

Summary
Lung parenchyma destruction represents a severe condition commonly found in chronic obstructive pulmonary disease (COPD), a leading cause of morbidity and mortality worldwide. Promoting lung regeneration is crucial for achieving clinical improvement. However, no therapeutic drugs are approved to improve the regeneration capacity due to incomplete understanding of the underlying pathogenic mechanisms. Here, we identify a positive feedback loop formed between adipose triglyceride lipase (ATGL)-mediated lipolysis and overexpression of CD36 specific to lung epithelial cells, contributing to disease progression. Genetic deletion of CD36 in lung epithelial cells and pharmacological inhibition of either ATGL or CD36 effectively reduce COPD pathogenesis and promote lung regeneration in mice. Mechanistically, disruption of the ATGL-CD36 loop rescues Z-DNA binding protein 1 (ZBP1)-induced cell necroptosis and restores WNT/β-catenin signaling. Thus, we uncover a crosstalk between lipolysis and lung epithelial cells, suggesting the regenerative potential for therapeutic intervention by targeting the ATGL-CD36-ZBP1 axis in COPD.
Keywords: lipolysis, ATGL, CD36, ZBP1, necroptosis, lung regeneration
Graphical abstract
Highlights
-
•
Lipolysis is increased in COPD, and ATGL inhibition alleviates COPD
-
•
CD36 is selectively upregulated in lung epithelial cells and promotes COPD
-
•
Lipolysis-CD36 positive feedback loop impairs lung regeneration via ZBP1
-
•
The ATGL-CD36-ZBP1 axis can be therapeutically exploited
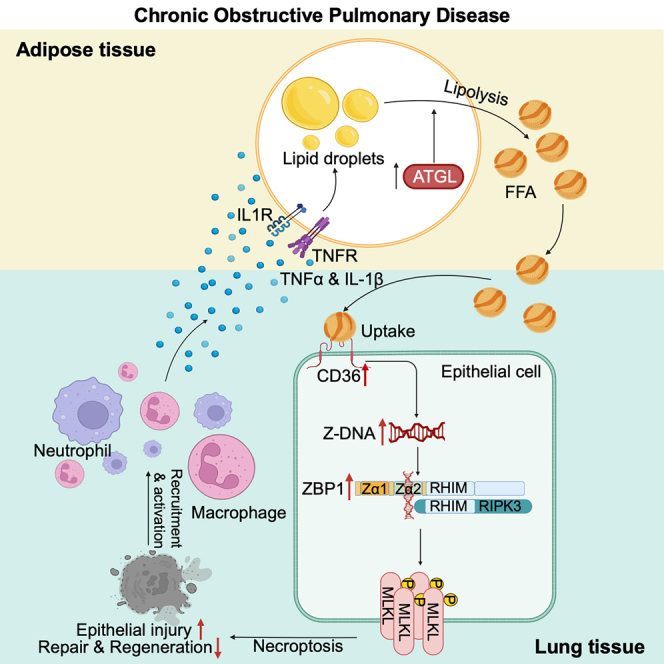
Wang et al. identify a positive feedback loop formed between ATGL-mediated lipolysis and the overexpression of CD36 in lung epithelial cells that dampens lung regeneration through ZBP1-induced necroptosis in chronic obstructive pulmonary disease (COPD) and suggest that the ATGL-CD36-ZBP1 axis can be therapeutically exploited.
Introduction
Chronic obstructive pulmonary disease (COPD) is a global health concern, accounting for over three million deaths annually.1 This disease is characterized by airway inflammation and lung tissue destruction, manifesting a spectrum of conditions ranging from bronchiolitis to emphysema, the latter being the most severe situation.2,3 Current treatments for COPD primarily focus on symptom management using bronchodilators and inhaled corticosteroids, etc., but have failed to halt disease progression due to their ineffectiveness in promoting lung regeneration, which underscores the urgent need for developing innovative therapeutic approaches.2,4
Lung regeneration defects, especially the emphysema lung featured by the enlargement of alveolar airspace and concomitant destruction of alveolar walls, lead to severe decline in respiratory function and unfavorable prognosis.3,5 Normally, a range of cells among distal lung epithelial cells possess the ability to regenerate alveoli, ensuring the restoration of both structure and function following injury, though AT2 cells are recognized as the primary regenerative responders.6,7,8,9,10,11,12,13,14,15,16 However, accumulating evidence indicates that within the context of COPD, alveolar regeneration is severely compromised, accompanied by reduced activation of WNT signaling.17,18 Alveolar regeneration hinges tightly on the intricate coordination of both internal and external factors,12,19,20,21,22,23,24,25,26,27,28 but the underlying mechanisms causing regeneration defects in emphysema lung remain unclear.
Patients with COPD often experience extrapulmonary complications, notably unintended weight loss and reductions in both fat and skeletal muscle mass, and the weight decline becomes more pronounced as COPD progresses, mirroring the cachexia observed in patients with cancer with poor prognosis.29,30,31,32 Adipose tissue loss often precedes muscle loss in cachexia.32 Consistently, the percentage of body fat is significantly lower in individuals with COPD compared to their healthy counterparts, and those with emphysema typically have a more reduced body mass index (BMI) resulting from decreased fat mass compared to patients with bronchitis.33,34 Lipolysis, wherein triglycerides are broken down by lipases to release free fatty acids (FFAs), is the driving factor for adipose tissue loss and can be intensified by pro-inflammatory cytokines.32 Approaches that counteract lipolysis through the suppression of the lipolysis rate-limiting enzyme adipose triglyceride lipase (ATGL) have shown promise in reducing both adipose and muscle tissue loss and demonstrating therapeutic potential for some diseases.32,35 However, it remains elusive whether lipolysis is involved in COPD, and how this process affects lung regeneration is yet to be determined.
CD36 is a multifunctional scavenger receptor, interacting with diverse ligands such as collagen, thrombospondin, various lipoproteins, and lipids.36 While its function in macrophages and T cells is well documented, its role in the maintenance of epithelial cell homeostasis remains largely unexplored.37,38,39 Here, we show that adipose lipolysis is increased, and elevated lipolysis is reciprocally regulated with lung epithelial cell-specific overexpression of CD36 in COPD. This synergistic interplay promotes Z-DNA binding protein 1 (ZBP1)-dependent cell death in lung epithelial cells, impairing lung regeneration. Pharmacological inhibition of CD36 effectively alleviates the pathogenesis of COPD and promotes lung regeneration, highlighting potential targets that can be therapeutically exploited.
Results
Lipolysis is increased in COPD, and lipolysis inhibition reduces acute cigarette-smoke-induced pulmonary inflammation
Given that patients with COPD with emphysema exhibit more body weight decline,29 we hypothesized that lipolysis might function in the pathogenesis of COPD. To test this possibility, we employed a well-established murine model of COPD induced by 4 months chronic cigarette smoke (CS) exposure (Figure 1A). Histopathological analysis revealed that mice exposed to 4 months chronic CS developed emphysema, as evidenced by an increased mean linear intercept (MLI) (Figure S1A). To ascertain whether lipolysis indeed increases in COPD, we selected gonadal adipose tissue (GAT), the largest visceral fat reservoir, for further analysis. We observed a notable reduction in the volume and weight of the GAT in the chronic CS-exposed mice (Figures 1B and S1B). Further examination of lipolysis-associated gene expression showed that, compared to the control group (air-exposed), mice exposed to chronic CS had elevated mRNA and protein expression levels of the rate-limiting lipolytic enzyme ATGL (Figures 1C and 1D). Concurrently, there was a decreased mRNA expression of negative regulators of lipolysis, lipoprotein lipase (Lpl), and cell death activator CIDE-A (Cidea), within the GAT of chronic CS-exposed mice (Figure S1C). In a time course assay of chronic CS-exposed mice, we observed a gradual increase of ATGL expression in GAT and a progressive loss of GAT weight (Figures S1D and S1E). The reduction in GAT weight was more pronounced at 4 months, which is when emphysema developed (Figures S1E and S1F). These results suggest that the lipolysis is activated in the COPD mouse model.
Figure 1.

Lipolysis is increased in COPD, and lipolysis inhibition reduces acute CS-induced pulmonary inflammation
(A) Experimental scheme for (B)–(H).
(B) Relative GAT weight of the indicated groups of mice (n = 5 mice per group).
(C) Quantitative reverse-transcription PCR (qRT-PCR) analysis showing Atgl mRNA expression in GAT of the indicated groups of mice (n = 5 mice per group, mean value of 3 technical replicates).
(D) Immunoblot showing ATGL expression in GAT, with GAPDH as loading control (n = 5 mice per group, representative one of 3 technical replicates).
(E) Relative FFA levels in culture supernatant of GAT of the indicated groups of mice (n = 5 mice per group).
(F) Relative FFA levels in serum of the indicated groups of mice (n = 5 mice per group).
(G) Relative FFA levels in culture supernatant of GAT stimulated with indicated cytokines (n = 5 mice per group).
(H) ELISA showing TNF-α and IL-1β expression in serum of the indicated groups of mice (n = 5 mice per group).
(I) RNA-seq heatmap of differentially expressed genes in PBS versus CSE-treated MLE-12 cells and associated top Gene Ontology (GO) terms. Mean signal of three biological replicates.
(J) Network analysis showed top 5 upregulated signaling pathways in CSE-treated MLE-12 cells. Links between nodes shows biological correlation extracted from String database.
(K) Experimental scheme for (L)–(O).
(L–O) Cell count analysis of total cells, neutrophils, macrophages, and lymphocytes from BALF of the indicated groups of mice (n = 5 mice per group). The number of biological replicates is indicated (each dot and immunoblot lane in graphs indicate data from individual mice); error bars denote mean ± SEM. ∗p < 0.05, ∗∗p < 0.01, and ∗∗∗p < 0.001 (t test).
See also Figure S1.
We subsequently investigated whether activated lipolysis in COPD leads to an increased release of FFAs. We cultured adipose tissues from GAT derived from both control mice and 4 months chronic CS-exposed mice (Figure 1A). As shown, GATs from the chronic CS-exposed mice released a significantly higher amount of FFAs in the culture supernatant compared to those from control mice (Figure 1E). We also found elevated FFA levels in the serum of chronic CS-exposed mice, mirroring the observed increase in FFAs in patients with severe COPD (Figure 1F).40 The increased FFAs release was also replicated in the time course study of chronic CS-exposed mice (Figures S1G and S1H). Meanwhile, we detected higher expression levels of inflammatory cytokines interleukin (IL)-1β and tumor necrosis factor alpha (TNF-α) released in the supernatant from GAT culture of 4 months chronic CS-exposed mice, while the expression level of IL-6 remained unaltered (Figures S1I–S1K). Inflammatory response is also a hallmark of COPD. This led us to assess if these inflammatory cytokines directly mediate lipolysis. We found that TNF-α and IL-1β, but not IL-6, could enhance the release of FFAs in ex vivo cultured GATs (Figures 1G and S1L). Additionally, there was a notable upregulation of TNF-α and IL-1β in the serum of 4 months chronic CS-exposed mice (Figure 1H). These findings indicate that COPD enhances lipolysis in adipose tissues, resulting in adipose tissue atrophy.
Fatty acids released from lipolysis can be utilized by other organs and are known to serve as potent modulators of inflammation.41,42 Consistently, our transcriptomic sequencing analysis revealed an augmented inflammatory response and fatty acid metabolism in the alveolar epithelial cells post-exposure to cigarette smoke extract (CSE) (Figure 1I). Interestingly, network analysis connected fatty acid metabolism to inflammatory response and cell death, the hallmark of COPD, implying a functional role of lipolysis in COPD (Figure 1J). Therefore, we first subjected an acute CS-induced pulmonary inflammation mouse model to treatment with the ATGL inhibitor, atglistatin, to test the functional role (Figure 1K). We found that acute CS exposure also increases ATGL protein expression in GAT, simultaneously in lung tissue and pulmonary lipofibroblasts, indicating a systemic lipolysis effects similar to those seen in cancer cachexia,35 despite our analyses described earlier focusing on GAT as a representative tissue (Figures S1M–S1P). Next, pulmonary inflammatory responses in the bronchoalveolar lavage fluid (BALF) were evaluated by cellular staining and differential cell counts. The vehicle-treated mice exposed to acute CS showed obviously increase in total leukocytes, neutrophils, and macrophages relative to the controls exposed to normal air (Figures 1L–1N). Interestingly, atglistatin treatment markedly blocked the increase in total leukocytes, neutrophils, and macrophages in mice exposed to acute CS (Figures 1L–1N). The lymphocyte counts showed a similar trend, though the differences were not statistically significant (Figure 1O). Together, our results demonstrate that lipolysis is augmented in COPD and links to the heightened inflammation observed in COPD.
Lipolysis inhibition ameliorates COPD pathogenesis in chronic CS-exposed mice
We further explored whether lipolysis inhibition could offer benefits against the pathological manifestations associated with chronic CS-induced COPD. We first examined whether the inhibition of lipolysis could prevent the onset of COPD (Figure 2A). The protein expression of ATGL in the GAT of chronic CS-exposed mice was substantially reduced following prophylactic atglistatin intervention (Figure 2B). Analysis of the BALF via staining and cell enumeration revealed that chronic CS-exposed mice treated with vehicle displayed notably elevated counts of total leukocytes, neutrophils, macrophages, and lymphocytes (Figures 2C–2F). In contrast, chronic CS-exposed mice administered with atglistatin exhibited significant reductions in these cell counts (Figures 2C–2F). Importantly, collagen deposition around the airways of chronic CS-exposed mice, indicative of airway remodeling, was significantly reduced after atglistatin treatment (Figure 2G). Moreover, histopathological quantitative assessments of airspace enlargement and alveolar surface area indicated that atglistatin conferred protection against chronic CS-induced emphysema (Figures 2H–2J).
Figure 2.

Lipolysis inhibition ameliorates COPD pathogenesis in chronic CS-exposed mice
(A) Experimental scheme for (B)–(J).
(B) Immunoblot showing ATGL expression in GAT of the indicated groups of mice (n = 4 mice per group, representative one of 3 technical replicates).
(C–F) Cell count analysis of total cells, neutrophils, macrophages, and lymphocytes in BALF of the indicated groups of mice (n = 5 mice per group).
(G) Representative images of Masson’s trichrome staining of lung of the indicated groups of mice (scale bars, 50 μm, n = 5 mice per group).
(H) Representative images of H&E staining of lung of the indicated groups of mice (scale bars, 50 μm, n = 5 mice per group).
(I) Mean linear intercept of alveolus of the indicated groups of mice (n = 5 mice per group).
(J) Alveolar area of the indicated groups of mice (n = 5 mice per group).
(K) Experimental scheme for (L)–(R).
(L) Representative images of Masson’s trichrome staining of lung of the indicated groups of mice (scale bars, 50 μm, n = 5 mice per group).
(M) Representative images of H&E staining of lung of the indicated groups of mice (scale bars, 50 μm, n = 5 mice per group).
(N) Mean linear intercept of alveolus of the indicated groups of mice (n = 5 mice per group).
(O) Alveolar area of the indicated groups of mice (n = 5 mice per group).
(P) Body weight of the indicated groups of mice (n = 5 mice per group).
(Q) Relative GAT weight of the indicated groups of mice (n = 5 mice per group).
(R) Relative FFA levels in serum of the indicated groups of mice (n = 5 mice per group). The number of biological replicates is indicated (each dot and immunoblot lane in graphs indicate data from individual mice); error bars denote mean ± SEM. ∗p < 0.05, ∗∗p < 0.01, and ∗∗∗p < 0.001 (t test).
Next, we aimed to determine whether the inhibition of lipolysis could have a therapeutic effect for COPD emphysema. Therapeutic treatment with atglistatin was started at the fifth month of chronic CS exposure, a time point when the emphysematous phenotype had already been established (Figure 2K). Histopathological quantitative assessments of airspace enlargement and alveolar surface area indicated that therapeutic atglistatin treatment significantly reversed the COPD emphysema phenotype. This was accompanied by reduced collagen deposition and FFA levels in serum, as well as the restoration of body weight and GAT weight (Figures 2L–2R). These data demonstrate that lipolysis is necessary to drive and maintain COPD pathogenesis.
CD36-specific overexpression in lung epithelial cells mediates COPD pathogenesis
The scavenger receptor CD36 mediates the transport of fatty acids into cells, and lipid transport is tightly associated with inflammatory responses.36 Our RNA sequencing (RNA-seq) data revealed an upregulation of CD36 expression in alveolar epithelial cells following CSE stimulation (Figure 1I), providing a potential mechanism link through which lipolysis promotes COPD development. To test this, we further interrogated CD36 expression in epithelial samples derived from bronchial brushings of patients with COPD from publicly available datasets.43,44,45 Remarkably, CD36 ranks among the top upregulated membrane proteins in patients with COPD (Figure 3A). We also validated the upregulated expression of CD36 lung epithelial cells in multiple datasets (Figures S2A–S2C). Simultaneously, we observed a positive correlation between the mRNA expression of CD36 and CXCL8, and TNF-α and IL-1β in epithelial cell of patients with COPD (Figures S2D–S2I). To further confirm these findings, we isolated lung epithelial cells from mice and found that the protein expression of CD36 was significantly upregulated in chronic CS-exposed mice and elastase-induced emphysematous mice (Figures 3B and S2J). Interestingly, there was no notable change of CD36 expression in lung non-epithelial cells between the control group mice and the chronic CS-exposed mice (Figure S2K). Fatty acids represent a mixture, and previous studies have shown that increased palmitoleic acid and oleic acid are lipid biomarkers within patients with COPD with uncertain roles.40,46,47 We tested if oleic acid could activate CD36 expression because it is often used to create lung injury models and found that intravenous injection of oleic acid significantly increased the expression of CD36 in pulmonary epithelial cells (Figure S2L). Together, these data suggest a lung epithelium-specific role of CD36 in mediating COPD pathogenesis.
Figure 3.

CD36 is a specific overexpression in lung epithelial cells and drives pathogenesis in COPD
(A) Venn diagram of differentially expressed genes overlapping with GO cell surface protein gene (top). The rank of differentially expressed cell surface protein gene (bottom left). Top upregulated cell surface protein genes in COPD are shown (bottom right).
(B) Immunoblot showing CD36 expression in lung epithelial cells from mice exposed to fresh air and CS for 4 months (n = 3 mice per group, representative one of 3 technical replicates).
(C) Experimental scheme for (D)–(M).
(D) Immunoblot showing CD36 expression in lung epithelial cells of the indicated groups of mice (representative one of n = 3 biological replicates).
(E) Measurement of relative lung compliance of the indicated groups of mice (n = 5 mice per group).
(F) Representative images of H&E staining of lung of the indicated groups of mice (scale bars, 50 μm, n = 5 mice per group).
(G) Mean linear intercept of alveolus of the indicated groups of mice (n = 5 mice per group).
(H) Alveolar area of lung of the indicated groups of mice (n = 5 mice per group).
(I) Representative images of Masson’s trichrome staining of lung of the indicated groups of mice (scale bars, 50 μm, n = 5 mice per group).
(J–M) Cell count analysis of total cells, neutrophils, macrophages, and lymphocytes in BALF of the indicated groups of mice (n = 5 mice per group).
(N) Experimental scheme for (O)–(S).
(O) Measurement of relative lung compliance of the indicated groups of mice (n = 5 mice per group).
(P) Representative images of H&E staining of lung of the indicated groups of mice (scale bars, 50 μm, n = 5 mice per group).
(Q) Mean linear intercept of alveolus of the indicated groups of mice (n = 5 mice per group).
(R) Alveolar area of the indicated groups (n = 5 mice per group).
(S) Representative images of Masson’s trichrome staining of lung of the indicated groups of mice (scale bars, 50 μm, n = 5 mice per group). The number of biological replicates is indicated (each dot and immunoblot lane in graphs indicate data from individual mice); error bars denote mean ± SEM. ∗p < 0.05, ∗∗p < 0.01, and ∗∗∗p < 0.001 (t test).
See also Figure S2.
To investigate the role of the highly expressed CD36 in lung epithelial cells in COPD pathogenesis, we utilized the adenoviral vector AAV6.2FF, which has high cell-type selectivity in directing transgene expression in the terminal bronchioles and alveoli epithelial cells following intratracheal administration.48,49 This enabled us to deliver Cre recombinase selectively to the distal lung epithelial cells to delete Cd36 in Cd36flox/flox strain (Figure S2M). For simplicity, AAV6.2FF-Cre-treated Cd36flox/flox mice will be referred to as CKO mice and AAV6.2FF-Vector-treated Cd36flox/flox mice as wild-type (WT) mice.
We first aimed to investigate whether Cd36 CKO could prevent the development of COPD. To this end, Cd36flox/flox mice were intratracheally administrated with AAV6.2FF-Cre after two months of chronic CS exposure (Figure 3C). As expected, we found that CD36 protein expression was selectively abolished in the lung epithelial cells of CKO mice, while CD36 expression in non-epithelial lung cells remained largely unaffected (Figures 3D and S2N–S2P). Interestingly, under normal air conditions, Cd36 CKO does not compromise the lung function of mice, confirmed by lung compliance measurements (Figure 3E). There were also no observed pathological changes in normal air-exposed Cd36 CKO mice, as evidenced through histopathological assessments of airway remodeling and emphysema parameters (Figures 3F–3I). Additionally, BALF differential cell counts showed no signs of airway inflammation in normal air-exposed Cd36 CKO mice (Figures 3J–3M). However, we found that, compared to chronic CS-exposed WT mice, Cd36 CKO significantly prevented emphysema development (Figures 3E–3H). Meanwhile, loss of CD36 also significantly suppressed airway remodeling and inflammation (Figures 3I–3M). Next, we investigated whether Cd36 CKO could benefit mice from the established emphysema, in which Cd36flox/flox mice were intratracheally administrated with AAV6.2FF-Cre after four months of chronic CS exposure (Figures 3N, S2O, and S2P). We found that Cd36 CKO significantly alleviated the established emphysema phenotype and improved lung function in mice (Figures 3O–3S). Together, these data indicate that the epithelium CD36 is both a driving force and a maintenance factor for COPD pathogenesis.
Reciprocal regulation between CD36 and lipolysis attenuates WNT/β-catenin signaling, impairing lung regeneration
Unexpectedly, when we examined the histology, we found that, compared to chronic CS-exposed WT mice, GAT size of chronic CS-exposed Cd36 CKO mice had significantly recovered, as shown by the GAT weight gain (Figures 4A and S3A). This prompted us to investigate whether there is a reciprocal regulation between CD36 and lipolysis in COPD. First, we cultured mouse GAT to detect the content of FFAs in the supernatant. We found that Cd36 CKO significantly reduced FFAs release of GAT from chronic CS-exposed mice (Figure 4B). Furthermore, the levels of lipolysis-promoting inflammatory factors in the supernatant of cultured GAT, namely IL-1β and TNF-α, were also markedly decreased (Figures 4C and 4D). Then, we examined the protein expression of CD36 in lung epithelial cells of chronic CS-exposed mice treated with or without ATGL inhibitor atglistatin. Our findings revealed that atglistatin treatment significantly reduced CD36 protein expression (Figures 4E and S3B). These data suggest that high expression of CD36 in lung epithelial cells is reciprocally regulated with lipolysis (Figure 4F).
Figure 4.

Reciprocal regulation between CD36 and lipolysis attenuates WNT/β-catenin signaling, impairing lung regeneration
(A) Relative GAT weight of the indicated groups of mice exposed to fresh air or CS for 4 months (n = 5 mice per group).
(B) Relative FFA levels in culture supernatant of GAT from the indicated groups of mice exposed to fresh air or CS for 4 months (n = 5 mice per group).
(C and D) ELISA showing TNF-α and IL-1β expression in culture supernatant of GAT from the indicated groups of mice exposed to fresh air or CS for 4 months (n = 5 mice per group).
(E) Immunoblot showing CD36 expression in lung epithelial cells from the indicated groups of mice exposed to fresh air or CS for 4 months (n = 4–5 mice per group, representative one of 3 technical replicates).
(F) The schematic diagram depicting the reciprocal regulation of CD36 high expression in pulmonary epithelial cells and lipolysis.
(G) Immunoblot showing AXIN2 and β-catenin expression in lung epithelial cells from the indicated groups of mice exposed to fresh air or CS for 4 months (n = 4–5 mice per group, representative one of 3 technical replicates).
(H and I) Representative immunofluorescent images of SFTPC (red) and DAPI (blue) from the indicated groups of mice exposed to fresh air or CS for 4 months (scale bars, 50 μm, n = 5 mice per group).
(J) Immunoblot showing AXIN2 and β-catenin expression in lung epithelial cells from the indicated groups of mice exposed to fresh air or CS for 4 months (n = 3–4 mice per group, representative one of 3 technical replicates).
(K) AT2 organoid-forming capacity of the indicated groups of mice exposed to fresh air or CS for 4 months (n = 3 mice per group, mean value of 3 technical replicates). The number of biological replicates is indicated (each dot and immunoblot lane in graphs indicate data from individual mice); error bars denote mean ± SEM. ∗p < 0.05, ∗∗p < 0.01, and ∗∗∗p < 0.001 (t test).
See also Figure S3.
We next sought to determine why inhibiting lipolysis or Cd36 CKO could promote the repair of emphysematous lungs. WNT/β-catenin signaling is essential for alveolar regeneration and activating WNT/β-catenin signaling can induce the repair of emphysematous lungs.50 We found that atglistatin treatment significantly restored the mRNA and protein expression of β-catenin and AXIN2 in chronic CS-exposed mice (Figures 4G, S3C, and S3D). Furthermore, loss of AT2 cells in chronic CS-exposed mice was dramatically restored after atglistatin treatment (Figure 4H). Atglistatin treatment also notably upregulated the mRNA expression of the distal lung epithelial cell marker Nkx2.1, the AT2 cell marker Sftpc, and the AT1 cell marker T1α (Figures S3E and S3F). A similar effect was observed in chronic CS-exposed Cd36 CKO mice (Figures 4I, 4J, and S3G–S3J). More importantly, by culturing organoids derived from mouse AT2 cells in vitro, we observed that Cd36 CKO significantly restored the organoid-forming capability of chronic CS-exposed mice (Figure 4K). Together, these findings suggest that the reciprocal regulation of CD36 and lipolysis impairs the function of alveolar stem cells, worsening lung regeneration.
ZBP1-mediated cell necroptosis downstream of CD36 and lipolysis impairs lung regeneration
To understand the mechanisms by which the reciprocal regulation of lipolysis and CD36 impairs lung regeneration, we conducted a comparative analysis between our transcriptional profiles of CSE-treated alveolar epithelial cells and transcriptional processes perturbed in patients with COPD. We found that the shared upregulated pathways again involved in interferon α and γ response, fat acid metabolism, TNF-α, and necrotic cell death pathways (Figures 5A, 5B, and S4A–S4E). Because necroptosis links cell death to inflammation, we reasoned that the reciprocal regulation of lipolysis and CD36 orchestrates necroptosis to worsen lung regeneration.
Figure 5.

ZBP1-mediated cell necroptosis downstream of CD36 and lipolysis impairs lung regeneration
(A) Bar plot showing shared upregulated pathways of DEGs in patients with COPD from GSE8545 and CSE-treated MLE-12 cells.
(B) Gene set enrichment analysis (GSEA) plot showing enrichment of gene set of necrotic signaling in the CSE group.
(C) Experimental scheme for (D)–(J).
(D and E) Immunoblot showing ZBP1, p-MLKL, MLKL (D), and AXIN2 and β-catenin (E) expression in lung epithelial cells of the indicated groups of mice (n = 4–5 mice per group, representative one of 3 technical replicates).
(F) AT2 organoid-forming capacity of the indicated groups of mice (n = 3 mice per group, mean value of 3 technical replicates).
(G–J) Cell count analysis of total cells, neutrophils, and macrophages in BALF of the indicated groups of mice (n = 5 mice per group).
(K) Immunoblot showing ZBP1, p-MLKL, and MLKL expression in lung epithelial cells from mice exposed to fresh air or CS for 4 months (n = 3 mice per group, representative one of 3 technical replicates).
(L) Immunoprecipitation (IP) results depicting the interaction between ZBP1 and RIPK3 in mice exposed to fresh air or CS for 4 months (representative one of n = 3 biological replicates).
(M and N) Immunoblot showing ZBP1 (M), p-MLKL, and MLKL expression (N) in epithelial cells from the indicated groups of mice exposed to fresh air or CS for 4 months (n = 3–4 mice per group, representative one of 3 technical replicates).
(O) Immunoblot showing ZBP1, p-MLKL, and MLKL expression in epithelial cells from the indicated groups of mice exposed to fresh air or CS for 4 months (n = 4–5 mice per group, representative one of 3 technical replicates).
(P) Representative immunofluorescent images of Z-DNA (red) and DAPI (blue) of the indicated groups in MLE-12 (scale bars, 20 μm, representative one of n = 3 biological replicates). The number of biological replicates is indicated (each dot and immunoblot lane in graphs indicate data from individual mice); error bars denote mean ± SEM. ∗p < 0.05 and ∗∗p < 0.01 (t test).
See also Figure S4.
Necroptosis can be executed through both receptor-interacting protein kinase 1 (RIPK1)-dependent and independent manner, with the later scenario involving ZBP1-induced necroptosis through interacting with receptor-interacting protein kinase 3 (RIPK3) via its RIP homotypic interaction motif (RHIM) domain.51 It has been reported that in established emphysema models, inhibiting RIPK1 did not ameliorate symptoms.52,53 In contrast, inhibiting RIPK3 but not RIPK1 two months after CS exposure protected mice from chronic CS-induced emphysema.52 Thus, we postulated that the reciprocal regulation of lipolysis and CD36 impairs lung regeneration through ZBP1-mediated necroptosis. To validate this, we first used Zbp1 knockout (KO) mice to ascertain whether ZBP1 mediates lung epithelial cell necroptosis (Figure S4F) after CS stimulation. We subjected the Zbp1 KO mice to acute CS exposure (Figure 5C). Compared to air control, acute CS exposure causes ZBP1 activation, accompanied by increased phosphorylated mixed-lineage kinase domain-like pseudokinase (MLKL), a hallmark of necroptosis (Figure 5D). Nevertheless, KO of Zbp1 completely inhibited the expression of phosphorylated MLKL (Figure 5D). We also observed a restoration in the protein expression of β-catenin and AXIN2 in mice exposed to acute CS after Zbp1 KO (Figure 5E). Importantly, Zbp1 KO restored the organoid-forming capability of AT2 cells (Figure 5F). Additionally, Zbp1 KO also suppressed acute CS-induced airway inflammation in mice (Figures 5G–5J). These data suggest that ZBP1 induced lung epithelial cell necroptosis after CS exposure.
Next, we investigated whether the reciprocal regulation of lipolysis and CD36 regulated ZBP1-mediated necroptosis in chronic CS-exposed mice. We observed a significant upregulation of ZBP1 and phosphorylated MLKL expression in the lung epithelial cells of chronic CS-exposed mice (Figures 5K and S4G). This effect was also observed in oleic acid-treated mice (Figure S4H). Importantly, ZBP1 knockdown in lung epithelia cells obviously alleviated COPD (Figures S4I–S4Q). ZBP1 promotes necroptosis by recruiting RIPK3. We found an obvious interaction between ZBP1 and RIPK3 in the lung epithelial cells of the chronic CS-exposed mice (Figure 5L). Interestingly, Cd36 CKO suppressed ZBP1 and phosphorylated MLKL expression in the lung epithelial cells of chronic CS-exposed mice (Figures 5M and 5N). A similar inhibitory effect was observed in mice treated with atglistatin (Figure 5O). The activation of ZBP1 depends on the presence of Z-form RNA or DNA (Z-RNA/DNA). We discovered that CSE could induce the formation of Z-form nucleic acids (Z-NAs), which was sensitive to DNase I treatment but not to RNase A, suggesting that CSE stimulates the formation of Z-DNA in lung epithelial cells (Figure S4R), consistent with previous reports.54 The increased Z-DNA was also found in AT2 cells of CS-exposed mouse (Figure S4S). However, knockdown of Cd36 expression significantly inhibited the formation of Z-DNA, restored Sftpc mRNA expression, and promoted cell growth (Figures 5P, S4T, and S4U). Together, these findings demonstrate that upregulation of CD36 impairs lung regeneration through promoting ZBP1-mediated lung epithelial cell necroptosis in COPD.
Therapeutic targeting of CD36 attenuates lung pathogenesis and promotes lung regeneration in COPD
We next sought to evaluate the potential benefits of targeting CD36 on COPD prevention and treatment using a CD36 monoclonal antibody and the small-molecule inhibitor salvianolic acid B (SAB), both of which specifically targets CD36.55,56 Firstly, we tested whether prophylactic treatment targeting CD36 could prevent the development of COPD (Figures 6A and S5A). Quantitative assessment of collagen deposition around the airways showed that both the CD36 monoclonal antibody and SAB treatments improved airway remodeling in chronic CS-exposed mice (Figures 6B and S5B). Additionally, histopathological quantification of the mean linear intercept and alveolar surface area revealed that CD36 inhibition significantly suppressed emphysema progression in mice (Figures 6C–6E and S5C–S5E). We further investigated whether prophylactic targeting of CD36 promotes lung regeneration. We found that prophylactic CD36 targeting inhibited ZBP1-mediated necroptosis and restored the expression levels of β-catenin and AXIN2 of the WNT signaling pathway in lung epithelial cells from chronic CS-exposed mice (Figures 6F–6H and S5F–S5J). We also found that blocking CD36 alleviated oleic acid-induced ZBP1-mediated necroptosis (Figure S5K). In addition, the expression levels of the distal lung epithelial marker Nkx2.1 and the alveolar epithelial cell markers Sftpc and T1α were also significantly restored after prophylactic CD36 targeting (Figures 6I and S5L–S5N).
Figure 6.

Therapeutic targeting of CD36 attenuates lung pathogenesis and promotes lung regeneration
(A) Experimental scheme for (B)–(I).
(B) Representative images of Masson’s trichrome staining of lung of the indicated groups of mice (scale bars, 50 μm, n = 5 mice per group).
(C) Representative images of H&E staining of lung of the indicated groups (scale bars, 50 μm, n = 5 mice per group).
(D) Mean linear intercept of alveolus of the indicated groups of mice (n = 5 mice per group).
(E) Alveolar area of the indicated groups of mice (n = 5 mice per group).
(F–H) Immunoblot showing CD36 (F), AXIN2 and β-catenin (G), and ZBP1, p-MLKL, and MLKL (H) expression in lung epithelial cells of the indicated groups of mice (n = 4–5 mice per group, representative one of 3 technical replicates).
(I) qRT-PCR analysis showing Sftpc and T1α mRNA expression in lung tissue of the indicated groups of mice (n = 5 mice per group, mean value of 3 technical replicates).
(J) Experimental scheme for (K)–(O).
(K) Representative images of H&E staining of lung of the indicated groups of mice (scale bars, 50 μm, n = 5 mice per group).
(L) Mean linear intercept of alveolus of the indicated groups of mice (n = 5 mice per group).
(M) Alveolar area of the indicated groups (n = 5 mice per group).
(N) Representative images of Masson’s trichrome staining of lung of the indicated groups of mice (scale bars, 50 μm, n = 5 mice per group).
(O) Representative immunofluorescent images of SFTPC (red) and DAPI (blue) of lung of the indicated groups of mice (scale bars, 50 μm, n = 5 mice per group). The number of biological replicates is indicated (each dot and immunoblot lane in graphs indicate data from individual mice); error bars denote mean ± SEM. ∗p < 0.05 and ∗∗p < 0.01 (t test).
See also Figure S5.
We then explored the therapeutic potential of CD36 in established COPD using CD36 monoclonal antibodies. Quantitative assessment of collagen deposition around the airways and histopathological quantification of the mean linear intercept and alveolar surface area showed that CD36 monoclonal antibody treatments improved airway remodeling and alleviated emphysema in chronic CS-exposed mice, respectively (Figures 6J–6N). Additionally, we found that therapeutic CD36 monoclonal antibody treatment also promoted the expression of regeneration-related markers (Figure 6O). These findings reveal that therapeutic targeting of CD36 not only improves the pathogenesis of COPD but also promotes lung regeneration.
Therapeutic targeting of CD36 attenuates lung pathogenesis and promotes lung regeneration in elastase-induced emphysema
We further assessed the therapeutic potential of CD36 using a porcine pancreatic elastase (PPE)-induced emphysema model, as CD36 is a multifunctional receptor implicated in various physiological and pathological processes.57 First, we investigated whether therapeutically targeting CD36 could inhibit the development of emphysema (Figures 7A and S6A). We found that both CD36 monoclonal antibody and SAB significantly suppressed the development of emphysema and improved lung function in mice, as evidenced by MLI quantification and compliance measurements (Figures 7B–7D and S6B–S6D). Concurrently, inhibiting CD36 reduced the expression of ZBP1 and phosphorylated MLKL while restoring the expression of AXIN2 and β-catenin (Figures 7E, 7F, S6E, and S6F). These findings suggest that therapeutic targeting of CD36 can halt the progression of elastase-induced emphysema.
Figure 7.

Therapeutic targeting of CD36 attenuates lung pathogenesis and promotes lung regeneration in elastase-induced emphysema
(A) Experimental scheme for (B)–(F).
(B) Representative images of H&E staining of lung of the indicated groups of mice (scale bars, 100 μm, n = 5 mice per group).
(C) Mean linear intercept of alveolus of the indicated groups of mice (n = 5 mice per group).
(D) Measurement of relative lung compliance of the indicated groups of mice (n = 5 mice per group).
(E and F) Immunoblot showing ZBP1, p-MLKL and MLKL (E), and AXIN2 and β-catenin (F) expression in lung epithelial cells of the indicated groups of mice (n = 4–5 mice per group, representative one of 3 technical replicates).
(G) Experimental scheme for (H)–(M).
(H) Representative images of H&E staining of lung of the indicated groups of mice (scale bars, 100 μm, n = 5 mice per group).
(I) Mean linear intercept of alveolus of the indicated groups of mice (n = 5 mice per group).
(J) Measurement of relative lung compliance of the indicated groups (n = 5 mice per group).
(K and L) Immunoblot showing ZBP1, p-MLKL and MLKL (K), and AXIN2 and β-catenin (L) expression in lung epithelial cells of the indicated groups of mice (n = 4–5 mice per group, representative one of 3 technical replicates).
(M) AT2 organoid-forming capacity of the indicated groups of mice (n = 3 mice per group, mean value of 3 technical replicates). The number of biological replicates is indicated (each dot and immunoblot lane in graphs indicate data from individual mice); error bars denote mean ± SEM. ∗p < 0.05 and ∗∗p < 0.01 (t test).
See also Figure S6.
Next, we asked whether therapeutic intervention of CD36 could benefit the established emphysema, with treatment starting 7 days post PPE instillation (Figures 7G and S6G). Through quantification of MLI and compliance measurements, we found that both the CD36 monoclonal antibody and SAB effectively promoted lung repair in mice with established emphysema (Figures 7H–7J and S6H–S6J). Similarly, both CD36 monoclonal antibody and SAB suppressed the expression of ZBP1 and phosphorylated MLKL, restoring the expression of AXIN2 and β-catenin in mice with established emphysema (Figures 7K–7L, S6K, and S6L). Additionally, we conducted organoid experiments on AT2 cells sorted from monoclonal antibody-treated and control groups of mice; we observed that blocking CD36 significantly restored the organoid-forming capability of AT2 cells (Figure 7M). Taken together, these results indicate that therapeutic targeting of CD36 can effectively prevent and treat emphysema, promoting lung regeneration.
Discussion
Understanding the mechanisms causing regenerative defects in COPD lungs is the key to halting disease progression. Here, we reveal that ATGL-induced lipolysis engages CD36-specific overexpression in lung epithelial cells to accelerate ZBP1-mediated lung epithelial cell necroptosis and impairment of lung regeneration in COPD. Specifically, we show that CD36 is indispensable for driving the pathogenesis of COPD but not required for maintaining normal lung function, providing a rationale for its potential therapeutic targeting.
BMI is a well-known prognostic marker, and higher BMI independently predicts improved long-term survival rates in COPD.58,59,60 Moreover, the unintentional weight loss other than the low fat-free mass index was independently linked to mortality.61 These cohort studies, coupled with evidence that lipolysis-induced adipose tissue atrophy precedes muscle wasting and that inhibiting lipolysis can counteract both adipose tissue and muscle atrophy, underscore the importance of maintaining adipose tissue homeostasis.32,35 However, while the significance of lipolysis is evident, its role in COPD had been largely unexplored. Our research reveals that ATGL-driven lipolysis contributes to the progression of COPD and hampers lung regeneration, rather than merely acting as a complication of COPD. However, lipolysis under COPD conditions may be a systemic process. Future studies should explore which adipose tissue or cell contributes most to the observed phenotype, as well as how lipolysis in these tissues or cells is temporally and spatially coordinated.
Prior studies indicated that overexpression of CD36 promotes lipotoxicity and ferroptosis in CD8+ T cells, undermining their cytotoxic effects against tumor cells.38,39 Similar to this detrimental influence, our findings suggest that there is a specific overexpression of CD36 in lung epithelial cells in COPD. Using the AAV6.2FF-Cre tool, a well-characterized vector that selectively delivers transgenes to distal lung epithelial cells with high EpCAM expression,48 we found that deletion of Cd36 inhibits lung epithelial cell necroptosis and promotes lung regeneration in COPD. Importantly, our study establishes a link between ATGL-mediated lipolysis and the overexpression of CD36 in lung epithelial cells, emphasizing their reciprocal regulation in impairing lung regeneration in COPD. Our research also indicates that CD36 can promote CS-induced pulmonary inflammation, which aligns with the understanding that CD36 can amplify pro-inflammatory reactions.62 Notably, we cannot completely exclude the possibility that AAV6.2FF-Cre may affect resident alveolar macrophages, as these cells also express EpCAM and CD36.63 Therefore, future research needs to develop more precise epithelial-specific Cre tools to eliminate this possibility. Muscle loss is another trait of patients with partial COPD, even though continuing decline in fat-free mass is uncommon in COPD, and fat tissue atrophy usually precedes muscle wasting in conditions of cachexia.30,64 A recent study revealed that CD36 blockade enhanced regeneration in both young and mature muscles.65 It would be interesting for future studies to determine whether CD36 inhibition might reverse muscle loss in the context of COPD.
Epithelial cell death is a hallmark of COPD.66 However, recent in-depth studies have indicated that inhibiting RIPK3-mediated necroptosis, rather than apoptosis or RIPK1, can effectively hinder the progression of late-stage COPD, validating RIPK3-mediated necroptosis as a primary determinant in the impairment of epithelial regeneration.52 While ZBP1 has been identified to facilitate RIPK1-independent necroptosis via its association with RIPK3,51 its contribution to COPD has yet to be experimentally confirmed. Our research revealed that the expression of ZBP1 is markedly upregulated under both acute and chronic CS exposure, and ZBP1 knockdown in lung epithelial cells significantly improves COPD pathogenesis. Furthermore, KO of ZBP1 mitigated epithelial cell necroptosis and restored the organoid-forming capacity of AT2 cells, suggesting its detrimental impact on lung regeneration, which aligns with a recent study indicating its role in promoting necroptosis in gut stem cells.67 Our data further suggest that inhibiting ATGL or CD36 can dampen ZBP1-mediated lung epithelial cell necroptosis. Thus, our findings also offer a deeper understanding of the upstream mechanisms governing ZBP1-mediated cell necroptosis. Collectively, our research uncovers a reciprocal regulation between CD36 and ATGL-mediated lipolysis, which promotes ZBP1-mediated epithelial cell necroptosis. This sheds light on the critical roles of CD36 and lipolysis in the progression and regenerative deficits of COPD and suggests the potential of CD36 as a therapeutic target.
Limitations of the study
Our findings mainly rely on murine samples and lack adequate validation in human patients with COPD due to challenges in specimen acquisition. Future studies should explore these findings in human samples. Additionally, while we suggest an epithelial cell-specific role of CD36 using AAV6.2FF-Cre and in vitro AT2 organoid culture, we cannot completely exclude the interventions of resident alveolar macrophages as it has been reported that these cells also express EpCAM and CD36 on their surface.63 Future work is needed to further clarify this by developing more epithelia-specific Cre tools. Moreover, the mechanism by which the lipolysis-CD36 axis induces Z-DNA formation following CS exposure remains unclear and needs to be further investigated.
Resource availability
Lead contact
Further information and requests for reagents should be directed to and will be fulfilled by the lead contact, Jiansheng Li (ljs2015@hactcm.edu.cn).
Materials availability
Cell lines, plasmids, and recombinant DNA used in this study are detailed in the key resources table. Cd36fl/fl and Zbp1 KO mice and all stable reagents generated in this study are available from the lead contact with a completed materials transfer agreement.
Data and code availability
-
•
RNA-seq data reported in this paper were deposited in GEO under the accession number GEO: GSE246501. This paper analyzes publicly available data GSE8545, GSE186359, and GSE11906.
-
•
This paper does not report the original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
Acknowledgments
We acknowledge the members of our laboratory for helpful discussion. This work was supported by grants from the National Key Research and Development Program of China (no. 2023YFC3502600 and 2023YFC3502602), the Scientific and Technological Research Project of Henan Province (232102311212), the China Postdoc Innovation Talent Support Program (BX2021093), the National Natural Science Foundation of China (81973822, 82230007, and 82325003), and the Chinese inheritance and innovation team of Chinese medicine (ZYYCXTD-C-202206).
Author contributions
Jiazhen Wang and J.L. conceived the project and designed the experiments. Jiazhen Wang and R.W. performed experiments with the help from J.H., Y.L., Jiayi Wang, P.X., and Y.Z. Y.L. and Y.Y. performed bioinformatic analyses. Jiazhen Wang, H.Z., and J.L. supervised the project. Jiazhen Wang, R.W., H.Z., and J.L. wrote the manuscript.
Declaration of interests
The authors declare no competing interests.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| CD36 antibody (C1C3) | GeneTex | Cat#GTX100642; RRID:AB_1949927 |
| ATGL Polyclonal Antibody | Proteintech | Cat#55190-1-AP; RRID:AB_11182818 |
| ADRP/Perilipin-2 Polyclonal antibody | Proteintech | Cat#15294-1-AP; RRID:AB_2878122 |
| Phospho-MLKL (Ser358) (D6H3V) | Cell Signaling Technology | Cat#91689S; RRID:AB_2732034 |
| MLKL(D6W1K) Rabbit mAb | Cell Signaling Technology | Cat#37705S; RRID:AB_2799118 |
| RIP3 (D4G2A) Rabbit mAb | Cell Signaling Technology | Cat#95702S; RRID:AB_2721823 |
| anti-ZBP1, mAb (Zippy-1) | Adipogon | Cat#AG-20B-0010-C100; RRID:AB_2490191 |
| GAPDH Polyclonal antibody | Proteintech | Cat#:10494-1-AP; RRID:AB_2263076 |
| Anti-Axin 2 antibody | Abcam | Cat# ab32197; RRID:AB_2290204 |
| β-Catenin (D10A8) XP® Rabbit mAb | Cell Signaling Technology | Cat# 8480S; RRID:AB_11127855 |
| Anti-Z-DNA/Z-RNAI (Z22) | Absoluteantibody | Cat#AB00783-3; RRID:AB_2820286 |
| IgG | Cell Signaling Technology | Cat#3900S; RRID:AB_1550038 |
| Anti Prosurfactant Protein C antibody | Abcam | Cat# ab211326; RRID:AB_2927746 |
| Scavenger Receptor B2/CD36 Monoclonal Antibody (Clone JC63.1) | cayman | Cat#10009893; RRID:AB_10613953 |
| Biotin anti-mouse CD45 Anitbody | Biolegend | Cat#103104; RRID:AB_312969 |
| Biotin anti-mouse CD31 Anitbody | Biolegend | Cat#102504; RRID:AB_312911 |
| Biotin anti-mouse CD24 Anitbody | Biolegend | Cat#101804; RRID:AB_312837 |
| Biotin anti-mouse Ly-6A/E(Sca-1) Anitbody | Biolegend | Cat#108104; RRID:AB_313341 |
| Mouse IgA,kappa [S107](Low endotoxin, azide free)-Isotype Control | Abcam | Cat#AB37354; RRID:AB_840445 |
| Chemicals, peptides, and recombinant proteins | ||
| DMEM | Gibco | Cat# 11995073 |
| DME/F-12 | Cytiva | Cat#SH30023.01 |
| Advanced DMEM/F12 | Gibco | Cat#12634010 |
| Fetal Bovine Serum | Sigma-Aldrich | Cat# F8687 |
| Bovine Serum Albumin | Sigma-Aldrich | Cat#A1933 |
| Hanks' Balanced Salt Solution | Gibco | Cat#14175095 |
| EasySEP™Biotin Positive Selection Kit II | STEMCELL | Cat#17683 |
| CD326(Epcam) MicroBeads, mouse | Miltenyi Biotec | Cat#130-105-958 |
| Corning® Matrigel® Basement Membrane Matrix Growth Factor Reduced | Corning | Cat#354230 |
| EGF | MedChemExpress | Cat#HY-P7067 |
| ITS Media Supplement (100X) | Beyotime Biotechnology | Cat# C0341 |
| FGF10 | MedChemExpress | Cat#HY-P7170 |
| CHIR99021 | GlpBio | Cat#GC16702 |
| SB431542 | MedChemExpress | Cat#HY-10432 |
| Y-27632 | MedChemExpress | Cat#HY-10071 |
| SB202190 | MedChemExpress | Cat#HY-10295 |
| B27 supplement minus vitamin A | Gibco | Cat#12587010 |
| N-Acetylcysteine | Sigma-Aldrich | Cat#A9165 |
| Nicotinamide | Sigma-Aldrich | Cat#N0636 |
| GlutaMax (100x) | Gibco | Cat#35050061 |
| HEPES | Gibco | Cat#15630130 |
| Penicillin/Streptomycin | Gibco | Cat#15070063 |
| Primocin | InvivoGen | Cat#ant-pm-1 |
| IL-6 | MedChemExpress | Cat#HY-P7063 |
| IL-1β | MedChemExpress | Cat#HY-P7073A |
| TNF-α | MedChemExpress | Cat#HY-P7090A |
| Elastase from porcine pancreas | Glpbio | Cat#GC61737 |
| Salvianolic Acid B | Glpbio | Cat# GN10344 |
| RNAiso Plus | TaKaRa | Cat#9109 |
| ReverTra Ace® qPCR RT Kit | TOYOBO | Cat#FSQ-101 |
| MS Mouse TNF-α ELISA Kit | Elabscience | Cat#E-MSEL-M0002 |
| MS Mouse IL-6 ELISA Kit | Elabscience | Cat#E-MSEL-M0001 |
| MS Mouse IL-1β ELISA Kit | Elabscience | Cat#E-MSEL-M0003 |
| Free Fatty Acid Quantitation Kit | Sigma-Aldrich | Cat#MAK044 |
| Deposited data | ||
| RNA-seq | This paper | GSE246501 |
| RNA-seq | Raman et al.43 | GSE11906 |
| RNA-seq | Tanosaki et al.44 | GSE186359 |
| RNA-seq | Ammous et al.45 | GSE8545 |
| Experimental models: Cell lines | ||
| HEK293T | ATCC | Cat#CRL-3216; RRID:CVCL_0063 |
| L-WRN | ATCC | Cat#CRL-3276; RRID:CVCL_DA06 |
| MLE-12 | ATCC | Cat#CRL-2110; RRID:CVCL_3751 |
| Experimental models: Organisms/strains | ||
| Mice:C57BL/6J | Vitonlihua | Cat#219 |
| Mice:Cd36 fl/fl | Jackson Laboratory | Cat#032276; RRID:IMSR_JAX:032276 |
| Mice:Zbp1 | Gempharmatech | Cat#T029037; RRID:IMSR_GPT:T029037 |
| Recombinant DNA | ||
| PLVX-shRNA1 | Clontech | Cat#632177 |
| pDGM6 | Addgene | Cat#110660; RRID:Addgene_110660 |
| AAV6.2FF | This study | N/A |
| Software and algorithms | ||
| GraphPad Prism 9 | GraphPad Software | https://www.graphpad.com/ |
| javaGSEA(v2.2.4) | Broad Institute | http://software.broadinstitute.org/gsea/index.jsp |
Experimental model and subject details
Mice
C57BL/6J background Cd36 fl/fl and Zbp1-KO mice were procured from the Jackson Laboratory and Gempharmatech Co., Ltd, respectively. C57BL/6J mice were acquired from Beijing Vitonlihua Experimental Animal Co., Ltd. The experiments were conducted using 8-10 weeks-old female mice, and mice were randomly assigned to experimental groups. No mouse was excluded from the analysis unless there were technical reasons, or the mouse died of an unknown cause prior to drug intervention. Animal experiments were conducted in accordance with the approval of the Ethical Committee for Animal Welfare of Wuhan university or Henan University of Chinese Medicine.
Primary cell isolation
Lung tissue was sectioned into small fragments. Subsequently, a gentle enzymatic digestion was carried out at 37°C, using a digestion solution containing Liberase (50 μg/mL) and DNase I (25 μg/mL). The digested tissue suspension was filtered through a 70-micron mesh to obtain a single-cell suspension. Red blood cells were removed with RBC Lysis Buffer before staining for lineage markers. After centrifugation at 300g for 5 min, the isolated single cells were resuspended in 1 mL of PBS supplemented with 2% FBS. For isolating mouse AT2 cells, cells were incubated with biotin-conjugated CD45, CD31, CD24, and Sca-1 antibodies and then processed using the EasySep Mouse Streptavidin RapidSpheres Isolation Kit to remove lineage-positive cells. Then, the lineage-negative cells were subjected to positive selection using CD326 magnetic beads from Miltenyi Biotec, according to the manufacturer’s instructions. Sorting mouse lung epithelial cells were the same as above, except that during the removal of lineage-positive cells, the CD24 and Sca-1 antibodies were omitted.
Organoids culture
Sorted mouse AT2 cells were mixed with 50 μL of Corning Matrigel (Corning, 354230). This mixture was then added to a 24-well culture plate and placed in a cell culture incubator, 37°C for 30 min, to allow the Corning Matrigel to solidify. Following gel solidification, Advanced DMEM/F12 mixed at a 1:1 volume ratio with L-WRN medium, supplementing with 50 ng/mL EGF, 20 ng/mL FGF10, 3 μM CHIR99021, 10 μM SB431542, 500 nM SB202190, 1×B27 supplement without vitamin A, 1.25 mM N-Acetylcysteine, 5 mM Nicotinamide, 1×GlutaMAX, 10 mM HEPES, 100 mg/L streptomycin, 100 U/ml penicillin, and 50 μg/mL primocin, was added to the well. For the first three days, 10 μM Y27632 was added. The cells were then cultured for additional days until organoids formed.
Cell lines
MLE-12 were cultured in DME/F12 (Hyclone), supplemented with 2% FBS, 1%ITS and 1% penicillin/streptomycin. HEK293T and L-WRN were cultured in DMEM (Hyclone), supplemented with 10% FBS and 1% penicillin/streptomycin.
Method details
Mice models and treatment
In the model of chronic cigarette smoke-induced COPD, the cigarettes were sourced from the Hongqiqu brand (Tar Content: 10 mg; Carbon Monoxide in Smoke: 12 mg). Each mouse was restrained in individual enclosure, remaining motionless, and underwent whole-body exposure to cigarette smoke with a total particulate matter (TPM) concentration of 500 mg/m3 for a duration of 50 min, twice per day, five days weekly. For lipolysis inhibition, mice were subjected to i.p. injections of either atglistatin (200 μmol/kg) dissolved in PBS containing 0.25% Cremophor EL (pH 7.1, MCE.) or Vehicle every other day for at least 1 h before smoke exposure. For the prophylactic COPD model, atglistatin was administered starting from the third month of smoke exposure and continued until the end of the fourth month. For the therapeutic COPD model, atglistatin was administered starting from the fifth month of smoke exposure and continued until the end of the sixth month. For targeting Cd36 gene knockout, Cd36 fl/fl mice underwent a single intratracheal administration of AAV6.2FF-Vector or AAV6.2FF-Cre, 1 × 1011 vg/mouse. For the prophylactic COPD model, AAV was administered after two months of smoke exposure. For the therapeutic COPD model, AAV was administered after four months of smoke exposure. For therapeutic targeting of CD36, mice were administered α-CD36 antibody (Cayman, 10009893) or IgA control antibody (Abcam, AB37354) via i.p., at least 1 h before smoke exposure, every other day. For the prophylactic COPD model, α-CD36 antibody was administered starting from the third month of smoke exposure and continued until the end of the fourth month. For the therapeutic COPD model, α-CD36 antibody was administered starting from the fifth month of smoke exposure and continued until the end of the sixth month. Similarly, an equivalent intervention approach was applied to another group of mice, involving i.p. injections of PBS (vehicle, 100 μL) or Salvianolic Acid B (Glpbio, GN10344) dissolved in PBS (10 mg/kg). For targeting Zbp1 gene knockdown, C56BL/6 mice underwent a single intratracheal administration of AAV6.2FF-shCONTROL or AAV6.2FF-shZBP1, 1 × 1011 vg/mouse, after four months exposure of cigarette smoke.
In the model of acute cigarette smoke-induced COPD, mice were subjected to cigarette smoke, four times daily, spanning five days. For targeting Zbp1 gene knockout, Zbp1-KO and WT mice were continuously exposed to acute cigarette smoke for 5 days. For lipolysis inhibition, mice received i.p. injections of atglistatin (200 μmol/kg) dissolved in PBS containing 0.25% Cremophor EL (pH 7.1, MCE.) or Vehicle at least 1 h before smoke exposure, on the day preceding smoking. This intervention regimen was maintained for a duration of six consecutive days.
In the model of elastase-induced pulmonary emphysema, mice were administered porcine pancreatic elastase (40 U/kg). For therapeutic targeting of CD36, mice received either α-CD36 antibody (Cayman, 10009893) or IgA control antibody (Abcam, AB37354) via i.p. In the prophylactic model, the α-CD36 antibody was administered daily starting from the day preceding elastase injection and continued until the end of the seventh day. In the therapeutic COPD model, the α-CD36 antibody was administered beginning on the seventh day after elastase injection and continued every other day until the end of the 21st day. Similarly, an equivalent intervention approach was applied to another group of mice, involving i.p. injections of PBS (vehicle, 100 μL) or Salvianolic Acid B (Glpbio, GN10344) dissolved in PBS (10 mg/kg).
Lung function measurements
After anesthesia, the mice underwent endotracheal intubation, followed by the measurement of pulmonary compliance in the mice. Respiratory function was measured using FinePointe PFT running DSI Buxco PFT software.
Airway inflammation
Airway inflammation was assessed by examining cytospin preparations of cells retrieved from bronchoalveolar lavage fluid (BALF). These specimens were obtained through lung lavages utilizing Hank’s Balanced Salt Solution (Life Technologies, Australia) via a cannula inserted into the trachea. Following collection, BALF underwent centrifugation (3000 rpm, 10 min, 4°C); subsequently, red blood cells were lysed using a lysis buffer and the remaining cells were sedimented. Total leukocyte counts were quantified using a hemocytometer. Subsequently, cells were cytocentrifuged and subjected to May-Grunwald-Giemsa staining for visualization. Differential leukocyte counts were determined based on morphological criteria. To ensure impartiality, all specimens were given concealed identifiers, and the quantifications were executed in a blinded manner.
Histopathology
The lung tissue was fixed in 4% paraformaldehyde solution for 24 h, and then dehydrated and embedded in paraffin. For H&E stain: After removing the paraffin from the section, immerse the tissue in Harris’s Hematoxylin solution for a duration as required, typically 2–5 min. Rinse the tissue once again under running water to remove excess stain. Apply Eosin solution, usually for 1–2 min, to stain the cytoplasm. Rinse the tissue once more under running water, and then dehydrate them in different concentrations of ethanol. Seal the tissue in a clearing and hydrophilic medium.
For Masson’s trichrome stain: lung tissue was stained with hematoxylin for 5∼10min, then rinsed with 1% hydrochloric acid for differentiation. Ponceau 2R, acid fuchsin and glacial acetic acid were prepared into Masson complex solution stain sections for 5∼10min. After rinse with running water, treat with 1% phosphorywood acid for about 5min. Then re-dyed with bright green dyeing solution for 5min and treated with 1% ice acetic acid solution and dehydrated with ethanol for observation.
Plasmids, adeno-associated virus, and lentivirus production
pDGM6 and PLVX-SHRNA1 were used. pDGM6 were first modified to AAV6.2FF using standard molecular cloning techniques. Adeno-associated virus was produced in AAV-293 cells using Lipofiter reagent (Hanbio, HB-LF-1000) with viral packaging constructs AAV6.2FF. After 72 h of transfection, cells containing AAV particles were harvested. Purification was carried out through column chromatography employing the Adeno-Associated Virus Purification Kit (Biomiga, V1469-01). Adeno-associated virus titer was determined using the SYBRGreen method. All the target sequences for shRNA were listed in the Table S1. The short hairpin RNA constructs against Cd36 were designed and cloned in PLVX-SHRANA1 according to instructions. Lentiviruses were produced in HEK293T cells using calcium phosphate with viral packaging constructs pMD2.G and pSPAX2. Viral supernatants were harvested at 48 and 72 h after transfection, and filtered through a 0.45 μm low protein binding membrane (Millipore).
Quantitative RT-PCR
Total RNA from lung tissue was purified using TRIzol (Life Technologies) according to the manufacturer’s instructions. One microgram of purified total RNA was retrotranscribed using the ReverTra Ace qPCR RT Kit (TOYOBO). The levels of specific RNAs were measured using Bio-Rad real-time PCR machine and Fast SybrGreen PCR mastermix according to the manufacturer’s instructions. Primer sequences are listed in the Table S1. The 2 -ΔΔCt method was used to normalize expression.
Immunoprecipitation and immunoblotting
For immunoprecipitation (IP), lung epithelial cell lysates were prepared, IP lysis buffer (Thermo Fisher Scientific) supplemented with both protease and protease inhibitor cocktail (Roche) was employed. The protocol followed the manufacturer’s instructions from Thermo Fisher Scientific. The cleavage product was incubated overnight at 4°C with ZBP1 antibody. The antibody-cleaved product mixture was then bound to A/G magnetic beads for 4 h. Subsequently, the sample was heated to 100°C in SDS loading buffer and boiled for 15 min. The magnetic beads were then washed and the supernatant containing the antigen-antibody complex was retained for further use. For immunoblotting, cells were lysed in RIPA buffer supplemented with a protease inhibitor cocktail (Roche). Subsequently, the total lysates were separated using SDS-PAGE gels. Membranes were blocked at room temperature for 30 min in TBS containing 5% non-fat dried milk. Following this, they were incubated overnight at 4°C with primary antibodies, suitably diluted in the same blocking buffer. After undergoing three washes in TBST, the membranes were exposed to horseradish peroxidase (HRP)-conjugated secondary antibodies, which were diluted in the blocking buffer, for a 1-h incubation at room temperature. This was followed by three additional washes with TBST before the membranes were incubated with ECL Western Blotting Substrate (Bio-Rad) and then exposed using X-ray Super RX Films (Fujifilm).
Elisa
The levels of IL-6, IL-1β, and TNF-α were quantified using enzyme-linked immunosorbent assay (ELISA). Specifically, ELISA kits designed for IL-6, IL-1β, and TNFα were employed following the manufacturer’s provided instructions. The absorbance (450nm) was then measured using a microplate reader (BIOTEK).
Measurement of free fatty acid
Free fatty acid (FFA) levels in medium and mouse serum were determined by Free fatty acids, half micro test kit (sigma) per manufacturer’s instructions. Culturing medium was span to remove residuals and supernatant was collected to determine FFA level. For measurement of FFA level in GAT explants culturing medium, equal amounts of GAT explants (100mg) were cultured in DMEM containing 1% BSA and then treated with indicated cytokines or chemokines for 24h. Culturing medium was then collected to determine FFA level.
Immunofluorescence
For cell immunofluorescence, cells were cultivated on glass-bottom cell culture dishes (Biosharp) and allowed to adhere for a minimum of 24 h prior to their utilization in experimental procedures. After treatment or viral infection, the cells were immobilized using freshly prepared 4% (w/v) paraformaldehyde, rendered permeable with 0.2% (v/v) Triton X-100, and subsequently obstructed using MAXblock Blocking Medium (Beyotime). Subsequent to this, the cells were incubated with primary antibodies Anti-Z-DNA/Z-RNA (Absolute antibody, AB00783-3, 1:200) at 4°C over the course of the night. After undergoing three washes with PBS, the slides were exposed to secondary antibodies conjugated with fluorophores for 1 h at room temperature. Following an additional three rounds of PBS washing, the slides were affixed using ProLong Gold antifade reagent (Thermo Fisher Scientific) and subjected to imaging using a ZEISS LSM 700 confocal microscopy apparatus.
For tissue immunofluorescence, paraffin-embedded tissues were sectioned at 3 mm thickness. In brief, sections were deparaffinized and rehydrated, followed by antigen retrieval using sodium citrate buffer under high temperature, and then allowed to cool naturally to room temperature. Permeabilization was performed with 0.2% Triton X-100 at room temperature for 1 h. The sections were then blocked with 5% goat serum at room temperature. Immunofluorescent staining was carried out with primary antibodies against proSPC (abcam, ab211326, 1:1000), ATGL (Santa Cruz Biotechnology, sc365278, 1:500), ADRP (Proteintech, 15294-1-AP, 1:500), Anti-Z-DNA/Z-RNA (Absolute antibody, AB00783-3, 1:200) with overnight incubation at 4°C. Subsequently, appropriate fluorescent secondary antibodies were applied, and nuclei were stained with DAPI. Fluorescent images were scanned and acquired using the PANNORAMIC MIDI II (3DHISTECH Ltd., Hungary). Image analysis, including the measurement of fluorescent expression area, was performed using ImageJ software.
RNA-seq and data analysis
Total RNA was isolated using Trizol reagent (Thermo Fisher). TruePrep RNA Library Prep Kit for Illumina (Vazyme, #TR502-01) was used for library preparation. RNA libraries were sequenced on an Illumina Hiseq X Ten platform with paired-end reads (150-bp read length). Quality control of the raw fastq files was performed with FastQC (v0.12.1). Data cleaning and adapter trimming were performed with fastp (v0.20.1). Pseudoalignment of the reads was performed with kallisto (v0.46.2). Gene level quantification and differential gene expression analysis was performed with an R package Deseq2 (v1.40.2). Heatmap plot was made with R package pheatmap (v1.0.12). Network analyses were performed with Cytoscape (v3.10.1). Gene set enrichment analysis was performed with GSEA software (4.3).
Public database analysis
Publicly released total RNA-seq data were downloaded from NCBI Gene Expression Omnibus (GEO) datasets. The GEOquery package was used to download the series matrix files of the databases above in R (v4.0.2). Soft formatted family files were downloaded to correctly map the probe ID to the gene symbol. The correlation between two gene expression was analyzed by Pearson’s correlation coefficient. For differentially expressed gene screening, we screened differentially expressed genes (DEGs) between COPD patients and controls using the R package of the Microarray Data Linear Model (limma, version 3.46.0). The significant DEGs were identified according to the thresholds of adjusted p < 0.05 and fold change (FC) > 1.5. The common DEGs in the datasets were visualized by ggVennDiagram. For Gene set enrichment analysis, we use the R Bioconductor package clusterProfiler to test which known pathways are significantly affected in COPD patients. To this end, the genes tested for differential expression between treated and untreated samples were first ranked by decreasing log fold expression changes, and then their enrichment was evaluated against the hallmark gene sets pathway annotations from the MSigDB database. Normalized enrichment scores and empirical p values were estimated using default parameters, and multiple testing correction was carried out using the Benjamini–Hochberg method.
Quantification and statistical analysis
The Student’s t test was used for significance testing. p values of less than 0.05 were considered statistically significant. Statistical analyses were performed using GraphPad Prism 9 0.0 or the R statistical environment. In the figures, asterisks indicate ∗p < 0.05, ∗∗p < 0.01 and ∗∗∗p < 0.001.
Published: September 9, 2024
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.xcrm.2024.101732.
Contributor Information
Haojian Zhang, Email: haojian_zhang@whu.edu.cn.
Jiansheng Li, Email: ljs2015@hactcm.edu.cn.
Supplemental information
References
- 1.Rabe K.F., Watz H. Chronic obstructive pulmonary disease. Lancet. 2017;389:1931–1940. doi: 10.1016/S0140-6736(17)31222-9. [DOI] [PubMed] [Google Scholar]
- 2.Barnes P.J., Burney P.G.J., Silverman E.K., Celli B.R., Vestbo J., Wedzicha J.A., Wouters E.F.M. Chronic obstructive pulmonary disease. Nat. Rev. Dis. Primers. 2015;1:15076. doi: 10.1038/nrdp.2015.76. [DOI] [PubMed] [Google Scholar]
- 3.Kheradmand F., Zhang Y., Corry D.B. Contribution of adaptive immunity to human COPD and experimental models of emphysema. Physiol. Rev. 2023;103:1059–1093. doi: 10.1152/physrev.00036.2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Barnes P.J. New anti-inflammatory targets for chronic obstructive pulmonary disease. Nat. Rev. Drug Discov. 2013;12:543–559. doi: 10.1038/nrd4025. [DOI] [PubMed] [Google Scholar]
- 5.Taraseviciene-Stewart L., Voelkel N.F. Molecular pathogenesis of emphysema. J. Clin. Invest. 2008;118:394–402. doi: 10.1172/JCI31811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Basil M.C., Cardenas-Diaz F.L., Kathiriya J.J., Morley M.P., Carl J., Brumwell A.N., Katzen J., Slovik K.J., Babu A., Zhou S., et al. Human distal airways contain a multipotent secretory cell that can regenerate alveoli. Nature. 2022;604:120–126. doi: 10.1038/s41586-022-04552-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Penkala I.J., Liberti D.C., Pankin J., Sivakumar A., Kremp M.M., Jayachandran S., Katzen J., Leach J.P., Windmueller R., Stolz K., et al. Age-dependent alveolar epithelial plasticity orchestrates lung homeostasis and regeneration. Cell Stem Cell. 2021;28:1775–1789.e5. doi: 10.1016/j.stem.2021.04.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kim C.F.B., Jackson E.L., Woolfenden A.E., Lawrence S., Babar I., Vogel S., Crowley D., Bronson R.T., Jacks T. Identification of bronchioalveolar stem cells in normal lung and lung cancer. Cell. 2005;121:823–835. doi: 10.1016/j.cell.2005.03.032. [DOI] [PubMed] [Google Scholar]
- 9.Liu Q., Liu K., Cui G., Huang X., Yao S., Guo W., Qin Z., Li Y., Yang R., Pu W., et al. Lung regeneration by multipotent stem cells residing at the bronchioalveolar-duct junction. Nat. Genet. 2019;51:728–738. doi: 10.1038/s41588-019-0346-6. [DOI] [PubMed] [Google Scholar]
- 10.Zuo W., Zhang T., Wu D.Z., Guan S.P., Liew A.A., Yamamoto Y., Wang X., Lim S.J., Vincent M., Lessard M., et al. p63(+)Krt5(+) distal airway stem cells are essential for lung regeneration. Nature. 2015;517:616–620. doi: 10.1038/nature13903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Vaughan A.E., Brumwell A.N., Xi Y., Gotts J.E., Brownfield D.G., Treutlein B., Tan K., Tan V., Liu F.C., Looney M.R., et al. Lineage-negative progenitors mobilize to regenerate lung epithelium after major injury. Nature. 2015;517:621–625. doi: 10.1038/nature14112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Basil M.C., Katzen J., Engler A.E., Guo M., Herriges M.J., Kathiriya J.J., Windmueller R., Ysasi A.B., Zacharias W.J., Chapman H.A., et al. The Cellular and Physiological Basis for Lung Repair and Regeneration: Past, Present, and Future. Cell Stem Cell. 2020;26:482–502. doi: 10.1016/j.stem.2020.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Barkauskas C.E., Cronce M.J., Rackley C.R., Bowie E.J., Keene D.R., Stripp B.R., Randell S.H., Noble P.W., Hogan B.L.M. Type 2 alveolar cells are stem cells in adult lung. J. Clin. Invest. 2013;123:3025–3036. doi: 10.1172/JCI68782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nabhan A.N., Brownfield D.G., Harbury P.B., Krasnow M.A., Desai T.J. Single-cell Wnt signaling niches maintain stemness of alveolar type 2 cells. Science. 2018;359:1118–1123. doi: 10.1126/science.aam6603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zacharias W.J., Frank D.B., Zepp J.A., Morley M.P., Alkhaleel F.A., Kong J., Zhou S., Cantu E., Morrisey E.E. Regeneration of the lung alveolus by an evolutionarily conserved epithelial progenitor. Nature. 2018;555:251–255. doi: 10.1038/nature25786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kathiriya J.J., Brumwell A.N., Jackson J.R., Tang X., Chapman H.A. Distinct Airway Epithelial Stem Cells Hide among Club Cells but Mobilize to Promote Alveolar Regeneration. Cell Stem Cell. 2020;26:346–358.e4. doi: 10.1016/j.stem.2019.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Conlon T.M., John-Schuster G., Heide D., Pfister D., Lehmann M., Hu Y., Ertüz Z., Lopez M.A., Ansari M., Strunz M., et al. Inhibition of LTbetaR signalling activates WNT-induced regeneration in lung. Nature. 2020;588:151–156. doi: 10.1038/s41586-020-2882-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Baarsma H.A., Skronska-Wasek W., Mutze K., Ciolek F., Wagner D.E., John-Schuster G., Heinzelmann K., Günther A., Bracke K.R., Dagouassat M., et al. Noncanonical WNT-5A signaling impairs endogenous lung repair in COPD. J. Exp. Med. 2017;214:143–163. doi: 10.1084/jem.20160675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wu H., Tang N. Stem cells in pulmonary alveolar regeneration. Development. 2021;148:dev193458. doi: 10.1242/dev.193458. [DOI] [PubMed] [Google Scholar]
- 20.Zepp J.A., Zacharias W.J., Frank D.B., Cavanaugh C.A., Zhou S., Morley M.P., Morrisey E.E. Distinct Mesenchymal Lineages and Niches Promote Epithelial Self-Renewal and Myofibrogenesis in the Lung. Cell. 2017;170:1134–1148.e10. doi: 10.1016/j.cell.2017.07.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shiraishi K., Shah P.P., Morley M.P., Loebel C., Santini G.T., Katzen J., Basil M.C., Lin S.M., Planer J.D., Cantu E., et al. Biophysical forces mediated by respiration maintain lung alveolar epithelial cell fate. Cell. 2023;186:1478–1492.e15. doi: 10.1016/j.cell.2023.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wu H., Yu Y., Huang H., Hu Y., Fu S., Wang Z., Shi M., Zhao X., Yuan J., Li J., et al. Progressive Pulmonary Fibrosis Is Caused by Elevated Mechanical Tension on Alveolar Stem Cells. Cell. 2020;180:107–121.e17. doi: 10.1016/j.cell.2019.11.027. [DOI] [PubMed] [Google Scholar]
- 23.Han S., Lee M., Shin Y., Giovanni R., Chakrabarty R.P., Herrerias M.M., Dada L.A., Flozak A.S., Reyfman P.A., Khuder B., et al. Mitochondrial integrated stress response controls lung epithelial cell fate. Nature. 2023;620:890–897. doi: 10.1038/s41586-023-06423-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang Z., Wei D., Bin E., Li J., Jiang K., Lv T., Mao X., Wang F., Dai H., Tang N. Enhanced glycolysis-mediated energy production in alveolar stem cells is required for alveolar regeneration. Cell Stem Cell. 2023;30:1028–1042.e7. doi: 10.1016/j.stem.2023.07.007. [DOI] [PubMed] [Google Scholar]
- 25.Liang J., Huang G., Liu X., Taghavifar F., Liu N., Wang Y., Deng N., Yao C., Xie T., Kulur V., et al. The ZIP8/SIRT1 axis regulates alveolar progenitor cell renewal in aging and idiopathic pulmonary fibrosis. J. Clin. Invest. 2022;132:e157338. doi: 10.1172/JCI157338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li X., Wu J., Sun X., Wu Q., Li Y., Li K., Zhang Q., Li Y., Abel E.D., Chen H. Autophagy Reprograms Alveolar Progenitor Cell Metabolism in Response to Lung Injury. Stem Cell Rep. 2020;14:420–432. doi: 10.1016/j.stemcr.2020.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yao C., Guan X., Carraro G., Parimon T., Liu X., Huang G., Mulay A., Soukiasian H.J., David G., Weigt S.S., et al. Senescence of Alveolar Type 2 Cells Drives Progressive Pulmonary Fibrosis. Am. J. Respir. Crit. Care Med. 2021;203:707–717. doi: 10.1164/rccm.202004-1274OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Liang J., Zhang Y., Xie T., Liu N., Chen H., Geng Y., Kurkciyan A., Mena J.M., Stripp B.R., Jiang D., Noble P.W. Hyaluronan and TLR4 promote surfactant-protein-C-positive alveolar progenitor cell renewal and prevent severe pulmonary fibrosis in mice. Nat. Med. 2016;22:1285–1293. doi: 10.1038/nm.4192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Celli B.R., Locantore N., Tal-Singer R., Riley J., Miller B., Vestbo J., Yates J.C., Silverman E.K., Owen C.A., Divo M., et al. Emphysema and extrapulmonary tissue loss in COPD: a multi-organ loss of tissue phenotype. Eur. Respir. J. 2018;51:1702146. doi: 10.1183/13993003.02146-2017. [DOI] [PubMed] [Google Scholar]
- 30.Houben-Wilke S., Augustin I.M., Vercoulen J.H., van Ranst D., Bij de Vaate E., Wempe J.B., Spruit M.A., Wouters E.F.M., Franssen F.M.E. COPD stands for complex obstructive pulmonary disease. Eur. Respir. Rev. 2018;27:180027. doi: 10.1183/16000617.0027-2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sanders K.J.C., Kneppers A.E.M., van de Bool C., Langen R.C.J., Schols A.M.W.J. Cachexia in chronic obstructive pulmonary disease: new insights and therapeutic perspective. J. Cachexia Sarcopenia Muscle. 2016;7:5–22. doi: 10.1002/jcsm.12062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Grabner G.F., Xie H., Schweiger M., Zechner R. Lipolysis: cellular mechanisms for lipid mobilization from fat stores. Nat. Metab. 2021;3:1445–1465. doi: 10.1038/s42255-021-00493-6. [DOI] [PubMed] [Google Scholar]
- 33.Takabatake N., Nakamura H., Abe S., Hino T., Saito H., Yuki H., Kato S., Tomoike H. Circulating leptin in patients with chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 1999;159:1215–1219. doi: 10.1164/ajrccm.159.4.9806134. [DOI] [PubMed] [Google Scholar]
- 34.Schols A.M., Creutzberg E.C., Buurman W.A., Campfield L.A., Saris W.H., Wouters E.F. Plasma leptin is related to proinflammatory status and dietary intake in patients with chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 1999;160:1220–1226. doi: 10.1164/ajrccm.160.4.9811033. [DOI] [PubMed] [Google Scholar]
- 35.Das S.K., Eder S., Schauer S., Diwoky C., Temmel H., Guertl B., Gorkiewicz G., Tamilarasan K.P., Kumari P., Trauner M., et al. Adipose triglyceride lipase contributes to cancer-associated cachexia. Science. 2011;333:233–238. doi: 10.1126/science.1198973. [DOI] [PubMed] [Google Scholar]
- 36.Silverstein R.L., Febbraio M. CD36, a scavenger receptor involved in immunity, metabolism, angiogenesis, and behavior. Sci. Signal. 2009;2:re3. doi: 10.1126/scisignal.272re3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wang H., Franco F., Tsui Y.C., Xie X., Trefny M.P., Zappasodi R., Mohmood S.R., Fernández-García J., Tsai C.H., Schulze I., et al. CD36-mediated metabolic adaptation supports regulatory T cell survival and function in tumors. Nat. Immunol. 2020;21:298–308. doi: 10.1038/s41590-019-0589-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Xu S., Chaudhary O., Rodriguez-Morales P., Sun X., Chen D., Zappasodi R., Xu Z., Pinto A.F.M., Williams A., Schulze I., et al. Uptake of oxidized lipids by the scavenger receptor CD36 promotes lipid peroxidation and dysfunction in CD8(+) T cells in tumors. Immunity. 2021;54:1561–1577.e1567. doi: 10.1016/j.immuni.2021.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ma X., Xiao L., Liu L., Ye L., Su P., Bi E., Wang Q., Yang M., Qian J., Yi Q. CD36-mediated ferroptosis dampens intratumoral CD8(+) T cell effector function and impairs their antitumor ability. Cell Metab. 2021;33:1001–1012.e5. doi: 10.1016/j.cmet.2021.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Li R., Adami A., Chang C.C., Tseng C.H., Hsiai T.K., Rossiter H.B. Serum Acylglycerols Inversely Associate with Muscle Oxidative Capacity in Severe COPD. Med. Sci. Sports Exerc. 2021;53:10–18. doi: 10.1249/MSS.0000000000002441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fritsche K. Fatty acids as modulators of the immune response. Annu. Rev. Nutr. 2006;26:45–73. doi: 10.1146/annurev.nutr.25.050304.092610. [DOI] [PubMed] [Google Scholar]
- 42.Goransson O., Kopietz F., Rider M.H. Metabolic control by AMPK in white adipose tissue. Trends Endocrinol Metab. 2023;34:704–717. doi: 10.1016/j.tem.2023.08.011. [DOI] [PubMed] [Google Scholar]
- 43.Raman T., O'Connor T.P., Hackett N.R., Wang W., Harvey B.G., Attiyeh M.A., Dang D.T., Teater M., Crystal R.G. Quality control in microarray assessment of gene expression in human airway epithelium. BMC Genom. 2009;10:493. doi: 10.1186/1471-2164-10-493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tanosaki T., Mikami Y., Shindou H., Suzuki T., Hashidate-Yoshida T., Hosoki K., Kagawa S., Miyata J., Kabata H., Masaki K., et al. Lysophosphatidylcholine Acyltransferase 1 Deficiency Promotes Pulmonary Emphysema via Apoptosis of Alveolar Epithelial Cells. Inflammation. 2022;45:1765–1779. doi: 10.1007/s10753-022-01659-4. [DOI] [PubMed] [Google Scholar]
- 45.Ammous Z., Hackett N.R., Butler M.W., Raman T., Dolgalev I., O'Connor T.P., Harvey B.G., Crystal R.G. Variability in small airway epithelial gene expression among normal smokers. Chest. 2008;133:1344–1353. doi: 10.1378/chest.07-2245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Callejon-Leblic B., Pereira-Vega A., Vazquez-Gandullo E., Sanchez-Ramos J.L., Gomez-Ariza J.L., Garcia-Barrera T. Study of the metabolomic relationship between lung cancer and chronic obstructive pulmonary disease based on direct infusion mass spectrometry. Biochimie. 2019;157:111–122. doi: 10.1016/j.biochi.2018.11.007. [DOI] [PubMed] [Google Scholar]
- 47.Nambiar S., Tan D.B.A., Clynick B., Bong S.H., Rawlinson C., Gummer J., Corte T.J., Glaspole I., Moodley Y.P., Trengove R. Untargeted metabolomics of human plasma reveal lipid markers unique to chronic obstructive pulmonary disease and idiopathic pulmonary fibrosis. Proteomics. Clin. Appl. 2021;15:e2000039. doi: 10.1002/prca.202000039. [DOI] [PubMed] [Google Scholar]
- 48.Kang M.H., van Lieshout L.P., Xu L., Domm J.M., Vadivel A., Renesme L., Mühlfeld C., Hurskainen M., Mižíková I., Pei Y., et al. A lung tropic AAV vector improves survival in a mouse model of surfactant B deficiency. Nat. Commun. 2020;11:3929. doi: 10.1038/s41467-020-17577-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Rindler T.N., Brown K.M., Stockman C.A., van Lieshout L.P., Martin E.P., Weaver T.E., Zacharias W.J., Wootton S.K., Whitsett J.A., Bridges J.P. Efficient Transduction of Alveolar Type 2 Cells with Adeno-associated Virus for the Study of Lung Regeneration. Am. J. Respir. Cell Mol. Biol. 2021;65:118–121. doi: 10.1165/rcmb.2021-0049LE. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hu Y., Ng-Blichfeldt J.P., Ota C., Ciminieri C., Ren W., Hiemstra P.S., Stolk J., Gosens R., Königshoff M. Wnt/beta-catenin signaling is critical for regenerative potential of distal lung epithelial progenitor cells in homeostasis and emphysema. Stem Cell. 2020;38:1467–1478. doi: 10.1002/stem.3241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Shan B., Pan H., Najafov A., Yuan J. Necroptosis in development and diseases. Genes Dev. 2018;32:327–340. doi: 10.1101/gad.312561.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chen D., Gregory A.D., Li X., Wei J., Burton C.L., Gibson G., Scott S.J., St Croix C.M., Zhang Y., Shapiro S.D. RIP3-dependent necroptosis contributes to the pathogenesis of chronic obstructive pulmonary disease. JCI Insight. 2021;6:e144689. doi: 10.1172/jci.insight.144689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Van Eeckhoutte H.P., Donovan C., Kim R.Y., Conlon T.M., Ansari M., Khan H., Jayaraman R., Hansbro N.G., Dondelinger Y., Delanghe T., et al. RIPK1 kinase-dependent inflammation and cell death contribute to the pathogenesis of COPD. Eur. Respir. J. 2023;61:2201506. doi: 10.1183/13993003.01506-2022. [DOI] [PubMed] [Google Scholar]
- 54.Szczesny B., Marcatti M., Ahmad A., Montalbano M., Brunyánszki A., Bibli S.I., Papapetropoulos A., Szabo C. Mitochondrial DNA damage and subsequent activation of Z-DNA binding protein 1 links oxidative stress to inflammation in epithelial cells. Sci. Rep. 2018;8:914. doi: 10.1038/s41598-018-19216-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Griffin M.F., Borrelli M.R., Garcia J.T., Januszyk M., King M., Lerbs T., Cui L., Moore A.L., Shen A.H., Mascharak S., et al. promotes hypertrophic skin scarring via CD36 in preclinical in vitro and in vivo models. Sci. Transl. Med. 2021;13:eabb3312. doi: 10.1126/scitranslmed.abb3312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Pascual G., Avgustinova A., Mejetta S., Martín M., Castellanos A., Attolini C.S.O., Berenguer A., Prats N., Toll A., Hueto J.A., et al. Targeting metastasis-initiating cells through the fatty acid receptor CD36. Nature. 2017;541:41–45. doi: 10.1038/nature20791. [DOI] [PubMed] [Google Scholar]
- 57.Shapiro S.D., Goldstein N.M., Houghton A.M., Kobayashi D.K., Kelley D., Belaaouaj A. Neutrophil elastase contributes to cigarette smoke-induced emphysema in mice. Am. J. Pathol. 2003;163:2329–2335. doi: 10.1016/S0002-9440(10)63589-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Landbo C., Prescott E., Lange P., Vestbo J., Almdal T.P. Prognostic value of nutritional status in chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 1999;160:1856–1861. doi: 10.1164/ajrccm.160.6.9902115. [DOI] [PubMed] [Google Scholar]
- 59.Celli B.R., Cote C.G., Marin J.M., Casanova C., Montes de Oca M., Mendez R.A., Pinto Plata V., Cabral H.J. The body-mass index, airflow obstruction, dyspnea, and exercise capacity index in chronic obstructive pulmonary disease. N. Engl. J. Med. 2004;350:1005–1012. doi: 10.1056/NEJMoa021322. [DOI] [PubMed] [Google Scholar]
- 60.Schols A.M., Slangen J., Volovics L., Wouters E.F. Weight loss is a reversible factor in the prognosis of chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 1998;157:1791–1797. doi: 10.1164/ajrccm.157.6.9705017. [DOI] [PubMed] [Google Scholar]
- 61.Kwan H.Y., Maddocks M., Nolan C.M., Jones S.E., Patel S., Barker R.E., Kon S.S.C., Polkey M.I., Cullinan P., Man W.D.C. The prognostic significance of weight loss in chronic obstructive pulmonary disease-related cachexia: a prospective cohort study. J. Cachexia Sarcopenia Muscle. 2019;10:1330–1338. doi: 10.1002/jcsm.12463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Stewart C.R., Stuart L.M., Wilkinson K., van Gils J.M., Deng J., Halle A., Rayner K.J., Boyer L., Zhong R., Frazier W.A., et al. CD36 ligands promote sterile inflammation through assembly of a Toll-like receptor 4 and 6 heterodimer. Nat. Immunol. 2010;11:155–161. doi: 10.1038/ni.1836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.McCowan J., Fercoq F., Kirkwood P.M., T'Jonck W., Hegarty L.M., Mawer C.M., Cunningham R., Mirchandani A.S., Hoy A., Humphries D.C., et al. The transcription factor EGR2 is indispensable for tissue-specific imprinting of alveolar macrophages in health and tissue repair. Sci. Immunol. 2021;6:eabj2132. doi: 10.1126/sciimmunol.abj2132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Tsoli M., Swarbrick M.M., Robertson G.R. Lipolytic and thermogenic depletion of adipose tissue in cancer cachexia. Semin. Cell Dev. Biol. 2016;54:68–81. doi: 10.1016/j.semcdb.2015.10.039. [DOI] [PubMed] [Google Scholar]
- 65.Moiseeva V., Cisneros A., Sica V., Deryagin O., Lai Y., Jung S., Andrés E., An J., Segalés J., Ortet L., et al. Senescence atlas reveals an aged-like inflamed niche that blunts muscle regeneration. Nature. 2023;613:169–178. doi: 10.1038/s41586-022-05535-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Sauler M., Bazan I.S., Lee P.J. Cell Death in the Lung: The Apoptosis-Necroptosis Axis. Annu. Rev. Physiol. 2019;81:375–402. doi: 10.1146/annurev-physiol-020518-114320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Wang R., Li H., Wu J., Cai Z.Y., Li B., Ni H., Qiu X., Chen H., Liu W., Yang Z.H., et al. Gut stem cell necroptosis by genome instability triggers bowel inflammation. Nature. 2020;580:386–390. doi: 10.1038/s41586-020-2127-x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
RNA-seq data reported in this paper were deposited in GEO under the accession number GEO: GSE246501. This paper analyzes publicly available data GSE8545, GSE186359, and GSE11906.
-
•
This paper does not report the original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.