

Abstract
Currently fielded treatments for nerve agent intoxication include atropine, an acetylcholine receptor antagonist, and pralidoxime (2PAM), a small molecule reactivator of acetylcholinesterase (AChE). 2PAM reactivates nerve agent-inhibited AChE via direct nucleophilic attack by the oxime moiety on the phosphorus center of the bound nerve agent. Due to a permanently charged pyridinium motif, 2PAM is not thought to cross the blood brain barrier and therefore cannot act directly in the neuronal junctions of the brain. In this study, a non-permanently charged, non-oxime molecule (ADOC) initially identified using pesticide-inhibited AChE was characterized in vitro against nerve agent inhibited recombinant human AChE. The inhibitory and reactivation potentials of ADOC were determined with native AChE and AChE inhibited with tabun, sarin, soman, cyclosarin, VX, or VR and then compared to those of 2PAM. Several structural analogs of ADOC were used to probe the reactivation mechanism of the molecule. Finally, guinea pigs were used to examine the protective efficacy of the compound after exposure to sarin. The results of both in vitro and in vivo testing will be useful in the design of future small molecule reactivators.
Keywords: Reactivator, Oxime, Organophosphorus Nerve Agent, Pralidoxime, Guinea Pig, Acetylcholinesterase
1. Introduction
Organophosphorus (OP) nerve agents, especially those known as chemical warfare agents (Figure 1), are highly toxic [1]. These compounds induce toxicity by irreversibly inhibiting acetylcholinesterase (AChE) via covalent bond formation (phosphonylation) with the active site serine (S200), which in turn allows the accumulation of the neurotransmitter acetylcholine [2]. This inhibition event occurs in nerve junctions in both the peripheral body systems and, once nerve agent has crossed the blood brain barrier, in the central nervous system (CNS) [3]. If left untreated, the excess acetylcholine swiftly causes a cholinergic crisis leading to disruption of respiratory and cardiac function, and subsequent death. Recent use of OP nerve agents against civilian populations [4, 5] underscores the need for novel treatments against OP intoxication as well as increased clinical knowledge of appropriate procedures for the management of nerve agent casualties. The current standard treatment for nerve agent intoxication includes administration of atropine (an antimuscarinic), diazepam (an anticonvulsant), and a pyridinium oxime (in the United States, pralidoxime (2PAM, Figure 1)) which acts to reactivate phosphonylated AChE [6].
Figure 1.

The chemical structures of six organophosphorus nerve agents and two reactivator compounds referenced in this work.
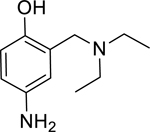
Pyridinium oximes are thought to reactivate phosphonylated AChE via a direct nucleophilic attack of the oximate anion on the phosphorus group [7, 8]. Cleavage of the phosphorus-serine bond releases the resulting phosphonylated oxime molecule which upon dissociation regenerates AChE in its active form. Unfortunately, the permanent positive charge of the pyridinium moiety found in 2PAM (and oximes such as 1–2-hydroxyiminomethyl-1-pyridino-3–4-carbamoyl-1-pyridino-2-oxapropane (HI6) and 1,1’-oxybis(methylene) bis4-(E)-hydroxyiminomethylpyridinium (obidoxime) which are fielded in other countries) prevents these molecules from efficiently crossing the blood brain barrier and reactivating inhibited AChE in the brain and CNS [6]. A tertiary oxime, monoisonitrosoacetone (MINA), which does not contain a quaternary amine motif, has been shown to enter the CNS and reactivate phosphonylated acetylcholinesterase in the brain [3]. However, the absence of the pyridinium motif in MINA means that the active oximate anion is formed much less frequently at physiological pH, likely accounting for the overall decreased reactivation potential at pH 7.4 of MINA as compared to 2PAM [9, 10]. A need exists for novel drugs which not only reactivate OP-inhibited AChE but also cross the blood brain barrier to act on inhibited enzyme found in the CNS. The permanent positive charge generally needed to lower the pKa of an oxime moiety to a level which will be more reactive at physiological pH does not lend itself to these needs. Initially, 4-amino-2-((diethylamino)methyl)phenol (1; ADOC; Figure 1) was discovered to have reactivation potential for AChE inhibited by the OP pesticide diisopropyl fluorophosphates (DFP) via a high thru-put screening method [11]. In this study, the novel reactivator ADOC, which does not utilize a standard oxime motif, is characterized for activity against OP nerve agent inhibited AChE. A series of structural analogs of ADOC were also tested, and experiments designed to probe the mechanism of reactivation were conducted. Finally, the in vivo activity of ADOC was explored using a standardized reactivation experiment [12] in guinea pigs.
2. Materials and methods
2.1. General Materials
All inhibition and reactivation assays were conducted using recombinant human acetylcholinesterase (RHuAChE, AA780, Chesapeake PERL, Inc. Savage, MD). Hydrolysis of acetylthiocholine iodide (AtCh, A5751, Sigma-Aldrich Co. LLC., St. Louis, MO) was measured using 5,5′-Dithiobis-2-nitrobenzoic acid (DTNB, D8130, Sigma-Aldrich Co. LLC., St. Louis, MO). Measurements were made using a SpectraMax Plus 384 Microplate Reader (Molecular Devices, Sunnyvale, CA) utilizing 96-well clear bottom plates (655101, Greiner Bio-One GmbH, Frickenhausen, Germany). Unless otherwise indicated, all assays were performed in 0.1 M potassium phosphate buffer at pH 7.0 with 1 mg/mL bovine serum albumin (BSA, A7906, Sigma-Aldrich Co. LLC., St. Louis, MO) included to help stabilize the RHuAChE. All organophosphorus nerve agents were obtained from the US Army Edgewood Chemical Biological Center (Aberdeen Proving Ground, MD) at a purity >98.5% as determined by 31P NMR.
2.2. Compounds
1: 4-amino-α-diethylamino-O-cresol (ADOC; Sigma-Aldrich Co. LLC Catalog # S561274)
2: 2-[(diethylamino)methyl]phenol (Princeton BioMolecular Research Catalog# OSSK_515806)
3: 4-aminophenol (Sigma-Aldrich Co. LLC Catalog # 60034)
4: (3-aminobenzyl)diethylamine (Sigma-Aldrich Co. LLC Catalog # CBR00121)
5: 3-[(diethylamino)methyl]-4-ethoxyaniline (Enamine Catalog# EN300–80977)
6: 4-amino-3-((diethylamino)methyl)phenol (Synthesized for this research; 1H NMR (600 MHz, Methanol-d4) δ 6.67 – 6.53 (m, 3H), 3.53 (s, 2H), 2.53 (q, J = 7.2 Hz, 4H), 1.06 (t, J = 7.2 Hz, 6H). 13C NMR (151 MHz, MeOD) δ 149.62, 138.39, 125.70, 117.45, 116.98, 114.30, 56.49, 46.25, 10.54.)
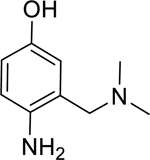
7: 4-amino-2-((dimethylamino)methyl)phenol (Synthesized for this research; 1H NMR (400 MHz, Methanol-d4) δ 6.65 – 6.54 (m, 3H), 3.57 (s, 2H), 2.32 (s, 6H). 13C NMR (101 MHz, MeOD) δ 149.91, 138.71, 122.48, 117.10, 116.42, 115.54, 61.00, 43.26.)
8: 4-amino-3-((dimethylamino)methyl)phenol (Synthesized for this research; 1H NMR (400 MHz, Methanol-d4) δ 6.69 – 6.50 (m, 3H), 3.37(s, 2H), 2.23 (s, 6H). 13C NMR (101 MHz, MeOD) δ 149.56, 138.31, 125.15, 117.56, 116.95, 114.65, 61.98, 43.89.)
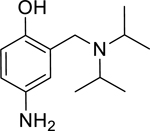
9: 4-amino-3-((diisopropylamino)methyl)phenol (Synthesized for this research; 1H NMR (400 MHz, Chloroform-d) δ 6.62 (d, J = 8.4 Hz, 1H), 6.54 (dd, J = 8.4, 2.8 Hz, 1H), 6.41 (d, J = 2.6 Hz, 1H), 3.78 (s, 2H), 3.17 (p, J = 6.7 Hz, 2H), 1.12 (d, J = 6.7 Hz, 12H). 13C NMR (101 MHz, CDCl3) δ 151.29, 138.04, 123.27, 116.36, 115.88, 115.48, 48.55, 47.70, 19.79.)
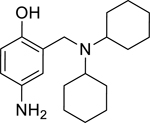
10: 4-amino-2-((dicyclohexylamino)methyl)phenol (Synthesized for this research; 1H NMR (400 MHz, Chloroform-d) δ 7.92 (d, J = 2.8 Hz, 1H), 7.28 (s, 1H), 6.75 (d, J = 9.0 Hz, 1H), 4.05 (s, 2H), 2.83 – 2.72 (m, 2H), 1.94 – 1.03 (multiple peaks, 20H). 13C NMR (101 MHz, CDCl3) δ 167.19, 138.94, 125.30, 124.32, 121.87, 116.65, 58.36, 49.50, 30.06, 25.89, 25.68.)
11: 2-((diethylamino)methyl)-4-(dimethylamino)phenol (Synthesized for this research; 1H NMR (600 MHz, Chloroform-d) δ 6.73 (d, J = 8.7 Hz, 1H), 6.66 (dd, J = 8.7, 3.0 Hz, 1H), 6.47 (d, J = 2.9 Hz, 1H), 3.73 (s, 2H), 2.82 (s, 6H), 2.61 (q, J = 7.2 Hz, 4H), 1.10 (t, J = 7.2 Hz, 6H). 13C NMR (151 MHz, CDCl3) δ 150.43, 144.44, 122.46, 116.30, 114.82, 114.48, 57.44, 46.32, 42.17.)
12: 4-acetamido-2-[(diethylamino)methyl]phenol (Sigma-Aldrich Co. LLC Catalog # S414026)
13: 2-amino-6-((diethylamino)methyl)phenol (Synthesized for this research; 1H NMR (400 MHz, Chloroform-d) δ 6.72 – 6.57 (m, 2H), 6.43 (dd, J = 7.0, 1.8, 1H), 5.32 (s, 1H), 3.75 (s, 2H), 2.64 (q, J = 7.2 Hz, 3H), 1.13 (t, J = 7.2 Hz, 6H). 13C NMR (101 MHz, Chloroform-d) δ 145.80 , 134.80 , 121.44 , 118.88 , 118.16, 114.39 , 56.83 , 46.26.)
14: 2-amino-6-((dimethylamino)methyl)phenol (Synthesized for this research; 1H NMR (400 MHz, Chloroform-d) δ 6.70 – 6.61 (m, 2H), 6.44 – 6.41 (m, 1H), 3.63 (s, 2H), 2.34 (s, 6H). 13C NMR (101 MHz, Chloroform-d) δ 145.54 , 134.78 , 121.34 , 119.00 , 118.11 , 114.59 , 62.77 , 44.51.)
Synthesis of compounds 6, 7, 8, 9, and 10 followed the general procedure of adding the amine (diethylamine for compound 6, dimethylamine for compound 7 and 8, diisopropylamine for compound 9, dicyclohexylamine for compound 10) to a solution of 2-hydroxy-5-nitrobenzaldehyde (7,9,10) or 5-hydroxy-2-nitrobenzaldehyde(6, 8) in DCM. The solution was cooled to 0ºC and 6 molar equivalents of sodium triactoxyborohydride were added followed by stirring at room temperature for 5 hours. Reactions were then quenched by the addition of aqueous sodium bicarbonate and extracted using ethyl acetate after which the combined extracts were washed with brine and dried with magnesium sulfate. Solvent was then removed via rotary evaporation and the crude products were isolated using flash chromatography (10% MeOH/DCM). Lithium aluminum hydride (10 molar equivalents) was added to the purified product in dichloromethane at 0ºC. The reaction was allowed to return to room temperature and was then stirred for 5 hours. Quenching with 15% aqueous sodium hydroxide at 0ºC was followed by a filtration through celite. Solvent was then removed as described earlier and the products were again purified using flash chromatography (20% MeOH/DCM).
Synthesis of compound 11: To a solution of ADOC in THF was added formaldehyde (10 molar equivalents), and then triactoxyborohydride(6 molar equivalents) was added at 0 ºC. The solution was allowed to return to room temperature and stirred for 5 hours. The reaction was then quenched by the addition of sodium bicarbonate and extracted with ethylacetate after which combined organic layers were washed with brine and dried with magnesium sulfate. Solvent was removed via rotary evaporation and the products were purified by flash chromatography (20% MeOH/DCM).
Synthesis of compounds 13 and 14: Into a solution of 3-nitrosalicylic acid was added 1-[3-(dimethylamino)propyl]-3-ethylcarbodiimide hydrochloride (EDC, 1.5 molar equivalent). The solution was stirred for 15 minutes before diethylamine (13) or dimethylamine (14) (1.2 molar equivalents) was added followed by addition of DMAP (2 molar equivalents). The reaction was stirred for 16 hours before 1N HCl was added and the aqueous layer extracted by DCM. The organic extracts were combined and dried over Na2SO4 and reduced in vacuo. The product was purified by flash chromatography (10% MeOH/DCM) to give 2-hydroxy-N,N-diethyl-3-nitrobenzamide as an oil. Into LiAlH4 (10 molar equivalents) in dry THF was added 2-hydroxy-N,N-diethyl-3-nitrobenzamide. The resulting mixture was refluxed for 5 hours then quenched using the Fieser method [13], dried over Na2SO4, and filtered. The filtered solution was reduced in vacuo to give the product as a brown oil.
2.3. Measurement of IC50
In 96-well plates, RHuAChE activity against AtCh was measured in the presence of the indicated compound at concentrations ranging from 60 nM to125 mM. 100% activity was determined using a negative control containing enzyme alone, and all activities were compared relative to this control. The resulting activities were plotted against the log value of the compound concentration and fit using a variable slope dose response curve (Prism, GraphPad Software, Inc., La Jolla, CA).
2.4. Inhibition
To determine the type of inhibition displayed by ADOC, the activity of RHuAChE was measured in the presence of several concentrations of that compound at varying concentrations of AtCh. Inverse velocities were plotted according to the inverse AtCh concentration and fit using a linear regression analysis (Prism, GraphPad Software, Inc., La Jolla, CA) to determine the nature of the inhibition.[11]
2.5. Reactivation
RHuAChE was inhibited with a molar excess of nerve agent for a minimum of 10 min at room temperature. Excess free nerve agent was removed using Centri-Sep spin columns (Life Technologies, Grand Island, NY) and the inhibited sample was diluted to an appropriate concentration for use in further assays. An uninhibited positive control was similarly treated using buffer in place of nerve agent. Enzyme samples were mixed with reactivator compound to initiate reactivation and aliquots were removed at various time points, diluted twenty-fold and measured for activity against AtCh. Percent reactivation was calculated by comparing each sample to a row matched positive control of uninhibited RHuAChE in the presence of reactivator. Residual activity at time zero was subtracted from each following time point to account for incomplete inhibition, and then triplicates were averaged. Nonlinear regression assuming maximal reactivation of 100% was used to determine kobs [14, 15] at each reactivator concentration. These kobs values were plotted against reactivator concentration and fit using a modified Michaelis-Menten equation.[16] Alternatively, the half-time of reactivation (t1/2) was calculated for many compounds at a single concentration of 1 mM using a one-phase association fit of the percent reactivation over time (Prism, GraphPad Software, Inc., La Jolla, CA), and these times were then compared to the spontaneous reactivation half-time of sample matched inhibited enzyme using a ratio paired t-test.
2.6. In vivo Experimental Procedure
All testing was done as previously described in Shih et al. (2009 [12]). In short, blood was collected at least 24 hours prior to experimentation from male Hartley guinea pigs (250 – 330g) from Charles River Labs (Kingston, NY). These samples were used to establish baseline acetylcholinesterase activity in red blood cells and whole blood for each animal using a modification of the assay described by Ellman et al. (1961 [17]). On the study day, each animal received a pretreatment of atropine methyl nitrate (2 mg/kg) intramuscularly followed fifteen minutes later with exposure to a 1.0 LD50 dose of sarin (GB), cyclosarin (GF), VX, or saline via subcutaneous injection. Five minutes later, when the inhibition of acetylcholinesterase activity by these nerve agents reached maximum levels in the blood, one of 5 doses of ADOC (1.8, 10.0, 18.0, 32.0, and 56.0 mg/kg) or saline were administered intramuscularly. One hour after nerve agent exposure and 45 minutes after reactivator administration, animals were humanely euthanized and all samples were collected and stored for later analysis.
3. Results
Initially, ADOC was tested in vitro for reactivation potential with recombinant human acetylcholinesterase (RHuAChE) inhibited by sarin (GB) or VX (data not shown). While reactivation was noted in both cases, full reactivation to uninhibited RHuAChE activity levels was not achieved under the conditions tested. To determine if the lack of complete reactivation was due to direct inhibition of the enzyme by ADOC, IC50 values for ADOC (17.8μM) and 2PAM (456μM) were determined at a fixed concentration of RHuAChE (Figure 2A). Because the high level of inhibition displayed by ADOC could interfere with further in vitro testing of reactivation using established discontinuous, indirect methods, subsequent assays were conducted to determine the type of inhibition caused by ADOC (Figure 2B).
Figure 2.

A) IC50 of ADOC(●) and 2PAM (▲) was determined with human recombinant acetylcholinesterase (RHuAChE). B) Competitive inhibition assay using ADOC in increasing concentrations as the inhibitor and AtCh as the competitive substrate.
ADOC displayed a mixture of competitive (Ki = 6.3±0.9μM) and noncompetitive (Ki’ = 37.4±2.1μM) reversible inhibition indicating the possibility of two binding sites in agreement with the findings of Katz et al [11]. Given these findings, the inhibition of RHuAChE by reactivator molecules was accounted for in all subsequent experiments, where the activity of the enzyme in the presence of each concentration of each reactivator (absent any OP) was used as the baseline activity level for the corresponding OP-inhibited sample. This was crucial to account for the experimental parameters used to test the reactivation potential of ADOC against nerve agent inhibited RHuAChE.
Under these controlled conditions, the kinetic parameters of reactivation by ADOC and 2PAM of RHuAChE inhibited by various OP nerve agents were determined using established reactivation methods (Figure 3) [16, 18–21]. In the cases of GB, GF, VX, and VR the rate of reactivation (kr, Scheme 1) by ADOC (0.34±0.14min−1, 0.13±0.03min−1, 0.50±0.20min−1, 4.01±2.37min−1 respectively) was greater than that mediated by 2PAM (0.06±0.02min−1, 0.04±0.02min−1, 0.06±0.01min−1, 0.06±0.04min−1 respectively). However, the dissociation constant (KD) of ADOC (7.61±4.07mM, 4.45±3.37mM, 2.08±0.94mM, 5.46±2.02mM respectively) relative to 2PAM (0.71±0.13mM, 2.43±0.65mM, 0.54±0.15mM, 1.34±0.79mM respectively) was similarly elevated. The overall rate of reactivation (kr/KD = kr2) by ADOC was thus only significantly increased relative to 2PAM for reactivation of RHuAChE inhibited by VR (Table 1). Of note, no evidence for cooperative behavior (as previously described during reactivation [11]) was observed under these experimental conditions, suggesting that the cooperativity reported before may be a condition-specific phenomenon related to the methods used in the prior study.
Figure 3.

Rates (kr, min−1) and dissociation constants (KD, μM) of the reactivation of inhibited RHuAChE by 2PAM (black) and ADOC (grey). Significant differences between 2PAM and ADOC values are indicated by *(t-test, p<0.05).
Scheme 1.

Reactivation from Worek et al. (2004 [19])
Table 1.
Overall reactivation rate for indicated agents and reactivators.
| Reactivator | kr2 (M−1 min−1) | |||||
|---|---|---|---|---|---|---|
| GA | GB | GD | GF | VX | VR | |
| 2-PAM | 7.0±4.4 | 81.0±16 | 17.6±16 | 18.4±4.2 | 121±29 | 46±1.2 |
| ADOC | 4.5±3.3 | 45.4±7.2 | 5.6±4.1 | 40.5±29 | 266±109 | 719*±386 |
Standard deviation of three independent experiments is indicated below value. Statistically significant increase of reactivation rate by ADOC as compared to 2PAM is indicated by
(t-test, p<0.05).
In an effort to expand upon the proposed mechanism of reactivation of ADOC [11], an initial structure-activity relationship study was conducted using several ADOC structural analogs. The importance of each substituent of the benzyl ring for reactivation of OP nerve agent inhibited enzyme was explored (Table 2). The removal of the aniline amine group (2) or the benzyl amine group (3) caused a decrease in the magnitude of reactivation as compared to ADOC but also substantially decreased the inhibitory effects on native RHuAChE. These combined results could indicate that these substituents play a role in binding rather than reactivation chemistry, especially in light of the fact that removal of the phenol substituent (4) obliterates all reactivation functionality. Similarly, blocking the phenol as an ether (5) causes the loss of reactivation functionality against all but one agent (GB), for which the function is greatly decreased as compared to ADOC. Taken together, these results indicate that the phenol substituent is essential for reactivation.
Table 2.
Initial Exploration of Active Moieties. Ratio (Spontaneous/compound) of half-times (t1/2) of reactivation for compounds (1mM) with RHuAChE after inhibition by the indicated agent.
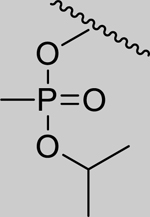
|
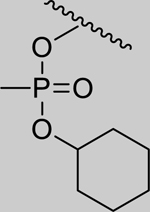
|
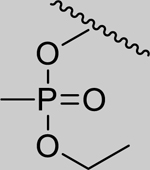
|
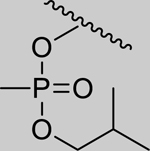
|
Inhibition of RHuAChE @ 1mM | ||
|---|---|---|---|---|---|---|
| GB | GF | VX | VR | |||
| Spontaneous Reactivation (t1/2 in days) | 2.01 (±0.07) | 1.25 (±0.14) | 1.18 (±0.21) | 0.35 (±0.07) | ||
| 1 |
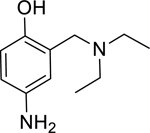
|
361* (158–824) | 164* (97.3–277) | 1,069* (393.6–2911) | 14,723* (6.46–33,573,761) | 60% (±1) |
| 2 |
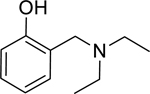
|
1.51* (1.12–2.03) | 1.54* (1.34–1.78) | 1.87* (1.24–2.84) | 3.65* (3.14–4.24) | 0% (±6) |
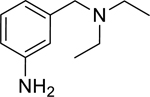
| 3 |
|
2.24* (1.98–2.53) | 1.48* (1.15–1.91) | 2.24* (1.16–4.32) | 14.3* (8.86–23.0) | 0% (±6) |
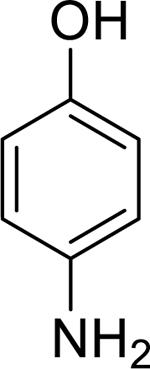
| 4 |
|
0.98 (0.81–1.20) | 0.93 (0.89–0.97) | 0.97 (0.66–1.43) | 0.96 (0.61–1.51) | 0% (±6) |
| 5 |
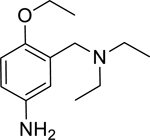
|
1.54* (1.28–1.84) | 0.89 (0.77–1.03) | 1.11 (0.53–2.34) | 1.25 (0.90–1.73) | 5% (±4) |
indicates that t1/2 in presence of compound is significantly less than spontaneous using a ratio t-test with p<0.05.
Standard deviation of at least three independent experiments is indicated for spontaneous reactivation half-times in days. 95% confidence intervals of ratios are indicated below each value.
All half-times were determined using RHuAChE in the presence of 1mM compound as the positive control. The far right column indicates the level of inhibition of RHuAChE caused directly by the compound at 1mM when compared to a buffer only control of RHuAChE at the same concentration. Standard deviation of this value determined using controls from all independent experiments is indicated.
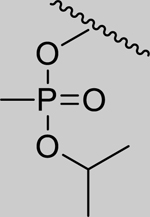
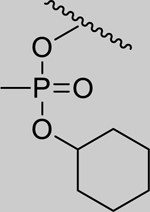
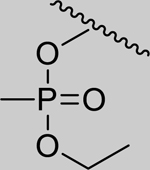
Given that the removal of the benzyl amine (3) had a slightly less deleterious effect on the reactivation potential of ADOC than removal of the aniline amine (4), additional analogs of ADOC were used to probe the importance of this moiety (Table 3). Changing the relative position of the benzylic diethylamine on the ring (6) reduced the reactivation potential of the molecule but also nearly abolished inhibitory potential against naive RHuAChE. Interestingly, reducing the bulk of the benzyl amine (7) greatly increases the inhibitory potential of the molecule against naive RHuAChE; however this affect can be negated by altering the relative position of this smaller benzyl amine (8) to a configuration similar to that of 6. In the hopes of counteracting the inhibitory effects of 7, an analog in which the benzyl amine bulk had been increased (9) was tested and, surprisingly, was found to also have increased inhibitory potential against naïve RHuAChE. Further increase in the bulk of the benzyl amine (10) resulted in loss of inhibitory potential and reactivation potential, most likely due to sterically driven decreased binding of the small molecule. The inhibition and reactivation results of 9 and 10 offer some insight into the mechanism of reactivation by helping to rule out the likelihood that quinone methide formation is involved as these compounds would be expected to form a common quinone methide intermediate with ADOC if this mechanism is operative [22]. Given all of these results and the high pKa (~9) of the benzyl amine, it seems likely that this portion of the molecule contributes mainly to the binding strength and selectivity of ADOC rather than the reactivation functionality of the molecule.
Table 3.
Exploring the Role of the Benzylamine Group. Ratio (spontaneous/compound) of half-times (t1/2) of reactivation for compounds (1mM) with RHuAChE after inhibition by the indicated agent.
|
|
|
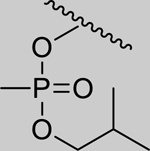
|
Inhibition of RHuAChE @ 1mM | ||
|---|---|---|---|---|---|---|
| GB | GF | VX | VR | |||
| 1 |
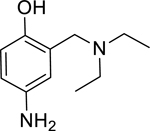
|
361* (158–824) | 164* (97.3–277) | 1,069* (393.6–2911) | 14,723* (6.46–33,573,761) | 60% (±1) |
| 6 |
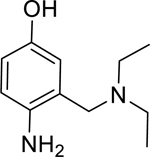
|
1.97* (1.54–2.52) | 1.80* (1.46–2.22) | 2.76* (1.95–3.93) | 4.06* (3.01–5.49) | 1% (±4) |
| 7 |
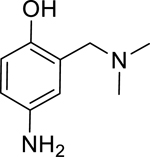
|
--- | --- | --- | --- | 95% (±1) |
| 8 |
|
2.13* (1.63–2.78) | 1.96* (1.92–2.01) | 2.72* (2.12–3.48) | 5.65* (1.20–26.5) | 18% (±6) |
| 9 |
|
--- | --- | --- | --- | 94% (±1) |
| 10 |
|
0.88 (0.65–1.20) | 0.74 (0.60–0.90) | 1.09 (0.60–1.98) | 0.93 (0.76–1.14) | 9% (±4) |
---Reactivation could not be determined due to high level of inhibition by compound.
Additional studies were conducted to analyze the contribution of the aniline amine substituent to the reactivity of ADOC (Table 4). The role of the electron donating potential of the aniline amine group was explored by both increasing (11) and decreasing (12) the electron donating potential as guided by the reported Hammett values for these substituents [23]. While both of these modifications displayed significantly increased reactivation compared to spontaneous levels for all agents, reactivation was markedly decreased compared to that of ADOC. Additionally, the alteration of the aniline amine in both cases abolished all inhibition of RHuAChE raising the question of whether the decreased reactivation activity of these molecules was a result of decreased binding or decreased reactivation rate. Determination of the dissociation constant (KD; 11: 0.39±0.23mM; 12: 0.92±0.12mM) and reactivation rate (kr; 11: 0.07±0.02min−1; 12: 0.03±0.01min−1) for each compound supports the conclusion that the loss of reactivation potential is due to a slowed reactivation rate rather than a decrease in binding.
Table 4.
Exploring the Role of the Aniline Amine Group. Ratio (spontaneous/compound) of half-times (t1/2) of reactivation for compounds (1mM) with RHuAChE after inhibition by the indicated agent.
|
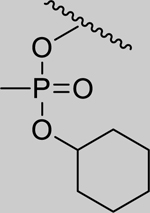
|
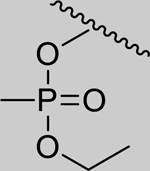
|
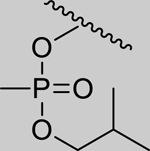
|
Inhibition of RHuAChE @ 1mM | Hammett Values[23] (σp) | ||
|---|---|---|---|---|---|---|---|
| GB | GF | VX | VR | ||||
| 1 |
|
361* (158–824) | 164* (97.3–277) | 1,069* (393.6–2911) | 14,723* (6.46–33,573,761) | 60% (± 1) | −0.66 |
| 11 |
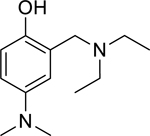
|
6.15* (4.98–7.60) | 3.07* (2.92–3.23) | 16.5* (13.0–20.8) | 32.7* (24.1–44.4) | 0% (± 6) | −0.83 |
| 12 |
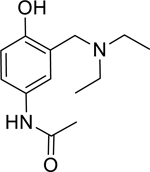
|
3.85* (3.22–4.61) | 1.72* (1.67–1.77) | 6.95* (5.03–9.59) | 19.0* (15.2–23.7) | 0% (± 5) | 0.00 |
| 13 |
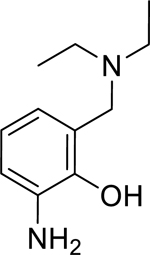
|
1.43* (1.37–1.50) | 1.09 (0.80–1.49) | 1.24 (0.99–1.56) | 1.65* (1.54–1.78) | 35% (± 4) | N/A |
| 14 |
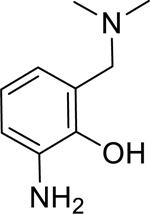
|
1.70* (1.40–2.06) | 1.30 (0.90–1.89) | 1.61* (1.17–2.21) | 1.59* (1.35–1.87) | 43% (± 11) | N/A |
Given these data, compounds which could still take advantage of the inductive electron donating affect but were more sterically hindered about the phenol (13 and 14) were tested for reactivation potential. While both of these compounds retained reactivation potential above that of spontaneous rates for some agents, reactivation rates in general were greatly decreased compared to ADOC. Taken together, the results suggest that while the para relationship of the aniline amine relative to the phenol is important, this relationship may have more to do with positioning the phenol upon binding to the inhibited AChE enzyme, than to any inductive activation effects.
Finally, ADOC was used in in vivo experiments to evaluate the compound as a potential therapeutic for nerve agent intoxication. Utilizing methodology previously developed to quantify the level of cholinesterase reactivation in guinea pigs after exposure to nerve agents [12], the dose-dependent reactivation potential of ADOC in red blood cells, whole blood, brainstem, cerebellum, cortex, mid-brain, hippocampus, striatum, spinal cord, diaphragm, heart and skeletal muscle from animals exposed to nerve agents was tested. While a small dose-dependent reactivation effect was noted in the red blood cells and whole blood after exposure to VX and GB, similar responses were not noted in any other tissue samples (Figure 4). In fact, no significant reactivation against any agent after administration of any dose of ADOC was noted in the tissue samples tested suggesting that ADOC may not readily leave the bloodstream or cross the blood brain barrier.
Figure 4.

Dose-dependence of In Vivo Reactivation by ADOC. A) Reactivation of red blood cell acetylcholinesterase by ADOC administered at doses of 1.8, 10.0, 18.0, 32.0, or 56.0 mg/kg to animals exposed to 1xLD50 GB (▲), GF (▼) or VX (●). Whole blood reactivation displayed similar trends though absolute reactivation values differed (data not shown). B) Reactivation of mid-brain tissue acetylcholinesterase by ADOC administered at the indicated doses to animals exposed to GB (▲), GF (▼) or VX (●). All other tissues tested (brainstem, cerebellum, cortex, hippocampus, striatum, spinal cord, diaphragm, heart and skeletal muscle) displayed similar reactivation trends though absolute reactivation values differed (data not shown).
4. Discussion
To evaluate the IC50 values in a clinically relevant manner, 2PAM is administered at a dose of 145 μmol/kg [12] which translates to a maximum concentration of ~2mM in human blood plasma, assuming that a 70kg adult has 5L of blood and the full dose of administered drug reaches the blood stream. A recent study showed that in fact only a maximum 2.5% to 3% of 2PAM can be detected in the plasma after intramuscular injection [24] which would translate to a plasma concentration of approximately 60 μM after administration of three autoinjector doses [12], the current recommended emergency treatment (Mark I Nerve Agent Antidote Kit) for a patient displaying symptoms of nerve agent exposure. While 2PAM at the above concentration does not cause toxic effects via acetylcholinesterase inhibition, supported by the IC50 determined in this study (456μM), ADOC (IC50 = 17.8μM) administered at a similar dose could result in toxic effects due to direct inhibition of acetylcholinesterase.
While the lack of apparent reactivation of AChE by ADOC in the tissues of OP-exposed guinea pigs was somewhat disappointing, several factors affecting in vivo reactivation can be explored. First, the pharmacokinetic properties of ADOC were not specifically tested and, as this compound is structurally different from the pyridinium oximes upon which the experimental paradigm was developed, could have led to the compound being administered at a time or via a route which was not conducive to maximal effects. Second, it is possible that ADOC is not as potent a reactivator of guinea pig acetylcholinesterase as it is for the human form of the enzyme. Both of these points could be explored by further testing both in vivo and in vitro, refining the animal testing parameters.
Despite the lack of evidence supporting the in vivo protective efficacy of ADOC as a treatment for nerve agent intoxication, full characterization of the mechanism of reactivation of this small molecule which has in vitro reactivation effects on par with 2PAM could lead to a new class of nerve agent intoxication treatments. The use of ADOC analogs to explore the mechanism yielded insight into the roles of the ring substituents with both the benzylic diethylamine and the aniline amine contributing to the binding and specificity of ADOC, while only the phenol group appears to be critical for reactivation. Further work to characterize the product of reactivation could lead to a definitive answer as to whether the phenol is essential for a direct nucleophilic attack of the phosphonylated active site or whether ADOC mediates an indirect action via a water molecule (as postulated by Katz et al [11]).
In conclusion, while ADOC may not prove to be an ideal drug candidate to address the issue of nerve agent intoxication, the characterization of ADOC and several analogs here can be used to inform the design of a new class of reactivators which do not rely upon an oxime as the primary reaction group for the regeneration of functional AChE after inhibition by organophosphorus nerve agents.
Acknowledgements
The views expressed in this manuscript are those of the author(s) and do not reflect the official policy of the Department of Army, Department of Defense, or the U.S. Government.
The experimental protocol was approved by the Animal Care and Use Committee at the United States Army Medical Research Institute of Chemical Defense and all procedures were conducted in accordance with the principles stated in the Guide for the Care and Use of Laboratory Animals (National Research Council, 2011), and the Animal Welfare Act of 1966 (P.L. 89-544), as amended.
This research was supported by the Defense Threat Reduction Agency – Joint Science and Technology Office, Medical S&T Division.
C.L.C. and J.A.K. were supported in part by an appointment to the Internship/Research Participation Program for the US Army Medical Research Institute of Chemical Defense, administered by the Oak Ridge Institute for Science and Education through an agreement between the US Department of Energy and the USAMRICD.
Footnotes
Conflict of Interest
None.
References
- 1.Dunn MA and Sidell FR, Progress in medical defense against nerve agents. JAMA, 1989. 262(5): p. 649–52. [PubMed] [Google Scholar]
- 2.Millard CB, et al. , Crystal structures of aged phosphonylated acetylcholinesterase: nerve agent reaction products at the atomic level. Biochemistry, 1999. 38(22): p. 7032–9. [DOI] [PubMed] [Google Scholar]
- 3.Skovira JW, et al. , Reactivation of brain acetylcholinesterase by monoisonitrosoacetone increases the therapeutic efficacy against nerve agents in guinea pigs. Chem Biol Interact, 2010. 187(1–3): p. 318–24. [DOI] [PubMed] [Google Scholar]
- 4.Rosman Y, et al. , Lessons learned from the Syrian sarin attack: evaluation of a clinical syndrome through social media. Ann Intern Med, 2014. 160(9): p. 644–8. [DOI] [PubMed] [Google Scholar]
- 5.Smithson A. and Levy L, Ataxia: The Chemical and Biological Terrorism Threat and the US Response. 2000, Washington, DC: Henry L Stimson Centre. [Google Scholar]
- 6.Masson P, Evolution of and perspectives on therapeutic approaches to nerve agent poisoning. Toxicol Lett, 2011. 206(1): p. 5–13. [DOI] [PubMed] [Google Scholar]
- 7.Ashani Y. and Cohen S, Nucleophilicity of some reactivators of phosphorylated acetylcholinesterase. J Med Chem, 1971. 14(17): p. 621–6. [DOI] [PubMed] [Google Scholar]
- 8.Hagedorn I, Gundel WH, and Schoene K, [Reactivation of phosphorylated acetylcholine esterase with oximes: contribution to the study of the reaction course]. Arzneimittelforschung, 1969. 19(4): p. 603–6. [PubMed] [Google Scholar]
- 9.Davies DR and Green AL, The kinetics of reactivation, by oximes, of cholinesterase inhibited by organophosphorus compounds. Biochem J, 1956. 63(4): p. 529–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Worek F. and Thiermann H, Reactivation of organophosphate-inhibited human acetylcholinesterase by isonitrosoacetone (MINA): a kinetic analysis. Chem Biol Interact, 2011. 194(2–3): p. 91–6. [DOI] [PubMed] [Google Scholar]
- 11.Katz FS, et al. , Discovery of New Classes of Compounds that Reactivate Acetylcholinesterase Inhibited by Organophosphates. Chembiochem, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shih TM, et al. , Evaluation of nine oximes on in vivo reactivation of blood, brain, and tissue cholinesterase activity inhibited by organophosphorus nerve agents at lethal dose. Toxicol Mech Methods, 2009. 19(6–7): p. 386–400. [DOI] [PubMed] [Google Scholar]
- 13.Ho T-L, et al. , Lithium aluminum hydride, in Fieser and Fieser’s Reagents for Organic Synthesis. 2006, John Wiley & Sons, Inc. [Google Scholar]
- 14.Worek F, et al. , Reactivation of organophosphate-inhibited human, Cynomolgus monkey, swine and guinea pig acetylcholinesterase by MMB-4: a modified kinetic approach. Toxicol Appl Pharmacol, 2010. 249(3): p. 231–7. [DOI] [PubMed] [Google Scholar]
- 15.Sit RK, et al. , New structural scaffolds for centrally acting oxime reactivators of phosphylated cholinesterases. J Biol Chem, 2011. 286(22): p. 19422–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Aurbek N, et al. , Analysis of inhibition, reactivation and aging kinetics of highly toxic organophosphorus compounds with human and pig acetylcholinesterase. Toxicology, 2006. 224(1–2): p. 91–9. [DOI] [PubMed] [Google Scholar]
- 17.Ellman G, et al. , A New and Rapid Colorimetric Determination of Acetylcholinesterase Activity. Biochemical Pharmacology, 1961. 7: p. 88–95. [DOI] [PubMed] [Google Scholar]
- 18.Worek F, et al. , Reactivation kinetics of acetylcholinesterase from different species inhibited by highly toxic organophosphates. Arch Toxicol, 2002. 76(9): p. 523–9. [DOI] [PubMed] [Google Scholar]
- 19.Worek F, et al. , Kinetic analysis of interactions between human acetylcholinesterase, structurally different organophosphorus compounds and oximes. Biochem Pharmacol, 2004. 68(11): p. 2237–48. [DOI] [PubMed] [Google Scholar]
- 20.Worek F, Szinicz L, and Thiermann H, Estimation of oxime efficacy in nerve agent poisoning: a kinetic approach. Chem Biol Interact, 2005. 157–158: p. 349–52. [DOI] [PubMed] [Google Scholar]
- 21.Worek F, et al. , Recent advances in evaluation of oxime efficacy in nerve agent poisoning by in vitro analysis. Toxicol Appl Pharmacol, 2007. 219(2–3): p. 226–34. [DOI] [PubMed] [Google Scholar]
- 22.Ercegovic SM, Synthesis and Characterization of Quinone Methide Precursors as Acetylcholinesterase Reactivators, in Department of Chemistry. 2011, Ohio State University. [Google Scholar]
- 23.Hansch C, Leo A, and Taft RW, A survey of Hammett substituent constants and resonance and field parameters. Chem Rev, 1991. 91(2): p. 165–195. [Google Scholar]
- 24.Abbara C, et al. , Pharmacokinetic analysis of pralidoxime after its intramuscular injection alone or in combination with atropine-avizafone in healthy volunteers. British Journal of Pharmacology, 2010. 161: p. 1857–1867. [DOI] [PMC free article] [PubMed] [Google Scholar]