

Abstract
Because of their role in the control of the topological state of DNA, topoisomerases are ubiquitous and vital enzymes, which participate in nearly all events related to DNA metabolism including replication and transcription. We show here that human topoisomerase I (Topo I) plays an unexpected role of ‘molecular matchmaker’ for G-quartet formation. G-quadruplexes are multi-stranded structures held together by square planes of four guanines (‘G-quartets’) interacting by forming Hoogsteen hydrogen bonds. Topo I is able to promote the formation of four-stranded intermolecular DNA structures when added to single-stranded DNA containing a stretch of at least five guanines. We provide evidence that these complexes are parallel G-quartet structures, mediated by tetrads of hydrogen-bonded guanine. In addition, Topo I binds specifically to pre-formed parallel and anti-parallel G4–DNA.
INTRODUCTION
DNA topoisomerases are a family of highly conserved and vital enzymes that control the topological state of DNA by transient cleavage of one (topoisomerase of type I) or two strands (type II) of the double helix (1). As a consequence, they maintain the integrity of the genetic material during replication, transcription, recombination and chromosome segregation and they have been shown to participate in most of the reactions that involve double-stranded DNA (2). Topoisomerase inhibitors have also gained wide clinical significance due to their efficacy as antitumour agents (1,3). Human DNA topoisomerase I (Topo I) is a type IB enzyme that cleaves and forms a transient 3′ phosphorotyrosyl bond with one strand of the double helix through a tyrosine residue in position 723 (Tyr723) (1,4). It is a monomeric protein of 100 kDa and contains four domains: the N-terminal, the ‘core’, the ‘linker’ and the C-terminal domain that contains the active site Tyr723.
On the other hand, it is well established that guanine-rich DNA sequences can form non B-DNA structures containing G-quartets (5). Recently, these structures have received great attention because sequences with a potential to form G-tetrads structures are often found in the human genome (6) and in particular the G-rich single-stranded 3′ overhangs of the telomeres of several species can adopt in vitro a quadruplex structure (7). In this paper, we show that Topo I induces G-quartet formation with oligonucleotides containing the polypurine tract (PPT) of the HIV-1 viral sequence (Table 1). Furthermore, we show that it also binds to pre-formed intermolecular and intramolecular G-quartet structures.
Table 1. Oligonucleotides sequences.
| Duplexes |
|
|
| 69R |
5′-GCTGTAGATCTTAGCCACTTTTTAAAAGAAA |
|
| |
AGGGGGGACTGGAAGGGCTAATTCACTCCCAACGAAGA-3′ |
|
| 69Y |
5′-TCTTCGTTGGGAGTGAATTAGCCCTTCCAGT |
|
| |
CCCCCCTTTTCTTTTAAAAAGTGGCTAAGATCTACAGC-3′ |
|
| 29R |
5′-CCACTTTTTAAAAGAAAAGGGGGGACTGG-3′ |
|
| 29Y |
5′-CCAGTCCCCCCTTTTCTTTTAAAAAGTGG-3′ |
|
| Oligonucleotide |
Sequence |
Complex |
| 16TCG | 5′-TTTTCTTTTGGGGGGT-3′a | Yes |
| 16TC |
5′-TTTTCTTTTCCCCCCT-3′ |
No |
| G6T |
5′-GGGGGGT-3′ |
Yes |
| T4CT4 |
5′-TTTTCTTTT-3′ |
No |
| acr-5′-16TCG |
Acr-5′-TTTTCTTTTGGGGGGT-3′ |
Yes |
| BPI-5′-16TCG |
BPI-5′-TTTTCTTTTGGGGGGT-3′ |
Yes |
| 16TCG-3′-BPI |
5′-TTTTCTTTTGGGGGGT-3′-BPI |
Yes |
| 16PCG7 |
5′-PPPPCPPPPXXXXXXT-3′b,c |
No |
| 16TCGgly |
5′-TTTTCTTTTYYYYYYT-3′d |
No |
| 15HG6T |
5′-TTTGGGGGGTCTTTT-3′ |
Yes |
| 16TCG5 |
5′-TTTTCTTGTTGGGGGT-3′ |
Yes |
| 16TCG4 |
5′-TTGTTCTTGTTGGGGT-3′ |
No |
| 13GA |
5′-GGAGGAGGAGGAG-3′ |
No |
| 21g |
5′-GGGTTAGGGTTAGGGTTAGGG-3′ |
n.d.e |
| 21gMet | 5′-GGGTTAGGGTTAGGGTTAGGG-3′f | n.d. |
The duplexes contain the 16 bp PPT oligopyrimidine·oligopurine sequence of the HIV-1 virus (underlined). Runs of four or more G are indicated in bold. The ability of different oligonucleotides to form the complex is also reported. Prior to treatment with Topo I, the different oligonucleotides were incubated as indicated in Materials and Methods with the target duplex 29R*/29Y, 3′ end-radiolabelled on the oligopurine strand, and then the samples were analysed on a 15% sequencing gel. The presence (>3% total radioactivity) or the absence of the band of slower mobility is reported in the column ‘Complex’.
aC = 5-methyl-2′-deoxycytidine.
bP = 5-propynyl-2′-deoxyuridine.
cX = N7-deaza-N9-deoxyriboguanine.
dY = N7-deoxyriboguanine.
en.d. = not determined.
fG = N7-methyl-2′-deoxyguanine.
MATERIALS AND METHODS
Oligonucleotides
The sequences of all oligonucleotides used in this study are reported in Table 1. Oligonucleotides were purchased from Eurogentec and purified using quick spin columns and Sephadex G-25 fine (Boehringer Mannheim). Concentrations were determined spectrophotometrically at 25°C using molar extinction coefficients at 260 nm calculated from a nearest-neighbour model (8). Prior to each experiment all oligonucleotides were treated for 30 min at 37°C in 10 mM NaOH followed by neutralisation at pH 7.0 and ethanol precipitation in order to disrupt the self-associated structures. After this treatment the oligonucleotides were stored for 1 week in a water solution containing 0.1 M lithium salt, in order to minimise quadruplex formation. The oligonucleotides were 3′ end-labelled with [α-32P]ddATP (Amersham, Arlington Heights, IL) by terminal transferase (Amersham) or 5′ end-labelled with [γ-32P]ATP (Amersham) by T4 polynucleotide kinase (New England Biolabs, Beverly, MA).
Topoisomerase enzymatic assays
To analyse the Topo I DNA cleavage products, the 69 bp fragment was 3′ end-labelled on the purine-rich strand. The 69 bp duplex was incubated for 1 h at 20°C, in 50 mM Tris–HCl, pH 7.5, 60 mM NaCl, 10 mM MgCl2, 0.5 mM DTT, 0.1 mM EDTA and 30 µg BSA (total reaction volume 20 µl). Approximately 0.4 nM of enzyme were added, followed by incubation for 1 h at 20°C. The DNA–Topo I cleavage complexes were dissociated by addition of SDS to a final concentration of 0.25% and of proteinase K (Sigma) to 250 µg/ml, followed by incubation for 35 min at 55°C. After ethanol precipitation, all samples were resuspended in 6 µl of formamide, heated at 90°C for 4 min and then chilled on ice for 4 min, before being loaded onto a denaturing 15% polyacrylamide gel [19:1 acrylamide:bisacrylamide] containing 7.5 M urea in TBE buffer (50 mM Tris base, 55 mM boric acid, 1 mM EDTA). For the experiment presented in Figure 1D, a longer heating time (20 min) at 90°C was required for proper denaturation of the complex. The gels were scanned with a Molecular Dynamics 445SI PhosphorImager. Human Topo I was purchased from TopoGEN, Inc. Mutants were obtained as described previously by Rossi et al. (9).
Figure 1.

Formation of a complex of slower mobility [S]. A weight marker was used (molecular weight MV, Boehringer Mannheim). The oligopurine-containing strand of the duplex is indicated by R and the oligopyrimidine sequence by Y. Oligonucleotide sequences are given in Table 1. (A) The 69 bp duplex was 3′ end-radiolabelled on the purine strand (lane 1) and was incubated for 1 h at 20°C as described in the presence of 3 µM CPT (lane 2) or in the presence of 2 µM 16TCG (lane 3). The samples were analysed on a denaturing 15% polyacrylamide gel. (B) 20 nM of 3′ end-radiolabelled 16TCG was incubated with 2 µM of unlabelled 16TCG oligonucleotide (lane 1) and after addition of Topo I (lane 2) and proteolyses, the samples were analysed on a sequencing gel. The band marked with an asterisk corresponds to a labelling artefact (radiolabelled dimer). (C) 20 nM of 3′ end-radiolabelled 29R was incubated with 2 µM of 15HG6T oligonucleotide (lanes 2 and 3) or 2 µM of G6T oligonucleotide (lanes 4 and 5) in the absence (lanes 2 and 4) or in the presence of Topo I (lanes 3 and 5). (D) The complex of slower mobility was cut, purified and heated for 20 min at 90°C prior to reloading on a sequencing gel.
Gel retardation assays
To study the binding of Topo I to pre-formed G4–DNA, 20 nM of radiolabelled G-rich oligonucleotide were incubated for 15 min at 20°C, in 50 mM Tris–HCl, pH 7.5, 60 mM KCl, 10 mM MgCl2, 0.5 mM DTT, 0.1 mM EDTA, 30 µg BSA, 10% sucrose, 1 µg/µl dT26 and 1 µg/µl of a 26 bp non-specific duplex, in the absence or in the presence of the enzyme. The samples were analysed by gel electrophoresis in 0.25× TBE onto a 6% polyacrylamide gel [29:1 acrylamide:bisacrylamide].
RESULTS AND DISCUSSION
Observation of an unusual DNA structure in the presence of Topo I
We investigated whether human Topo I could induce the formation of unusual DNA structures. The oligonucleotide 16TCG containing a stretch of six contiguous guanines (G6) was used in this study (Table 1). Two duplexes, containing the PPT 16 bp oligopyrimidine·oligopurine sequence present in the integrated HIV-1 virus, were used in this study: a 69 and 29 bp long duplex (see Table 1 for sequences). The 69 bp duplex was radiolabelled on the oligopurine strand (Fig. 1A, lane 1). The duplex was incubated with 0.4 nM Topo I in the presence of camptothecin (3 µM, lane 2) or 2 µM oligonucleotide 16TCG (lane 3) and the products, after protein digestion, were analysed on a sequencing gel (Fig. 1A). When the oligonucleotide was added to the duplex prior to addition of Topo I, the presence of a band of slower mobility [S] than the 69 nt radiolabelled oligopurine-containing strand (R) was observed (Fig. 1A, lane 3).
We investigated whether the enzyme would also be able to catalyse the reaction between single-stranded DNA fragments (Fig. 1B and C). As a matter of fact, upon addition of Topo I to the radiolabelled 16TCG oligonucleotide (20 nM) in the presence of 2 µM of unlabelled 16TCG, a band of slower mobility [S] was observed (Fig. 1B, lane 2); this band was not present in the absence of the enzyme (lane 1). Furthermore, Topo I induced the formation of this complex with the 29mer oligopurine strand of the duplex (Fig. 1C, 29R*) in the presence of 2 µM of two oligonucleotides containing six contiguous guanines: 15HG6T (lane 3) and G6T (lane 5) (see Table 1 for sequences). In the absence of Topo I, no band of slower mobility was observed (Fig. 1C, lanes 2 and 4, respectively).
The [S] band observed in Figure 1A, lane 3, was cut out, eluted, purified and heated at 90°C for 20 min in the presence of formamide prior to reloading on a sequencing gel. We found a band corresponding to the 69 nt oligopurine strand of the target DNA (Fig. 1D, arrow), which implies that the integrity of the 3′ end-labelled oligonucleotide was conserved and no covalent complex was formed in the slow running band. This finding ruled out the hypothesis of a complex being the product of a Topo I-mediated religation of the cleaved radiolabelled duplex to multiple oligonucleotides (10,11). It is also important to notice that the band [S] of low mobility was observed after protease digestion and electrophoresis on a denaturing gel. Furthermore, it was independent of the protease used [proteinase K (Sigma), pronase and trypsin (Roche Molecular Biochemicals)], implying that the slow mobility was not due to a protein fragment bound to the target DNA.
Characterisation of the complex
The partners. To characterise the complex, we first examined which strands were present in the slower band by radiolabelling one at a time the different oligonucleotides present in the reaction: the oligopyrimidine-containing strand of the duplex (Y), the oligopurine-containing strand of the duplex (R) and the oligonucleotides. We observed that R (Fig. 1C) and the oligonucleotide 16TCG (Fig. 1B), which have only six contiguous guanines in common, were present in the complex. When the oligopyrimidine-containing strand of the duplex (Y) was labelled (either by T4-kinase at the 5′-end or by terminal transferase at the 3′-end), no retarded band was observed (not shown). This observation strongly argues against the presence of the pyrimidine (Y) strand in the complex. Furthermore, radiolabelling of the R and 16TCG oligonucleotides at either the 3′- or the 5′-end gave the same result; as did oligonucleotides 15HG6T and G6T.
Role of the contiguous guanines stretch. It is well known that guanine-rich oligomers may adopt non B-DNA structures held together by G-quartets (Fig. 2A). A G-quartet consists of four guanine bases arranged in a square planar array by a cyclic hydrogen-bonding pattern, and these square planes of guanines stack to form quadruple-helical structures (5). A variety of G-quadruplex motifs exist in vitro and can be classified in terms of both molecularity and strand orientation (12).
Figure 2.

The complex is four-stranded. (A) Model structure of a guanine quartet and a four-stranded intermolecular complex. (B) Determination of the stoichiometry of the complex induced by Topo I between radiolabelled oligonucleotide 29R (A) and oligonucleotide G6T (B). The tetrameric species are indicated on the right. The arrows indicate the free probe and the G4–DNA structures.
To elucidate the structure of the complex, we tested different oligonucleotides (Table 1). The substitution of the guanines by cytosines (oligonucleotide 16TC) prevented the formation of the complex. When the 16TCG oligonucleotide was split in two parts: T4CT4 (oligonucleotide T4CT4) and G6T (oligonucleotide G6T), only the latter was able to form the complex ([S], Fig. 1C, lane 5), suggesting that guanine bases are essential to the formation of the complex. We also used oligonucleotides linked at their 3′- or 5′-end to DNA intercalators (such as acridine, oligonucleotide acr-5′-16TCG) or to other ligands (benzopyridoindole BPI, oligonucleotides BPI-5′-16TCG and 16TCG-3′-BPI) (13,14). The retarded band was still observed in the presence of Topo I independently of the presence of a 3′- or 5′-end substituent. Once more this observation confirmed that the complex was not the product of the religation step of the catalytic cycle of Topo I using the oligonucleotide as substrate, because the 3′ or 5′ OH-extremities (potential nucleophilic groups of the religation reaction) were not necessary to form the complex.
In order to ascertain the role of the guanine stretch, the guanine bases of the third strand were substituted by guanine analogues where the deoxyribose was attached to the N7 position instead of the N9 (oligonucleotide 16TCGgly), or by N7-deazaguanines (oligonucleotide 16PCG7, P = 5-propynyl-2′-deoxyuridine). Topo I-treated samples, run on a sequencing gel after protease digestion, revealed that no complex of slow mobility was formed (Table 1). The unavailability of the N7s of these guanines for Hoogsteen hydrogen bonding strongly suggested that the complex could form only when formation of G-quartets was possible (5).
It is well established that, at 20°C, pH 7.5, and in the presence of 10 mM of MgCl2, all the above oligonucleotides are able to bind to the target PPT DNA and form a local triple helix (15). As such, we tested the oligonucleotide 15HG6T (Table 1), which is no longer able to form the triple-helical structure but still has six contiguous guanines, in order to exclude any possible role of the formation of a triple helix on the target duplex. As shown in Figure 1C (lane 3), Topo I is still able to form the complex of lower mobility [S] with this oligonucleotide, confirming the role of the guanine stretch and showing that triplex formation was not required.
To answer the question of how long the stretch of consecutive guanines must be, two 16mers bearing the same base composition as the 16TCG oligonucleotide, but differing in the number of contiguous guanines, five for 16TCG5 and four for 16TCG4, respectively, were used. In order to observe the formation of the complex under our experimental conditions, at least five consecutive guanines were necessary (Table 1). We cannot rule out the possibility that the complex formed by oligonucleotide 16TCG4 was formed in solution in the presence of Topo I but that it was not stable enough to survive heat treatment before electrophoresis. Finally, the oligonucleotide 13GA, which has the ability to self-associate as a parallel homoduplex structure (16), does not form, in the presence of human Topo I, any band of slower mobility compared to the target DNA (Table 1).
Stoichiometry of the complex. We determined the stoichiometry of the complex by mixing the 29 nt long oligopurine strand of the duplex (A) with the 7 nt long oligonucleotide of sequence G6T (B) in the presence of Topo I (Fig. 2B). If similar amounts of oligomer A are mixed with B, then the stoichiometry of a given complex can be determined from the number of mixed species that are produced (17). For example, tetramer formation would result in five different complexes resolved on a native gel: the AAAA, AAAB, AABB, ABBB and BBBB complexes. In the presence of Topo I, when only oligomer A is radiolabelled, four species are observed (the BBBB complex is not radiolabelled), confirming the formation of a four-stranded complex. If an excess of A is added, the intensity of the species containing multiple copies of this strand increases (Fig. 2B, lane 2). All these findings suggested that G-quartets are formed and that Topo I induces a four-stranded complex.
Furthermore, this experiment allowed us to calculate that 0.4 nM of protein were able to convert 0.1–0.2 µM (~5–10%) of single-stranded oligonucleotide into its tetrameric structure.
Role of Topo I
To investigate the role of Topo I, other proteins such as Topo II (TopoGEN, Inc.), BSA (Biolabs, New England), histone H2A (Sigma) and different mutants of Topo I were used (Fig. 3). Neither Topo II, nor BSA nor histone H2A favoured the formation of the complex (data not shown). Preheating of Topo I for 20 min at 90°C inactivated the enzyme and the G-quartet complex was no longer formed (data not shown). Wild-type Topo I and its mutant deleted of the N-terminal fragment (ΔN), which still contains the catalytic region of the enzyme (4), induced the formation of the complex (Fig. 3, lanes 3 and 5). In contrast, the ΔC mutant, deleted of the C-terminal fragment, which contains the catalytic site, and the Y723F mutant, where the catalytic tyrosine Y723 is substituted by a phenylalanine, did not form the complex (Fig. 3, lanes 2 and 4). This finding indicates a specific role of Topo I and a requirement for an intact tyrosine residue in the active site.
Figure 3.
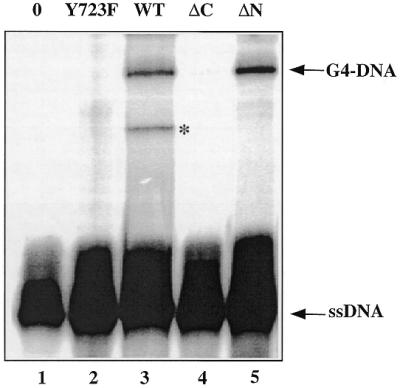
Role of Topo I. The 69 bp duplex after preincubation with 2 µM 16TCG in the described buffer was treated with different Topo I mutants: lane 1, no enzyme; lane 2, Topo I mutant Y723F; lane 3, Topo I wild-type; lane 4, Topo I mutant ΔC; lane 5, Topo I mutant ΔN. After protease digestion, the samples were loaded on a sequencing gel. The presence of the band marked with an asterisk was dependent on the Topo I preparation in the experiment and was not further characterised.
Topo I binding to G4 DNA structures
It was also interesting to investigate whether human Topo I binds to preformed G4–DNA structures. The radiolabelled 16TCG was incubated in the presence of 60 mM KCl at room temperature for 24 h in order to favour the formation of the G4–DNA complex. Then it was incubated in the absence or in the presence of Topo I or Topo II and the samples were analysed on a non-denaturing polyacrylamide gel (Fig. 4A). Topo I bound all the 16TCG oligonucleotide engaged in a G4–DNA structure and a diffuse band that may correspond to a Topo I/G4–DNA complex appeared (lane 2). In contrast Topo I did not shift the band corresponding to the monomeric species (single-stranded 16TCG). Topo II did not displace the G4–DNA band (lane 3) and thus did not recognise the G-quadruplex.
Figure 4.

Topo I binds to intermolecular and intramolecular G4–DNA. 20 nM of radiolabelled 16TCG and 2 µM of non-radiolabelled 16TCG preincubated in KCl (A) or 20 nM of radiolabelled 21g (B) were incubated for 15min at 20°C in the absence (lane 1) or in the presence of 0.4 nM Topo I (lane 2) or Topo II (lane 3). The samples were analysed by gel electrophoresis in 0.25× TBE onto a 6% polyacrylamide gel [29:1 acrylamide:bisacrylamide]. The arrows indicate the free probe, the G4–DNA structure and the Topo I/G4–DNA complex (Topo/G4 complex). (C) Competition experiments. 20 nM of radiolabelled 21g were incubated in the above buffer (lane 1) and in the presence of Topo I (lane 2) with a 1000-fold excess of: 21g (lane 3), 16TCG (lane 4), 21gMet (lane 5) and 16PCG7 (lane 6). The 21gMet oligonucleotide was obtained by DMS treatment for 15 min at 37°C from the 21g oligonucleotide; after this treatment the oligonucleotide does not form G-quartets structures. Only the quadruplex-forming oligonucleotides (lanes 3 and 4) are therefore able to compete. The oligonucleotides sequences are reported in Table 1.
In all these experiments, the G-quadruplex was formed by the association of four strands. An important question was whether Topo I was able to bind to intramolecular G-quartet-containing structures. The 21g oligonucleotide (see Table 1 for sequence), derived from the human telomeric G-rich strand, forms an intramolecular G-quartet structure under the conditions used in Topo I binding experiments (18). The oligonucleotide was incubated in the absence or in the presence of Topo I or II (Fig. 4B). We observed that Topo I bound to this intramolecular G-quadruplex (lane 2). Again Topo II had no effect (lane 3). Competition experiments, performed with 20 nM of radiolabelled 21g and a 1000-fold excess of different oligonucleotides (Fig. 4C), confirmed that Topo I binds only to the oligonucleotides that may form a quadruplex such as the 21g (lane 3) and the 16TCG oligonucleotide (lane 4). While the 16PCG7 oligonucleotide (lane 5) and the methylated form of the 21g oligonucleotide on the N7 of the guanines (lane 6) did not compete for binding of the enzyme to the 21g oligonucleotide.
CONCLUSIONS
Here we have demonstrated that human Topo I is able to induce the formation of a four-stranded complex held together by formation of G-quartets in the case of ssDNA containing a stretch of six or five guanines, and therefore it acts as a molecular matchmaker for G4–DNA formation (19). It has been shown that Topo I recognises and binds with different affinities cruciform DNA, mismatched duplexes, single-stranded DNA and hairpins (20), but to our knowledge, no study on the interaction of Topo I with G-quadruplexes has been reported as yet. Although the in vivo existence of G-quadruplex structure has not yet been proven, several regions of the human genome with the potential to form such a structure have been described: telomeres (12), the immunoglobulin switch region (5), the c-myc promoter (21) and the fragile-X-syndrome triplet repeats (22). Furthermore, conserved G-rich sequences are also found in retroviruses, such as the PPT of HIV-1 used in the present work (15), and it has been shown that G-quartet-forming oligonucleotides are able to inhibit the integrase activity of this retrovirus (23). The prevalence of G-rich regions implies that G-quadruplexes may indeed have a role in several biological events; and proteins, which interfere with the formation of these structures, could participate in these biological events. Several proteins have been shown to interact specifically with G-quartet-containing complexes (24,25); for example RAP 1, a yeast telomere binding protein (TBP) (26), and the β subunit of Oxytricha TBP (27); they both promote the formation of G-quartets by telomeric DNA. These proteins increase the rate at which the quadruplexes are formed and the authors have suggested a possible mechanism where the protein increases the effective local DNA concentration (26). Concerning other topoisomerases and G-quadruplexes, it has been shown that Topo II is able to cleave a DNA duplex containing hundreds of repeats of the human telomeric sequence (G3T2A) (28), and oligonucleotides derived from the immunoglubulin switch region complexed in a G-quartet structure (29). Yeast Topo III interacts with SGS1 helicase (30), which specifically unwinds G-quartet structures (31). More recently, the human RecQ helicases, Bloom’s syndrome and Werner’s syndrome DNA helicases have also been shown to unwind G-tetrads (32,33), and to interact with Topo III (34). A G4–DNA resolvase activity has also been identified in human placental tissue (35). Recent studies in eukaryotic cells have led to the discovery of new topoisomerases and new roles for Topo I in cellular processes (2). For example an RNase activity has been described for the vaccinia Topo I (36) and a kinase activity for the human Topo I (9).
We have shown that human Topo I binds to preformed intermolecular and intramolecular G-tetrads structures (Fig. 4), and induces the formation of intermolecular ones (Figs 1 and 2). It also promotes the opening of a 69 bp duplex to form a stable G-quartet-containing structure. The mutant with a phenylalanine instead of tyrosine in the active site of Topo I was unable to promote G-quartet formation. Although we have not demonstrated that the enzymatic activity of Topo I is required for G-quartet formation, this result might indicate that the active site of the protein is involved in nucleating the association of the four strands. In addition, we have shown that Topo I acts as a catalyst to promote the formation of intermolecular quadruplex species. We observed that 0.4 nM of Topo I is sufficient to produce ≥0.1 µM of parallel G4 DNA (i.e., 250 quadruplexes per protein). It should be noted that parallel quadruplex formation in the absence of protein requires a much higher oligonucleotide concentration (millimolar strand concentration range, instead of micromolar) in the presence of potassium ions and after an extensive incubation at 37–95°C (37,38).
These results open a new field of investigation concerning the roles of Topo I in the control of G-quartet formation together with other enzymatic processes (helicases, resolvases, recombinases) that are able to destabilise such structures. Different parts of the genome, such as telomeres, promoters or rDNA, may be the sites of such regulatory events.
Acknowledgments
ACKNOWLEDGEMENTS
The authors thank Prof. Frank Seela (University of Osnabrueck) for the gift of the N7-deaza-N9-deoxyriboguanines- and N7-deoxyriboguanines-containing oligonucleotides; and L. Lacroix, C. Giovannangeli and D. Praseuth for helpful discussions. This work was supported by grants (to P.B.A.) from the European Community and (to J.T.) from the Association pour le Recherche sur le Cancer (ARC).
REFERENCES
- 1.Wang J.C. (1996) DNA topoisomerases. Annu. Rev. Biochem., 65, 635–692. [DOI] [PubMed] [Google Scholar]
- 2.Nitiss J.L. (1998) Investigating the biological functions of DNA topoisomerases in eukaryotic cells. Biochim. Biophys. Acta, 1400, 63–81. [DOI] [PubMed] [Google Scholar]
- 3.Pommier Y., Pourquier,P., Fan,Y. and Strumberg,D. (1998) Mechanism of action of eukaryotic DNA topoisomerase I and drugs targeted to the enzyme. Biochim. Biophys. Acta, 1400, 83–105. [DOI] [PubMed] [Google Scholar]
- 4.Redinbo M.R., Stewart,L., Kuhn,P., Champoux,J.J. and Hol,W.G. (1998) Crystal structures of human topoisomerase I in covalent and noncovalent complexes with DNA. Science, 279, 1504–1513. [DOI] [PubMed] [Google Scholar]
- 5.Sen D. and Gilbert,W. (1988) Formation of parallel four-stranded complexes by guanine-rich motifs in DNA and its applications for meiosis. Nature, 334, 364–366. [DOI] [PubMed] [Google Scholar]
- 6.Gilbert D.E. and Feigon,J. (1999) Multistranded DNA structures. Curr. Opin. Struct. Biol., 9, 305–314. [DOI] [PubMed] [Google Scholar]
- 7.Williamson J.R., Raghuraman,M.K. and Cech,T.R. (1989) Monovalent cation-induced structure of telomeric DNA: the G-quartet model. Cell, 59, 871–880. [DOI] [PubMed] [Google Scholar]
- 8.Cantor C.R., Warshaw,M.M. and Shapiro,H. (1970) Oligonucleotides interactions. III. Circular dichroism studies of the conformation of deoxyoligonucleotides. Biopolymers, 9, 1059–1077. [DOI] [PubMed] [Google Scholar]
- 9.Rossi F., Labourier,E., Gallouzi,I.E., Derancourt,J., Allemand,E., Divita,G. and Tazi,J. (1998) The C-terminal domain but not the tyrosine 723 of human DNA topoisomerase I active site contributes to kinase activity. Nucleic Acids Res., 26, 2963–2970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shuman S. (1994) Novel approach to molecular cloning and polynucleotide synthesis using vaccinia DNA topoisomerase. J. Biol. Chem., 269, 32678–32684. [PubMed] [Google Scholar]
- 11.Henningfeld K.A., Arslan,T. and Hecht,S.M. (1996) Alteration of DNA primary structure by DNA topoisomerase I. Isolation of the covalent topoisomerase I-DNA binary complex in enzymatically competent form. J. Am. Chem. Soc., 118, 11701–11714. [Google Scholar]
- 12.Williamson J.R. (1994) G-quartet structures in telomeric DNA. Annu. Rev. Biophys. Biomol. Struct., 23, 703–730. [DOI] [PubMed] [Google Scholar]
- 13.Mergny J.L., Duval-Valentin,G., Nguyen,C.H., Perrouault,L., Faucon,B., Rougée,M., Montenay-Garestier,T., Bisagni,E. and Hélène,C. (1992) Triple Helix-Specific Ligands. Science, 256, 1681–1684. [DOI] [PubMed] [Google Scholar]
- 14.Silver G.C., Sun,J.S., Nguyen,C.H., Boutorine,A.S., Bisagni,E. and Hélène,C. (1997) Stable triple-helical DNA complexes formed by benzopyridoindole- and benzopyridoquinoxaline-oligonucleotide conjugates. J. Am. Chem. Soc., 119, 263–268. [Google Scholar]
- 15.Giovannangeli C., Diviacco,S., Labrousse,V., Gryaznov,S., Charneau,P. and Hélène,C. (1997) Accessibility of nuclear DNA to triplex-forming oligonucleotides: the integrated HIV-1 provirus as a target. Proc. Natl Acad. Sci. USA, 94, 79–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Arimondo P.B., Barcelo,F., Sun,J.S., Maurizot,J.C., Garestier,T. and Hélène,C. (1998) Triple helix formation by (G,A)-containing oligonucleotides: asymmetric sequence effect. Biochemistry, 37, 16627–16635. [DOI] [PubMed] [Google Scholar]
- 17.Sen D. and Gilbert,W. (1990) A sodium-potassium switch in the formation of four-stranded G4-DNA. Nature, 344, 410–414. [DOI] [PubMed] [Google Scholar]
- 18.Mergny J.L., Phan,A.T. and Lacroix,L. (1998) Following G-quartet formation by UV-spectroscopy. FEBS Lett., 435, 74–78. [DOI] [PubMed] [Google Scholar]
- 19.Portman D.S. and Dreyfuss,G. (1994) RNA annealing activities in HeLa nuclei. EMBO J., 13, 213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Thiyagarajan M.M., Waldman,S.A., Noe,M. and Kmiec,E.B. (1998) Binding characteristics of Ustilago maydis topoisomerase I to DNA containing secondary structures. Eur. J. Biochem., 255, 347–355. [DOI] [PubMed] [Google Scholar]
- 21.Simonsson T., Pecinka,P. and Kubista,M. (1998) DNA tetraplex formation in the control region of c-myc. Nucleic Acids Res., 26, 1167–1172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fry M. and Loeb,L.A. (1994) The fragile X syndrome d(CGG)n nucleotide repeats form a stable tetrahelical structure. Proc. Natl Acad. Sci. USA, 91, 4950–4954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mazumder A., Neamati,N., Ojwang,J.O., Sunder,S., Rando,R.F. and Pommier,Y. (1996) Inhibition of the human immunodeficiency virus Type 1 integrase by guanosine quartet structures. Biochemistry, 35, 13762–13771. [DOI] [PubMed] [Google Scholar]
- 24.Mergny J.L., Mailliet,P., Lavelle,F., Riou,J.F., Laoui,A. and Hélène,C. (1999) The development of telomerase inhibitors: the G-quartet approach. Anticancer Drug Des., 14, 327–339. [PubMed] [Google Scholar]
- 25.Muniyappa K., Anuradha,S. and Byers,B. (2000) Yeast meiosis-specific protein Hop1 binds to G4 DNA and promotes its formation. Mol. Cell. Biol., 20, 1361–1369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Giraldo R., Suzuki,M., Chapman,L. and Rhodes,D. (1994) Promotion of parallel DNA quadruplexes by a yeast telomere binding protein: a circular dichroism study. Proc. Natl Acad. Sci. USA, 91, 7658–7662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fang G. and Cech,T.R. (1993) The beta subunit of Oxytricha telomere-binding protein promotes G-quartet formation by telomeric DNA. Cell, 74, 875–885. [DOI] [PubMed] [Google Scholar]
- 28.Yoon H.J., Choi,I.Y., Kang,M.R., Kim,S.S., Muller,M.T., Spitzner,J.R. and Chung,I.K. (1998) DNA topoisomerase II cleavage of telomeres in vitro and in vivo. Biochim. Biophys. Acta, 1395, 110–120. [DOI] [PubMed] [Google Scholar]
- 29.Chung I.K., Mehta,V.B., Spitzner,J.R. and Muller,M.T. (1992) Eukaryotic topoisomerase II cleavage of parallel stranded DNA tetraplexes. Nucleic Acids Res., 20, 1973–1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gangloff S., McDonald,J.P., Bendixen,C., Arthur,L. and Rothstein,R. (1994) The yeast type I topoisomerase Top3 interacts with Sgs1, a DNA helicase homolog: a potential eukaryotic reverse gyrase. Mol. Cell. Biol., 14, 8391–8398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sun H., Bennett,R.J. and Maizels,N. (1999) The Saccharomyces cerevisiae Sgs1 helicase efficiently unwinds G-G paired DNAs. Nucleic Acids Res., 27, 1978–1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sun H., Karow,J.K., Hickson,I.D. and Maizels,N. (1998) The Bloom’s syndrome helicase unwinds G4 DNA. J. Biol. Chem., 273, 27587–27592. [DOI] [PubMed] [Google Scholar]
- 33.Fry M. and Loeb,L.A. (1999) Human werner syndrome DNA helicase unwinds tetrahelical structures of the fragile X syndrome repeat sequence d(CGG)n. J. Biol. Chem., 274, 12797–12802. [DOI] [PubMed] [Google Scholar]
- 34.Wu L., Karow,J.K. and Hickson,I.D. (1999) Genetic recombination: Helicases and topoisomerases link up. Curr. Biol., 9, R518–R520. [DOI] [PubMed] [Google Scholar]
- 35.Harrington C., Lan,Y. and Akman,S.A. (1997) The identification and characterization of a G4-DNA resolvase activity. J. Biol. Chem., 272, 24631–24636. [DOI] [PubMed] [Google Scholar]
- 36.Sekiguchi J. and Shuman,S. (1997) Site-specific ribonuclease activity of eukaryotic DNA topoisomerase I. Mol. Cell, 1, 89–97. [DOI] [PubMed] [Google Scholar]
- 37.Han H., Cliff,C.L. and Hurley,L.H. (1999) Accelerated assembly of G-quadruplex structures by a small molecule. Biochemistry, 38, 6981–6986. [DOI] [PubMed] [Google Scholar]
- 38.Hanakahi L.A., Sun,H. and Maizels,N. (1999) High affinity interactions of nucleolin with G-G-paired rDNA. J. Biol. Chem., 274, 15908–15912. [DOI] [PubMed] [Google Scholar]