

Abstract
Age data are essential for estimating life history parameters and are thus critical for population assessment, management, and conservation. Traditional vertebrae-based age estimation in elasmobranchs can be costly, time intensive, of low accuracy, and is by necessity lethal. Herein, epigenetic clocks were developed for an elasmobranch, the cownose ray (Rhinoptera bonasus), using aquarium-born individuals (n = 42) with known dates of birth (age range: 7−7,878 days or 0−21 years) and two tissue types (fin clips and whole blood) that can be sampled in a relatively non-invasive manner. Enzymatically-converted restriction site-associated DNA sequencing (ECrad-seq) was used to identify CpG sites that exhibited age-correlated DNA methylation. The epigenetic clocks developed were highly accurate (mean absolute error, MAE, < 0.75 years) and precise (R2 > 0.98). Age-associated CpG sites were identified across tissues, and a multi-tissue clock was also highly accurate (MAE < 1 year) and precise (R2 = 0.97). Using the developed fin clip clock, three wild-caught individuals of unknown age but managed in aquariums for > 22 years were predicted to be 22.10−23.49 years old. Overall, the results have important implications for future epigenetic clock development and noninvasive age estimation in elasmobranchs.
Keywords: Ageing, Epigenetic clock, DNA methylation, Multi-tissue, Sharks, Rays
Subject terms: DNA methylation, Population dynamics, Marine biology
Introduction
Age data provide the basis for calculating growth and mortality rates, age-at-maturity, and estimates of longevity, all of which are essential for population assessment, management, and conservation of fishes. Accurate age estimation is particularly critical for elasmobranchs (sharks, skates, and rays), which are often less resilient and highly vulnerable to human-induced mortality1due to their relatively K-selected life history characteristics (i.e., low fecundity, late maturity, slow growth, and high longevity2). Age estimation in elasmobranchs has traditionally involved counting growth zones in vertebral centra or spines3, which can be costly and time intensive4, of low precision and/or accuracy for some species3,5, and is necessarily lethal. Given that one third of elasmobranchs are threatened with extinction6, and the importance of age data for understanding the ecology of species and conducting population assessment for conservation and management planning, the development of an accurate, noninvasive ageing technique would be of great utility for elasmobranch species.
The development of DNA methylation-based epigenetic ageing techniques has received increasing attention in recent years7, due in large part to the fact that epigenetic ageing is non-lethal, inexpensive relative to traditional age estimation using hard structures, and well-suited for rapid age estimation8. DNA methylation is an epigenetic mechanism which refers primarily to the addition of methyl groups (CH3) to cytosines located within CpG dinucleotides (cytosines followed by guanines)9, and recent studies have demonstrated that changes in DNA methylation levels at certain CpG sites exhibit strong correlations with chronological age7. This has led to the development of age-predictive models based on changes in DNA methylation, referred to as epigenetic clocks10. Epigenetic clocks summarize age-associated increases (hypermethylation) or decreases (hypomethylation) in DNA methylation across a select group of CpG sites throughout the genome which can be used collectively to estimate chronological age in wild animals7. While the utility of epigenetic age estimation has now been demonstrated in a handful of teleosts, including but not limited to the European sea bass (Dicentrarchus labrax)11, zebrafish (Danio rerio)12, red snapper (Lutjanus campechanus)13, red grouper (Epinephelus morio)13, golden perch (Macquaria ambiguaspp.)8, and blackbelly rosefish (Helicolenus daactylopterus)14, the application of this ageing technique to an elasmobranch species has not yet been fully investigated (see Beal et al15.).
The cownose ray (Rhinoptera bonasus) is a benthopelagic batoid that occurs in the western Atlantic Ocean, from New England in the United States of America (U.S.A.) to Argentina16, and is known to migrate in large schools along the U.S. East Coast and Gulf of Mexico17. As is the case for many other elasmobranchs, cownose rays exhibit late maturity (6−8 years), long gestation (11−12 months), and low reproductive potential (typically one offspring per reproductive cycle), making them highly susceptible to human-induced mortality17. Maximum longevity in the wild is reported to be 21 years17. Cownose rays are one of the most common elasmobranch species displayed in aquariums and zoos worldwide18, and because individuals born in aquariums have known dates of birth, the need for validated age estimates used to train epigenetic clocks can be eliminated. In addition, potential metabolic effects of environmental heterogeneity on levels of DNA methylation are minimized in controlled conditions (e.g., laboratories, aquariums), making cownose rays an ideal candidate for the development of age-predictive epigenetic clocks in an elasmobranch species.
Therefore, the objectives of the present study were to: (1) develop epigenetic clocks for the cownose ray, using two tissues types (fin clips and whole blood) and aquarium-born individuals with known dates of birth, through the de novo identification of CpG sites exhibiting age-correlated DNA methylation; (2) assess the influence of sex on epigenetic clock accuracy; and (3) assess the accuracy of a multi-tissue epigenetic clock, developed through the identification of age-associated CpG sites that are shared across tissue types (fin clips and whole blood).
Methods
Fin clips and whole blood were obtained in 2023 from cownose rays that were born and reared at Ripley’s Aquarium of Myrtle Beach, Ripley’s Aquarium of the Smokies, and Ripley’s Aquarium of Canada. Cownose rays were individually captured using dip nets, weighed, and measured before a small piece of the trailing edge of the pelvic fin was removed using a sterile scalpel blade or scissors, in accordance with ARRIVE guidelines. A blood sample was obtained through wing venipuncture using a 1.5 inch, 18–21-gauge needle. Whole blood samples (150 uL) were added to 1 mL RNAlater, gently inverted to mix, and stored at −80 °C until DNA extraction, while fin clips (approximately 0.5 cm2) were immersed in 20% DMSO-0.25 M EDTA NaCl-saturated buffer19 and stored at −80 °C until DNA extraction. All animal handling and procedures were approved by the Ripley’s Aquariums Research Committee and Georgia Aquarium IACUC protocol (GAI-24-06). All methods were performed in accordance with the relevant guidelines and regulations.
Genomic DNA was extracted from fin clips using the Mag–Bind Blood & Tissue DNA Kit (Omega Bio-tek, Inc., Norcross, USA) and from whole blood using the DNeasy PowerSoil Pro Kit (Qiagen, Hilden, Germany), and a library was prepared for enzymatically-converted restriction site-associated DNA sequencing (ECrad-seq). Briefly, restriction digests were performed using MfeI-HF, and unique hemi-methylated barcoded adaptors were ligated to each sample20. Samples were then sheared (Covaris M220 Ultrasonicator, Covaris, Inc., Woburn, USA), pooled, and size selected using a Pippin Prep Size Selection System (Sage Science, Inc., Beverly, USA). The library was then split into two portions, and one portion was treated with a methylation-sensitive enzyme using the NEBNext Enzymatic Methyl-seq Kit (New England Biolabs, Ipswich, USA). This enzyme treatment converts unmethylated cytosines into uracils through chemical deamination, and uracils are subsequently replaced by thymines during subsequent PCR. This results in predictable base substitutions at all unmethylated cytosines, which can be identified by comparing sequences from the treated portion to the untreated portion. The library (including both the treated and untreated portion) was sequenced across a single lane on an Illumina NovaSeq 6000 (Illumina, Inc., San Diego, USA). Enzyme-treated reads were mapped to a reference genome constructed from the untreated reads using the dDocentpipeline21 with c = 0.88, K1 = 2, and K2 = 1. Mapped reads were then filtered to retain primary alignments, proper pairs, and those with a mapping quality ≥40.
CpG sites that could not be successfully genotyped in the untreated portion or that were identified as potential single nucleotide polymorphisms (SNPs; defined as sites where > 5% of total untreated reads across individuals displayed a cytosine to thymine substitution on the forward strand or guanine to adenine substitution on the reverse strand)13,14were removed from the dataset. CpG sites were then filtered to retain only sites present in ≥ 80% of individuals per tissue type. Percent methylation was estimated as the number of methylated reads divided by the total number of reads, and per-site 95% confidence intervals were calculated around the estimate for each individual22. Only those sites with confidence intervals < 0.85 in at least 80% of individuals per tissue type were retained, which was roughly equivalent to a mean of 10 reads per site13,14.
To identify CpG sites exhibiting age-correlated DNA methylation, a Bayesian framework was used to estimate the parameters of a generalized linear model (GLM) that included known age in days, sex, and tissue type as fixed factors and individual as a random factor, using the package rstanarmversion 2.19.323. The response variable was the binomial expression of the number of methylated reads (n) and the total number of reads for each sample (k) at a given CpG site (n/k). GLMs were considered to have converged if the effective sample size (n_eff), was greater than 2,000 and the Gelman-Rubin convergence diagnostic was less than 1.0124,25. CpG sites with a 95% credible interval that did not include zero for the slope of known age versus methylation were considered to exhibit significant age-correlated methylation. For each CpG site that exhibited age-correlated methylation, individuals with over-dispersed confidence intervals (> 0.85) were entered as missing data. Because downstream analysis does not allow for missing data, methylation frequencies at missing individuals/sites were imputed using the package miceversion 3.9.026.
The relationship between known age and percent methylation across CpG sites was characterized using elastic net penalized regression modeling, as implemented in the package glmnetversion 4.0.227, in a leave-one-out cross validation context. Prior to penalized regression modeling, methylation data were normalized using the Yeo-Johnson transformation28 in the package bestNormalize version 1.9.1. Independent models were constructed for each tissue type (i.e., fin clips and whole blood), for each tissue type by sex, and for both tissue types combined. Prior to constructing the combined (multi-tissue) model, potential tissue-specific signal in percent methylation was removed by filtering out any CpG sites that exhibited age-correlated methylation and a relationship with tissue type or an interaction between tissue type and age. An internally cross-validated version of glmnet(‘cv.glmnet’) was utilized to automatically select the optimal penalty parameter (λ), and models were run with an initial alpha parameter (α) of 0, to assess model slope coefficients for all CpG sites included. To reduce model complexity, the number of CpG sites included in each of the three models was reduced based upon independent Pearson correlation values29, obtained using the ‘corr.test’ function from the package psychversion 2.2.9, and model slope coefficients from ‘cv.glmnet’14. The subsets of CpG sites which minimized mean absolute error (MAE) in each of the final predictive models were then identified through the use of a custom R script which randomly selected up to 75 CpG sites over 10,000 iterations of ‘cv.glmnet’ (available at https://github.com/marinegenomicslab/CownoseRay_EpigeneticAgeing).
Performance of the models was assessed using linear regressions and by computing MAE between known ages and ages predicted by the epigenetic clock(s). Relative error was calculated by dividing the absolute error by the known age, and linear regressions were conducted on known age versus relative error to determine if error in the models increased with increasing age. Finally, to investigate the potential application of an epigenetic clock developed using aquarium-born individuals to wild-caught individuals of unknown age, the female-specific fin clip clock was tested against three individuals captured in Tampa Bay, FL in November of 2000 and subsequently maintained at one of the three Ripley’s Aquariums until the time of sampling in September of 2023. All analyses were conducted in R version 3.6.0.
Results
Sampling and data filtering
Fin clips and whole blood were obtained from 42 aquarium-born cownose rays, with ages ranging from 7 to 7,878 days (0 to 21 years) and from three wild-caught cownose rays reared in aquariums from the time of capture (year 2000) to the time of sampling (year 2023; Table 1). Across all individuals and both tissue types, 8,042,910 CpG sites were recovered. A total of 515,753 sites either could not be successfully genotyped in the untreated portion of the library or were identified as potential SNPs and removed from the dataset. A total of 99,509 CpG sites had sufficiently narrow confidence intervals in at least 80% of individuals per tissue type.
Table 1.
Sample summary and morphometrics for all cownose rays (Rhinoptera bonasus) included in epigenetic clock development and testing.
| Source | n | Sex (M: F) |
Age (days) |
Age (years) |
Disc width (cm) |
Mass (kg) |
|---|---|---|---|---|---|---|
| Aquarium | 42 | 16:26 | 7−7,878 | 0−21 | 34.0−92.0 | 1.36−16.40 |
| Wild | 3 | 0:3 | > 8,333 | > 22 | 75.5−77.0 | 9.10−10.40 |
Tissue-specific epigenetic clocks
Across all fin clip samples, Bayesian GLMs identified 7,813 CpG sites that exhibited significant age-correlated methylation, and penalized regression analysis retained 32 CpG sites in the age-predictive model. Strong agreement was observed between known age and predicted age (R2 = 0.98), with an MAE of 246.6 days (0.68 years; Fig. 1A; Table 2). Relative error did not increase with increasing age (p = 0.13).
Figure 1.

Epigenetic age predictions versus known age for epigenetic clocks developed using fin clip samples (A) and whole blood samples (B). Dashed lines indicate lines of 1:1 agreement between predicted and known ages. Solid lines represent linear regression fits to the data. The coefficient of determination (R2) and mean absolute error (MAE) reported in days are displayed.
Table 2.
Summary statistics for epigenetic clocks developed for the cownose ray (Rhinoptera bonasus), including the number and age range of individuals used, the number of CpG sites included in the model, the coefficient of determination (R2), and the mean absolute error (MAE) reported in days.
| Model | n | Age (days) | CpG sites | R 2 | MAE (days) |
|---|---|---|---|---|---|
| Fin Clip | 42 | 7−7,878 | 32 | 0.98 | 246.6 |
| Fin Clip by Sex | |||||
| Males | 16 | 247−7,878 | 10 | 0.99 | 172.6 |
| Females | 26 | 7−7,514 | 19 | 0.98 | 234.3 |
| Whole Blood | 42 | 7−7,878 | 30 | 0.98 | 269.8 |
| Whole Blood by Sex | |||||
| Males | 16 | 247−7,878 | 8 | 0.99 | 121.1 |
| Females | 26 | 7−7,514 | 17 | 0.98 | 216.1 |
| Combined Tissue | 42 | 7−7,878 | 62 | ||
| Fin Clip | 0.97 | 309.0 | |||
| Whole Blood | 0.97 | 354.4 |
Across all whole blood samples, Bayesian GLMs identified 7,630 CpG sites that exhibited significant age-correlated methylation, and penalized regression analysis retained 30 CpG sites in the age-predictive model. As above for the fin clip clock, strong agreement was observed between known age and predicted age (R2 = 0.98), with an MAE of 269.8 days (0.74 years; Fig. 1B; Table 2). Relative error did not increase with increasing age (p = 0.07).
The inclusion of sex data enhanced the accuracy and precision of both the fin clip clock (R2 = 0.99 and MAE = 172.6 days for males; R2 = 0.98 and MAE = 234.3 days for females; Fig. 2A; Table 2) and whole blood clock (R2 = 0.99 and MAE = 121.1 days for males; R2 = 0.98 and MAE = 216.1 days for females; Fig. 2B; Table 2). Sex-specific epigenetic clocks developed also included fewer CpG sites (Table 2).
Figure 2.

Epigenetic age predictions versus known age for epigenetic clocks developed using fin clip samples (A) and whole blood samples (B) and separately for females and males. Dashed lines indicate lines of 1:1 agreement between predicted and known ages. Solid lines represent linear regression fits to the data. The coefficient of determination (R2) and mean absolute error (MAE) reported in days are displayed.
Multi-tissue epigenetic clock
Across both tissue types, Bayesian GLMs identified 5,212 CpG sites that exhibited significant age-correlated methylation (Fig. 3A), of which 2,372 CpG sites exhibited significant age-correlated methylation and no relationship with tissue type or interaction between tissue type and age (Fig. 3B). Penalized regression analysis retained 62 CpG sites in the age-predictive model. The multi-tissue clock performed well for both the fin clip samples (R2 = 0.97 and MAE = 309.0 days or 0.85 years; Fig. 4; Table 2) and the whole blood samples (R2 = 0.97 and MAE = 354.4 days or 0.97 years; Fig. 4; Table 2). Relative error did not increase with increasing age for the fin clip samples (p = 0.06) or the whole blood samples (p = 0.09). No CpG sites were shared between the multi-tissue clock and the single-tissue clocks.
Figure 3.

Principal component analysis (PCA) of percent methylation across CpG sites, depicting the removal of the tissue-specific signal in percent methylation for the development of the multi-tissue epigenetic clock. Panel A includes all CpG sites exhibiting age-correlated DNA methylation across both tissue types (n = 5,212 CpG sites). Panel B includes only the CpG sites that exhibited age-correlated DNA methylation and no CpG sites that exhibited a relationship with tissue type or interaction between tissue type and age (n = 2,372 CpG sites).
Figure 4.
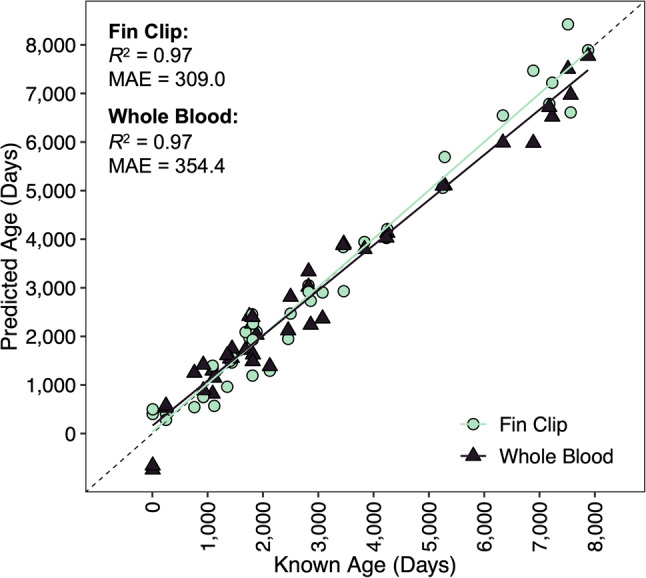
Epigenetic age predictions versus known age for the multi-tissue epigenetic clock. Dashed lines indicate lines of 1:1 agreement between predicted and known ages. Solid lines represent linear regression fits to the data. The coefficient of determination (R2) and mean absolute error (MAE) reported in days are displayed.
Predicting age of wild-caught individuals
Based on the time of initial capture (November 2000) to the time of tissue sampling for the present study (September 2023), the three wild-caught individuals had a minimum age of 22.83 years. Because all three individuals were female, the female-specific fin clip clock was used to estimate age, and the three wild-caught individuals were predicted to be 22.10, 22.22, and 23.49 years old (± 0.76 years; standard error of estimate).
Discussion
The development of accurate and precise epigenetic clocks for the cownose ray, demonstrated herein, suggests that epigenetic age estimation could provide an effective, non-lethal ageing approach for elasmobranch species of management and/or conservation concern. Given the relatively K-selected life histories of many elasmobranchs2, and their associated low resiliency and high vulnerability to human-induced mortaltity1, the ability to collect and incorporate age-specific biological data into population assessments without the need for lethal sampling would be particularly beneficial for elasmobranch species.
Single-tissue epigenetic clocks developed for the cownose ray were both highly accurate and precise. While previous research into DNA methylation-based age estimation in elasmobranchs demonstrated differences in global DNA methylation in lemon sharks (Negaprion brevirostris) from three different age groups (juveniles, subadults, adults)15, the epigenetic clocks developed in the present study can predict age with accuracy levels under a year. In addition, epigenetic clock accuracy did not decrease with increasing age, which is particularly important for long-lived species like many elasmobranchs, and is likely the result of characterizing a large number of independent CpG sites exhibiting age-correlated methylation specifically within the cownose ray genome14. In addition, the use of aquarium-born individuals with known dates of birth likely contributed to the high accuracy and precision observed. However, the relatively low number of individuals available for this study precluded the application of a more rigorous predictive approach, involving training and testing datasets to minimize model overfitting30, and a larger number of age-validated individuals will be necessary in the future to more thoroughly validate the predictive capabilities of these epigenetic clocks.
The inclusion of sex, and the development of sex-specific epigenetic clocks, enhanced the accuracy and precision of both the fin clip and whole blood clocks. Given the presence of external morphological features in males, sex data can be easily and non-invasively obtained for elasmobranchs and could thus be included in epigenetic clock design and epigenetic age assessment with minimal effort. The sex-specific epigenetic clocks also included fewer CpG sites, which is important to consider when generating multiplex assays for production ageing, such as via genotyping-in-thousands by sequencing (GT-seq)31, because multiplex PCR assays are easier to design with fewer sets of primer pairs32. These results suggest that patterns of age-associated DNA methylation are somewhat sex-specific, consistent with results reported for the blackbelly rosefish14and with previous research conducted on humans33 and roe deer (Capreolus capreolus)34, where distinct patterns in epigenetic ageing were identified between sexes.
While previous studies have failed to develop accurate multi-tissue epigenetic clocks for fishes11,14, the combined fin clip and whole blood clock was both highly accurate and precise. The ability to develop a multi-tissue clock could be due to a variety of factors, including the use of an enzyme-based alternative to bisulfite conversion (which has been shown to provide increased DNA sequencing quality35), the effective removal of tissue-specific patterns in DNA methylation through the Bayesian generalized linear models (GLMs), the choice of tissue types, or some combination of these factors. The multi-tissue epigenetic clocks developed in both Anastasiadi and Piferrer11and Weber et al14. involved the use of muscle tissue, which is recognized as one of the worst predictors of age in humans, due potentially to the rejuvenation of DNA methylation profiles in muscle tissue via muscle stem cells following injury or disease36. By contrast, whole blood has been the preferred target tissue in a variety of mammal clocks, including mice (Mus musculus)37, chimpanzees (Pan troglodytes)38, and short-tailed shearwaters (Ardenna tenuirostris)39. While the development of multi-tissue epigenetic clocks would increase the diversity of data sources (i.e., tissue types) from which age estimates could be obtained, the single-tissue epigenetic clocks developed in the present study were more accurate and precise, and the simultaneous development of tissue-specific clocks required similar time and effort as the development of the multi-tissue clock.
The degree to which environmental heterogeneity influences the epigenome and potential subsequent effects on epigenetic clock performance remain understudied7. Epigenetic clock development in the present study involved the use of known-age aquarium-born individuals reared in controlled environments. Individuals reared in controlled environments (e.g., laboratories, aquariums) are not exposed to the same degree of environmental variation experienced by fishes in the wild, and thus may not exhibit the same degree or type of environmentally induced epigenetic change13. Thus, the ability to predict the age of wild-caught individuals using an epigenetic clock developed with aquarium-raised individuals, and vice versa, remains unknown. However, the female-specific fin clip clock developed was able to predict wild-caught individuals raised in aquariums for > 22.83 years as being 22.10, 22.22, and 23.49 years old. While age estimates for at least two of the three wild-caught individuals were underpredicted to some unknown degree, all three individuals were accurately predicted to be older than any individual included in clock development. Similarly, using an Australian lungfish (Neoceratodus forsteri) clock developed using a mix of wild-caught and aquarium-born individuals30, Mayne et al40. predicted the age of a wild-caught Australian lungfish raised in aquariums for > 84 years to be 109 years old, suggesting that clocks designed from aquarium-born individuals may be useful for age assessment in the wild. Finally, age estimates for the wild-caught but aquarium-raised individuals (minimum age of 22.83 years) exceed the maximum longevity previously reported for wild-caught cownose rays (21 years17). This could be due to a variety of factors, including the potential that animals can live longer in managed care than in the wild41, the presence of systemic age underestimation in wild-caught elasmobranchs42, and/or the notion that exceptionally old animals are rarely encountered in the wild.
The present study demonstrates the ability to develop accurate, non-invasive, epigenetic clocks for an elasmobranch species, which are often less resilient and highly vulnerable to fishing mortality1and can be difficult to age using traditional ageing techniques3,5. Moreover, traditional vertebrae-based age estimation is necessarily lethal, which is particularly problematic for elasmobranch species of conservation concern. In addition, the present study demonstrates the successful development of an accurate and precise multi-tissue epigenetic clock. Given that various tissue types are commonly, non-lethally sampled during fisheries research (e.g., fin clips, whole blood, muscle tissue), the ability to use a single epigenetic clock for multiple tissue types would increase the ease with which age estimates can be obtained, with important implications for future epigenetic clock development. Overall, through applying epigenetic age estimation to an elasmobranch, the present study adds to a growing body of literature suggesting that epigenetic clocks have the potential to greatly advance the generation of age estimates for research, management, and conservation of fish species.
Acknowledgements
The authors thank Dr. Andrew Fields, Dr. William Patterson III, and Dr. Zachary Siders for helpful discussions related to study design and statistical analyses. Husbandry and veterinary professionals at Ripley’s Aquariums, including Dr. Stephen Anderson, Stacia White, Ben Skeen, Cethlynn Weatherly, Shannon Krieg, Phil Morabito, Kelsey Remmes, Richard Brown, Sarah Maroney, Cameron Snell, Tyler Ruhs, David Fraser, Michael Alexander, and Tami West, provided expert care of the cownose rays and documented reproductive histories making it possible to collect samples from individuals ranging in age from 1 week to more than 20 years. The authors thank Chandran Sabanayagam and Joanne Cooper at University of Delaware. Funding for this research was provided by Ripley’s Aquariums. This is publication 42 of the Marine Genomics Laboratory and 134 of Genetic Studies in Fishes (Genetic Studies in Marine Fishes).
Author contributions
DNW, JTW, KL, and DSP conceived of and designed this study. JTW, RHG, CB, VL, and FEL oversaw the collection of samples and their associated metadata. DNW conducted all molecular laboratory work and bioinformatics. The first draft of this manuscript was written by DNW and was revised with input from all co-authors. All authors have reviewed and approved the manuscript.
Data availability
Datasets and data analysis scripts are available at https://github.com/marinegenomicslab/CownoseRay_EpigeneticAgeing. Raw NovaSeq reads are available in the NCBI SRA (BioProject PRJNA1148050).
Declarations
Competing interests
The authors declare no competing interests.
Correspondence and requests for materials should be addressed to DNW or DSP.
Footnotes
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
D. Nick Weber, Email: dweber@islander.tamucc.edu
David S. Portnoy, Email: david.portnoy@tamucc.edu
References
- 1.Winemiller, K. O. Life history strategies, population regulation, and implications for fisheries management. Can. J. Fish. Aquat. Sci.62, 872–885 (2005). [Google Scholar]
- 2.Musick, J. A. Ecology and conservation of long-live marine animals. Am. Fish. Soc. Sym. 23, 1–10 (1999). [Google Scholar]
- 3.Cailliet, G. M. Perspectives on elasmobranch life-history studies: a focus on age validation and relevance to fishery management. J. Fish. Biol.87, 1271–1292 (2015). [DOI] [PubMed] [Google Scholar]
- 4.Helser, T. E., Benson, I. M. & Barnett, B. K. Proceedings of the research workshop on the rapid estimation of fish age using Fourier transform near infrared spectroscopy (FT-NIRS). AFSC Processed Rep. 2019-06. Alaska Fisheries Science Center, NOAA, National Marine Fisheries Service (2019).
- 5.Matta, M. E., Tribuzio, C. A., Ebert, D. A., Goldman, K. J. & Gburski, C. M. Age and growth of elasmobranchs and applications to fisheries management and conservation in the Northeast Pacific Ocean. Adv. Mar. Biol.77, 179–220 (2017). [DOI] [PubMed] [Google Scholar]
- 6.Dulvy, N. K. et al. Overfishing drives over one-third of all sharks and rays toward a global extinction crisis. Curr. Biol.31, 4773–4787 (2021). [DOI] [PubMed] [Google Scholar]
- 7.Piferrer, F. & Anastasiadi, D. Age estimation in fishes using epigenetic clocks: applications to fisheries management and conservation biology. Front. Mar. Sci.10.3389/fmars.2023.1062151 (2023).
- 8.Mayne, B. et al. Accurate, non-destructive, and high-throughput age estimation for golden perch (Macquaria ambigua spp.) using DNA methylation. Sci. Rep.13, 9547 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Moore, L. D., Le, T. & Fan, G. DNA methylation and its basic function. Neuropsychopharmacol. 38, 23–38 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Anastasiadi, D. & Piferrer, F. A. Bioinformatic analysis for age prediction using epigenetic clocks: application to fisheries management and conservation biology. Front. Mar. Sci.10, 1096909 (2023). [Google Scholar]
- 11.Anastasiadi, D. & Piferrer, F. A. Clockwork fish: age prediction using DNA methylation-based biomarkers in the European seabass. Mol. Ecol. Resour.20, 387–397 (2020). [DOI] [PubMed] [Google Scholar]
- 12.Mayne, B. et al. A DNA methylation age predictor for zebrafish. Aging. 12, 24817–24835 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Weber, D. N. et al. Novel epigenetic age estimation in wild-caught Gulf of Mexico reef fishes. Can. J. Fish. Aquat. Sci.79, 1–5 (2022). [Google Scholar]
- 14.Weber, D. N., Fields, A. T., Chamberlin, D. W., Patterson, W. F. III & Portnoy, D. S. Epigenetic age estimation in a long-lived, deepwater scorpionfish: insights into epigenetic clock development. Can. J. Fish. Aquat. Sci.81, 620–631 (2024). [Google Scholar]
- 15.Beal, A. P., Hackerott, S., Feldheim, K., Gruber, S. H. & Eirin-Lopez, J. M. Age group DNA methylation differences in lemon sharks (Negaprion brevirostris): implications for future age estimation tools. Ecol. Evol.12, e9226 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Last, P.R., White, W.T., de Carvalho, M.R., Seret, B., Stehmann, M.F.W., & Naylor, G.J.P. Rays of the World. CSIRO Publishing, Clayton South, Victoria, Australia (2016).
- 17.Fisher, R. A., Call, G. C. & Grubbs, R. D. Age, growth, and reproductive biology of cownose rays in Chesapeake Bay. Mar. Coast Fish.5, 224–235 (2013). [Google Scholar]
- 18.Ferreira, C. M., Field, C. L. & Tuttle, A. D. Hematological and plasma biochemical parameters of aquarium-maintained cownose rays. J. Aquat. Anim. Health. 22, 123–128 (2010). [DOI] [PubMed] [Google Scholar]
- 19.Seutin, G., White, B. N. & Boag, P. T. Preservation of avian blood and tissue samples for DNA analyses. Can. J. Zool.69, 82–90 (1991). [Google Scholar]
- 20.Peterson, B. K., Weber, J. N., Kay, E. H., Fisher, H. S. & Hoekstra, H. E. Double digest RADseq: an inexpensive method for de novo SNP discovery and genotyping in model and non-model species. PLoS ONE. 7, e37135 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Puritz, J. B., Hollenbeck, C. M. & Gold, J. R. dDocent: a RADseq, variant-calling pipeline designed for population genomics of non-model organisms. PeerJ. 10.7717/peerj.431 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Clopper, C. J. & Pearson, E. S. The use of confidence or fiducial limits illustrated in the case of the binomial. Biometrika. 26, 404–413 (1934). [Google Scholar]
- 23.Goodrich, B., Gabry, J., Ali, I. & Brilleman, S. rstanarm: Bayesian applied regression modeling via Stan. R package version 2.19.3 (2020).
- 24.Lunn, D., Barrett, J., Sweeting, M. & Thompson, S. Fully bayesian hierarchical modeling in two stages, with application to meta-analysis. J. R Stat. Soc. Ser. C Appl. Stat.62, 551–572 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Muth, C., Oravecz, Z. & Gabry, J. User-friendly bayesian regression modelling: a tutorial with rstanarm and shinystan. Quant. Meth Psychol.14, 99–119 (2018). [Google Scholar]
- 26.van Buuren, S. & Groothuis-Oudshoorn, K. Mice: Multivariate imputation by chained equations in R. J. Stat. Softw.45, 1–67 (2011). [Google Scholar]
- 27.Friedman, J., Hastie, T. & Tibshirani, R. Regularization paths for generalized linear models via coordinate descent. J. Stat. Softw.33, 1–22 (2010). [PMC free article] [PubMed] [Google Scholar]
- 28.Yeo, I. K. & Johnson, R. A. A new family of power transformations to improve normality or symmetry. Biometrika. 87, 954–959 (2000). [Google Scholar]
- 29.Bertucci, E. M., Mason, M. W., Rhodes, O. E. & Parrot, B. J. Exposure to ionizing radiation disrupts normal epigenetic aging in Japanese medaka. Aging. 13, 22752–22771 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mayne, B. et al. Nonlethal age estimation of three threatened fish species using DNA methylation: Australian lungfish, Murray Cod and Mary River Cod. Mol. Ecol. Res.21, 2324–2332 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Campbell, N. R., Harmon, S. A. & Narum, S. R. Genotyping-in-thousands by sequencing (GT-seq): a cost effective SNP genotyping method based on custom amplicon sequencing. Mol. Ecol. Res.15, 855–867 (2015). [DOI] [PubMed] [Google Scholar]
- 32.Meek, M. H. & Larson, W. A. The future is now: Amplicon sequencing and sequence capture usher in the conservation genomics era. Mol. Ecol. Res.19, 795–803 (2019). [DOI] [PubMed] [Google Scholar]
- 33.Yusipov, I. et al. Age-related DNA methylation changes are sex-specific: a comprehensive assessment. Aging. 12, 24057–24080 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lemaitre, J. et al. DNA methylation as a tool to explore ageing in wild roe deer populations. Mol. Ecol. Res.22, 1002–1015 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mayne, B. et al. Increased scalability and sequencing quality of an epigenetic age prediction assay. PLoS ONE. 19, e0297006 (2024). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Horvath, S. DNA methylation age of human tissues and cell types. Genome Biol.14, R115 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Han, Y. et al. Epigenetic age-predictor for mice based on three CpG sites. eLife. 7, 1–10 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ito, H., Udono, T., Hirata, S. & Inoue-Murayama, M. Estimation of chimpanzee age based on DNA methylation. Sci. Rep.8, 9998l (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Paoli-Iseppi, R. et al. Age estimation in a long-lived seabird (Ardenna Tenuirostris) using DNA methylation-based biomarkers. Mol. Ecol. Res.19, 411–425 (2019). [DOI] [PubMed] [Google Scholar]
- 40.Mayne, B., Espinoza, T. & Roberts, D. Tell us a story granddad: age and origin of an iconic Australian lungfish. Front. Environ. Sci.10, 931467 (2022). [Google Scholar]
- 41.Tidiere, M. et al. Comparative analyses of longevity and senescence reveal variable survival benefits of living in zoos across mammals. Sci. Rep.6, 36361 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Harry, A. V. Evidence for systemic age underestimation in shark and ray ageing studies. Fish. Fish.19, 185–200 (2018). [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Datasets and data analysis scripts are available at https://github.com/marinegenomicslab/CownoseRay_EpigeneticAgeing. Raw NovaSeq reads are available in the NCBI SRA (BioProject PRJNA1148050).