

Abstract
Background
Deep learning-based drug-target affinity (DTA) prediction methods have shown impressive performance, despite a high number of training parameters relative to the available data. Previous studies have highlighted the presence of dataset bias by suggesting that models trained solely on protein or ligand structures may perform similarly to those trained on complex structures. However, these studies did not propose solutions and focused solely on analyzing complex structure-based models. Even when ligands are excluded, protein-only models trained on complex structures still incorporate some ligand information at the binding sites. Therefore, it is unclear whether binding affinity can be accurately predicted using only compound or protein features due to potential dataset bias.
In this study, we expanded our analysis to comprehensive databases and investigated dataset bias through compound and protein feature-based methods using multilayer perceptron models. We assessed the impact of this bias on current prediction models and proposed the binding affinity similarity explorer (BASE) web service, which provides bias-reduced datasets.
Results
By analyzing eight binding affinity databases using multilayer perceptron models, we confirmed a bias where the compound-protein binding affinity can be accurately predicted using compound features alone. This bias arises because most compounds show consistent binding affinities due to high sequence or functional similarity among their target proteins. Our Uniform Manifold Approximation and Projection analysis based on compound fingerprints further revealed that low and high variation compounds do not exhibit significant structural differences. This suggests that the primary factor driving the consistent binding affinities is protein similarity rather than compound structure. We addressed this bias by creating datasets with progressively reduced protein similarity between the training and test sets, observing significant changes in model performance. We developed the BASE web service to allow researchers to download and utilize these datasets. Feature importance analysis revealed that previous models heavily relied on protein features. However, using bias-reduced datasets increased the importance of compound and interaction features, enabling a more balanced extraction of key features.
Conclusions
We propose the BASE web service, providing both the affinity prediction results of existing models and bias-reduced datasets. These resources contribute to the development of generalized and robust predictive models, enhancing the accuracy and reliability of DTA predictions in the drug discovery process. BASE is freely available online at https://synbi2024.kaist.ac.kr/base.
Supplementary Information
The online version contains supplementary material available at 10.1186/s12859-024-05968-3.
Keywords: Drug-target affinity prediction, Drug discovery, Deep learning, Dataset bias, Protein similarity
Background
Drug-target affinity (DTA) prediction is critical in the early stages of drug discovery, as it enables the identification of potential drug candidates that effectively bind to target proteins. Due to the high cost and time-consuming nature of in vitro and in vivo experiments, in silico DTA prediction methods have been developed [1]. Molecular docking, a conventional computational simulation method, uses protein 3D structures to generate binding poses and predicts binding affinity through scoring functions. However, its application has been limited by the availability of accurate protein 3D structures [2]. The advent of AlphaFold2 and its successor AlphaFold3 have revolutionized this field by generating high-resolution protein 3D structures and protein-ligand complexes [3, 4]. Additionally, deep learning approaches have been developed to enhance scoring functions for binding affinity prediction. These methods can be broadly classified into structure-based and feature descriptor-based methods.
Structure-based deep learning methods often represent binding pockets as rectangular grids and use Convolutional Neural Networks (CNNs) to extract features or Graph Neural Networks (GNNs) to model protein-ligand interactions [5–8]. Despite these advances, the number of experimentally validated protein-ligand complexes, such as those in the PDBbind database, remains limited [9]. This limitation has raised concerns about whether deep learning models truly learn protein-ligand interactions from the available data. Previous studies have shown that models trained solely on protein or ligand structures can achieve comparable performance in predicting binding affinities to those using complex structures. This finding suggests that such models might not effectively capture critical information from protein-ligand interactions [10, 11]. However, these studies predominantly used the PDBbind database, which focuses primarily on protein-ligand complexes. Therefore, even models trained solely on protein or ligand structures might still reflect information specific to these complexes. For example, a protein-only model trained on complex structures might still retain implicit information about the ligand conformation and configuration within the binding site, even after the ligand has been removed. Therefore, these methods do not clearly show whether there is a bias toward predicting binding affinity based solely on protein or ligand structures. Hence, it is necessary to evaluate whether recent deep learning-based methods for predicting compound-protein binding affinities are truly learning the intended interactions and to identify potential biases where predictions may depend solely on features of the compounds or proteins.
In addition to structure-based deep learning methods, feature descriptor-based methods have been developed to predict binding affinity because of their wide applicability and low computational cost. These methods utilize compound Simplified Molecular Input Line Entry System (SMILES), fingerprints, and protein amino acid sequences to represent molecular information [12, 13]. However, these methods still struggle to address the vast chemical space because of the limited binding affinity datasets. Recent efforts have been made to improve feature representation and address data scarcity. For example, SSM-DTA [14] employs semi-supervised learning by incorporating both labeled and unlabeled compound and protein data, whereas ColdDTA [15] uses data augmentation through subgraph removal. Despite these advancements, the complexity of these models remains high relative to the number of labeled DTA data available.
In this study, we comprehensively analyzed eight datasets, including PDBbind, BindingDB [16], ChEMBL [17], IUPHAR [18], GPCRdb [19], GLASS [20], Davis [21], and NR-DBIND [22], to investigate whether feature-based methods can accurately predict binding affinity using only the properties of compounds or proteins. We identified potential biases within these datasets, investigated their causes, and evaluated the impact of these biases on deep learning models. As a result, we observed that recent models relied on protein similarity within the datasets, rather than effectively learning the intended interactions. To address this issue, we developed datasets with reduced protein similarity bias. These datasets are provided through a web service called BASE (Binding Affinity Similarity Explorer). By utilizing BASE, researchers can access more balanced datasets and receive training and test sets split according to user-defined similarity types and cutoffs, which can potentially enhance the robustness and generalizability of binding affinity prediction models in future studies. A schematic overview of our analytical approach, from identifying potential biases to addressing them through bias-reduced datasets, is shown (Fig. 1).
Fig. 1.

Overview of the analytical process for addressing dataset bias in compound-protein binding affinity predictions. The process consists of five steps: (1) analyzing model complexity relative to data availability; (2) identifying bias in eight binding affinity datasets; (3) understanding the bias by linking it to high protein similarity; (4) discovering the problem as reduced protein similarity leads to performance declines; and (5) addressing the bias by creating datasets with reduced protein similarity.
Materials and methods
Dataset construction
We collected data from several public databases to analyze the characteristics of the DTA datasets. The main sources included PDBbind (v2020) refined set [9], BindingDB (v202401) [16], ChEMBL (v33) [17], and IUPHAR (v2024.1) [18]. Additionally, protein class-specific binding information was gathered from GPCRdb [19], GLASS [20], Davis [21], and NR-DBIND [22].
Binding affinity data were included if they were measured as dissociation constant () values, or as , , or metrics only when the assay description explicitly specified a binding affinity experiment. This careful selection ensured that we excluded data where these metrics might represent inhibition or efficacy measurements unrelated to direct binding affinity. For example, we included entries with descriptions such as “Binding affinity against human melatonin receptor type 1B by using 2-[125I]iodomelatonin as radioligand,” and excluded those such as “Inhibition of EGF-dependent EGFR autophosphorylation.” We converted the concentration values measured in nanomolar () units to a logarithmic scale for normalization using the following formula:
The dataset was restricted to human proteins, excluding protein complexes. For multiple binding affinities reported for a single compound-protein pair, the lowest affinity value (i.e., the weakest binding) was selected to ensure a conservative and robust analysis. The statistics of the databases are summarized in Table 1.
Table 1.
Statistics of the constructed dataset
| Database | Number of compounds | Number of human proteins | Number of compound-protein relations |
|---|---|---|---|
| PDBbind | 789 | 174 | 819 |
| BindingDB | 149,307 | 1443 | 232,001 |
| ChEMBL | 79,915 | 1605 | 193,104 |
| IUPHAR | 517 | 341 | 763 |
| GPCRdb | 51,283 | 183 | 78,086 |
| GLASS | 2671 | 143 | 3438 |
| Davis | 72 | 372 | 26,784 |
| NR-DBIND | 7000 | 28 | 12,455 |
| Total (Unique) | 230,194 | 1854 | 419,971 |
Regression analysis based on the compound and protein indices
We hypothesized that the high test performance of existing binding affinity prediction models might be due to the possibility that binding affinity could be predicted using only compound or protein information. To test this hypothesis, we created indices for the compounds and proteins on the basis of their average binding affinities. We indexed the compounds in ascending order of their average binding affinities, starting at one for the lowest affinity. Similarly, we indexed proteins according to the average binding affinity of compounds targeting each protein.
To evaluate whether binding affinity could be predicted using these indices alone, we performed linear regression analyses via the stats package in R [23]. Scatter plots of binding affinity against these indices were examined, and based on their patterns, we selected a third-degree polynomial model (cubic regression) to fit the data. The performance of the regression was assessed using the Pearson correlation coefficient (PCC). This analysis was conducted separately for each database and for the combined dataset to identify common patterns.
In summary, we assessed whether binding affinity could be predicted using only compound or protein indices, rather than direct compound-protein interaction information.
Calculating the coefficient of variation and categorizing compounds
To determine the proportion of compounds with consistent binding affinities, we calculated the coefficient of variation (CV) for each compound. We computed the CV as the standard deviation of the binding affinity values divided by the mean binding affinity: CV . We excluded compounds with binding affinity data for only one protein, as the standard deviation could not be calculated. We used CV instead of variance to account for the wide range in binding affinity values.
We categorized the compounds based on a CV threshold. The compounds with a CV above this threshold were considered to have a wide range of binding affinity values, whereas those with a CV below this threshold were deemed to have a more concentrated range. This threshold was determined using the mean CV value of approved drugs collected from DrugBank [24], as approved drugs are expected to have standard binding affinity variations across their targets. The compounds were thus classified into single-target compounds (affinity data existing for only one protein) and multi-target compounds. The multi-target compounds were further categorized into low and high variation groups based on their CV values relative to the threshold.
Development of a neural network model for binding affinity prediction using compound structural features
To determine whether the binding affinity of low variation (CV) and high variation compounds could be predicted solely based on structural features, we developed a neural network model using Extended Connectivity Fingerprints (ECFPs) [25]. SMILES information for the compounds was obtained from the PubChem [26] database and converted into ECFPs via the RDKit [27] package, with a radius of 2 and hashed to 1024 bits.
The neural network model was a multilayer perceptron (MLP) with three hidden layers: 128 neurons in the first layer, 64 in the second layer, and 32 in the third layer, all using ReLU activation functions. The output layer consisted of a single neuron to predict binding affinity. The model was trained using a learning rate of 0.001 and the Adam optimizer, with mean squared error as the loss function. The dataset was split into a training set (80%) and a test set (20%) for model training and evaluation. Performance was evaluated using the PCC. Model implementation and training were carried out using the Keras [28] framework.
Development of a binding affinity prediction model using compound, protein, and interaction features
To investigate whether binding affinity for low CV and high CV compounds could be predicted using structural features of compounds, proteins, and their interactions, we developed prediction models for each feature type. Our goal was to evaluate the improvement in prediction accuracy when combining features from compounds, proteins, and their interactions.
For the compound features, we utilized the 1024-bit ECFP4. Protein features were encoded by converting individual amino acids into integers, which represent sequences as 1200-length vectors. Shorter sequences were zero-padded, and longer sequences were truncated. Interaction features were derived using Extended Connectivity Interaction Features (ECIFs) [29], a set of 1540 integer-valued features developed specifically for binding affinity prediction. ECIF counts the types of protein-ligand atom interactions within a specified distance of 6 Å and considers various chemical and structural properties of each atom including element type, valence, heavy atoms, number of hydrogens, aromaticity, and ring membership. ECIF features were generated using a Python package provided by the original authors. Protein 3D structural data were obtained in PDB format from the AlphaFold2 database [30], and compound structures were converted from SMILES to SDF format via RDKit. The compounds were aligned near the active sites of the proteins using PyMOL [31] to ensure accurate positioning for ECIF generation.
The neural network model for predicting binding affinity was an MLP with three hidden layers: 128, 64, and 32 neurons, all of which use ReLU activation functions. For the combined feature model, embeddings from each feature type (compound, protein, and interaction) were passed through two hidden layers before being concatenated. This combined embedding was then processed through an additional dense layer before the final output layer.
The model was compiled using the Adam optimizer with a learning rate of 0.001 and trained with the mean squared error as the loss function. The training and test sets were consistent across all feature type models, with an 80:20 split. Model implementation and training were performed using the Keras framework, as with the ECFP4 model.
SHAP-based feature importance analysis of the combined feature model
To determine the most important feature type in predicting the binding affinity of low CV and high CV compounds, we analyzed the combined model which includes compound, protein, and interaction features using SHapley Additive exPlanations (SHAP) [32]. SHAP quantifies the contributions of each feature to the predictions, indicating how much individual features shift the predictions.
We used the SHAP Python package, specifically employing Deep SHAP, an enhanced version of the DeepLIFT [33] algorithm. Deep SHAP approximates SHAP values for deep learning models by integrating over many background samples. This method ensures that the contributions of features to model predictions sum to the difference between the expected model output on the background samples and the current model output
The SHAP analysis process involved the following steps:
Model loading: We loaded the trained model for predicting binding affinity using combined features (compounds, proteins, and interactions).
Data sampling: Due to limitations of computational resources, we selected a subset of the training data (10,000 samples), including ECFP4, protein sequence, and ECIF features, and combined them into a single dataset.
Explainer creation: We created a SHAP explainer using the DeepExplainer class, with the sampled training data as background samples.
SHAP value computation: We computed SHAP values for a subset of the test data (1000 samples) to gain insights into feature importance for the model predictions on unseen data.
Mathematically, the SHAP values for a feature are calculated as follows:
where is the set of all features, is any subset of features not containing , and is the model prediction when only features in are present. The SHAP value represents the contribution of feature to the difference between the model predictions with and without that feature. By aggregating SHAP values across all samples, we estimated the overall importance of each feature, identifying the most influential features in predicting binding affinity.
UMAP analysis of structural feature differences between compound variation types
To determine whether structural differences between low CV and high CV compounds contribute to their variation in binding affinity, we performed uniform manifold approximation and projection (UMAP) [34] analysis based on ECFP4 fingerprints via the UMAP package in Python.
We configured UMAP with 15 neighbors to define the local neighborhood size for embedding, a minimum distance of 0.1 to maintain the local structure, and the Jaccard metric to measure similarity between the ECFP4.
For visualization, we used the first and second UMAP components to enable a clear comparison of the structural characteristics of low CV and high CV compounds in two-dimensional space.
Sequence and functional similarity analysis among target proteins of compounds
To assess whether the binding affinities of low CV compounds are associated with higher similarity among their target proteins, we calculated sequence and functional similarities.
For sequence similarity, we used the Smith-Waterman algorithm [35] with the BLOSUM62 substitution matrix [36] to compute local alignment scores between amino acid sequences. We performed this calculation using the Biopython [37] package, with all other parameters set to default values. Normalized Smith-Waterman scores were calculated for each pair of sequences by adjusting the alignment scores with the geometric mean of the self-alignment scores.
We calculated functional similarity based on the Gene Ontology Biological Process (GOBP) [38] terms associated with each protein. We employed the GOSemSim [39] package in R, utilizing the semantic similarity method proposed by Wang et al. [40]. This method consists of three key steps:
Semantic values of GO terms: Each GO term is represented as part of a directed acyclic graph (DAG). The semantic value of a GO term is calculated by aggregating the contributions of all its ancestor terms.
Semantic similarity of GO terms: The semantic similarity between two GO terms is based on the contributions of their shared ancestor terms. The closer the shared ancestors are, the higher the similarity between the terms.
Functional similarity of genes (proteins): For two genes (or proteins), the functional similarity is computed by averaging the highest semantic similarity scores from all pairs of GO terms associated with each gene.
We then compared these similarity metrics between low CV and high CV compounds to identify significant differences in protein similarity.
Training data selection based on average similarity constraints
To create training sets with progressively decreasing similarity to the test set, we calculated an integrated similarity metric combining sequence and functional similarities. We adopted the integrated similarity calculation methods from STITCH [41], and followed the train-test split methodologies described by Li and Yang [42].
The integrated similarity between two proteins and was calculated using the following formula: where represents the sequence similarity and where represents the functional similarity.
In our approach, we fixed the test set by randomly selecting 20% of the entire dataset. We then selected training samples based on their average similarity to the test set samples. This process involved calculating the average similarity for each sample in the training set relative to all samples in the test set, and then filtering out samples that exceeded a specified similarity cutoff. The detailed splitting process is as follows:
Similarity calculation: Let denote the similarity between any two samples where belongs to the training set and where belongs to the test set
Average similarity: Define the average similarity for a sample in the training set as follows: , where is the number of samples in the test set.
New training set definition: The new training set is then defined as: which means that consists of samples from such that the average similarity between sample and all samples in is less than or equal to the similarity cutoff .
State-of-the-art model implementation
ColdDTA
ColdDTA [15] uses data augmentation to improve predictions for “cold” proteins and compounds not present in the training set. Compounds are represented as graphs, with atoms as nodes and bonds as edges. During augmentation, subgraphs are randomly removed to create new training samples while keeping the binding affinity information unchanged. For feature extraction, ColdDTA utilizes a standard message-passing GNN [43] for compounds. Atomic features include atom type, total number of hydrogen atoms, hybridization mode, and atomic valence. Proteins are represented by amino acid sequences, embedded into 128-dimensional vectors and processed using a 1D CNN [44] to extract features. The final prediction is made by feeding the fused features into multiple fully connected layers. Model performance was evaluated using PCC and other relevant metrics. Hyperparameters and additional training details followed the specifications in the original ColdDTA paper and its associated GitHub repository.
MMD-DTA
MMD-DTA [45] predicts binding affinity using a multimodal framework that integrates molecular graphs, atomic fingerprints, protein sequences, and 2D distance maps of amino acids. The features of the molecular graph are extracted using a graph isomorphism network [46]. Molecular fingerprints are processed by MLPs. Protein encoding involves extracting sequence information using a 1D CNN, and structural information is extracted from 2D pairwise distance maps, processed with a 2D CNN [47]. The feature fusion module combines drug and protein features through a residual connection and an attention mechanism. The fused features are then fed into an interaction module composed of an MLP and max pooling layers, enabling the final prediction of binding affinity. Model performance was evaluated using PCC and other metrics. Hyperparameters and training details followed the specifications provided in the original publication and its GitHub repository.
Evaluation metrics
We primarily used the PCC to measure the performance of the regression models. PCC assesses the linear correlation between the measured binding affinity and the predicted binding affinity. Additionally, we utilized mean squared error (MSE) and concordance index (CI) [48] as supplementary metrics. The MSE measures the error between the measured and predicted values, whereas the CI calculates the probability of concordance between the measured and predicted values, providing a generalized area under the ROC curve (AUROC). We also evaluated the binary classification performance of the prediction model with a binding affinity threshold of , using precision, recall, and balanced accuracy as metrics. The formulas for these metrics are as follows:
- Mean squared error (MSE)
where is the measured binding affinity, and where is the predicted binding affinity. - Pearson correlation coefficient (PCC)
where and are the means of the measured and predicted binding affinities, respectively. -
Concordance index (CI)
where and denote the measured binding affinity and model prediction of the -th sample, respectively, and denotes the normalization constant representing the total number of data pairs with differing affinity values. is an indicator function that returns a value of 1 if is greater than , and 0 otherwise. is the Heaviside step function defined as:For binary classification with a threshold, we used:
- Precision
where is the number of true positives, and where is the number of false positives. - Recall
where is the number of false negatives. - Balanced accuracy
where is the number of true negatives.
Web service development and computing environment
To facilitate the reproduction of our findings regarding similarity bias and to support the development of models that overcome these challenges, we developed a web service named BASE. We built BASE on the Shiny [49] framework, providing a user-friendly, web-based platform. Users can create and download training sets based on three types of similarity: protein sequence, functional, and integrated similarity. For each type, the training set is defined by a specific similarity threshold relative to the test set. Additionally, BASE offers regression prediction results for ColdDTA and MMD-DTA, which users can download for further analysis.
All data processing and computations were performed using R version 4.2.1 [23] and Python 3.9.19 [50]. We executed CPU-based calculations on a system equipped with 10 Intel® Xeon® Gold 6230R processors. We conducted deep learning model development on machines with eight NVIDIA® V100 GPUs.
Results and discussion
Investigation of model complexity relative to data size in binding affinity prediction methods
We began our analysis with the suspicion, based on a review of existing studies, that the high accuracy of current models might be due to an underlying bias in the datasets. To explore this, we examined the number of trainable parameters and the amount of input data used in the latest compound-protein binding affinity prediction methods.
Table 2 presents the number of trainable parameters, input data size, and performance of six state-of-the-art binding affinity prediction models: MMD-DTA [45], ProSmith [51], NHGNN-DTA [52], SSM-DTA [14], ColdDTA [15], and FusionDTA [53]. The results show high model complexity, with parameter-to-data ratios ranging from approximately 42 to 866. Despite these high parameter-to-data ratios, the models achieved impressive test performances, with CI values often exceeding 0.90.
Table 2.
Number of trainable parameters, data size, and performance of existing DTA prediction models
| Model | Trainable parameters | Data points | Parameters/data ratio | Test performance (CI) |
|---|---|---|---|---|
| MMD-DTA | 3,018,065 | 30056 | 100.415 | 0.905 |
| ProSmith | 44,120,897 | 1,039,565 | 42.442 | 0.911 |
| NHGNN-DTA | 1,857,665 | 30,056 | 61.807 | 0.914 |
| SSM-DTA | 326,284,800 | 37,6751 | 866.049 | 0.890 |
| ColdDTA | 1,609,441 | 30,056 | 53.548 | 0.819 |
| FusionDTA | 2,013,537 | 30,056 | 66.993 | 0.913 |
This unexpectedly high performance under challenging training conditions suggests the presence of bias within the datasets. This implies that the models might predict binding affinity without truly utilizing the interaction information between compounds and proteins.
Revealing structural bias in datasets through compound and protein indices
To investigate the potential bias in datasets further, we conducted a comprehensive analysis using eight databases: PDBbind [9], BindingDB [16], ChEMBL [17], IUPHAR [18], GPCRdb [19], GLASS [20], Davis [21], and NR-DBIND [22]. We generated compound and protein indices by ordering the average binding affinity values and used these indices to perform regression analyses.
Database-specific analysis
We analyzed each database individually to determine whether binding affinity could be predicted using compound and protein indices. For each database, predictions were fitted with a third-degree polynomial function. The results revealed that in almost all the databases (7 out of 8), predictions using the compound index achieved a PCC of 0.8 or higher (Fig. 2). The exception was the Davis dataset, where the binding affinity values varied significantly for each compound, preventing the bias of prediction solely based on the compound index. However, the Davis dataset included only 72 compounds and focused exclusively on kinase targets, making it less suitable for generalization because of its limited scope. Predictions using the protein index showed greater variability and generally lower performance compared to the compound index. Although we restricted our final dataset to human proteins for consistency, we also repeated the analysis on the entire PDBbind dataset without filtering. The results showed that, in both the filtered and unfiltered datasets, binding affinity predictions based solely on compound information achieved a PCC exceeding 0.95, indicating that the observed bias persisted regardless of the filtering for human proteins.
Fig. 2.
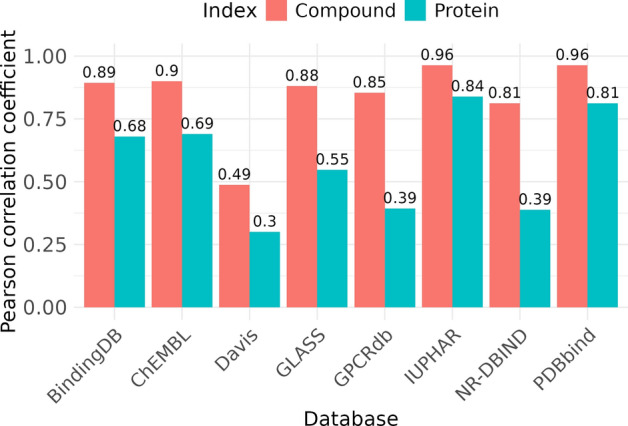
PCCs for binding affinity prediction using compound indices (orange) and protein indices (cyan) across various databases: PDBbind, BindingDB, ChEMBL, IUPHAR, GPCRdb, GLASS, Davis, and NR-DBIND. For each database, two bars represent the correlation strength, with specific values annotated above each bar for clarity.
These findings suggest that the datasets allow for accurate binding affinity predictions using only compound features, without fully capturing interaction information. Previous studies have suggested that binding affinity can be predicted using only compound or protein structures through the analysis of complex structures in the PDBbind dataset [10], although this was not clearly demonstrated. Our analysis confirmed the bias that binding affinity can be predicted using only compound features. However, the ability to predict binding affinity using protein features varied across databases. When all the databases were combined, predictions using compound features remained accurate, whereas those using protein features were less accurate. This finding can be expressed as follows: compound-protein binding affinity ≅ f(rank(compound)), where f is a polynomial function, and rank is the mean order of binding affinity. From a machine learning perspective, using a label (binding affinity) to create a feature (compound index) introduces data leakage [54], making accurate performance evaluation impossible. While this data leakage prevents full trust in the current regression performance, our results uncovered a critical characteristic of the data: compound-protein binding affinity can be fitted based on the ranking of compound binding affinities, indicating an inherent bias in the dataset structure.
Integrated database analysis
By integrating the databases, we analyzed inherent biases across the entire dataset and identified common bias patterns. This analysis revealed that binding affinity predictions based on the compound index fit well with a third-degree polynomial function, achieving a PCC of 0.921 (Fig. 3a). In contrast, the protein index was less accurate, with a PCC of 0.663 (Fig. 3b).
Fig. 3.
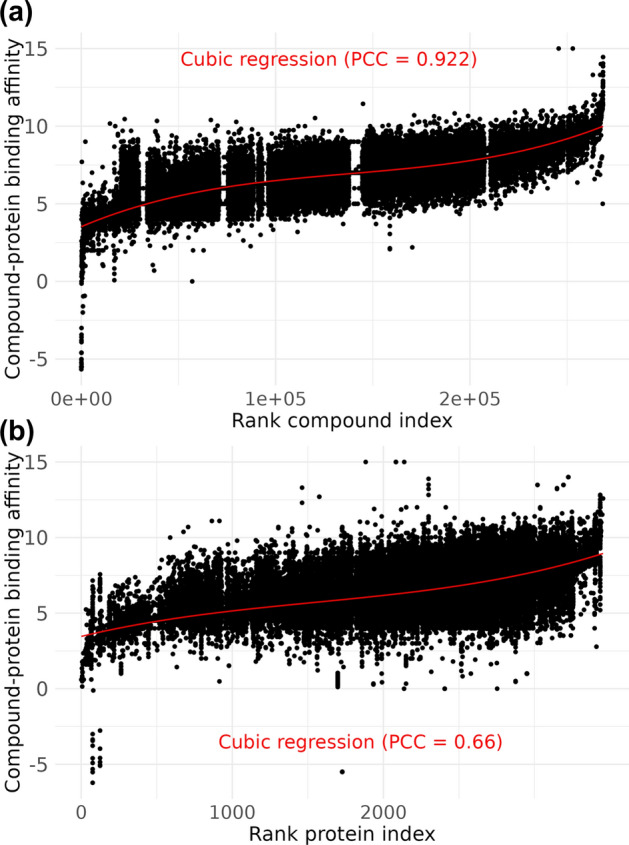
Cubic regression of binding affinity using compound and protein indices. Scatter plots show binding affinity against A the rank compound index and B the rank protein index with a third-degree polynomial (cubic) regression line (red) fitted. The PCCs for these regressions are displayed in red text. Data from all the databases were combined to calculate these PCC values.
Binding affinity prediction using structural features of low and high variation compounds
To further explore the observation that the compound index can predict binding affinity, we examined whether the binding affinities for each compound were concentrated within a specific range. We calculated the CV for each compound and categorized them as either high or low variation based on the mean CV of the approved drugs. Among the compounds that bind to multiple proteins, 81.9% (50,432 out of 61,603 compounds) were low variation compounds (Fig. 4a).
Fig. 4.
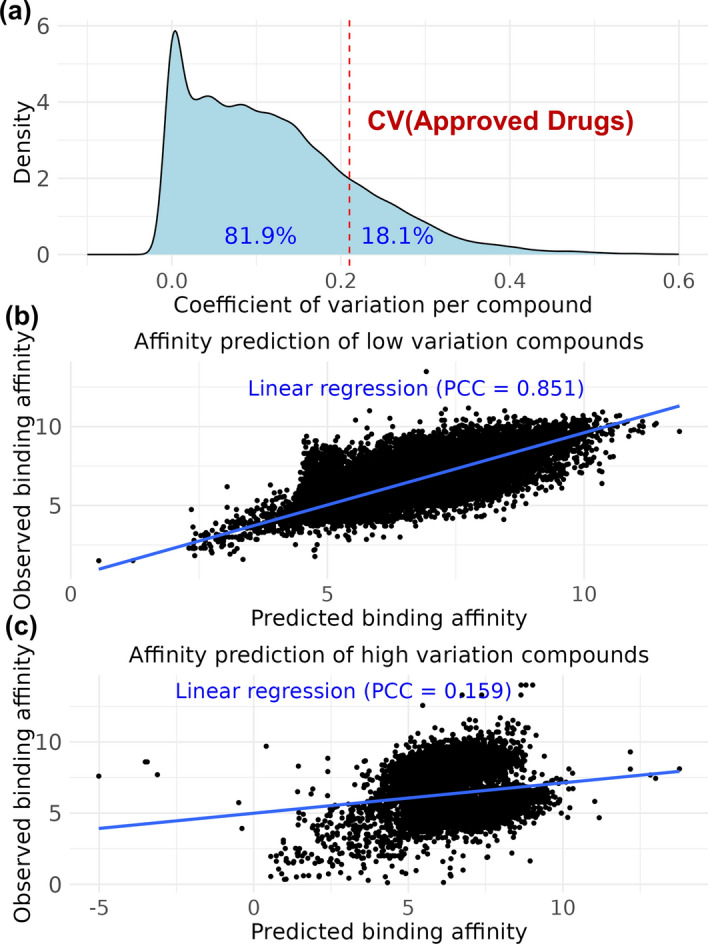
Predicting binding affinity using ECFP4 and CV per compound. A Density plot of the CV per compound, with the mean CV of approved drugs marked by a red dashed line. The proportions of compounds classified as having low variation (81.9%) and high variation (18.1%) based on this CV threshold are shown in blue text. Regression plots for binding affinity prediction of B low variation compounds and C high variation compounds using ECFP4. The scatter plots compare the predicted binding affinity (X-axis) versus the observed binding affinity (Y-axis), with the linear regression line in blue. The PCCs are displayed in blue text, with PCC = 0.851 for low variation compounds and PCC = 0.159 for high variation compounds.
For these low variation compounds, we investigated whether binding affinity could be predicted solely using compound features. We developed a regression model using ECFP4 [25] features and a standard MLP neural network. The model achieved a PCC of 0.851 on the test set, demonstrating that the structural features of the compounds alone could accurately predict the binding affinity of low variation compounds (Fig. 4b). This relationship can be expressed as follows: low variation compound-protein binding affinity ≅ f(ECFP4(compound)), where f is an artificial neural network.
In contrast, for high variation compounds, which constitute 18.1% of the dataset, the binding affinity predictions were significantly less accurate, with a PCC of 0.159 (Fig. 4c). This finding indicates that for high variation compounds, the binding affinities are more dispersed across different proteins, making it difficult to predict using only the compound features.
This discovery revealed a paradox: for low variation compounds, binding affinity could be predicted using only the compound structures without considering the protein targets. However, high variation compounds require additional factors, such as protein features. This highlights a critical characteristic of the dataset: the inherent bias toward compounds whose binding affinities are concentrated within a narrow range.
Evaluating binding affinity prediction using combined compound, protein, and interaction features
We aimed to evaluate whether binding affinity predictions for low CV compounds could be improved by incorporating protein and interaction features along with compound features, using a standard MLP model. Among the models using individual features, the compound model using ECFP4 achieved a PCC of 0.851, outperforming the protein (Seq) and interaction (IFP) [29] feature models, which had PCCs of approximately 0.7 (Fig. 5).
Fig. 5.
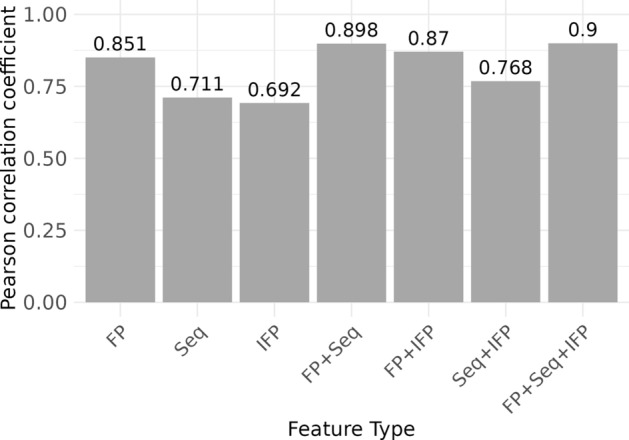
Predicting binding affinity for low variation compounds using different feature types and their combinations. The bar plot presents PCCs for predicting the binding affinity of low variation compounds using various feature types: FP (ECFP4), Seq (protein sequence encoding), and IFP. The first three bars represent individual features: FP, Seq, and IFP. The following three bars show combinations of two features: FP + Seq, FP + IFP, and Seq + IFP. The rightmost bar represents the combination of all three features: FP + Seq + IFP. The height of each bar indicates the PCC value, with specific values annotated above each bar for clarity.
Combining different pairs of features resulted in moderate improvements in prediction performance. The model combining compound and protein features achieved a PCC of 0.898, whereas the model combining compound and interaction features had a PCC of 0.87. The combination of protein and interaction features resulted in a PCC of 0.768. When all three features—compound, protein, and interaction—were combined, the model achieved a PCC of 0.9, which was comparable to the performance of the compound and protein combination.
This trend is consistent with previous studies analyzing binding affinity prediction based on complex structures [11]. Among our feature-based models, the compound feature model was the most effective among the individual feature models. Combining two features generally improved the prediction accuracy, but incorporating all three features did not result in significant performance gains compared with the model using compound and protein features. In conclusion, for low CV compounds, compound features played a decisive role in binding affinity prediction, with protein features providing a slight improvement. A similar analysis was conducted for high CV compounds, with the results included in the supplementary material (Fig. S1), revealing that combining features also improved prediction performance for this group.
Feature importance in the combined feature model using SHAP analysis
To determine whether the prediction results of the combined compound, protein, and interaction feature model depended mainly on compound features for low CV compounds, we analyzed feature importance using SHAP [32] values. Figure 6 shows the most important features based on the average absolute SHAP values. The higher a feature is on the plot, the greater its impact on the model predictions.
Fig. 6.
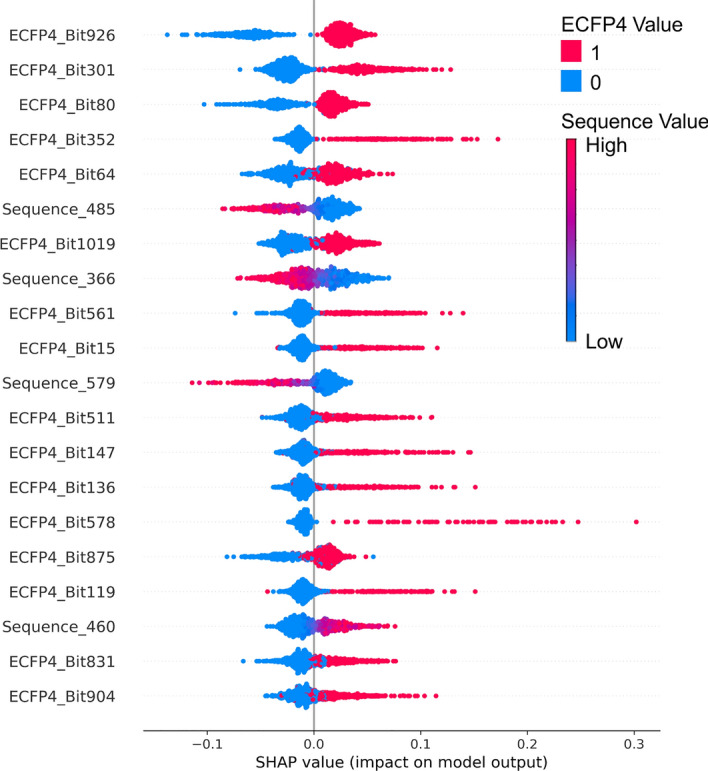
Key features in the combined ECFP4, Sequence, and IFP model based on SHAP values. This plot displays the top 20 most influential features in the combined model, ranked by their mean absolute SHAP values. Each dot represents an instance in the test set, positioned on the X-axis by its SHAP value. ECFP4 bits are color-coded: red for "On" (value = 1) and blue for "Off" (value = 0). Sequence values are indicated by a gradient color scale, with blue for lower values and red for higher values.
The results indicate that ECFP4 features dominate the top 20 important features for low CV compounds, suggesting that the model relies heavily on ECFP4 bits. Sequence features also appear in the top 20 but are less prevalent. The interaction fingerprint (IFP) features did not fall into the top 20, indicating that they have less impact than ECFP4 and sequence features.
The distribution of dots along the x-axis for each feature indicates how consistent or variable its influence is across samples. For instance, ECFP4_Bit_926 shows a significant spread in SHAP values, indicating both positive and negative impacts depending on the sample. However, the cluster of red dots on the positive side suggests a generally greater positive impact. The high ranking of ECFP4 bits, which have the smallest feature dimensions, suggests that for low CV compounds, binding affinity can indeed be predicted using only compound features. In contrast, for high CV compounds, a similar SHAP analysis revealed that protein features play a more prominent role, as shown in the supplementary material (Fig. S2), highlighting the differences in feature importance between the low and high CV groups.
ECFP4-based UMAP analysis of structural differences between low CV and high CV compounds
To understand why many compounds exhibit low CV, we hypothesized that low CV compounds possess distinct structural features leading to consistent binding affinities across various targets. To test this hypothesis, we used ECFP4 to perform UMAP [34] embedding to visualize and compare structural differences between low and high CV compounds. We calculated the ECFP4 embeddings for each compound and then applied UMAP to reduce the dimensionality for visualization. The resulting UMAP plot (Fig. 7) shows the distribution of low CV (blue) and high CV (red) compounds based on their structural features.
Fig. 7.
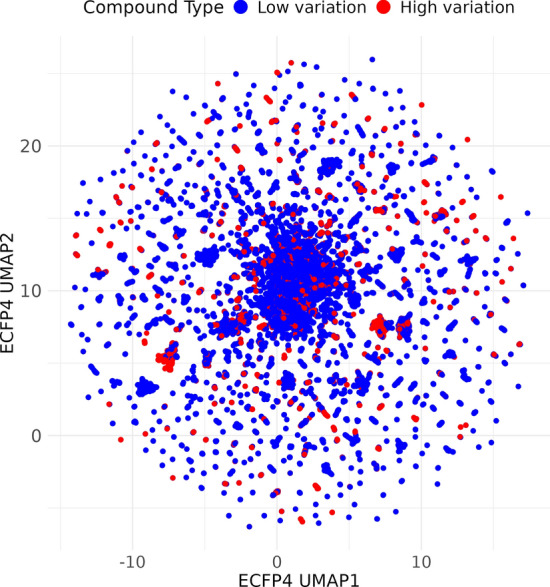
UMAP analysis of structural differences between low and high CV compounds. This UMAP plot visualizes the structural differences between low CV (blue dots) and high CV (red dots) compounds based on their ECFP4 features. The two UMAP dimensions, UMAP1 and UMAP2, are plotted on the X and Y axes, respectively.
The UMAP results indicate that high CV and low CV compounds do not form distinct clusters, suggesting that there is no significant structural differentiation between the two groups. This implies that structural features are not the primary factor contributing to the observed variations in binding affinity.
Comparing the similarity among target proteins for each compound in low and high affinity variation groups
We speculated that the consistent binding affinities observed for low CV compounds could be attributed to the high similarity among their target proteins. To test this hypothesis, we calculated the amino acid sequence similarity [35, 36] among the target proteins of each compound and compared the low variation group to the high variation group.
The results showed that the sequence similarity among the target proteins of low variation compounds was significantly higher than that of high variation compounds (Fig. 8a). Additionally, for low variation compounds with an average sequence similarity below 0.5, we calculated the functional similarity based on Gene Ontology [38, 40]. The functional similarity among the target proteins of low variation compounds was also significantly higher than that of high variation compounds (Fig. 8b).
Fig. 8.
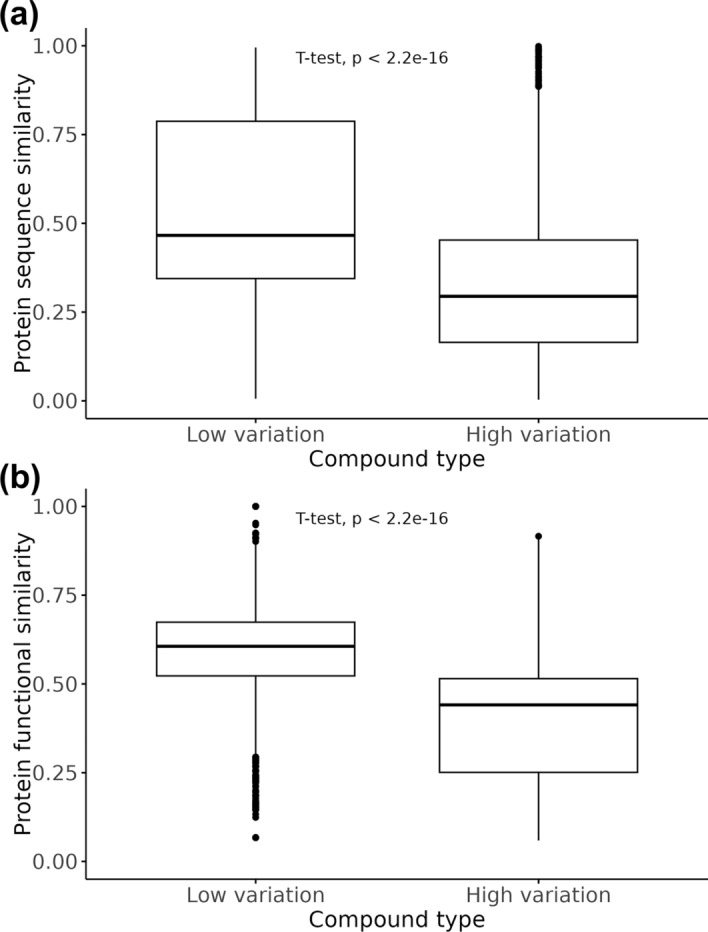
Comparison of similarity among target proteins for low and high affinity variation groups. Boxplots comparing A sequence similarity and B functional similarity among target proteins for each compound in the low and high variation groups. Functional similarity (B) is calculated using only data with average sequence similarity below 0.5. Similarity scores are displayed, with statistical significance of mean differences indicated by p values from t-tests.
These findings suggest that the consistent binding affinities observed for low CV compounds were due to the high sequence or functional similarity of their target proteins. This finding indicates that while structural differences in compounds did not account for the variation, the similarity in the target proteins was the key factor.
Evaluating the effect of similarity bias on the performance of binding affinity prediction models
Given the nature of these datasets, we realized that properly controlling the similarity between the training and test sets is essential for a fair evaluation of binding affinity prediction models. Randomly splitting the data often results in test sets containing proteins highly similar to those in the training set, leading to overoptimistic performance estimates.
To address this issue, we fixed the test sets and gradually lowered the average protein similarity between the training and test sets using an integrated similarity value that accounts for both sequence and functional similarities. As similar data were progressively removed, the size of the training set decreased accordingly. To distinguish the effects of decreasing training data from those of similarity reduction, we also conducted a control experiment by randomly subsampling the same number of data points from the training set at each similarity cutoff.
First, we evaluated the simple MLP model combining ECFP4, protein sequence encoding, and IFP. The results revealed a significant decrease in the PCC as the average integrated similarity between the training and test sets decreased from a similarity cutoff of 1, where similarity was not considered. The PCC decreased from 0.867 to 0.328, indicating that the model performance was heavily influenced by similarity bias (Fig. 9a). As the similarity decreased, both the CI [48] and the classification performance based on a threshold of also significantly declined (Table 3).
Fig. 9.
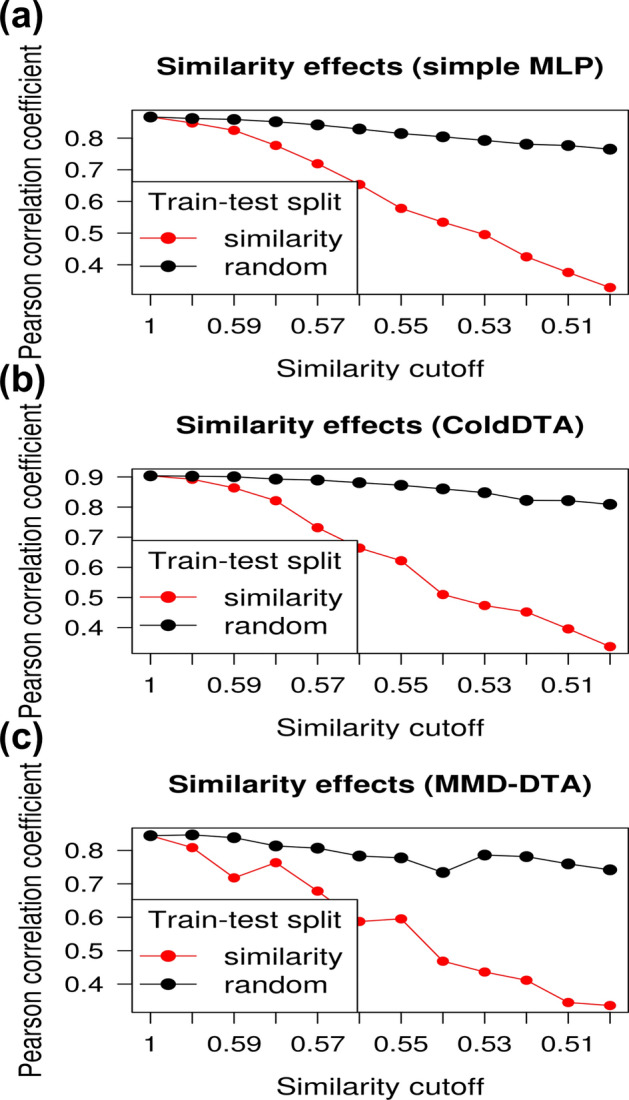
Effect of decreasing protein similarity between the training and test sets on binding affinity prediction performance. Line plots display test set PCCs for the custom-developed A simple MLP model, and the state-of-the-art models B ColdDTA and (C) MMD-DTA, as the similarity cutoff between the training and test sets is adjusted. The red line represents the performance excluding samples above the similarity cutoff, whereas the black line represents the performance of control datasets generated by random sampling to match the number of samples at each similarity cutoff.
Table 3.
Regression and classification performance of simple MLP with varying similarity cutoffs
| Similarity cutoff | Number of train sets | PCC | MSE | CI | Prec | Recall | BACC |
|---|---|---|---|---|---|---|---|
| 1 | 313,414 | 0.867 | 0.691 | 0.854 | 0.858 | 0.908 | 0.862 |
| 0.6 | 289,107 | 0.848 | 0.780 | 0.843 | 0.886 | 0.843 | 0.855 |
| 0.59 | 259,290 | 0.824 | 0.872 | 0.829 | 0.872 | 0.841 | 0.845 |
| 0.58 | 226,521 | 0.777 | 1.097 | 0.806 | 0.847 | 0.832 | 0.824 |
| 0.57 | 183,555 | 0.719 | 1.380 | 0.780 | 0.839 | 0.778 | 0.797 |
| 0.56 | 148,252 | 0.653 | 1.722 | 0.750 | 0.828 | 0.704 | 0.762 |
| 0.55 | 113,519 | 0.578 | 2.027 | 0.715 | 0.786 | 0.655 | 0.718 |
| 0.54 | 81,362 | 0.534 | 2.163 | 0.704 | 0.789 | 0.622 | 0.709 |
| 0.53 | 61,755 | 0.495 | 2.572 | 0.687 | 0.795 | 0.497 | 0.670 |
| 0.52 | 46,652 | 0.425 | 2.971 | 0.660 | 0.788 | 0.372 | 0.624 |
| 0.51 | 36,447 | 0.376 | 3.096 | 0.639 | 0.760 | 0.366 | 0.612 |
| 0.5 | 29,831 | 0.328 | 3.462 | 0.624 | 0.750 | 0.291 | 0.586 |
The number of test data points is fixed at 80,578. As the similarity to the test set diminishes, indicated by lower cutoff values, the performance metrics (PCC: Pearson correlation coefficient, MSE: mean squared error, CI: concordance Index, Prec: precision, Recall, and BACC: balanced Accuracy) generally decline.
For ColdDTA, the regression PCC on the test sets decreased from 0.9 to approximately 0.3 as the average integrated similarity between the training and test sets decreased (Fig. 9b). This indicates that the high prediction performance was due to the similarity bias inherent in the dataset. Similarly, performance metrics including the CI and classification based on a threshold also declined (Table 4).
Table 4.
Regression and classification performance of ColdDTA with varying similarity cutoffs
| Similarity cutoff | Number of train sets | PCC | MSE | CI | Prec | Recall | BACC |
|---|---|---|---|---|---|---|---|
| 1 | 313,414 | 0.904 | 0.495 | 0.880 | 0.882 | 0.925 | 0.887 |
| 0.6 | 289,107 | 0.892 | 0.548 | 0.872 | 0.869 | 0.928 | 0.878 |
| 0.59 | 259,290 | 0.864 | 0.683 | 0.854 | 0.867 | 0.902 | 0.866 |
| 0.58 | 226,521 | 0.821 | 0.878 | 0.832 | 0.845 | 0.889 | 0.844 |
| 0.57 | 183,555 | 0.732 | 1.279 | 0.789 | 0.824 | 0.817 | 0.801 |
| 0.56 | 148,252 | 0.664 | 1.584 | 0.756 | 0.802 | 0.774 | 0.770 |
| 0.55 | 113,519 | 0.622 | 1.753 | 0.735 | 0.783 | 0.737 | 0.743 |
| 0.54 | 81,362 | 0.510 | 2.199 | 0.694 | 0.759 | 0.657 | 0.700 |
| 0.53 | 61,755 | 0.473 | 2.330 | 0.677 | 0.753 | 0.617 | 0.684 |
| 0.52 | 46,652 | 0.452 | 2.383 | 0.668 | 0.737 | 0.636 | 0.679 |
| 0.51 | 36,447 | 0.396 | 2.704 | 0.647 | 0.735 | 0.496 | 0.638 |
| 0.5 | 29,831 | 0.337 | 2.960 | 0.629 | 0.722 | 0.486 | 0.628 |
The number of test data points is fixed at 80,578
MMD-DTA showed a similar trend, with some variability in performance decline as the similarity between the training and test sets decreased (Fig. 9c, Table 5). These results demonstrate that current models rely heavily on the similarity between the training and test sets and fail to reliably predict the binding affinity for targets that are not similar to the targets in the training set.
Table 5.
Regression and classification performance of MMD-DTA with varying similarity cutoffs
| Similarity cutoff | Number of train sets | PCC | MSE | CI | Prec | Recall | BACC |
|---|---|---|---|---|---|---|---|
| 1 | 313,414 | 0.844 | 0.946 | 0.838 | 0.890 | 0.789 | 0.836 |
| 0.6 | 289,107 | 0.809 | 0.881 | 0.819 | 0.820 | 0.876 | 0.822 |
| 0.59 | 259,290 | 0.718 | 1.298 | 0.800 | 0.836 | 0.824 | 0.815 |
| 0.58 | 226,521 | 0.763 | 1.083 | 0.793 | 0.834 | 0.793 | 0.801 |
| 0.57 | 183,555 | 0.678 | 1.531 | 0.749 | 0.826 | 0.665 | 0.748 |
| 0.56 | 148,252 | 0.587 | 1.940 | 0.707 | 0.814 | 0.566 | 0.705 |
| 0.55 | 113,519 | 0.595 | 1.756 | 0.708 | 0.776 | 0.628 | 0.705 |
| 0.54 | 81362 | 0.468 | 2.358 | 0.663 | 0.774 | 0.449 | 0.646 |
| 0.53 | 61,755 | 0.436 | 2.536 | 0.656 | 0.769 | 0.407 | 0.630 |
| 0.52 | 46,652 | 0.411 | 2.719 | 0.645 | 0.770 | 0.292 | 0.593 |
| 0.51 | 36,447 | 0.345 | 2.739 | 0.611 | 0.722 | 0.277 | 0.574 |
| 0.5 | 29,831 | 0.336 | 3.310 | 0.616 | 0.804 | 0.133 | 0.547 |
The number of test data points is fixed at 80,578
These findings align with existing research on machine learning-based scoring functions for estimating binding affinity using complex structures, where a decrease in protein similarity between the training and test sets also led to performance degradation [42]. This consistency underscores the importance of considering protein similarity when evaluating binding affinity prediction models. Additionally, it highlights the necessity of training and evaluating models on datasets where such biases are minimized to ensure reliable and generalizable predictions.
Web service for providing bias-reduced datasets and its impact on binding affinity prediction
We developed a web-based platform named Binding Affinity Similarity Explorer (BASE), which provides datasets that can be used to develop more robust and generalizable binding affinity prediction models by addressing the similarity bias between training and test sets. BASE allows users to create customized training sets by excluding proteins similar to those in the test set, thereby reducing bias. Users can define similarity thresholds based on three types of similarities—protein sequence, gene ontology, and integrated similarity—relative to the test set. The training and test sets used in the evaluation results for the MLP, ColdDTA, and MMD-DTA models (reported in Tables 3, 4, and 5) can be accessed and downloaded from the Data Browser tab on the BASE website. Specifically, these datasets can be found under the “integrated” similarity type, with options to select different similarity cutoffs (Fig. 10). In addition, we provide prediction results for each model under various similarity cutoffs through the Running Examples tab, allowing users to visualize how prediction performance changes as similarity thresholds are adjusted.
Fig. 10.
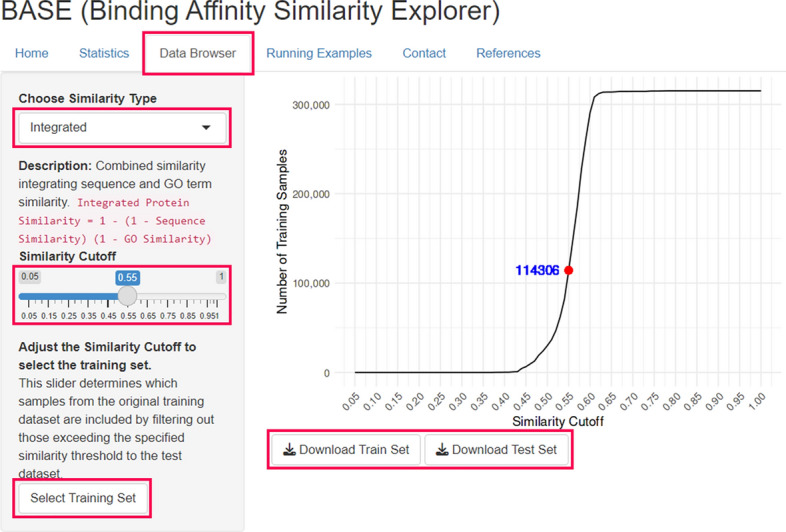
BASE web service data browser tab interface. This interface of BASE allows users to split training and test sets by protein similarity. Users can select similarity types and adjust the similarity cutoff, which updates the number of selected training samples displayed in blue on the line graph. The “Select Training Set” button shows the dataset information in table form, and datasets can be downloaded as CSV files using the “Download Train Set” and “Download Test Set” buttons. Clickable and selectable items are highlighted with red lines.
To validate the effectiveness of these bias-reduced training sets, we conducted a SHAP analysis on the feature importance of a simple MLP model combining compound, protein, and interaction features. Using ECFP4 (1024 bits), sequence encoding (1200 length), and IFP (1540 length), we created a feature set of 3764 dimensions. We then extracted the top 500 features based on their mean absolute SHAP values to understand the overall distribution of feature types. Initially, without considering similarity (similarity cutoff = 1), more than 75% of the top 500 features were protein features. As the similarity cutoff decreased to 0.5, the proportion of protein features decreased to less than 50%, whereas the proportions of compound and interaction features increased from 22.2% and 2% to 37.4% and 12.8%, respectively (Fig. 11). This shift indicated that models trained on our proposed bias-reduced dataset began to balance the importance of various features, especially increasing the significance of interaction features.
Fig. 11.
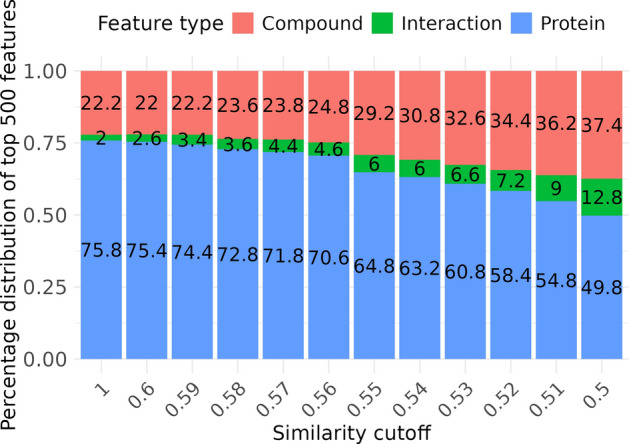
Proportion of the top 500 features by type across different similarity cutoffs. This plot illustrates the distribution of feature types among the top 500 features ranked by mean absolute SHAP values: compound (ECFP4), interaction (IFP), and protein (sequence). The features are identified from models trained on datasets filtered by different similarity cutoffs and evaluated on a consistent test set. The X-axis represents the similarity cutoff values, whereas the Y-axis represents the percentage distribution of each feature type. The colors indicate the feature types: orange for compounds, green for interactions, and blue for proteins.
Although the test performance decreased when these bias-reduced datasets were used, the models began to balance the importance of various features. By reducing the reliance on protein similarity, the models were able to develop a more comprehensive understanding of the factors that contribute to binding affinity.
Conclusions
We extended previous studies focused on complex structures within the PDBbind dataset by analyzing a broader database using compound and protein feature-based methods. As a result, we identified a dataset bias that suggests that compound-protein binding predictions may rely on compound or protein features rather than learning the intended interactions.
Specifically, we revealed that low CV compounds, which constitute the majority of compounds with known binding affinities to multiple proteins, had binding affinities that could be predicted using ECFP4 features alone. We found no structural differences between low and high CV compounds; instead, the consistent binding affinities of low CV compounds were due to the high sequence and functional similarity among their target proteins. This finding underscores the importance of controlling protein similarity between training and test sets for accurate evaluation. By progressively reducing the protein similarity between the training and test sets, we observed a significant decrease in prediction performance across our simple MLP model and state-of-the-art models such as ColdDTA and MMD-DTA, confirming that high accuracy was largely due to similarity bias.
Therefore, we developed BASE, a web service that provides bias-reduced datasets by mitigating the influence of protein similarity. BASE offers training and test sets split according to user-defined similarity types and cutoffs, and it provides the binding affinity prediction results of existing methods based on these similarity cutoffs. BASE promotes a more balanced use of compound, protein, and interaction features, reducing reliance on protein similarity. The next step is to develop predictive models that can achieve higher performance using these bias-reduced datasets, leading to more robust and generalizable binding affinity predictions.
Supplementary Information
Acknowledgements
The authors thank the anonymous reviewers for their valuable suggestions.
Abbreviations
- DTA
Drug-target affinity
- CV
Coefficient of variation
- SMILES
Simplified molecular input line entry system
- ECFP
Extended connectivity fingerprint
- IFP
Interaction fingerprint
- ECIF
Extended connectivity interaction features
- MLP
Multilayer perceptron
- CNN
Convolutional neural network
- GNN
Graph neural network
- PCC
Pearson correlation coefficient
- MSE
Mean squared error
- CI
Concordance index
- AUROC
Area under the receiver operating characteristic curve
- UMAP
Uniform manifold approximation and projection
- SHAP
SHapley Additive exPlanations
- GOBP
Gene ontology biological process
Author contributions
Conceptualization and design: H.S.; G.-S.Y.; Data collection and statistical analysis: H.S.; Investigation of previous studies and data processing: H.S.; Writing the manuscript draft: H.S.; Critical review and editing of the manuscript: H.S.; S.L.; J.K.; H.P.; M.-H.H.; G.-S.Y.; Funding acquisition and supervision: G.-S.Y.
Funding
This research was supported by the BK21 FOUR funded by the Ministry of Education (MOE, Korea) and the National Research Foundation (NRF) of Korea.
Availability of data and materials
All the raw data used in this study can be accessed and downloaded online. The processed data can be downloaded from BASE. The code used in the analysis was deposited at https://github.com/hyojin0912/HJ-DTA-DataBias.
Declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Zhang H, Liu X, Cheng W, Wang T, Chen Y. Prediction of drug-target binding affinity based on deep learning models. Comput Biol Med. 2024;174:108435. [DOI] [PubMed] [Google Scholar]
- 2.Saikia S, Bordoloi M. Molecular docking: challenges, advances and its use in drug discovery perspective. Curr Drug Targets. 2019;20(5):501–21. [DOI] [PubMed] [Google Scholar]
- 3.Jumper J, Evans R, Pritzel A, Green T, Figurnov M, Ronneberger O, et al. Highly accurate protein structure prediction with AlphaFold. Nature. 2021;596(7873):583–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Abramson J, Adler J, Dunger J, Evans R, Green T, Pritzel A, et al. Accurate structure prediction of biomolecular interactions with AlphaFold 3. Nature. 2024;630:1–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gomes J, Ramsundar B, Feinberg EN, Pande VS. Atomic convolutional networks for predicting protein-ligand binding affinity. 2017. arXiv preprint arXiv:170310603.
- 6.Ragoza M, Hochuli J, Idrobo E, Sunseri J, Koes DR. Protein–ligand scoring with convolutional neural networks. J Chem Inf Model. 2017;57(4):942–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lim J, Ryu S, Park K, Choe YJ, Ham J, Kim WY. Predicting drug–target interaction using a novel graph neural network with 3D structure-embedded graph representation. J Chem Inf Model. 2019;59(9):3981–8. [DOI] [PubMed] [Google Scholar]
- 8.Son J, Kim D. Development of a graph convolutional neural network model for efficient prediction of protein-ligand binding affinities. PLoS ONE. 2021;16(4):e0249404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Liu Z, Su M, Han L, Liu J, Yang Q, Li Y, et al. Forging the basis for developing protein–ligand interaction scoring functions. Acc Chem Res. 2017;50(2):302–9. [DOI] [PubMed] [Google Scholar]
- 10.Yang J, Shen C, Huang N. Predicting or pretending: artificial intelligence for protein-ligand interactions lack of sufficiently large and unbiased datasets. Front Pharmacol. 2020;11:508760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Volkov M, Turk J-A, Drizard N, Martin N, Hoffmann B, Gaston-Mathé Y, et al. On the frustration to predict binding affinities from protein–ligand structures with deep neural networks. J Med Chem. 2022;65(11):7946–58. [DOI] [PubMed] [Google Scholar]
- 12.Öztürk H, Özgür A, Ozkirimli E. DeepDTA: deep drug–target binding affinity prediction. Bioinformatics. 2018;34(17):i821–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lee I, Keum J, Nam H. DeepConv-DTI: Prediction of drug-target interactions via deep learning with convolution on protein sequences. PLoS Comput Biol. 2019;15(6):e1007129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pei Q, Wu L, Zhu J, Xia Y, Xie S, Qin T, et al. Breaking the barriers of data scarcity in drug–target affinity prediction. Brief Bioinform. 2023;24(6):386. [DOI] [PubMed] [Google Scholar]
- 15.Fang K, Zhang Y, Du S, He J. ColdDTA: utilizing data augmentation and attention-based feature fusion for drug-target binding affinity prediction. Comput Biol Med. 2023;164:107372. [DOI] [PubMed] [Google Scholar]
- 16.Gilson MK, Liu T, Baitaluk M, Nicola G, Hwang L, Chong J. BindingDB in 2015: a public database for medicinal chemistry, computational chemistry and systems pharmacology. Nucleic Acids Res. 2016;44(D1):D1045–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zdrazil B, Felix E, Hunter F, Manners EJ, Blackshaw J, Corbett S, et al. The ChEMBL Database in 2023: a drug discovery platform spanning multiple bioactivity data types and time periods. Nucleic Acids Res. 2024;52(D1):D1180–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Harding SD, Armstrong JF, Faccenda E, Southan C, Alexander SP, Davenport AP, et al. The IUPHAR/BPS guide to pharmaCOLOGY in 2024. Nucleic Acids Res. 2024;52(D1):D1438–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pándy-Szekeres G, Caroli J, Mamyrbekov A, Kermani AA, Keserű GM, Kooistra AJ, et al. GPCRdb in 2023: state-specific structure models using AlphaFold2 and new ligand resources. Nucleic Acids Res. 2023;51(D1):D395–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chan WK, Zhang H, Yang J, Brender JR, Hur J, Özgür A, et al. GLASS: a comprehensive database for experimentally validated GPCR-ligand associations. Bioinformatics. 2015;31(18):3035–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Davis MI, Hunt JP, Herrgard S, Ciceri P, Wodicka LM, Pallares G, et al. Comprehensive analysis of kinase inhibitor selectivity. Nat Biotechnol. 2011;29(11):1046–51. [DOI] [PubMed] [Google Scholar]
- 22.Réau M, Lagarde N, Zagury J-F, Montes M. Nuclear receptors database including negative data (NR-DBIND): a database dedicated to nuclear receptors binding data including negative data and pharmacological profile: miniperspective. J Med Chem. 2018;62(6):2894–904. [DOI] [PubMed] [Google Scholar]
- 23.Team RC. RA language and environment for statistical computing, R Foundation for Statistical. Computing; 2020.
- 24.Knox C, Wilson M, Klinger CM, Franklin M, Oler E, Wilson A, et al. Drugbank 6.0: the drugbank knowledgebase for 2024. Nucleic Acids Res. 2024;52(1):D1265–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rogers D, Hahn M. Extended-connectivity fingerprints. J Chem Inf Model. 2010;50(5):742–54. [DOI] [PubMed] [Google Scholar]
- 26.Kim S, Chen J, Cheng T, Gindulyte A, He J, He S, et al. PubChem 2023 update. Nucleic Acids Res. 2023;51(D1):D1373–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Landrum G. RDKit: open-source cheminformatics. Zenodo; 2006.
- 28.Chollet F. Keras: the python deep learning library. Astrophysics source code library. 2018. ascl: 1806.022.
- 29.Sánchez-Cruz N, Medina-Franco JL, Mestres J, Barril X. Extended connectivity interaction features: improving binding affinity prediction through chemical description. Bioinformatics. 2021;37(10):1376–82. [DOI] [PubMed] [Google Scholar]
- 30.Varadi M, Bertoni D, Magana P, Paramval U, Pidruchna I, Radhakrishnan M, et al. AlphaFold protein structure database in 2024: providing structure coverage for over 214 million protein sequences. Nucleic Acids Res. 2024;52(D1):D368–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schrodinger L. The PyMOL molecular graphics system. Version. 2015;1:8. [Google Scholar]
- 32.Lundberg SM, Lee S-I. A unified approach to interpreting model predictions. Adv Neural Inf Process Syst. 2017;30:4765. [Google Scholar]
- 33.Shrikumar A, Greenside P, Kundaje A, editors. Learning important features through propagating activation differences. In: International conference on machine learning, PMlR; 2017
- 34.McInnes L, Healy J, Melville J. Umap: uniform manifold approximation and projection for dimension reduction. arXiv preprint arXiv:180203426. 2018.
- 35.Smith TF, Waterman MS. Identification of common molecular subsequences. J Mol Biol. 1981;147(1):195–7. [DOI] [PubMed] [Google Scholar]
- 36.Henikoff S, Henikoff JG. Amino acid substitution matrices from protein blocks. Proc Natl Acad Sci. 1992;89(22):10915–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cock PJ, Antao T, Chang JT, Chapman BA, Cox CJ, Dalke A, et al. Biopython: freely available Python tools for computational molecular biology and bioinformatics. Bioinformatics. 2009;25(11):1422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, et al. Gene ontology: tool for the unification of biology. Nat Genet. 2000;25(1):25–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yu G, Li F, Qin Y, Bo X, Wu Y, Wang S. GOSemSim: an R package for measuring semantic similarity among GO terms and gene products. Bioinformatics. 2010;26(7):976–8. [DOI] [PubMed] [Google Scholar]
- 40.Wang JZ, Du Z, Payattakool R, Yu PS, Chen C-F. A new method to measure the semantic similarity of GO terms. Bioinformatics. 2007;23(10):1274–81. [DOI] [PubMed] [Google Scholar]
- 41.Szklarczyk D, Santos A, Von Mering C, Jensen LJ, Bork P, Kuhn M. STITCH 5: augmenting protein–chemical interaction networks with tissue and affinity data. Nucleic Acids Res. 2016;44(D1):D380–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Li Y, Yang J. Structural and sequence similarity makes a significant impact on machine-learning-based scoring functions for protein–ligand interactions. J Chem Inf Model. 2017;57(4):1007–12. [DOI] [PubMed] [Google Scholar]
- 43.Gilmer J, Schoenholz SS, Riley PF, Vinyals O, Dahl GE. Message passing neural networks. Mach Learn Meets Quantum Phys. 2020;968:199–214. [Google Scholar]
- 44.Kiranyaz S, Avci O, Abdeljaber O, Ince T, Gabbouj M, Inman DJ. 1D convolutional neural networks and applications: a survey. Mech Syst Signal Process. 2021;151:107398. [Google Scholar]
- 45.Qi Z, Liu L, Wei Y, Zhang S, Liao B. MMD-DTA: a multi-modal deep learning framework for drug-target binding affinity and binding region prediction. bioRxiv. 2023:2023.09.19.558555. [DOI] [PubMed]
- 46.Xu K, Hu W, Leskovec J, Jegelka S. How powerful are graph neural networks? arXiv preprint arXiv:181000826. 2018.
- 47.Zheng S, Li Y, Chen S, Xu J, Yang Y. Predicting drug–protein interaction using quasi-visual question answering system. Nat Mach Intell. 2020;2(2):134–40. [Google Scholar]
- 48.Gönen M, Heller G. Concordance probability and discriminatory power in proportional hazards regression. Biometrika. 2005;92(4):965–70. [Google Scholar]
- 49.Chang W, Cheng J, Allaire J, Sievert C, Schloerke B, Xie Y, et al. Shiny: web application framework for R. 2023. URL: https://github.com/rstudio/shiny
- 50.Van Rossum G, editor. Python programming language. In: USENIX annual technical conference, Santa Clara, CA; 2007.
- 51.Kroll A, Ranjan S, Lercher MJ. Drug-target interaction prediction using a multi-modal transformer network demonstrates high generalizability to unseen proteins. bioRxiv. 2023:2023.08.21.554147.
- 52.He H, Chen G, Chen CY-C. NHGNN-DTA: a node-adaptive hybrid graph neural network for interpretable drug–target binding affinity prediction. Bioinformatics. 2023;39(6):355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Yuan W, Chen G, Chen CY-C. FusionDTA: attention-based feature polymerizer and knowledge distillation for drug-target binding affinity prediction. Brief Bioinform. 2022;23(1):506. [DOI] [PubMed] [Google Scholar]
- 54.Kaufman S, Rosset S, Perlich C, Stitelman O. Leakage in data mining: formulation, detection, and avoidance. ACM Trans Knowl Discov Data (TKDD). 2012;6(4):1–21. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All the raw data used in this study can be accessed and downloaded online. The processed data can be downloaded from BASE. The code used in the analysis was deposited at https://github.com/hyojin0912/HJ-DTA-DataBias.