

Summary
Atypical teratoid/rhabdoid tumors (ATRTs) are highly malignant embryonal tumors of the central nervous system with a dismal prognosis. Using a newly developed and validated patient-derived ATRT culture and xenograft model, alongside a panel of primary ATRT models, we found that ATRTs are selectively sensitive to the nucleoside analog gemcitabine. Gene expression and protein analyses indicate that gemcitabine treatment causes the degradation of sirtuin 1 (SIRT1), resulting in cell death through activation of nuclear factor κB (NF-κB) and p53. Furthermore, we discovered that gemcitabine-induced loss of SIRT1 results in a nucleus-to-cytoplasm translocation of the sonic hedgehog (SHH) signaling activator GLI2, explaining the observed additional gemcitabine sensitivity in SHH-subtype ATRT. Treatment of ATRT xenograft-bearing mice with gemcitabine resulted in a >30% increase in median survival and yielded long-term survivors in two independent patient-derived xenograft models. These findings demonstrate that ATRTs are highly sensitive to gemcitabine treatment and may form part of a future multimodal treatment strategy for ATRTs.
Keywords: atypical teratoid/rhabdoid tumor, sirtuin 1, patient-derived models, therapy development, pediatric oncology, gemcitabine, ATRT, neuro oncology, p53
Graphical abstract
Highlights
-
•
VUMC-ATRT-03 is a newly developed SHH-1B ATRT culture and xenograft model
-
•
ATRTs are highly sensitive to gemcitabine treatment both in vitro and in vivo
-
•
Gemcitabine kills ATRT cells through SIRT1 depletion and p53 activation
-
•
Gemcitabine interferes with hedgehog signaling in SHH-subtype ATRT
Metselaar et al. identify gemcitabine as a highly potent drug for the treatment of atypical teratoid/rhabdoid tumors (ATRTs). Using a panel of ATRT cell cultures and mouse models, the authors reveal how gemcitabine kills ATRT cells and why it could form a centerpiece for future treatment of patients with ATRT.
Introduction
Atypical teratoid/rhabdoid tumors (ATRTs) are highly malignant embryonal tumors of the central nervous system (CNS), often found in infants and young children. ATRTs have historically been considered incurable tumors, and even though outcomes have recently improved slightly due to multimodal therapy, standardized treatment regimens are often absent, and between 57% and 70% of all patients with ATRT succumb within 5 years post-diagnosis.1,2,3,4,5 ATRTs are characterized by bi-allelic loss of function of the tumor suppressor SMARCB1, or in rare cases, SMARCA4, which are core subunits of the SWItch/Sucrose Non-Fermentable (SWI/SNF) chromatin remodeling complex.6 Genome-wide methylation and RNA sequencing experiments have distinguished three distinct molecular subgroups of ATRT, all typically characterized by low mutational burden and bland chromosomal copy-number variation profiles: sonic hedgehog (SHH), Myc (MYC), and tyrosinase (TYR).7,8 More recently, the SHH group has been further divided into the subgroups SHH-1A, SHH-1B, and SHH-2.9 This subdivision of ATRT into specific molecular groups is an important step in understanding these clinically heterogenic malignancies, enabling researchers and clinicians to seek novel tailored therapeutic strategies. SHH-ATRT is the most common of the three subgroups and is characterized by activation of the SHH signaling pathway, as demonstrated by strong overexpression and activation of GLI family zinc finger 2 (GLI2) and MYCN.7,10 SHH-ATRTs harbor a compact, hypermethylated chromatin structure with a strong epigenetically dysregulated expression profile, including overexpression of EHMT2 (G9a), EZH2, and several bromodomain-containing proteins.7,8 Aside from the differences between each subgroup, most ATRTs share the similarity of having intact, but epigenetically repressed, tumor suppressor genes, like p53.11,12
In this study, we developed a new SHH-1B subtype ATRT culture and xenograft model and found, in multiple primary models, that ATRTs are specifically sensitive to treatment with the clinically registered chemotherapeutic agent gemcitabine. Using RNA sequencing, protein analysis, knockdown, and knockout experiments, we established that this ATRT-specific gemcitabine sensitivity results from suppression of sirtuin 1 (SIRT1), which causes p53 activation. We validated our findings in ATRT xenograft mouse models that showed significant increases in survival upon gemcitabine treatment. Furthermore, we discovered additional tumor-specific toxicity of gemcitabine treatment in SHH-subgroup ATRT, caused by gemcitabine-induced inhibition of the SHH signaling pathway. The results of our study warrant further clinical investigation of gemcitabine as a potential therapy for patients with ATRT.
Results
Establishment of a patient-derived ATRT culture and xenograft model
Tumor tissue was obtained through resection from an 11-year-old girl, suffering from a high-grade malignant, embryonal brain tumor. MRI imaging showed a large diffuse mass in the right fronto-temporal part of the cerebral hemisphere (Figure 1A). Immunohistochemistry revealed a complete loss of SMARCB1 in the nuclei of all tumor cells, corroborating the diagnosis of ATRT (Figure 1B). Genome-wide DNA methylation profiling confirmed the diagnosis and assigned the tumor to the SHH subtype (Figure S1). Fresh tumor tissue was put into culture (VUMC-ATRT-03) and maintained as neurospheres. Upon stable growth, VUMC-ATRT-03 cells were orthotopically injected into athymic nude mice. These mice developed tumors that, like the primary tumor, were characterized by a loss of SMARCB1 as shown by immunohistochemistry (Figure 1C). Genome-wide DNA methylation profiling on VUMC-ATRT-03 cells harvested from mice confirmed the maintenance of the SHH subtype within the model (Figure S2). In addition, a panel of immunohistochemical stainings confirmed that high proliferation (Ki-67), multilineage differentiation (GFAP, S100, and vimentin), and loss of blood-brain barrier (BBB) integrity (GLUT1, BCRP, P-gp, and CD31) were present in both the original tumor tissue and mouse orthotopic xenografts of this neoplasm (Figures 1D and 1E). Furthermore, chromosomal copy-number variations as detected based on the genome-wide methylome analysis revealed that the VUMC-ATRT-03 mouse passage model and original tumor tissue are largely overlapping, revealing that chromosomal alterations were maintained stable through modeling (Figure S2). Further clustering of the VUMC-ATRT-03 DNA methylation profiles against 130 other ATRT cases from two previous studies7,9 confirmed that the VUMC-ATRT-03 model clusters to the SHH-1B subgroup, which explains the relatively old age of onset and rough copy-number profile of this tumor (Figures 1F and 1G). Another ATRT model (VUMC-ATRT-01), that was recently established and presented by our team, clustered within the SHH-1A subgroup (Figures 1F and 1G).
Figure 1.

Establishment of a novel patient-derived SHH-ATRT culture and xenograft model
(A) Diagnostic T2-weighted MRI of the patient from which VUMC-ATRT-03 was derived (top, coronal plane; bottom, sagittal plane).
(B) Immunohistochemistry of patient-derived resection material depicting SMARCB1 expression in brown, revealing typical loss of nuclear SMARCB1 exclusively in tumor cells. The positive nuclei of non-neoplastic and microvascular cells serve as a positive internal control.
(C) Immunohistochemistry for SMARCB1 in a mouse brain carrying a VUMC-ATRT-03 xenograft confirming the absence of SMARCB1 in the nuclei of tumor cells.
(D) Panel of immunohistochemical images depicting ATRT hallmarks in patient-derived resection material (high proliferation: Ki-67; patches of multilineage differentiation: GFAP, S100, vimentin, and keratin; loss of BBB integrity: GLUT-1, BCRP, P-gp, and CD31). Loss of GLUT1 (indicated by the red arrow) indicates intratumor vascular malformations and loss of BBB integrity.
(E) Panel of immunohistochemical images depicting ATRT hallmarks in a mouse brain carrying VUMC-ATRT-03 xenografts (high proliferation: Ki-67; patches of multilineage differentiation: GFAP, S100, vimentin, and keratin; loss of BBB integrity: GLUT-1, BCRP, P-gp, and CD31).
(F) Unsupervised t-distributed stochastic neighbor embedding (t-SNE) clustering of DNA methylation profiles of 134 ATRT samples. Cases are annotated based on previously described subgrouping analysis.9 VUMC-ATRT-01 and VUMC-ATRT-03 methylation profiles annotate to the SHH cluster.
(G) Unsupervised t-SNE clustering of DNA methylation profiles of 94 SHH-ATRT samples. Here, VUMC-ATRT-01 clusters with the SHH-A1 group and the VUMC-ATRT-03 with the SHH-1B group.
Gemcitabine is an effective ATRT-specific chemotherapeutic in vitro
We conducted an epigenetic compound library screen (#11076, Cayman Chemical, USA) using the VUMC-ATRT-03 cells and a panel of ten other primary pediatric high-grade brain tumor neurosphere cultures. This screen indicated that only the ATRT model responded strongly to the clinically registered nucleoside analog gemcitabine (Figure 2A; Table S1).13 To study if this sensitivity applied to other ATRT cultures, we tested the effect of gemcitabine on a panel of seven primary ATRT models and found all ATRTs to be over a 10-fold more sensitive to gemcitabine compared to non-ATRT pediatric CNS tumor models (Figure 2B). Additionally, we observed that SHH-subtype ATRT models respond significantly more sensitive to gemcitabine treatment compared to MYC-subtype ATRT models.
Figure 2.

Epigenetic compound screening identifies gemcitabine as an effective therapeutic in ATRT
(A) Compound library screen cell viability readout in a panel of primary pediatric CNS tumor models. Lower: the gemcitabine-treated cell viability readout is highlighted, revealing that the only ATRT model among the panel shows a complete loss of viability upon gemcitabine treatment (1,000 nM, 96 h).
(B) Cell viability IC50 curves of gemcitabine treatment in a panel of seven primary ATRT culture models; two pediatric high-grade glioma (HGG) cell cultures are used as controls. SHH-subtype ATRT models show higher gemcitabine sensitivity compared to an MYC-subtype ATRT model, while controls show no sensitivity.
Gemcitabine treatment activates p53 signaling through the depletion of ATRT-specific SIRT1 overexpression
To study the mechanisms underlying the ATRT-specific gemcitabine sensitivity, we investigated if known gemcitabine targets are upregulated in ATRT compared to other tumors. Using this approach, we identified that the gemcitabine target SIRT1 is significantly overexpressed in a dataset consisting of 49 ATRT samples compared to healthy brain, cerebellum, and other high-grade brain tumor tissues (Figure 3A). Consequently, we validated SIRT1 protein expression in ATRT neurosphere models in vitro and found that gemcitabine treatment depletes SIRT1 protein expression in these cultures (Figures 3B and S3). To further assess the impact of SIRT1 inhibition in ATRT, we performed differential expression analysis on six ATRT cultures treated with gemcitabine and mapped the differential expression of SIRT1 downstream targets (Figure 3C). We found that gemcitabine activates components of the nuclear factor κB (NF-κB) and p53 pathways, which are known to be suppressed by SIRT1.14,15,16 Conversely, differential expression analysis revealed a highly significant downregulation of SHH pathway components after gemcitabine treatment (Figure 3C). In parallel, we found that gemcitabine treatment did not upregulate mRNA expression of NF-κB subunits in other pediatric brain tumor cultures (Figure 3D). Furthermore, RNA expression data revealed that gemcitabine upregulates transcription of the full NF-κB signaling pathway (Figures 3E and S4). Using western blotting, we confirmed that gemcitabine treatment activates the NF-κB signaling pathway by increasing the phosphorylation of the p65 NF-κB subunit in our primary ATRT models (Figure 3F). Since SIRT1 and NF-κB are both modulators of p53 and the majority of ATRTs do not harbor a TP53 mutation, we hypothesized that p53 may play a role in ATRT-specific gemcitabine sensitivity.17,18,19,20,21 Therefore, we assessed p53 protein expression in five primary ATRT models, before and after gemcitabine treatment, and found that all ATRT models show elevated p53 protein expression upon gemcitabine treatment (Figure 3G). Furthermore, RNA expression data revealed that gemcitabine treatment activates the overall (Kyoto Encyclopedia of Genes and Genomes [KEGG] defined) p53 signaling machinery, as well as p53 transcriptional targets in our ATRT models, while several cell cycle regulators are downregulated (Figures 3H and S5). Since p53 activation causes apoptosis over other forms of cell death, we assessed cleaved poly [ADP-ribose] polymerase (PARP) protein expression in our ATRT cell lines and found that gemcitabine indeed increases this apoptosis hallmark in four of five tested ATRT cell cultures, while cleaved PARP did not increase in two diffuse midline glioma (DMG) cell lines (Figure S6). Additionally, since patients with ATRT often receive NF-κB-suppressing corticosteroids, we tested if dexamethasone would suppress gemcitabine effectivity in our ATRT models. No anti-synergy was found between dexamethasone and gemcitabine at clinically relevant doses (Figure S7).
Figure 3.

ATRT-specific SIRT1 upregulation suppresses NF-κB and p53 activity, which can be reversed through gemcitabine treatment
(A) mRNA expression levels of SIRT1 in normal brain, cerebellum, and various tissues (in green, GSE: 11882, GSE: 3526, GSE: 7307) compared to adult and pediatric CNS tumor tissues (in blue, GSE: 7696, GSE: 16011, GSE: 26576, GSE: 19578, GSE: 74195, GSE: 64415) and ATRT tissues (in red, GSE: 70678).
(B) Western blot analysis depicting SIRT1 expression in VUMC-ATRT-01 and VUMC-ATRT-03 cells after gemcitabine treatment (0, 5, and 10 nM) for 24 h.
(C) Volcano plot depicting −log10 false discovery rate (FDR)-corrected p value of differential expressed genes between DMSO (n = 6) and gemcitabine-treated (n = 6) ATRT cell cultures VUMC-ATRT-01, VUMC-ATRT-03, CHLA-02, CHLA-05, CHLA-06, and CHLA-266. Downstream targets of SIRT1 (SHH, PTCH1, TP73, CDKN1A, NFKB2, RELB, RELA, NFKB1, FOXO3, TP53, E2F1, XRCC6, FOXO1, PPARG, LXR, and PPARGC1A) are marked in red.
(D) Untreated (blue) vs. gemcitabine (red)-treated mRNA expression of NFKB1 (black) and NFKB2 (magenta) in VUMC-ATRT-03 and a panel of pediatric HGG models.
(E) Heatmap representation illustrating mRNA expression of the KEGG NF-κB gene set in VUMC-ATRT-01 and VUMC-ATRT-03 cells before and after gemcitabine treatment (PAGE FDR p < 0.05).
(F) Western blot analysis depicting p65-NF-κB and phospho-p65-NF-κB (pNF-κB) expression in VUMC-ATRT-01 and VUMC-ATRT-03 cells after gemcitabine treatment (0, 5, and 10 nM) for 24 h.
(G) Western blot analysis depicting p53 expression in VUMC-ATRT-01, VUMC-ATRT-03, CHLA-ATRT-02, CHLA-ATRT-04, CHLA-ATRT-05, and CHLA-ATRT-06 cells after gemcitabine treatment (0, 10, and 20 nM) for 24 h.
(H) Volcano plot depicting −log10 FDR-corrected p value of differential expressed genes between DMSO (n = 6) and gemcitabine-treated (n = 6) ATRT cell cultures VUMC-ATRT-01, VUMC-ATRT-03, CHLA-02, CHLA-05, CHLA-06, and CHLA-266. All 69 genes of the “KEGG p53_signaling_pathway” are highlighted in red.
Gemcitabine-induced SIRT1 depletion causes p53-mediated cell death in ATRT
To study the role of SIRT1 and p53 in ATRT cells, we engineered VUMC-ATRT-01 and VUMC-ATRT-03 cells in which SIRT1 is downregulated by stable short hairpin RNA (shRNA) expression (Figure 4A) and used CRISPR-Cas9 to make stable p53 knockouts of these models (Figure 4C). The SIRT1 knockdown in ATRT cells did not show loss of cell viability in culture, but when treated with gemcitabine, we observed a 2- to 10-fold increased sensitivity compared to wild-type ATRT (Figure 4B). Adversely, TP53 knockout in ATRT cells caused a complete loss of sensitivity to gemcitabine (Figure 4D), demonstrating the pivotal role of p53 in gemcitabine toxicity in ATRT (Figure 4E).
Figure 4.

Gemcitabine treatment induces p53-mediated cell death, through SIRT1 depletion in ATRT
(A) Western blot analysis depicting SIRT1 expression in VUMC-ATRT-01 and VUMC-ATRT-03 cells transduced with a variety of SIRT1 shRNA (Sh#1, 2, and 3) or a control construct (wild type [WT]).
(B) IC50 viability curves of VUMC-ATRT-01 and VUMC-ATRT-03 cells transduced with control (scrambled) or one of three SIRT1 shRNA treated with different gemcitabine concentrations for 96 h.
(C) Western blot analysis depicting p53 expression in VUMC-ATRT-01 WT and p53 knockout cells (KO1 and KO2), as established through CRISPR-Cas9.
(D) IC50 viability curves of CRISPR-Cas9-modified VUMC-ATRT-01 WT, scrambled, and p53 knockout cells, treated with different gemcitabine concentration for 96 h.
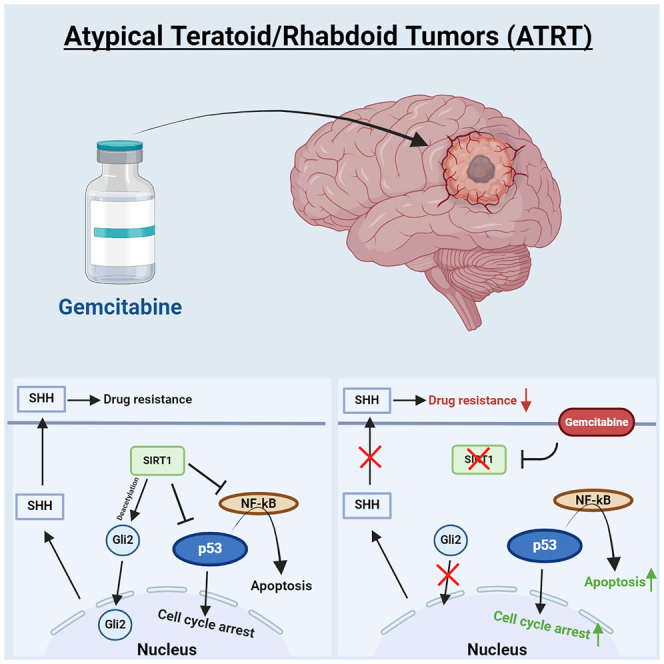
(E) Illustration of the proposed mechanism through which gemcitabine causes tumor toxicity in ATRT.
Gemcitabine blocks SHH activity through the loss of GLI2 nuclear localization
As described earlier, we observed differential gemcitabine sensitivity between the SHH-ATRT and MYC-ATRT models and found downregulation of SHH pathway components upon gemcitabine treatment. Therefore, we investigated differential mRNA expression between SHH-ATRT and non-SHH-ATRT in a publicly available expression dataset consisting of 49 ATRT samples (Figure 5A). As anticipated, we identified SIRT1 as being significantly upregulated in SHH- compared to non-SHH-ATRT (Figure 5B). No significant differences in SIRT1 expression were observed between SHH-1A and SHH-2 groups, although data are limited (Figure S8). Besides its role as a p53 suppressor, SIRT1 is also known to be an activator of GLI2, the hallmark protein for SHH signaling activity, through GLI deacetylation, which allows for nuclear translocation.22 As such, we hypothesized that gemcitabine-induced loss of SIRT1 would inhibit GLI2 deacetylation, thus preventing nuclear localization of GLI2. Therefore, we performed GLI2 immunofluorescence stainings in VUMC-ATRT-01 and VUMC-ATRT-03 cells, before and after gemcitabine treatment, and found that this treatment causes a significant loss of GLI2 nuclear localization, indicating lost SHH signaling activity (Figures 5C–5E). Furthermore, parametric analysis of gene set enrichment (PAGE) revealed negative regulation of expression of genes involved in SHH signaling in our cultures upon treatment with gemcitabine (Table S2). These results indicate that gemcitabine-induced loss of SIRT1 acts as a double-edged sword in SHH-ATRT through both activation of p53 and inhibition of SHH signaling activity (Figure 5F).
Figure 5.

Gemcitabine deactivates SHH signaling in SHH-ATRTs through loss of SIRT1, increasing SHH-ATRT-specific drug sensitivity
(A) t-SNE clustering of 49 individual tumor samples of patients with ATRT (dataset GSE: 70678) based on overall mRNA expression profiles (perplexity: 12). SHH subgroup is shown as a distinct group from other ATRT samples in upper-left corner as confirmed by overall high GLI2 expression (blue to red) versus low GLI2 expression in the non-SHH ATRT (yellow to green).
(B) mRNA expression levels of SIRT1 in SHH ATRT (n = 16) versus non-SHH ATRT (n = 33) (Wilcoxon rank-sum: p = 0.028).
(C) Immunofluorescent stainings of GLI2 (green), α-tubulin (red), and DAPI (blue) in VUMC-ATRT-03 cells treated with 20 nM gemcitabine for 24 h show the loss of nuclear localization of GLI2. Upper: an average depiction of all wells. Lower: zoomed depictions of the indicated area in the upper panels.
(D) Quantification of percentage nuclear (DAPI) overlap with GLI2 between DMSO and gemcitabine-treated VUMC-ATRT-01 (n = 20) and VUMC-ATRT-03 cells (n = 20) (one-way ANOVA: ∗∗∗∗p < 0.0001).
(E and F) Quantification of total GLI2-positive nuclei per well between DMSO and gemcitabine-treated VUMC-ATRT-01 (n = 20) and VUMC-ATRT-03 cells (n = 20) (one-way ANOVA: ∗∗∗∗p < 0.0001). (F) Illustration of the proposed mechanism through which gemcitabine causes tumor toxicity in ATRT, including the mechanisms that cause extra sensitivity in SHH-subgroup ATRT.
Gemcitabine treatment prolongs survival in ATRT xenograft models
To evaluate the therapeutic efficacy of gemcitabine in vivo, we tested this compound in athymic nude mice carrying patient-derived ATRT xenografts (VUMC-ATRT-03). Treatment (14 days) started 8 days after tumor transplantation, at which time f mice were stratified into two equivalent groups (control and gemcitabine) based on the average trend in body weight. No treatment-related toxicities were observed, and all mice developed ATRT, as determined by postmortem immunohistochemistry. We observed over 30% prolonged survival (p = 0.003) in the gemcitabine group compared to control mice, based on >20% weight loss or severe neurological symptoms as exclusion criteria (Figures 6A and S9A). Besides the frontotemporal ATRT xenograft model VUMC-ATRT-03, we also tested therapeutic efficacy of gemcitabine in an orthotopic xenograft of a cerebellar ATRT model (VUMC-ATRT-01). To this second in vivo trial, we added a doxorubicin group as positive treatment control. Here, we found a similar 30% prolonged survival (p = 0.0008) of gemcitabine-treated mice compared to both control and doxorubicin-treated mice (Figures 6B and S9B). Additionally, in both trials, we found one long-term survivor among gemcitabine-treated mice, which were sacrificed upon the determined endpoint of each trial (day 120 and 180, respectively). To evaluate the hypothesized underlying mechanisms of gemcitabine efficacy, we collected brains from five pre-selected mice at the end of their treatment cycles on which we conducted immunohistochemistry. In line with our in vitro data, we found that gemcitabine treatment activates p53 (Figure 6C), while causing a loss of SIRT1, concordant with our previous findings (Figure 6D). Immunohistochemistry also confirmed that all mice, including both long-term survivors, developed ATRT.
Figure 6.

Gemcitabine treatment reveals prolonged survival in two SHH-ATRT xenograft models
(A) Survival analysis of VUMC-ATRT-03 orthotopic xenograft-bearing mice treated with vehicle (black line, n = 8) and gemcitabine (red line, n = 8). The gemcitabine-treated group shows significant survival benefit over vehicle-treated mice (p = 0.003, log rank test).
(B) Survival analysis of VUMC-ATRT-01 orthotopic xenograft-bearing mice treated with vehicle (black line, n = 9), doxorubicin (blue line, n = 7), and gemcitabine (red line, n = 10). The gemcitabine-treated group shows significant benefit over vehicle and doxorubicin-treated mice (p = 0.0008, log rank test).
(C) p53 immunohistochemical staining (in brown) in VUM-ATRT-01 xenograft patches, isolated at final day of treatment. Gemcitabine-treated mice show a higher ratio of p53-positive nuclei in their tumors compared to vehicle-treated animals.
(D) SIRT1 immunohistochemical staining (in brown) in VUM-ATRT-01 xenograft patches, isolated at final day of treatment. Gemcitabine-treated mice show lower SIRT1 expression compared to vehicle-treated animals.
Discussion
In the current study, we show that gemcitabine selectively acts as a potent chemotherapeutic agent in ATRT compared to non-ATRT in vitro and in vivo pediatric brain tumor models. From expression data, we observed that this sensitivity correlates to the expression of SIRT1 mRNA, which is thought to act as a promoter of tumorigenesis and drug resistance by hypermethylating the promoter regions of several tumor suppressor genes.23,24,25,26
Gemcitabine is a clinically well-known chemotherapeutic that was approved for medical use in 1995 and was even added to the World Health Organization’s List of Essential Medicines.27 Several subsequent studies investigated the brain penetration of gemcitabine and found that gemcitabine does cross the BBB, albeit to a limited extend. However, the gemcitabine concentrations found in glioblastoma biopsies, taken from patients treated with a clinically relevant dose of 1,000 mg/m2, exceeded the lethal gemcitabine dose for our SHH-ATRT in vitro models.28 In pediatric patients, weekly gemcitabine doses up to 2,100 mg/m2 are well tolerated, suggesting that relevant gemcitabine brain concentrations in these patients are feasible.29 Additionally, in a previous study, we found that ATRTs share significant physiological BBB deficiencies and that even non-BBB-penetrable drugs can penetrate ATRT in xenograft models.30 As such, we do expect that gemcitabine penetrates the brain and can be used for potential clinical treatment of ATRT.
The tumor suppressor p53 is considered the main inducer of cell-cycle arrest and apoptosis, and the vast majority of cancers harbor either a TP53 mutation or have lost the ability to induce p53. In this study, we show that gemcitabine mainly exerts its cytotoxic effect on ATRT cells through activation of p53. These results indicate that gemcitabine would only be effective in p53 wild-type tumors, which underlines the clinical relevance of this study, since ATRTs rarely carry TP53 mutations.12 Furthermore, we show that the p53 inhibitor SIRT1 is overexpressed in ATRT compared to other CNS tumors, which might explain why p53 is often suppressed in ATRT, contributing to its highly malignant behavior.
In correlation with p53, we found that NF-κB is activated upon gemcitabine treatment in our ATRT models, and we suggest that this activation works synergistically with p53 to induce apoptosis. For decades, there has been discussion on the pro-apoptotic versus the anti-apoptotic roles of NF-κB signaling. Despite contradictory studies that have shown both anti- and pro-apoptotic functioning of NF-κB activity, even within the same cell lines or cancers, today there is an overall consensus that NF-κB is a context-dependent apoptosis regulator.19,31,32 As a modulator of apoptosis, excessive NF-κB activation has been described to contribute to p53-regulated apoptosis in several cancers, corresponding to our observations in this study.33
Since some other aggressive tumors like acute myeloid leukemia, colon cancer, and pancreatic cancer also overexpress SIRT1, gemcitabine treatment may also be applicable to those tumor types. This is confirmed by the observation that a direct link between SIRT1 expression and gemcitabine treatment efficacy has been found in pancreatic cancers.34,35,36 These thorough and exclusive studies show that pancreatic ductal adenocarcinoma rely on high SIRT1 expression for their proliferation and migration capacities and that the inhibition of SIRT1 directly sensitizes these tumors to gemcitabine treatment.
Interestingly, like SHH-ATRT, pancreatic cancers often are characterized by highly active hedgehog signaling.36,37 Several recent studies show that gemcitabine resistance in pancreatic tumors is the result of reactivation of SHH signaling, which induces stemcellness and embryonal development.38,39 These results support our findings that show how loss of SHH signaling in SHH-subtype ATRT causes strong gemcitabine sensitivity.
In this study, systemic treatment of murine ATRT orthotopic xenograft models with gemcitabine prolonged survival significantly. As such, in the second in vivo trial, we compared the efficacy of monotherapy gemcitabine with that of doxorubicin, a currently used chemotherapeutic agent in ATRT treatment regimens.2,4 Our results show that gemcitabine monotherapy causes a significantly stronger antitumoral effect in ATRT than doxorubicin, without causing any adverse effects in vivo, which alludes to the potential of gemcitabine as part of multimodal therapy in ATRT.
Several studies showed that gemcitabine can cause significant immunomodulatory effects on the tumor microenvironment.40,41,42 Therefore, the therapeutic response of gemcitabine may be different in immune-competent in vivo models. Unfortunately, there is still a lack of immunocompetent ATRT syngeneic mouse models, especially those that do not harbor a TP53 mutation. Even though gemcitabine is not known to cause adverse immune-associated symptoms in patients—and this manuscript aims to support rapid translation of gemcitabine to patients with ATRT—future studies with gemcitabine in immunocompetent syngeneic models may help to improve its efficacy and identify synergistic gemcitabine strategies with immunotherapies, as has recently been found for other malignancies.43,44
In conclusion, we demonstrate that ATRTs are highly sensitive to the clinically registered chemotherapeutic gemcitabine. We found that gemcitabine treatment hampers ATRT-specific SIRT1 overexpression, causing p53 activation and cell death in these tumors. Additionally, we demonstrate that gemcitabine sensitivity is more pronounced in SHH-subtype ATRT, supposedly because of the GLI-activating role of SIRT1, which is diminished upon gemcitabine treatment. Subsequently, we demonstrate that gemcitabine significantly prolongs the survival of mice with orthotopic ATRT xenografts compared to both control and conventional doxorubicin treatment. Overall, gemcitabine treatment may offer an easily translatable opportunity to improve outcomes for patients with ATRT, for whom effective treatment options are still scarce.
Limitations of the study
In this study, we did not include any TYR-subtype ATRT cell cultures, as these are notoriously difficult to grow. Thus, while ample TYR-ATRT samples are present in the presented expression datasets, gemcitabine drug sensitivity may be different in TYR-ATRT cells in vitro. Furthermore, the presented mechanistic rationale for SIRT1-mediated gemcitabine sensitivity has not yet been validated in vivo due to limited funds and the translational scope if this study. Finally, this study does not investigate the impact of gemcitabine on the tumor immune-microenvironment, while there is strong interest from the scientific community to use immunocompetent tumor models for drug intervention studies. Due to the absence of clear driver mutations, we are still awaiting access to validated immunocompetent syngeneic ATRT mouse models.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse anti- SIRT1 | Abcam | Cat# ab110304, RRID:AB_10864359 |
| Rabbit anti- NF-kB p65 (D14E12) | Cell Signaling Technology | Cat# 8242, RRID:AB_10859369 |
| Rabbit anti- phospho-NF-kB p65 (Ser536) | Cell Signaling Technology | Cat# 3033, RRID:AB_331284 |
| Mouse anti- beta Actin (C4) | Millipore | Cat# MAB1501, RRID:AB_2223041 |
| IRDye® 680RD Goat anti-Mouse IgG | LI-COR Biosciences | Cat# 926–68070, RRID:AB_10956588 |
| IRDye® 800CV Goat anti- rabbit IgG | LI-COR Biosciences | Cat# 926–32211, RRID:AB_621843 |
| Mouse anti- BAF47/SMARCB1 | BD Biosciences | Cat# 612111, RRID: AB_399481 |
| Rabbit anti- Ki-67 | Abcam | Cat# ab15580, RRID:AB_443209 |
| Mouse anti- GFAP | BioTrend | Cat# BT46-5002-80, RRID:AB_2278911 |
| Rabbit anti- S100 | DakoCytomation | Cat# Z0311, RRID:AB_10013383 |
| Rat anti- BCRP | Abcam | Cat# ab24115, RRID:AB_447879 |
| Rabbit anti- ABCB1 (P-gp) | Cell Signaling Technology | Cat# 13978, RRID:AB_2798357 |
| Rabbit anti- CD31 | Thermo Fisher Scientific | Cat# PA5-16301, RRID:AB_10981955 |
| Mouse anti- p53 (D07) | DAKO/Agilent | Cat# M7001, RRID:AB_2206626 |
| Rabbit anti- GLUT-1 | Millipore | Cat# 07–1401, RRID:AB_1587074 |
| Rabbi anti- Gli2 (9HCLC) | Abcam | Cat# ab277800, RRID:AB_449559 |
| Mouse anti- α-Tubulin | Sigma-Aldrich | Cat# T9026, RRID:AB_477593 |
| Alexa Fluor 488 Goat anti- rabbit IgG | Thermo Fisher Scientific | Cat# A-11008, RRID:AB_143165 |
| Alexa Fluor 568 Goat anti- mouse IgG | Thermo Fisher Scientific | Cat# A-11004, RRID:AB_2534072 |
| Bacterial and virus strains | ||
| NEB® 5-alpha Competent E. coli | New England Biolabs | C2987H |
| pRSV-Rev lentiviral packaging plasmid (3rd gen) | Dull et al.45 | Addgene: #12253 |
| pMDLg/pRRE lentiviral packaging plasmid (3rd gen) | Dull et al.45 | Addgene: #12251 |
| pMD2.G lentiviral envelope plasmid (3rd gen) | Dull et al.45 | Addgene: #12259 |
| Biological samples | ||
| ATRT tissues obtained through surgical resection | Pathology dept. Amsterdam UMC/Netherlands Brain bank | Protcol: METC VUmc 2009/237 |
| Chemicals, peptides, and recombinant proteins | ||
| Epigenetic Screening Library | Cayman Chemical Company | #11076 |
| Gemcitabine (CAS: 95058-81-4) | MedChemExpress LLC | HY-17026 |
| Doxorubicin (CAS: 23214-92-8) | MedChemExpress LLC | HY-15142 |
| Critical commercial assays | ||
| CellTiter-Glo® 3D Luminescent Cell Viability Assay | Promega | #G9683 |
| GeneJET Gel Extraction Kit | Thermo Fisher Scientific | #K0691 |
| Deposited data | ||
| Raw RNA-seq data belonging to the 'Gemcitabine therapeutically disrupts essential SIRT1-mediated p53 repression in Atypical Teratoid/Rhabdoid Tumors' manuscript. | Mendeley Data | https://doi.org/10.17632/gfx5dwt2tg.1 |
| Experimental models: Cell lines | ||
| HEK293T/17 | ATCC | RRID:CVCL_1926 |
| CHLA-ATRT-02 | ATCC | RRID:CVCL_B045 |
| CHLA-ATRT-04 | ATCC | RRID:CVCL_0F38 |
| CHLA-ATRT-05 | ATCC | RRID:CVCL_AQ41 |
| CHLA-ATRT-06 | ATCC | RRID:CVCL_AQ42 |
| CHLA-ATRT-266 | Gift from Dr Alonso | RRID:CVCL_M149 |
| VUMC-ATRT-01 | Own lab | N/A |
| VUMC-ATRT-03 | Own lab | N/A |
| VUMC-DIPG-A | Own lab | RRID:CVCL_IT43 |
| VUMC-DIPG-F | Own lab | N/A |
| VUMC-DIPG-10 | Own lab | RRID:CVCL_IT43 |
| VUMC-DIPG-11 | Own lab | N/A |
| VUMC-HGG-09 | Own lab | N/A |
| JHH-DIPG-01 | Gift from Dr Raabe | RRID:CVCL_IT47 |
| HSJD-DIPG-07 | Gift from Dr Montero Carcaboso | RRID:CVCL_VU70 |
| SU-pcGBM-02 | Gift from Dr Monje | RRID:CVCL_IT42 |
| Experimental models: Organisms/strains | ||
| Athymic nude mice (Crl:NU(NCr)-Foxn1nu BALB/c outbred) | Envigo | #069 |
| Oligonucleotides | ||
| shSIRT1 #1: GCAAAGCCTTTCTGAATCTAT in pLKO.1 vector | GE Healthcare | N/A |
| shSIRT1 #2: CCTCGAACAATTCTTAAAGAT in pLKO.1 vector | GE Healthcare | N/A |
| shSIRT1 #3: GCGGGAATCCAAAGGATAATT in pLKO.1 vector | GE Healthcare | N/A |
| TP53 exon4 FWD primer: caccGAAGGGACAGAAGATGACAG | This paper | N/A |
| TP53 exon4 REV primer: aaacCTGTCATCTTCTGTCCCTTC | This paper | N/A |
| hSpCas9 U6 Seq FWD: GAGGGCCTATTTCCCATGATTCC | This paper | N/A |
| Recombinant DNA | ||
| LentiCRISPR v2 vector | Addgene | #52961 |
| pLKO.1 vector | Addgene | #136035 |
| Software and algorithms | ||
| R2: Genomics Analysis and Visualization Platform | Established by Jan Koster | https://hgserver2.amc.nl/ |
| MARS Data Analysis Software | BMG LABTECH | ID: # 81306 |
| LAS X Life Science Microscope Software Platform | Leica | https://www.leica-microsystems.com |
| RStudio: Integrated Development for R | RStudio, PBC, | http://www.rstudio.com/ |
| Image Studio Lite v5.7 | Li-Cor | https://www.licor.com/bio/image-studio-lite/ |
| GraphPad Prism 10.0.0 | GraphPad Software | www.graphpad.com |
| ImageJ (FIJI) | ImageJ Software | https://doi.org/10.1038/nmeth.2019 |
| Other | ||
| Tecan D300e picoliter dispenser | Tecan Group | N/A |
| Cell-repellent 96-well F-bottom plates | Greiner Bio-one | #650971 |
| SCREENSTAR® 96-well plates | Greiner Bio-One | #655-866 |
| Illumina Nextseq 500 sequencing system | Illumina | N/A |
| LI-COR© Odyssey xF fluorescent imager model 9120 | Surplus Solutions | N/A |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, dr. Esther Hulleman (e.hulleman@prinsesmaximacentrum.nl).
Materials availability
VUMC-ATRT-03 cells (SHH-1B ATRT) generated in this study are available from the lead contact with a completed Materials Transfer Agreement.
Data and code availability
-
•
Raw and analyzed RNA-seq data are publicly available at Mendeley Data Repository under DOI number: https://doi.org/10.17632/gfx5dwt2tg.1.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
Experimental model and subject details
Patient material
Tumor tissue was obtained through surgical resection from an 11-year old patient that underwent surgery in the Amsterdam University Medical Center (Amsterdam, the Netherlands) for a tumor of which the clinical pathological diagnosis was ATRT. All patient material was collected according to national and institutional guidelines (Research Ethics Committee approval: #METC VUmc 2009/237) and in accordance with the declaration of Helsinki and put into culture as described previously.46
Cell cultures
CHLA-ATRT-02 (SHH-ATRT), CHLA-ATRT-04 (SHH-ATRT), CHLA-ATRT-05 (SHH-ATRT) and CHLA-ATRT-06 (MYC-ATRT) cultures were obtained from the American Type Culture Collection (ATCC). VUMC-ATRT-01 (SHH-ATRT)30 and VUMC-ATRT-03 (SHH-ATRT) were established from tumor tissue obtained by surgical resection at the Amsterdam UMC (Amsterdam, the Netherlands) as described previously.46 CHLA-ATRT-266 (MYC-ATRT) was a kind gift from Dr Alonso (University of Navarra, Pamplona, Spain). Using Sanger sequencing and expression analysis, no TP53 mutations were observed in any of the ATRT cultures.
Non-ATRT pediatric glioma models VUMC-DIPG-A, VUMC-DIPG-F, VUMC-DIPG-10, VUMC-DIPG-11, and VUMC-HGG-09, were established through autopsy or resection material and characterized as described previously.46 Non-ATRT models JHH-DIPG-01, HSJD-DIPG-07, and SU-pcGBM-02 were kind gifts from Dr Raabe (Johns Hopkins Hospital, Baltimore, USA), Dr Montero Carcaboso (Hospital San Joan de Déu, Barcelona, Spain) and Dr Monje (Stanford University, Stanford, USA), respectively.
All cells were cultured as neurospheres as described previously.46,47,48 Cells were only used when confirmed mycoplasm negative and short-tandem repeat (STR) analysis was used to ensure cell line identities.
Mice
Both xenograft modeling and in vivo efficacy studies were performed in 4-week old female athymic nude mice (BALB/c outbred background, Envigo). Animals were provided food and water ad libitum for the entire duration of the experiments.
Animal ethics statement
All animal experiments were approved by local and governmental animal experimental committees and carried out according to national and institutional guidelines (national project permit: AVD114002017841) (protocol 0841-NCH17-01A1 and 0841-NCH20-13 animal welfare committee, Vrije Universiteit, Amsterdam).
Method details
Human mRNA expression datasets
Expression datasets that were used in this study to compare mRNA between patients were the following: Healthy brain (GSE: 11882),49 healthy cerebellum (GSE: 3526),50 healthy various (GSE: 7307, Human body index project; accession code: PRJNA98081), ATRT (GSE: 70678),7 glioblastoma (GSE: 7696),51 glioma (GSE: 16011),52 pediatric DIPG (GSE: 26576),53 pediatric glioma (GSE: 19578).54
Chemicals
The epigenetic compound library (#11076) was purchased from Cayman Chemical Company (Ann Arbor, Michigan, USA). Gemcitabine (CAS: 95058-81-4) and doxorubicin (CAS: 23214-92-8) were purchased from MedChemExpress LLC (Monmouth Junction, New Jersey, USA). For in vitro studies, all chemicals were dissolved in DMSO and stored as 10mM stock concentration at −20°C. For in vivo studies, both doxorubicin and gemcitabine where freshly dissolved in 0.9% saline before infusion.
Compound screening and cell viability assays
For compound screening assays, cells were plated in TSM-medium at a density of 5000 cells/well in cell-repellent 96-well F-bottom plates (#650971, Greiner Bio-one, Kremsmünster, Austria). Compounds were dispersed 24h after cell seeding, using a Tecan D300e picoliter dispenser (Tecan Group Ltd, Switzerland) and incubated at 37°C and 5% CO2 for 96h. The number of viable cells was measured using CellTiter-Glo 3D Luminescent Cell Viability Assay (#G9683, Promega, Madison, USA) according to the manufacturers protocol. The concordant luminescence was measured using a Tecan Infinite 200 reader using iControl 1.10 software.
Methylation profiling
DNA methylation arrays and copy number profiles were performed and analyzed using the Heidelberg classifier, as described previously.7
RNA-sequencing
For the first batch, VUMC-ATRT-03, VUMC-DIPG-10, VUMC-DIPG-11, VUMC-HGG-09, JHH-DIPG-01, HSJD-DIPG-07, and SU-pcGBM-2 neurospheres were treated with 10nM gemcitabine or DMSO as control. For the second batch, VUMC-ATRT-01, VUMC-ATRT-03, CHLA-02, CHLA-05, CHLA-06, and CHLA-266 neurospheres were treated with 10nM gemcitabine or DMSO as control. After 24h, cells were collected and processed as described previously.48 Sequencing, performed on an Illumina Nextseq 500 sequencer, data processing in the R2 platform (R2.amc.nl), and statistical analysis were also assessed as previously described.48,55
Western blotting
Immunoblotting was performed as described previously.56 Protein isolation was conducted using RIPA lysis buffer supplemented with protease and phosphatase inhibitors. Membranes were incubated with mouse anti-SIRT1 (1:1000, #ab110304, Abcam, Cambridge, UK), rabbit anti-NF-kB p65 (D14E12) (1:1000, #8242, Cell Signaling Technology, Danvers, Massachusetts, USA), Rabbit anti-phospho-NF-kB p65 (Ser536, 93H1) (1:2000, #3033. Cell Signaling Technology), mouse anti-p53 (Clone DO-7) (1:1000, #M7001, DAKO, Agilent, Santa Clara, California, USA) and mouse anti-beta Actin (Clone C4) (1:10.000, MAB1501, Millipore, Burlington, Massachusetts, USA). Subsequently, membranes were incubated with secondary goat anti-mouse IRDye©680RD antibody (1:10.000, LI-COR©, Lincoln, NA, USA) and/or goat anti-rabbit IRDye©800CV antibody (1:10.000, LI-COR©). Signals were detected using an LI-COR© Odyssey fluorescent imager (model 9120, Surplus Solutions, LLC).
Immunohistochemistry and immunofluorescent imaging
For immunohistochemistry, fresh tissues were fixed in 4% PFA for 48h, followed by paraffin embedding. Embedded tissues were cut into 4.5μm coronal sections and stained for SMARCB1 (1:100; 612111, BD Biosciences), Ki-67 (1:3000; ab15580, Abcam), glial fibrillary acidic protein (GFAP) (1:500; BT46-5002–04, BioTrend), S100 (1:1000; Z0311, DakoCytomation), human vimentin (1:4000; M0725, DakoCytomation), breast cancer resistance protein (BCRP) (1:1000; ab24115, Abcam), permeability glycoprotein (P-gp) (1:1000, #13978, Cell Signaling Technologies), CD31 (1:200, #PA5-16301, Thermo Fisher Scientific), p53 (1:500, Clone D07, M7001, DAKO), NF-kB (1:500, #D14E12, Cell Signaling technology), Sirtuin 1 (SIRT1) (1:100, #ab110304, Abcam, Cambridge, UK), glucose transporter 1 (GLUT1) (1:100–2000; 07–1401, Millipore) and hematoxylin for background contrast. Primary antibody detection was achieved through secondary peroxidase/DAB+ staining using the Dako REAL EnVision Detection System (Agilent, DAKO, #K5007).
For immunofluorescence, VUMC-ATRT-01 and VUMC-ATRT-03 cells were cultured in TSM supplemented with FCS for 12h, to ensure adherence, in Greiner SCREENSTAR 96-well plates (#655–866) specialized for fluorescent imaging. Cells were treated with gemcitabine for 24h, fixed and stained for GLI2 (1:200, Abcam, #ab277800) and α-Tubulin (1:500, Merck & Co. Inc., #T9026) as previously described.48 As secondary step Alexa Fluor 488 (goat anti-rabbit, 1:10,000, Invitrogen, #A-11008) and Alexa Fluor 568 (goat anti-mouse, 1:10,000, Invitrogen, #A-11004) were incubated for 1h at RT. Imaging was performed using a Leica DMi8 inverted microscope (Leica Camera AG, Wetzlar, Germany), including automated stage and 3D Thunder deconvolution, using a 20× dry objective equipped with a Leica DFC700GT camera. 10 wells per condition were scanned and imaged were deconvoluted and stitched by LASX (Leica). Subsequently, images were analyzed in ImageJ (FIJI): DAPI and Gli2 positive areas were mapped using the Threshold and ROI selection and Watershed functions. Subsequently, ROI regions that overlap were quantified within ROI manager.
Establishment of stable shRNA expressing cells
SIRT1 knockdown cells were established using the pLKO.1-shSIRT1.1, pLKO.1-shSIRT1.2, and pLKO.1-shSIRT1.3 (GE Healthcare, Chicago, USA) plasmids as described previously.47 pLKO.1-Scrambled plasmids (Addgene, #136035) were used as negative control. shRNA sequences are described in Table S3.
Establishment of stable TP53 knockout cells
VUMC-ATRT-01 cells were transduced (lentiviral) with a LentiCRISPR v2 plasmid (#52961, Addgene) that was cloned with a p53-sgRNA (Table S4) according to the applied protocol (#rev20140208, Zhang Lab). Using puromycin (2.0μg/mL) selection for 7 days, successfully transduced cells were selected. Knockout efficiency and location of the indel were confirmed through sanger sequencing, using TIDE analysis (tide.nki.nl) (Figure S10). Loss of p53 protein was assessed through western blotting.
Xenograft modeling and in vivo efficacy studies
For modeling, mice were stereotactically injected with 500.000 patient-derived VUMC-ATRT-03 cells into the frontotemporal region of the left cerebral hemisphere (from bregma: X(-2), Y(0.5), Z(-3)mm), concordant with the original location of the tumor. Cells were injected in an injection volume of 5 μL at a flow rate of 2.5 μL/min to minimize the neurological side effects of the procedure, as well as potential backflow of cells. Mice were euthanized upon developing severe neurological symptoms or >20% weight loss. Brains were fixed in 4% PFA for 48h for immunohistochemistry, or tumor cells were harvested and put in culture to establish VUMC-ATRT-03 mose passage cultures. For in vivo efficacy studies, mice were stereotactically injected (as mentioned above) with 500.000 VUMC-ATRT-01 cells into the cerebellum (from lambda: X(-1), Y(-2.3), Z(-2.3)mm) or with 500.000 VUMC-ATRT-03 cells into the frontotemporal region of the left cerebral hemisphere, concordant with the original location in the patients. After eight days, mice were randomized into groups based on average body weight and received gemcitabine (200 mg/kg), doxorubicin (20 mg/kg) or 0.9% saline once per week for three weeks via I.P. injection. All animals developed tumors as confirmed by immunohistochemistry. Humane endpoints were set as either >20% weight loss or severe neurological symptoms.
Quantification and statistical analysis
mRNA expression between groups from RNA-sequencing data was assessed using the one-way analysis of variance (ANOVA). Also quantification of ROI overlap between groups in immunofluorescent analyses was assessed using one-way ANOVA. In vitro cell survival percentages were compared using the independent t-test (two-sided). In vivo survival differences between groups were tested using the log rank (Mantel-Cox) test. The statistical analyzes were performed using GraphPad Prism (version 6). A p-value <0.05 was considered statistically significant.
Acknowledgments
All research described has been funded by the Children Cancer Free Foundation (KiKa) (https://www.kika.nl/), a non-profit organization that funds pediatric oncology research (KiKa projects 210 and 431). We thank Senna Visser for her kind technical and laboratory support.
Author contributions
D.S.M. and E.H. conceived and designed the project. D.S.M., M.H.M., J.R.G., M.C.d.G., and P. Waranecki developed and validated the in vitro and in vivo models used in the study. D.S.M., M.H.M., J.R.G., A.d.C., L.R., and P. Waranecki performed the functional in vitro and western blotting experiments. D.S.M., M.C.d.G., M. Breur, and M. Bugiani performed the immunohistochemistry experiments. D.S.M., M.H.M., and P. Waranecki designed and performed the in vivo trials. S.E.M.V.v.Z. performed and analyzed clinical imaging. P. Wesseling and N.G. acquired patient material and performed and validated methylation profiling. N.G. and J.K. provided bioinformatical and statistical expertise and support. M. Bugiani and P. Wesseling provided pathological expertise and support. A.R. and N.E.F. provided patient imaging data and clinical treatment results. G.J.L.K. and E.H. acquired funding and supervised the study. All authors contributed to writing the manuscript.
Declaration of interests
The authors declare no competing interests.
Published: August 28, 2024
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.xcrm.2024.101700.
Supplemental information
References
- 1.Reddy A.T., Strother D.R., Judkins A.R., Burger P.C., Pollack I.F., Krailo M.D., Buxton A.B., Williams-Hughes C., Fouladi M., Mahajan A., et al. Efficacy of High-Dose Chemotherapy and Three-Dimensional Conformal Radiation for Atypical Teratoid/Rhabdoid Tumor: A Report From the Children's Oncology Group Trial ACNS0333. J. Clin. Oncol. 2020;38:1175–1185. doi: 10.1200/jco.19.01776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chi S.N., Zimmerman M.A., Yao X., Cohen K.J., Burger P., Biegel J.A., Rorke-Adams L.B., Fisher M.J., Janss A., Mazewski C., et al. Intensive multimodality treatment for children with newly diagnosed CNS atypical teratoid rhabdoid tumor. J. Clin. Oncol. 2009;27:385–389. doi: 10.1200/jco.2008.18.7724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ginn K.F., Gajjar A. Atypical teratoid rhabdoid tumor: current therapy and future directions. Front. Oncol. 2012;2:114. doi: 10.3389/fonc.2012.00114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Slavc I., Chocholous M., Leiss U., Haberler C., Peyrl A., Azizi A.A., Dieckmann K., Woehrer A., Peters C., Widhalm G., et al. Atypical teratoid rhabdoid tumor: improved long-term survival with an intensive multimodal therapy and delayed radiotherapy. The Medical University of Vienna Experience 1992-2012. Cancer Med. 2014;3:91–100. doi: 10.1002/cam4.161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Frühwald M.C., Hasselblatt M., Nemes K., Bens S., Steinbügl M., Johann P.D., Kerl K., Hauser P., Quiroga E., Solano-Paez P., et al. Age and DNA methylation subgroup as potential independent risk factors for treatment stratification in children with atypical teratoid/rhabdoid tumors. Neuro Oncol. 2020;22:1006–1017. doi: 10.1093/neuonc/noz244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hasselblatt M., Gesk S., Oyen F., Rossi S., Viscardi E., Giangaspero F., Giannini C., Judkins A.R., Frühwald M.C., Obser T., et al. Nonsense mutation and inactivation of SMARCA4 (BRG1) in an atypical teratoid/rhabdoid tumor showing retained SMARCB1 (INI1) expression. Am. J. Surg. Pathol. 2011;35:933–935. doi: 10.1097/PAS.0b013e3182196a39. [DOI] [PubMed] [Google Scholar]
- 7.Johann P.D., Erkek S., Zapatka M., Kerl K., Buchhalter I., Hovestadt V., Jones D.T.W., Sturm D., Hermann C., Segura Wang M., et al. Atypical Teratoid/Rhabdoid Tumors Are Comprised of Three Epigenetic Subgroups with Distinct Enhancer Landscapes. Cancer Cell. 2016;29:379–393. doi: 10.1016/j.ccell.2016.02.001. [DOI] [PubMed] [Google Scholar]
- 8.Torchia J., Golbourn B., Feng S., Ho K.C., Sin-Chan P., Vasiljevic A., Norman J.D., Guilhamon P., Garzia L., Agamez N.R., et al. Integrated (epi)-Genomic Analyses Identify Subgroup-Specific Therapeutic Targets in CNS Rhabdoid Tumors. Cancer Cell. 2016;30:891–908. doi: 10.1016/j.ccell.2016.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Federico A., Thomas C., Miskiewicz K., Woltering N., Zin F., Nemes K., Bison B., Johann P.D., Hawes D., Bens S., et al. ATRT-SHH comprises three molecular subgroups with characteristic clinical and histopathological features and prognostic significance. Acta Neuropathol. 2022;143:697–711. doi: 10.1007/s00401-022-02424-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ho B., Johann P.D., Grabovska Y., De Dieu Andrianteranagna M.J., Yao F., Frühwald M., Hasselblatt M., Bourdeaut F., Williamson D., Huang A., Kool M. Molecular subgrouping of atypical teratoid/rhabdoid tumors-a reinvestigation and current consensus. Neuro Oncol. 2020;22:613–624. doi: 10.1093/neuonc/noz235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Abro B., Kaushal M., Chen L., Wu R., Dehner L.P., Pfeifer J.D., He M. Tumor mutation burden, DNA mismatch repair status and checkpoint immunotherapy markers in primary and relapsed malignant rhabdoid tumors. Pathol. Res. Pract. 2019;215:152395. doi: 10.1016/j.prp.2019.03.023. [DOI] [PubMed] [Google Scholar]
- 12.Gröbner S.N., Worst B.C., Weischenfeldt J., Buchhalter I., Kleinheinz K., Rudneva V.A., Johann P.D., Balasubramanian G.P., Segura-Wang M., Brabetz S., et al. The landscape of genomic alterations across childhood cancers. Nature. 2018;555:321–327. doi: 10.1038/nature25480. [DOI] [PubMed] [Google Scholar]
- 13.de Sousa Cavalcante L., Monteiro G. Gemcitabine: metabolism and molecular mechanisms of action, sensitivity and chemoresistance in pancreatic cancer. Eur. J. Pharmacol. 2014;741:8–16. doi: 10.1016/j.ejphar.2014.07.041. [DOI] [PubMed] [Google Scholar]
- 14.Yi J., Luo J. SIRT1 and p53, effect on cancer, senescence and beyond. Biochim. Biophys. Acta. 2010;1804:1684–1689. doi: 10.1016/j.bbapap.2010.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Solomon J.M., Pasupuleti R., Xu L., McDonagh T., Curtis R., DiStefano P.S., Huber L.J. Inhibition of SIRT1 catalytic activity increases p53 acetylation but does not alter cell survival following DNA damage. Mol. Cell Biol. 2006;26:28–38. doi: 10.1128/mcb.26.1.28-38.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kauppinen A., Suuronen T., Ojala J., Kaarniranta K., Salminen A. Antagonistic crosstalk between NF-κB and SIRT1 in the regulation of inflammation and metabolic disorders. Cell. Signal. 2013;25:1939–1948. doi: 10.1016/j.cellsig.2013.06.007. [DOI] [PubMed] [Google Scholar]
- 17.Vaziri H., Dessain S.K., Ng Eaton E., Imai S.I., Frye R.A., Pandita T.K., Guarente L., Weinberg R.A. hSIR2(SIRT1) functions as an NAD-dependent p53 deacetylase. Cell. 2001;107:149–159. doi: 10.1016/s0092-8674(01)00527-x. [DOI] [PubMed] [Google Scholar]
- 18.Webster G.A., Perkins N.D. Transcriptional cross talk between NF-kappaB and p53. Mol. Cell Biol. 1999;19:3485–3495. doi: 10.1128/mcb.19.5.3485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lin B., Williams-Skipp C., Tao Y., Schleicher M.S., Cano L.L., Duke R.C., Scheinman R.I. NF-kappaB functions as both a proapoptotic and antiapoptotic regulatory factor within a single cell type. Cell Death Differ. 1999;6:570–582. doi: 10.1038/sj.cdd.4400528. [DOI] [PubMed] [Google Scholar]
- 20.Lin Z., Fang D. The Roles of SIRT1 in Cancer. Genes Cancer. 2013;4:97–104. doi: 10.1177/1947601912475079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Carrà G., Lingua M.F., Maffeo B., Taulli R., Morotti A. P53 vs NF-κB: the role of nuclear factor-kappa B in the regulation of p53 activity and vice versa. Cell. Mol. Life Sci. 2020;77:4449–4458. doi: 10.1007/s00018-020-03524-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Xie Y., Liu J., Jiang H., Wang J., Li X., Wang J., Zhu S., Guo J., Li T., Zhong Y., et al. Proteasome inhibitor induced SIRT1 deacetylates GLI2 to enhance hedgehog signaling activity and drug resistance in multiple myeloma. Oncogene. 2020;39:922–934. doi: 10.1038/s41388-019-1037-6. [DOI] [PubMed] [Google Scholar]
- 23.Sasca D., Hähnel P.S., Szybinski J., Khawaja K., Kriege O., Pante S.V., Bullinger L., Strand S., Strand D., Theobald M., Kindler T. SIRT1 prevents genotoxic stress-induced p53 activation in acute myeloid leukemia. Blood. 2014;124:121–133. doi: 10.1182/blood-2013-11-538819. [DOI] [PubMed] [Google Scholar]
- 24.Jiang K., Lyu L., Shen Z., Zhang J., Zhang H., Dong J., Yan Y., Liu F., Wang S. Overexpression of SIRT1 is a poor prognostic factor for advanced colorectal cancer. Chin. Med. J. 2014;127:2021–2024. [PubMed] [Google Scholar]
- 25.Stenzinger A., Endris V., Klauschen F., Sinn B., Lorenz K., Warth A., Goeppert B., Ehemann V., Muckenhuber A., Kamphues C., et al. High SIRT1 expression is a negative prognosticator in pancreatic ductal adenocarcinoma. BMC Cancer. 2013;13:450. doi: 10.1186/1471-2407-13-450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang Z., Chen W. Emerging Roles of SIRT1 in Cancer Drug Resistance. Genes Cancer. 2013;4:82–90. doi: 10.1177/1947601912473826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.World Health Organization . World Health Organization; 2019. World Health Organization Model List of Essential Medicines: 21st List 2019.https://apps.who.int/iris/handle/10665/325771 [Google Scholar]
- 28.Sigmond J., Honeywell R.J., Postma T.J., Dirven C.M.F., de Lange S.M., van der Born K., Laan A.C., Baayen J.C.A., Van Groeningen C.J., Bergman A.M., et al. Gemcitabine uptake in glioblastoma multiforme: potential as a radiosensitizer. Ann. Oncol. 2009;20:182–187. doi: 10.1093/annonc/mdn543. [DOI] [PubMed] [Google Scholar]
- 29.Reid J.M., Qu W., Safgren S.L., Ames M.M., Krailo M.D., Seibel N.L., Kuttesch J., Holcenberg J. Phase I trial and pharmacokinetics of gemcitabine in children with advanced solid tumors. J. Clin. Oncol. 2004;22:2445–2451. doi: 10.1200/jco.2004.10.142. [DOI] [PubMed] [Google Scholar]
- 30.Meel M.H., Guillén Navarro M., de Gooijer M.C., Metselaar D.S., Waranecki P., Breur M., Lagerweij T., Wedekind L.E., Koster J., van de Wetering M.D., et al. MEK/MELK inhibition and blood-brain barrier deficiencies in atypical teratoid/rhabdoid tumors. Neuro Oncol. 2020;22:58–69. doi: 10.1093/neuonc/noz151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kaltschmidt B., Kaltschmidt C., Hofmann T.G., Hehner S.P., Dröge W., Schmitz M.L. The pro- or anti-apoptotic function of NF-kappaB is determined by the nature of the apoptotic stimulus. Eur. J. Biochem. 2000;267:3828–3835. doi: 10.1046/j.1432-1327.2000.01421.x. [DOI] [PubMed] [Google Scholar]
- 32.Radhakrishnan S.K., Kamalakaran S. Pro-apoptotic role of NF-kappaB: implications for cancer therapy. Biochim. Biophys. Acta. 2006;1766:53–62. doi: 10.1016/j.bbcan.2006.02.001. [DOI] [PubMed] [Google Scholar]
- 33.Ryan K.M., Ernst M.K., Rice N.R., Vousden K.H. Role of NF-kappaB in p53-mediated programmed cell death. Nature. 2000;404:892–897. doi: 10.1038/35009130. [DOI] [PubMed] [Google Scholar]
- 34.Gong D.J., Zhang J.M., Yu M., Zhuang B., Guo Q.Q. Inhibition of SIRT1 combined with gemcitabine therapy for pancreatic carcinoma. Clin. Interv. Aging. 2013;8:889–897. doi: 10.2147/cia.S45064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhao G., Cui J., Zhang J.G., Qin Q., Chen Q., Yin T., Deng S.C., Liu Y., Liu L., Wang B., et al. SIRT1 RNAi knockdown induces apoptosis and senescence, inhibits invasion and enhances chemosensitivity in pancreatic cancer cells. Gene Ther. 2011;18:920–928. doi: 10.1038/gt.2011.81. [DOI] [PubMed] [Google Scholar]
- 36.Oon C.E., Strell C., Yeong K.Y., Östman A., Prakash J. SIRT1 inhibition in pancreatic cancer models: contrasting effects in vitro and in vivo. Eur. J. Pharmacol. 2015;757:59–67. doi: 10.1016/j.ejphar.2015.03.064. [DOI] [PubMed] [Google Scholar]
- 37.Bai Y., Bai Y., Dong J., Li Q., Jin Y., Chen B., Zhou M. Hedgehog Signaling in Pancreatic Fibrosis and Cancer. Medicine (Baltim.) 2016;95:e2996. doi: 10.1097/md.0000000000002996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jia Y., Gu D., Wan J., Yu B., Zhang X., Chiorean E.G., Wang Y., Xie J. The role of GLI-SOX2 signaling axis for gemcitabine resistance in pancreatic cancer. Oncogene. 2019;38:1764–1777. doi: 10.1038/s41388-018-0553-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jia Y., Xie J. Promising molecular mechanisms responsible for gemcitabine resistance in cancer. Genes Dis. 2015;2:299–306. doi: 10.1016/j.gendis.2015.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhang X., Wang D., Li Z., Jiao D., Jin L., Cong J., Zheng X., Xu L. Low-Dose Gemcitabine Treatment Enhances Immunogenicity and Natural Killer Cell-Driven Tumor Immunity in Lung Cancer. Front. Immunol. 2020;11:331. doi: 10.3389/fimmu.2020.00331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dammeijer F., De Gooijer C.J., van Gulijk M., Lukkes M., Klaase L., Lievense L.A., Waasdorp C., Jebbink M., Bootsma G.P., Stigt J.A., et al. Immune monitoring in mesothelioma patients identifies novel immune-modulatory functions of gemcitabine associating with clinical response. EBioMedicine. 2021;64:103160. doi: 10.1016/j.ebiom.2020.103160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Suzuki E., Sun J., Kapoor V., Jassar A.S., Albelda S.M. Gemcitabine has significant immunomodulatory activity in murine tumor models independent of its cytotoxic effects. Cancer Biol. Ther. 2007;6:880–885. doi: 10.4161/cbt.6.6.4090. [DOI] [PubMed] [Google Scholar]
- 43.Koh E.K., Lee H.R., Son W.C., Park G.Y., Kim J., Bae J.H., Park Y.S. Combinatorial immunotherapy with gemcitabine and ex vivo-expanded NK cells induces anti-tumor effects in pancreatic cancer. Sci. Rep. 2023;13:7656. doi: 10.1038/s41598-023-34827-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Salewski I., Henne J., Engster L., Schneider B., Lemcke H., Skorska A., Berlin P., Henze L., Junghanss C., Maletzki C. Combined Gemcitabine and Immune-Checkpoint Inhibition Conquers Anti-PD-L1 Resistance in Low-Immunogenic Mismatch Repair-Deficient Tumors. Int. J. Mol. Sci. 2021;22:5990. doi: 10.3390/ijms22115990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Dull T., Zufferey R., Kelly M., Mandel R.J., Nguyen M., Trono D., Naldini L. A third-generation lentivirus vector with a conditional packaging system. J Virol. 1998;72:8463–8471. doi: 10.1128/JVI.72.11.8463-8471.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Meel M.H., Sewing A.C.P., Waranecki P., Metselaar D.S., Wedekind L.E., Koster J., van Vuurden D.G., Kaspers G.J.L., Hulleman E. Culture methods of diffuse intrinsic pontine glioma cells determine response to targeted therapies. Exp. Cell Res. 2017;360:397–403. doi: 10.1016/j.yexcr.2017.09.032. [DOI] [PubMed] [Google Scholar]
- 47.Meel M.H., Metselaar D.S., Waranecki P., Kaspers G.J.L., Hulleman E. An efficient method for the transduction of primary pediatric glioma neurospheres. MethodsX. 2018;5:173–183. doi: 10.1016/j.mex.2018.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Metselaar D.S., Meel M.H., Benedict B., Waranecki P., Koster J., Kaspers G.J.L., Hulleman E. Celastrol-induced degradation of FANCD2 sensitizes pediatric high-grade gliomas to the DNA-crosslinking agent carboplatin. EBioMedicine. 2019;50:81–92. doi: 10.1016/j.ebiom.2019.10.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Berchtold N.C., Cribbs D.H., Coleman P.D., Rogers J., Head E., Kim R., Beach T., Miller C., Troncoso J., Trojanowski J.Q., et al. Gene expression changes in the course of normal brain aging are sexually dimorphic. Proc. Natl. Acad. Sci. USA. 2008;105:15605–15610. doi: 10.1073/pnas.0806883105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Roth R.B., Hevezi P., Lee J., Willhite D., Lechner S.M., Foster A.C., Zlotnik A. Gene expression analyses reveal molecular relationships among 20 regions of the human CNS. Neurogenetics. 2006;7:67–80. doi: 10.1007/s10048-006-0032-6. [DOI] [PubMed] [Google Scholar]
- 51.Murat A., Migliavacca E., Gorlia T., Lambiv W.L., Shay T., Hamou M.F., de Tribolet N., Regli L., Wick W., Kouwenhoven M.C.M., et al. Stem cell-related "self-renewal" signature and high epidermal growth factor receptor expression associated with resistance to concomitant chemoradiotherapy in glioblastoma. J. Clin. Oncol. 2008;26:3015–3024. doi: 10.1200/jco.2007.15.7164. [DOI] [PubMed] [Google Scholar]
- 52.Gravendeel L.A.M., Kouwenhoven M.C.M., Gevaert O., de Rooi J.J., Stubbs A.P., Duijm J.E., Daemen A., Bleeker F.E., Bralten L.B.C., Kloosterhof N.K., et al. Intrinsic gene expression profiles of gliomas are a better predictor of survival than histology. Cancer Res. 2009;69:9065–9072. doi: 10.1158/0008-5472.Can-09-2307. [DOI] [PubMed] [Google Scholar]
- 53.Paugh B.S., Broniscer A., Qu C., Miller C.P., Zhang J., Tatevossian R.G., Olson J.M., Geyer J.R., Chi S.N., da Silva N.S., et al. Genome-wide analyses identify recurrent amplifications of receptor tyrosine kinases and cell-cycle regulatory genes in diffuse intrinsic pontine glioma. J. Clin. Oncol. 2011;29:3999–4006. doi: 10.1200/jco.2011.35.5677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Paugh B.S., Qu C., Jones C., Liu Z., Adamowicz-Brice M., Zhang J., Bax D.A., Coyle B., Barrow J., Hargrave D., et al. Integrated molecular genetic profiling of pediatric high-grade gliomas reveals key differences with the adult disease. J. Clin. Oncol. 2010;28:3061–3068. doi: 10.1200/jco.2009.26.7252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Meel M.H., de Gooijer M.C., Metselaar D.S., Sewing A.C.P., Zwaan K., Waranecki P., Breur M., Buil L.C.M., Lagerweij T., Wedekind L.E., et al. Combined Therapy of AXL and HDAC Inhibition Reverses Mesenchymal Transition in Diffuse Intrinsic Pontine Glioma. Clin. Cancer Res. 2020;26:3319–3332. doi: 10.1158/1078-0432.Ccr-19-3538. [DOI] [PubMed] [Google Scholar]
- 56.Meel M.H., de Gooijer M.C., Guillén Navarro M., Waranecki P., Breur M., Buil L.C.M., Wedekind L.E., Twisk J.W.R., Koster J., Hashizume R., et al. MELK Inhibition in Diffuse Intrinsic Pontine Glioma. Clin. Cancer Res. 2018;24:5645–5657. doi: 10.1158/1078-0432.Ccr-18-0924. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
Raw and analyzed RNA-seq data are publicly available at Mendeley Data Repository under DOI number: https://doi.org/10.17632/gfx5dwt2tg.1.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.