min in lanes 1, 2, 9 and 10, 2 min in lanes 3 and 11, 5 min in lanes 4 and 12, 8 min in lanes 5 and 13, 10 min in lanes 6 and 14, 15 min in lanes 7 and 15, and 20 min in lanes 8 and 16. F, the region from which the radioactivity of fragmented DNA was counted; U, the region from which the radioactivity of unfragmented DNA was counted. The ratio of fragmentation was calculated by dividing the signal from region F by the total signal (regions F + U) from the sample in each lane. The extent of the T4 endoV-dependent fragmentation (filled circles) was calculated by subtracting the control ratio of fragmentation by S1 nuclease treatment alone (T4 endoV treatment omitted; open squares) from the ratio of fragmentation by T4 endoV treatment followed by S1 nuclease treatment (open circles). These data are plotted against the period of alloxan treatment in (C).
To compensate for the nicking and fragmentation that unavoidably occurred during DNA isolation from yeast cells (see Discussion), signals obtained in this way were corrected by subtraction of the signals from a control sample that had not been subjected to T4 endoV treatment.
We examined the validity of the S1-assisted T4 endoV DNA damage assay for use in the measurement of damage in mtDNA. We chose experimental conditions published previously where alloxan introduces oxidative damage in DNA (19,20). Thus, we treated isolated mitochondria with alloxan for various periods and then the purified mtDNA was subjected to control experiments. To reduce the background signal (from mtDNA from untreated mitochondria) we grew wild-type yeast cells under anaerobic conditions (where the cells were cultured in YPD overlaid with liquid paraffin without shaking to keep oxidative damage in the mtDNA to a minimum level). The variation in the total radioactivity level recovered from each gel lane was ±25% (standard deviation) amongst all samples in Figure 1B. As shown in Figure 1C, the signal from samples treated with S1 nuclease only was constant (variation ±3%) and the signals from samples treated with both T4 endoV and S1 nuclease increased linearly with treatment time with alloxan up to 12 min. After treatment for >20 min all radioactivity was recovered from region F in the gel (as shown in Fig. 1B), i.e. the signals were saturated (Fig. 1C). The corrected signals (those dependent on the T4 endoV treatment) are proportional to the amount of oxidative damage (induced by the in vivo treatment with alloxan) up to 0.3, indicating that this assay produces reliable signals proportional to the amount of oxidative damage (Fig. 3C). It should be noted that signal values give no information on the absolute number of lesions, rather they are an evaluation of the relative amounts of DNA lesions.
Figure 3.

Effects of an inhibitor (malonic acid) of the TCA cycle on induction of vegetative petite cells and accumulation of oxidative mtDNA damage at a non-permissive temperature (37°C). (A) The suppression of temperature-induced petite production in a mhr1-1 culture. mhr1-1 and MHR1 cells were grown in YPD containing various amounts of malonic acid at 37°C with aeration for 48 h. The cells from each culture were diluted and spread on YPD plates and incubated at 30°C for 4 days to form colonies. The respiration proficiency of each colony was determined by a replica plating method. Open circles, MHR1; closed circles, mhr1-1. (B) A decrease in the temperature shift-induced increase in the amount of oxidative mtDNA damage in mhr1-1 cells. Whole-cell DNA was prepared from MHR1 or mhr1-1 cells grown in YPGly containing various concentrations of malonic acid at 37°C for 5 h with aeration. The extracted DNA was subjected to the DNA damage assay. Average values of two independent experiments were plotted against the concentration of malonic acid. Open circles, MHR1; closed circles, mhr1-1. Controls were DNA from cells grown at a permissive temperature (30°C); open squares, MHR1; closed squares, mhr1-1.
The accumulation of DNA damage to mtDNA in cells growing under aerobic conditions
First we compared the amount of mtDNA damage between aerobically and anaerobically grown cells. It has been proposed that in mitochondria formation of oxidative DNA damage and its repair are in equilibrium, but with a level much higher than that of nuclear DNA (34; see also reviews cited in Introduction). Then we isolated mtDNA from MHR1 and mhr1-1 yeast cells cultured at 30°C under anaerobic and aerobic conditions. We found that the signals from the DNA damage assay were much higher for the DNA samples obtained from cells grown under standard (aerobic) conditions compared with the signals from mtDNA from cells cultured under anaerobic conditions (Fig. 2A). Thus, mtDNA in both MHR1 and mhr1-1 cells grown at 30°C under standard aerobic conditions accumulate damage spontaneously. The difference in signal between mtDNA obtained from MHR1 cells and that obtained from mhr1-1 cells grown at 30°C was not significant (Fig. 2A).
Figure 2.


The accumulation of lesions in mtDNA during growth. (A) mtDNA from cells grown under anaerobic and aerobic conditions. For preparation of whole-cell DNA from anaerobically grown cells, MHR1 and mhr1-1 cells were inoculated into 200 ml of YPD overlaid with 75 ml of liquid paraffin in a 500 ml flask at 1 × 105 cells/ml and allowed to grow without shaking at 30°C for 2–3 days. Cell numbers increased up to 1 × 108 cells/ml during cultivation. mtDNA was isolated and the amount of oxidative damage was assayed as described above. The calculated results were plotted along with values obtained for aerobically grown cells sampled just before the temperature shift, shown in (B) and (C). (B and C) The increase in the amount of mtDNA damage in mhr1-1 cells induced by a temperature shift to 37°C. MHR1 or mhr1-1 cells grown in YPGly at 30°C were diluted into YPGly followed by cultivation with aeration at 37°C. At various time points an aliquot was withdrawn. Whole-cell DNA was prepared and subjected to the DNA damage assay. The hybridization profile is shown in (B). Lanes 1–4, controls of mtDNA from MHR1 cells treated with S1 nuclease alone; lanes 5–8, controls of mtDNA from mhr1-1 cells treated with S1 nuclease alone; lanes 9–12, mtDNA from MHR1 cells treated with T4 endoV followed by treatment with S1 nuclease; lanes 13–16, mtDNA from mhr1-1 cells treated with T4 endoV followed by treatment with S1 nuclease. The incubation periods after the temperature shift were 0 h in lanes 1, 5, 9 and 13, 1 h in lanes 2, 6, 10 and 14, 3 h in lanes 3, 7, 11 and 15 and 5 h in lanes 4, 8, 12 and 16. F, the region from which the radioactivity of fragmented DNA was counted; U, the region from which the radioactivity of unfragmented DNA was counted. The extent of the T4 endoV-dependent fragmentation was calculated and is plotted in (C). Average values of two independent experiments are plotted against period after the temperature shift. Open circles, MHR1; closed circles, mhr1-1.
Enhanced accumulation of damage in mtDNA of mhr1-1 mutant cells cultivated at 37°C
Previously we have shown that mhr1-1 cells maintain functional mtDNA at 30°C but >80% of mhr1-1 cells became vegetative petite (either ρ– or ρ0) during 40 h cultivation in YPD medium at 37°C, whereas MHR1 cells did not (16). If the petite induction at 37°C in mhr1-1 culture were caused, at least partially, by reduced repair activity, accumulation of spontaneous mtDNA damage would be enhanced. Then we examined whether DNA damage increased in the mtDNA of mhr1-1 mutant cells compared to MHR1 cells at 37°C. To design the experiments we considered the following possibility: when the amount of mtDNA damage exceeds a critical level the mitochondria lose respiratory functions before the establishment of vegetative petite cells. During subsequent growth mtDNA will not be attacked by reactive oxygen species, resulting in decreased damage levels. This may cause ambiguous experimental results. Therefore, we decided to examine the amount of mtDNA damage only during the period (5 h at 37°C) in which petite production was insignificant in both MHR1 and mhr1-1 cultures (16). In addition, we selected cells that retained respiratory function by growing cells in non-fermentable medium (YPGly).
We shifted the temperature of aerobic cultures of mhr1-1 and MHR1 cells in YPGly from 30 to 37°C followed by incubation for the indicated time, then whole-cell DNA samples were prepared and subjected to DNA damage assay. We found that the amount of DNA damage quickly increased in the mtDNA of mhr1-1 cells within 1 h to a level 2-fold higher after the temperature shift, but did not increase any further during the following 4 h incubation period (Fig. 2B and C). On the other hand, in MHR1 cells the increase in mtDNA damage after the temperature shift was much smaller. This result indicates that MHR1 function is responsible for maintaining the extent of spontaneous DNA damage in mtDNA within a certain level and is consistent with the assumption that mtDNA does not accumulate damage above a certain level without loss of respiratory function.
Suppression of vegetative petite induction and mtDNA damage by a reduction in oxidative stress in mhr1-1 cells at a non-permissive temperature (37°C)
To confirm that enhanced petite induction and mtDNA damage in mhr1-1 cells at 37°C was caused by oxidation of mtDNA, we tested the effects of an inhibitor of the TCA cycle on petite induction in mhr1-1 cells grown in glucose medium (YPD) at 37°C. Malonic acid is a substrate analog of succinate and an inhibitor of succinate dehydrogenase, an enzyme of the TCA cycle. It decreases the flow of electrons in the electron transport chain and results in a decrease in oxidative stress in mitochondria, as shown previously (see 35). Addition of malonic acid to cultures partially suppressed high level petite induction during prolonged incubation of mhr1-1 cells at 37°C (Fig. 3A). Malonic acid suppressed the temperature-induced increase in mtDNA damage after the temperature shift to 37°C, as shown by the DNA damage assay (Fig. 3B). Thus, temperature-dependent petite induction in mhr1-1 cultures is at least partly due to enhanced accumulation of damage in mtDNA, which is likely to be oxidative, and MHR1 functions maintain the amount of damage below the threshold capable of destructive effects on functional mtDNA.
Enhancement of petite induction and mtDNA damage by an inhibitor of cytochrome oxidase in mhr1-1 cells at a permissive temperature (30°C)
One might wonder if the observed 2-fold increase in the amount of oxidative damage in mtDNA of mhr1-1 (Fig. 2C) just after a temperature shift to 37°C could explain the observed effects of the mhr1-1 mutation on petite induction after prolonged incubation at the same temperature. Therefore, we tested the effects of azide, an inhibitor of cytochrome oxidase (36,37), on petite induction after prolonged incubation and accumulation of mtDNA damage just after addition of the inhibitor. Inhibition of cytochrome oxidase, the enzyme at the last step of the electron transport chain of mitochondria, increases intracellular oxidative stress by causing increased leakage of electrons from the electron transport system. After mhr1-1 cells were cultivated in the presence of 100 µM sodium azide for 48 h at a permissive temperature (30°C) ∼80% of cells were petite, while in MHR1 cultures only 5% of the cells were petite (Fig. 4A). During incubation of mhr1-1 cells for 5 h after addition of 100 µM sodium azide at 30°C mtDNA damage signals increased 2-fold compared to signals prior to the addition of azide, while only a slight increase in signal was observed with MHR1 cells under the same set of conditions (Fig. 4B). This result is consistent with the idea that there is a correlation between the 2-fold increase in the level of mtDNA damage just after a change in culture conditions or addition of the inhibitor and extensive petite induction after prolonged cultivation (48 h).
Figure 4.

Effects of an inhibitor (NaN3) of cytochrome oxidase on induction of vegetative petite cells and accumulation of oxidative mtDNA damage. (A) The enhancement of petite induction. mhr1-1 and MHR1 cells were grown in YPD containing various amounts of NaN3 at 30°C with aeration for 48 h. The cells from each culture were diluted and spread on YPD plates at 30°C for 4 days to form colonies. The respiration proficiency of each colony was determined by a replica plating method. Open circles, MHR1; closed circles, mhr1-1. (B) Enhanced mtDNA damage. Whole-cell DNA was prepared from MHR1 or mhr1-1 cells grown in YPGly containing various concentrations of NaN3 at 30°C for 5 h with aeration. The extracted DNA was subjected to the DNA damage assay. Average values of two independent experiments are plotted against the concentration of NaN3. Open circles, MHR1; closed circles, mhr1-1.
DISCUSSION
In this study we tested the possibility that the MHR1 gene is involved in a DNA repair system to protect mtDNA from spontaneous DNA damage.
A PCR-based assay has been successfully used to measure DNA damage in animal mitochondria (38). Because yeast mtDNA has a much larger size (∼80 kb) than animal mtDNA (16 kb) and possesses various repeat sequences, the PCR method is not suitable for the measurement of damage in entire yeast mtDNA. Techniques based on nicking at lesion sites by an endonuclease specific for DNA lesions, such as Fpg protein (29,39) and Ogg1 protein (40), followed by gel electrophoresis of DNA under denaturing conditions have been used to measure lesions in mtDNA in mammalian cells. Nicking and fragmentation of mtDNA during isolation disturb the results of PCR- and endonuclease-based techniques. This problem seriously affects the accuracy and reproducibility of DNA damage assays in yeast mitochondria because of a structural feature of yeast cells. Yeast cells have a cell wall and isolation of sufficient mtDNA to reflect the whole mtDNA population in a culture in these assays requires vigorous treatment of the cells, resulting in nicking and fragmentation of the DNA. In the method described here (the S1-assisted T4 endoV DNA damage assay) mtDNA nicked at the sites of damage by a DNA lesion-specific endonuclease were converted to double-stranded fragments by cleavage at the nick by S1 nuclease. Gel electrophoresis of double-stranded DNA fragments improved the reproducibility of the assay, as shown in this study. Because the migration of double-stranded DNA (linear) in gel electrophoresis is determined simply by the size of the fragment, while that of single-stranded fragments is determined by the size and their DNA sequence, a double-stranded DNA fragment with a given size forms a sharper band under native conditions than under denaturing conditions. Nicking and fragmentation of mtDNA during extraction from cells was corrected for using controls that omitted the DNA lesion-specific endonuclease treatment, as indicated by the linearity and small deviations of the data (Fig. 1C).
T4 endoV is believed to have an N-glycosylase activity specific for pyrimidine dimers, DNA lesions induced by UV irradiation. However, as described in Results, this endonuclease can be used in the detection of oxidative damage in DNA. We have shown here that the S1-assisted T4 endoV DNA damage assay successfully measures oxidative damage in DNA induced by alloxan (Fig. 1). It should be noted that the signals obtained in this study (Figs 2–4) were all within the range in which the signal was proportional to the amount of DNA damage (Fig. 1C).
Using this assay we have shown that cells grown under standard aerobic conditions accumulated damage in mtDNA at a much higher level than cells grown under anaerobic conditions (Fig. 2A). The 2-fold increase in the amount of mtDNA damage in mhr1-1 cells observed just after a temperature shift to 37°C is associated with extensive petite induction at later stages of cultivation (Fig. 2C; 16). Suppression of the temperature shift-induced increase in mtDNA damage by malonic acid, an inhibitor of the TCA cycle (see Results), correlated with a reduction in petite induction during cultivation of mhr1-1 cells at 37°C (Fig. 3). After addition of an inhibitor of cytochrome oxidase (NaN3 at 100 µM) at 30°C we again observed, as for mhr1-1 cells, a correlation between the 2-fold increase in the amount of mtDNA damage (just after addition) and extensive production of petite cells (after prolonged incubation) using the same set of conditions. On the other hand, in MHR1 cells under the same culture conditions the amount of mtDNA damage was maintained at the original level and no significant petite induction was observed. These observations indicate that MHR1 is required for the maintenance of cellular respiratory function through maintenance of the amount of spontaneous mtDNA damage within a tolerable level. There is a threshold level of initial DNA damage for vegetative petite production. For example, knowing that the number of DNA lesions induced by UV radiation is proportional to the dose, we previously reported that colonies of respiration-deficient cells were not formed when exposed up to a certain dose of UV radiation (mhr1-1 and another repair-deficient mutant cell), while above a threshold level of UV a large fraction of the mutants produced respiration-deficient cells after prolonged incubation (16).
The decrease in the amount of mtDNA damage under anaerobic cultivation, the suppression of the temperature shift-induced accumulation of mtDNA damage in mhr1-1 cells by addition of malonic acid and the increase in mtDNA damage only in mhr1-1 cells by addition of an inhibitor of cytochrome oxidase suggests that the observed damage, spontaneously introduced in mtDNA and removed or suppressed by a function of MHR1, is oxidative.
It is possible that Mhr1 protein is responsible for reducing oxidative stress in mitochondria, however, it is unlikely. Mhr1 protein is a mitochondrial matrix protein that binds single- or double-stranded DNA directly and this activity is completely abolished at 37°C by the mhr1-1 mutation, a single amino acid substitution mutation (F.Ling, to be published). Therefore, Mhr1 protein is more likely to play a role by acting directly on DNA rather than by playing a role in the reduction of oxidative stress.
After a temperature shift of mhr1-1 cultures to 37°C or addition of NaN3 to mhr1-1 cultures at 30°C we observed that mtDNA damage never exceeded a level above a 2-fold increase over the amount prior to the temperature shift or prior to addition of the inhibitor. This could mean that, as in the assumption for the design of the experiments (see Results), cells in which mtDNA damage accumulates to such a level lose the ability for oxidative respiration and therefore the accumulation of damage ceases in these cells. Another possibility is that mitochondria have another repair system which does not have sufficient capacity to maintain the original level of mtDNA damage in the absence of fully functional Mhr1 protein, resulting in a higher steady-state level of mtDNA damage.
Various studies have shown that DNA damage in the mitochondria of mammals, yeast and other organisms can apparently be removed from the DNA (see Introduction). These observations have been taken as evidence for the existence of DNA repair in mitochondria. However, since each cell contains multiple copies of mtDNA (10–104, depending on the organism and organ), this apparent removal of DNA damage could be explained in other ways, such as selective replication of undamaged mtDNA and/or selective elimination (such as degradation) of damaged DNA. These possibilities were addressed by previous studies on mammals and the apparent removal of mtDNA damage was confirmed to be due to mtDNA repair rather than through other processes (see for example 41,42). The absence of metabolic turnover of the components of DNA has been reported for yeast mitochondria (43). Considering these previous studies, it is likely that the MHR1 gene product plays an important role in the repair of mtDNA damage.
Cells with the mhr1-1 mutation show a clear deficiency in gene conversion-type recombination (16). A computer simulation has suggested that in multicopy systems, such as mitochondria, gene conversion tends to eliminate new mutations in DNA (44). Considering this possibility, the clear defects of mhr1-1 cells in gene conversion and in maintaining the amount of mtDNA lesions within a tolerable level suggest that gene conversion might play an important role in mtDNA repair. However, at present we are unsure of whether Mhr1 protein plays a direct or indirect role in mitochondrial recombination. Therefore, we need to consider other possibilities, for example that Mhr1 protein is indirectly involved in various types of DNA repair, like single strand-binding protein of E.coli. Further studies are required before we understand the molecular function of Mhr1 protein in the repair of mtDNA damage.
Acknowledgments
ACKNOWLEDGEMENTS
The authors express thanks to Dr D. Kang (Department of Biochemistry, Kyushu University School of Medicine) for providing us with information relating to the assay of DNA damage. This study was supported partly by a grant from Genome Exploration Research of RIKEN to F.L. and T.S., by a grant from the Biodesign Research Program of RIKEN to F.L. and T.S., by a grant-in-aid (08280102) for Scientific Research on Priority Areas from the Ministry of Education, Science, Sports and Culture of Japan to F.L. and by a grant for CREST from JST to T.S.
REFERENCES
- 1.Richter C. (1992) Reactive oxygen and DNA damage in mitochondria. Mutat. Res., 275, 249–255. [DOI] [PubMed] [Google Scholar]
- 2.Ames B.N., Shigenaga,M.K. and Hagen,T.M. (1993) Oxidants, antioxidants and the degenerative diseases of aging. Proc. Natl Acad. Sci. USA, 90, 7915–7922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Linnane A.W., Marzuki,S., Ozawa,T. and Tanaka,M. (1989) Mitochondrial DNA mutations as an important contributor to ageing and degenerative diseases. Lancet, 25, 642–645. [DOI] [PubMed] [Google Scholar]
- 4.Hayakawa M., Hattori,K., Sugiyama,S. and Ozawa,T. (1992) Age-associated oxygen damage and mutations in mitochondrial DNA in human hearts. Biochem. Biophys. Res. Commun., 189, 979–985. [DOI] [PubMed] [Google Scholar]
- 5.Lenaz G. (1998) Role of mitochondria in oxidative stress and ageing. Biochim. Biophys. Acta, 1366, 53–67. [DOI] [PubMed] [Google Scholar]
- 6.Wallace D.C. (1999) Mitochondrial diseases in man and mouse. Science, 283, 1482–1488. [DOI] [PubMed] [Google Scholar]
- 7.Croteau D.L., Stierum,R.H. and Bohr,V.A. (1999) Mitochondrial DNA repair pathways. Mutat. Res., 434, 137–148. [DOI] [PubMed] [Google Scholar]
- 8.LeDoux S.P., Driggers,W.J., Hollensworth,B.S. and Wilson,G.L. (1999) Repair of alkylation and oxidative damage in mitochondrial DNA. Mutat. Res., 434, 149–159. [DOI] [PubMed] [Google Scholar]
- 9.Sawyer D.E. and Van Houten,B. (1999) Repair of DNA damage in mitochondria. Mutat. Res., 434, 161–176. [DOI] [PubMed] [Google Scholar]
- 10.Cox M.M. (1999) Recombinational DNA repair in bacteria and the RecA protein. Prog. Nucleic Acid Res. Mol. Biol., 63, 311–366. [DOI] [PubMed] [Google Scholar]
- 11.Thacker J. (1999) The role of homologous recombination processes in the repair of severe forms of DNA damage in mammalian cells. Biochimie, 81, 77–85. [DOI] [PubMed] [Google Scholar]
- 12.Taylor E.M. and Lehmann,A.R. (1998) Conservation of eukaryotic DNA repair mechanisms. Int. J. Radiat. Biol., 74, 277–286. [DOI] [PubMed] [Google Scholar]
- 13.Dujon B. (1981) Mitochondrial genetics and function. In Strathern,J.N., Jones,E.W. and Broach,J.R. (eds), The Molecular Biology of the Yeast Saccharomyces: Life Cycle and Inheritance. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY, pp. 505–635.
- 14.Lahaye A., Stahl,H., Thines-Sempoux,D. and Foury,F. (1991) PIF1: a DNA helicase in yeast mitochondria. EMBO J., 10, 997–1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Foury F. and Kolodynski,J. (1983) pif mutation blocks recombination between mitochondrial ρ+ and ρ– genomes having tandemly arrayed repeat units in Saccharomyces cerevisiae. Proc. Natl Acad. Sci. USA, 80, 5345–5349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ling F., Makishima,F., Morishima,N. and Shibata,T. (1995) A nuclear mutation defective in mitochondrial recombination in yeast. EMBO J., 14, 4090–4101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Inaoka T., Ishida,M. and Ohtsuka,E. (1989) Affinity of single- or double-stranded oligodeoxyribonucleotides containing a thymine photodimer for T4 endonuclease V. J. Biol. Chem., 264, 2609–2614. [PubMed] [Google Scholar]
- 18.Philippsen P., Stotz,A. and Scherf,C. (1991) DNA of Saccharomyces cerevisiae. Methods Enzymol., 194, 169–182. [DOI] [PubMed] [Google Scholar]
- 19.Driggers W.J., Ledoux,S.P. and Wilson,G.L. (1993) Repair of oxidative damage within the mitochondrial DNA of RINr 38 cells. J. Biol. Chem., 268, 22042–22045. [PubMed] [Google Scholar]
- 20.Bromme H.J., Ebelt,H., Peschke,D. and Peschke,E. (1999) Alloxan acts as a prooxidant only under reducing conditions: influence of melatonin. Cell. Mol. Life Sci., 55, 487–493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Daum G., Bohni,P.C. and Schatz,G. (1982) Import of proteins into mitochondria. Cytochrome b2 and cytochrome c peroxidase are located in the intermembrane space of yeast mitochondria. J. Biol. Chem., 257, 13028–13033. [PubMed] [Google Scholar]
- 22.Sharp P.A., Sugden,B. and Sambrook,J. (1973) Detection of two restriction endonuclease activities in Haemophilus parainfluenzae using analytical agarose. Ethidium bromide electrophoresis. Biochemistry, 12, 3055–3063. [DOI] [PubMed] [Google Scholar]
- 23.Southern E.M. (1975) Detection of specific sequences among DNA fragments separated by gel electrophoresis. J. Mol. Biol., 98, 503–517. [DOI] [PubMed] [Google Scholar]
- 24.Taketo A., Yasuda,S. and Sekiguchi,M. (1972) Initial step of excision repair in Escherichia coli: replacement of defective function of uvr mutants by T4 endonuclease V. J. Mol. Biol., 70, 1–14. [DOI] [PubMed] [Google Scholar]
- 25.Seawell P.C., Smith,C.A. and Ganesan,A.K. (1980) den V gene of bacteriophage T4 determines a DNA glycosylase specific for pyrimidine dimers in DNA. J. Virol., 35, 790–796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.McMillan S., Edenberg,H.J., Radany,E.H., Friedberg,R.C. and Friedberg,E.C. (1981) den V gene of bacteriophage T4 codes for both pyrimidine dimer-DNA glycosylase and apyrimidinic endonuclease activities. J. Virol., 40, 211–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nakabeppu Y. and Sekiguchi,M. (1981) Physical association of pyrimidine dimer DNA glycosylase and apurinic/apyrimidinic DNA endonuclease essential for repair of ultraviolet-damaged DNA. Proc. Natl Acad. Sci. USA, 78, 2742–2746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dizdaroglu M., Zastawny,T.H., Carmical,J.R. and Lloyd,R.S. (1996) A novel DNA N-glycosylase activity of E. coli T4 endonuclease V that excises 4,6-diamino-5-formamidopyrimidine from DNA, a UV-radiation- and hydroxyl radical-induced product of adenine. Mutat. Res., 362, 1–8. [DOI] [PubMed] [Google Scholar]
- 29.Boiteux S., Gajewski,E., Laval,J. and Dizdaroglu,M. (1992) Substrate specificity of the Escherichia coli Fpg protein (formamidopyrimidine-DNA glycosylase): excision of purine lesions in DNA produced by ionizing radiation or photosensitization. Biochemistry, 31, 106–110. [DOI] [PubMed] [Google Scholar]
- 30.Dizdaroglu M., Bauche,C., Rodriguez,H. and Laval,J. (2000) Novel substrates of Escherichia coli nth protein and its kinetics for excision of modified bases from DNA damaged by free radicals. Biochemistry, 39, 5586–5592. [DOI] [PubMed] [Google Scholar]
- 31.Senturker S., Auffret van der Kemp,P., You,H.J., Doetsch,P.W., Dizdaroglu,M. and Boiteux,S. (1998) Substrate specificities of the ntg1 and ntg2 proteins of Saccharomyces cerevisiae for oxidized DNA bases are not identical. Nucleic Acids Res., 26, 5270–5276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shishido K. and Ando,T. (1975) Site-specific fragmentation of bacteriophage T5 DNA by single-strand-specific S1 endonuclease. Biochim. Biophys. Acta, 390, 125–132. [DOI] [PubMed] [Google Scholar]
- 33.Wiegand R.C., Godson,G.N. and Radding,C.M. (1975) Specificity of the S1 nuclease from Aspergillus oryzae. J. Biol. Chem., 250, 8848–8855. [PubMed] [Google Scholar]
- 34.Ames B.N. (1989) Endogenous oxidative DNA damage, aging and cancer. Free Radic. Res. Commun., 7, 121–128. [DOI] [PubMed] [Google Scholar]
- 35.Senbongi H., Ling,F. and Shibata,T. (1999) A mutation in a mitochondrial ABC transporter results in mitochondrial dysfunction through oxidative damage of mitochondrial DNA. Mol. Gen. Genet., 262, 426–436. [DOI] [PubMed] [Google Scholar]
- 36.Li W. and Palmer,G. (1993) Spectroscopic characterization of the interaction of azide and thiocyanate with the binuclear center of cytochrome oxidase: evidence for multiple ligand sites. Biochemistry, 32, 1833–1843. [DOI] [PubMed] [Google Scholar]
- 37.Bennett M.C., Mlady,G.W., Kwon,Y.H. and Rose,G.M. (1996) Chronic in vivo sodium azide infusion induces selective and stable inhibition of cytochrome c oxidase. J. Neurochem., 66, 2606–2611. [DOI] [PubMed] [Google Scholar]
- 38.Yakes F.M. and Van Houten,B. (1997) Mitochondrial DNA damage is more extensive and persists longer than nuclear DNA damage in human cells following oxidative stress. Proc. Natl Acad. Sci. USA, 94, 514–519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Michaels M.L., Cruz,C., Grollman,A.P. and Miller,J.H. (1992) Evidence that mutY and mutM combine to prevent mutations by an oxidatively damaged form of guanine in DNA. Proc. Natl Acad. Sci. USA, 89, 7022–7025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.van der Kemp P.A., Thomas,D., Barbey,R., de Oliveira,R. and Boiteux,S. (1996) Cloning and expression in Escherichia coli of the OGG1 gene of Saccharomyces cerevisiae, which codes for a DNA glycosylase that excises 7,8-dihydro-8-oxoguanine and 2,6-diamino-4-hydroxy-5-N- methylformamidopyrimidine. Proc. Natl Acad. Sci. USA, 93, 5197–5202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Anson R.M., Croteau,D.L., Stierum,R.H., Filburn,C., Parsell,R. and Bohr,V.A. (1998) Homogenous repair of singlet oxygen-induced DNA damage in differentially transcribed regions and strands of human mitochondrial DNA. Nucleic Acids Res., 26, 662–668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Driggers W.J., Holmquist,G.P., LeDoux,S.P. and Wilson,G.L. (1997) Mapping frequencies of endogenous oxidative damage and the kinetic response to oxidative stress in a region of rat mtDNA. Nucleic Acids Res., 25, 4362–4369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Williamson D.H. and Fennell,D.J. (1974) Apparent dispersive replication of yeast mitochondrial DNA as revealed by density labelling experiments. Mol. Gen. Genet., 131, 193–207. [DOI] [PubMed] [Google Scholar]
- 44.Birky C.W. Jr and Skavaril,R.V. (1976) Maintenance of genetic homogeneity in systems with multiple genomes. Genet. Res., 27, 249–265. [DOI] [PubMed] [Google Scholar]