

Abstract
Aim: To identify and raise awareness of healthcare service gaps for individuals with adult-onset leukoencephalopathy with axonal spheroids and pigmented glia (ALSP).
Materials & methods: An ALSP patient journey map from symptom onset throughout disease course was developed using existing literature, patient and clinician feedback from a structured workshop and community survey data regarding attitudes toward genetic testing.
Results: ALSP diagnosis is frequently delayed due to low awareness of this rare condition and symptom overlap with more common neurological conditions. Multiple factors impact patients’ decision-making regarding genetic testing for ALSP, symptom management and participation in research studies.
Conclusion: These results highlight the challenges faced by individuals with ALSP and should support program development to improve patient care.
Keywords: : ALSP, CSF1R, genetic testing, neurodegenerative disease, patient journey map
Plain Language Summary
Adult-onset leukoencephalopathy with axonal spheroids and pigmented glia (ALSP) is a rare and fatal neurological disease. Symptoms generally appear between the ages of 40 and 50 years and worsen rapidly. Many patients have changes in memory, personality, behavior, and movement.
A patient journey map shows important events that patients experience over the course of a disease. The map can be used to identify challenges that patients face and where the quality of medical care can be improved. This patient journey map for ALSP is based on published scientific articles, a workshop where patients and others who have experience with ALSP talked about their challenges, and answers to questions from a survey on genetic testing for ALSP.
The map shows that diagnosis of ALSP is often delayed. Because ALSP is a rare disease, few medical providers have experience managing it. Medical providers may not suspect it because many ALSP symptoms overlap with symptoms of other common neurologic diseases. Genetic testing is needed to be sure that a person has ALSP, but it can be hard for patients to get tested since it is expensive and there may not be a testing center nearby. Finally, some people might not want to get tested if they are fearful of getting a diagnosis.
This patient journey map should be used to raise awareness of ALSP and the challenges faced by patients and those who care for them. It may also help researchers and medical providers know how to better support patients who have ALSP.
Tweetable Abstract
This patient journey map, developed to identify gaps in the care of patients with ALSP, highlights diagnostic challenges and barriers to genetic testing. It should be used to guide future research and advocacy to address challenges and better meet the needs of the ALSP community.
Plain language summary
Article highlights.
Introduction
Patient journey maps illustrate typical disease stages, milestones and touchpoints between the patient, care partner(s) and healthcare provider(s) and can be used to identify areas where patient care can be improved.
Methods
The patient journey map for adult-onset leukoencephalopathy with axonal spheroids and pigmented glia (ALSP) was based on a literature search, findings from an interactive workshop with representatives of the ALSP community, and the results of a survey on genetic testing.
Results
ALSP symptom heterogeneity, overlap with more common neurological conditions, and medical providers lack of disease awareness were highlighted as contributing reasons for diagnostic delays.
Cost, insurance coverage, and geographic location were identified as barriers to genetic testing for ALSP.
Discussion
A standardized care model for ALSP including genetic testing and a multidisciplinary clinical care team may begin to address the challenges identified by the ALSP patient journey map.
The number of confirmed ALSP cases is lower than expected, and it is possible that diagnostic accuracy could be improved if a standardized care model were to be implemented.
Shortening the time to diagnosis is important since ALSP progresses rapidly and the window for considering potential experimental treatments can be missed if symptoms have become too severe.
Conclusion
These findings should be used to increase awareness of ALSP among healthcare providers and guide future research efforts.
1. Introduction
Adult-onset leukoencephalopathy with axonal spheroids and pigmented glia (ALSP) is a fatal, rare, rapidly progressive neurological disease primarily caused by autosomal dominant pathogenic variants in the CSF1R gene [1,2]. In contrast to primary neurodegenerative diseases such as Alzheimer’s disease and dementia with Lewy bodies that occur in older adults [3], ALSP has a mean age of symptom onset between 40 and 50 years [4]. Symptom presentation is heterogeneous and can include personality changes and progressive cognitive and motor impairments such as weakness, impaired gait, bradykinesia, rigidity and/or tremor [1,5]. The average time from the onset of ALSP symptoms to death is 6–8 years, with relatively rapid and dramatic disease progression that quickly diminishes quality of life for patients, families and their care partner(s). End-stage disease results in complete loss of speech, self-awareness and voluntary movement such that most patients are bedridden and completely dependent on care partners [1,5].
The differential diagnosis of ALSP is complex due to the overlapping presentation of symptoms and radiological findings with multiple neurodegenerative disorders such as frontotemporal dementia (FTD), early-onset Alzheimer’s disease and multiple sclerosis (MS) [1,6]. Because ALSP is a rare disease, medical providers may lack awareness of or have limited experience with it, and, thus, may not initially suspect it at the first presentation of symptoms. This is further confounded by the nonspecificity of the presenting symptoms of ALSP, as evidenced by the variety of medical specialty clinics (e.g., movement disorder, dementia, psychiatric, adult leukodystrophy and MS) to which patients present for initial consultation [7]. As such, misdiagnosis of ALSP is common [8], which delays genetic counseling and the identification of patients who may be eligible to participate in research and clinical trials for new treatments.
Diagnostic criteria have been shown to be ≥96% sensitive for correctly identifying ALSP cases, although the specificity of these criteria for distinguishing between ALSP and other similar diseases was lower [9]. Therefore, accurate clinical suspicion should prompt genetic testing for confirmation; however, access to testing is not widely available to all patients. Geography and cost have been identified globally as barriers to genetic testing for neurodegenerative diseases, since genetic clinics are disproportionately located in large urban academic medical centers [10,11]. Lack of insurance coverage for genetic testing or counseling, limited awareness by healthcare providers or access to healthcare providers with experience in rare genetic diseases, and lack of awareness of personal risk or family medical history are also known barriers to genetic testing and thus to subsequent steps in a patient's journey. Individual attitudes, beliefs and emotional factors can also impact decisions regarding genetic testing [11].
Patient journey mapping is a way to facilitate understanding of a disease from the perspective of what patients experience including symptom onset to diagnosis, treatment and management of daily life. Typical disease stages, milestones and touchpoints between the patient, care partner(s), healthcare provider(s) and others engaged in care are often illustrated in a patient journey map. Describing the patient journey can also serve as a roadmap to facilitate effective communication and collaboration among healthcare providers, patients and families. The map can be used as a tool to align on areas where the quality of care should be improved. Studies have demonstrated the value of patient journey mapping to identify gaps in healthcare services and to improve the quality of those services [12,13]. The unmet medical needs of patients with progressive neurologic disorders such as MS, amyotrophic lateral sclerosis (ALS) and FTD have been described based on research questionnaires [1], but little has been reported on the severe physical, psychological, emotional and financial impacts of ALSP on patients, or associated diagnostic and care challenges. As such, the patient journey mapping approach may be particularly useful for raising awareness of this rapidly progressive and fatal disease.
We developed a patient journey map for ALSP informed by existing literature, patient and clinician experiences of ALSP collected during a collaborative workshop, and results from a survey of the ALSP community that assessed attitudes and behaviors around genetic testing.
2. Materials & methods
This work was an extension of a broader, retrospective review of the ALSP literature that was conducted by MEDLINE search via PubMed [1,8]. Eligible case reports and series published from 1 January 1980 through 22 March 2022, were identified for patients with an ALSP diagnosis that was confirmed by testing for the presence of CSF1R pathogenic variants. Detailed clinical data from the resulting cohort of 291 patients with genetically confirmed ALSP from 93 published reports were extracted to support the development of a preliminary patient journey map. The preliminary map was then refined based on outcomes from a virtual, structured ALSP workshop sponsored by Vigil Neuroscience, Inc. (Watertown, MA, USA), in collaboration with members of the ALSP community (neurologists, neuropsychologists, primary care physicians and patient advocacy group representatives). To further inform development of the patient journey map, members of the ALSP community were surveyed to better understand behaviors and attitudes surrounding genetic testing for ALSP.
2.1. Structured ALSP workshop
A diverse group of ALSP community representatives were invited to participate in a structured ALSP patient journey workshop, including 13 patients and care partners from the United States (six states), Ireland and Australia; and nine healthcare providers (clinicians and genetic counselors) from the United States (five states), Canada and the UK. A participant agreement was executed between each participant and Vigil Neuroscience, Inc. In advance of the workshop, participants were provided with a preliminary ALSP patient journey map to provide context for workshop discussions and to introduce the topics to be addressed during the workshop. Four breakout groups were formed by expressed preference of the participants, each assigned to discuss a stage of the patient journey including the path to a symptomatic diagnosis, the path to a genetic diagnosis, potential therapeutic options and living with ALSP.
To refine each stage of the patient journey, workshop participants contributed to an open discussion about their experiences with ALSP. Discussions were moderated by consultants from Imbue Partners, LLC (Middleton, MA, USA). Each breakout group met for 65 minutes followed by a 75 min debrief with all other workshop participants to share key insights. Semi-structured questions were used to guide the discussion for each topic. Based on workshop findings on each theme, salient moments in the patient journey representing key opportunities to improve patients’ and care partners’ experiences, address emotions and/or palliate the disease impact were identified to refine the preliminary patient journey map. The map was then revised by prioritizing these key moments and visually emphasizing challenges and gaps in care that exist within each stage of the journey for patients with ALSP.
2.2. ALSP community genetic testing survey
The genetic testing survey (Supplementary Figure S1) was developed and distributed electronically by patient advocacy groups and by Open Health™ (New York, NY, USA) via social media to US residents aged ≥18 years on the Vigil Neuroscience, Inc., distribution list, which included individuals who were diagnosed with ALSP and were living with symptoms, presymptomatic carriers of the CSF1R gene, or the biological family members of patients or carriers. Participation was strictly voluntary. To assure anonymity, respondents’ exact location (i.e., US state of residence) was not included. The survey comprised 21 questions focused on respondent age, family information related to genetic testing for ALSP, respondents’ testing status and attitudes toward and experiences with genetic testing. Question types included drop-down menu response, multiple choice (e.g., single response or “select all that apply”), precoded scaled response options (e.g., “significant impact,” “somewhat of an impact” or “no impact”) and open-ended.
All qualified responses were compiled, including responses to individual questions from incomplete surveys. Responses were analyzed descriptively and categorized by topic to identify respondent trends.
3. Results
Based on a recent comprehensive, retrospective analysis of ALSP cases from the existing literature [1,8], three steps along the disease course of ALSP were identified for inclusion in the preliminary (preworkshop) patient journey map (Supplementary Figure S2):
Path to diagnosis
Genetic testing
Living with ALSP
3.1. Key findings from the ALSP workshop
A wide variety of early ALSP symptoms were described, including apathy or social disengagement, personality changes and aggression, gait apraxia, cognitive decline and anxiety (Table 1). Symptoms such as distinctive gait changes, extreme slowing down, shuffling, or uncoordinated arms and legs were often first noticed by family members, whereas cognitive changes, such as impaired comprehension and forgetfulness, were more apparent in the workplace. For some patients, cognitive limitations caused frustration and triggered fear of dementia. Other patients lacked symptom recognition and self-awareness; in such cases, family members or coworkers were the first to recognize symptoms.
Table 1.
Early signs of adult-onset leukoencephalopathy with axonal spheroids and pigmented glia as reported by patients, care partners and healthcare professionals.
| Symptom category | Symptoms as reported by patients and care partners | Symptoms as reported by healthcare professionals |
|---|---|---|
| Cognitive impairment | Problems in the workplace “My coworker told me she thought I had MS” “My brother was asking me for advice on how to do his job” Forgetfulness “[…] being forgetful on the job” “Then aggression came along, […] not being able to remember causes that (i.e., aggression)” Impaired comprehension “She was starting to put things up [on social media] that just didn't make sense” |
Problems in the workplace “It's when people start often making mistakes in their job, that's when people know that something is seriously wrong” Impaired decision-making |
| Behavioral and psychiatric | Apathy “She just wasn't engaged anymore” Aggression Depression and/or anxiety “Early on, we could say it's not the disease: it's the stress of work, it's the stress of being a mom, raising children, financial stress” Impaired/out-of-character decision-making |
Depression and/or anxiety “[Regarding mood changes] It was clear that what she was presenting was completely disproportionate to […] menopause” |
| Motor | Impaired mobility “I noticed this extreme slowing down, they couldn't pick up the pace” “My coworker told me she thought I had MS” Shuffling gait “Why is nanny walking like a penguin?” |
Gait apraxia “I remember some patients saying, “I don't know how to walk anymore’” Incoordination of upper and lower limbs |
| Speech difficulty | Difficulty breathing and speaking | Speech disturbance/difficulty with words |
| Other | Loss of vision | — |
MS: Multiple sclerosis.
A key theme that emerged during the workshop was that it is challenging to diagnose ALSP due to symptom heterogeneity. Overlapping symptoms with more common neurological conditions such as MS, FTD, Alzheimer’s disease or Parkinson’s disease frequently prolong the path to an ALSP diagnosis. Workshop participants reported that prior to diagnosis, early observed symptoms tended to be attributed to stress, life changes or more common causes before the less common cause of ALSP was ever considered. Because each patient presents differently, it is difficult to recognize a genetic disease pattern even within a family with a history of ALSP or another neurodegenerative disease. Before a diagnosis of ALSP is made, patients without a family medical history of ALSP often receive referrals to several different kinds of medical specialists, many of whom have limited experience or knowledge of ALSP due to its rarity, often delaying diagnosis. Participants emphasized the importance of referral to a neurologist with expertise in leukodystrophy, white matter pathology, behavioral neurology or movement disorder neurology, since diagnosis and disease management are needlessly delayed until the right specialists are found.
Some participants reported feeling relief when a diagnosis of ALSP was finally made. Following a long period of uncertainty, the diagnosis allowed them to move forward, plan for the future, do their own research, and arrange for support. However, despite this sense of relief, participants also expressed simultaneous feelings of fear, anxiety, isolation, and anger as they processed the terminal nature of ALSP, the stigma associated with dementia, and the possibility that other members of their family could be affected. Fear and anxiety were also associated with a realization that the healthcare resources available to patients with ALSP are limited, owing to the disease's relatively young age of onset compared with other progressive neurological conditions such as Alzheimer's disease. Many of the services that those with significant cognitive and motor impairment qualify for, such as assisted living or memory care facilities, do not accept patients younger than 60 years. Such scenarios complicate disease management and contribute to family and care partner strain. This complexity is further compounded by the autosomal dominant nature of this disease whereby multiple generations can be impacted at the same time.
Workshop participants also discussed how the incremental loss of independence that accompanies symptom progression adds a considerable emotional component to the disease burden. Care partners may be at risk for job loss due to increasing patient dependency, the often overwhelming nature of caregiving responsibilities, and rapid, unpredictable changes in patient care needs as the disease progresses. In addition to managing the emotional and financial repercussions of daily life with ALSP, care partners reported that navigating the healthcare system and finding trustworthy, meaningful information and resources were added responsibilities and that they had to do their own research to find specialists and advocate for disease management. Without any kind of formal or structured care model in place, workshop participants reported having to take on the responsibility of assembling their own “make-shift” multidisciplinary care team to provide symptomatic treatments, including cognitive and/or emotional treatments (e.g., behavior therapy, antipsychotics and antidepressants), physical therapy, hydrotherapy, speech therapy and nutrition.
Generally, participants were committed to advancing ALSP research. Hematopoietic stem cell transplant (HSCT) was recognized as an experimental treatment option, although one that is an option only for a subset of patients who meet stringent criteria. In addition, participants felt a sense of urgency to shorten the time from symptom onset to diagnosis, since HSCT cannot be considered once symptoms have become too advanced. Participating in clinical trials was also of interest, although some participants had concerns that studies’ recruitment criteria may limit trial participation, and access to the full spectrum of clinical research opportunities can be limited for patients whose medical providers are inexperienced with or unaware of ALSP.
The personal and emotional nature of deciding to obtain genetic testing was another key theme that emerged from the workshop. Patients and family members may feel isolated. Positive results may lead to feelings of anger or guilt for passing along the disease-causing genetic variant to their children, and family members who test negative may feel survivor’s guilt. Some patients prepared themselves by going into the test expecting the result to be positive, as if they were only getting tested to confirm a diagnosis. Both patients and genetic counselors felt that it was important to have family present, since receiving the test results can be overwhelming even for patients who feel they are well prepared.
Initially, some individuals decide not to proceed with genetic testing. Workshop participants shared concerns that a diagnosis would affect their way of life. Some did not perceive a benefit to knowing, and others felt unprepared for the impact of the results. Reasons for changing one's mind about genetic testing – deciding to get tested after initially choosing not to – included learning about eligibility for HSCT or clinical trials, wanting to alleviate the anxiety of not knowing, witnessing other family members receive an ALSP diagnosis, approaching the age of potential symptom onset, symptom appearance, realizing that knowing might impact major life decisions such as going to medical school or saving for retirement, and wanting to help advance research for other patients, including family members. Other participants cited the possibility of intervention, enrollment in clinical trials or the impact that positive results could have on family and life planning as reasons why they had always planned to be tested.
Workshop attendees also described the sense of relief they felt after they received confirmation of an ALSP diagnosis through genetic testing and how this helped them to gain access to symptomatic therapies or potential experimental therapeutic options. Far-reaching implications of receiving the results of genetic testing, such as the ability/inability to purchase life insurance and other financial considerations, were also discussed. The realization that some patients were not aware of these ramifications until after testing highlights a valuable role for genetic counselors in raising awareness of these issues prior to testing.
Genetic counselors who participated in the workshop explained that the concept of early or predictive testing is fairly new and that generating awareness and improving access to testing has been challenging. Particularly in the United States, access to testing can be affected by medical insurance coverage and other financial factors. Workshop participants who did have access to genetic counseling reported widely variable experiences, with some finding it highly beneficial and others reporting no benefit at all.
3.2. Key findings from the genetic testing surveys
A total of 58 surveys with qualifying responses were received, with complete results from 39 respondents (Table 2). In terms of respondent age distribution, 31/58 (53.4%) were above 45 years of age and 27/58 (46.6%) were below. Of the 58 respondents, 22 (37.9%) had been diagnosed with ALSP. Overall, 40/58 (69.0%) reported that at least 2 members of their family had experienced symptoms (including 12/22 respondents who personally had an ALSP diagnosis who had at least one other family member diagnosed with ALSP). Fifty respondents provided additional information about their personal and family genetic testing status. Almost all respondents (46/50 [92.0%]) had personally decided not to test or had at least one family member who had decided not to test. At the time of the survey, 20/50 (40.0%) respondents had not received genetic testing and 30/50 (60.0%) had been tested.
Table 2.
Characteristics of genetic survey respondents.
| Characteristic | Totala | ||
|---|---|---|---|
| Personally diagnosed with ALSP, n/N (%) | 22/58 (37.9) | ||
| Number of family members (including the survey respondent) who experienced ALSP symptoms n/N (%) | |||
| 1 | 10/50 (20.0) | ||
| 2 | 12/50 (24.0) | ||
| 3 | 20/50 (40.0) | ||
| 4 | 1/50 (2.0) | ||
| 5 or more | 7/50 (14.0) | ||
| Had genetic testing | Did not have genetic testing | Total | |
|---|---|---|---|
| Respondent age group (years), n/N (%) | |||
| 18–25 | 2/30 (6.7) | 2/20 (10.0) | 4/58 (6.99) |
| 26–35 | 4/30 (13.3) | 6/20 (30.0) | 10/5858 (17.22) |
| 36–45 | 5/30 (16.7) | 6/20 (30.0) | 13/58 (22.44) |
| 46–55 | 11/30 (3.7) | 3/20 (15.0) | 18/58 (31.00) |
| 56–65 | 5/30 (16.7) | 3/20 (15.0) | 10/58 (17.22) |
| 66–75 | 2/30 (6.7) | 0 | 2/58 (3.4) |
| 76 or older | 1/30 (3.3) | 0 | 1/58 (1.7) |
Fifty-eight surveys with qualifying responses were received, and surveys with complete results were received from 39 respondents.
ALSP: Adult-onset leukoencephalopathy with axonal spheroids and pigmented glia.
Among those who had not received genetic testing, 18 provided responses about their future testing plans. Eight of 18 (44.4%) had no plans to pursue testing in the future, 4/18 (22.2%) were undecided about future plans, and 6/18 (33.3%) indicated that they planned to pursue testing in the future (of these, 4/6 [66.7%] planned to test within the next 6 months).
Respondents who had not received testing reported that they wanted to avoid the mental burden of knowing genetic testing results (10/18 [55.6%]) and that they wanted to have financial preparations such as life insurance in place first (9/18 [50.0%]; Figure 1A).
Figure 1.
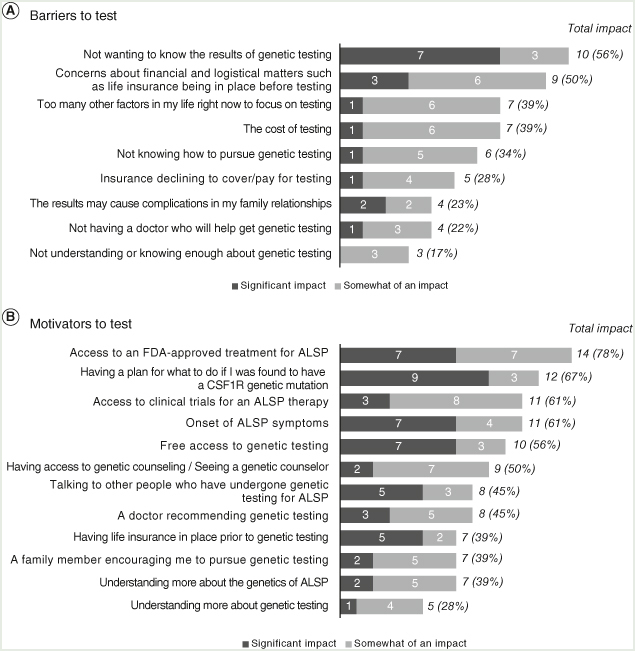
Individuals who did not receive genetic testing. Respondents could have selected “significant” or “somewhat significant” for more than one response to each question.
ALSP: Adult-onset leukoencephalopathy with axonal spheroids and pigmented glia.
“There is no cure and I want to live my [life] and wake up every day not feeling like I am living a death sentence.”
“I don't want the weight of knowing if I have a degenerative disease on my mind.”
The cost of testing (7/18 [38.9%]) and not knowing how to pursue testing (6/18 [33.3%]) were also barriers for this group. Fifteen of 18 (83.3%) respondents said that multiple factors contributed to their decision not to test. Access to treatment approved by the US FDA (14/18 [77.8%]) was reported most frequently by this group to be a factor that may motivate them to pursue testing in the future, followed by wanting to have a plan for the future (12/18 [66.7%]) and awareness of clinical trial opportunities (11/18 [61.1%]; Figure 1B).
Among the 30 respondents who had already been tested for ALSP, 23 provided insight into barriers they encountered prior to testing and the factors that motivated them to follow through with it. Seven of 23 (30.4%) said they were originally undecided or uninterested in genetic testing. Similar to the nontested group, the tested group reported concerns about getting life insurance in place (10/23 [43.5%]) and the emotional toll of results (10/23 [43.5%]; Figure 2A). Cost had less impact on the decision to test (3/23 [13.0%]), and family history seemed to motivate a substantial portion of respondents in this group to follow through with testing: 18/23 (78.3%) had a family member with a pathogenic CSF1R variant, a family member who had developed symptoms, and/or a known family history of ALSP.
Figure 2.
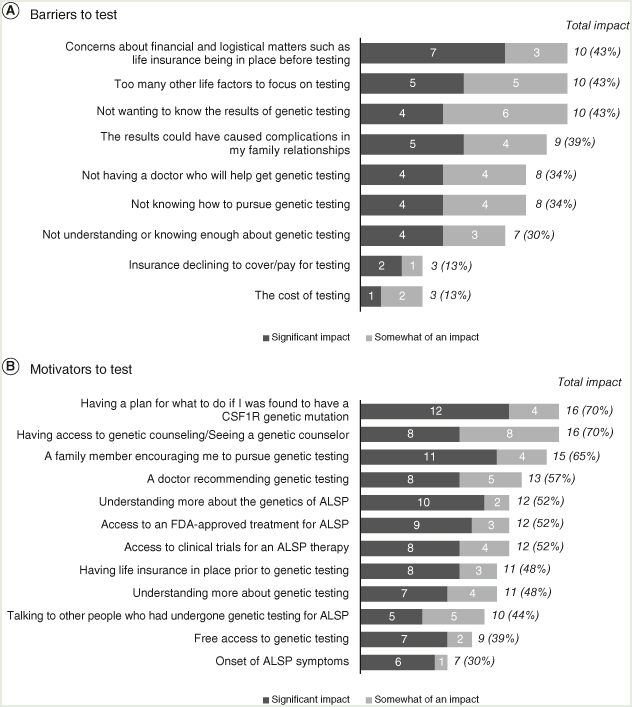
Individuals who received genetic testing. Respondents could have selected “significant” or “somewhat significant” for more than one response to each question.
ALSP: Adult-onset leukoencephalopathy with axonal spheroids and pigmented glia.
“To determine if eligible to be a bone marrow donor for my brother.”
“Lost a few family members. I have 5 children and 14 grandchildren and knew it was the only thing to do.”
“I am my sister’s caregiver. I am the older sister, just dealing with her on a daily basis is a constant reminder of what can happen to me and more of the family.”
Smaller proportions of patients in this group decided to get tested due to symptoms (3/23 [13.0%), doctor recommendation (3/23 [13.0%]), or to contribute to ALSP research (3/23 [13.0%]). Many respondents received genetic testing for life planning purposes as they approached the age when a sibling or other family members had previously been diagnosed. Thus, respondents in the tested group tended to be older than those who were not tested (aged >45 years: tested 19/30 [63.3%]; nontested 6/20 [30.0%]). Key factors that motivated them (7 of whom had been originally undecided or uninterested in genetic testing) to pursue testing included the desire to have a plan in place if they tested positive (16/23 [70.0%]), having access to genetic counseling (16/23 [70.0%]), and encouragement from a family member (15/23 [65.2%]; Figure 2B).
3.3. Mapping the ALSP patient journey
Based on key themes that emerged during the workshop, key moments in the patient journey were identified and prioritized and the preliminary patient journey map was revised to align with the workshop findings. Therefore, the revised patient journey map (Figure 3) starts from the time of ALSP symptom onset or discovery of a family history of ALSP and extends to diagnosis and genetic testing, emphasizing the iterative process of reaching a diagnosis and the emotional considerations and barriers associated with genetic testing. Decision-making regarding potential experimental therapeutic and symptom management options and the burden of living with ALSP are also illustrated.
Figure 3.
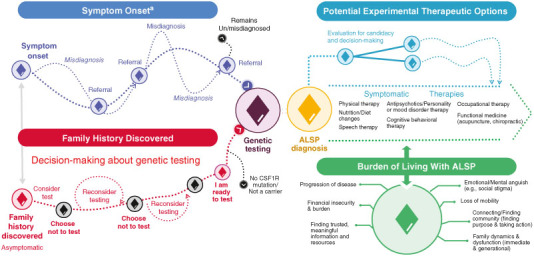
ALSP patient journey map.
aSymptoms include but are not limited to cognitive impairment, psychiatric symptoms, pain, motor impairment, gait disorder, urinary incontinence, speech disturbance, seizures and sleep disorders.
ALSP: Adult-onset leukoencephalopathy with axonal spheroids and pigmented glia.
4. Discussion
This patient journey map, initially (Supplementary Figure S2) informed by a literature review and refined based on the outcomes of the ALSP workshop and community survey (Figure 3), illustrates the challenging path for people living with ALSP, from symptom onset through genetic testing, symptom management and life adjustments. The path to a symptomatic ALSP diagnosis is often prolonged and iterative, owing to the heterogeneity of presenting symptoms. Referrals are often made to multiple specialists before a definitive diagnosis is reached. There is a lack of disease knowledge among many medical professionals, and a limited number of clinicians (including neurologists) specialize in the treatment and management of ALSP. As such, misdiagnosis is common, delaying the initiation of clinical care, symptomatic treatments and research enrollment. Although many individuals with ALSP are interested in research, if they are seeing medical providers with limited experience with or awareness of ALSP, they may never learn of available treatments to potentially manage symptoms or clinical research opportunities.
These findings are consistent with a previous analysis of published cases of patients with ALSP in which an accurate initial diagnosis was made in only approximately 25% of cases [8] and ALSP case studies in which long diagnostic delays due to overlapping symptoms with other more common neurodegenerative diseases were described [14–19]. One such study highlighted the heterogeneity of symptoms in a 63-year-old female who presented with 4 years of repetitive scratching followed by 10 years of progressive behavioral, cognitive and motor decline that for a long time was misdiagnosed as FTD [16]. In another family, members initially presented with Parkinsonism and eventually displayed clinical features that presented similar to corticobasal degeneration such as apraxia, seizures, and pyramidal tract signs [15].
The revised ALSP patient journey map also reflects challenges and barriers associated with the path to a genetic diagnosis. Emotional sentiments reported during the workshop, such as the need for extensive mental preparation before an individual chooses to be tested, fear of diagnosis, denial and guilt about passing a genetic mutation on to children, were similar to emotional barriers to genetic testing that have been reported for other autosomal dominant diseases [20]. Workshop results, largely corroborated by data from the community genetic survey, also provided valuable context regarding the influence of age, family dynamics and the perceived actionability of a genetic diagnosis on decision-making around genetic testing. Similar to genetic testing studies in ALS in which high percentages of patients with a family history of ALS receive genetic testing [21], the presence of a family history and/or encouragement from family members seems to influence the decision to get tested for ALSP.
Cost, insurance coverage and access to genetic counseling were also identified as important barriers to testing among survey respondents who had not yet completed genetic testing. Prior studies have shown that access to genetic counselors, who can help prepare individuals for the practical and emotional consequences of test results, is not equitable and is often driven by medical coverage, knowledge, awareness among healthcare providers, and other financial factors [10,20,22]. Particularly for cases in which the benefits of a genetic diagnosis include the potential for treatment or where clinical trials may be genotype driven, as is the case for some patients with ALS/FTD spectrum disorders [21,23], genetic testing is increasingly recommended and efforts to overcome these barriers should be made. Increasing awareness within the ALSP community of the role of genetic counselors and increasing awareness of ALSP within the genetic counseling community may improve the experience of those living with ALSP through facilitation of earlier diagnosis and access to symptomatic and/or experimental therapies.
We propose that a standardized care model for ALSP, including genetic counseling as part of a collaborative multidisciplinary clinical care team, is essential to begin to address the diagnostic and care challenges illustrated by the ALSP patient journey map. Multidisciplinary care models to organize and improve the quality of care have been proposed for other neurodegenerative diseases with similar disease burden to ALSP (e.g., leukodystrophies, Alzheimer’s disease and Parkinson’s disease) [24,25], but guidance for finding experts, resources and research to support the benefits of a multidisciplinary care team specifically for ALSP is lacking. Thus, studies of these conditions can serve as proxies in support of a management model that accounts for the considerable physical, psychological, emotional and financial burdens that ALSP imposes upon patients, families and care partners (Box 1) [26,27]. Without a standardized care model in place, the burden of coordinating care across providers often falls on care partners, and the lack of a clear pathway for referrals and inequities in access to healthcare providers with ALSP experience are fundamental gaps in care that likely contribute to treatment delays. In addition to improving the logistics and quality of care, it is possible that diagnostic accuracy could be improved with the development and dissemination of diagnostic criteria that could indicate and prompt the need for genetic counseling. The prevalence of ALSP in the United States is estimated to be at least 10,000 [8], but only a small number of patients with confirmed ALSP have been identified [1], possibly owing to high initial misdiagnosis rates. No disease-modifying treatment options are currently approved; case studies of HSCT in patients with ALSP have been published [28–30], but no prospective controlled studies of HSCT in this population have been conducted. Therefore, shortening the time to diagnosis remains important. ALSP progresses rapidly and the window for considering HSCT or any potential experimental treatments can be missed if symptoms have become too severe by the time a diagnosis is made.
Box 1. The treatment centers of excellence model.
A center of excellence is a multidisciplinary healthcare delivery network that brings together medical teams that are experienced in diagnosing and treating complex diseases. Centers of excellence are an increasingly favored clinical care approach for complex diseases due to the importance of integrating patient and provider experiences and identifying and validating practice parameters and treatment protocols [26]. General centers of excellence are now particularly encouraged for rare diseases, offering research opportunities for diseases in which clinical care options may be dependent upon or limited to cutting-edge therapies.
Modeled after networks that focus on the care and research of single or small groups of rare diseases, the NORD Rare Disease Centers of Excellence Program is a national network of medical institutions in the United States committed to the diagnosis, treatment and research of rare diseases.
Goals of each participating center are to:
Shorten the time to diagnosis.
Improve quality and access to care.
Accelerate research to develop new treatments.
Increase the number of multisite clinical trials.
Train more rare disease specialists.
The complex challenges experienced by those living with adult-onset leukoencephalopathy with axonal spheroids and pigmented glia (ALSP) and the larger medical community require attention and proposed solutions. To facilitate diagnosis and management of ALSP, the centers of excellence model may be adapted, or even reimagined within a virtual framework to potentially include experts from movement disorder, dementia, psychiatric/behavioral and adult leukodystrophy specialty clinics and genetic testing centers. Through this model, patients living with ALSP, as well as their families and care partners, can be connected with the limited number of providers with expertise in diagnosing and treating rare leukodystrophies such as ALSP as well as in interpretation of genetic results.
It should be noted that because ALSP is a rare disease, participation in both the patient journey workshop and community genetic survey was limited. Sample sizes were relatively small, and selection bias may exist due to sampling procedures, the requirement for participants to speak English during the workshop, and survey distribution to only US residents. Although patient, care partner, family member, and clinician participation in the workshop facilitated the sharing of a variety of perspectives and experiences, these findings may not apply to all individuals impacted by ALSP and similar future initiatives should be undertaken internationally to confirm the generalizability of these results. In addition, although quantitative methods were not used to compile ALSP workshop data, the collective results from both the workshop and the genetic survey represent a robust mixed methods approach for developing the ALSP patient journey map.
5. Conclusion
For patients with ALSP, the journey from symptom onset to ALSP diagnosis and management of daily life is lengthy, frequently iterative, costly and deeply emotional. The typical age of onset of ALSP is between 40 and 50 years, when many patients are actively employed, physically active and have not yet reached an age to qualify for many support services and resources available to older individuals. The dramatic, rapid symptom progression and loss of function in this age group contribute to the heavy disease burden observed with ALSP.
Despite the highly variable experiences of individual ALSP patients, these results highlight common diagnostic challenges faced by members of the ALSP community and provide valuable context regarding barriers to genetic testing. The goal of developing this patient journey map was to increase awareness of ALSP among healthcare providers, identify gaps in care and opportunities for improvements, and guide future research efforts and advocacy work to better meet the needs of those impacted by ALSP. It is hoped that these results will be used to support the development of programs designed to facilitate networking among ALSP providers and researchers, similar to programs that have been successfully implemented for other rare diseases [26].
Supplementary Material
Funding Statement
The authors declare financial support was received for the research, writing, and/or publication of this article. This analysis was supported by Vigil Neuroscience, Inc.
Supplemental material
Supplemental data for this article can be accessed at https://doi.org/10.1080/17582024.2024.2404378
Author contributions
BK Rush, C Cassandro, CM Rubio, HA Rutherford and A Toumadj were involved in design and conceptualization of the work. C Cassandro and CM Rubio were involved in data collection. All authors were involved in data interpretation, critically reviewed and revised the manuscript, and approved the final version for submission.
Financial disclosure
The authors declare financial support was received for the research, writing, and/or publication of this article. This analysis was supported by Vigil Neuroscience, Inc. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
Competing interests disclosure
BK Rush has received financial support from Vigil Neuroscience, Inc. for work as a consultant. A Smith and E Sullivan have nothing to disclose; C Cassandro, CM Rubio and A Toumadj are full-time employees of Vigil Neuroscience, Inc. HA Rutherford is a consultant with Vigil Neuroscience, Inc., and is in receipt of studentship from MRC Discovery Medicine North Doctoral Training Partnership (UK) and Sir Jules Thorn Scholarship. RL Piana has received consulting fees from Vigil Neuroscience, Inc., not directly related to this manuscript. The authors have no other competing interests or relevant affiliations with any organization or entity with the subject matter or materials discussed in the manuscript apart from those disclosed.
Writing disclosure
Medical writing and editorial support were provided by Allyson Lehrman, DPM, and Melissa Austin of Apollo Medical Communications, part of Helios Global Group, and funded by Vigil Neuroscience, Inc.
Ethical conduct of research
Clinical Research Ethics Committee or Independent Review Board approval was not required for the workshop or community survey.
References
Papers of special note have been highlighted as: • of interest
- 1.Papapetropoulos S, Pontius A, Finger E, et al. Adult-onset leukoencephalopathy with axonal spheroids and pigmented glia: review of clinical manifestations as foundations for therapeutic development. Front Neurol. 2022;12788168. doi: 10.3389/fneur.2021.788168 [DOI] [PMC free article] [PubMed] [Google Scholar]; • A position paper on adult-onset leukoencephalopathy with axonal spheroids and pigmented glia (ALSP), including sections on the multistep diagnosis process and patient and caregiver perspectives of disease burden due to unmet medical need.
- 2.Rush BK, Tipton PW, Strongosky A, et al. Neuropsychological profile of CSF1R-related leukoencephalopathy. Front Neurol. 2023;14:1155387. doi: 10.3389/fneur.2023.1155387 [DOI] [PMC free article] [PubMed] [Google Scholar]; • A study to document the neuropsychological profile of CSF1R-related leukoencephalopathy (CRL), providing support for the benefits of a multidisciplinary team for the care of patients with CRL.
- 3.Chin KS, Teodorczuk A, Watson R. Dementia with Lewy bodies: challenges in the diagnosis and management. Aust NZJ Psychiatry. 2019;53(4):291–303. doi: 10.1177/0004867419835029 [DOI] [PubMed] [Google Scholar]
- 4.Konno T, Kasanuki K, Ikeuchi T, et al. CSF1R-related leukoencephalopathy: a major player in primary microgliopathies. Neurology. 2018;91(24):1092–1104. doi: 10.1212/WNL.0000000000006642 [DOI] [PMC free article] [PubMed] [Google Scholar]; • A review providing epidemiological background on CSF1R-related leukoencephalopathy, highlighting its young mean age of onset (43 years), average duration of disease (6.8 years) and heterogeneous combination of motor and neuropsychiatric symptoms that overlap with other neurologic diseases. This paper also includes a genetic perspective section describing a variety of CSF1R mutations that have been previously reported.
- 5.Sundal C, Wszolek ZK. CSF1R-related adult-onset leukoencephalopathy with axonal spheroids and pigmented glia. In Adam MP, Feldman J, Mirzaa GM, et al., editors. GeneReviews®. Seattle, WA: University of Washington, Seattle; 2012. Aug 30 [Updated 2017 Oct 5] 1993–2023. [Google Scholar]
- 6.Solomon AJ, Arrambide G, Brownlee WJ, et al. Differential diagnosis of suspected multiple sclerosis: an updated consensus approach. Lancet Neurol. 2023;22(8):750–768. doi: 10.1016/s1474-4422(23)00148-5 [DOI] [PubMed] [Google Scholar]
- 7.Papapetropoulos S, Marsh A, Meier A, et al. A retrospective, natural history of adult-onset leukoencephalopathy with axonal spheroids and pigmented glia (ALSP): untangling the phenotype of a commonly misdiagnosed autosomal dominant neurodegenerative disease. National Society of Genetic Counselors (NSGC). Nashville, TN: 41st Annual Conference; November 16–22, 2022. [Google Scholar]
- 8.Papapetropoulos S, Gelfand JM, Konno T, et al. Clinical presentation and diagnosis of adult-onset leukoencephalopathy with axonal spheroids and pigmented glia: a literature analysis of case studies. Front Neurol. 2024;15:1320663. doi: 10.3389/fneur.2024.1320663 [DOI] [PMC free article] [PubMed] [Google Scholar]; • A literature analysis showing that ALSP is frequently misdiagnosed, due to symptoms that overlap with other more common disorders. Genetic testing can provide a definitive diagnosis but use in clinical practice may be limited by cost and unequal access.
- 9.Konno T, Yoshida K, Mizuta I, et al. Diagnostic criteria for adult-onset leukoencephalopathy with axonal spheroids and pigmented glia due to CSF1R mutation. Eur J Neurol. 2018;25(1):142–147. doi: 10.1111/ene.13464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Roberts JS, Patterson AK, Uhlmann WR. Genetic testing for neurodegenerative diseases: ethical and health communication challenges. Neurobiol Dis. 2020;141:104871. doi: 10.1016/j.nbd.2020.104871 [DOI] [PMC free article] [PubMed] [Google Scholar]; • A review of neurodegenerative diseases for which genetic testing is available, summarizing ethical considerations and communication challenges involved with genetic testing for severe life-threatening conditions.
- 11.Srinivasan S, Won NY, Dotson WD, et al. Barriers and facilitators for cascade testing in genetic conditions: a systematic review. Eur J Hum Genet. 2020;28(12):1631–1644. doi: 10.1038/s41431-020-00725-5 [DOI] [PMC free article] [PubMed] [Google Scholar]; • A review of 229 articles to identify barriers and facilitators that may affect decision-making around genetic testing of at-risk relatives of individuals who have been diagnosed with a genetic condition (i.e., cascade testing). The identification of factors that impact the uptake of cascade testing is important for timely and accurate diagnosis of autosomal dominant conditions that remain underdiagnosed in the population.
- 12.Benson M, Albanese A, Bhatia KP, et al. Development of a patient journey map for people living with cervical dystonia. Orphanet J Rare Dis. 2022;17(1):130. doi: 10.1186/s13023-022-02270-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.O'Regan D, Garcia-Borreguero D, Gloggner F, et al. Mapping the insomnia patient journey in Europe and Canada. Front Public Health. 2023;11:1233201. doi: 10.3389/fpubh.2023.1233201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Arshad F, Vengalil S, Maskomani S, et al. Novel CSF1R variant in adult-onset leukoencephalopathy masquerading as frontotemporal dementia: a follow-up study. Neurocase. 2021;27(6):484–489. doi: 10.1080/13554794.2021.2022704 [DOI] [PubMed] [Google Scholar]
- 15.Baba Y, Ghetti B, Baker MC, et al. Hereditary diffuse leukoencephalopathy with spheroids: clinical, pathologic and genetic studies of a new kindred. Acta Neuropathol. 2006;111(4):300–311. doi: 10.1007/s00401-006-0046-z [DOI] [PubMed] [Google Scholar]
- 16.Friedberg A, Ramos EM, Yang Z, et al. Case report: novel CSF1R variant in a patient with behavioral variant frontotemporal dementia syndrome with prodromal repetitive scratching behavior. Front Neurol. 2022;13:909944. doi: 10.3389/fneur.2022.909944 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gore E, Manley A, Dees D, et al. A young-onset frontal dementia with dramatic calcifications due to a novel CSF1R mutation. Neurocase. 2016;22(3):257–262. doi: 10.1080/13554794.2016.1175635 [DOI] [PubMed] [Google Scholar]
- 18.Lynch DS, Jaunmuktane Z, Sheerin UM, et al. Hereditary leukoencephalopathy with axonal spheroids: a spectrum of phenotypes from CNS vasculitis to parkinsonism in an adult onset leukodystrophy series. J Neurol Neurosurg Psychiatry. 2016;87(5):512–519. doi: 10.1136/jnnp-2015-310788 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sundal C, Fujioka S, Van Gerpen JA, et al. Parkinsonian features in hereditary diffuse leukoencephalopathy with spheroids (HDLS) and CSF1R mutations. Parkinsonism Relat Disord. 2013;19(10):869–877. doi: 10.1016/j.parkreldis.2013.05.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bernhardt BA, Zayac C, Pyeritz RE. Why is genetic screening for autosomal dominant disorders underused in families?The case of hereditary hemorrhagic telangiectasia. Genet Med. 2011;13(9):812–820. doi: 10.1097/GIM.0b013e31821d2e6d [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Roggenbuck J. C9orf72 and the care of the patient with ALS or FTD: progress and recommendations after 10 years. Neurol Genet. 2021;7(1):e542. doi: 10.1212/nxg.0000000000000542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Delikurt T, Williamson GR, Anastasiadou V, et al. A systematic review of factors that act as barriers to patient referral to genetic services. Eur J Hum Genet. 2015;23(6):739–745. doi: 10.1038/ejhg.2014.180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Crook A, Jacobs C, Newton-John T, et al. Toward genetic counseling practice standards for diagnostic testing in amyotrophic lateral sclerosis and frontotemporal dementia. Amyotroph Lateral Scler Frontotemporal Degener. 2022;23(7–8):562–574. doi: 10.1080/21678421.2022.2051553 [DOI] [PubMed] [Google Scholar]
- 24.Adang LA, Sherbini O, Ball L, et al. Revised consensus statement on the preventive and symptomatic care of patients with leukodystrophies. Mol Genet Metab. 2017;122(1–2):18–32. doi: 10.1016/j.ymgme.2017.08.006 [DOI] [PMC free article] [PubMed] [Google Scholar]; • A consensus statement summarizing the systems affected by leukodystrophies and management options for the wide range of symptoms experienced by affected patients. A framework to address the multifaceted needs of patients with leukodystrophies, including a wide network of care providers is proposed.
- 25.van der Marck MA, Fultz BA, Callahan CM. Geriatric models of care for neurodegenerative disorders. Handb Clin Neurol. 2019;167:57–72. doi: 10.1016/b978-0-12-804766-8.00004-2 [DOI] [PubMed] [Google Scholar]
- 26.NORD Rare Disease Centers of Excellence . 2024. Available from: https://rarediseases.org/rare-disease-centers-of-excellence/ [Google Scholar]
- 27.Duis J, van Wattum PJ, Scheimann A, et al. A multidisciplinary approach to the clinical management of Prader–Willi syndrome. Mol Genet Genomic Med. 2019;7(3):e514. doi: 10.1002/mgg3.514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Eichler FS, Li J, Guo Y, et al. CSF1R mosaicism in a family with hereditary diffuse leukoencephalopathy with spheroids. Brain. 2016;139(Pt 6):1666–1672. doi: 10.1093/brain/aww066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gelfand JM, Greenfield AL, Barkovich M, et al. Allogeneic HSCT for adult-onset leukoencephalopathy with spheroids and pigmented glia. Brain. 2020;143(2):503–511. doi: 10.1093/brain/awz390 [DOI] [PubMed] [Google Scholar]
- 30.Tipton PW, Stanley ER, Chitu V, et al. Is pre-symptomatic immunosuppression protective in CSF1R-related leukoencephalopathy? Mov Disord. 2021;36(4):852–856. doi: 10.1002/mds.28515 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.