

Abstract
While evolutionary biology traditionally focuses on the spread of mutations within populations, the dynamics of mutational spread within individuals, particularly in long-lived clonally, spreading organisms remain poorly understood. Here we examine the genetic structure of ‘Pando’, Earth’s largest known quaking aspen (Populus tremuloides) clone. We sequenced over 500 samples across Pando and neighboring clones, including multiple tissue types. At fine spatial scales, we detected significant genetic structure, particularly in leaf tissue, but this signal weakened across larger distances, suggesting either rapid root growth homogenizes the system over time or mechanisms exist that prevent widespread mutation transmission. Phylogenetic analyses date Pando between ~12,000 and 37,000 years old, supported by continuous aspen pollen presence in nearby lake sediments. Tissues accumulated mutations at different rates, with leaves showing significantly higher mutation loads than roots or branches. This work provides the first quantitative age estimate for this remarkable organism and reveals how massive clonal plants maintain genetic integrity while accumulating potentially adaptive variation over millennia. Our findings illuminate evolutionary processes in long-lived modular organisms and demonstrate how within-organism selection might operate in species lacking regular unicellular bottlenecks.
Introduction
Understanding how mutations arise and spread through a population is essential to understanding biological evolution. The advent of high-throughput genome sequencing has allowed us to study mutational dynamics in a vast array of previously intractable non-model organisms [1], but most prior work has focused on how mutations spread among largely well-individuated organisms (i.e., a life cycle that includes regular genetic bottlenecks), ignoring the effects of within-organism somatic mutations. This is a reasonable assumption for animals, in which germ cells segregate early during ontogeny, but many multicellular organisms (i.e., plants [2–4], fungi [5], red algae [6], brown algae [7]) grow clonally, and do not have germline sequestration.
In contrast to organisms with discrete generations, many organisms grow via clonal extension of a simple modular body plan. This is common within plants and fungi, and can result in the colonization of some habitats over vast distances and exceptionally long periods. For example, seagrasses grow via rhizomal extension, and can have genets that span large areas (e.g., a 47 km transect) [8]. Similarly, a 2,500-year-old clone of the fungus Armillaria gallica has spread over 75 hectares of forest rhizosphere [9]. Clonal proliferation may be especially resilient in the face of periodic disturbance. For instance in the Quaking Aspen, P. tremuloides, the growth of new ramets is stimulated by nutrients and light availability in areas recently damaged by fire [10, 11]. It is thus not particularly surprising that clonally-expanding organisms can be both very large and long-lived.
Clonally-expanding organisms may evolve quite differently than canonical multicellular organisms that undergo regular unicellular genetic bottlenecks. In the absence of regular bottlenecks, somatic mutations can accumulate and be carried to new tissue as these organisms grow [12]. While the emergence of somatic mutations in animals can lead to lethal cellular proliferation, clonal organisms might be less susceptible to the morbidity and mortality of cancer. Indeed, in organisms like plants, which have permanent cell-cell bonds and a modular growth plan, cancer poses little systemic risk. Notably, clonally-spreading plants and fungi have some of the longest documented lifespans. For example, in seagrasses, such as Posidonea australis [13], P. oceanica [14], Thalassia testudinum [8], or Zostera marina L. [15], estimates suggest that individual clones may be more than 6000 years old. With indeterminate growth, the longevity of modules is decoupled from that of the genet (and, in cases where genets remain connected, the organism), making clonally-expanding organisms potentially immortal. In such organisms, novel mutations may even provide a benefit, facilitating adaptive evolution via among-module competition. While some work has examined the dynamics of within-individual somatic mutations [8, 10, 16–18], we still know little about how long-lived clonally-spreading organisms evolve.
Here, we examine the genetic demographic history of Pando, one of the largest genets of the clonally-spreading Quaking Aspen (P. tremuloides) [19]. This species can reproduce vegetatively by growing roots, from which new ramets grow. While individual ramet lifespan averages 110 years [20], clones can regenerate from the root stock such that the organism can be far older than its parts. This clone has gathered particular attention for its size (42.6 hectares comprising ~47,000 individual stems [21]), and is generally thought to be ancient [22]. To investigate its evolutionary history, we sequenced over 500 samples from across Pando and neighboring clones, including leaves, roots, and bark tissues, analyzing patterns of somatic mutations to understand how genetic variation accumulates and spreads within this massive organism. Our phylogenetic analyses, corroborated by local pollen records, date Pando to between 12,000 and 37,000 years old, offering key insights into the evolutionary dynamics of long-lived clonal organisms while providing the first quantitative age estimate for this remarkable organism.
Materials and methods
Sampling
To describe the evolutionary history of the Pando clone, we generated four sets of data using different spatial scales and sequencing strategies (Table 1). The Pando clone (Populus tremuloides) is located in the Fishlake National Forest, Utah, USA (38°31’N, 111°45’W), and ranges in altitude from 2,700–2,790 m. We generated a large-scale dataset by sampling leaves from the whole Pando stand, including the neighboring non-Pando clones, on a 50-m grid in June 2006 and November 2007 (see [21] for more details), sampling from both a smaller (younger) and a taller (older) tree at each location (“large-scale dataset”, 184 samples, Supplementary figure S1, left panel). To test for fine-scale within-clone genetic structure, we sampled leaves, roots, bark from the trunk and branches from two additional subsections from within the Pando clone in June 2022 (“fine-scale dataset”, 101 samples, Supplementary figure S1, right panel). One of the sampling sites chosen for this additional sampling is in an area that was clear-cut 30 years ago and the other one is in an older area (Supplementary figures S1 and S2). To avoid batch effects and possible confounding effects of the two different spatial scales, the large and fine-scale datasets were analyzed separately (see ordination plots in Supplementary figure S3). 100 additional leaf samples were collected from P. tremuloides in the USA’s Intermountain region (Colorado, Wyoming, Nevada, Idaho) to generate the ‘panel of normals’ (see “Identifying somatic mutations” section). Leaves were kept in paper coin envelope and placed in desiccant. Root and bark samples were placed in polyethylene bags and kept at cool temperatures before long-term storage at −20°C. Finally, to test our ability to accurately identify somatic mutations, we re-sequenced 12 samples from the fine-scale dataset 8 times (same DNA extraction sequenced 8 times, “replicate dataset”, 96 total individual reactions, 80 kept after filtering).
Table 1.
To study the evolutionary history of the Pando clone, we generated datasets at different spatial scales and using different sequencing strategies. The large-scale and fine-scale datasets have the same initial number of mutations as the variant calling was done on both sets at once. Nb. = number.
| Dataset | Nb. of samples | Nb. of variants (all/somatic) |
|---|---|---|
| large-scale | 184 (Pando + neighboring clones) 89 (Pando only) | 22,888/-15,925/3,942 |
| fine-scale | 101 | 15,925/3,034 |
| replicate | 80 | 4,607/101 |
| panel of normals | 100 | 99,234 |
Sequencing
The 296 leaf samples from the Pando and surrounding clones, and the 45 root samples, 45 leaves and 27 bark samples from trunk and branches were prepared for Genotyping-By-Sequencing (GBS). Woody tissues were powdered using a pester and mortal and further lysed using Tissue Lyzer II (TissueLyser II, Qiagen). Genomic DNA was extracted using the DNeasy Plant Pro Kit (Cat. No. 69204, Qiagen). To generate a reduced complexity DNA library, the genome was digested using MseI and EcoR1 enzymes. The fragments were labelled and prepared for sequencing using oligonucleotides consisting of Illumina adaptors and unique 8–10 base pair (bp) sequences. The fragments were amplified and size-selected to only keep fragments between 300 and 400 bp-long, before sequencing. Library preparation and sequencing were done in three batches, with 367 samples sequenced with an Illumina HiSeq 4000 (1 × 100 base pair reads) in 2018, 126 and 96 samples sequenced on a NovaSeq (1 × 100 base pair reads) in 2022 and 2024, respectively (one lane each). The 2018 and 2022 samples were sequenced at the University of Texas Genomic Sequencing and Analysis Facility (Austin, TX, USA). The 2024 samples were sequenced at the Utah State Genomics Core facility. The total number of reads was 1,027,955,624. The mean sequencing depth was 13.5x for the large-scale dataset, 18.1x for the fine-scale dataset and 43.4x for the replicates.
Genome alignment and variant calling
The raw fastq files were filtered by removing PhiX and adapter sequences. We matched each barcode to their corresponding sample ID and split the reads to their individual sample files.
We used the mem algorithm from bwa (default options, version 0.7.17-r1188, [23]) to align the reads to the published reference genome for P. tremuloides [24], and samtools to compress, sort and index the alignments (Version: 1.16 [23]). We called the variants using samtools mpileup algorithm (Version: 1.16) and bcftools (Version: 1.16). The large-scale and fine-scale datasets were pooled for variant calling, and the replicate and “panel of normals” datasets were kept separate. We kept mapped reads with a quality >30, skipped bases with base quality >30 and ignored insertion–deletion polymorphisms. At this step, we separated the fine-scale and large-scale samples. We then filtered our set of SNPs for each data set by keeping the sites for which we had data (mapped reads) in no fewer than 60% of individuals, a mean coverage per sample of 4× minimum, and at least one read supporting the non-reference allele. We also removed SNPs failing the base quality rank-sum test (P < 0.005), mapping-quality rank-sum test (P < 0.005), and the read position rank-sum test (P < 0.01).
Delineating the Pando clone
In order to differentiate between the samples pertaining to the Pando clone and the surrounding clones in the large-scale dataset, we obtained Bayesian estimates of genotypes. We specifically computed the posterior mean genotype as a point estimate based on the genotype likelihood from bcftools and a binomial prior based on the allele frequency estimates from the vcf file. We used principal component analysis (PCA) to ordinate the samples; this was performed on the matrix of centered but not scaled genotype estimates. The PCA separated the Pando clone samples, from the surrounding clone samples (Figure 1) in PCA space. We used k-means clustering (R kmeans function, with K=2) to identify and label the different clusters of samples and further split the variant file into two files: the Pando variant file and the surrounding clones variant file, with 9,424 and 20,178 SNPs, respectively.
Fig 1. Parsing out the Pando samples from the surrounding clone samples.

(A) The projection of genotypes (22,888 variants) form three distinct clusters: two clusters with negative PC1 values and one cluster with positive PC1 values. Points are labeled with a color proportional to their PC1 value. (B) Plotting the PC1 value into the sampling space delineates the Pando cluster (positive PC1 values) from the surrounding clone clusters (negative PC1 values).
Identifying Somatic Mutations
Germline mutations are inherited and should be common to Pando as a whole. Somatic mutations, however, are mutations that appeared after seed formation and during the organism’s growth, potentially capturing the evolutionary history of the organism. To identify somatic mutations in the Pando clone, we developed a comparative analysis pipeline using neighboring clones and regional samples as reference points. We created a “panel of normals” [25] reference dataset comprising variants from neighboring clones and 100 P. tremuloides samples collected across the USA’s Intermountain region (Colorado, Wyoming, Nevada, Idaho). We implemented multiple filtering steps to ensure accurate mutation detection. We removed variants present in both the Pando samples and the “panel of normals” reference dataset, as these likely represented germline mutations or highly mutable sites. To minimize false positives from sequencing errors given the inherent per base pair error rate of approximately 0.31% for Illumina reads [26], we excluded mutations detected in only one sample. To validate our mutation detection approach, we performed technical replication by sequencing 12 samples eight times each from the same DNA extraction. In the replicate dataset, we classified mutations as somatic if they appeared in at least two replicates per sample but in no more than 80% of total samples, with this threshold chosen to exclude potential germline mutations that might not be detected in all samples due to technical limitations. In the fine-scale and large-scale datasets, we classified mutations as somatic if they appeared in at least two samples, but in no more than 80% of total samples. For a full description of our mutation filtering pipeline, see Supplementary Methods.
Spatial analyses
To detect spatial structure in the dataset, we applied the same set of analyses on two different datasets: (1) a large-scale, and (2) a fine-scale dataset. We first compared the proportion of shared variants per pair of samples to their physical distance (number of shared mutations between a pair of samples, divided by the mean number of mutations for the same pair of samples). We then computed the mean geographic distance between groups of samples sharing a mutation (with the mean taken over all mutations). We used Vincenty ellipsoid method (distVincentyEllipsoid function in R) to calculate the shortest spatial distance between two samples. For each analysis, we compared the empirical values to values obtained from a randomized dataset to assess the significance of the results. To generate null distributions, we randomized either the genotypes or the pair of spatial coordinates (latitude and longitude) and ran the same analysis as ran on the non-permuted data (500 or 1,000 permutations).
Coalescent model using BEAST
We used the software package BEAST (version 2.7.5) to estimate the height of the phylogenetic tree for the Pando samples based on the accumulated somatic mutations; this was done using a coalescent Bayesian skyline model for effective population size [27–29]. We chose the GTR nucleotide-substitution model to account for unequal substitutions rates between bases [30], starting with equal rates for all substitutions. We selected an optimized relaxed clock with a mean clock rate of 1. To generate the nexus file, we assigned genotypes based on the point estimate value (the sample is heterozygote, coded “A”, for the mutation if the point estimate value > 0.5, homozygote otherwise, coded “T”). A single chain was run for 7×107 states. To estimate the age of the tree, we converted the phylogeny height to years a posteriori following this calculation:
with being the phylogenetic tree height as given by BEAST, the total number of mutations, , the total number of base pairs sequenced, the leaf somatic mutation rate (1.33 * 10−10 per base per haploid genome per year [31]), taking into account that the Pando clone is triploid [32, 33]. The total number of base pairs sequenced (129,194,577) was estimated using angsd [34], and reduced following the proportion of base pairs that we filtered out because of low coverage (48%).
Accounting for missing mutations
To get an estimate of the number of somatic mutations that we might have missed in genomic regions we sequenced, we combined the 12 samples from the fine scale dataset, with the replicate dataset (these same 12 samples sequenced 8 times). We called variants on this combined dataset and identified 55 somatic mutations. We then compared the number of times we found a mutation in the replicate set for one sample, and in the same sample from the reference set. On average, if a mutation was in the replicate set, it was detected 20% of the times in the initial sample. This result was robust to permuting the reference and replicate set samples (Supplementary figure S9). It however implies that (1) we may be missing somatic mutations, or that (2) only 20% of the mutations we detect are real. We take this into account when calculating the age of the Pando clone by considering three different scenarii : (i) all mutations detected are real, (ii) only 20% of the mutations we detect are real and (iii) we are missing 80% of the mutations. To also account for how the phylogenetic tree height might be affected by missing mutations, we calculated the relationship between the number of missing mutations and the phylogeny height. To do so, we randomly removed an increasing percentage of mutations, simulated the phylogeny in BEAST and found a linear relationship between the proportion of missing mutations and the phylogenetic tree height. We used this regression to estimate the Pando clone age.
Pollen analysis
Pollen analysis followed standard acid digestion procedures [35]. Pollen residues were classified and tabulated using light microscopy at 40x until a minimum of 300 terrestrial grains were counted. Pollen identification was assisted by relevant keys and literature (e.g., Kapp et al. 2000 [36]). We assume that the Populus pollen type, which is generally not diagnostic to species-level assignment, reflects quaking aspen in this environmental setting.
Data accessibility
Scripts and detailed steps can be found on the paper repository paper repository.
Results
Delineating the Pando clone
Pando samples (89 out of 184 samples) formed a distinct cluster in PCA space (Figure 1A) with spatial boundaries for Pando that were consistent with previously defined clone boundaries based on morphological differences [19], and microsatellite markers [21, 32] (Figure 1B and Supplementary Table 1). We thus verified the spatial extent, 42.6 ha, of Pando.
Identifying somatic mutations
We generated a replicate dataset to quantify our ability to recover rare mutations between samples. Our filtering approach (see Methods for details) identified a set of 101 putative somatic mutations present in less than 40% of samples, with no mutations detected between 40% and our 80% threshold (Figure 2A,B). We found that that when a mutation was detected in two replicates of a sample, it appeared on average in 3.5 replicates total (44% of replicates), significantly higher than expected by chance (randomization test, null expectation = 0.37 with 1,000 permutations, P < 0.001, Figure 2C). The detection of mutations remained consistent across varying coverage depths (Supplementary Figure Supplementary figure S4). While these results validate our ability to detect somatic mutations, they also suggest our approach may be missing some true mutations, a limitation we address in our age estimation analyses. Having established our mutation detection methodology, we applied these filtering criteria to both our large-scale and fine-scale datasets for subsequent analyses.
Fig 2. Replication power for somatic mutations.
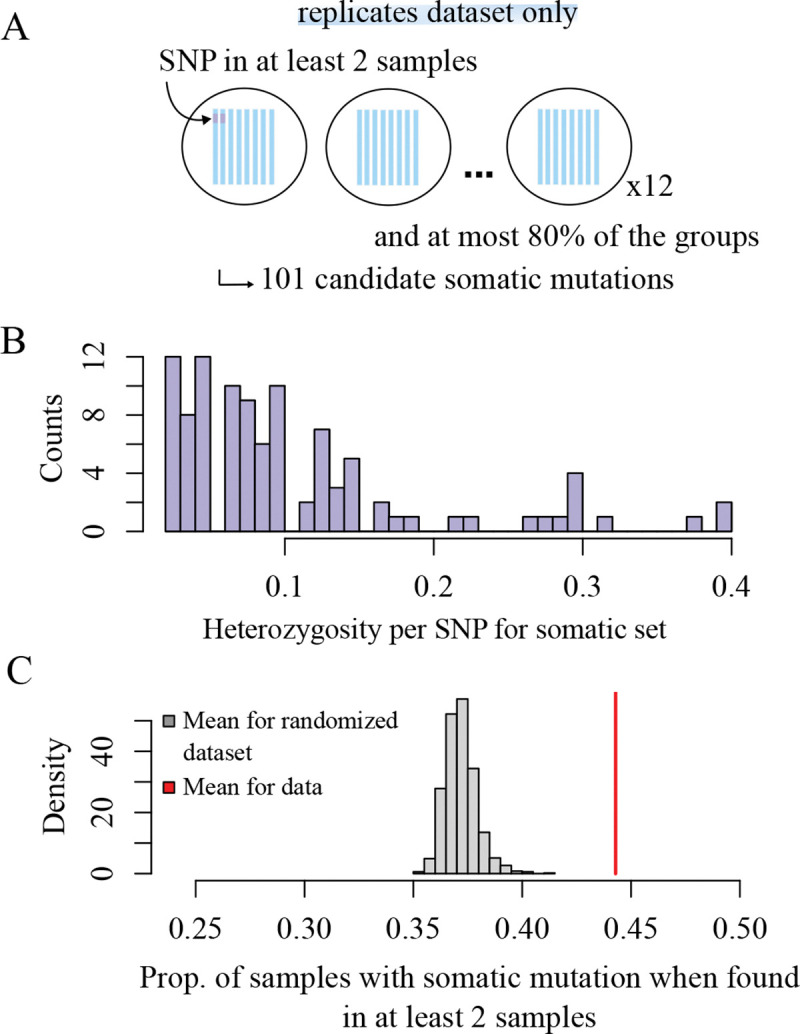
(A) To filter for somatic mutations, we labeled the mutations that were found in at least two samples per replicate group, and at most 80% of the samples (see methods for details on the filters). We identified 101 somatic mutations, (B) found in less than 40% of the individuals. (C) If a mutation is present in two samples in a group, it is found on average in 44% of the samples total.
Patterns of spatial genetic structure for somatic mutations - large-scale
We identified 3,942 putative somatic mutations from the 89 Pando ramet samples (large-scale dataset, Table 1). On average, samples shared 26.8% somatic mutations (range = 583 to 1,679). Due to clonal reproduction and spatial restriction in dispersal, we expected to observe a non-random spatial distribution of somatic mutations [37]. More specifically, we expected ramets that are close in space to share more mutations than ramets that are further apart from each other. However, there was only a weak correlation between the proportion of shared variants and the physical distance between pairs of ramets (Figure 3A, Pearson correlation coefficient = −0.02, 95%CI = [−0.05, 0.00], Figure 3B, null expectation = −0.001 with 1,000 permutations of the somatic mutation set, P < 0.001). We uncovered further spatial structure when focusing on spatial distribution of each somatic mutation. The mean distance between all samples sharing a mutation, averaged over all mutations, is smaller than expected by chance (Figure 3C&D, mean distance for groups sharing a somatic mutations is 264.28 m, as compared to the mean distance (null expectation) of 279.93 m for a randomized dataset with 500 permutations of the sample coordinates, P < 0.002). Given that a single root can extend up to 15 m [38], and our grid sampling had a minimum distance of 50 m, we hypothesized that we might be missing spatial signals at finer scales. Additionally, focusing solely on leaves could overlook somatic mutation signals, as clonal aspen expand through their roots (Figure 4). To better understand the spread of somatic mutations within and between ramets and tissue types, we conducted our analyses at a finer spatial scale by comparing samples from sub-sections of the clone and from different tissues within ramets.
Fig 3. Detecting spatial genetic structure at large scale.

(A) We use the set of 3,942 somatic mutations identified in the Pando clone samples to test for spatial genetic structure. Focusing on the sample-level, we observe that the number of shared variants between pairs of samples decreases with the physical distance between sample pairs (Pearson correlation coefficient between number of variants and spatial distance is −0.02, 95% CI = [−0.05, 0.00]), which is significantly different from a randomized distribution (P < 0.001) (B). (C & D) Focusing on the variant-level, we find that the mean distance within a group of samples sharing the variant is significantly less than expected by chance (mean distance for data is 264.28 m and mean distance for randomized dataset is 279.93 m, P < 0.001).
Fig 4. Conceptual model of somatic mutation inheritance between ramets within an aspen clone.

When a mutation arises, we expect it to propagate down to the new tissues as the clone continues to grow. New mutations are symbolized with the lightning bolt. The mutation identity is marked as a colored star and the dark marks corresponds to where samples could be collected from the clone.
Patterns of spatial genetic structure for somatic mutations - fine-scale
To detect fine-scale spatial structure and differences between tissue types, we focused on a smaller spatial scale, sampling ramets 1–15 m apart in a circular scheme at two locations within the Pando clone (~120 m apart, see Supplementary figures S1 and S2), as well as tissues within ramets (roots, shoots, branches, and leaves).
Overall, we found significant evidence of genetic structure in our set of 3,034 mutations, with genetic differences increasing with spatial distance (Figure 5A, Pearson correlation coefficient = −0.1, 95%CI= [−0.12, −0.07], null expectation = 0.00 with 500 permutations, P = 0.006). The signal was especially strong for leaves (Pearson correlation coefficient −0.44, 95% CI = [−0.49, −0.38]), with more somatic mutations shared between spatially close leaves compared to random (P < 0.001). The roots also shared significantly more mutations than expected under a null distribution (Pearson correlation coefficient −0.11, 95% CI = [0.18, −0.03], P = 0.026). This signal was not observed in the branches and the shoots (Pearson correlation coefficient −0.06, 95% CI = [−0.24, 0.11] for branches and −0.05, 95% CI = [−0.37, 0.28] for shoots).
Fig 5. Detecting spatial genetic structure at the finer scale.

We use the set of 3034 somatic mutations detected in the finer scale dataset to test for smaller-scale and within tissues spatial genetic structure. (A) Focusing at the sample-level, we observe an overall significantly negative correlation between genetic and physical distance (thick lines, Pearson correlation coefficient = −0.097, [CI] = [−0.12, 0.07]), driven mostly by the leaves and the roots (compared to null distributions, P < 0.001 and P = 0.026, respectively). (B) Focusing on the variant-level, we find that the mean distance within a group of samples sharing the variant (thick line, mean distance for the data is 46.33 m) is significantly less than expected by chance when considering all tissue types together (mean distance for the null distribution is 55.31 m, P < 0.001), signal that is mostly driven by the leaves (mean distance for leaves only is 39.28 m, as compared to 53.36 m expected under the null distribution, see S7 for means and p-values).
A variant-level approach showed that the number of shared somatic mutations per pair of samples decreased with increasing spatial distance (Figure 5B, mean distance for groups sharing a somatic mutations is 46.33 m, as compared to the mean distance (null expectation) of 55.31 m for a randomized dataset with 500 permutations, P = 0.002). The leaves showed the strongest spatial structure signal using this metric (Figure 5B and Supplementary figure S5), while other tissue types did not differ from the null expectation. The absence of signal in the shoots and branches may be partly explained by the significantly higher number of mutations recovered in leaves compared to other tissues (Supplementary figure S6).
Age of the Pando clone
We reconstructed the phylogenetic history of the Pando samples with a variable population size coalescent model (BEAST2 [27]). Because the somatic mutations are rare, they can be harder to detect using Illumina technology when the read depth is not exceptionally high (mean read depth is 13.5×). To estimate the proportion of missed mutations, we leveraged our replicates dataset (Figure 2). We pooled the set of replicates with the samples they originated from, and called variants on this joint dataset (108 samples before filtering). We then compared the mutations found in the set of replicates, to the mutation in their single sample. We find that on average, if a mutation is found at least once in the group of replicates, it is detected 20% (SE = 1%) of the times in the original sample. The single samples were not less likely to replicate than the set of replicates : there was no difference when swapping the single sample being compared to the set of replicates (Supplementary figure S9).
To take into account the effect of missing mutations on the phylogenetic tree height and thus the Pando clone age, we empirically estimated the relationship between the proportion of missing mutations and the phylogenetic tree height (Figure 6A). This relationship was linear, which we extrapolated to take into account false negatives or positives (i.e. mutations that we either missed, or called but are not real). This scaled tree height was then converted to years (see Methods for more details).
Fig 6. Pando is between 17,000 and 36,000 years old based on the large-scale dataset.

(A) We use the relationship between the proportion of missing mutations from a simulated dataset and the phylogenetic tree height to take into account the somatic mutations that we might be missing in the Pando clone (linear regression y = 0.10 + 0.11x, P < 2.2 * 1016, R2 = 0.92). (B) With this correction, we calculate the Pando clone age based on three different assumptions: (1) if the mutations we detect are all real, the Pando clone would be about 34,055 years old (± sd = 1,255 years); (2) if we are missing 80% of the mutations, then the clone would on average be 36,351 years old (± sd = 733 years); (3) finally, if only 20% of the mutations we detect are real somatic mutations, the Pando clone would be 17,238 years old (± sd = 29 years). (C) The Bayesian skyline plot suggests a steady population increase followed by a plateau. Note that this example was scaled for assumption 1 (all the mutations that we detect are real somatic mutations). (D) Despite thousands of years of evolutionary history, the Pando clone shows minimal phylogenetic structure (points colored according to PC1 score). (E) Pollen records from the Fish Lake show Populus was consistently present during the last 15,000 years, and generally well-represented over the last 60,000 years.
We calculated three different estimates of Pando’s age based on three different assumptions (Figure 6B). First, if the mutations we detected are all true positives and we did not miss any somatic mutations in the proportion of the genome we sequenced, we do not have to apply any correction to the phylogeny height conversion and the Pando clone would be about 34,055 years old (assumption 1, sd = 1,255 years). Second, if we take into account that we only detected 20% of the somatic mutations present in the samples and use the linear relationship (Figure 6A) to account for false negatives, then the clone would on average be 36,351 years old (assumption 2, sd = 733 years). Finally, if only 20% of the mutations we detect are true positives, the Pando clone would be 17,238 years old (assumption 3, sd = 29 years). The population dynamics reconstruction suggest a slow and steady increase during the first half of Pando’s life, followed by a steadier population size (Figure 6C). The unit of effective population size here can be thought of in terms of cellular lineages giving rise to new tissues (as compared to individuals when working with germline mutations). Despite its thousands of years of history, the phylogeny of the Pando clone samples suggests only minimal structure (Figure 6D). The same analysis of the fine-scale dataset suggests results of a similar range, that is, an age for Pando between ~12,000 and 37,000 years if we merge the range estimation given from both fine-scale and large-scale datasets (Supplementary figure S10). Interestingly, pollen records from the Fish Lake support a continuous presence of Populus during the last 15,000 years, potentially up to 60,000 years ago, which generally coincides with our age estimates for Pando (Figure 6E).
Discussion
In this study, we explored the evolutionary and developmental history of one of Earth’s largest clonal organisms, confirming that the Pando clone of quaking aspen consists of a single genet spanning 42.6 hectares. We detected thousands of somatic mutations distributed across this organism, providing novel insight into how genetic variation accumulates and spreads within long-lived, clonally expanding trees. While mutations show some spatial structure, particularly within leaves and roots, the signal was weaker than typically observed in clonal organisms. By analyzing the accumulation and distribution of these mutations, and accounting for technical uncertainties in mutation detection, we estimate the Pando clone is at least 12,000 years old (Figure 6).
Pando could not have persisted if glaciers occupied Fish Lake Plateau. Boulder exposure ages from the plateau suggest a local last glacial maximum of 21,100 years ago, raising the possibility of mountain glacier coverage in the area [39]. However, models of Fish Lake Plateau glaciers show they occurred at elevations between 2,950 and 3,190 m — notably higher than Pando’s current location of 2,700 m [39]. This suggests that vegetation could have persisted at Pando’s location throughout this glacial period [40, 41]. Supporting this interpretation, subfossil pollen analysis from nearby Fish Lake sediment cores demonstrates a continuous presence of Populus pollen over the last 15,000 years, with evidence of its presence extending back approximately 60,000 years (Figure 6E, upper panel).
To explore the spatial genetic demography of Pando, we sequenced leaves across a 50-m grid covering the entire Pando area as well as leaves, branches, shoots and roots at a finer scale, with samples collected 1–15 m apart in two locations within the clone. Our findings reveal spatial genetic structure within the clone, with samples sharing more mutations when geographically closer (Figure 3 & 5). While we were able to detect this spatial signal at a fine-scale in the leaves and roots, it was weaker at larger scales than expected and usually observed in clonal organisms [37, 42, 43]. Although we can clearly distinguish Pando samples from neighboring clones (Figure 1) and detect some internal genetic structure within the clone (Figures 3 & 5), the relatively low number of shared mutations between closely related tissues (roots, shoots and branches, Figure 5) suggests an intriguing underlying dynamic.
The weak spatial genetic structure we observed could be explained by two non-mutually exclusive hypotheses. First, given that aspen roots can grow up to 15 meters per generation [38], and Pando spans roughly 2 kilometers at its widest point, it would take only about 133 generations of ramet growth for a lineage to traverse the entire clone. Over thousands of years, this relatively rapid growth potential could lead to Pando functioning as a well-mixed system, effectively homogenizing genetic variation across the organism. Alternatively, research on within-clone mutation diversity suggests a different explanation: somatic mutations may not always be passed on asexually-produced offspring [44]. This pattern has been observed in Fragaria vesca (woodland strawberry), where mutations present in mother plants were absent in daughter plants propagated via stolons [16]. Such observations suggest that somatic mutations occurring in local tissues are not always passed down to the next generation of cells. As roots grow, the meristematic island that will give rise to new ramets gets pushed by waves of cells, protecting the stem cells from mutation accumulation [45]. This mechanism of mutation restriction is further supported by the remarkably low number of somatic SNPs differences detected between two oak leaf genomes sampled from the same individual (17 mutations differed between two leaves in a 236-year-old oak tree [18]). Despite prolonged lifespan and exposure to significant environmental changes, plants seem to have evolved mechanisms that protect the meristems from accumulating mutations. When sequencing entire tissues, we might be observing the localized accumulation of somatic mutations rather than the cell lineages contributing to organismal evolution, which would explain the relatively weak spatial genetic structure.
Our results suggest differing rates of somatic mutations between tissues that constitute the ”germline” (i.e., contribute to future generations of ramets) versus those that do not, and between annual and perennial tissues. We found that leaves accumulate more mutations than branches, shoots, and roots. This aligns with findings from other studies, where longer-lived organs (leaves) showed lower mutation rates compared to more short-lived structures (petals) [16]. Similarly, in Prunus mira (peach trees), mutation accumulation in branches — tissues involved in sexual reproduction — was lower than in roots [16].
These tissue-specific differences in mutation accumulation have important implications for our age estimates of the Pando clone. Our dating approach necessarily relied on mutation rates derived from leaf tissue to estimate mutation accumulation across the entire organism. However, given that leaves show higher mutation rates compared to other tissues, as demonstrated both in our results and in other organisms where mutation rates can vary by an order of magnitude across tissues [46], our age estimates likely represent conservative lower bounds. The challenge of accurate age estimation is further complicated by cellular mosaicism within tissues. While emerging single-cell sequencing methods [47] may eventually enable more precise tracking of mutation lineages in non-model organisms like aspen, current technical limitations necessitate working with tissue-level data that potentially masks finer-scale genetic variation.
This study provides new insights into the evolutionary history of Pando, one of Earth’s oldest and largest known organisms. By analyzing somatic mutations across different spatial scales and tissue types, we estimate the clone’s age to be at least 12,000 years old, with upper estimates reaching up to 37,000 years. Our findings reveal a weaker-than-expected spatial genetic structure within the clone, suggesting that Pando is either well mixed due to rapid growth, or that mutations accumulate locally rather than dispersing consistently along tissue lineages. The latter would support the hypothesis that mutation-protected pools of cells help maintain the genetic integrity of an indefinitely growing organism. In addition, this work underscores the technical challenges of studying rare mutations in non-model organisms, and paves the way for future studies on mutational dynamics in clonally-spreading long-lived perennials.
Supplementary Material
Acknowledgments
We would like to thank the GT QBioS Graduate Program for its support and the Society for the Study of Evolution for granting a Rosemary Grant Advanced Award to Rozenn M. Pineau that helped with pushing this work forward. This work was initiated by a seed grant from AV and JM. The work was further supported by grants from the NIH (Grant No. 5R35GM138030), the NSF Division of Environmental Biology (Grant No. DEB-1845363) to WCR and (Grant No. DEB-1844941) to ZG, and the NSF grant Paleo Perspectives on Climate Change (P2C2) Program (Grant No. 2102997) to JM and AB. The Utah Agricultural Experiment station provided support to KM and this publication is UAES journal paper No. 9835. The support and resources from the Center for High Performance Computing at the University of Utah are gratefully acknowledged.
References
- 1.Ekblom R, Galindo J. Applications of next generation sequencing in molecular ecology of non-model organisms. Heredity. 2011;107(1):1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lanfear R. Do plants have a segregated germline? PLoS biology. 2018;16(5):e2005439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Berger F, Twell D. Germline specification and function in plants. Annual review of plant biology. 2011;62:461–484. [Google Scholar]
- 4.Hallmann A. Evolution of reproductive development in the volvocine algae. Sexual plant reproduction. 2011;24(2):97–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Aanen DK. Germline evolution: sequestered cells or immortal strands? Current Biology. 2019;29(16):R799–R801. [DOI] [PubMed] [Google Scholar]
- 6.Monro K, Poore AG. The potential for evolutionary responses to cell-lineage selection on growth form and its plasticity in a red seaweed. The American Naturalist. 2009;173(2):151–163. [Google Scholar]
- 7.Collado-Vides L. Clonal architecture in marine macroalgae: ecological and evolutionary perspectives. Evolutionary Ecology. 2001;15(4):531–545. [Google Scholar]
- 8.Bricker E, Calladine A, Virnstein R, Waycott M. Mega clonality in an aquatic plant—a potential survival strategy in a changing environment. Frontiers in plant science. 2018;9:435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Anderson JB, Bruhn JN, Kasimer D, Wang H, Rodrigue N, Smith ML. Clonal evolution and genome stability in a 2500-year-old fungal individual. Proceedings of the Royal Society B. 2018;285(1893):20182233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang GG. Early regeneration and growth dynamics of Populus tremuloides suckers in relation to fire severity. Canadian Journal of Forest Research. 2003;33(10):1998–2006. [Google Scholar]
- 11.Johnstone JF. Effects of aspen (Populus tremuloides) sucker removal on postfire conifer regeneration in central Alaska. Canadian Journal of Forest Research. 2005;35(2):483–486. [Google Scholar]
- 12.Tomimoto S, Satake A. Modelling somatic mutation accumulation and expansion in a long-lived tree with hierarchical modular architecture. Journal of Theoretical Biology. 2023;565:111465. [DOI] [PubMed] [Google Scholar]
- 13.Edgeloe JM, Severn-Ellis AA, Bayer PE, Mehravi S, Breed MF, Krauss SL, et al. Extensive polyploid clonality was a successful strategy for seagrass to expand into a newly submerged environment. Proceedings of the Royal Society B. 2022;289(1976):20220538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Arnaud-Haond S, Duarte CM, Diaz-Almela E, Marba N, Sintes T, Serrao EA. Implications of extreme life span in clonal organisms: millenary clones in meadows of the threatened seagrass Posidonia oceanica. PloS one. 2012;7(2):e30454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Reusch TB, Boström C. Widespread genetic mosaicism in the marine angiosperm Zostera marina is correlated with clonal reproduction. Evolutionary Ecology. 2011;25:899–913. [Google Scholar]
- 16.Wang L, Ji Y, Hu Y, Hu H, Jia X, Jiang M, et al. The architecture of intra-organism mutation rate variation in plants. PLoS biology. 2019;17(4):e3000191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Anderson RP, Macdonald FA, Jones DS, McMahon S, Briggs DE. Doushantuo-type microfossils from latest Ediacaran phosphorites of northern Mongolia. Geology. 2017;45(12):1079–1082. [Google Scholar]
- 18.Schmid-Siegert E, Sarkar N, Iseli C, Calderon S, Gouhier-Darimont C, Chrast J, et al. Low number of fixed somatic mutations in a long-lived oak tree. Nature Plants. 2017;3(12):926–929. [DOI] [PubMed] [Google Scholar]
- 19.Barnes BV. The clonal growth habit of American aspens. Ecology. 1966;47(3):439–447. [Google Scholar]
- 20.DeByle NV, Winokur RP. Aspen: ecology and management in the western United States. vol. 119. US Department of Agriculture, Forest Service, Rocky Mountain Forest and; ...; 1985. [Google Scholar]
- 21.DeWoody J, Rowe CA, Hipkins VD, Mock KE. “Pando” lives: molecular genetic evidence of a giant aspen clone in central Utah. Western North American Naturalist. 2008;68(4):493–497. [Google Scholar]
- 22.Grant MC. The trembling giant. Discover. 1993;14(10):82. [Google Scholar]
- 23.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, et al. The sequence alignment/map format and SAMtools. bioinformatics. 2009;25(16):2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lin YC, Wang J, Delhomme N, Schiffthaler B, Sundström G, Zuccolo A, et al. Functional and evolutionary genomic inferences in Populus through genome and population sequencing of American and European aspen. Proceedings of the National Academy of Sciences. 2018;115(46):E10970–E10978. [Google Scholar]
- 25.Dou Y, Gold HD, Luquette LJ, Park PJ. Detecting somatic mutations in normal cells. Trends in Genetics. 2018;34(7):545–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schirmer M, D’Amore R, Ijaz UZ, Hall N, Quince C. Illumina error profiles: resolving fine-scale variation in metagenomic sequencing data. BMC bioinformatics. 2016;17:1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Drummond AJ, Rambaut A, Shapiro B, Pybus OG. Bayesian coalescent inference of past population dynamics from molecular sequences. Molecular biology and evolution. 2005;22(5):1185–1192. [DOI] [PubMed] [Google Scholar]
- 28.Bouckaert R, Heled J, Kühnert D, Vaughan T, Wu CH, Xie D, et al. BEAST 2: a software platform for Bayesian evolutionary analysis. PLoS computational biology. 2014;10(4):e1003537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bouckaert R, Vaughan TG, Barido-Sottani J, Duchêne S, Fourment M, Gavryushkina A, et al. BEAST 2.5: An advanced software platform for Bayesian evolutionary analysis. PLoS computational biology. 2019;15(4):e1006650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Waddell PJ, Steel M. General time-reversible distances with unequal rates across sites: mixing Γ and inverse Gaussian distributions with invariant sites. Molecular phylogenetics and evolution. 1997;8(3):398–414. [DOI] [PubMed] [Google Scholar]
- 31.Hofmeister BT, Denkena J, Colomé-Tatché M, Shahryary Y, Hazarika R, Grimwood J, et al. A genome assembly and the somatic genetic and epigenetic mutation rate in a wild long-lived perennial Populus trichocarpa. Genome Biology. 2020;21(1):1–27. [Google Scholar]
- 32.Mock KE, Rowe C, Hooten MB, Dewoody J, Hipkins V. Clonal dynamics in western North American aspen (Populus tremuloides). Molecular Ecology. 2008;17(22):4827–4844. [DOI] [PubMed] [Google Scholar]
- 33.Mock KE, Callahan CM, Islam-Faridi MN, Shaw JD, Rai HS, Sanderson SC, et al. Widespread triploidy in western North American aspen (Populus tremuloides). PLoS One. 2012;7(10):e48406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Korneliussen TS, Albrechtsen A, Nielsen R. ANGSD: analysis of next generation sequencing data. BMC bioinformatics. 2014;15(1):1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fagri K, Iversen J. Textbook of Pollen Analysis (3rd version); 1989. [Google Scholar]
- 36.Kapp RO. Guide to Pollen and Spores. 2nd ed. College Station, Texas: The American Association of Stratigraphic Palynologists Foundation; 2000. [Google Scholar]
- 37.Vekemans X, Hardy OJ. New insights from fine-scale spatial genetic structure analyses in plant populations. Molecular ecology. 2004;13(4):921–935. [DOI] [PubMed] [Google Scholar]
- 38.Day MW. The root system of aspen. American Midland Naturalist. 1944; p. 502–509. [Google Scholar]
- 39.Marchetti DW, Harris MS, Bailey CM, Cerling TE, Bergman S. Timing of glaciation and last glacial maximum paleoclimate estimates from the Fish Lake Plateau, Utah. Quaternary Research. 2011;75(1):183–195. [Google Scholar]
- 40.Clark PU, Dyke AS, Shakun JD, Carlson AE, Clark J, Wohlfarth B, et al. The last glacial maximum. science. 2009;325(5941):710–714. [DOI] [PubMed] [Google Scholar]
- 41.Marshall SJ, James TS, Clarke GK. North American ice sheet reconstructions at the Last Glacial Maximum. Quaternary Science Reviews. 2002;21(1–3):175–192. [Google Scholar]
- 42.Chybicki IJ, Trojankiewicz M, Oleksa A, Dzialuk A, Burczyk J. Isolation-by-distance within naturally established populations of European beech (Fagus sylvatica). Botany. 2009;87(8):791–798. [Google Scholar]
- 43.Kuss P, Pluess AR, Ægisdóttir HH, Stöcklin J. Spatial isolation and genetic differentiation in naturally fragmented plant populations of the Swiss Alps. Journal of Plant Ecology. 2008;1(3):149–159. [Google Scholar]
- 44.Reusch TB, Boström C, Stam WT, Olsen JL. An ancient eelgrass clone in the Baltic. Marine Ecology Progress Series. 1999;183:301–304. [Google Scholar]
- 45.Burian A, Barbier de Reuille P, Kuhlemeier C. Patterns of Stem Cell Divisions Contribute to Plant Longevity. Current Biology. 2016;26(11):1385–1394. doi: 10.1016/j.cub.2016.03.067. [DOI] [PubMed] [Google Scholar]
- 46.Moore L, Cagan A, Coorens TH, Neville MD, Sanghvi R, Sanders MA, et al. The mutational landscape of human somatic and germline cells. Nature. 2021;597(7876):381–386. [DOI] [PubMed] [Google Scholar]
- 47.Abascal F, Harvey LM, Mitchell E, Lawson AR, Lensing SV, Ellis P, et al. Somatic mutation landscapes at single-molecule resolution. Nature. 2021;593(7859):405–410. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Scripts and detailed steps can be found on the paper repository paper repository.