

Abstract
Analysis of single cancer cells is critical to obtain accurate patient diagnosis and prognosis. In this work, we report the selective dielectrophoretic capture and electrochemical analysis of single melanoma cells at an array of interlocked spiral bipolar electrodes (iBPEs). Following dielectrophoretic capture, individual melanoma cells are hydrodynamically transferred into picoliter-scale chambers for subsequent analysis. The interlocked spiral end of the iBPE (the sensing pole) is utilized to read out an electrochemical enzyme-linked immunosorbent assay (eELISA), which quantifies the expression of a cell surface antigen, melanoma cell adhesion marker (MCAM). The opposite pole of each BPE is located in a fluidically isolated compartment containing reagents for electrogenerated chemiluminescence (ECL), such that luminescence reports iBPE current. In a preliminary device design, the ECL intensity was insufficient to detect MCAM expression on single cells. To achieve single-cell analysis, we decreased the gap size between the interlocked spirals tenfold (5.0 μm to 0.5 μm), thereby creating a more sensitive biosensor by enhanced redox cycling of the product of eELISA. This work is significant because it allows for the selective isolation and sensitive analysis of individual melanoma cells in a device amenable to point-of-care (POC) application by combining dielectrophoresis (DEP) with interdigitated bipolar electrodes (IDBPEs).
Graphical Abstract
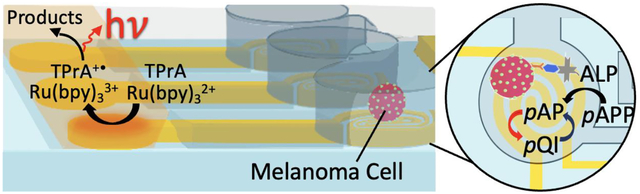
Cell-to-cell differences drive cancer spread and the acquisition of therapeutic resistance. Therefore, there is a need for widely accessible methods for analysis of tumor cells individually. Here, single melanoma cells are captured by dielectrophoresis (DEP) and evaluated in parallel for the expression of melanoma cell adhesion marker (MCAM) by electrochemical enzyme linked immunosorbent assay (eELISA) at an array of 30 wireless bipolar electrodes (BPEs). These BPEs utilize an interlocked spiral design to accomplish redox cycling of the product of ELISA for signal enhancement. This enhancement allows for the signal from individual cells to be reported by electrochemiluminescence (ECL). The reported integration of parallel single-cell isolation and redox cycling with electrochemical sensing at BPEs achieves the sensitivity and single-cell resolution needed for cancer diagnostics.
Introduction
Metastasis occurs when cells from the primary tumor site enter the bloodstream and then extravasate to form secondary tumors in distant tissues.[1] In melanoma, these cells that have entered the blood stream are called circulating melanoma cells (CMCs) and if detected and analyzed, have immense diagnostic and prognostic value.[2–5] However, CMCs pose a difficult challenge for cell isolation and analysis as less than ten CMCs can be found in one milliliter of blood.[6] Further, melanoma cells lack reliable surface antigens with sufficiently high expression to ensure their isolation by prevailing methods based on immunoaffinity interactions.[7]
Adding to this challenge is the fact that traditional cell analysis techniques take an ensemble measurement of a population of cells. However, tumor cells, especially CMCs, are heterogeneous. Therefore, ensemble measurements can fail to accurately characterize important subpopulations of cells.[8] Single-cell isolation and analysis allows cell-to-cell variations to be identified thus providing a more complete picture of the drivers of disease and response to therapy.[9–10] Current single-cell isolation and analysis approaches involve fluorescence-activated cell sorting (FACS),[11–13] magnetic activated cell sorting (MACS),[8, 14] laser capture microdissection (LCM),[15–17] and many microfluidic techniques.[18–21] Microfluidic methods offer many benefits including portability, low cost, microliter-scale sample consumption, ability to observe and image the cells, and amenability to multistep or multi-endpoint analyses.
Microfluidic devices utilize separation methods that leverage cell immunoaffinity as well as magnetic[19], hydrodynamic[22–23], or dielectrophoretic forces[24–26] (among others) in combination with geometric features of the device (pockets, wells, pillars, etc.) to accomplish single-cell isolation. Moscovici et al. devised a biosensor comprising an array of circular gold electrodes with anti-epithelial cell adhesion molecule (EpCAM) antibody immobilized onto the surface for the electrochemical detection of tumor cells.[27] After cells were captured by the anti-EpCAM antibodies, ferricyanide was introduced into the biosensor as a redox active probe, and the accessibility of the electrode surface to ferricyanide was measured by differential pulse voltammetry. Steric blocking of the electrode by a cell led to a decrease in the measured current – a sensing strategy similar to that previously used in electrochemical detection of mRNA.[28] Moscovici et al. were successfully able to demonstrate the electrochemical detection of model prostate cancer cells and achieved an LOD of 125 cells per sensor. This device was further optimized for multiplexing, cell separation, and even rare circulating tumor cell (CTC) capture by utilizing bio-conjugated magnetic nanospheres and similar electrochemical detection methods.[29–30] These methods all rely on cell surface biomarkers, but CMCs do not express surface antigens highly enough to be amenable for sensitive detection by these strategies – especially at the single-cell level.
Li et al. developed an array of bipolar electrodes (BPEs) for the high throughput and selective capture of model CTCs (breast adenocarcinoma) in the presence of model white blood cells, demonstrating the utility of combining BPEs with dielectrophoresis (DEP) for cell capture and sorting.[25] Later Li et al. added nanoliter-scale chambers adjacent to each pocket. The chambers were utilized to retain and individually sequester each cell after DEP capture.[31] The chambers were further isolated with an immiscible phase, creating distinct environments for parallel analysis of single CTCs. By utilizing a hydrophobic ionic liquid (IL) for this isolation step, high conductivity was maintained across the device, allowing electrical lysis of the isolated cells to be achieved at the BPEs through application of an increased voltage at the driving electrodes. The IL also provides an avenue to introduce functionality, such as formation of solid plugs at the entrance to each chamber by electropolymerization.[32] The resulting platform was utilized for isolation of melanoma cells from patient-derived blood samples and, separately, for analysis of a cytosolic enzyme linked to cellular senescence.[33–34] In these applications, the BPE array was utilized both for DEP-based cell capture and for electrical cell lysis. However, the ability of the BPE array to carry out electrochemical reactions useful for cell analysis was not leveraged, largely owing to the high sensitivity required to analyze single cells.
To address this issue, Borchers et al. developed interdigitated bipolar electrodes (IDBPEs), which facilitate redox cycling of electroactive species for signal amplification. By decreasing the gap between the digits of the IDBPEs from 35-μm to 5-μm, the authors reported a 6.3-fold increase in the electrochemiluminescence (ECL) intensity at the reporting pole of the IDBPE.[35] Furthermore, the authors demonstrated sensitive electrochemical analysis of model CMCs at an array of IDBPEs. Microwells were fabricated on top of the sensing end of each IDBPE to facilitate DEP-based cell capture and retention in the wells. IDBPEs, with a 5-μm gap between digits, possessed the sensitivity to produce an ECL signal at the reporting pole of the IDBPE for quantitation of the cell surface markers on the CMCs captured at the sensing end of the biosensor.[36] This work done by Borchers et al. demonstrated the advantages of IDBPEs and DEP for multi-cell capture and quantification. However, the approach was unable to accomplish single-cell capture, and the microwells were subject to crosstalk.
Here, we report label-free dielectrophoretic capture and individual electrochemical analysis of melanoma cells in a microfluidic device consisting of an array of interlocked spiral BPEs (iBPEs). The interlocked spiral end of each iBPE (the sensing pole) is housed within a picoliter-scale chamber where melanoma cells are captured individually by DEP. The iBPE is utilized for an electrochemical enzyme linked-immunosorbent assay (eELISA) to quantify the expression of a surface antigen, melanoma cell adhesion marker (MCAM) at the single-cell level. The current at each iBPE is correlated to the degree of antigen expression on the corresponding cell and is reported as light, through ECL at the reporting pole. To further enhance the sensitivity of the biosensor, the gap size between interlocking spirals was decreased from 5.0 μm to 0.5 μm to facilitate redox cycling of the electroactive product of eELISA. The results of this study are significant because they demonstrate the sensitive analysis of individual melanoma cells in a device amenable to point-of-care application. This advancement is achieved by combining selective enrichment of malignant cells by DEP, the facile arraying of BPEs, and the amplification afforded by interdigitation.
Theoretical Background
DEP is a label-free cell capture mechanism. DEP describes the movement of a polarizable particle with an induced dipole (p) in a nonuniform electric field () as describe by eq 1.[37–38]
| (1) |
DEP cell isolation and capture techniques rely on the morphology and composition of the cell, which determine its dielectric properties and their relationship to those of the surrounding medium, to allow for label-free cell capture and detection.[24] The polarizability of a cell is frequency dependent and is described by the Clausius-Mossoti (CM) factor (),[39] which appears as a term in the DEP force equation (eq 2) when the induced dipole term is expanded. The CM factor is determined by the complex permittivities of the cell and the surrounding medium (eq 3). The time-averaged DEP force () is described by the following equations.
| (2) |
| (3) |
where is the permittivity of the medium, R is the particle’s radius, and is the real part of the CM factor. The effective complex permittivity () of the cell and the complex permittivity of the medium () govern whether the cell will experience positive DEP (pDEP) or negative DEP (nDEP). In a medium of sufficiently low conductivity, at lower frequencies, the cell is shielded from the electric field by the cell membrane (a capacitor) and behaves as an insulator (). Therefore, the cell moves from a region of high electric field strength to a one of low electric field strength. At higher frequencies, the permittivity of the cell is higher than that of the medium, and the cell moves from low to high electric field strength (pDEP). Since the DEP force is not reliant on a specific biomarker, only the frequency of the applied electric field needs to be tuned to select for distinct cell types.
Cell capture in a microfluidic device depends on the applied AC voltage, frequency, and both the device and electrode geometry. Li et al. used DEP to selectively capture model circulating tumor cells (CTCs) in a microfluidic device comprised of an array bipolar electrodes (BPEs).[25] At 40 kHz, the MDA-MB-231 cells (model breast cancer cells) experienced pDEP and were attracted to the tips of the electrodes, while Jurkat cells (model white blood cells) remained in the microchannels. This frequency dependence illustrates the selectivity of DEP as a capture mechanism. Combining BPEs with the selectivity afforded by dielectrophoretic capture mechanisms, as in this work, has proven to be a powerful tool for single-cell analysis.
A BPE is a conductive material immersed in an electrolyte solution.[40–42] BPEs are considered “wireless” electrodes because a potential is not directly applied to them but rather to two driving electrodes placed across the electrolyte in which the BPE is situated. When a potential difference is applied between the driving electrodes, the BPE floats to a potential intermediate to that of the electrolyte solution it contacts, generating the highest interfacial potential differences at the cathodic and anodic ends of the BPE. These interfacial potential differences, the anodic and cathodic overpotentials, if sufficiently large, can drive faradaic reactions at either end of the BPE. Because of their wireless nature, BPEs can be facilely arrayed for electrochemical imaging,[43–45] electrokinetic separation,[46] point-of-need sensing,[47] biosensing,[48] and single-cell analysis.[33, 49–50] An ECL reaction is a typical reporting mechanism utilized to transduce current to a visible signal for BPEs, particularly when employed in sensing applications.[51] ECL that proceeds by oxidation occurs at the anodic (reporting) pole of the BPE and thus requires a cathodic sensing reaction at the cathodic (sensing) pole. The Crooks group demonstrated the advantages of ECL as a reporting reaction. They report a range over which the relationship between ECL intensity and analyte concentration is linear once a minimum current threshold was met.[52] While researchers have demonstrated the advantages of ECL as a reporting mechanism, in BPEs, the reaction is not sensitive enough to optically report the detection of many biological analytes at concentrations relevant to clinical diagnostics and many basic research applications.
To meet the need for low detection limits for biological analytes, researchers have developed methods for enhancing the ECL intensity at the reporting pole of BPEs including increasing the surface area at the sensing side of BPEs,[53–58] limiting the BPE location to the maximum potential drop area,[59–60] and redox cycling.[61] Mao et al. fabricated glassy carbon electrodes (GCE) conjugated with Pt/Ru nanoparticles to increase the ECL signal in their IgG immunosensor. They report a 4-fold increase in ECL intensity when their Pt/Ru nanoparticles are present and a lower limit of detection than the competing IgG immunosensor.[55] Voci et al. developed a one-channel system where a rhombus-shaped BPE was located in the thinnest part of the channel. Due to the increased potential drop in the thin channel, or micropore, researchers observed a 100-fold decrease in the potential needed to observe ECL in their system.[59] While these techniques result in signal enhancement of the ECL reaction, they are not necessarily universal for sensing applications and require an additional cost and fabrication step to the BPE system. Redox cycling can be achieved by utilizing closely spaced electrodes that can be easily fabricated with standard photolithographic techniques.
Redox cycling is the repeated transfer of electrons between individual redox molecules and electrodes. This situation is often achieved by closely-spaced oppositely-polarized electrodes known as interdigitated electrodes. When an anode and cathode are closely spaced, the generated diffusion layers overlap so that the supply of both oxidized and reduced species are constantly replenished. When the space between these electrodes is decreased, there is a proportional increase in the current as redox species diffuse more rapidly across the steep concentration gradient. Thus, interdigitation of the sensing side of a BPE increases the current and in turn, the luminescence intensity of the reporting reaction. In our previous work, we demonstrated the advantages of interdigitating the sensing side of an array of closed BPEs. A 6.3-fold increase in ECL intensity was observed by decreasing the gap size between electrodes from 15 μm to 5 μm. These results were obtained using Fe(CN)63-/ Fe(CN)64- and ECL solutions in the sensing and reporting channels, respectively.[35] This work demonstrated that interdigitated bipolar electrodes (IDBPEs) significantly enhance the ECL signal, making them useful for biosensing applications.
Our recent work combined DEP and interdigitated bipolar electrodes (IDBPEs) for multi-cell capture in open microwells.[36] After dielectrophoretic capture, an alkaline phosphatase (ALP)-tagged antibody-labeled melanoma cell underwent an electrochemical enzyme-linked immunosorbent assay (eELISA), which produced p-aminophenol (pAP). The current that arose from redox cycling of pAP and its oxidized form, p-quinone imine (pQI), noticeably increased over time at a rate correlated to the expression level of the target cell surface antigen. Instead of capturing cells based on immunoaffinity,[62–63] or size,[64–67] we leveraged the dielectrophoretic properties of these cells for capture. These results are significant because they demonstrate the advantages of combining IDBPEs with DEP for multiple cell capture and analysis.
Results and Discussion
Development and Characterization of an Interlocked Spiral Bipolar Electrode.
The microfluidic devices in this work comprise a linear array of 30 circular microchambers each overlying the sensing (cathodic) pole of a gold interlocked spiral BPE. These microchambers are addressed by a microfluidic channel (the “sensing” channel), which is 10 mm long, 84 μm wide, and 25 μm tall. The sensing channel further encapsulates driving electrode 1 and driving electrode 2, which are positioned on either side of the narrow entrance (micropocket) of each chamber and work in concert to drive DEP-based cell capture. Driving electrodes, unlike the iBPEs, have wire leads that provide direct ohmic connection to an external power supply. The reporting channel (10 mm long × 84 μm wide × 25 μm tall) contains the anodic pole of each BPE and driving electrode 3 (Scheme 1b). Driving electrodes 2 and 3 apply the potential difference required for faradaic reactions at the iBPEs and in this way facilitate electrochemical sensing. The center-to-center spacing between iBPEs (along the channel length) is 305 μm. Driving electrode 2 and each iBPE’s sensing pole make up the interlocking spirals. These are considered interdigitated electrodes and facilitate the redox cycling of pAP and pQI, as displayed in Scheme 1c. Each spiral electrode in a single interlocked system has a width of 10 μm and is spaced 5 μm apart. Together, the interlocked spirals make up a circle of 100 μm in diameter, with 101 μm-diameter chambers surrounding each interlocked spiral. Driving electrode 2 is spaced 20 μm from driving electrode 1, a 21 μm-wide gold microband that extends the length of the sensing channel. The sensing pole and reporting pole of each iBPE are interconnected by a 112 μm long and 15 μm wide gold microband, which extends underneath an insulating wall that prevents intermixing of reagents required for sensing and reporting. The reporting end of the BPEs are closed circles (75 μm in diameter) that facilitate the co-oxidation of ruthenium (III) bipyridine (Ru(bpy)32+) and tripropylamine (TPA) for ECL. The reporting poles are situated 100 μm from Driving electrode 3, a 35 μm wide driving electrode that extends the length of the reporting channel.
Scheme 1.
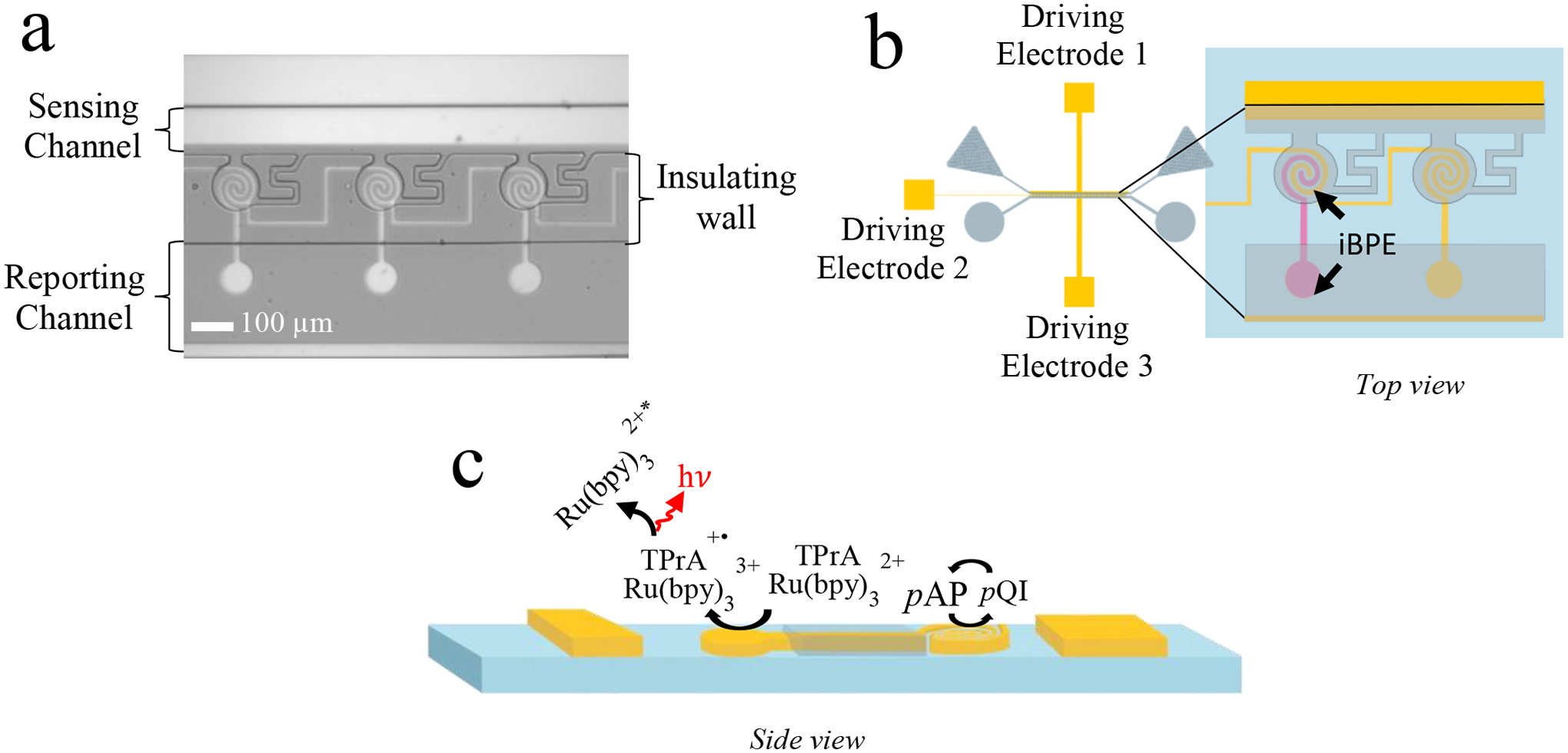
(a) Brightfield image of the 5 μm-gap device. (b) Illustration of 5 μm-gap device that zooms into a close-up of driving electrode 2 and BPEs. (c) Side view of the device displaying the reactions occurring at each BPE pole under an applied voltage bias.
To investigate the electrochemical response of this device, we obtained linear sweep voltammograms (LSVs) and chronoamperograms (CAs) by applying a potential difference between driving electrodes 2 and 3 while simultaneously monitoring the resulting current and corresponding luminescence intensity generated at the reporting pole of each iBPE. In this experiment, the reporting channel contained ECL solution (no flow). The sensing channel and chambers were filled sequentially with 0.1, 0.25, 0.5, 0.75, and then 1.0 mM pAP in 0.5 M phosphate buffer (pH 7.4). During LSVs, the voltage applied between driving electrodes 2 and 3 was swept from +1.00 V to + 3.00 V (with driving electrodes 2 connected to the positive lead), and a cathodic peak was observed at +1.73 V. This peak is attributed to the redox cycling of pAP and pQI and ECL at the opposing poles of the iBPEs.
Subsequently, for chronoamperometric measurements, a constant potential of this same magnitude (+1.73 V) was applied between driving electrodes 2 and 3 for 30 s and the current for the entire BPE array was monitored. Simultaneously, a series of micrographs was obtained to monitor the ECL response at the reporting poles of 5 iBPEs. This experiment was repeated 5 times in each of three devices for a total of 15 replicates. Figure 1a is a plot of the chronoamperometric current, averaged over three trials, at each pAP concentration. A linear relationship (R2 = 0.97) was observed between current (from the full array) and pAP concentration with a slope of 0.65 μA mM−1. The large variability (represented by the error bar) observed at the highest pAP concentration is attributed to fouling of the electrodes by pAP over the five replicates carried out in each device. Figure 1b is a plot of the spatially averaged ECL intensity (averaged across all iBPEs) integrated over the period of t = 20 s to 30 s following initiation of each potential step. A linear correlation (R2 = 0.92) was observed between ECL intensity and pAP concentration with a slope of 3.0 counts μm−2 mM−1. Figure 1c is a plot of this averaged ECL intensity at each of these five concentrations of pAP (0.10, 0.25, 0.50, 0.75, and 1.0 mM) versus the corresponding current. The plot was fitted linearly (R2 = 0.94) with a slope of 4.7 counts μm−2 μA−1. Figure 1d is a brightfield micrograph of the luminescence obtained at the iBPE reporting poles at three distinct concentrations of pAP in the sensing channel (in ascending order, 0.1, 0.5, and 1.0 mM). This result is important because it demonstrates the linear relationship between ECL intensity and current over the range of pAP concentrations employed in this work, while enabling the quantification of pAP at biologically relevant concentrations in the interlocked spiral BPE device. Additionally, the LOD for current and ECL intensity was found to be 0.09 μA (0.14 mM pAP) and 1.75 counts μm−2 (0.38 mM pAP), respectively. However, luminescence was not observed at the lower concentrations (0.1- and 0.25-mM pAP), limiting the range at which analyte detection can be optically reported at the 5.0 μm-gap interlocked spiral.
Figure 1.
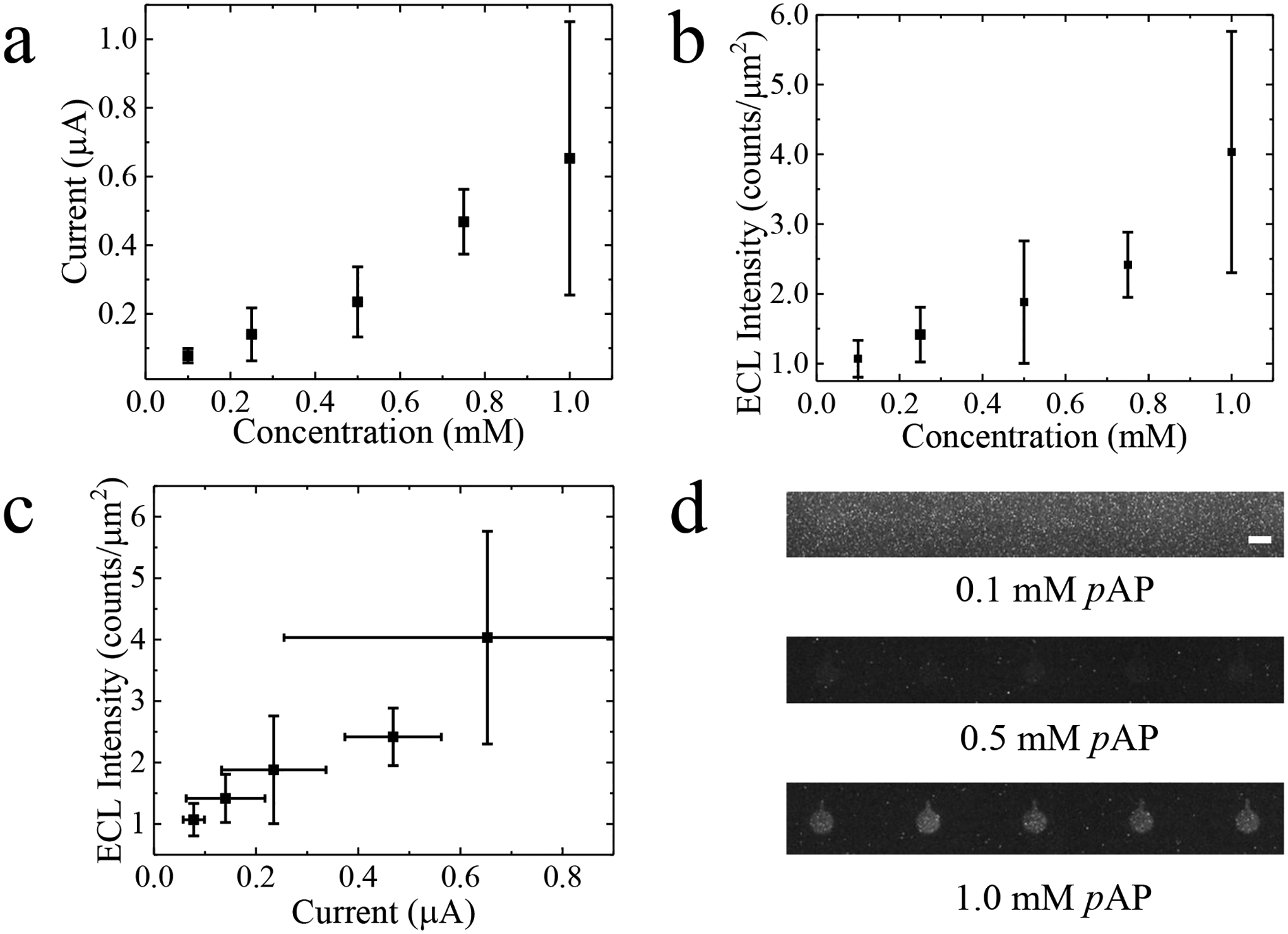
Plot of (a) current measured at the driving electrodes and (b) ECL intensity versus concentration for five concentrations of pAP (0.1, 0.25, 0.5, 0.75, or 1.0 mM) under an applied voltage of +1.73 V. (c) Current versus ECL intensity plot for the same results reported in (a) and (b). (d) Brightfield micrographs of the ECL response when 0.1, 0.5-, or 1.0-mM pAP is in the sensing channel. Scale bar, 100 m.
Dielectrophoretic Capture and Electrochemical Analysis of a Melanoma-Specific Cell-Surface Antigen.
First, the conditions required for cell capture in the micropockets and their transfer into the chambers were identified. The electric field distribution created by application of a voltage between driving electrodes 1 and 2 was characterized by a numerical simulation using commercial software (COMSOL 5.2a). This simulation allows for the dielectrophoretic response of A375 melanoma cells in a low conductivity buffer (LCB) under an applied AC voltage of 5.0 Vpp to be estimated. According to simulation results in Figure 2a, the electric field strength increases from the channel to the interior of the micropocket (3.34 × 105 V m−1) – the entry to the chamber. The narrowness of the micropocket constricts the electric field lines (Figure 2b), thereby potentiating dielectrophoretic capture of cells at that location by insulating DEP (iDEP).[68–69] In addition to creating a high electric field point for pDEP to occur, the dimensions of the micropocket allow for one cell, with a diameter of less than 20 μm, to be captured. Experimentally, we observed that under these electric field conditions, cells experience pDEP and are attracted to the micropockets.
Figure 2.
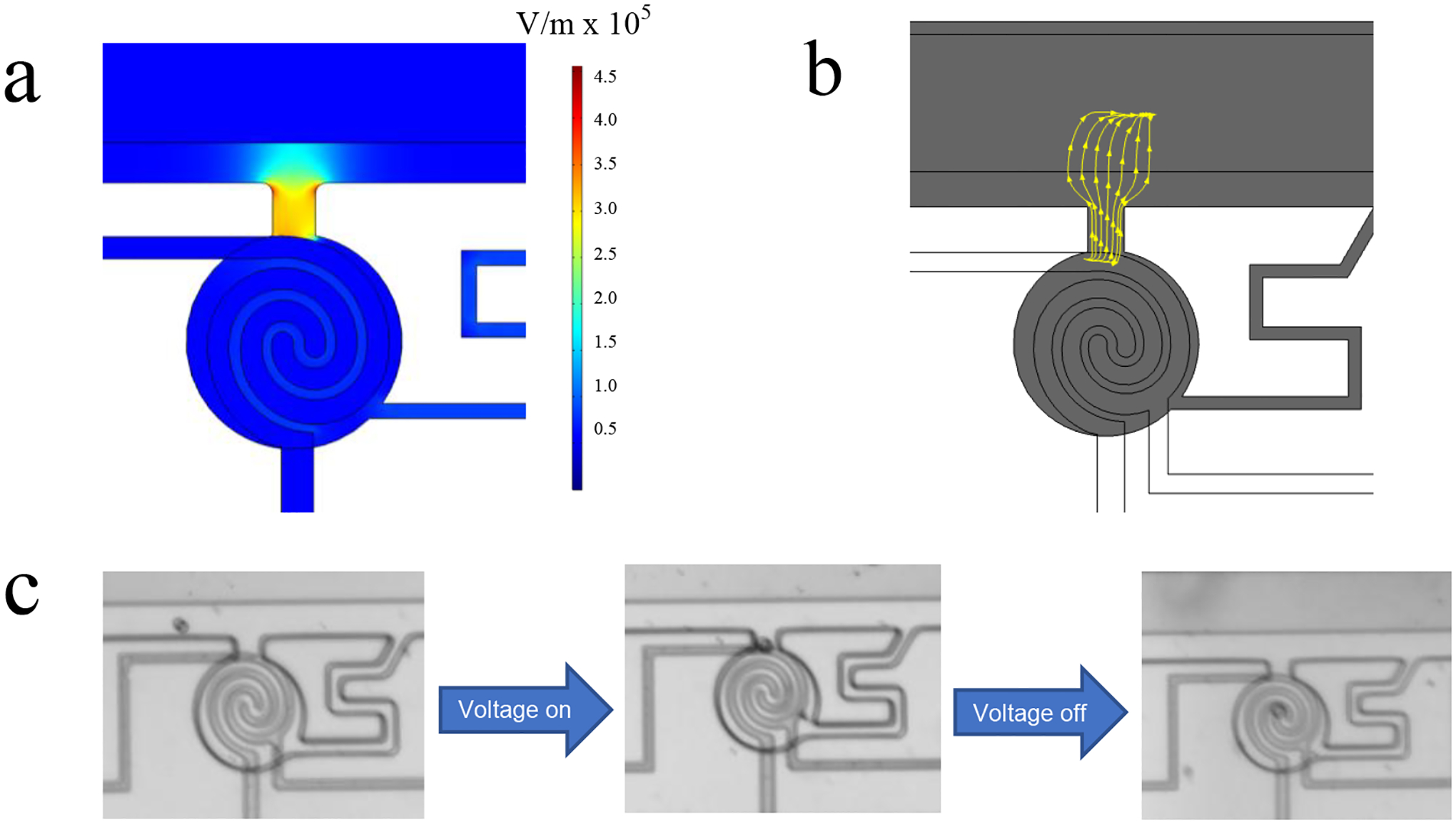
(a) COMSOL simulation of the electric field distribution in the device. (b) COMSOL simulation of the electric field lines in the micro-pocket of the chamber at 100 kHz and an AC voltage of +5.0 Vpp. (c) Brightfield images of an A375 melanoma cell in the sensing channel in response to turning ON and OFF the AC voltage. Scale bar, 100 m.
This electrochemical analysis was utilized to determine MCAM expression produced by melanoma cells. MCAM, a protein over-expressed by circulating melanoma cells (CMCs), has become a viable biomarker for early cancer detection, as their expression level directly correlates with cancer progression.[70–72] Here, A375 melanoma cells are employed as model CMCs. These cells were labeled off-chip with biotinylated anti-human MCAM and streptavidin-conjugated ALP, as described in the Experimental Section. These cells were then suspended in DEP buffer at a concentration of about 1.0 × 106 cells/mL. Cell solution was flowed into the sensing channel of the device, which contained DEP buffer, at a linear flow velocity of 110–150 μm s−1 and were allowed to flow through the channel for 5 min before the capture voltage was applied (+7.0 Vpp). Upon voltage application, cells were captured at the micropockets and subsequently transferred into the chambers after the AC voltage was turned off. The narrow serpentine leak channel (7.0 m wide × 300 m long × 25 m tall) creates a hydrodynamic force that drags the cells into the chambers.[25] Figure 2c is a time series of brightfield micrographs showing each step of this process. Once cells were captured and transferred into chambers, according to the cell capture protocol in the Experimental Section, the low conductivity buffer was exchanged for the ELISA substrate (10 mM pAPP in 0.5 M phosphate buffer (pH 7.4)), and the chambers were sealed with heavy mineral oil to prevent crosstalk via diffusion of the assay product (pAP).
Both electrochemical and optical detection were utilized to report the results of ELISA. Once microchannels and chambers were filled with the redox inactive substrate, pAPP, the ALP label on the cell cleaved the phosphate group on the pAPP to generate the redox active product, pAP. After 30 min, a 30-s CA was obtained at +1.73 V every 10 min (to 90 min total incubation time), and the resulting ECL intensity was simultaneously recorded. Figure 3a is a plot of the average current generated from the assay product over this 90-min incubation period at 5 distinct time points. It was observed that the current increased with a sigmoidal shape over time. Note that in our previous work, it was demonstrated that no increase in current is observed for unlabeled cells (in the absence of ALP) at an interdigitated BPE.[36] This result indicates that the current does not arise from redox active species generated by the cells (for example, reactive oxygen species). Figure 3b is a plot of the average ECL intensity from the reporting pole of 5 BPEs over time. Luminescence was not visible at this pAP concentration and the ECL intensity integrated over 20 s to 30 s did not significantly increase over time (range [1.2 to 1.38 counts μm−2]). We report a peak current at 0.16 μA and a peak ECL intensity of 1.34 counts μm−2. While the peak current is above the LOD for this device, the ECL intensity is below the LOD and cannot be detected with this 5.0-m gap device. Utilizing the current versus pAP concentration calibration curve in Figure 1a, the measured current corresponds to a pAP concentration of about 0.25 mM.
Figure 3.
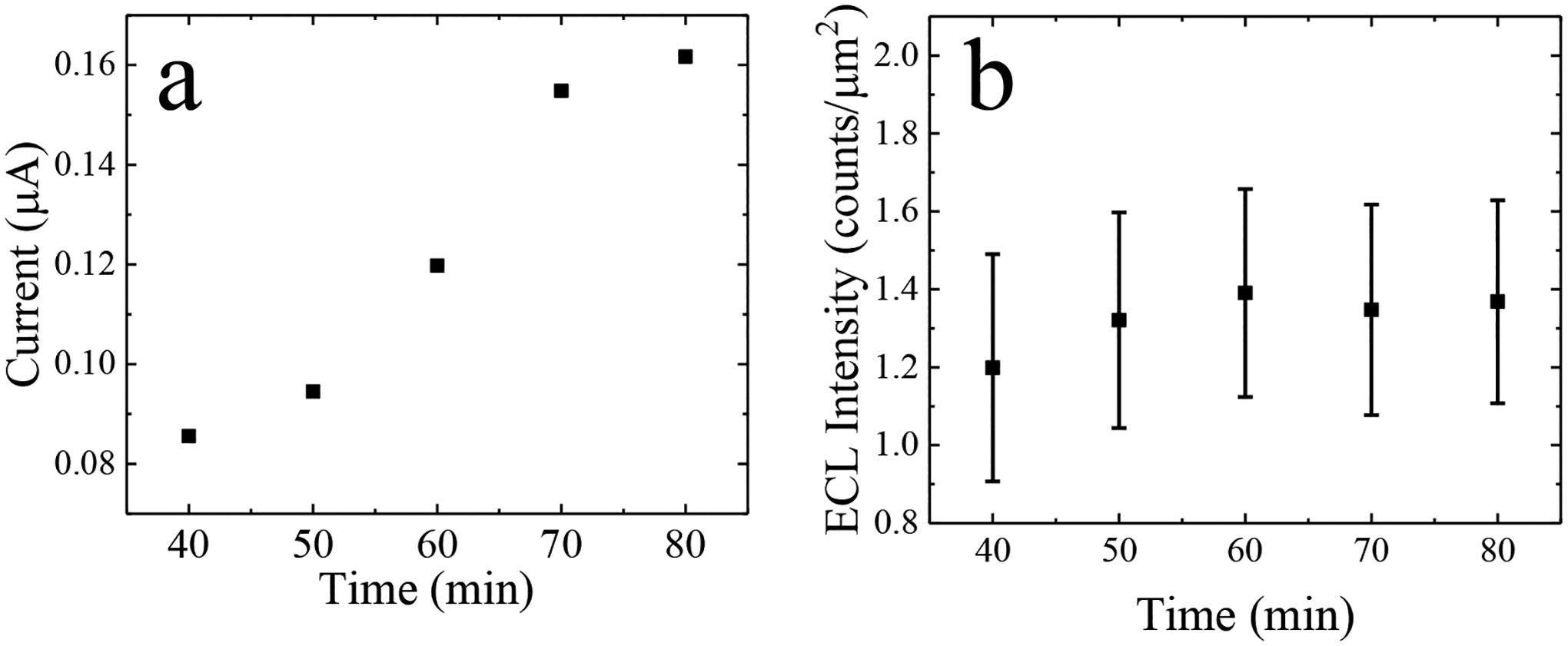
(a) Plot of current measured between driving electrodes 2 and 3 versus incubation time. CAs were obtained at a potential step of +1.73 V for 30 s at each 40, 60, 70, 80, 90 min incubation times. (b) Average ECL intensity obtained at 5 iBPEs versus incubation time plot. ECL intensity was integrated for 10 s from t = 20s to 30s after the applied potential step.
From the calibration curve of known pAP concentrations and the corresponding current, the concentration of pAP produced by the cells, in the device presented here, can be determined at each incubation time point. Michaelis-Menten kinetics[73] (Eq 1) was used to relate pAP concentration with MCAM concentration.
| (1) |
This equation relates the reaction concentration of the product [P], which is here pAP, to that of the enzyme [E0], here ALP, and substrate [S] = 10 mM, here pAPP, using the Michaelis constant (KM = 0.16 mM)[74] and enzymatic reaction rate (kcat = 441 s−1). The average concentration of ALP in the chambers was determined by integrating equation (1) with respect to time and solving for enzyme concentration (Eq 2),
| (2) |
Assuming the ALP to MCAM ratio is 1:1, the number of ALP molecules is equivalent to the number of MCAM surface antigens expressed on the cell surface.[75] With a pAP concentration of 0.072 mM, an ALP concentration of 0.049 pM was calculated. This ALP concentration corresponds to 495,000 MCAM surface antigens expressed by all of the cells in the array combined, with an average of 16,000 MCAMs per cell – an expression level that is reasonable for this cell line. This result is important because it demonstrates the quantification of MCAM expressed by an A375 cell in a single-cell capture and analysis device. While a detectable signal is obtained electrically (via current), an optical signal is not. As is, this device is not sufficiently sensitive to optically report the pAP concentrations produced in the assay. An increase in the optical signal would be necessary for clinical samples because of the low concentration of CTCs in a blood sample.
Development of a submicron gap interlocked spiral BPE.
In order to increase the ECL signal at the reporting pole, we developed a 0.5 μm-gap device. The decreased gap size was implemented to facilitate faster diffusion of redox species along the steep concentration gradient generated by closely spaced electrodes. This newly made device has all the same dimensions as the 5.0 μm-gap interlocked spiral BPE, except for the width of spirals and the gap between the spirals. Each spiral microband has a width of 14 μm and the gap between them is 0.5 μm, or 500 nm. Figure 4a is a close-up image of the sensing side of one iBPE. Figure 4b is a brightfield micrograph of the PDMS chambers atop the 0.5 μm-gap device. The same PDMS master mold as in the 5.0 μm-gap device was utilized for this narrower gap device.
Figure 4.
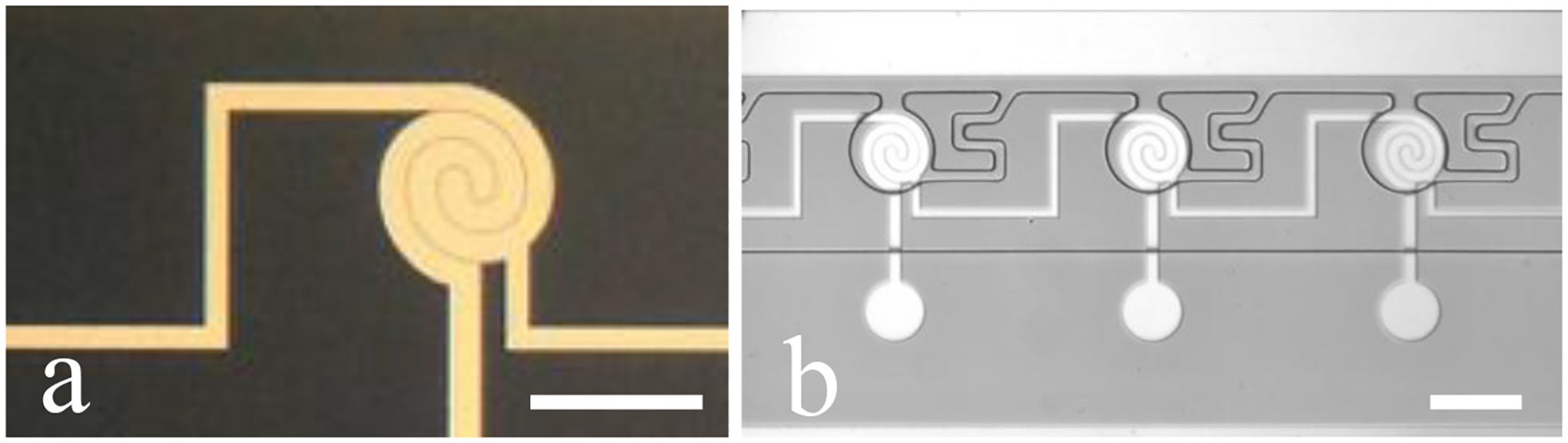
(a) Image of the sensing side of a 0.5 μm-gap interlocked spiral BPE. (b) Brightfield micrograph of interlocked spiral device with 0.5 μm gaps. Scale bar, 100 m.
LSVs and CAs were performed to electrochemically characterize the 0.5 μm-gap devices. The peak cathodic current at which pAP redox cycling occurred was the same as in the previous device at +1.73 V, when the voltage was swept from +1.00 V to +3.00 V. CAs were performed at +1.73 V, and the ECL signal was simultaneously recorded. Figure 5a is a plot of the average ECL intensity, in two devices, across a range of pAP concentrations (0.05, 0.1, 0.25, 0.3, and 0.4 mM). A linear relationship (R2 = 0.96) was observed between average ECL intensity and pAP concentration with a slope of 29.7 counts μm−2 mM−1 (10-fold greater than that observed for the 5.0 μm-gap device). Figure 5b is a plot of average ECL intensity versus concentration for both the 0.5 μm-gap and 5.0 μm-gap devices. The plot compares the ECL intensities of both interlocked spiral BPE devices. The signal enhancement afforded by the narrower gap is more pronounced at greater pAP concentrations. A 2.2-fold increase in ECL intensity was observed at 0.1 mM pAP and a 5.3-fold increase at 0.25 mM pAP in the 0.5 μm-gap devices compared to the 5.0 μm-gap devices. Figure 5c is a plot of the CA current versus pAP concentration for both devices. The plot compares the current of the 0.5 μm-gap device (Figure S1) and the 5.0 μm-gap device. A 2.9-fold increase in current was observed at 0.1 mM pAP and a 4.5-fold increase at 0.25 mM pAP. These results are significant because they demonstrate the enhancement of ECL signal in a concentration range that is relevant to the pAP production in a single cell capture device. In the 5.0 μm-gap devices, concentrations below 0.3 mM pAP were not optically detectable. However, in the 0.5 μm-gap device, a LOD of 0.1 mM pAP was achieved, which is 3 times lower than in the 5.0 μm-gap device.
Figure 5.
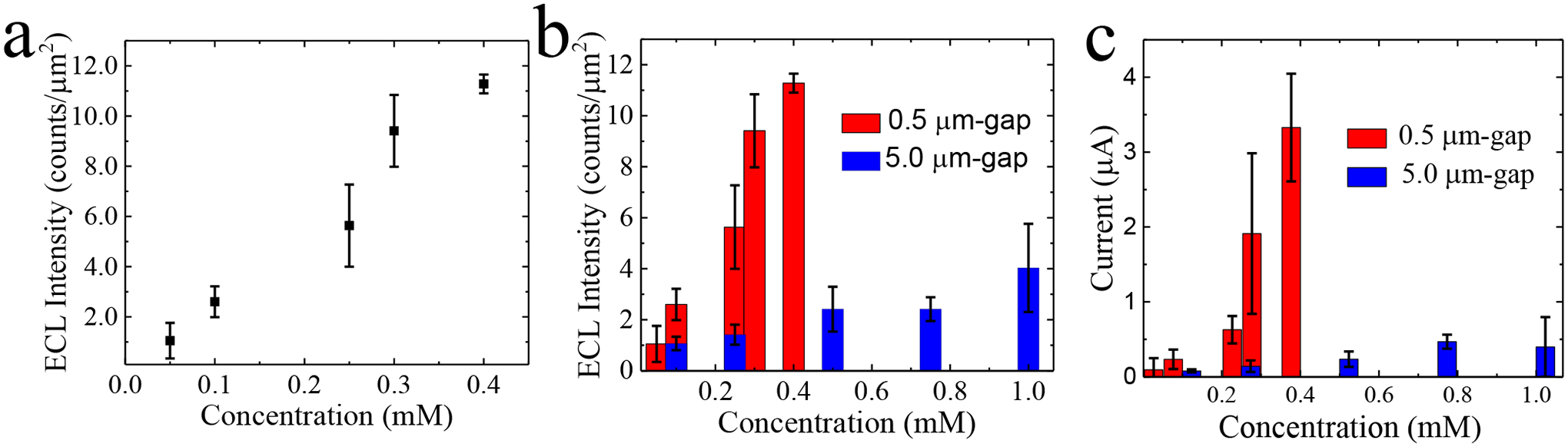
(a) ECL intensity versus concentration plot for pAP concentrations: 0.05, 0.1, 0.25, 0.3, and 0.4 mM. Plots of (b) ECL intensities and (c) currents obtained by chronoamperometry in the 0.5 μm and 5 μm-gap devices over the same concentration range as the data presented in (a).
Performance of the 0.5 μm-gap device.
Following electrochemical characterization, the assay, previously run in the 5.0 μm-gap devices, was performed in the 0.5 μm-gap device. Cells were captured in the device at 5 Vpp, 100 kHz, and an average linear flow velocity of 110–150 μm s−1. Figure 6a is a brightfield micrograph of the chamber with captured cells. One cell is captured in the second chamber (from left), while the rest remain empty. Figure 6b is a darkfield image of the reporting pole that corresponds to the chambers in Figure 6a. The reporting pole with the brightest luminescence is the BPE pole that corresponds to the chamber containing the cell. The contrast was increased by 1.0% in the darkfield image at the 70 min incubation time point in order to clearly depict the results. Figure 6c is a plot of the resulting ECL intensity produced over time upon the application of +1.73 V for 30 s for 6 separate cell environments. For the chambers that contain one cell, the ECL intensity increases linearly with incubation time (R2 = 0.86). Figure 6d displays the ECL intensity at the anodic pole of the BPE for chambers that contain a cell versus chambers that do not contain a cell. The ECL intensity for the chambers without a cell remains below 0.01 counts μm−2. According to the calibration curve, 0.099 mM pAP was produced by one cell in chamber 2. This concentration of pAP corresponds to 5801 molecules of MCAM, on an A375 cell. Using flow cytometry (results in Figure S3), we determined that there is an average of 8017 MCAM molecules per cell. The 0.5 μm device can detect MCAM on the same order of magnitude as the known amount of MCAM. These results are significant because they demonstrate a device that can optically and electrochemically report the expression of a cell surface antigen at the single-cell level. Cell selection and capture by DEP is independent of the expression of this marker, and the method does not require fluorescence imaging capabilities. The combination of electrochemical ELISA with an array of interdigitated electrodes (here wireless iBPEs) and physical confinement creates an avenue for further signal amplification.
Figure 6.
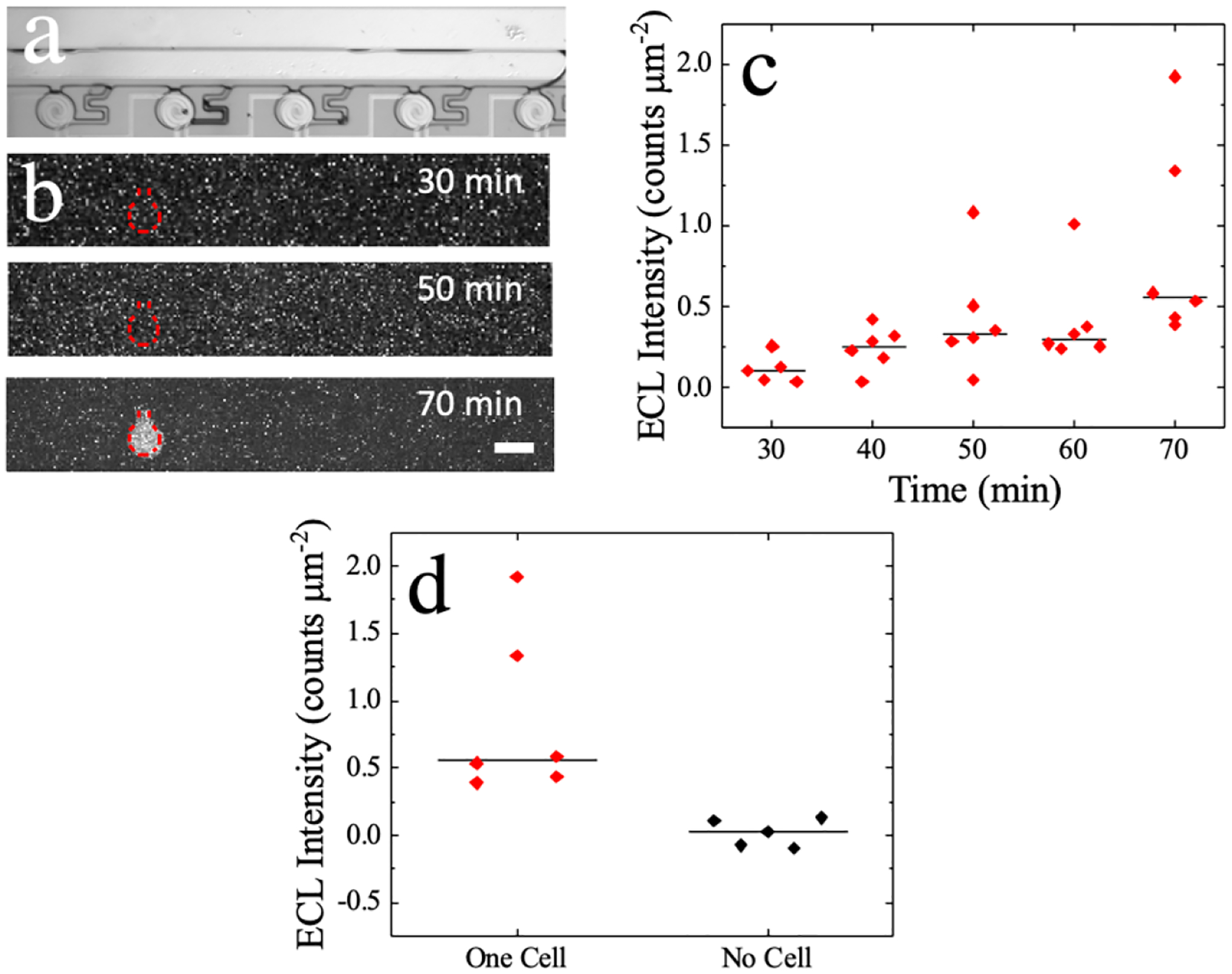
(a) Brightfield image of cells captured in chamber. (b) Darkfield image showing ECL at the BPE reporting pole at 30-, 50-, and 70- min of eELISA. Scale bar, 100 m. (c) Plot of ECL intensity over a 70 min incubation time for five different cell-containing chambers. (d) Plot of ECL intensity at 70 min incubation time for chambers containing one cell compared to empty chambers (no cell).
Conclusion
In this work, we developed an interlocked spiral bipolar electrode for the selective capture and electrochemical analysis of melanoma-specific cell surface antigens in single melanoma cells. A key point is that cells are selected by DEP based on biophysical properties that are independent of the level of expression of the surface marker that is analyzed. The original 5.0 μm-gap device displayed a linear relationship between current and pAP concentration, ECL intensity, and current. An electrochemical enzyme-linked immunosorbent assay revealed that these 5.0 μm-gap devices are sufficiently sensitive to electrically detect pAP production from single melanoma cells. However, the concentration of pAP produced during the assay was not detectable optically via ECL. E-beam lithography was utilized to develop a 0.5 μm-gap device. The 0.5 μm-gap device demonstrated a linear relationship between ECL intensity and concentration. This submicron-gap device produced an order-of-magnitude increase in sensitivity. These results demonstrate the quantification of pAP in a concentration range suitable for single-cell analysis. Additionally, these results indicate that this device is amenable for optical detection of single cells with distinguishable ECL intensity measurements. In this work each device contains an array of 30 BPEs, however bipolar electrochemistry affords the use of up to hundreds of BPEs controlled by the same set of driving electrodes. A larger BPE array would increase the chances of CTC capture and has the potential to provide insight into heterogeneous cell populations.
Experimental Methods
Cell labeling.
A375 melanoma cells (ATCC, Manassas, VA) were sub-cultured and suspended in 5 mL of Dulbecco’s Phosphate Buffered Saline (DPBS, Corning, Manassas, VA) at 1.0 × 106 cells/mL concentration. Cell concentration and viability were monitored via a Countess 3 Automated Cell Counter (Life Technologies, Thermo Fisher Scientific, Inc., Waltham, MA). One hundred milliliters of biotinylated anti-human MCAM (Human MCAM/CD146 Biotinylated Antibody R&D Systems, Biotechne, Minneapolis, MN) suspended in cell labeling buffer (DPBS with 1% fetal bovine serum (FBS, Corning, Manassas, VA)) was added to the cells for a final concentration of 1.0 × 106 cells/ 100 μL of antibody solution and allowed to incubate for 1 h. Following incubation, the cells were centrifuged and washed with 1 mL of DPBS three times. One hundred milliliters of streptavidin-conjugated alkaline phosphatase (ALP) (Invitrogen, Thermo Fisher Scientific, Inc., Waltham, MA) suspended in cell labeling buffer was added to the antibody-labeled cells for a final concentration of 1.0 × 106 cells/ 100 μL of cell labeling solution and incubated for 30 min. After incubation, cells were centrifuged and washed with 1 mL of DPBS two times. Cells were then rinsed and suspended in 1 mL of “DEP buffer” and considered ready to be inserted into the device and used for experiments. DEP buffer is comprised of 8.0% sucrose (Sigma Aldrich, St. Louis, MO), 0.3% dextrose (D-glucose, Sigma Aldrich, St. Louis, MO), 0.1% bovine serum albumin (Thermo Fisher Scientific, Inc., Waltham, MA), and 100 units/mL catalase (C30, Sigma Aldrich, St. Louis, MO). in 1.0 mM Tris Buffer (Sigma Aldrich, St. Louis, MO).
Device fabrication for an iBPE with a microscale gap.
The 5.0 μm-gap device was fabricated using standard photolithographic and etching methods. Gold/chromium coated glass microscope slides (5 nm-thick Cr adhesion layer and 100 nm-thick Au top layer, EMF Corp. Ithaca, NY) were spin-coated with AZ P4620 photoresist (Integrated Micro Materials (IMM), Argyle, TX) and exposed to a UV light source under a chrome electrode mask. Slides were developed in AZ 400 K developer, wet etched with gold etchant (4% KI/1% I2) and chromium etchant (Sigma Aldrich, St. Louis, MO), followed by a DDI water rinse. For further AZ photoresist removal, the slides were then mechanically agitated in 1-methyl-2-pyrrolidinone (NMP) at 85 °C for 10 min and dried with N2 gas. Slides were then placed in a UV Ozone cleaner (HELIOS-500, UVFab, Walnut Creek, AZ) for 1 h. The NMP cleaning process was repeated, and the slides were placed back in the UV ozone cleaner for another 30 min. After sufficiently cleaning the slides, they were rinsed with isopropanol and dried with N2 gas.
A polydimethylsiloxane (PDMS) monolith with microfluidic channels and chambers was made by casting Sylgard 184 (Dow corning, Midland, MI) to a master mold made of SU-8 2025 (Kayakli Advanced Materials, Westborough, MA) that was patterned on a Si wafer by standard photolithographic methods. A 10:1 ratio of PDMS base to crosslinker was poured onto the master mold and cured at room temperature for a minimum of 3 d. The PDMS was then cut from the mold, and inlets and outlets for each channel were punched (4 mm-diameter punches for the top inlet, 1 mm punches for the top outlet, and 3 mm punches for the bottom inlets and outlets). The PDMS and the gold patterned slide were thoroughly rinsed with ethanol and dried with N2 gas. The gold patterned slide was further cleaned in the UV/ozone cleaner for 10 min, then rinsed with isopropanol and dried with N2. To induce permanent bonding between each substrate, the slide and PDMS were placed in a plasma cleaner (PDC-001, Harrick Scientific, Ithaca, NY) and exposed to air plasma at medium power for 60 s. After plasma treatment, ethanol was added to the slide for ease of alignment and the PDMS design was aligned with the gold electrode features under a microscope (Nikon AZ100 Zoom). The whole device was post-baked for at least 12 h in an oven at 65 °C. At least 3 h before experimentation, the flags of the device were painted with silver paint (Cl 1001, Engineered Materials Systems, Delaware). Before performing experiments, the top channel of the microfluidic device was filled with 3.0 μM Pluronic F-127 (Sigma Aldrich, St. Louis, MO) for 1 h to increase the hydrophilicity of the PDMS surface. The bottom channel was filled with water during this time. The device was then used for electrochemical measurements or dielectrophoretic capture of cells.
Device fabrication for an iBPE with a submicron gap.
An iBPE with a 0.5 m gap between the interlocking spiral electrodes was fabricated using electron-beam (e-beam) lithography. E-beam lithography is a lithographic technique, developed from scanning electron microscope (SEM) technology, that uses a beam of electrons to pattern a computer-generated design on a wafer. First, an adhesion promoter, hexamethyldisilazane (HMDS, Sigma Aldrich, St. Louis, MO), was spin-coated onto a 4-in borosilicate glass wafer to ensure optimal adhesion of the e-beam resist to the glass. After spin coating, the wafer was dried with nitrogen. E-beam resist, ZEP 250a (Zeon Corporation, Tokyo, Japan), a resist easily disturbed by the e-beam at lower currents which results in higher resolution nano-features, was spin-coated onto the HMDS-coated wafer. The post bake of the Zep 520a wafer was performed on a hotplate at 180 °C for 3 min. Once the wafer cooled, 50 nm of gold was sputtered onto the wafer with an AJA sputterer (AJA International Inc, Scituate, MA) to create a reflective and conductive layer used to reduce surface charging when the e-beam strikes the wafer. After the wafer was completely prepped, it was inserted into the Vistec EBPG 5000+ e-beam, and the AutoCad designed pattern was written with a beam energy of 200 μC.
Once the design was finished writing, the thin gold layer was developed in gold etchant (KI/I2) for 15 s, and the e-beam resist was developed in amyl acetate (Sigma Aldrich, St. Louis, MO) for 30 s. A CHA e-beam evaporator (CHA industries, Livermore, CA) was utilized to deposit a 5.0 nm-layer of chromium and a 100 nm-layer of gold onto the wafer. The thin Cr layer was used to assist in the adhesion of Au to the glass wafer. After fully depositing both layers, the wafer was left in a warm (80 °C) lift-off solvent, N-methyl-pyrrolidone (NMP, Sigma Aldrich, St. Louis, MO), for at least 12 h. The wafer was rinsed with acetone and isopropanol, then dried with N2 gas. If Cr/Au was still present in unwanted areas of the wafer after rinsing, the wafer was placed in a warm ultrasonic bath of NMP to remove any remaining pieces of gold. The complete wafer was diced with a wafer saw (Disco DAC 552 Automatic Cutting Saw, Tokyo, Japan) into four 40 mm by 25 mm devices. The procedure for device assembly is the same as that for the 5.0 μm-gap devices, as described in the preceding subsection.
Electrochemical Measurements.
Prior to electrochemical measurements, the sensing channel was filled with a solution composed of 0.5 M phosphate buffer (pH 7.4) and varying concentrations of pAP (Sigma Aldrich, St. Louis, MO) as specified in each experiment. Before electrochemical assay measurements, the sensing channel was filled with pAPP (GoldBio, St. Louis, MO). For both scenarios, the reporting channel was filled with a 25 mM Ru(bpy)32+/5 mM tripropylamine (TPA) (ECL solution). Linear sweep voltammetry and chronoamperometry were performed on a Pine Research WaveDriver 10 Potentiostat (Durham, NC). Electrical leads were connected to the side and bottom driving electrode with silver-painted copper tape. The lead of the working electrode of the potentiostat was attached to the anodic driving electrode (side electrode; first spiral). A combined auxiliary and reference lead was connected to the cathodic driving electrode (bottom electrode).
Linear sweep voltammograms (LSVs) were obtained over a potential range of +1.00 V to +3.00 V at a rate of 25 mV s−1. After the linear sweep voltammograms (LSVs), chronoamperograms (CAs) were run at +1.73 V for 30 s at 30,000 intervals. The sCMOS camera (Andor Zyla 4.2, Oxford Instruments, Abingdon, Oxfordshire, England) monitored and acquired the luminescence intensity in dark room conditions. The camera was set at 4 × 4 binning, 20 s exposure, 540 GHz, 1100 LUTs, and 0.05 fps.
Cell capture.
The sensing channel of all devices was filled with 3 μM F-127 Pluronic (Sigma Aldrich, St. Louis, MO) for 1 h. After being rinsed three times with DDI water, the top and bottom channels were filled with DEP buffer. A Pico Plus Elite syringe pump (Harvard Apparatus, Holliston, MA) paired with a 500 μL glass syringe (Hamilton Company, Reno, NV), in withdraw mode, was utilized to induce flow rate at 25 nL/min during all steps after DEP buffer was introduced into the device. Using a micropipette, 35 μL of DEP buffer was removed from the sensing channel inlet and replaced with 35 μL of A375 cell solution. Cells were allowed to flow throughout the top channel of the device for 5 min until cells were present throughout the entire array. DEP was used to capture a single cell at the micropocket of each chamber under the following conditions: 50 kHz, 7 Vpp and a 110 – 150 μm s−1 linear flow velocity. The frequency and AC voltage were controlled by a Tektronic AFG3011C waveform generator (Tektronic, Beaverton, OR). Leads of the waveform generator were connected by silver-painted copper tape to the top and side driving electrodes for DEP cell capture. After 5 min, to ensure cells capture, the AC voltage was turned off, and subsequently, cells were hydrodynamically transferred into the chamber. After cell capture and transfer were complete, the capture voltage was turned on while the substrate for eELISA, pAPP in 0.5 M phosphate buffer (pH 7.4), was introduced. The substrate was allowed to flow for 5 min before the AC voltage was turned off and the solution in the channel was displaced with heavy mineral oil, sealing the chambers. After the first 30 min of cell incubation with pAPP, electrochemical analysis was performed, as described above, for 90 min.
Data Analysis.
ECL intensities were summed within a region of interest (ROI) around the anodic pole of the device using Nikon Imaging software. Values were reported after background subtraction and division by the total area of the anodic pole. LODs are reported as three times the standard error in y-values of the calibration curve divided by the slope.
Supplementary Material
Acknowledgements
We would like to acknowledge the National Institute of Health for funding this project through the R21 Trailblazer award number R21EB028583.
References
- [1].Siegel RL, Miller KD, Fuchs HE, Jemal A, CA Cancer J. Clin 2022, 72, 7–33. [DOI] [PubMed] [Google Scholar]
- [2].Xu X, Zhong JF, J. Invest. Dermatol 2010, 130, 2349–2351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Triozzi P, Zborowski M, Joshi P, Front. Oncol 2013, 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Kiniwa Y, Nakamura K, Mikoshiba A, Ashida A, Akiyama Y, Morimoto A, Okuyama R, BMC Cancer 2021, 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Aya-Bonilla CA, Morici M, Hong X, McEvoy AC, Sullivan RJ, Freeman J, Calapre L, Khattak MA, Meniawy T, Millward M, Ziman M, Gray ES, Br. J. Cancer 2020, 122, 1059–1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Mumford BS, Robertson GP, Mol. Diagn. Ther 2014, 18, 175–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Gray ES, Reid AL, Bowyer S, Calapre L, Siew K, Pearce R, Cowell L, Frank MH, Millward M, Ziman M, J. Invest. Dermatol 2015, 135, 2040–2048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Hu P, Zhang W, Xin H, Deng G, Front. Cell Dev. Biol 2016, 4, 116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Hodzic E, Bosn J Basic Med. Sci 2016, 16, 313–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Perkel JM, Nature 2021, 595, 614–616. [Google Scholar]
- [11].Li M, Liu H, Zhuang S, Goda K, RSC Adv 2021, 11, 20944–20960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Galbusera L, Bellement-Theroue G, Urchueguia A, Julou T, Van Nimwegen E, PLOS ONE 2020, 15, e0240233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Ahl PJ, Hopkins RA, Xiang WW, Au B, Kaliaperumal N, Fairhurst A-M, Connolly JE, Commun. Biol 2020, 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Labib M, Philpott DN, Wang Z, Nemr C, Chen JB, Sargent EH, Kelley SO, Acc. Chem. Res 2020, 53, 1445–1457. [DOI] [PubMed] [Google Scholar]
- [15].Espina V, Wulfkuhle JD, Calvert VS, Vanmeter A, Zhou W, Coukos G, Geho DH, Petricoin EF, Liotta LA, Nat. Protoc 2006, 1, 586–603. [DOI] [PubMed] [Google Scholar]
- [16].Keays KM, Owens GP, Ritchie AM, Gilden DH, Burgoon MP, Immunol J. Methods 2005, 302, 90–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Suarez-Quian CA, Goldstein SR, Pohida T, Smith PD, Peterson JI, Wellner E, Ghany M, Bonner RF, Biotechniques 1999, 26, 328–335. [DOI] [PubMed] [Google Scholar]
- [18].Jammes FC, Maerkl SJ, Microsyst. Nanoeng 2020, 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Labib M, Wang Z, Ahmed SU, Mohamadi RM, Duong B, Green B, Sargent EH, Kelley SO, Nat. Biomed. Eng 2021, 5, 41–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Coles BLK, Labib M, Poudineh M, Innes BT, Belair-Hickey J, Gomis S, Wang Z, Bader GD, Sargent EH, Kelley SO, van der Kooy D, Lab Chip 2021, 21, 4464–4476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Shaner SW, Allen JK, Felderman M, Pasko ET, Wimer CD, Cosford NDP, Kassegne S, Teriete P, AIP Adv 2019, 9, 065313. [Google Scholar]
- [22].Matuła K, Rivello F, Huck WTS, Adv. Biosyst 2020, 4, 1900188. [DOI] [PubMed] [Google Scholar]
- [23].Brouzes E, Humana Press, 2012, pp. 105–139. [Google Scholar]
- [24].Banovetz JT, Li M, Pagariya D, Kim S, Ganapathysubramanian B, Anand RK, Micromachines 2019, 10, 271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Li M, Anand RK, J. Am. Chem. Soc 2017, 139, 8950–8959. [DOI] [PubMed] [Google Scholar]
- [26].Bai Z, Deng Y, Kim D, Chen Z, Xiao Y, Fan R, ACS Nano 2020, 14, 7412–7424. [DOI] [PubMed] [Google Scholar]
- [27].Moscovici M, Bhimji A, Kelley S, Lab Chip 2013, 13. [DOI] [PubMed] [Google Scholar]
- [28].Fang Z, Kelley SO, Anal. Chem 2009, 81, 612–617. [DOI] [PubMed] [Google Scholar]
- [29].Wan Y, Zhou Y-G, Poudineh M, Safaei TS, Mohamadi RM, Sargent EH, Kelley SO, Angew. Chem. Int. Ed 2014, 53, 13145–13149. [DOI] [PubMed] [Google Scholar]
- [30].Mohamadi RM, Besant JD, Mepham A, Green B, Mahmoudian L, Gibbs T, Ivanov I, Malvea A, Stojcic J, Allan AL, Lowes LE, Sargent EH, Nam RK, Kelley SO, Angew. Chem. Int. Ed 2015, 54, 139–143. [DOI] [PubMed] [Google Scholar]
- [31].Li M, Anand RK, Chem. Sci 2019, 10, 1506–1513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Chen H, Anderson JL, Anand RK, ACS Appl. Mater. Interfaces 2022, 14, 18087–18096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Chen H, Osman SY, Moose DL, Vanneste M, Anderson JL, Henry MD, Anand RK, Lab Chip 2023, 23, 2586–2600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Banovetz JT, Manimaran S, Schelske BT, Anand RK, Anal. Chem 2023, 95, 7880–7887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Borchers JS, Campbell CR, Van Scoy SB, Clark MJ, Anand RK, ChemElectroChem 2021, 8, 3482–3491. [Google Scholar]
- [36].Borchers JS, Clark MJ, Van Scoy SB, Anand RK, ChemElectroChem 2024. [Google Scholar]
- [37].Gagnon ZR, Electrophoresis 2011, 32, 2466–2487. [DOI] [PubMed] [Google Scholar]
- [38].Abd Rahman N, Ibrahim F, Yafouz B, Sensors 2017, 17, 449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Pethig R, The Clausius-Mossotti Factor. In Dielectrophoresis, 2017, pp. 119–144. Wiley Online Library. DOI: 10.1002/9781118671443 [DOI] [Google Scholar]
- [40].Rahn KL, Anand RK, Anal. Chem 2021, 93, 103–123. [DOI] [PubMed] [Google Scholar]
- [41].Mavré F, Anand RK, Laws DR, Chow K-F, Chang B-Y, Crooks JA, Crooks RM, Anal. Chem 2010, 82, 8766–8774. [DOI] [PubMed] [Google Scholar]
- [42].Ulrich C, Andersson O, Nyholm L, Björefors F, Anal. Chem 2009, 81, 453–459. [DOI] [PubMed] [Google Scholar]
- [43].Hsueh A-J, Mutalib NAA, Shirato Y, Suzuki H, ACS Omega 2022, 7, 20298–20305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Ding H, Su B, Jiang D, ChemistryOpen 2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Anderson TJ, Defnet PA, Zhang B, Anal. Chem 2020, 92, 6748–6755. [DOI] [PubMed] [Google Scholar]
- [46].Berzina B, Anand RK, Anal. Chem 2018, 90, 3720–3726. [DOI] [PubMed] [Google Scholar]
- [47].Salve M, Mandal A, Amreen K, Rao BVVSNP, Pattnaik PK, Goel S, IEEE Trans. Instrum. Meas 2021, 70, 1–10.33776080 [Google Scholar]
- [48].Yang X-Y, Bai Y-Y, Huangfu Y-Y, Guo W-J, Yang Y-J, Pang D-W, Zhang Z-L, Anal. Chem 2021, 93, 1757–1763. [DOI] [PubMed] [Google Scholar]
- [49].Tian Z, Wu Y, Shao F, Tang D, Qin X, Wang C, Liu S, Anal. Chem 2021, 93, 5114–5122. [DOI] [PubMed] [Google Scholar]
- [50].Wang Y, Jin R, Sojic N, Jiang D, Chen HY, Angew. Chem. Int. Ed 2020, 59, 10416–10420. [DOI] [PubMed] [Google Scholar]
- [51].Zhang N, Gao H, Xu C-H, Cheng Y, Chen H-Y, Xu J-J, Anal. Chem 2019, 91, 12553–12559. [DOI] [PubMed] [Google Scholar]
- [52].Mavré F, Chow K-F, Sheridan E, Chang B-Y, Crooks JA, Crooks RM, Anal. Chem 2009, 81, 6218–6225. [Google Scholar]
- [53].Qi H, Peng Y, Gao Q, Zhang C, Sensors 2009, 9, 674–695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Jie GF, Huang HP, Sun XL, Zhu JJ, Biosens. Bioelectron 2008, 23, 1896–1899. [DOI] [PubMed] [Google Scholar]
- [55].Mao L, Yuan R, Chai YQ, Zhuo Y, Jiang W, Analyst 2011, 136, 1450–1455. [DOI] [PubMed] [Google Scholar]
- [56].Kim Y, Kim J, Anal. Chem 2014, 86, 1654–1660. [DOI] [PubMed] [Google Scholar]
- [57].Wu M-S, Yuan D-J, Xu J-J, Chen H-Y, Chem. Sci 2013, 4, 1182–1188. [Google Scholar]
- [58].Matlock-Colangelo L, Baeumner AJ, Lab Chip 2012, 12, 2612–2620. [DOI] [PubMed] [Google Scholar]
- [59].Voci S, Ismail A, Pham P, Yu J, Maziz A, Mesnilgrente F, Reynaud L, Livache T, Mailley P, Buhot A, Leichle T, Kuhn A, Leroy L, Bouchet-Spinelli A, Sojic N, J. Electrochem. Soc 2020, 167, 137509. [Google Scholar]
- [60].Rizwan M, Mohd-Naim N, Ahmed M, Sensors 2018, 18, 166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Ma C, Zaino Iii LP, Bohn PW, Chem. Sci 2015, 6, 3173–3179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Chen K, Amontree J, Varillas J, Zhang J, George TJ, Fan ZH, Sci. Rep 2020, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Wang Z, Ahmed S, Labib M, Wang H, Hu X, Wei J, Yao Y, Moffat J, Sargent EH, Kelley SO, Nat. Biomed. Eng 2022, 6, 108–117. [DOI] [PubMed] [Google Scholar]
- [64].Bankó P, Lee SY, Nagygyörgy V, Zrínyi M, Chae CH, Cho DH, Telekes A, J. Hematol. Oncol 2019, 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Takagi H, Dong L, Kuczler MD, Lombardo K, Hirai M, Amend SR, Pienta KJ, Int. J. Mol. Sci 2020, 21, 9031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Ribeiro-Samy S, Oliveira MI, Pereira-Veiga T, Muinelo-Romay L, Carvalho S, Gaspar J, Freitas PP, López-López R, Costa C, Diéguez L, Sci. Rep 2019, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Chen H, Zhang Z, Wang B, AIP Adv 2018, 8, 120701. [Google Scholar]
- [68].Lapizco‐Encinas BH, Electrophoresis 2019, 40, 358–375. [DOI] [PubMed] [Google Scholar]
- [69].Benhal P, Quashie D, Kim Y, Ali J, Sensors 2020, 20, 5095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Li J, King M, Front. Oncol 2012, 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Guezguez B, Vigneron P, Lamerant N, Kieda C, Jaffredo T, Dunon D, J. Immunol 2007, 179, 6673–6685. [DOI] [PubMed] [Google Scholar]
- [72].Smart JA, Oleksak JE, Hartsough EJ, Mol. Cancer Res 2021, 19, 25–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Le H. AS, Tan E, Vol. 2022, LibreTexts, 2021. [Google Scholar]
- [74].Khalid W, Göbel G, Hühn D, Montenegro J-M, Rivera-Gil P, Lisdat F, Parak WJ, J. Nanobiotechnology 2011, 9, 46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Lin AV, Indirect ELISA. In: Hnasko R (ed) ELISA: methods and protocols Springer; New York, 2015, pp. 51–59. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.