


Abstract
Nucleotide excision repair (NER) and transcription are intimately related. First, TFIIH has a dual role in transcription initiation and NER and, secondly, transcription leads to more efficient repair of damage present in transcribed sequences. It is thought that elongating RNAPII, stalled at a DNA lesion, is used for the loading of the NER machinery in a process termed transcription-coupled repair (TCR). Non-transcribed regions are repaired by the so-called global genome repair (GGR). We have previously defined a number of yeast genes, whose deletions confer transcription-dependent hyper-recombination phenotypes. As these mutations cause impairment of transcription elongation we have assayed whether they also affect DNA repair. We show that null mutations of the HPR1 and THO2 genes, encoding two prominent proteins of the THO complex, increase UV sensitivity of yeast cells lacking GGR. Consistent with this result, molecular analyses of DNA repair of the RPB2 transcribed strand using T4 endo V show that hpr1 and tho2 do indeed impair TCR. However, this effect is not confined to TCR alone because the mutants are slightly affected in GGR. These results indicate that THO affects both transcription and NER. We discuss different alternatives to explain the effect of the THO complex on DNA repair.
INTRODUCTION
Nucleotide excision repair (NER) is an evolutionarily conserved DNA repair pathway that maintains the genome free of a wide variety of lesions, such as those induced by UV light and some chemical agents (1,2). In yeast, more than 25 proteins participate in this multi-step process (2).
One of the most intriguing aspects of all forms of DNA repair is the mechanism by which a DNA lesion is detected. In NER, even though the proteins that recognize the lesion (UvrA in Escherichia coli and Rad4/Rad23 and XPC/HHR23B in yeast and humans) have been identified, it is not clear how such proteins are targeted to the site of the lesion. According with the mechanism of lesion detection, NER can be divided into two subpathways: global genome repair (GGR) and transcription-coupled repair (TCR). The first one acts in all regions of the genome irrespective of whether or not they are transcribed. In yeast, GGR requires Rad7, a protein carrying leucine-rich repeats and Rad16, a member of the SWI2/SNF2 subfamily of putative helicases. These proteins presumably act in a complex (3,4) that might be required in chromatin remodeling to facilitate damage detection by Rad4/Rad23 (5–7). TCR depends on the product of the Rad26 gene and is confined to the transcribed strands (TS) of active genes (8,9).
TCR takes advantage of the incapacity of the RNA polymerases (RNAP) to pass particular DNA lesions such as UV-induced cyclobutane-pyrimidine dimers (CPDs). RNAPII, stalled at a DNA lesion, is used for the loading of the NER machinery at the site of the lesion. It has been shown that a stalled bacterial RNAP at sites of base damage in the TS inhibits the repair of CPDs by the UvrABC NER system in vitro (10). This suggests that TCR requires the displacement of RNAP from the damaged site to allow NER, a reaction mediated by a transcription-repair coupling factor (TRCF) (11).
In eukaryotes, in which TCR was first discovered (12,13), TRCF candidates are the human Cockayne syndrome B and A proteins, or their respective yeast orthologs Rad26 and Rad28. While human cells deficient in CSB and CSA and yeast rad26 mutants show TCR defects (9,14), rad28 mutants are TCR proficient (15). CSB and Rad26 belong to the SWI2/SNF2 subfamily of putative helicases. CSB, as well as bacterial TRCF, have been shown to have ATPase activity (16,17), but no helicase activity has been detected for either protein. However, the function of CSB/Rad26 has not yet been established. The observations that CSB-deficient cells display reduced transcription levels (18), that purified CSB enhances RNAPII-mediated elongation in vitro (19,20) and that CSB resides in a RNA polymerase II (RNAPII)-containing complex (21) have raised the possibility that CSB/Rad26 is a transcription elongation factor. The TCR phenotype of CSB/Rad26-deficient cells could be a consequence of impaired transcription elongation. The observations that α-amanitin, an inhibitor of transcription elongation, inhibits TCR in CHO cells (22) and that the absence of the Spt4 elongation factor bypasses the requirement for Rad26 in yeast TCR would be consistent with the idea that CSB/Rad26 is not the eukaryotic TRCF (23).
We have identified four genes, HPR1, THO2, MFT1 and THP2, whose null mutations confer a strong hyper-recombination phenotype between repeats that is dependent on transcription elongation (24–26). These genes encode proteins that belong to the same protein complex, termed THO (24), which connects transcription with mitotic recombination. Mutations in these genes impair transcription elongation (27,28). Such an impairment is particularly strong in long and G+C-rich DNA sequences (29). As a consequence of the transcription elongation defect, mutants of the THO complex show an increased recombination frequency between direct repeats (24–31), and a high frequency of chromosome and plasmid loss (26,27,30). We have hypothesized that THO may have a role in transcription elongation. In the mutants of the THO complex, transcription elongation could be impaired leading to the formation of recombinogenic structures (27,28).
Considering the existing connection between transcription and repair and the connection between transcription and genetic instability provided by the THO complex, we have asked whether mutation of HPR1 and THO2, the two most prominent components of the THO complex, impair NER. We show that this is the case. hpr1 and tho2 are affected in TCR, but additionally also slightly in GGR. Altogether, our results support a general effect of the THO complex in NER.
MATERIALS AND METHODS
Yeast strains
The yeast strains used in this study, all isogenic, are listed in Table 1.
Table 1. Saccharomyces cerevisiae strains used in this study.
| Strain | Genotype | Source/reference |
|---|---|---|
| W303-1A | MATa ade2-1 can1-100 his3-11,15 leu2-3,112 trp1-1 ura3-1 | R. Rothstein (New York) |
| W839-5D | MATα ade2-1 can1-100 his3-11,15 leu2-3,112 trp1-1 ura3-1 rad1Δ::LEU2 | R. Rothstein (New York) |
| U768-1C | MATα ade2-1 can1-100 his3-11,15 leu2-3,112 trp1-1 ura3-1 hpr1Δ::HIS3 | R. Rothstein (New York) |
| W303-236 | MATα ade2-1 can1-100 his3-11,15 leu2-3,112 trp1-1 ura3-1 rad16Δ::URA3 | (37) |
| MGSC97 | MATα ade2-1 can1-100 his3-11,15 leu2-3,112 trp1-1 ura3-1 rad7Δ::URA3 | (37) |
| MGSC102 | MATα ade2-1 can1-100 his3-11,15 leu2-3,112 trp1-1 ura3-1 rad26Δ::HIS3 | (9) |
| SChY58a | MATa ade2-1 can1-100 his3-11,15 leu2-3,112 trp1-1 ura3-1 hpr1Δ::kanMX4 | S. Chávez (Seville) |
| RK2-6C | MATa ade2-1 can1-100 his3-11,15 leu2-3,112 trp1-1 ura3-1 tho2Δ::kanMX4 | (26) |
| TR16-11A | MATa ade2-1 can1-100 his3-11,15 leu2-3,112 trp1-1 ura3-1 tho2Δ::kanMX4 rad16Δ::URA3 | This work |
| TR16-9B | MATα ade2-1 can1-100 his3-11,15 leu2-3,112 trp1-1 ura3-1 tho2Δ::kanMX4 rad16Δ::URA3 | This work |
| TR7-8A | MATa ade2-1 can1-100 his3-11,15 leu2-3,112 trp1-1 ura3-1 tho2Δ::kanMX4 rad7Δ::URA3 | This work |
| TR7-1B | MATα ade2-1 can1-100 his3-11,15 leu2-3,112 trp1-1 ura3-1 tho2Δ::kanMX4 rad7Δ::URA3 | This work |
| TR26-1C | MATa ade2-1 can1-100 his3-11,15 leu2-3,112 trp1-1 ura3-1 tho2Δ::kanMX4 rad26Δ::HIS3 | This work |
| HR7-7B | MATa ade2-1 can1-100 his3-11,15 leu2-3,112 trp1-1 ura3-1 hpr1Δ::HIS3 rad7Δ::URA3 | This work |
| HR7-5D | MATα ade2-1 can1-100 his3-11,15 leu2-3,112 trp1-1 ura3-1 hpr1Δ::HIS3 rad7Δ::URA3 | This work |
| HR16-3D | MATa ade2-1 can1-100 his3-11,15 leu2-3,112 trp1-1 ura3-1 hpr1Δ::HIS3 rad16Δ::URA3 | This work |
| HR16-2C | MATα ade2-1 can1-100 his3-11,15 leu2-3,112 trp1-1 ura3-1 hpr1Δ::HIS3 rad16Δ::URA3 | This work |
| R267-10A | MATα ade2-1 can1-100 his3-11,15 leu2-3,112 trp1-1 ura3-1 rad26Δ::HIS3 rad7Δ::URA3 | This work |
| MU26-10B | MATa ade2-1 can1-100 his3-11,15 leu2-3,112 trp1-1 ura3-1 hpr1Δ::HIS3 rad26Δ::HIS3 | This work |
| HR2616-2A | MATα ade2-1 can1-100 his3-11,15 leu2-3,112 trp1-1 ura3-1 hpr1Δ::HIS3 rad26Δ::HIS3 rad16Δ::URA3 | This work |
| HR2616-7C | MATa ade2-1 can1-100 his3-11,15 leu2-3,112 trp1-1 ura3-1 hpr1Δ::HIS3 rad26Δ::HIS3 rad16Δ::URA3 | This work |
| HR267-2B | MATa ade2-1 can1-100 his3-11,15 leu2-3,112 trp1-1 ura3-1 hpr1Δ::HIS3 rad26Δ::HIS3 rad7Δ::URA3 | This work |
| HR267-6A | MATα ade2-1 can1-100 his3-11,15 leu2-3,112 trp1-1 ura3-1 hpr1Δ::HIS3 rad26Δ::HIS3 rad7Δ::URA3 | This work |
UV survival curves
Yeast cells were grown in YEPD-rich medium (32) to an OD600 of 1. Different dilutions were plated on YEPD and irradiated with the different UV doses with a 254 nm UV lamp (Philips T UV 30 W). Plates were incubated for 3 days at 30°C in the dark before colonies were counted and survival was calculated. All survival curves shown represent the average of three independent experiments.
UV irradiation and DNA isolation
Yeast cultures were grown in 500 ml of YEPD overnight to a final OD600 of 1.8. Cells were collected by centrifugation, resuspended in 1.6 l chilled phosphate-buffered saline (PBS) and irradiated with a 254 nm UV lamp at 25 J/m2. Subsequently, cells were incubated in YEPD for various time courses in the dark at 30°C. After this, cell pellets collected by centrifugation were frozen in liquid nitrogen and maintained at –80°C until DNA was isolated.
For DNA isolation we followed a modified version of previously published procedures (33). Pellets were resuspended to a final concentration of 1.5–2 × 109/ml in NIB pH 7.2 (17% glycerol, 50 mM MOPS, 150 mM KAc, 2 mM MgCl, 0.5 mM spermidine and 0.15 mM spermine) with 3 mg/ml Zymoliase-20T and incubated at room temperature for 20 min. Cellular suspensions were diluted seven times with chilled water and centrifuged to collect nuclei. Pellets were resuspended in 5.4 ml of TE (50 mM Tris–HCl pH 8, 20 mM EDTA) and mixed with 600 µl of 10% SDS. After phenol extraction and ethanol precipitation, DNA was finally purified on CsCl gradients and dialyzed in TE (34).
Gene- and strand-specific DNA repair assays
Five micrograms of genomic DNA were digested with PvuI and PvuII, generating a 5.2 kb RPB2 fragment (35) and a 4.7 kb GAL7 fragment (36), with HaeII, which generates a 3.5 kb MATα fragment and a 4.1 kb HMLα fragment, or with HindIII, which generates a 4.3 kb MATa and a 5.1 kb HMRa fragment (37). In each case, DNA samples were divided in two halves. One was incubated with T4 endonuclease V (T4 endo V) at 37°C for 2 h. After electrophoresis and Southern blot transfer to Hybond N+, samples were hybridized at 65°C (7% SDS and 0.5 M phosphate buffer pH 7) with 32P-labeled DNA probes. The same filters were rehybridized up to four times, after removing probes by harsh treatment with boiling 0.1% SDS. Bands were quantified in a Fujix FLA3000. All values of the bands corresponding to the T4-untreated samples were divided by the T4-mock sample of zero time. These values were multiplied by their corresponding values of the T4-treated sample and subtracted from the T4-treated zero time to eliminate the basal background. The final percentage of repair was calculated by dividing these resulting values by the value of the T4-mock-treated zero time.
RPB2 and GAL7 strand-specific probes were obtained by primer extension from M13 derivative vectors as described (37). 634 bp Yα-specific probes were obtained by PCR with oligonucleotides GCCAAACTGTGAGTAATATGC and TCATCTGTGATTTGTGGATTT and labeled with 32P-dCTP. 602 bp Ya-specific probes were generated by PCR with primers ACCCGACTATGCTATTTTAAT and GGGGAAACTGTATAAAACTTC. All results shown correspond to the average of two to four independent experiments.
Northern analyses
For determination of RNA levels after UV treatment, yeast cells were grown at 30°C to an OD600 of ∼0.5 in 100 ml YEP–3% glycerol–2% lactate, harvested by centrifugation and resuspended in 100 ml of PBS pH 7. Half of the culture was irradiated with a 254 nm UV light lamp at 70 J/m2 and the other half was used as the unirradiated control. Each half was then grown at 30°C in 60 ml YEP–2% galactose medium. At the indicated time 10 ml aliquots were taken, collected by centrifugation, frozen in liquid nitrogen and maintained at –80°C until RNA extraction. Ten micrograms of each RNA sample were subjected to northern analysis following previously published procedures (27). Filters were first hybridized with a 0.75 kb PvuII/AvaI GAL1 probe and then re-hybridized with a 589 bp DNA internal fragment obtained by PCR as described (27). mRNA levels were quantified in a Fujix FLA3000 analyzer and are given as arbitrary units. All values were normalized with respect to the 28S rRNA. Northern analysis of RPB2 was performed with RNA isolated from mid-log phase cultures according to standard procedures (34).
RESULTS
Increased UV sensitivity of hpr1 and tho2 mutant strains that also lack GGR
The hpr1 and tho2 mutations confer a defect in transcription elongation that causes a strong transcription-dependent hyper-recombination phenotype between DNA repeats. As NER consists of two subpathways, one of which, TCR, relies on an elongating RNAPII to detect DNA lesions, we decided to assay if hpr1 and tho2 mutations cause a defect in TCR. These mutations by themselves do not confer a DNA repair defect detectable as a UV-sensitivity phenotype (see Fig. 1) (25). As can be seen in Figure 1, the same UV-sensitivity curves were obtained in wild-type, hpr1, tho2 and rad26 strains, impaired in TCR, as well as in double mutant combination hpr1 rad26 and tho2 rad26. As the TCR defect of rad26 strains is only observed as a UV-sensitivity phenotype when GGR is also abolished, we decided to construct double mutant strains hpr1 rad7, tho2 rad7, hpr1 rad16 and tho2 rad16. As shown in Figure 1 the viability upon UV irradiation of these double mutants was significantly reduced below the levels of the single mutants rad7 and rad16. The survival curves were similar to those of rad7 rad26 and rad16 rad26 double mutants. This result indicates that both hpr1 and tho2 impair NER in the absence of GGR. In addition, triple mutants affected in rad26, hpr1/tho2 and rad7/rad16 display an increased UV sensitivity as compared with the double mutants hpr1/tho2 rad7/rad16 or rad26 rad7/rad16 (Fig. 2). Thus, Rad26 and Hpr1/Tho2 play different roles in TCR.
Figure 1.

UV sensitivity curves of isogenic W303 yeast strains carrying different single and double combinations of the hpr1, tho2, rad26, rad7 and rad16 null mutations. The isogenic rad1 strain W839-5D lacking NER was used as a negative control. Strains used are those listed in Table 1. The experiments were repeated two or three times for each genotype, giving similar results with standard deviations below 10% (data not shown). Only average values are shown.
Figure 2.

UV sensitivity curves of isogenic W303 yeast strains carrying different combinations of the hpr1, tho2, rad26, rad7 and rad16 null mutations compared with the appropriate controls. Other details as in Figure 1 legend.
RNA synthesis is significantly reduced in wild-type, rad26 and tho2 cells after UV irradiation
In yeast, RNA synthesis of induced genes is initially inhibited after UV irradiation (38), probably a consequence of the inability of the RNAPII to pass through CPDs accumulated in the TS of genes. To determine whether the ability of resuming RNA synthesis after UV irradiation in cells affected in TCR is impaired, we determined the ability of rad26 and tho2 cells to transcribe the endogenous GAL1 gene after irradiation with 70 J/m2 UV. As can be seen in Figure 3, even in the absence of UV irradiation the rad26 and tho2 mutants display reduced transcription rates. Furthermore, after UV irradiation, there was a significant retardation in the kinetics of activation of GAL1 in all strains. Whereas non-irradiated rad26 cells reached 59% of the RNA levels of wild-type cells, this value was only 11% in non-irradiated tho2 cells. Consistent with previous observations (26,39), our results reflect an intrinsic defect of both types of mutants in transcription, regardless of UV damage.
Figure 3.

Kinetics of transcription activation of the endogenous GAL1 gene upon UV irradiation in wild-type, rad26 and tho2 strains. (A) Northern analysis of GAL1 at different times of transcription activation by addition of 2% galactose to YEP–3% glycerol–2% lactate mid-log phase cultures whether or not irradiated with 70 J/m2. (B) Kinetics of transcription activation of GAL1 as determined by quantification of the northern analysis. All data were normalized with respect to rRNA values.
T4 endo V analysis of repair in the TS and non-transcribed strands (NTS) of the constitutively expressed gene RPB2
To determine, at the molecular level, whether hpr1 and tho2 impair TCR we analyzed the ability of hpr1 and tho2 cells to repair the TS and NTS of the constitutively expressed RPB2 gene. The kinetics of repair of UV-induced lesions was followed by T4 endo V treatment. This methodology previously established that both wild-type and rad7 cells show the same rates of repair in the TS of RPB2, whereas rad26 cells show reduced rates (9,37). In addition, it is known that wild-type and rad26 cells show the same kinetics of repair in the NTS of RPB2, whereas rad7 cells show no repair (9,37). We have confirmed these results in our W303 isogenic strains (data not shown).
Figure 4 shows that in both the hpr1 and tho2 mutants there is a defect in the repair of the TS, especially observed as a reduction in the initial rate of repair. The defect in the repair of the TS of RPB2 in both the hpr1 and tho2 mutants is as strong as in rad26, although in rad26 cells this effect is persistent and never reaches wild-type levels of DNA repair.
Figure 4.

T4 endo V analysis of DNA repair at the endogenous RPB2 gene. (A) Analysis of repair of the TS and NTS of RPB2. Cells obtained from mid-log phase YEPD cultures were irradiated with 25 J/m2 UV and allowed to recover in YEPD-rich medium for different times to permit removal of lesions by NER. DNA was mocked (–) or T4 endo V treated (+), electrophoresed, blotted and hybridized with the appropriate RPB2 single stranded probes obtained by primer extension. (B) Kinetics of repair as determined from the quantification analysis of the hybridization experiments. Strains rad7 and rad26 are shown as controls for wild-type TCR and wild-type GGR, respectively. The experiments were repeated four times for each genotype, giving similar results with standard deviations below 20%. Only average values are shown.
Surprisingly, hpr1 and tho2 mutants showed also reduced kinetics of repair of the NTS of RPB2, although less pronounced than that displayed by rad7 cells. Only at 120 min, both hpr1 and tho2 mutants reached wild-type levels of repair (Fig. 4). Therefore, hpr1 and tho2 mutants show delayed kinetics of repair of both the TS and the NTS of RPB2.
Transcription levels of RPB2 are reduced in rad26, rad7, hpr1 and tho2 cells
Given that hpr1 and tho2 mutants are affected in DNA repair and transcription we decided to determine the levels of transcription of RPB2 in all mutants analyzed (Fig. 5). Northern analysis shows that in all strains, including rad7, there was a 40–50% reduction in the overall levels of RPB2 RNA. As expected, transcription of RPB2 was impaired in hpr1 and tho2 cells, as well as in rad26. The unexpected reduced levels of transcription of RPB2 observed in rad7 cells suggest that the delayed kinetics of TCR observed in hpr1 and tho2 might not be caused by a defect in transcription. With similar reduction in transcription efficiency, rad7 has no effect in the repair of the TS of RPB2.
Figure 5.
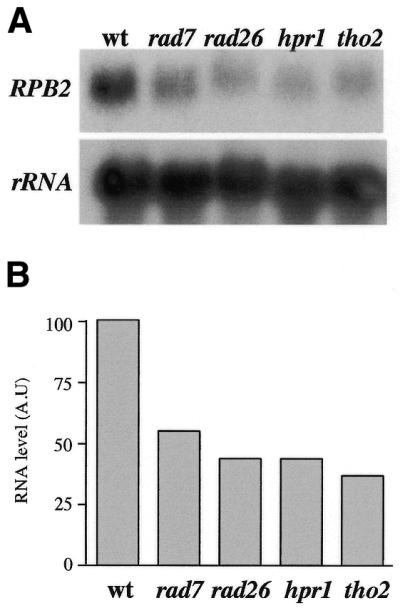
(A) Northern analysis of the endogenous RPB2 gene in wild-type, rad7, rad26, hpr1 and tho2 strains. (B) Transcription levels of RPB2 as determined from the quantification analysis of the northern analysis. Details as in Figure 3.
Besides the transcriptional effect of hpr1 and tho2 on transcription elongation (26,27), it is worth noting that the expression levels of RPB2, GAL1 and GAL7 genes in rad26 mutants were below 40% of the wild-type levels (Figs 3 and 5; data not shown). This result is consistent with a putative role in transcription elongation of Rad26/CSB (16,22,39). However, transcription of RPB2 is also affected in rad7 mutants (Fig. 5), but there is no evidence that the role of Rad7 is related to transcription. Therefore, DNA lesions might cause the low transcriptional levels observed in untreated rad26 and rad7 cells. If the assumption is correct, it could also imply that Rad26 may not necessarily be directly involved in transcription elongation, in contrast to what has been recently suggested (39).
T4 endo V analysis of UV-damage repair in GAL7 and HMLα and HMRa loci under repressed conditions
As the analysis of RPB2, a constitutively expressed gene, indicates that hpr1 and tho2 cells are affected in the kinetics of repair of both the TS and the NTS, we analyzed whether repair of non-transcribed genes was also affected. We first determined the kinetics of repair of both strands of the GAL7 gene under repressed conditions. As expected, the efficiency of TCR proficient rad7 cells to repair the TS of the repressed GAL7 gene was reduced as compared with its ability to repair the TS of the constitutively expressed RPB2 gene (Fig. 6; compare with Fig. 4). Interestingly, there was still some repair of the TS of GAL7 in rad7 cells (Fig. 6) (40). This repair is dependent on RAD26 and it has been interpreted as residual transcription at repressed condition (40), though we have not been able to detect any transcript by northern analysis (data not shown). The hpr1 and tho2 mutants show a kinetics of repair of the TS and NTS strands of GAL7 similar to that observed in RPB2 (compare Figs 4 and 6).
Figure 6.

Repair analysis at the endogenous GAL7 gene under repressed conditions, that is in cells grown in YEPD (2% glucose) cultures. (A and B) as in Figure 4. The experiments were repeated three times for each genotype, giving similar results with standard deviations below 20%. Only average values are shown.
We also analyzed GGR in the silenced HMLα and HMRa loci that lack transcription due to silencing. As can be seen in Figure 7, repair is completely abolished in rad7 cells, supporting a lack of transcription and associated TCR in these loci and the dependence of GGR on Rad7. As in the RPB2 and GAL7 genes, hpr1 and tho2 mutants show a delay in their capability to repair the silenced HMLα and HMRa loci. These results suggest that in addition to an effect in TCR, observed at both genetic and physical levels, hpr1 and tho2 are also affected in GGR. This effect, although detectable at the molecular level, apparently does not result in an UV-sensitivity phenotype of hpr1/tho2 single mutants or rad26 hpr1/tho2 double mutants.
Figure 7.

Physical analysis of DNA repair at the endogenous HMLα and HMRa silenced loci. (A and B) as in Figure 4. As the HML and HMR probes used also hybridize with the MAT locus, the corresponding MAT band is also detected. The experiments were repeated twice for each genotype, giving similar results with standard deviations below 20%. Only average values are shown.
DISCUSSION
We have shown that hpr1 and tho2 cells, impaired in transcription elongation and having a strong transcription-dependent hyper-recombination phenotype, are impaired in NER. The hpr1 and tho2 mutations confer UV sensitivity when combined with either rad7 or rad16, which abolish GGR. Molecular analyses of DNA repair with T4 endo V indicate that hpr1 and tho2 cells are impaired in their ability to repair the TS of RPB2 similarly to rad26 cells. In contrast to rad26 cells, hpr1 and tho2 also show repair defects of non-transcribed DNA, although at levels that are only detected at the molecular level.
Hpr1 and Tho2 are part of the THO complex, which connects transcription to genomic instability. Our observations that hpr1 and tho2 reduce UV survival only in strains impaired in GGR, and impair the repair of the constitutively expressed RPB2 gene upon UV irradiation, as determined by T4 endo V analysis (Fig. 4) indicate that the THO complex modulates or has a role in TCR. However, in contrast to rad26 mutations, hpr1 and tho2 have a slight effect on GGR. Thus, defective repair is also physically detected in the NTS of RPB2, in the repressed GAL7 gene and in the silenced HMLα and HMRa loci (Figs 4, 6 and 7). Such a defect in GGR cannot be observed genetically as a UV-sensitivity phenotype of rad26 hpr1/tho2 double mutants. In addition, in GGR-less strains the double mutations rad26 hpr1/tho2 confer a more severe UV-sensitivity phenotype than each single mutation separately. These results indicate that the putative role that Hpr1 and Tho2 might have in NER is not exclusive of TCR and is different to that of Rad26. Indeed, this is not the only difference between Rad26 and Hpr1/Tho2, because in contrast to hpr1 and tho2 cells, rad26 cells do not show a detectable hyper-recombination phenotype and are able to transcribe the bacterial lacZ sequences (S.González-Barrera, A.G.Rondón and A.Aguilera, unpublished results).
The negative effect of rad7 on the accumulation of RPB2 mRNA (Fig. 5) indicates that defective transcription does not necessarily imply defective TCR. However, it may not be surprising that the transcription elongation defect of hpr1 and tho2 is responsible for the TCR defect. The NER defects of hpr1 and tho2 cells are unlikely to be caused by lack of expression of a particular NER gene, because global transcription analysis of tho2 cells using DNA microarrays shows that all known NER genes are transcribed (M.Gallardo, K.Ohta and A.Aguilera, unpublished results).
It is interesting to note that hpr1 and tho2 cells show a low rate of DNA repair, that is, a low amount of UV lesions repaired per minute as inferred by the slope of the curves of Figure 4. However, such slopes are similar in wild-type and hpr1 and tho2 mutants after 60–80 min upon UV irradiation (Fig. 4), suggesting that after this time, mutant cells recover the wild-type capacity of repair. In any case, the overall levels of removed UV lesions are still below wild-type levels at 120 min upon UV irradiation. This might reflect a process of adaptation. It is known that pre-irradiation of yeast cells enhances the removal of lesions induced by a second UV dose (1). Therefore, we cannot exclude the possibility that a slower adaptation response of hpr1 and tho2 cells to UV damage could make the DNA repair defects of hpr1 and tho2 mutants more severe.
At first, a transient sequestering of NER proteins could explain the NER defects of hpr1 and tho2 mutants. If the defects of hpr1 and tho2 in transcription elongation lead to frequent stalling of the elongating RNAPII, independently of UV lesions, a recruitment of the NER machinery could occur at many sites. As a consequence, NER proteins would be transiently sequestered, leading to a reduction of available NER components, and a concomitant defect in repair. Thus, a stalled RNAPII could recruit TFIIH and other NER factors regardless of the presence of a DNA lesion. Alternatively, the NER defects of hpr1 and tho2 could be explained by a putative unknown role of THO in NER.
Proteins connecting DNA repair and transcription could include, besides TFIIH, the transcription terminator factors 2 from Drosophila and humans (DmF2 and HuF2) and XAB2, a human protein identified by its ability to bind the NER protein XPA. DmF2 and HuF2 are members of the SWI2/SNF2 family (41,42) and dissociate RNAPI and RNPAII stalled at a CPD (43). XAB2 interacts with CSB, CSA and RNAPII and anti-XAB2 antibodies inhibit TCR and transcription when micro- injected in fibroblasts (44). Interestingly, Saccharomyces cerevisiae and Schizosaccharomyces pombe orthologs of XAB2 seem to have a role in splicing (45,46), opening the possibility that RNA-processing proteins associated with elongation might influence TCR.
In this sense it is important to note that hpr1 mutants are affected in RNA export (47) and that overexpression of Sub2, a putative RNA helicase involved in RNA splicing and export (48,49), suppresses the hyper-recombination phenotype of hpr1 cells (50). In addition, sub2 mutants show similar hyper-recombination phenotypes to hpr1 (50). Furthermore, a genetic interaction between Sub2 and Rad3, a component of TFIIH involved in RNAPII-dependent transcription and NER, has recently been inferred from the isolation of rad3 alleles that suppress the growth defects of sub2 (51). Our study, therefore, not only extends the possible functional role of THO to NER, but opens the possibility of a connection between DNA repair, transcription and the processing of the nascent RNA. The identification of the biochemical function of the THO complex will help to understand the mechanisms connecting these processes.
Acknowledgments
ACKNOWLEDGEMENTS
We thank Lars E. T. Jansen and Ralf E. Wellinger for critical reading of the manuscript and D. Haun for style correction. This work was supported by grants PB96-1350 and BIO98-1363-CE from the Ministry of Education and Culture of Spain and RG0075/1999-M from the Human Frontier Science Program to A.A. and grants of IRS to J.B. S.G.-B. was a recipient of a pre-doctoral training grant from the Ministry of Education and Culture of Spain.
REFERENCES
- 1.Waters R., Zhang,R. and Jones,N.J. (1993) Inducible removal of UV-induced pyrimidine dimers from transcriptionally active and inactive genes of Saccharomyces cerevisiae. Mol. Gen. Genet., 239, 28–32. [DOI] [PubMed] [Google Scholar]
- 2.Prakash S. and Prakash,L. (2000) Nucleotide excision repair in yeast. Mutat. Res., 451, 13–24. [DOI] [PubMed] [Google Scholar]
- 3.Reed S.H., You,Z. and Friedberg,E.C. (1998) The yeast RAD7 and RAD16 genes are required for postincision events during nucleotide excision repair. In vitro and in vivo studies with rad7 and rad16 mutants and purification of a Rad7/Rad16-containing protein complex. J. Biol. Chem., 273, 29481–29488. [DOI] [PubMed] [Google Scholar]
- 4.Wang Z., Wei,S., Reed,S.H., Wu,X., Svejstrup,J.Q., Feaver,W.J., Kornberg,R.D. and Friedberg,E.C. (1997) The RAD7, RAD16, and RAD23 genes of Saccharomyces cerevisiae: requirement for transcription-independent nucleotide excision repair in vitro and interactions between the gene products. Mol. Cell. Biol., 17, 635–643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.de Laat W.L., Jaspers,N.G. and Hoeijmakers,J.H. (1999) Molecular mechanism of nucleotide excision repair. Genes Dev., 13, 768–785. [DOI] [PubMed] [Google Scholar]
- 6.Guzder S.N., Sung,P., Prakash,L. and Prakash,S. (1999) Synergistic interaction between yeast nucleotide excision repair factors NEF2 and NEF4 in the binding of ultraviolet-damaged DNA. J. Biol. Chem., 274, 24257–24262. [DOI] [PubMed] [Google Scholar]
- 7.Jansen L.E., Verhage,R.A. and Brouwer,J. (1998) Preferential binding of yeast Rad4.Rad23 complex to damaged DNA. J. Biol. Chem., 274, 33111–33114. [DOI] [PubMed] [Google Scholar]
- 8.Tijsterman M., Verhage,R.A., van de Putte,P., Tasseron-de Jong,J.G. and Brouwer,J. (1997) Transitions in the coupling of transcription and nucleotide excision repair within RNA polymerase II-transcribed genes of Saccharomyces cerevisiae. Proc. Natl Acad. Sci. USA, 94, 8027–8032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.van Gool A.J., Verhage,R., Swagemakers,S.M., van de Putte,P., Brouwer,J., Troelstra,C., Bootsma,D. and Hoeijmakers,J.H. (1994) RAD26, the functional S. cerevisiae homolog of the Cockayne syndrome B gene ERCC6. EMBO J., 13, 5361–5369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Selby C.P. and Sancar,A. (1990) Transcription preferentially inhibits nucleotide excision repair of the template DNA strand in vitro. J. Biol. Chem., 265, 21330–21336. [PubMed] [Google Scholar]
- 11.Selby C.P. and Sancar,A. (1993) Molecular mechanism of transcription-repair coupling. Science, 260, 53–58. [DOI] [PubMed] [Google Scholar]
- 12.Bohr V.A., Smith,C.A., Okumoto,D.S. and Hanawalt,P.C. (1985) DNA repair in an active gene: removal of pyrimidine dimers from the DHFR gene of CHO cells is much more efficient than in the genome overall. Cell, 40, 359–369. [DOI] [PubMed] [Google Scholar]
- 13.Mellon I., Spivak,G. and Hanawalt,P.C. (1987) Selective removal of transcription-blocking DNA damage from the transcribed strand of the mammalian DHFR gene. Cell, 51, 241–249. [DOI] [PubMed] [Google Scholar]
- 14.Venema J., Mullenders,L.H., Natarajan,A.T., van Zeeland,A.A. and Mayne,L.V. (1990) The genetic defect in Cockayne syndrome is associated with a defect in repair of UV-induced DNA damage in transcriptionally active DNA. Proc. Natl Acad. Sci. USA, 87, 4707–4711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bhatia P.K., Verhage,R.A., Brouwer,J. and Friedberg,E.C. (1996) Molecular cloning and characterization of Saccharomyces cerevisiae RAD28, the yeast homolog of the human Cockayne syndrome A (CSA) gene. J. Bacteriol., 178, 5977–5988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Selby C.P. and Sancar,A. (1997) Human transcription-repair coupling factor CSB/ERCC6 is a DNA-stimulated ATPase but is not a helicase and does not disrupt the ternary transcription complex of stalled RNA polymerase II. J. Biol. Chem., 272, 1885–1890. [DOI] [PubMed] [Google Scholar]
- 17.Citterio E., Rademakers,S., van der Horst,G.T., van Gool,A.J., Hoeijmakers,J.H. and Vermeulen,W. (1998) Biochemical and biological characterization of wild-type and ATPase-deficient Cockayne syndrome B repair protein. J. Biol. Chem., 273, 11844–11851. [DOI] [PubMed] [Google Scholar]
- 18.Balajee A.S., May,A., Dianov,G.L., Friedberg,E.C. and Bohr,V.A. (1997) Reduced RNA polymerase II transcription in intact and permeabilized Cockayne syndrome group B cells. Proc. Natl Acad. Sci. USA, 94, 4306–4311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Selby C.P. and Sancar,A. (1997) Cockayne syndrome group B protein enhances elongation by RNA polymerase II. Proc. Natl Acad. Sci. USA, 94, 11205–11209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tantin D., Kansal,A. and Carey,M. (1997) Recruitment of the putative transcription-repair coupling factor CSB/ERCC6 to RNA polymerase II elongation complexes. Mol. Cell. Biol., 17, 6803–6814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.van Gool A.J., Citterio,E., Rademakers,S., van Os,R., Vermeulen,W., Constantinou,A., Egly,J.M., Bootsma,D. and Hoeijmakers,J.H. (1997) The Cockayne syndrome B protein, involved in transcription-coupled DNA repair, resides in an RNA polymerase II-containing complex. EMBO J., 16, 5955–5965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Christians F.C. and Hanawalt,P.C. (1992) Inhibition of transcription and strand-specific DNA repair by alpha-amanitin in Chinese hamster ovary cells. Mutat. Res., 274, 93–101. [DOI] [PubMed] [Google Scholar]
- 23.Jansen L.E., den Dulk,H., Brouns,R.M., de Ruijter,M., Brandsma,J.A. and Brouwer,J. (2000) Spt4 modulates Rad26 requirement in transcription-coupled nucleotide excision repair. EMBO J., 19, 6498–6507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chavez S., Beilharz,T., Rondon,A.G., Erdjument-Bromage,H., Tempst,P., Svejstrup,J.Q., Lithgow,T. and Aguilera,A. (2000) A protein complex containing Tho2, Hpr1, Mft1 and a novel protein, Thp2, connects transcription elongation with mitotic recombination in Saccharomyces cerevisiae. EMBO J., 19, 5824–5834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Aguilera A. and Klein,H.L. (1990) HPR1, a novel yeast gene that prevents intrachromosomal excision recombination, shows carboxy-terminal homology to the Saccharomyces cerevisiae TOP1 gene. Mol. Cell. Biol., 10, 1439–1451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Piruat J.I. and Aguilera,A. (1998) A novel yeast gene, THO2, is involved in RNA pol II transcription and provides new evidence for transcriptional elongation-associated recombination. EMBO J., 17, 4859–4872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chavez S. and Aguilera,A. (1997) The yeast HPR1 gene has a functional role in transcriptional elongation that uncovers a novel source of genome instability. Genes Dev., 11, 3459–3470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Prado F., Piruat,J.I. and Aguilera,A. (1997) Recombination between DNA repeats in yeast hpr1Δ cells is linked to transcription elongation. EMBO J., 16, 2826–2835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chavez S., Garcia-Rubio,M., Prado,F. and Aguilera,A. (2001) Hpr1 is preferentially required for transcription of either long or G+C-rich DNA sequences in Saccharomyces cerevisiae. Mol. Cell. Biol., 21, 7054–7064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Santos-Rosa H. and Aguilera,A. (1994) Increase in incidence of chromosome instability and non-conservative recombination between repeats in Saccharomyces cerevisiae hpr1Δ strains. Mol. Gen. Genet., 245, 224–236. [DOI] [PubMed] [Google Scholar]
- 31.Aguilera A. and Klein,H.L. (1989) Genetic and molecular analysis of recombination events in Saccharomyces cerevisiae ocurring in the presence of the hyper-recombination mutation hpr1.Genetics, 122, 503–517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sherman F., Fink,G.R. and Hicks,J.B. (1986) Methods in Yeast Genetics. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- 33.Huberman J.A., Spotila,L.D., Nawotka,K.A., el-Assouli,S.M. and Davis,L.R. (1987) The in vivo replication origin of the yeast 2 microns plasmid. Cell, 51, 473–481. [DOI] [PubMed] [Google Scholar]
- 34.Sambrook J., Fritsch,E.F. and Maniatis,T. (1989) Molecular Cloning: A Laboratory Manual. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- 35.Sweder K.S. and Hanawalt,P.C. (1992) Preferential repair of cyclobutane pyrimidine dimers in the transcribed strand of a gene in yeast chromosomes and plasmids is dependent on transcription. Proc. Natl Acad. Sci. USA, 89, 10696–10700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Leadon S.A. and Lawrence,D.A. (1992) Strand-selective repair of DNA damage in the yeast GAL7 gene requires RNA polymerase II. J. Biol. Chem., 267, 23175–23182. [PubMed] [Google Scholar]
- 37.Verhage R., Zeeman,A.M., de Groot,N., Gleig,F., Bang,D.D., van de Putte,P. and Brouwer,J. (1994) The RAD7 and RAD16 genes, which are essential for pyrimidine dimer removal from the silent mating type loci, are also required for repair of the nontranscribed strand of an active gene in Saccharomyces cerevisiae. Mol. Cell. Biol., 14, 6135–6142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Reagan M.S. and Friedberg,E.C. (1997) Recovery of RNA polymerase II synthesis following DNA damage in mutants of Saccharomyces cerevisiae defective in nucleotide excision repair. Nucleic Acids Res., 25, 4257–4263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lee S.K., Yu,S.L., Prakash,L. and Prakash,S. (2001) Requirement for yeast RAD26, a homolog of the human CSB gene, in elongation by RNA polymerase II. Mol. Cell. Biol., 21, 8651–8656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Verhage R.A., van Gool,A.J., de Groot,N., Hoeijmakers,J.H., van de Putte,P. and Brouwer,J. (1996) Double mutants of Saccharomyces cerevisiae with alterations in global genome and transcription-coupled repair. Mol. Cell. Biol., 16, 496–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Xie Z. and Price,D.H. (1996) Purification of an RNA polymerase II transcript release factor from Drosophila. J. Biol. Chem., 271, 11043–11046. [DOI] [PubMed] [Google Scholar]
- 42.Xie Z. and Price,D. (1997) Drosophila factor 2, an RNA polymerase II transcript release factor, has DNA-dependent ATPase activity. J. Biol. Chem., 272, 31902–31907. [DOI] [PubMed] [Google Scholar]
- 43.Hara R., Selby,C.P., Liu,M., Price,D.H. and Sancar,A. (1999) Human transcription release factor 2 dissociates RNA polymerases I and II stalled at a cyclobutane thymine dimer. J. Biol. Chem., 274, 24779–24786. [DOI] [PubMed] [Google Scholar]
- 44.Nakatsu Y., Asahina,H., Citterio,E., Rademakers,S., Vermeulen,W., Kamiuchi,S., Yeo,J.P., Khaw,M.C., Saijo,M., Kodo,N. et al. (2000) XAB2, a novel tetratricopeptide repeat protein involved in transcription-coupled DNA repair and transcription. J. Biol. Chem., 275, 34931–34937. [DOI] [PubMed] [Google Scholar]
- 45.McDonald W.H., Ohi,R., Smelkova,N., Frendewey,D. and Gould,K.L. (1999) Myb-related fission yeast cdc5p is a component of a 40S snRNP-containing complex and is essential for pre-mRNA splicing. Mol. Cell. Biol., 19, 5352–5362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Russell C.S., Ben-Yehuda,S., Dix,I., Kupiec,M. and Beggs,J.D. (2000) Functional analyses of interacting factors involved in both pre-mRNA splicing and cell cycle progression in Saccharomyces cerevisiae. RNA, 6, 1565–1572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Schneiter R., Guerra,C.E., Lampl,M., Gogg,G., Kohlwein,S.D. and Klein,H.L. (1999) The Saccharomyces cerevisiae hyperrecombination mutant hpr1Δ is synthetically lethal with two conditional alleles of the acetyl coenzyme A carboxylase gene and causes a defect in nuclear export of polyadenylated RNA. Mol. Cell. Biol., 19, 3415–3422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Luo M.L., Zhou,Z., Magni,K., Christoforides,C., Rappsilber,J., Mann,M. and Reed,R. (2001) Pre-mRNA splicing and mRNA export linked by direct interactions between UAP56 and Aly. Nature, 413, 644–647. [DOI] [PubMed] [Google Scholar]
- 49.Strasser K. and Hurt,E. (2001) Splicing factor Sub2p is required for nuclear mRNA export through its interaction with Yra1p. Nature, 413, 648–652. [DOI] [PubMed] [Google Scholar]
- 50.Fan H.Y., Merker,R.J. and Klein,H.L. (2001) High-copy-number expression of Sub2p, a member of the RNA helicase superfamily, suppresses hpr1-mediated genomic instability. Mol. Cell. Biol., 21, 5459–5470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Jensen T.H., Boulay,J., Rosbash,M. and Libri,D. (2001) The DECD box putative ATPase Sub2p is an early mRNA export factor. Curr. Biol., 11, 1711–1715. [DOI] [PubMed] [Google Scholar]