

Summary
Given expanding studies in epidemiology and disease-oriented human studies offering hundreds of associations between the human “ome” and disease, prioritizing molecules relevant to disease mechanisms among this growing breadth is important. Here, we link the circulating proteome to human heart failure (HF) propensity (via echocardiographic phenotyping and clinical outcomes) across the lifespan, demonstrating key pathways of fibrosis, inflammation, metabolism, and hypertrophy. We observe a broad array of genes encoding proteins linked to HF phenotypes and outcomes in clinical populations dynamically expressed at a transcriptional level in human myocardium during HF and cardiac recovery (several in a cell-specific fashion). Many identified targets do not have wide precedent in large-scale genomic discovery or human studies, highlighting the complementary roles for proteomic and tissue transcriptomic discovery to focus epidemiological targets to those relevant in human myocardium for further interrogation.
Keywords: heart failure, transcriptomics, proteomics, snRNA-seq, multiomics
Graphical abstract
Highlights
-
•
Expanding studies in epidemiology identify many molecular targets in heart failure
-
•
Prioritizing among these targets for downstream studies is critical
-
•
We link a proteome to cardiac structure, function, outcomes, and transcription
-
•
This offers a complementary proteomic-transcriptional path for discovery
Studies in epidemiology have identified many targets in heart failure, fueling the need to identify ways to prioritize among them for discovery. Here, Perry et al. enlist a combined proteomic-transcriptional approach to identify biological targets in human heart failure. This approach offers a complementary path for target discovery for future studies.
Introduction
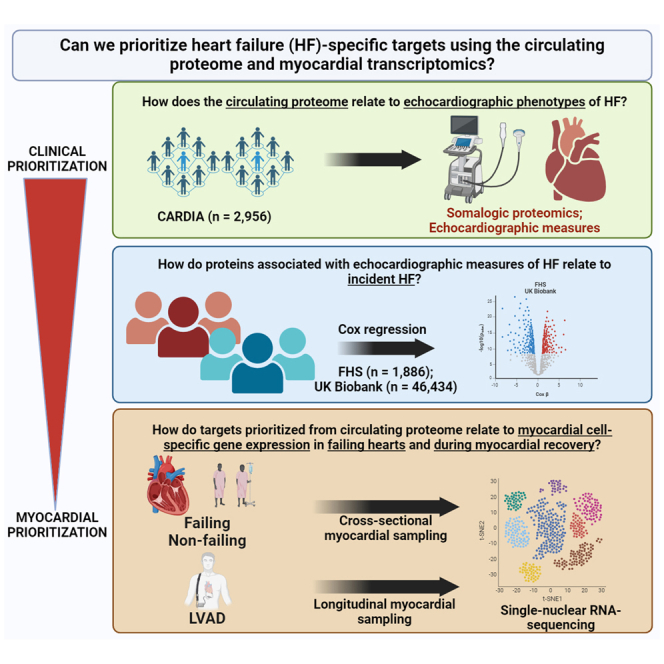
Despite advances in heart failure (HF) care and prevention efforts, the lifetime risk of HF at age 50 years is nearly 1 in 4.1 HF has a dismal prognosis once diagnosed despite guideline-directed medical therapy.2 While the pathophysiology of HF can be studied in preclinical animal models through genetic manipulation and phenotyping, critical genetic and physiological differences between these model systems and human biology limit the translatability of findings (e.g., lack of consistency in effect, off-target effects, confounding, etc.). Obtaining human myocardial tissue in many patients with HF is challenging, given that endomyocardial biopsy is rarely indicated clinically and not without risk. Thus, there is a need to develop non-invasive methods in humans that (1) identify molecular surrogates of cellular physiology that are (2) relevant to HF and (3) exist early in its progression to identify and prioritize pathways causally implicated in pathogenesis for targeting.3,4,5,6,7
For the last nearly two decades, large human population-based studies have leveraged increasingly broad profiling efforts from circulation (“omics”) to identify prognostic and diagnostic biomarkers of HF, its phenotypes, and genetic susceptibility.4,5,8 Nevertheless, HF is a phenotypically heterogeneous, acquired condition with contributions from multiple organ systems, limiting studies of genetic susceptibility. Furthermore, sifting through hundreds of clinical phenotype-to-molecule associations from large populations presents a “needle-in-a-haystack” problem, given the time and cost inefficiency of gain- and loss-of-function screens in model systems. In addition, mapping to extant resources (e.g., Tabula Sapiens,9 GTEx10) is limited, as the included samples are not specifically targeted to HF. This represents a fundamental problem in the next phase in HF studies (and cardiovascular omics more broadly): are there any plausible methods to “filter” the circulating biomarker space to prioritize targets of particular interest in HF?
We addressed this question by using successive layers of prioritization from the proteome, phenome, and myocardial transcriptome at single-nucleus resolution in HF and during myocardial recovery as an approach to parse targets from cohort epidemiology of relevance in human HF. We first studied nearly 50,000 individuals across three population studies (Coronary Artery Risk Development in Young Adults [CARDIA], Framingham Heart Study [FHS], UK Biobank) to identify robust cross-cohort circulating proteomic signatures of key clinical measures, including echocardiographic pathophenotypes that predispose to HF and long-term incident HF (“clinical prioritization”). We mapped these targets onto (1) cell-specific gene expression in human hearts with and without HF (including across cardiac chambers) and (2) during myocardial recovery (under ventricular assist device unloading) using single-nucleus RNA sequencing (snRNA-seq). Our primary hypothesis was that—despite the various tissue sources of the human proteome and potential RNA-protein discordance—overlaying circulating proteins related to clinical phenotypes on cell-specific myocardial gene expression across HF states and with HF therapy might offer a way to prioritize targets for future study that demonstrate relevance within both populations and human myocardial tissue.
Results
Characteristics of included study participants
The characteristics of participants from the population-based cohorts are shown in Tables 1 and 2. CARDIA participants had a mean age of 50 years (56% women, 46% Black), compared to a slightly older distribution of age and a similar balance across gender in FHS and UK Biobank participants (who were primarily White). There was an overall low prevalence of cardiovascular disease (CVD) and moderate to high prevalence of risk factors (e.g., BMI, hypertension) across cohorts.
Table 1.
Characteristics of participants from the CARDIA study
| Covariate | Derivation set (N = 2,075) |
Validation set (N = 890) |
p value | ||
|---|---|---|---|---|---|
| Mean (SD)/N (%) | Missing data | Mean (SD)/N (%) | Missing data | ||
| Age at year 25 | 50.1 (3.7) | 0 | 50.2 (3.6) | 0 | 0.45 |
| 40–49 years | 845 (40.7%) | N/A | 354 (39.8%) | N/A | N/A |
| 50–59 years | 1,230 (59.3%) | N/A | 536 (60.2%) | N/A | N/A |
| Female (n, %) | 1,176 (56.7%) | 0 | 481 (54.0%) | 0 | 0.19 |
| Black (n, %) | 966 (46.6%) | 0 | 410 (46.1%) | 0 | 0.81 |
| Body mass index, kg/m2 | 30.2 (7.2) | 4 | 30.3 (7.1) | 0 | 0.88 |
| BMI <25 | 511 (24.7%) | N/A | 206 (23.1%) | N/A | N/A |
| BMI 25–30 | 661 (31.9%) | N/A | 278 (31.2%) | N/A | N/A |
| BMI >30 | 899 (43.4%) | N/A | 406 (45.6%) | N/A | N/A |
| Systolic blood pressure, mmHg | 118.2 (15.1) | 3 | 119.6 (15.9) | 1 | 0.021 |
| SBP <130 | 1,681 (81.1%) | N/A | 696 (78.3%) | N/A | N/A |
| SBP ≥130 | 391 (18.9%) | N/A | 193 (21.7%) | N/A | N/A |
| Smoking status (n, %) | N/A | 15 | N/A | 4 | 0.96 |
| Current smoker | 551 (26.7%) | N/A | 235 (26.5%) | N/A | N/A |
| Quit within a year | 121 (5.9%) | N/A | 50 (5.6%) | N/A | N/A |
| Never or quit over a year | 1,388 (67.4%) | N/A | 601 (67.8%) | N/A | N/A |
| Diabetes (fasting glucose ≥126 mg/dL or on medications) (n, %) | 169 (8.1%) | 0 | 69 (7.8%) | 0 | 0.72 |
| History of heart failure hospitalization | 39 (1.9%) | 0 | 8 (0.9%) | 0 | 0.07 |
| Any cardiovascular disease by year 25 (n, %) | 70 (3.4%) | 0 | 27 (3.0%) | 0 | 0.63 |
| Total cholesterol (mg/dL) | 191.7 (37.1) | 0 | 192.8 (36.3) | 0 | 0.43 |
| Total high-density lipoprotein cholesterol (mg/dL) | 57.5 (17.1) | 0 | 58.3 (19.4) | 0 | 0.29 |
| LV end-diastolic volume (mL) | 111.3 (29.4) | 150 | 112.9 (31.4) | 65 | 0.18 |
| Normal | 1,835 (95.3%) | N/A | 786 (95.3%) | N/A | N/A |
| LV end-systolic volume (mL) | 43.7 (17.8) | 151 | 44.3 (18.5) | 65 | 0.41 |
| Normal | 1,810 (94.1%) | N/A | 782 (94.8%) | N/A | N/A |
| LV mass (g) | 170.1 (53.2) | 158 | 171.3 (52.8) | 66 | 0.61 |
| Normal | 1,368 (71.4%) | N/A | 589 (71.5%) | N/A | N/A |
| E/e′ ratio | 8.1 (2.4) | 54 | 8.0 (2.2) | 26 | 0.74 |
| Normal | 1,670 (82.6%) | N/A | 730 (84.5%) | N/A | N/A |
| E/A ratio | 1.3 (0.4) | 34 | 1.3 (0.4) | 15 | 0.94 |
| Normal | 1,989 (97.5%) | N/A | 849 (97.0%) | N/A | N/A |
| LV ejection fraction (%) | 61.4 (7.5) | 151 | 61.4 (6.9) | 65 | 1.00 |
| Normal | 1,831 (95.2%) | N/A | 803 (97.3%) | N/A | N/A |
| Peak longitudinal strain (%) | −15.0 (2.4) | 263 | −15.1 (2.4) | 111 | 0.52 |
| Normal | 1,665 (91.9%) | N/A | 711 (91.3%) | N/A | N/A |
| Left atrium internal dimensions (cm) | 3.7 (0.5) | 56 | 3.7 (0.5) | 29 | 0.43 |
| Normal | 1,244 (61.6%) | N/A | 515 (59.8%) | N/A | N/A |
| Left atrial volume (mL) | 49.6 (16.0) | 28 | 50.4 (16.0) | 6 | 0.25 |
| Normal | 1,253 (67.8%) | N/A | 538 (68.7%) | N/A | N/A |
| Tricuspid annular plane systolic excursion (cm) | 2.5 (0.5) | 169 | 2.6 (0.5) | 53 | 0.25 |
| Normal | 1,816 (95.3%) | N/A | 796 (95.1%) | N/A | N/A |
| Estimated RV systolic pressure (mmHg) | 31.1 (5.9) | 1,286 | 30.9 (5.8) | 554 | 0.54 |
| Normal | 615 (77.9%) | N/A | 265 (78.9%) | N/A | N/A |
SD, standard deviation; BMI, body mass index; SBP, systolic blood pressure; LV, left ventricular; RV, right ventricular; NA, not available.
Table 2.
Characteristics of participants from the Framingham Heart Study and UK Biobank
| Covariate | UK Biobank (N = 46,434) Mean (SD)/N (%) | FHS (N = 1,886) Mean (SD)/N (%) |
|---|---|---|
| Age, years | 57 (8) | 55 (10.0) |
| <30 | 0 (0%) | 1 (0.1%) |
| 30–39 | 1 (<0.1%) | 108 (5.7%) |
| 40–49 | 10,145 (22%) | 506 (27%) |
| 50–59 | 14,597 (31%) | 601 (32%) |
| 60–69 | 21,381 (46%) | 527 (28%) |
| 70–79 | 310 (0.7%) | 139 (7.4%) |
| ≥80 | 0 (0%) | 4 (0.2%) |
| Female, n (%) | 25,361 (55) | 1,009 (54) |
| Non-Hispanic White, n (%) | 43,414 (93) | 1,864 (98.8) |
| Hispanic White, n (%) | N/A | 6 (0.32) |
| Black, n (%) | 1,009 (2.2) | 1 (0.05) |
| Asian, n (%) | 969 (2.1) | 1 (0.05) |
| Other race, n (%) | 1,042 (2.2) | 14 (0.75) |
| Body mass index, kg/m2 | 27.5 (4.8) | 27.4 (5.1) |
| BMI <25 | 14,756 (32%) | 662 (35%) |
| BMI ≥25 and <30 | 11,619 (25%) | 762 (40%) |
| BMI ≥30 | 19,817 (43%) | 462 (25%) |
| Current smoker, n (%) | 4,970 (11%) | 368 (20%) |
| Systolic blood pressure, mmHg | 140 (20) | 126 (19) |
| Systolic blood pressure ≥130 | 29,548 (64%) | 746 (40%) |
| Diabetes, n (%) | 2,686 (5.8%) | 145 (7.7%) |
| Cardiovascular disease, n (%) | 3,015 (6.5%) | 109 (5.8%) |
| Total cholesterol, mg/dL | N/A | 205 (36) |
| Total high-density lipoprotein cholesterol, mmol/L: UK Biobank, mg/dL: FHS | 1.44 (0.38) | 50 (15) |
Identifying a structural-functional proteome of the human heart
Our first step was to define cross-sectional proteomic correlates of echocardiographic measures capturing a broad myocardial phenome, across left ventricular (LV) systolic function, LV diastolic function, LV morphology, and right ventricular (RV) function to assemble a “structural-functional” proteome of human HF susceptibility (Figure 1A; full regression summaries in Data S1). Across both derivation and replication sets within CARDIA, we identified the greatest number of proteomic associations with LV diastolic function (2,492 SomaLogic aptamers), followed by LV systolic function and morphology (1,125 and 808 aptamers, respectively; Figure 1B). Of note, we did not replicate associations with measures of RV function identified in our derivation sample, likely owing to the limited sample size for both TAPSE and RV measures. For individual phenotypes and proteins, we observed a broad concordance in derivation and validation protein-phenotype associations (r = 0.6; p < 0.001; Figure 1C), suggesting internal validity. We validated the associations with two key HF-related echocardiographic phenotypes (LV mass and left atrial dimension) available alongside aptamer-based proteomics in the FHS (Figure S1), demonstrating broad concordance in the directionality of protein-to-phenotype correlations between CARDIA and the FHS. In total, 2,663 aptamers significantly associated with any echocardiographic phenotype (representing 2,454 unique proteins) in both derivation and validation samples, of which 413 were jointly associated with at least one measure of LV systolic function, LV diastolic function, and LV morphology. These 413 aptamers (378 unique proteins) were carried forward for further analysis.
Figure 1.

Proteomic architecture of cardiac structure and function in CARDIA
(A) Protein associations that are directionally consistent in both CARDIA derivation and validation samples and are significant at a 5% FDR in both after adjusting for age, gender, and race. “Pearson” reports the Pearson correlation between beta coefficients from models in CARDIA derivation and validation samples for each respective phenotype. Displayed effect sizes (beta) are from regression models performed in the derivation sample.
(B) An UpSet plot with the number of proteins related to each echocardiographic domain, demonstrating broadly shared proteins implicated across cardiac phenotypes.
(C) Beta coefficients from linear models of echocardiographic measures as a function of circulating proteins from the derivation (x axis) and validation (y axis) CARDIA samples (with Pearson r).
Prioritization of targets based on HF association in two diverse cohorts
To further enrich for proteins related to the eventual clinical phenotype of interest (HF), we next performed regression in two independent cohorts—the FHS and UK Biobank—to identify proteins associated with incident HF. This approach addresses potential issues around reverse causation of protein-phenotype associations in CARDIA by examining the relation of a protein to an incident HF outcome. Of the 2,663 aptamers in our cardiac structural-functional proteome from CARDIA, we filtered to those aptamers related to LV systolic function, LV diastolic function, and LV morphology parameters (N = 413) and tested their associations with clinical HF in either the FHS or UK Biobank. To capture the broadest related proteome for discovery, we adjusted for age and gender (and race in the UK Biobank) in Cox models, electing not to adjust for potential mediators for this initial step (hypertension and obesity; regression summaries in Data S1).
In the FHS, we observed 230 incident HF events (median: 25 years of follow-up; 25th–75th percentile: 18–27 years), with 48 aptamers (out of 74 tested; Figure 2A) associated with incident HF. In the UK Biobank (1,092 events; median: 13.4 years of follow-up, 25th–75th percentile: 12.7–14.1 years), we identified 177 proteins (out of 378 tested; Figure 2B) associated with incident HF. Despite differences in proteomic coverage and cohort characteristics, effect sizes in the FHS and UK Biobank (where protein quantification was present in both cohorts) were generally consistent (r = 0.67, p < 0.001). We did not observe a statistically significant interaction between any proteomic aptamers and age in regression models for incident HF in the FHS, but we did find 24 proteins with weak interactions with age in the UK Biobank (Figure S2). Of the 378 unique proteins (from 413 SomaScan aptamers) in our cardiac structural-functional proteome from CARDIA related to LV systolic function, LV diastolic function, and LV morphology, 134 proteins were associated with incident HF in either the FHS or UK Biobank. In pathway analysis (using the entire CARDIA assayed proteome as a background), these 134 targets specified enrichment in pathways of coagulation and cell adhesion, among others (Figure 2C).
Figure 2.

Proteomics of incident HF in two independent studies (the FHS and UK Biobank)
(A) Volcano plots in FHS (adjusted for age and gender) and UK Biobank (adjusted for age, gender, race). Blue color indicates <5% FDR. Note that in the FHS where SomaScan proteomics was used, there can be multiple aptamers for the same protein (e.g., F9).
(B) Pathway analysis of the 134 targets (KEGG, Reactome) prioritized by echocardiographic associations with LV systolic function, LV diastolic function, and LV morphology, which were associated with incident HF in FHS or UK Biobank. The assayed proteome from CARDIA was used as background. p.adjust indicates FDR-corrected p values.
Chamber enrichment of targets related to varying HF physiology
To determine whether proteins associated with echocardiographic traits were enriched transcriptionally in any given cardiac chamber, we interrogated a previously published snRNA-seq dataset11 to evaluate gene expression in the left and right atria and ventricles of the normal human heart.11 We filtered differentially expressed genes (at 10% false discovery rate [FDR]) in a stepwise fashion—(1) atria versus ventricles, (2) left versus right ventricle, and (3) left versus right atrium, identifying genes most enriched in the left atrium and left ventricle (165 unique genes in the left atrium and 81 unique genes in the left ventricle). This filtering method was chosen to highlight chamber enrichment of prioritized genes (and does not imply chamber-specific gene expression). Gene symbols of proteins associated with echocardiographic measures (at 5% FDR in CARDIA; Figure 1A) were intersected with those identified from the myocardium as above (Figure 3). HSPA1A was enriched in left atrial cardiomyocytes and associated with LA dimension, mean E/e′ ratio, and transmitral E/A ratio, consistent with a demonstrated role for heat shock proteins in atrial pathophysiology (atrial fibrillation).12,13,14,15 ROBO2, ERP29, and ZNF483 were enriched in cardiomyocytes of both the left atrium and left ventricle and were associated with multiple phenotypes. GEM, associated with the transmitral E/A ratio, was enriched in left atrial fibroblasts and cardiomyocytes, consistent with putative roles in cardiac contraction and electrical activity.16,17
Figure 3.

Four-chamber deconvolution of proteomic associations with echocardiographic measures of HF
Made in BioRender. Genes of significant proteins (FDR <0.05 in both CARDIA derivation and validation sets) associated with echocardiographic measures of HF were filtered to those significantly differentially expressed in the left atrium and LV as published.11
Prioritizing targets via cell-specific myocardial RNA expression in human HF
The final step of our approach was to identify which targets prioritized by structural, functional, and prognostic associations from the circulating proteome would exhibit dynamic expression in human heart tissue in two major states: (1) failing (versus non-failing) myocardium and (2) myocardial recovery with LV assist device (LVAD) support.18,19
In this first approach, we studied 11 individuals with HF18 (age: 54.9 ± 9.3 years; 36% female; LV ejection fraction: 12.8% ± 3.3%) and 16 heart transplant donors (age: 56.8 ± 10.0 years; 63% female; LV ejection fraction: 59.1% ± 6.8%; Data S1). We identified 64 genes reported in the failing and non-failing myocardial snRNA-seq data (from the 134 encoding unique proteins prioritized by structure/function and clinical association after filters as discussed in the initial report), of which 60 genes were significantly differentially expressed in failing versus non-failing hearts in at least one cell type or across cell types (pseudobulk). We observed a generally higher myocardial expression for those genes from our prioritized list relative to all expressed genes (Figure 4A). A preponderance of differentially expressed genes were concentrated in fibroblasts, cardiomyocytes, and pericytes, cell types putatively implicated in myocardial fibrosis and contractile dysfunction (Figure 4B). Of note, cell-specific differential expression displayed patterns consistent with known cell-specific roles in HF: for example, hypertrophy markers were differentially expressed in cardiomyocytes (IL1RAP, IL1R1, ADAM23, DKK3, etc.), while fibrosis signaling mediators and collagen were more commonly differentially expressed in pericytes and fibroblasts (IGFBP4, MMP2, SERPINE1, and POSTN, among others; Figures 4C and 4D). Collectively, this approach identified genes implicated in described mechanisms of myocardial injury and hypertrophy, including those involved in hypertrophy (IL1R1,20 IL1RAP,21,22 DKK323,24), fibrosis and inflammation (MMP2, SERPINE1,25,26,27 POSTN,28,29,30 IGFBP431,32), metabolic function (PTGR1,33 TALDO1,34,35 FABP336), and ischemia (SERPINF137,38). In addition, we identified a host of other genes across different cell types not widely described in human HF but with supporting evidence from model systems (Table 3). Intersecting our prioritized targets with the results of large-scale murine phenotyping efforts39 yielded multiple genes whose knockout induced cardiovascular phenotypes relevant to HF (Data S1). These phenotypes included enlargement of the heart (TNXB, IL1R1, MMP2, NOTCH3), dilated LV and reduced contractility (NCAM2), and abnormalities in electrical conduction (SMPDL3A, CHRDL1, CHL1).
Figure 4.

Single-nucleus myocardial prioritization of clinical targets in human HF
(A) The mean count expression (pseudobulked) between the full list of genes expressed versus those 134 prioritized by our approach, demonstrating a higher myocardial expression for those prioritized genes.
(B) The number of differentially expressed genes by cell type in single-nucleus RNA-seq in HF (dilated cardiomyopathy) versus non-failing (donor) myocardium.18 “Unique” represents genes uniquely differentially expressed in that cell type; “Shared_Once” and “Shared_Multi” represent genes differentially expressed in one other cell type or multiple, respectively. The number of differentially expressed genes in the pseudobulk population is shown in black as a reference and not considered in the shared or unique count for the cell type differentially expressed genes. Fibroblasts, pericytes, and cardiomyocytes are among the top 3 cell types by number of differentially expressed genes.
(C) A heatmap of the number of shared differentially expressed genes across cell types. Fibroblast and pericytes had the most frequent shared differentially expressed genes from our prioritized set.
(D) A heatmap of the log2 fold change for genes differentially expressed in the three prioritized cell types (now visualized across cell types). Genes with blanks signify the absence of reporting of differential expression in the parent data. “lfc” signifies log fold change, and “mean” signifies mean expression.
(E–G) Individual genes across failing and non-failing myocardial samples in fibroblasts, pericytes, and cardiomyocytes by single-nucleus RNA-seq for responders and non-responders across the three cell types. The y axis represents the fold change in individuals who experienced cardiac recovery (more positive = greater expression with hemodynamic unloading with assist device). The asterisks reflect an FDR < 10% (executed only across the displayed transcripts).
Table 3.
Prior evidence for select gene targets differentially expressed in cardiomyocytes, fibroblasts, and pericytes
| Gene/protein | Cell type | FC | Molecular evidence |
|---|---|---|---|
| Fibrosis and inflammation | |||
| DKK3 (Dickkopf WNT signaling pathway inhibitor 3) | CM | ↓ | implicated in upregulation of canonical Wnt signaling; higher levels protect from cardiomyopathy in mice24,40 |
| MMP2 (matrix metalloproteinase 2) | FB | ↓ | decreased MMP2 leads to increased hypertrophy to hypertension in mice41 |
| IGFBP4 (insulin-like growth factor binding protein 4) | FB, PC | ↓/↓ | implicated in canonical Wnt signaling31,42 and protective angiogenic responses post-infarct43; increased circulating levels with SGLT2i therapy8 |
| SERPINE1 (plasminogen activator inhibitor 1) | FB, PC | ↓/↓ | human loss-of-function mutations in SERPINE1 associated with greater fibrosis, with unclear mechanism25 |
| ROR1 (receptor tyrosine kinase-like orphan receptor 1) | FB | ↑ | involved in myofibroblast differentiation; induced in fibroblasts after transverse aortic constriction in mice, potentially a protective mechanism against unchecked inflammation44 |
| IL1RAP (interleukin-1 receptor accessory protein) | CM | ↑ | component of the IL-1 receptor complex; implicated in LV dysfunction; IL-1 blockade improves peak aerobic capacity45,46,47 |
| POSTN (periostin) | FB | ↑ | known marker of activated fibroblasts48; genetic knockout of Postn in mice showed less fibrosis in pressure overload and following myocardial infarction28 |
| CHRDL1 (chordin-like 1) | FB | ↑ | increased expression protective against adverse cardiac remodeling and myocardial fibrosis28 |
| EDIL3 (epidermal growth factor-like repeats and discoidin I-like domains 3) | FB | ↑ | implicated in inflammation and angiogenesis; EDIL3-deficient mice protected against adverse cardiac remodeling49 and cardiac dysfunction50 |
| Metabolism | |||
| FABP3 (fatty acid binding protein 3) | CM | ↓ | lower levels worsen response to aortic construction in mice; can lead to metabolic transition in failing heart (fatty acid oxidation defect); can be rescued with PPAR-alpha agonist36 |
| TALDO1 (transaldolase) | FB | ↓ | a key enzyme in the pentose phosphate pathway35; homozygous deficiency of this gene in humans includes phenotypic manifestations of liver failure, aortic coarctation, and cardiomegaly34,51 |
| NRP1 (neuropilin 1) | FB, PC | ↑/↑ | involved in angiogenesis; rodents without Nrp1 expressed in cardiomyocytes and vascular smooth muscle cells exhibited cardiomyopathy52 |
| Hypertrophy and contractile function | |||
| UNC5D (unc-5 netrin receptor D) | CM | ↑ | receptor for netrin-1; netrin-1 signaling mitigates cardiac dysfunction during pressure overload53 |
| NCAM1 (neural cell adhesion molecule) | FB | ↑ | related to fibrosis, dilatation, and dysfunction in human dilated cardiomyopathy; cardiac expression correlates with circulating natriuretic peptide levels54 |
| IL-1R1 (interleukin-1 receptor, type 1) | CM, FB | ↑/↓ | involved in IL-1 signaling55; IL-1R1-null mice are protected against LV hypertrophy, myocardial fibrosis, and diastolic dysfunction associated with aging56 and LV dilation and dysfunction following myocardial infarction57 |
| ADAM23 (a disintegrin and metalloprotease domain-containing protein 23) | CM | ↓ | reduced expression in failing hearts; protective against LV hypertrophy58 |
| DCC (netrin-1) | CM | ↑ | netrin-1 signaling mitigates cardiac dysfunction and hypertrophy during pressure overload53 |
| GHR (growth hormone receptor) | FB, PC | ↑/↑ | receptor for growth hormone; mixed data suggesting possible improved contractility59 with GH administration; acromegalic patients can exhibit LV hypertrophy60,61,62 |
| ROBO2 (roundabout homolog 2) | CM, FB | ↑/↑ | involved in Slit-Robo signaling; implicated in cardiomyopathy susceptibility62 and congenital heart defects63,64 |
| Ischemia, thrombosis, and hypoxia | |||
| CHL1 (cell adhesion molecule L1-like) | CM | ↓ | no large studies in HF; expressed in the carotid body and may be relevant to hypoxia responses65 |
| Complement (C3) | FB | ↓ | C3-null mice show decreased fibrosis following ischemia-reperfusion injury66; no large HF studies. |
| SERPINF1 (pigment epithelium-derived factor [PEDF]) | FB | ↓ | circulating levels associated with vascular inflammation67; administration of PEDF in a rat model of acute myocardial infraction was protective against adverse cardiac remodeling68 |
| CCN4 (WNT1-inducible-signaling pathway protein 1) | PC | ↓ | induces vascular smooth muscle cell migration and proliferation69; unclear role in HF |
| Other | |||
| EGFR (epidermal growth factor receptor) | PC | ↓ | inhibitors of EGFR increase risk of HF in humans70; broad implications of EGFR on endothelium and cardiac development71 |
| SPARCL1 (SPARC-like protein 1) | FB, PC | ↓/↓ | most studies in the right ventricle; some evidence that SPARCL deficiency associated with greater susceptibility to cardiac rupture post-infarct72 |
| RGMB (repulsive guidance molecule B) | FB | ↑ | higher circulating level associated with lower incident HF3; appears protective against acute kidney injury via inhibition of necroptosis73; directionally consistent in genetic MR analyses in HF5 |
| IDS (iduronate 2-sulfatase) | FB | ↑ | lysosomal catabolism of glycosaminoglycans (Hunter syndrome); unclear connection to HF |
| BOC (brother of CDO) | FB, PC | ↑/↑ | elevated plasma levels associated with HF74 |
| NRXN3 (neurexin 3) | CM, FB | ↑/↑ | neurexin 3 involved in cell adhesion; may be implicated in congenital heart disease75 but role in HF unclear |
CM, cardiomyocyte, FB, fibroblast, PC, pericyte, FC, fold change (up arrow indicates higher in failing versus non-failing myocardium).
In addition, many targets had precedent for involvement in HF from literature. For example, we observed decreased expression of DKK3 (implicated in upregulation in canonical Wnt signaling and protection from cardiomyopathy in mice24,40), decreased IGFBP4 (implicated in canonical Wnt signaling42), increased POSTN (implicated in myocardial fibrosis28,29,30), increased expression of components of the interleukin-1 receptor (IL1R1, IL1RAP; elevated levels of IL-1 signaling have been implicated in LV hypertrophy and HF20,21,22), and increased expression of EDIL3 (implicated in angiogenesis48,50,76), among many others (Table 3). Importantly the clinical-myocardial approach prioritized several targets implicated in HF by traditional and associative genetic approaches (e.g., IDS in Hunter syndrome; RGMB in genome-wide association studies [GWASs]5), as well as many targets with a null finding (or no report) in human HF GWASs but with implicated mechanisms central to HF pathogenesis. Finally, the approach also captured several genes with broad cellular processes central to cardiac function, including cell cycle regulation (DPY30, NUDC) and development (CHRDL1, BOC, NOTCH3), without clearly previously outlined mechanisms in HF. We found model system mechanistic evidence for many targets identified by our tiered approach (Table 3).
Prioritization of targets via dynamic cell-specific expression during recovery from HF under mechanical circulatory support
Identifying whether genes encoding proteins discovered in epidemiologic approaches are dynamic in longitudinal studies during HF treatment—potentially even in those individuals who recover cardiac function with treatment—may provide even more compelling evidence to link those genes and proteins to HF disease mechanism. Therefore, we next studied 14 heart transplant donors19 (age: 51 ± 15 years, 21% female, LV ejection fraction: 62% ± 7% [1 missing value]) and 13 individuals with HF (age: 51 ± 16 years; 23% female; LV ejection fraction: 16% ± 8%; Data S1). Of the 13 individuals with HF, 5 were “responders” to ventricular assist device-mediated recovery (increasing LV ejection fraction 18 ± 12% to 45% ± 5%; decreasing LV end-diastolic dimension 6.9 ± 1.5 to 4.9 ± 0.6 cm) and 8 were “non-responders” (increasing LV ejection fraction 15% ± 6% to 21% ± 5%; decreasing LV end-diastolic dimension 7.4 ± 1.5 to 6.5 ± 1.5 cm). Changes in transcripts (encoding proteins from our clinical prioritization strategy) in recovered versus non-recovered LV function in cardiomyocytes, fibroblasts, and pericytes are shown in Figures 4E–4G. We observed broad dynamic alterations in prioritized transcripts in a cell-specific manner with different patterns by recovery status. For example, DKK3 displayed increased cardiomyocyte expression in individuals who experienced myocardial recovery, directionally consistent with a lower expression in failing hearts and higher levels associated with a reduced hazard of incident HF in the UK Biobank (connecting myocardial and clinical data). Analogously, in fibroblasts and pericytes, we observed a dramatic shift in genes implicated in multiple processes central to recovery, including hypertrophy, metabolism (PTGR1, TALDO1), cell injury (RGMB), and fibrosis/inflammation (MMP2, ROR1, SERPINE1).
Discussion
Prioritizing among hundreds to thousands of identified associations from molecular profiling studies in human populations for downstream mechanistic and therapeutic studies remains a challenging part of translational medicine in HF. Here, we leveraged layers of clinical and molecular information from human studies to begin to offer a scheme to address this pressing problem, spanning proteomics and phenotyping of cardiac remodeling in at-risk populations to single-nucleus transcriptomics within myocardial tissue in failing and non-failing states to map these proteins at an RNA level within the human heart. As anticipated based on prior studies,3,6,77,78,79 the clinical association step generated a broad array of diverse relations between a protein target and phenotype or outcome. Filtering these targets through the myocardial transcriptome achieved a >50-fold reduction in targets, many of which—despite not previously being implicated in human HF by genetics—grouped along canonical pathways of fibrosis and extracellular matrix remodeling, metabolism, hypertrophy signaling, and ischemia. In addition, several displayed mechanistic evidence in animal and cellular models of HF. Finally, the addition of a therapeutic perturbation (ventricular assist device) highlighted several targets of interest with both regulatory/protective and counter-regulatory/non-protective transcription consistent with function in model systems. These results suggest the potential for a complementary paradigm to translate epidemiologic catalogs of association in HF toward potentially mechanistically interesting, functional targets for further study.
The concept of tying the proteome to the transcriptome to prioritize targets has recently garnered attention,80,81,82 given the advent of applying single-cell genomics to sort targets. Despite this growing interest, two major criticisms have surfaced around this approach: (1) potential discordances between protein and RNA abundance may be gene specific and epigenetically influenced (thereby potentially excluding relevant genes) and (2) concordance between circulating protein abundance and myocardial transcription does not necessarily mean that the source of the protein is exclusively cardiac. Nevertheless, targets that emerge as connected to clinical phenotypes and outcomes and differentially regulated across HF states and with therapy are more likely to comprise functionally relevant actors within the human heart. Supporting the validity of this approach, this prioritization strategy identified genes historically implicated in hypertrophy and fibrosis signaling (IL1RAP, IL1R1, ADAM23, POSTN, etc.) and a set of other genes not previously widely noted in human HF spanning similar pathways in models (e.g., DKK324,40 and RGMB, among many others). Interestingly, several of these genes exhibited dynamic expression during hemodynamic unloading (LVAD) in a directionally consistent manner (e.g., DKK3, IL1RAP, etc.), more dramatically in myocardium from those individuals who experienced recovery. These targets may be particularly appealing for further study, given that a primary goal in HF therapy is improvement in LV function. Certainly, by construction, this approach does not prioritize targets and mechanisms that are outside the heart (e.g., trans-organ, such as adipose- or immune-cardiac signaling), and integration of larger proteomic and transcriptomic atlases in patients with HF (as suggested recently in other conditions82) would expand this approach. Indeed, recent reports in aging suggest that mapping of the human circulating proteome using tissue-specific RNA-seq atlases may yield “organ-specific” proteomic profiles that are highly prognostic.82 Nevertheless, given costs inherent in large gain- and loss-of-function screening approaches across multiple targets (e.g., CRISPR-based screens18), these results suggest that utilizing epidemiologic and transcriptional information to winnow targets to those directly relevant to human myocardium and clinical HF offers an important first step.
In addition, we recognize that this epidemiologic-translational approach does not confirm that the abundance of a given HF-associated protein reflects secretion by myocardium. Given the tissue ubiquity of proteins, it is currently nearly impossible in human studies to deconvolve myocardial-specific proteins from the circulating proteome, and confirming tissue-specific effects will require additional model system inquiry (e.g., via inducible cell-tissue-type specific Cre-lox approaches, CRISPR-based approaches in vitro). Stating that clinically prioritized proteins are definitively cardiac specific via myocardial transcription was not the goal of our study. Targets in functional pathways of HF that display dynamic RNA expression across HF states and with perturbation and serial sampling during disease recovery are more likely to identify molecules of importance for downstream studies in the myocardium, even if these molecules are ubiquitous. As a prime example, early studies of the renin-angiotensin system (a critical pharmacologic target in HF) demonstrated concordant induction of renin mRNA expression in both the heart and kidney in HF83 with plasma and myocardial renin levels correlated in cardiomyopathy,84 despite a primarily renal source of human renin in normal conditions. Linking epidemiologic data with transcriptional states in diseases is even more pressing in the context of prevention: obtaining myocardial tissue at early states in HF progression—where participants in the epidemiologic studies in this work fall and where disease-modifying therapies may have particular benefit—is clinically inaccessible.
Finally, only a limited number of targets identified here (e.g., RGMB5) had been previously identified by genome-wide studies of HF, despite a wider array of targets displaying support from model system systems. In general, HF GWASs have had limited success in identifying targets in HF, likely due to the heterogeneous nature of HF, gene-environment interactions, and pleiotropy with underlying driving risk factors related to acquired conditions and poor instrumental variables: indeed, major HF-related loci have been strongly linked to other comorbid diseases on the pathway to HF (e.g., hyperlipidemia, diabetes, obesity, and blood pressure).85 In contrast, the proteome and transcriptome directly reflect a variation in phenotype and exposure relevant to HF: coupling proteomics, phenotyping, and single-nucleus human myocardial transcription is a complementary approach that leverages the disease heterogeneity itself to prioritize targets.
To our knowledge, the approach here presents an emerging paradigm to connect findings from a large, well-phenotyped, observational human setting to dynamic states within human myocardium. The key innovation—prioritization through structural, clinical, and single-nucleus-based dynamic transcriptional states—implicated proteins in cardiomyocytes, pericytes, and fibroblasts in both canonical and other pathways of metabolism, ischemia, hypertrophy, and fibrosis, extending results from transcriptional approaches in HF with reduced or preserved ejection fraction.18,86,87,88,89 We envision that this proteo-transcriptional approach offers a method (complementary to human genetics) to deliver associations developed from large epidemiologic efforts for diagnosis and prognosis to the functional space to prioritize targets for mechanistic interrogation. Despite concerns around RNA-protein concordance and cardiac specificity, the findings here suggest the plausibility of this approach to identify biologically relevant targets (several previously described and across important pathways in HF) at a time when functional prioritization is urgently needed. The emergence of tissue and circulating proteomic, transcriptional, and epigenetic atlases during disease is likely to increase the specificity and richness of these approaches.
Limitations of the study
Despite the promising proteo-transcriptional linkage demonstrated here, our results have several important limitations. First, we have focused our myocardial snRNA-seq analyses on end-stage HF and donor tissue not used in transplantation. This is due to the clinical accessibility of these tissues (in the operating theater, at the time of LVAD and/or transplant) and appropriate ethical limitations in endomyocardial biopsy at earlier stages of HF outside of acute severe HF states.90 Second, we chose to focus primarily on non-ischemic cardiomyopathy for our snRNA-seq analyses, a different area from early HF states (likely represented in the community, from where our circulating proteomic data are derived) and certainly different from ischemic or other causes of HF. We recognize that this choice may limit the reporting of targets perturbed early in the heart or in other disease states. Nevertheless, the success of proteo-transcriptional mapping for several targets here underscores the importance of conducting these studies in a wide array of HF conditions (e.g., HF with preserved ejection fraction) and other forms (e.g., viral cardiomyopathy). While discovery in increasingly prevalent HF subtypes with clinical morbidity (like the preserved ejection fraction) are emerging,86 parsing the cause of HF in epidemiologic cohorts (either via administrative codes in the UK Biobank or via chart review in the FHS) is fraught, given incomplete data. Future efforts focused on these cohorts (e.g., National Institutes of Health HeartShare program) will yield fruitful data in this regard. Third, transcript-to-protein levels may not be concordant, with post-transcriptional and post-translational modifications, protein kinetics in circulation, and secretory-associated modifications having influence on circulating levels. Given the limitations with single-sample collection in a cohort setting, these remain important challenges to any work in this space. Finally, the proteomics panel used in the FHS cohort was an older version of the SomaScan platform (≈1,000 aptamers) compared to the CARDIA cohort, and the UK Biobank cohort utilized a modern Olink panel (≈3,000 proteins, with 378 overlapping with significant targets from CARDIA). Nevertheless, the targets that were selected by the cross-platform, cross-cohort approach (which has proven useful in recent reports82,91) included those targets that were significant across CARDIA to the FHS or UK Biobank, including those with proven biological plausibility (DKK3,24,40 FABP3,36 UNC5D53). Future studies across harmonized proteomics are likely to continue to develop this method.
Resource availability
Lead contact
The lead contact for this work for requests for materials is Dr. Sadiya Khan (s-khan-1@northwestern.edu).
Materials availability
This study did not generate new reagents.
Data and code availability
Data from CARDIA used in these analyses are available through the Coronary Artery Risk Development in Young Adults study (CARDIA; cardia.dopm.uab.edu). Data from the Framingham Heart Study (FHS) is available at dbGaP (dbGaP identifier pht006013) or via contact with the FHS coordinating center (www.framinghamheartstudy.org). UK Biobank data is available at UK Biobank Research Access Portal (analyses here conducted under proposal approval 57492). This paper does not report any original code. Any additional information required to reanalyze the data reported in this work paper is available from the lead contact upon request.
Acknowledgments
The authors would like to acknowledge the participants of the FHS, UK Biobank, and CARDIA studies, as well as the participants in the studies of myocardial transcriptomics. S.D. has received grant support from Abbot Laboratories and Bristol Myers Squib. S.G.D. is funded by the NIH, US Department of Veterans Affairs, the American Heart Association (AHA), the Nora Eccles Treadwell Foundation, and the Foundation of the NIH. R.V.S. is funded by the NIH and the AHA. A.S.P. is funded by the AHA. K.A. is funded by the NIH (K23HL166960), the International Society for Heart and Lung Transplantation Enduring Hearts Transplant Longevity Award, the Red Gates Foundation, and an AHA Career Development Award (#929347). R.K., S.J.S., and M.N. are funded by the NIH. S.S.K. is supported by the NIH (R01HL159250). K.A.W., Z.P., and T.T. are funded by the NIA Intramural Research Program at the NIH. Proteomics in CARDIA were funded by a grant to R.K. (R01HL122477). N.T. is supported by the NIH (R01HL170051). K.A.W., Z.P., T.T., and L.F. are supported by the NIA Intramural Research Program at the NIH. S.J.S is supported by research grants from the NIH (U54 HL160273, R01 HL140731, R01 HL149423), American Heart Association (24SFRNPCN1291224), AstraZeneca, Boston Scientific, Corvia, Pfizer, and Tempus. K.J.L. is supported by the NIH (R01 HL138466, R01 HL139714, R01 HL151078, R01 HL161185, and R35 HL161185), the Leducq Foundation Network (#20CVD02), the Burroughs Welcome Fund (1014782), the Children’s Discovery Institute of Washington University and St. Louis Children's Hospital (CH-II-2015-462, CH-II-2017-628, and PM-LI-2019-829), the Foundation for Barnes-Jewish Hospital (8038-88), and generous gifts from the Washington University School of Medicine. X.H. is supported by AHA grant 24CDA1260466. CARDIA is conducted and supported by the National Heart, Lung, and Blood Institute in collaboration with the University of Alabama at Birmingham (75N92023D00002 and 75N92023D00005), Northwestern University (75N92023D00004), the University of Minnesota (75N92023D00006), and the Kaiser Foundation Research Institute (75N92023D00003). This manuscript has been reviewed by CARDIA for scientific content. The FHS is supported by the National Heart, Lung, and Blood Institute at the National Institutes of Health (contracts N01-HC-25195, HHSN268201500001I, and 75N92019D00031), and proteomics were funded by R01-HL132320.
Author contributions
Conceptualization, A.S.P., K.A., R.V.S., and S.S.K.; formal analysis, A.S.P., K.A., X.H., E.F.-E., M.L.L., P.G., J.A., Z.P., C.M.J., L.S., S.Z., and Q.S.; writing and revision, A.S.P., K.A., R.V.S., T.T., S.G.D., K.J.L., C.S., J.R.V., T.S.S., L.F., S.D., J.W., R.B.P., R.K., S.J.S., K.A.W., Q.W., N.T., and M.N.; funding acquisition, Q.W., S.S.K., R.K., and R.V.S.
Declaration of interests
S.G.D. is a consultant for Abbott, and S.G.D. and K.J.L. have industry research support from Novartis. R.V.S. has served as a consultant for Amgen, Cytokinetics, and Thryv Therapeutics (with options ownership in Thryv). R.V.S. is a co-inventor on a patent for ex-RNA signatures of cardiac remodeling and a pending patent on proteomic signatures of fitness and lung and liver diseases. R.V.S. also has stock options with Thryv Therapeutics. A.S.P. is a co-inventor on a pending patent for proteomic signatures of fitness, lung, and liver diseases. K.A. does ad hoc consulting for Tegus and Guidepoint and serves on a DSMB for Fortrea. M.N. has received speaking honoraria from Cytokinetics. K.J.L. serves as a consultant for Medtronic, Implicit Biosciences, Kiniksa Pharmaceuticals, and Cytokinetics. K.J.L. receives grant support from Amgen, Novartis, Kiniksa Pharmaceuticals, and Implicit Biosciences. S.D. is a co-founder and has equity in Thryv therapeutics and Switch Therapeutics. S.D. has received grant support from Abbot Laboratories and Bristol Myers Squib. S.D. is a co-inventor on a patent on tissue-specific EV-RNAs. R.K. has received personal fees from GSK, Astrazeneca, Boehringer Ingelheim, and CVS Caremark. S.J.S. has received consulting fees from 35Pharma, Abbott, Alleviant, AstraZeneca, Amgen, Aria CV, Axon Therapies, BaroPace, Bayer, Boehringer-Ingelheim, Boston Scientific, BridgeBio, Bristol Myers Squibb, Corvia, Cyclerion, Cytokinetics, Edwards Lifesciences, Eidos, eMyosound, Imara, Impulse Dynamics, Intellia, Ionis, Lilly, Merck, MyoKardia, Novartis, Novo Nordisk, Pfizer, Prothena, ReCor, Regeneron, Rivus, SalubriusBio, Sardocor, Shifamed, Tectonic, Tenax, Tenaya, Ulink, and Ultromics. J.M.A. was employed by Amgen. J.W. reports consulting/scientific advisory board for Abiomed, Abbott, Astra Zeneca, Boehringer Ingelheim, and Cytokinetics within the last 24 months.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Deposited data | ||
| CARDIA study proteomics and clinical data | CARDIA study, dbGaP | Deposited at CARDIA Coordinating Center cardia.dopm.uab.edu; to be posted to dbGaP phs003491.v1.p1, proteomics |
| FHS study proteomics | dbGaP | pht006013 |
| UK Biobank study proteomics | UK Biobank | Approved access to UK Biobank Research Access Portal |
| Single nuclear RNA-seq from dilated cardiomyopathy and non-failing hearts | Single Cell Portal (Broad Institute) | https://singlecell.broadinstitute.org/single_cell/study/SCP1303/single-nuclei-profiling-of-human-dilated-and-hypertrophic-cardiomyopathy |
| Single nuclear RNA-seq, four-chamber, human heart | Single Cell Portal (Broad Institute) | https://singlecell.broadinstitute.org/single_cell/study/SCP498/transcriptional-and-cellular-diversity-of-the-human-heart |
| Single nuclear RNA-seq from cardiac recovery | NCBI GEO | GEO: GSE226314, GEO: GSE183852 |
| Software and algorithms | ||
| R studio | R project | https://www.r-project.org |
| Python | Python | https://www.python.org |
| STATA | STATA (Version 17) | https://www.stata.com |
Experimental model and subject details
CARDIA
The Coronary Artery Risk Development in Young Adults (CARDIA) study is a population-based, prospective longitudinal cohort of Black and White participants across four centers in the United States. Recruitment for CARDIA began in 1985-86 and strove to build a cohort representative of the Black and White populations in the United States. To participate in CARDIA, individuals had to be between 18 and 30 years old at the time of initial interview and free from long-term disease or disability (including deafness, blindness, inability to walk on a treadmill).92 Data from CARDIA for this analysis were collected from 2965 participants who attended a follow-up visit 25 years after baseline (2010–2011) with proteomics and echocardiographic measurements available. Clinical information and questionnaires were collected by standardized assessment (study review in ref.93). Procedures for two-dimensional echocardiography in CARDIA have been described previously.94 Measures were selected given their putative importance as pre-clinical markers of HF susceptibility: (1) apical four-chamber view LV end diastolic volume (ml, indexed by height and log-transformed); (2) apical four-chamber view LV end systolic volume (ml, indexed by height and log-transformed); (3) M-mode LV mass (grams, indexed by height2.7 and log-transformed); (4) mean E/e’ ratio (log-transformed); (5) mitral E/A ratio (log-transformed); (6) apical four-chamber view LV ejection fraction (%); (7) apical four-chamber peak longitudinal strain (%); (8) M-mode left atrium dimensions (cm); (9) two-dimensional left atrial volume (ml; end systolic LAV in 4-chamber view); (10) tricuspid annular plane systolic excursion; (11) estimated RV systolic pressure (mmHg). Study participants provided written informed consent, and CARDIA was approved by each site’s respective Institutional Review Board.
FHS
The Framingham Heart Study (FHS) is a population-based study which initially began in 1948 with 5209 participants from Framingham, Massachusetts. In the 1970s, the Framingham Offspring Study (now known as the Generation 2 cohort) began with recruitment of the children of the original Framingham participants, with a subset specifically invited due to the presence of coronary heart disease in their parent.95 We utilized clinical and proteomic data from 1886 participants in the FHS Generation 2 (“Offspring”) cohort95 at the 5th examination cycle without prevalent HF and who had complete data on covariates and incident HF.3 Methods for clinical and biochemical measures have been previously reported.96,97,98 HF was defined by 2 major or 1 major and 2 minor criteria (major: paroxysmal nocturnal dyspnea or orthopnea, increased venous pressure, distended neck veins, rales, cardiomegaly by radiograph, pulmonary edema, a third heart sound, hepatojugular reflux, and weight loss on diuretic therapy; minor: ankle edema, nocturnal cough, hepatomegaly, dyspnea on exertion, pleural effusion, decrease in vital capacity and tachycardia), as reported previously.3 All participants provided informed written consent, and all study protocols were approved by the Boston University Medical Center Institutional Review Board.
UK Biobank
We used data from the UK Biobank Pharma Proteomics Project wherein ≈52,000 participants had proteomic profiling with the Olink Explore 3072 platform.99 UK Biobank is broadly inclusive of the general population between the ages of 40–69, with sampling from the National Health Service with stratification on age, gender, and postcode.100 We excluded participants who reported a history of heart failure (UK Biobank Data Field 20002) and those with confounding ICD codes that prohibit identification of cases and controls. History of CVD was defined as physician diagnosed angina, myocardial infarction, or stroke (UK Biobank Data Field 6150). Incident HF cases were defined using ICD-code based methods (PheWAS package101). Time-to-event was defined as the time to first qualifying ICD code, and non-event participants were censored at the date of death (if applicable) or region-specific dates (UK Biobank Data Field 54): 31 October 2022 for England; 31 July 2021 for Scotland; and 28 February 2018 for Wales.
Method details
Proteomics
Proteomic quantification was performed on collected plasma samples using aptamer (FHS, CARDIA; SomaScan; Somalogic, Boulder, CO) or antibody-based (UK Biobank; Olink; Uppsala, Sweden) using standard methods.99,102 CARDIA utilized the SomaScan 7K platform for protein quantification. In CARDIA, data from sixty-eight participants with >1 sample run were averaged for use in models. We did not detect batch effects or outlier values (by principal components analysis). We excluded proteins with a coefficient of variation >20%. A final 7228 aptamers were included in the analysis for CARDIA. Proteins were log-transformed and standardized (mean 0, variance 1) prior to use in models.
FHS included an earlier version of the SomaScan platform (≈1000 aptamers), and we used published methods to normalize this data.3,103 Briefly, given differences in collection batch in FHS, protein levels were standardized within each of the two batches, pooled, and rank normalized across all samples with subsequent residualization against assay plate before analysis to remove plate-based effects, as described previously.3
UK Biobank used the Olink Explore 3072 platform (≈3000 proteins). Prior to public release, proteomics data underwent quality control and review by UK Biobank investigators.99 We used the proteomics data as provided and scaled the normalized protein expression (NPX) values to mean 0 and unit variance prior to models. To match proteins across platforms from CARDIA (SomaScan) to UK Biobank (Olink) we matched on UniProt identifier, and manual review of gene symbol and assay names for those not matched by UniProt.
Single-nuclear RNA sequencing
Publicly available datasets were utilized for single-nuclear RNA-sequencing (snRNA-seq) analyses.11,18,19 Myocardial tissue was obtained as described in the respective studies.18,19 Briefly, myocardial tissue was obtained from left ventricular apical cores at the time of left ventricular assist device (LVAD) implantation,18 adjacent sections during LVAD explant18 or transplant,18,19 or the left ventricle from organ donors without a history of heart failure.11,18,19 Single nuclear suspensions were generated through a combination of gentle physical dissociation (e.g., mincing of tissue or cryosectioning, dounce homogenization, sequential filtration of debris, etc.) and buffer solutions. Nuclei were loaded onto the 10X Genomics microfluidic platform using either the 3′ v3 or 5′ v1.1 single-cell kits. Downstream processing, QC, and analyses were performed as described below and in the respective manuscripts.11,18,19
Quantification and statistical analysis
Clinical prioritization (population-based studies)
Our clinical prioritization strategy involved selecting proteins significantly associated with both any echocardiographic phenotype (in both derivation and validation samples) and with incident HF in FHS and UK Biobank. To identify circulating proteomic correlates of echocardiographic measures, we used linear regression models with individual aptamers as the predictor, adjusted for age, gender, and race. We randomly split the CARDIA sample to a derivation (70%) and validation sample (30%). We performed linear regression analyses to assess the association between each log-transformed, standardized aptamer (continuous) and echocardiographic traits (continuous, transformed as noted above and standardized). In FHS and UK Biobank, we estimated Cox regression models for incident HF, with individual aptamers (FHS) or assays (UK Biobank) as independent variables, adjusted for age and gender in FHS, and adjusted for age, gender, and race in UK Biobank. An FDR of 5% (Benjamini-Hochberg) was used across all models to control type 1 error. Aptamers or assays associated with incident HF (after FDR correction), were examined for effect modification by age in Cox regression models in FHS and UK Biobank. Aptamers that passed a 5% FDR for association with echocardiographic features in derivation and validation samples in CARDIA and were associated with incident HF in either UK Biobank or FHS at the same FDR were selected for evaluation in single nucleus myocardial transcriptomics. Pathway analysis was performed on prioritized proteins (total 134 proteins) using ClusterProfiler in R104 on KEGG and Reactome databases. Hypergeometric tests were used to evaluate enrichment level for each pathway comparing to all the proteins measured.
Myocardial prioritization (single nucleus RNA-seq)
We used single-nuclear RNA-sequencing (snRNA-seq) data from 11 dilated cardiomyopathy (DCM) and 16 non-failing hearts18 as well as four-chamber expression from a normal human heart for deconvolution approaches.11 In comparing DCM (“heart failure”) and non-failing hearts, pseudo-bulk and cell-specific differential expression analyses were performed using limma-voom105 and two-sided P-values were corrected for multiple testing using Benjamini-Hochberg FDR. We focused our analysis on genes encoding proteins that filtered through clinical prioritization (see above; CellBender-corrected counts, 5% FDR threshold). We identified the number of differentially expressed genes per annotated cell types, and focused subsequent cell-specific expression analysis on 3 cell types (cardiomyocytes, fibroblasts, and pericytes) given their putative role in cardiac remodeling. For four-chamber deconvolution approaches, we identified genes that were differentially expressed using a generalized linear mixed model between (1) atria and ventricles, (2) between left and right atria, and (3) between left and right ventricles. Those differentially expressed genes (at a 10% FDR) most expressed in the left atrium and left ventricle were intersected with proteins associated with aforementioned echocardiographic measures indicative of HF risk, significant in both derivation and validation cohorts (at a 5% FDR).
As a final step, we studied transcript dynamics during ventricular unloading, an intervention that specifically impacts myocardial structure and function. We studied dynamic, cell-specific changes in expression during hemodynamic unloading with a left ventricular assist device (LVAD) in 27 individuals (14 non-failing donors, 13 advanced HF patients), as described.19 We only studied genes prioritized by the clinical filtering that exhibited cell-specific differential expression in failing versus non-failing myocardium as above and passed quality control filters as in the primary report.19 In brief, myocardium from the LVAD core (at the time of LVAD insertion) and from areas adjacent to the core at the time of LVAD removal were used for single nucleus expression profiling. “Responders” were defined using turn-down echocardiography during low speed assist device support (responders: final LVEF >40% and LV end-diastolic dimension [LVEDD] ≤5.9 cm; non-responders: final LVEF <35% with <50% relative improvement in LVEF regardless of the final LVEDD), as described.106 Individuals who did and did not experience cardiac structural and functional improvement by this definition were selected so that LVEF at the time of LVAD insertion was similar between groups. Cell-specific differential expression between the pre-LVAD state and responders or non-responders post-LVAD was performed using DESeq2. Specifically, cell type annotations from snRNA-seq were used to select cardiomyocytes, fibroblasts, and pericytes. Subsequently, counts were aggregated within each cell type to the sample level, and DESeq2 was used for differential expression analysis for two conditions: (1) pre-LVAD heart failure versus responders post-LVAD (paired analysis); and (2) pre-LVAD heart failure versus non-responders post-LVAD (paired analysis). We executed a type 1 error correction only over the transcripts that passed our initial snRNA-seq step in each of the three cell types (DCM vs. donor).
Published: September 2, 2024
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.xcrm.2024.101704.
Contributor Information
Ravi V. Shah, Email: ravi.shah@vumc.org.
Sadiya S. Khan, Email: s-khan-1@northwestern.edu.
Supplemental information
Results of analyses for clinical prioritization. Sheet “SD01” represents results of regressions in CARDIA. SomaLogic aptamer meta-data (aptname, seqid, somaid) and target identification (targetfullname, target, uniprot, entrezgeneid, entrezgenesymbol) are provided. “Outcome” refers to specific echocardiographic phenotype (dependent variable), and “physio” refers to the overall physiology into which that outcome was grouped. Columns for beta (estimated regression coefficient), se (standard error of the beta), N (sample size for model), p (P-value) and its FDR are provided for the 70% derivation (“der_”) and the 30% hold-out replication/validation samples (“val_”) are provided. These models were adjusted for age, gender, and race. Sheet “SD02” provides results from regression models for left atrial dimension and LV mass (log) in FHS for overlapping proteins with an FDR <5% in both CARDIA derivation and validation samples (SD01). These models were adjusted for age, gender, and height. Of note, the original manuscript detailing a subset of these associations has been reported3. Sheet “SD03” represents results of survival models in two cohorts (specified under “cohort” as UK Biobank or FHS) for each protein. Models in FHS displayed here were adjusted for age and gender. Models for UK Biobank displayed here were adjusted for age, gender, and race. Sheet “SD04” reports Cox model results for incident HF in UK Biobank including an age by protein interaction term. Sheet “SD05” represents clinical characteristics of individuals evaluated in snRNA-seq datasets. Sheet “SD06” represents results from the International Mouse Phenotypoing Consortium
References
- 1.Vasan R.S., Enserro D.M., Beiser A.S., Xanthakis V. Lifetime Risk of Heart Failure Among Participants in the Framingham Study. J. Am. Coll. Cardiol. 2022;79:250–263. doi: 10.1016/j.jacc.2021.10.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.McMurray J.J.V., Packer M., Desai A.S., Gong J., Lefkowitz M.P., Rizkala A.R., Rouleau J.L., Shi V.C., Solomon S.D., Swedberg K., et al. Angiotensin-neprilysin inhibition versus enalapril in heart failure. N. Engl. J. Med. 2014;371:993–1004. doi: 10.1056/NEJMoa1409077. [DOI] [PubMed] [Google Scholar]
- 3.Nayor M., Short M.I., Rasheed H., Lin H., Jonasson C., Yang Q., Hveem K., Felix J.F., Morrison A.C., Wild P.S., et al. Aptamer-Based Proteomic Platform Identifies Novel Protein Predictors of Incident Heart Failure and Echocardiographic Traits. Circ. Heart Fail. 2020;13 doi: 10.1161/CIRCHEARTFAILURE.119.006749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Henry A., Gordillo-Marañón M., Finan C., Schmidt A.F., Ferreira J.P., Karra R., Sundström J., Lind L., Ärnlöv J., Zannad F., et al. Therapeutic Targets for Heart Failure Identified Using Proteomics and Mendelian Randomization. Circulation. 2022;145:1205–1217. doi: 10.1161/CIRCULATIONAHA.121.056663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Moncla L.H.M., Mathieu S., Sylla M.S., Bossé Y., Thériault S., Arsenault B.J., Mathieu P. Mendelian randomization of circulating proteome identifies actionable targets in heart failure. BMC Genom. 2022;23:588. doi: 10.1186/s12864-022-08811-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Egerstedt A., Berntsson J., Smith M.L., Gidlöf O., Nilsson R., Benson M., Wells Q.S., Celik S., Lejonberg C., Farrell L., et al. Profiling of the plasma proteome across different stages of human heart failure. Nat. Commun. 2019;10:5830. doi: 10.1038/s41467-019-13306-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rasooly D., Peloso G.M., Pereira A.C., Dashti H., Giambartolomei C., Wheeler E., Aung N., Ferolito B.R., Pietzner M., Farber-Eger E.H., et al. Genome-wide association analysis and Mendelian randomization proteomics identify drug targets for heart failure. Nat. Commun. 2023;14:3826. doi: 10.1038/s41467-023-39253-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zannad F., Ferreira J.P., Butler J., Filippatos G., Januzzi J.L., Sumin M., Zwick M., Saadati M., Pocock S.J., Sattar N., et al. Effect of empagliflozin on circulating proteomics in heart failure: mechanistic insights into the EMPEROR programme. Eur. Heart J. 2022;43:4991–5002. doi: 10.1093/eurheartj/ehac495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tabula Sapiens Consortium∗. Jones R.C., Karkanias J., Krasnow M.A., Pisco A.O., Quake S.R., Salzman J., Yosef N., Bulthaup B., Brown P., et al. The Tabula Sapiens: A multiple-organ, single-cell transcriptomic atlas of humans. Science. 2022;376 doi: 10.1126/science.abl4896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.GTEx Consortium The GTEx Consortium atlas of genetic regulatory effects across human tissues. Science. 2020;369:1318–1330. doi: 10.1126/science.aaz1776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tucker N.R., Chaffin M., Fleming S.J., Hall A.W., Parsons V.A., Bedi K.C., Jr., Akkad A.D., Herndon C.N., Arduini A., Papangeli I., et al. Transcriptional and Cellular Diversity of the Human Heart. Circulation. 2020;142:466–482. doi: 10.1161/CIRCULATIONAHA.119.045401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.van Wijk S.W., Ramos K.S., Brundel B.J.J.M. Cardioprotective Role of Heat Shock Proteins in Atrial Fibrillation: From Mechanism of Action to Therapeutic and Diagnostic Target. Int. J. Mol. Sci. 2021;22 doi: 10.3390/ijms22010442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Decker R.S., Decker M.L., Nakamura S., Zhao Y.S., Hedjbeli S., Harris K.R., Klocke F.J. HSC73-tubulin complex formation during low-flow ischemia in the canine myocardium. Am. J. Physiol. Heart Circ. Physiol. 2002;283:H1322–H1333. doi: 10.1152/ajpheart.00062.2002. [DOI] [PubMed] [Google Scholar]
- 14.Yang M., Tan H., Cheng L., He M., Wei Q., Tanguay R.M., Wu T. Expression of heat shock proteins in myocardium of patients with atrial fibrillation. Cell Stress Chaperones. 2007;12:142–150. doi: 10.1379/csc-253r.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mandal K., Torsney E., Poloniecki J., Camm A.J., Xu Q., Jahangiri M. Association of high intracellular, but not serum, heat shock protein 70 with postoperative atrial fibrillation. Ann. Thorac. Surg. 2005;79:865–871. doi: 10.1016/j.athoracsur.2004.08.018. [DOI] [PubMed] [Google Scholar]
- 16.Tan F.L., Moravec C.S., Li J., Apperson-Hansen C., McCarthy P.M., Young J.B., Bond M. The gene expression fingerprint of human heart failure. Proc. Natl. Acad. Sci. USA. 2002;99:11387–11392. doi: 10.1073/pnas.162370099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Murata M., Cingolani E., McDonald A.D., Donahue J.K., Marbán E. Creation of a genetic calcium channel blocker by targeted gem gene transfer in the heart. Circ. Res. 2004;95:398–405. doi: 10.1161/01.RES.0000138449.85324.c5. [DOI] [PubMed] [Google Scholar]
- 18.Chaffin M., Papangeli I., Simonson B., Akkad A.D., Hill M.C., Arduini A., Fleming S.J., Melanson M., Hayat S., Kost-Alimova M., et al. Single-nucleus profiling of human dilated and hypertrophic cardiomyopathy. Nature. 2022;608:174–180. doi: 10.1038/s41586-022-04817-8. [DOI] [PubMed] [Google Scholar]
- 19.Amrute J.M., Lai L., Ma P., Koenig A.L., Kamimoto K., Bredemeyer A., Shankar T.S., Kuppe C., Kadyrov F.F., Schulte L.J., et al. Defining cardiac functional recovery in end-stage heart failure at single-cell resolution. Nat. Cardiovasc. Res. 2023;2:399–416. doi: 10.1038/s44161-023-00260-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nishikawa K., Yoshida M., Kusuhara M., Ishigami N., Isoda K., Miyazaki K., Ohsuzu F. Left ventricular hypertrophy in mice with a cardiac-specific overexpression of interleukin-1. Am. J. Physiol. Heart Circ. Physiol. 2006;291:H176–H183. doi: 10.1152/ajpheart.00269.2005. [DOI] [PubMed] [Google Scholar]
- 21.Orn S., Ueland T., Manhenke C., Sandanger O., Godang K., Yndestad A., Mollnes T.E., Dickstein K., Aukrust P. Increased interleukin-1beta levels are associated with left ventricular hypertrophy and remodelling following acute ST segment elevation myocardial infarction treated by primary percutaneous coronary intervention. J. Intern. Med. 2012;272:267–276. doi: 10.1111/j.1365-2796.2012.02517.x. [DOI] [PubMed] [Google Scholar]
- 22.Shang L., Yue W., Wang D., Weng X., Hall M.E., Xu Y., Hou M., Chen Y. Systolic overload-induced pulmonary inflammation, fibrosis, oxidative stress and heart failure progression through interleukin-1beta. J. Mol. Cell. Cardiol. 2020;146:84–94. doi: 10.1016/j.yjmcc.2020.07.008. [DOI] [PubMed] [Google Scholar]
- 23.Zhai C.G., Xu Y.Y., Tie Y.Y., Zhang Y., Chen W.Q., Ji X.P., Mao Y., Qiao L., Cheng J., Xu Q.B., Zhang C. DKK3 overexpression attenuates cardiac hypertrophy and fibrosis in an angiotensin-perfused animal model by regulating the ADAM17/ACE2 and GSK-3beta/beta-catenin pathways. J. Mol. Cell. Cardiol. 2018;114:243–252. doi: 10.1016/j.yjmcc.2017.11.018. [DOI] [PubMed] [Google Scholar]
- 24.Zhang Y., Liu Y., Zhu X.H., Zhang X.D., Jiang D.S., Bian Z.Y., Zhang X.F., Chen K., Wei X., Gao L., et al. Dickkopf-3 attenuates pressure overload-induced cardiac remodelling. Cardiovasc. Res. 2014;102:35–45. doi: 10.1093/cvr/cvu004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Khan S.S., Shah S.J., Strande J.L., Baldridge A.S., Flevaris P., Puckelwartz M.J., McNally E.M., Rasmussen-Torvik L.J., Lee D.C., Carr J.C., et al. Identification of Cardiac Fibrosis in Young Adults With a Homozygous Frameshift Variant in SERPINE1. JAMA Cardiol. 2021;6:841–846. doi: 10.1001/jamacardio.2020.6909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Baumeier C., Escher F., Aleshcheva G., Pietsch H., Schultheiss H.P. Plasminogen activator inhibitor-1 reduces cardiac fibrosis and promotes M2 macrophage polarization in inflammatory cardiomyopathy. Basic Res. Cardiol. 2021;116:1. doi: 10.1007/s00395-020-00840-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ghosh A.K., Kalousdian A.A., Shang M., Lux E., Eren M., Keating A., Wilsbacher L.D., Vaughan D.E. Cardiomyocyte PAI-1 influences the cardiac transcriptome and limits the extent of cardiac fibrosis in response to left ventricular pressure overload. Cell. Signal. 2023;104 doi: 10.1016/j.cellsig.2022.110555. [DOI] [PubMed] [Google Scholar]
- 28.Oka T., Xu J., Kaiser R.A., Melendez J., Hambleton M., Sargent M.A., Lorts A., Brunskill E.W., Dorn G.W., 2nd, Conway S.J., et al. Genetic manipulation of periostin expression reveals a role in cardiac hypertrophy and ventricular remodeling. Circ. Res. 2007;101:313–321. doi: 10.1161/CIRCRESAHA.107.149047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Snider P., Standley K.N., Wang J., Azhar M., Doetschman T., Conway S.J. Origin of cardiac fibroblasts and the role of periostin. Circ. Res. 2009;105:934–947. doi: 10.1161/CIRCRESAHA.109.201400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhao S., Wu H., Xia W., Chen X., Zhu S., Zhang S., Shao Y., Ma W., Yang D., Zhang J. Periostin expression is upregulated and associated with myocardial fibrosis in human failing hearts. J. Cardiol. 2014;63:373–378. doi: 10.1016/j.jjcc.2013.09.013. [DOI] [PubMed] [Google Scholar]
- 31.Wo D., Peng J., Ren D.N., Qiu L., Chen J., Zhu Y., Yan Y., Yan H., Wu J., Ma E., et al. Opposing Roles of Wnt Inhibitors IGFBP-4 and Dkk1 in Cardiac Ischemia by Differential Targeting of LRP5/6 and beta-catenin. Circulation. 2016;134:1991–2007. doi: 10.1161/CIRCULATIONAHA.116.024441. [DOI] [PubMed] [Google Scholar]
- 32.Su Y., Nishimoto T., Hoffman S., Nguyen X.X., Pilewski J.M., Feghali-Bostwick C. Insulin-like growth factor binding protein-4 exerts antifibrotic activity by reducing levels of connective tissue growth factor and the C-X-C chemokine receptor 4. FASEB Bioadv. 2019;1:167–179. doi: 10.1096/fba.2018-00015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gagestein B., von Hegedus J.H., Kwekkeboom J.C., Heijink M., Blomberg N., van der Wel T., Florea B.I., van den Elst H., Wals K., Overkleeft H.S., et al. Comparative Photoaffinity Profiling of Omega-3 Signaling Lipid Probes Reveals Prostaglandin Reductase 1 as a Metabolic Hub in Human Macrophages. J. Am. Chem. Soc. 2022;144:18938–18947. doi: 10.1021/jacs.2c06827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Verhoeven N.M., Huck J.H., Roos B., Struys E.A., Salomons G.S., Douwes A.C., van der Knaap M.S., Jakobs C. Transaldolase deficiency: liver cirrhosis associated with a new inborn error in the pentose phosphate pathway. Am. J. Hum. Genet. 2001;68:1086–1092. doi: 10.1086/320108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Banki K., Eddy R.L., Shows T.B., Halladay D.L., Bullrich F., Croce C.M., Jurecic V., Baldini A., Perl A. The human transaldolase gene (TALDO1) is located on chromosome 11 at p15.4-p15.5. Genomics. 1997;45:233–238. doi: 10.1006/geno.1997.4932. [DOI] [PubMed] [Google Scholar]
- 36.Zhuang L., Mao Y., Liu Z., Li C., Jin Q., Lu L., Tao R., Yan X., Chen K. FABP3 Deficiency Exacerbates Metabolic Derangement in Cardiac Hypertrophy and Heart Failure via PPARalpha Pathway. Front. Cardiovasc. Med. 2021;8 doi: 10.3389/fcvm.2021.722908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Simonson B., Chaffin M., Hill M.C., Atwa O., Guedira Y., Bhasin H., Hall A.W., Hayat S., Baumgart S., Bedi K.C., Jr., et al. Single-nucleus RNA sequencing in ischemic cardiomyopathy reveals common transcriptional profile underlying end-stage heart failure. Cell Rep. 2023;42 doi: 10.1016/j.celrep.2023.112086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yamagishi S.I., Matsui T. Pigment epithelium-derived factor (PEDF) and cardiometabolic disorders. Curr. Pharmaceut. Des. 2014;20:2377–2386. doi: 10.2174/13816128113199990473. [DOI] [PubMed] [Google Scholar]
- 39.Groza T., Gomez F.L., Mashhadi H.H., Muñoz-Fuentes V., Gunes O., Wilson R., Cacheiro P., Frost A., Keskivali-Bond P., Vardal B., et al. The International Mouse Phenotyping Consortium: comprehensive knockout phenotyping underpinning the study of human disease. Nucleic Acids Res. 2023;51:D1038–D1045. doi: 10.1093/nar/gkac972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lu D., Bao D., Dong W., Liu N., Zhang X., Gao S., Ge W., Gao X., Zhang L. Dkk3 prevents familial dilated cardiomyopathy development through Wnt pathway. Lab. Invest. 2016;96:239–248. doi: 10.1038/labinvest.2015.145. [DOI] [PubMed] [Google Scholar]
- 41.Wang X., Berry E., Hernandez-Anzaldo S., Takawale A., Kassiri Z., Fernandez-Patron C. Matrix metalloproteinase-2 mediates a mechanism of metabolic cardioprotection consisting of negative regulation of the sterol regulatory element-binding protein-2/3-hydroxy-3-methylglutaryl-CoA reductase pathway in the heart. Hypertension. 2015;65:882–888. doi: 10.1161/HYPERTENSIONAHA.114.04989. [DOI] [PubMed] [Google Scholar]
- 42.Zhu W., Shiojima I., Ito Y., Li Z., Ikeda H., Yoshida M., Naito A.T., Nishi J.i., Ueno H., Umezawa A., et al. IGFBP-4 is an inhibitor of canonical Wnt signalling required for cardiogenesis. Nature. 2008;454:345–349. doi: 10.1038/nature07027. [DOI] [PubMed] [Google Scholar]
- 43.Wo D., Chen J., Li Q., Ma E., Yan H., Peng J., Zhu W., Fang Y., Ren D.N. IGFBP-4 enhances VEGF-induced angiogenesis in a mouse model of myocardial infarction. J. Cell Mol. Med. 2020;24:9466–9471. doi: 10.1111/jcmm.15516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chavkin N.W., Sano S., Wang Y., Oshima K., Ogawa H., Horitani K., Sano M., MacLauchlan S., Nelson A., Setia K., et al. The Cell Surface Receptors Ror1/2 Control Cardiac Myofibroblast Differentiation. J. Am. Heart Assoc. 2021;10 doi: 10.1161/JAHA.120.019904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Van Tassell B.W., Seropian I.M., Toldo S., Mezzaroma E., Abbate A. Interleukin-1beta induces a reversible cardiomyopathy in the mouse. Inflamm. Res. 2013;62:637–640. doi: 10.1007/s00011-013-0625-0. [DOI] [PubMed] [Google Scholar]
- 46.Van Tassell B.W., Abouzaki N.A., Oddi Erdle C., Carbone S., Trankle C.R., Melchior R.D., Turlington J.S., Thurber C.J., Christopher S., Dixon D.L., et al. Interleukin-1 Blockade in Acute Decompensated Heart Failure: A Randomized, Double-Blinded, Placebo-Controlled Pilot Study. J. Cardiovasc. Pharmacol. 2016;67:544–551. doi: 10.1097/FJC.0000000000000378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Van Tassell B.W., Canada J., Carbone S., Trankle C., Buckley L., Oddi Erdle C., Abouzaki N.A., Dixon D., Kadariya D., Christopher S., et al. Interleukin-1 Blockade in Recently Decompensated Systolic Heart Failure: Results From REDHART (Recently Decompensated Heart Failure Anakinra Response Trial) Circ. Heart Fail. 2017;10 doi: 10.1161/CIRCHEARTFAILURE.117.004373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Niu X., Han Q., Li X., Li J., Liu Y., Li Y., Wu Y., Zhang K. EDIL3 influenced the alphavbeta3-FAK/MEK/ERK axis of endothelial cells in psoriasis. J. Cell Mol. Med. 2022;26:5202–5212. doi: 10.1111/jcmm.17544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wei X., Zou S., Xie Z., Wang Z., Huang N., Cen Z., Hao Y., Zhang C., Chen Z., Zhao F., et al. EDIL3 deficiency ameliorates adverse cardiac remodelling by neutrophil extracellular traps (NET)-mediated macrophage polarization. Cardiovasc. Res. 2022;118:2179–2195. doi: 10.1093/cvr/cvab269. [DOI] [PubMed] [Google Scholar]
- 50.Zhao M., Zheng Z., Peng S., Xu Y., Zhang J., Liu J., Pan W., Yin Z., Xu S., Wei C., et al. Epidermal Growth Factor-Like Repeats and Discoidin I-Like Domains 3 Deficiency Attenuates Dilated Cardiomyopathy by Inhibiting Ubiquitin Specific Peptidase 10 Dependent Smad4 Deubiquitination. J. Am. Heart Assoc. 2024;13 doi: 10.1161/JAHA.123.031283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Verhoeven N.M., Wallot M., Huck J.H.J., Dirsch O., Ballauf A., Neudorf U., Salomons G.S., van der Knaap M.S., Voit T., Jakobs C. A newborn with severe liver failure, cardiomyopathy and transaldolase deficiency. J. Inherit. Metab. Dis. 2005;28:169–179. doi: 10.1007/s10545-005-5261-6. [DOI] [PubMed] [Google Scholar]
- 52.Wang Y., Cao Y., Yamada S., Thirunavukkarasu M., Nin V., Joshi M., Rishi M.T., Bhattacharya S., Camacho-Pereira J., Sharma A.K., et al. Cardiomyopathy and Worsened Ischemic Heart Failure in SM22-alpha Cre-Mediated Neuropilin-1 Null Mice: Dysregulation of PGC1alpha and Mitochondrial Homeostasis. Arterioscler. Thromb. Vasc. Biol. 2015;35:1401–1412. doi: 10.1161/ATVBAHA.115.305566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wang N., Cao Y., Zhu Y. Netrin-1 prevents the development of cardiac hypertrophy and heart failure. Mol. Med. Rep. 2016;13:2175–2181. doi: 10.3892/mmr.2016.4755. [DOI] [PubMed] [Google Scholar]
- 54.Nagao K., Sowa N., Inoue K., Tokunaga M., Fukuchi K., Uchiyama K., Ito H., Hayashi F., Makita T., Inada T., et al. Myocardial expression level of neural cell adhesion molecule correlates with reduced left ventricular function in human cardiomyopathy. Circ. Heart Fail. 2014;7:351–358. doi: 10.1161/CIRCHEARTFAILURE.113.000939. [DOI] [PubMed] [Google Scholar]
- 55.Dinarello C.A. Overview of the IL-1 family in innate inflammation and acquired immunity. Immunol. Rev. 2018;281:8–27. doi: 10.1111/imr.12621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Narayan P., Trikantzopoulos E., Mezzaroma E., Mauro A.G., Vohra H., Abbate A., Toldo S. The interleukin-1 receptor type I promotes the development of aging-associated cardiomyopathy in mice. Cytokine. 2022;151 doi: 10.1016/j.cyto.2022.155811. [DOI] [PubMed] [Google Scholar]
- 57.Abbate A., Salloum F.N., Van Tassell B.W., Vecile E., Toldo S., Seropian I., Mezzaroma E., Dobrina A. Alterations in the interleukin-1/interleukin-1 receptor antagonist balance modulate cardiac remodeling following myocardial infarction in the mouse. PLoS One. 2011;6 doi: 10.1371/journal.pone.0027923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Xiang M., Luo H., Wu J., Ren L., Ding X., Wu C., Chen J., Chen S., Zhang H., Yu L., et al. ADAM23 in Cardiomyocyte Inhibits Cardiac Hypertrophy by Targeting FAK - AKT Signaling. J. Am. Heart Assoc. 2018;7 doi: 10.1161/JAHA.118.008604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Yang R., Bunting S., Gillett N., Clark R., Jin H. Growth hormone improves cardiac performance in experimental heart failure. Circulation. 1995;92:262–267. doi: 10.1161/01.cir.92.2.262. [DOI] [PubMed] [Google Scholar]
- 60.Hradec J., Kral J., Janota T., Krsek M., Hana V., Marek J., Malik M. Regression of acromegalic left ventricular hypertrophy after lanreotide (a slow-release somatostatin analog) Am. J. Cardiol. 1999;83:1506–1509.A8. doi: 10.1016/s0002-9149(99)00135-6. [DOI] [PubMed] [Google Scholar]
- 61.Tilkens B., Galazka P., Solis J., Crouch J.D., Tajik A.J. Massive Left Ventricular Hypertrophy With Acromegaly: Hypertrophic Obstructive Cardiomyopathy Phenocopy or Phenotype? JACC. Case Rep. 2023;25 doi: 10.1016/j.jaccas.2023.102033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hradec J., Marek J., Petrásek J. The nature of cardiac hypertrophy in acromegaly: an echocardiographic study. Cor Vasa. 1988;30:186–199. [PubMed] [Google Scholar]
- 63.Zhao J., Bruche S., Potts H.G., Davies B., Mommersteeg M.T.M. Tissue-Specific Roles for the Slit-Robo Pathway During Heart, Caval Vein, and Diaphragm Development. J. Am. Heart Assoc. 2022;11 doi: 10.1161/JAHA.121.023348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.MacMullin A., Jacobs J.R. Slit coordinates cardiac morphogenesis in Drosophila. Dev. Biol. 2006;293:154–164. doi: 10.1016/j.ydbio.2006.01.027. [DOI] [PubMed] [Google Scholar]
- 65.Huang X., Sun J., Rong W., Zhao T., Li D.H., Ding X., Wu L.Y., Wu K., Schachner M., Xiao Z.C., et al. Loss of cell adhesion molecule CHL1 improves homeostatic adaptation and survival in hypoxic stress. Cell Death Dis. 2013;4 doi: 10.1038/cddis.2013.284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Fang Z., Li X., Liu J., Lee H., Salciccioli L., Lazar J., Zhang M. The role of complement C3 in the outcome of regional myocardial infarction. Biochem. Biophys. Rep. 2023;33 doi: 10.1016/j.bbrep.2023.101434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Tahara N., Yamagishi S.i., Tahara A., Nitta Y., Kodama N., Mizoguchi M., Mohar D., Ishibashi M., Hayabuchi N., Imaizumi T. Serum level of pigment epithelium-derived factor is a marker of atherosclerosis in humans. Atherosclerosis. 2011;219:311–315. doi: 10.1016/j.atherosclerosis.2011.06.022. [DOI] [PubMed] [Google Scholar]
- 68.Ueda S.i., Yamagishi S.i., Matsui T., Jinnouchi Y., Imaizumi T. Administration of pigment epithelium-derived factor inhibits left ventricular remodeling and improves cardiac function in rats with acute myocardial infarction. Am. J. Pathol. 2011;178:591–598. doi: 10.1016/j.ajpath.2010.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Liu H., Dong W., Lin Z., Lu J., Wan H., Zhou Z., Liu Z. CCN4 regulates vascular smooth muscle cell migration and proliferation. Mol. Cell. 2013;36:112–118. doi: 10.1007/s10059-013-0012-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Piper-Vallillo A.J., Costa D.B., Sabe M.A., Asnani A. Heart Failure Associated With the Epidermal Growth Factor Receptor Inhibitor Osimertinib. JACC. CardioOncol. 2020;2:119–122. doi: 10.1016/j.jaccao.2020.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Makki N., Thiel K.W., Miller F.J., Jr. The epidermal growth factor receptor and its ligands in cardiovascular disease. Int. J. Mol. Sci. 2013;14:20597–20613. doi: 10.3390/ijms141020597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Schellings M.W.M., Vanhoutte D., Swinnen M., Cleutjens J.P., Debets J., van Leeuwen R.E.W., d'Hooge J., Van de Werf F., Carmeliet P., Pinto Y.M., et al. Absence of SPARC results in increased cardiac rupture and dysfunction after acute myocardial infarction. J. Exp. Med. 2009;206:113–123. doi: 10.1084/jem.20081244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Liu W., Chen B., Wang Y., Meng C., Huang H., Huang X.R., Qin J., Mulay S.R., Anders H.J., Qiu A., et al. RGMb protects against acute kidney injury by inhibiting tubular cell necroptosis via an MLKL-dependent mechanism. Proc. Natl. Acad. Sci. USA. 2018;115:E1475–E1484. doi: 10.1073/pnas.1716959115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Ahmed S., Ahmed A., Bouzina H., Lundgren J., Rådegran G. Elevated plasma endocan and BOC in heart failure patients decrease after heart transplantation in association with improved hemodynamics. Heart Ves. 2020;35:1614–1628. doi: 10.1007/s00380-020-01656-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Landis B.J., Helvaty L.R., Geddes G.C., Lin J.H.I., Yatsenko S.A., Lo C.W., Border W.L., Wechsler S.B., Murali C.N., Azamian M.S., et al. A Multicenter Analysis of Abnormal Chromosomal Microarray Findings in Congenital Heart Disease. J. Am. Heart Assoc. 2023;12 doi: 10.1161/JAHA.123.029340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Tabasum S., Thapa D., Giobbie-Hurder A., Weirather J.L., Campisi M., Schol P.J., Li X., Li J., Yoon C.H., Manos M.P., et al. EDIL3 as an Angiogenic Target of Immune Exclusion Following Checkpoint Blockade. Cancer Immunol. Res. 2023;11:1493–1507. doi: 10.1158/2326-6066.CIR-23-0171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Stenemo M., Nowak C., Byberg L., Sundström J., Giedraitis V., Lind L., Ingelsson E., Fall T., Ärnlöv J. Circulating proteins as predictors of incident heart failure in the elderly. Eur. J. Heart Fail. 2018;20:55–62. doi: 10.1002/ejhf.980. [DOI] [PubMed] [Google Scholar]
- 78.Wells Q.S., Gupta D.K., Smith J.G., Collins S.P., Storrow A.B., Ferguson J., Smith M.L., Pulley J.M., Collier S., Wang X., et al. Accelerating Biomarker Discovery Through Electronic Health Records, Automated Biobanking, and Proteomics. J. Am. Coll. Cardiol. 2019;73:2195–2205. doi: 10.1016/j.jacc.2019.01.074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Ngo D., Sinha S., Shen D., Kuhn E.W., Keyes M.J., Shi X., Benson M.D., O'Sullivan J.F., Keshishian H., Farrell L.A., et al. Aptamer-Based Proteomic Profiling Reveals Novel Candidate Biomarkers and Pathways in Cardiovascular Disease. Circulation. 2016;134:270–285. doi: 10.1161/CIRCULATIONAHA.116.021803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Govaere O., Hasoon M., Alexander L., Cockell S., Tiniakos D., Ekstedt M., Schattenberg J.M., Boursier J., Bugianesi E., Ratziu V., et al. A proteo-transcriptomic map of non-alcoholic fatty liver disease signatures. Nat. Metab. 2023;5:572–578. doi: 10.1038/s42255-023-00775-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Chan M.Y., Efthymios M., Tan S.H., Pickering J.W., Troughton R., Pemberton C., Ho H.H., Prabath J.F., Drum C.L., Ling L.H., et al. Prioritizing Candidates of Post-Myocardial Infarction Heart Failure Using Plasma Proteomics and Single-Cell Transcriptomics. Circulation. 2020;142:1408–1421. doi: 10.1161/CIRCULATIONAHA.119.045158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Oh H.S.H., Rutledge J., Nachun D., Pálovics R., Abiose O., Moran-Losada P., Channappa D., Urey D.Y., Kim K., Sung Y.J., et al. Organ aging signatures in the plasma proteome track health and disease. Nature. 2023;624:164–172. doi: 10.1038/s41586-023-06802-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Pieruzzi F., Abassi Z.A., Keiser H.R. Expression of renin-angiotensin system components in the heart, kidneys, and lungs of rats with experimental heart failure. Circulation. 1995;92:3105–3112. doi: 10.1161/01.cir.92.10.3105. [DOI] [PubMed] [Google Scholar]
- 84.Danser A.H., van Kesteren C.A., Bax W.A., Tavenier M., Derkx F.H., Saxena P.R., Schalekamp M.A. Prorenin, renin, angiotensinogen, and angiotensin-converting enzyme in normal and failing human hearts. Evidence for renin binding. Circulation. 1997;96:220–226. doi: 10.1161/01.cir.96.1.220. [DOI] [PubMed] [Google Scholar]
- 85.Shah S., Henry A., Roselli C., Lin H., Sveinbjörnsson G., Fatemifar G., Hedman Å.K., Wilk J.B., Morley M.P., Chaffin M.D., et al. Genome-wide association and Mendelian randomisation analysis provide insights into the pathogenesis of heart failure. Nat. Commun. 2020;11:163. doi: 10.1038/s41467-019-13690-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Hahn V.S., Knutsdottir H., Luo X., Bedi K., Margulies K.B., Haldar S.M., Stolina M., Yin J., Khakoo A.Y., Vaishnav J., et al. Myocardial Gene Expression Signatures in Human Heart Failure With Preserved Ejection Fraction. Circulation. 2021;143:120–134. doi: 10.1161/CIRCULATIONAHA.120.050498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Ye B., Bradshaw A.D., Abrahante J.E., Dragon J.A., Häußler T.N., Bell S.P., Hirashima F., LeWinter M., Zile M.R., Meyer M. Left Ventricular Gene Expression in Heart Failure With Preserved Ejection Fraction-Profibrotic and Proinflammatory Pathways and Genes. Circ. Heart Fail. 2023;16 doi: 10.1161/CIRCHEARTFAILURE.123.010395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Yang X., Cheng K., Wang L.Y., Jiang J.G. The role of endothelial cell in cardiac hypertrophy: Focusing on angiogenesis and intercellular crosstalk. Biomed. Pharmacother. 2023;163 doi: 10.1016/j.biopha.2023.114799. [DOI] [PubMed] [Google Scholar]
- 89.Koenig A.L., Shchukina I., Amrute J., Andhey P.S., Zaitsev K., Lai L., Bajpai G., Bredemeyer A., Smith G., Jones C., et al. Single-cell transcriptomics reveals cell-type-specific diversification in human heart failure. Nat. Cardiovasc. Res. 2022;1:263–280. doi: 10.1038/s44161-022-00028-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Heidenreich P.A., Bozkurt B., Aguilar D., Allen L.A., Byun J.J., Colvin M.M., Deswal A., Drazner M.H., Dunlay S.M., Evers L.R., et al. 2022 AHA/ACC/HFSA Guideline for the Management of Heart Failure: Executive Summary: A Report of the American College of Cardiology/American Heart Association Joint Committee on Clinical Practice Guidelines. Circulation. 2022;145:e876–e894. doi: 10.1161/CIR.0000000000001062. [DOI] [PubMed] [Google Scholar]
- 91.Perry A.S., Farber-Eger E., Gonzales T., Tanaka T., Robbins J.M., Murthy V.L., Stolze L.K., Zhao S., Huang S., Colangelo L.A., et al. Proteomic analysis of cardiorespiratory fitness for prediction of mortality and multisystem disease risks. Nat. Med. 2024;30:1711–1721. doi: 10.1038/s41591-024-03039-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Friedman G.D., Cutter G.R., Donahue R.P., Hughes G.H., Hulley S.B., Jacobs D.R., Jr., Liu K., Savage P.J. CARDIA: study design, recruitment, and some characteristics of the examined subjects. J. Clin. Epidemiol. 1988;41:1105–1116. doi: 10.1016/0895-4356(88)90080-7. [DOI] [PubMed] [Google Scholar]
- 93.Lloyd-Jones D.M., Lewis C.E., Schreiner P.J., Shikany J.M., Sidney S., Reis J.P. The Coronary Artery Risk Development In Young Adults (CARDIA) Study: JACC Focus Seminar 8/8. J. Am. Coll. Cardiol. 2021;78:260–277. doi: 10.1016/j.jacc.2021.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Armstrong A.C., Ricketts E.P., Cox C., Adler P., Arynchyn A., Liu K., Stengel E., Sidney S., Lewis C.E., Schreiner P.J., et al. Quality Control and Reproducibility in M-Mode, Two-Dimensional, and Speckle Tracking Echocardiography Acquisition and Analysis: The CARDIA Study, Year 25 Examination Experience. Echocardiography. 2015;32:1233–1240. doi: 10.1111/echo.12832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Kannel W.B., Feinleib M., McNamara P.M., Garrison R.J., Castelli W.P. An investigation of coronary heart disease in families. The Framingham offspring study. Am. J. Epidemiol. 1979;110:281–290. doi: 10.1093/oxfordjournals.aje.a112813. [DOI] [PubMed] [Google Scholar]
- 96.Levy D., Garrison R.J., Savage D.D., Kannel W.B., Castelli W.P. Left ventricular mass and incidence of coronary heart disease in an elderly cohort. The Framingham Heart Study. Ann. Intern. Med. 1989;110:101–107. doi: 10.7326/0003-4819-110-2-101. [DOI] [PubMed] [Google Scholar]
- 97.Levy D., Garrison R.J., Savage D.D., Kannel W.B., Castelli W.P. Prognostic implications of echocardiographically determined left ventricular mass in the Framingham Heart Study. N. Engl. J. Med. 1990;322:1561–1566. doi: 10.1056/NEJM199005313222203. [DOI] [PubMed] [Google Scholar]
- 98.Shah R.V., Yeri A.S., Murthy V.L., Massaro J.M., D'Agostino R., Sr., Freedman J.E., Long M.T., Fox C.S., Das S., Benjamin E.J., et al. Association of Multiorgan Computed Tomographic Phenomap With Adverse Cardiovascular Health Outcomes: The Framingham Heart Study. JAMA Cardiol. 2017;2:1236–1246. doi: 10.1001/jamacardio.2017.3145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Sun B.B., Chiou J., Traylor M., Benner C., Hsu Y.H., Richardson T.G., Surendran P., Mahajan A., Robins C., Vasquez-Grinnell S.G., et al. Plasma proteomic associations with genetics and health in the UK Biobank. Nature. 2023;622:329–338. doi: 10.1038/s41586-023-06592-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.UK Biobank Protocol for a large-scale prospective epidemiological resource. 2006. www.ukbiobank.ac.uk/resources/
- 101.Wu P., Gifford A., Meng X., Li X., Campbell H., Varley T., Zhao J., Carroll R., Bastarache L., Denny J.C., et al. Mapping ICD-10 and ICD-10-CM Codes to Phecodes: Workflow Development and Initial Evaluation. JMIR Med. Inform. 2019;7 doi: 10.2196/14325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Carrasco-Zanini J., Pietzner M., Lindbohm J.V., Wheeler E., Oerton E., Kerrison N., Simpson M., Westacott M., Drolet D., Kivimaki M., et al. Proteomic signatures for identification of impaired glucose tolerance. Nat. Med. 2022;28:2293–2300. doi: 10.1038/s41591-022-02055-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Katz D.H., Tahir U.A., Ngo D., Benson M.D., Gao Y., Shi X., Nayor M., Keyes M.J., Larson M.G., Hall M.E., et al. Multiomic Profiling in Black and White Populations Reveals Novel Candidate Pathways in Left Ventricular Hypertrophy and Incident Heart Failure Specific to Black Adults. Circ. Genom. Precis. Med. 2021;14 doi: 10.1161/CIRCGEN.120.003191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Wu T., Hu E., Xu S., Chen M., Guo P., Dai Z., Feng T., Zhou L., Tang W., Zhan L., et al. clusterProfiler 4.0: A universal enrichment tool for interpreting omics data. Innovation. 2021;2 doi: 10.1016/j.xinn.2021.100141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Law C.W., Chen Y., Shi W., Smyth G.K. voom: Precision weights unlock linear model analysis tools for RNA-seq read counts. Genome Biol. 2014;15:R29. doi: 10.1186/gb-2014-15-2-r29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Drakos S.G., Badolia R., Makaju A., Kyriakopoulos C.P., Wever-Pinzon O., Tracy C.M., Bakhtina A., Bia R., Parnell T., Taleb I., et al. Distinct Transcriptomic and Proteomic Profile Specifies Patients Who Have Heart Failure With Potential of Myocardial Recovery on Mechanical Unloading and Circulatory Support. Circulation. 2023;147:409–424. doi: 10.1161/CIRCULATIONAHA.121.056600. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Results of analyses for clinical prioritization. Sheet “SD01” represents results of regressions in CARDIA. SomaLogic aptamer meta-data (aptname, seqid, somaid) and target identification (targetfullname, target, uniprot, entrezgeneid, entrezgenesymbol) are provided. “Outcome” refers to specific echocardiographic phenotype (dependent variable), and “physio” refers to the overall physiology into which that outcome was grouped. Columns for beta (estimated regression coefficient), se (standard error of the beta), N (sample size for model), p (P-value) and its FDR are provided for the 70% derivation (“der_”) and the 30% hold-out replication/validation samples (“val_”) are provided. These models were adjusted for age, gender, and race. Sheet “SD02” provides results from regression models for left atrial dimension and LV mass (log) in FHS for overlapping proteins with an FDR <5% in both CARDIA derivation and validation samples (SD01). These models were adjusted for age, gender, and height. Of note, the original manuscript detailing a subset of these associations has been reported3. Sheet “SD03” represents results of survival models in two cohorts (specified under “cohort” as UK Biobank or FHS) for each protein. Models in FHS displayed here were adjusted for age and gender. Models for UK Biobank displayed here were adjusted for age, gender, and race. Sheet “SD04” reports Cox model results for incident HF in UK Biobank including an age by protein interaction term. Sheet “SD05” represents clinical characteristics of individuals evaluated in snRNA-seq datasets. Sheet “SD06” represents results from the International Mouse Phenotypoing Consortium
Data Availability Statement
Data from CARDIA used in these analyses are available through the Coronary Artery Risk Development in Young Adults study (CARDIA; cardia.dopm.uab.edu). Data from the Framingham Heart Study (FHS) is available at dbGaP (dbGaP identifier pht006013) or via contact with the FHS coordinating center (www.framinghamheartstudy.org). UK Biobank data is available at UK Biobank Research Access Portal (analyses here conducted under proposal approval 57492). This paper does not report any original code. Any additional information required to reanalyze the data reported in this work paper is available from the lead contact upon request.