

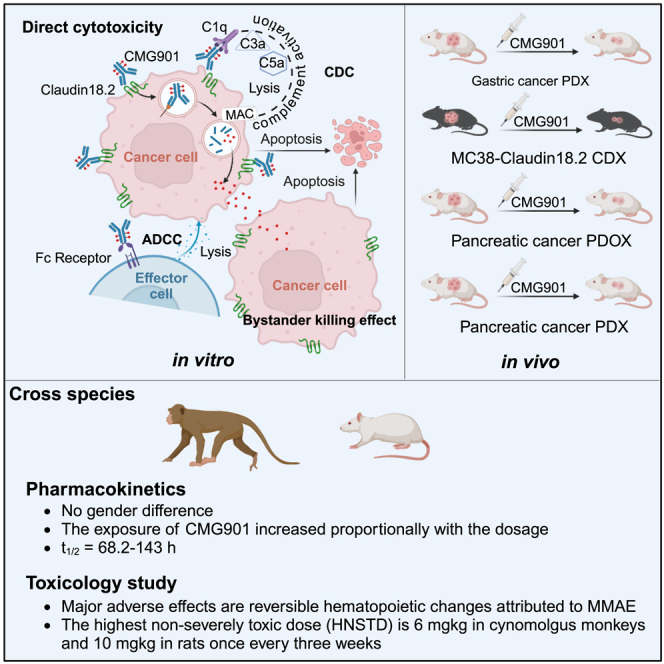
Summary
Claudin18.2 has been recently recognized as a potential therapeutic target for gastric/gastroesophageal junction or pancreatic cancer. Here, we develop a Claudin18.2-directed antibody-drug conjugate (ADC), CMG901, with a potent microtubule-targeting agent MMAE (monomethyl auristatin E) and evaluate its preclinical profiles. In vitro studies show that CMG901 binds specifically to Claudin18.2 on the cell surface and kills tumor cells through direct cytotoxicity, antibody-dependent cellular cytotoxicity (ADCC), complement-dependent cytotoxicity (CDC), and bystander killing activity. In vivo pharmacological studies show significant antitumor activity in patient-derived xenograft (PDX) models. Toxicity studies show that the major adverse effects related to CMG901 are reversible hematopoietic changes attributed to MMAE. The highest non-severely toxic dose (HNSTD) is 6 mg/kg in cynomolgus monkeys and 10 mg/kg in rats once every 3 weeks. CMG901’s favorable preclinical profile supports its entry into the human clinical study. CMG901 is currently under phase 3 investigation in patients with advanced gastric/gastroesophageal junction adenocarcinoma expressing Claudin18.2 (NCT06346392).
Keywords: CMG901, Claudin18.2, ADC, solid tumors
Graphical abstract
Highlights
-
•
CMG901 is a Claudin18.2-specific ADC
-
•
CMG901 kills tumor cells via direct cytotoxicity, ADCC, CDC, and bystander killing effects
-
•
The HNSTD is 6 mg/kg in cynomolgus monkeys and 10 mg/kg in rats once every 3 weeks
-
•
CMG901 is being investigated in a phase 3 clinical trial
Xu et al. develop a Claudin18.2-directed antibody-drug conjugate, CMG901, with a potent microtubule-targeting agent MMAE and evaluate its preclinical profiles. The preclinical profiles support its entry into human study. CMG901 is being investigated in a phase 3 clinical trial in patients with advanced gastric/gastroesophageal junction adenocarcinoma expressing Claudin18.2 (NCT06346392).
Introduction
There remains a substantial unmet clinical need for patients with gastric/gastroesophageal junction (GEJ) cancer and pancreatic cancer despite recent therapeutic advances in treatments, including programmed cell death protein 1 (PD-1) inhibitors or liposomal irinotecan. Antibody-drug conjugates (ADCs), a now well-established targeted therapeutic modality, in which an antibody is conjugated to a cytotoxic agent via a chemical linker, may improve patient outcomes with usually less systemic toxicities than standard-of-care chemotherapies. In comparison to traditional chemotherapies, ADCs are designed to specifically deliver a potent cytotoxic agent to tumor-associated antigens (TAAs) expressed on cancer cells, generally leading to a favorable therapeutic window.1
Claudin18.2, a member of the tight junction protein family, is a suitable TAA for ADC targeting. In healthy tissue, Claudin18.2 expression is restricted to epithelial cells of the gastric mucosa.2 However, Claudin18.2 expression is frequently abnormal during the onset and progression of various malignant tumors. Claudin18.2 is highly and stably expressed in multiple solid tumors, including gastric cancer, pancreatic cancer, esophageal cancer, lung cancer, colorectal carcinoma, and hepatocellular carcinoma.2,3,4 When malignant transformation of epithelial cells occurs, loss of cell polarity leads to the exposure of Claudin18.2 epitopes on the cell surface.5 In addition, Claudin18.2 has been shown to be involved in the proliferation, differentiation, and migration of tumor cells.6
Claudin18.2 is a clinically validated target for antibody-based therapies for gastric cancer. Zolbetuximab is a chimeric immunoglobulin G subtype 1 (IgG1) monoclonal antibody that specifically binds to Claudin18.2,7 and it is currently being investigated in multiple clinical trials in combination with chemotherapies. Initial results from 2 phase 3 studies showed that zolbetuximab in combination with either CAPOX or mFOLFOX6 significantly improved progression-free and overall survival compared to chemotherapy alone in patients with Claudin18.2-positive, human epidermal growth factor receptor-2 (HER2)-negative, locally advanced unresectable or metastatic gastric, and gastroesophageal junction cancer.8,9 In addition, CT041, a chimeric antigen receptor T cell modality, also demonstrated promising efficacy with an acceptable safety profile in heavily pretreated patients with Claudin18.2-positive digestive system cancers, specifically in those with gastric cancer.10
CMG901 is a Claudin18.2-targeting ADC consisting of a humanized anti-Claudin18.2 IgG1 antibody (CM311), a highly cytotoxic microtubule-disrupting agent monomethyl auristatin E (MMAE), and a protease-cleavable valine-citrulline (vc) dipeptide linker that covalently attaches MMAE to CM311 with a drug-to-antibody ratio of approximately 4. The aim of this study is to investigate the preclinical characteristics of CMG901, including the mechanisms of action, pharmacological activity, pharmacokinetics, and safety profiles in cynomolgus monkeys and rats.
Results
Claudin18.2 expression
Differential expression analysis for the specific isoform revealed that Claudin18.2 was upregulated in stomach adenocarcinoma, pancreatic cancer, lung adenocarcinoma, esophageal cancer, colon cancer, and bile duct cancer (Figure S1A). Claudin18.1 was expressed in esophageal carcinoma, lung adenocarcinoma, lung squamous cell carcinoma, stomach adenocarcinoma, and pancreatic adenocarcinoma (Figure S1B). In-depth mRNA expression data analysis demonstrated a positive ratio of Claudin18.2 expression in stomach and pancreatic cancer in comparison to corresponding normal tissues of 13.39% (60/448) and 30.73% (55/179) (Figure S1C), respectively. In normal tissues, Claudin18.2 mRNA was high mainly in stomach and gallbladder (Figures S1A and S1D). To identify the Claudin18.2 protein expression in solid tumors and normal tissues, we used different Claudin18.2 antibody clones, including a proprietary antibody 8-1 v8 and two from commercial sources, EPR19202-244 and 43-14A. The results showed that clones 8-1 v8 and EPR19202-244 were specific for Claudin18.2, while 43-14A detected both Claudin18.1 and Claudin18.2 isoforms (Figures S1E and S1F). Next, we investigated the Claudin18.2 expression in several normal vital organs, including heart, liver, spleen, lung, kidney, pancreas, and stomach, and demonstrated a significantly high and restricted expression of Claudin18.2 in stomach (Figure S1G). Claudin18.2 expression was then evaluated in a human cancer tissue microarray, and the results showed a significant heterogeneous expression pattern in gastric and pancreatic cancer (Figures S1H and S1I). Prevalence of Claudin18.2 in gastric cancer (≥20% tumor cell positivity [TC+], ≥2+ staining intensity) and pancreatic cancer (≥20% TC+ ≥2+) was 39.29% and 31%, respectively. These results suggest that Claudin18.2 represents a promising target for solid tumors due to its selective expression patterns.
Next, we also tested the effects of Claudin18.2 on cancer cell proliferation; knockdown of Claudin18.2 was conducted in human pancreatic cancer cell lines (BxPC-3 and CFPAC-1) (Figures S1J and S1K). As CCK-8 assays showed, knockdown of Claudin18.2 displayed no effects on cell proliferation (Figures S1L and S1M), suggesting that Claudin18.2 may not play a direct role in regulating cancer cell proliferation.
Binding, internalization, and trafficking into lysosome of CMG901
To provide a valuable therapeutic option for patients with Claudin18.2-positive tumors, we generated a Claudin18.2-ADC, CMG901, composed of a Claudin18.2-specific antibody CM311, a cleavable linker, and the cytotoxic payload MMAE, for the treatment of GEJ and pancreatic cancer.
We constructed Claudin 18.1 and Claudin18.2-expressing stable cell lines, including HEK293-hCLDN18.1, KATOIII-hCLDN18.2, 3T3-hCLDN18.2, and HEK293-hCLDN18.2. Unfortunately, the flow cytometry-compatible antibodies that specifically recognize Claudin18.1 are currently unavailable. Therefore, Claudin 18.1-specific reverse-transcription PCR was used to confirm its expression in HEK293-hCLDN18.1 cells. Both the specific 780 bp Claudin18.2 band and the 504 bp Claudin18 band were detected from HEK293-Claudin18.2 and KATOIII cells. In contrast, HEK293-Claudin18.1 cells only amplified the 504 bp Claudin18 fragment (Figure S2A). The results showed that HEK293-hCLDN18.1 cells specifically expressed Claudin18.1, not Claudin18.2. Claudin18.2 expression on the engineered stable cell lines was detected using flow cytometry with Claudin18.2-specific antibody.
To assess the ADC’s binding activity and specificity, we investigated the binding of CMG901 to cell lines KATOIII-hCLDN18.2, HEK293-hCLDN18.2, and HEK293-hCLDN18.1 using flow cytometry. CMG901 bound specifically to KATOIII-hCLDN18.2 and HEK293-hCLDN18.2 in a concentration-dependent manner with high affinity (half maximum effective concentration [EC50] = 2.44 to 6.43 nM) (Figures 1A and 1B). CM311 also showed similar binding ability than CMG901 (Figures 1A and 1B). However, no binding was observed with HEK293-hCLDN18.1 cells (Figure S2B). Comparing the binding to Claudin18.2 of CMG901 and CM311 revealed that MMAE conjugation had no significant impact on its binding affinity. Moreover, we also assessed the binding activities of CM311 and zolbetuximab analog to 3T3 cells expressing different levels of human Claudin18.2. For target cells with high expression level, CM311 bound to target cells with comparable affinity in comparison with zolbetuximab analog (EC50 1.2 vs. 2.2 nM) (Figure 1C). In contrast, for Claudin18.2-low cells, CM311 showed much higher affinity to target cells (Figure 1D). These results confirm that CMG901 binds specifically to Claudin18.2-positive, but not to Claudin18.1-positive, cells and with higher affinity than zolbetuximab analog.
Figure 1.

Binding, internalization, and trafficking into lysosome of CMG901
(A and B) Binding of CMG901 (anti-CLDN18.2 ADC), CM311 (anti-CLDN18.2 mAb), anti-KLH hIgG1, and anti-KLH hIgG1 ADC on human Claudin18.2-expressing KATOIII (A) or HEK293 (B) cells. Data are represented as mean ± SD (n = 2).
(C and D) Binding of CM311 and zolbetuximab on 3T3-hCLDN18.2High cells with high Claudin18.2 expression (C) or 3T3-hCLDN18.2Dim cells with low Claudin18.2 expression (n = 1).
(E) Endocytosis rate of CMG901 or CM311 (n = 1).
(F) Tracing of CMG901 or CM311 entering into the lysosome of KATOIII-hCLDN18.2 (n = 1). (G) Colocalization of CMG901 (red) and the lysosome marker LAMP-1 (green).
The antitumor activity of ADCs is not only dependent on their high affinity to the target but also on their efficient internalization and trafficking to the lysosome for linker cleavage and payload release.11 Therefore, the next step was to assess CMG901 intracellular internalization. At 2 h after incubation with cells, the surface of CMG901 or CM311 decreased in a time-lapse manner (Figures 1E and S2C) and internalized rapidly into the cells with a ratio of nearly 25% (Figure 1E), and a saturation limit was reached at around 4 to 6 h. We also used pHrodo, a highly specific endocytosis detection dye, to quantitatively assess the temporal changes of CMG901 or CM311 entering lysosomes. The results showed that an increasing amount of CMG901 or CM311 was internalized into the lysosomes over time (Figures 1F and S2D). Furthermore, the subcellular localization of CMG901 or CM311 to the lysosome was detected by confocal microcopy, and the results showed that CMG901 or CM311 colocalized with the lysosome marker lysosomal-associated membrane protein 1 (LAMP-1) (Figure 2E). The aforementioned results demonstrate that CMG901 and CM311 are being internalized and trafficked into lysosomes upon specific binding to Claudin18.2-positive cells.
Figure 2.

In vitro cytotoxicity of CMG901
(A and B) Cytotoxicity of CMG901, CM311, anti-KLH hIgG1, and anti-KLH hIgG1 ADC on KATOIII-hCLDN18.2 (A) and HEK293-hCLDN18.2 (B). Data are represented as mean ± SD (n = 3).
(C and D) ADCC of CMG901, CM311, anti-KLH hIgG1, and anti-KLH hIgG1 ADC on KATOIII-hCLDN18.2 (C) and HEK293-hCLDN18.2 (D). Data are represented as mean ± SD (n = 2).
(E and F) CDC of CMG901, CM311, anti-KLH hIgG1, and anti-KLH hIgG1 ADC on KATOIII-hCLDN18.2 (E) and HEK293-hCLDN18.2 (F). Data are represented as mean ± SD (n = 2).
(G) Bystander killing effect of CMG901 on Claudin18.2-negative organoids.
(H) Bystander killing effect of CMG901 on Claudin18.2-negative cell lines.
To investigate the binding affinity of CMG901 to Fcγ receptors (FcγRs), including FcγRI, FcγRIIAH131, FcγRIIBC, FcγRIIIAV158, FcγRIIIAF158, and FcRn, C1q, and evaluate the impact of payload conjugation by comparing binding activity to FcγR, FcRn, and C1q of CMG901 and CM311, binding affinities were measured by surface plasmon resonance. KD values of CMG901 binding to FcγRI, FcγRIIAH131, FcγRIIIAV158, and FcγRIIIAF158 were 5.95 nM, 6.29 μM, 125.6 nM, and 489.9 nM, respectively, and CMG901 bound weakly to FcγRIIBC; KD values of CM311 binding to FcγRI, FcγRIIAH131, FcγRIIIAV158, and FcγRIIIAF158 were 7.23 nM, 2.77 μM, 78.93 nM, and 330.30 nM, respectively, and CM311 bound weakly to FcγRIIBC. At pH 6.0, KD value of CMG901 binding to FcRn was 1.73 μM; KD value of CM311 binding to FcRn was 1.53 μM. At pH 7.4, both CMG901 and CM311 bound weakly to FcRn. Affinity of CMG901 to C1q was 116.4 nM, and affinity of CM311 to C1q was 47.92 nM (Table S1). The aforementioned results show that both CMG901 and CM311 are capable of binding to FcγRI, FcγRIIAH131, FcγRIIIAV158, FcγRIIIAF158, and C1q and bind weakly to FcγRIIB. The binding of CMG901 or CM311 to FcRn is pH dependent. Furthermore, the binding activities of FcγR, FcRn, and C1q are comparable before and after the payload conjugation.
In vitro cytotoxicity of CMG901
Following internalization and trafficking into lysosomes, the ADC releases its cytotoxic payload upon cleavage by the protease cathepsin B and induces cell death.11 To assess the cytotoxic activity of our ADC, serial dilutions of CMG901 or CM311 were incubated with Claudin18.2- or Claudin18.1-expressing cells, and cell viability was measured. The results showed a concentration-dependent cytotoxicity of CMG901 on KATOIII-hCLDN18.2 and HEK293-hCLDN18.2, with a half-maximum inhibitory concentration (IC50) of 0.13 and 0.14 nM, respectively; however, CM311 did not show any cytotoxicity in Claudin18.2-expressing cells up to the highest concentration of 66.67 nM (Figures 2A and 2B). CMG901 was not cytotoxic to HEK293-hCLDN18.1 (Figure S3A). In addition to MMAE, another payload DXd (exatecan derivative) with a different mechanism of antitumor activity was also conjugated to CM311 (CM311-DXd) to test its cytotoxicity. Compared with CM311-DXd, CMG901 showed similar potency on HEK293-hCLDN18.2 but more potency on KATOIII-hCLDN18.2 (Figures S3B and S3C). These findings indicate that CMG901 is a potent and target-specific anti-Claudin18.2 ADC.
The parental antibody CM311 is a humanized IgG1, with a wild-type-constant fragment (Fc) region and full effector functionalities such as , antibody-dependent cellular cytotoxicity (ADCC) and complement-dependent cytotoxicity (CDC) with the potential to inhibit cancer cell growth.12 To test the ADCC activity of CMG901 and CM311, KATOIII-hCLDN18.2 and HEK293-hCLDN18.2 cells were incubated with effector cells (peripheral blood mononuclear cells [PBMCs]). CMG901 and CM311 induced comparable concentration-dependent ADCC activity against both KATOIII-hCLDN18.2 (IC50 value of 0.80 vs. 0.37 nM) and HEK293-hCLDN18.2 (IC50 value of 4.77 vs. 2.66 nM) (Figures 3C and 3D); however, both CMG901 and CM311 did not show any ADCC activity on HEK293-hCLDN18.1 cell (Figure S3D). We also compared ADCC activity with zolbetuximab analog, and CM311 showed more potent ADCC activity on KATOIII-hCLDN18.2 cells (Figure S3E). CDC activity of CMG901 and CM311 was also assessed on Claudin18.2- and Claudin18.1-expressing cells, and lysis (%) was quantified. CMG901 and CM311 showed comparable concentration-dependent CDC activity on KATOIII-hCLDN18.2 (IC50 value of 3.66 vs. 3.71 nM) and HEK293-hCLDN18.2 (IC50 value of 4.77 vs. 2.66 nM) (Figures 2E and 2F), not on HEK293-hCLDN18.1 cell (Figure S3F). Compared with zolbetuximab analog, CM311 also showed more potent CDC activity on KATOIII-hCLDN18.2 cells (Figure S3G).
Figure 3.

In vivo antitumor activity of CMG901
(A) Tumor volume of the PDX model STO#025. Data are represented as mean ± SEM (n = 10).
(B) Tumor volume of the PDX model PAN#026. Data are represented as mean ± SEM (n = 10).
(C) Tumor volume of the PDX model STO#580. Data are represented as mean ± SEM (n = 10).
(D) Tumor volume of the PC-PDOX model. Data are represented as mean ± SEM (n = 6).
(E) Efficacy study of the combination of CMG901 and anti-PD-1 on HSC-NPG humanized PDX model STO#025. Data are represented as mean ± SEM (n = 6).
(F) Efficacy study of the combination of CMG901 and anti-PD-1 on MC38 syngeneic mouse model. MC38-hCLDN18.2 xenografts were grown subcutaneously in C57BL/6 mice and treated with different doses of CMG901 and anti-PD-1. Data are represented as mean ± SEM (n = 5).
(G) CD8+T cell infiltration in tumors. Data are represented as mean ± SEM (n = 3–4).
(H) NK cell infiltration in tumors. Data are represented as mean ± SEM (n = 3–5). ∗indicates statistical significance compared to the vehicle group, #denotes significance compared to the CM311 group. ∗p & #p < 0.05, ∗∗p & ##p < 0.01, ∗∗∗p & ###p < 0.001.
The valine-citrulline (Val-Cit, vc) dipeptide linker, which is cleaved preferentially by lysosomal proteases, has been commonly used to conjugate the microtubule inhibitor payload MMAE.13 The released payload MMAE can enter into surrounding CLDN18.2-negative cells via passive diffusion and exert bystander killing activity.14 To confirm the bystander activity of CMG901, we incubated Claudin18.2-positive organoids and Claudin18.2-negative organoids with serially diluted CMG901 for 6 days and measured the remaining live cell number to analyze the ratio between positive and negative cells using flow cytometry. The results showed that CMG901 killed both Claudin18.2-positive and Claudin18.2-negative organoids (Figure 2G; Figure S3H). In comparison, anti-KLH hIgG1 ADC had no effect on the viability of Claudin18.2-positive or negative cells. Furthermore, we also confirmed bystander killing in Claudin18.2-positive KATOIII-hCLDN18.2 and Claudin18.2-negative KATOIII cocultures (Figure 2H). All these results demonstrated CMG901 kills tumor cells through the release of the cytotoxic payload, Fc-mediated ADCC and CDC activities, and bystander killing.
To further verify the specificity and ADC cytotoxicity of CMG901, ADCC, and CDC effect of the antibody CM311, pancreatic cancer organoids with Claudin18.2 endogenously expression (Claudin18.2 wild type [WT]) and its knockout strain (Claudin18.2 KO) were used (Figure S3I). CMG901 was given to both strains of organoids after 48 h from seeding procedure; after 5 days of incubation, cell viability was tested. The results showed that CMG901 exerted robust tumor cell killing effect on Claudin18.2 WT organoids, while it has minor effect on Claudin18.2 KO organoids (Figure S3J). We also evaluated the ADCC effects of CM311, the antibody part of CMG901, in a PBMC-organoid co-culture system. The relative cell inhibition rates for Claudin18.2 WT organoids on different concentrations of 0.1, 1, and 10 μg/mL were 11.82%, 18.65%, and 29.62%, respectively, while the inhibition rate was below 10% on Claudin18.2 KO organoids (Figure S3K). For the CDC effects, CM311 also showed robust CDC activity on Claudin18.2 WT organoids, while no CDC effect was observed on Claudin18.2 KO organoids (Figure S3L). To sum up, CMG901 exerts potent in vitro antitumor activity by ADC cytotoxicity, but ADCC and CDC killing effect is only on Claudin18.2-positive organoids.
In vivo antitumor activity of CMG901
Next, we tested the in vivo antitumor activity of CMG901 in gastric STO#025 and pancreatic patient-derived xenograft (PDX) PAN#026 models. The detected CLDN18 RPKM values for STO#25 and PAN#026 were 721.43 and 731.178, respectively. Tumor fragments of about 15–30 mm3 in size were implanted subcutaneously into nude mice, and vehicle, CM311 (3 mg/kg), CMG901 (0.3, 1, and 3 mg/kg), and anti-KLH hIgG1 ADC (3 mg/kg) were intravenously injected once a week for 3 weeks. CMG901 treatment resulted in dose-dependent tumor growth inhibition (TGI) or regression while anti-KLH hIgG1 ADC had no antitumor activity in the tested models. In a gastric PDX model, 3 mg/kg of CMG901 led to tumor regression (−87.9% growth), while 1 mg/kg of CMG901 resulted in a significant TGI of 68% (p < 0.0001; Figure 3A). In a pancreatic PDX model, CMG901 treatment at 3 mg/kg led to tumor stasis (−9.1% growth), while 1 mg/kg led to a significant TGI of 59.6% (p < 0.0001; Figure 3B). In these studies, the unconjugated antibody CM311 was not effective at comparable doses.
In addition, we also explored the antitumor effect of CMG901 on gastric cancer PDX model STO#580, with a CLDN18 RPKM of 217.07, which is lower than that in STO#25. In this model, administration of CMG901 at 3 mg/kg resulted in significant TGI, while treatment with CMG901 at doses of 0.3 and 1 mg/kg and CM311 at 3 mg/kg showed modest TGI compared with the vehicle group (Figure 3C). The results from tumor suppression effects of CMG901 and CM311 at the same dosage on two PDX models STO#580 and STO#025 with different levels of Claudin18.2 expression demonstrated that for tumors with high Claudin18.2 expression, the antitumor efficacy of CMG901 and CM311 is superior to that in tumors with low Claudin18.2 expression (Table S2). This observation suggests a correlation between the expression level of Claudin18.2 and the efficacy of CMG901. No body weight loss was observed in animals following CMG901 treatment in all models (Figures S4A–S4C), indicating a favorable tolerability.
To further test the in vivo ADCC and CDC effects of CM311 and zolbetuximab, we generated hematopoietically humanized NPG mice xenografted with tissues from Claudin18.2-positive pancreatic cancer patient-derived organoid xenograft (PC-PDOX) model (Figure S4D). It was shown that CMG901 significantly suppressed tumor growth. Furthermore, marked reductions of tumor volume in CM311 with 48.05% and zolbetuximab with 47.96% were observed (Figure 3D), respectively. However, mild weight loss was observed in this model, possibly due to the transplant rejection (Figure S4E). These findings indicate that both zolbetuximab and CM311 exert robust antitumor activity in Claudin18.2 patient-derived organoid xenograft model through ADCC or CDC effects, and the dosage and administration frequency are critical for the antitumor effects.
Microtubule-depolymerizing agents like dolastatins or MMAE have been reported to induce antitumor immunity.15 The therapeutic efficacy of CMG901 and its induction of antitumor immunity were explored in PDX STO#025 inoculated into humanized Hu-HSC-NPG mice. Compared with CMG901 monotherapy, the combination of CMG901 (0.5 mg/kg) and anti-PD-1 (10 mg/kg) showed a marginal improvement in antitumor effects (TGI −66.7% vs. −59.0%; Figure 3E). To exclude any potential artificial effects in immunocompromised mice, we next investigated the combination antitumor effect in an immunocompetent mouse model by stably expressing human Claudin18.2 in a murine cancer cell line. CMG901 (at doses of 0.1, 0.3, and 1 mg/kg) suppressed tumor growth in a dose-dependent manner (TGI −35.42%, −72.34%, and −92.50%; Figure 3F). Compared to CMG901 monotherapy at a dose of 1 mg/kg, the combination with anti-PD-1 (0.1 or 0.3 mg/kg) showed no significant enhanced antitumor effect (0.1 mg/kg TGI −86.62%, p = 0.3543; 0.3 mg/kg TGI −103.93%, p = 0.2921). However, the combination of 0.3 mg/kg CMG901 and 0.3 mg/kg anti-PD-1 induced tumor regression (TGI −126.80%, p = 0.0035; Figure 3F). At the end of experiment, the tumors from control, anti-PD-1 0.3 mg/kg, CMG901 0.3 mg/kg, and the combination of CMG901 0.3 mg/kg and anti-PD-1 0.3 mg/kg, were excised, and the infiltrated immune cells were analyzed using flow cytometry. Compared to CMG901 monotherapy, the combination increased the ratio of intra-tumoral CD8+ T cells and natural killer (NK) cells (Figures 3G and 3H). The proportion of other immune cells, including CD3+, CD4+, and Foxp3+CD4+, was not significantly different between CMG901 monotherapy and the combination group (Figures S4F–S4I). The findings provide valuable insights into the effectiveness of CMG901 monotherapy and offer a rationale for drug combinations involving immunotherapy.
Pharmacokinetic assessment of CMG901 in cynomolgus monkeys and rats
Cross-species binding of CMG901 to human, cynomolgus monkey, and rat Claudin18.2-expressing HEK293 cells showed comparable binding affinities across the different species (Figures 1A, 1B, S5A, and S5B). A tissue cross-reactivity study revealed binding of CMG901 and CM311 to stomach tissues from human, cynomolgus monkey, and rat (Figure 4A; Table S3). It was found that there was specific binding of test article CMG901 and CM311 to stomach tissues (3/3 donors) from human, cynomolgus monkey, and Sprague-Dawley (SD) rat. In addition, test article-related staining was observed in 1 human liver tissue sample (1/3 donors). No specific staining on other human, cynomolgus monkey, or SD rat tissues was observed for CMG901 and CM311. These results indicated that CMG901 was pharmacologically active in both cynomolgus monkey and rat as per ICHS6 R (1) guideline requirements. Pharmacokinetic/toxicokinetics (TK) assessment of CMG901 was performed in cynomolgus monkeys and rats as part of Good Laboratory Practice (GLP)-compliant 10-week repeat-dose toxicity with 8-week recovery period studies.
Figure 4.

Pharmacokinetics study of CMG901 in cynomolgus monkeys and rats
(A) Tissue cross-reactivity (TCR) study of CMG901 or CM311 in human, cynomolgus monkey, and rat.
(B) Mean concentration-time curves of CMG901, total antibody (TAb), and MMAE in the CMG901 dose groups (1, 3, and 6 mg/kg) after the first dose by intravenous injection in cynomolgus monkeys for 10 weeks. Data are represented as mean ± SD (n = 5).
(C) Mean concentration-time curves of CMG901, TAb, and MMAE in the CMG901 dose groups (2.5, 5, and 10 mg/kg) after the first dose by intravenous injection in rats for 10 weeks. Data are represented as mean ± SD (n = 5).
(D) Precipitated radioactivity distributions in different tissues/body fluids after a single intravenous dose of 125I-CMG901 in rats (n = 6). (E) Plasma stability of CMG901 in human, cynomolgus monkey, and rat plasma.
CMG901 at 1, 3, and 6 mg/kg, CM311 at 6 mg/kg, or MMAE at 0.06 mg/kg (molar equivalent to 3 mg/kg CMG901) were intravenously injected into cynomolgus monkeys, and no significant gender-related difference was observed in mean systemic exposure (AUC0-t) and mean peak concentration (Cmax) of CMG901, TAb (total antibody), CM311, and MMAE. After the first dose, the exposure (AUC0-t and Cmax) of CMG901 and TAb increased proportionally with the dose levels, while that of MMAE increased greater than the dose level increment (the power exponent β for AUC0-t was 1.240 and 90% confidence interval was 0.889–1.590; the power exponent β for Cmax was 1.149 and 90% confidence interval was 0.914–1.384; Figure 4B). The half-life (t1/2) of CMG901 between dosages was 68.2–143 h. After the third dose, AUC0-t of CMG901, TAb, and MMAE increased greater than the dose level increase; Cmax of CMG901 and TAb increased dose proportionally, and that of MMAE increased greater than the dose level increment (Figure S5C). After 4 repeated injection every 3 weeks (q3w), no obvious accumulation was observed by comparing exposure parameters on day 1 and day 43. Compared with the exposure parameters (Cmax and AUC0-t) of 6 mg/kg CM311, Cmax of TAb at 6 mg/kg CMG901 was similar to that of 6 mg/kg CM311, but AUC0-t was significantly lower at 6 mg/kg CMG901 on day 1 and day 43 (Figure S5D). In addition, the t1/2 and MRTlast of 6 mg/kg CMG901 were shorter than those of 6 mg/kg CM311, suggesting the clearance is faster at 6 mg/kg CMG901. Compared with the exposure parameters (Cmax and AUC0-t) of 0.06 mg/kg MMAE, Cmax of MMAE at 3 mg/kg CMG901 was approximately 2% of that at 0.06 mg/kg MMAE, and AUC0-t of MMAE at 3 mg/kg CMG901 was approximately 30%–40% of that in the MMAE group (Figure S5E). In addition, the Tmax of MMAE at 3 mg/kg CMG901 (24–48 h) was significantly longer compared to that at 0.06 mg/kg MMAE (immediately after dosing), which indicated that MMAE was slowly released from CMG901 in cynomolgus monkeys.
After repeated intravenous injection of either CMG901 at 2.5, 5, and 10 mg/kg, CM311 at 10 mg/kg, or MMAE at 0.2 mg/kg (molar equivalent to 10 mg/kg CMG901) in rats, no significant gender-related difference was observed in AUC0-t and Cmax of CMG901, TAb, CM311, and MMAE, while statistical gender difference was observed in AUC0-t of MMAE in the low-dose group (2.5 mg/kg) after the last dose (p < 0.05, the ratio was 0.415). After the first dose, Cmax and AUC0-t of CMG901 and TAb, and Cmax of MMAE increased proportionally with the dose level increase, and AUC0-t of MMAE increased greater than the dose level increase (the power exponent β for AUC0-t was 1.512 and 90% confidence interval was 1.402–1.621; Figure 4C). After the last dose, Cmax of CMG901 increased dose proportionally, and Cmax of TAb and MMAE increased greater than the dose level increase (Figure S5F); AUC0-t of CMG901, TAb, and MMAE increased in an approximate dose-proportional manner or greater than the dose level increase. After 4 q3w doses, no obvious accumulation was observed. Compared with the exposure parameters (Cmax and AUC0-t) of 10 mg/kg CM311, Cmax of TAb in 10 mg/kg CMG901 was similar to that of 10 mg/kg CM311 (Figure S5G). However, AUC0-t was significantly lower in 10 mg/kg CMG901 in the first and last doses, suggesting the elimination rate is faster in the CMG901 group. Compared with the main TK parameters at 0.2 mg/kg MMAE, Cmax of MMAE in 10 mg/kg CMG901 was approximately 2.5% of that at 0.2 mg/kg MMAE (Figure S5H). The average Tmax of MMAE in 10 mg/kg CMG901 (24 h) was significantly delayed compared to the MMAE group (immediately after dosing).
A tissue distribution and excretion study in rats was conducted utilizing 1 mg/kg of 125I CMG901 and 125I-CM311. The results showed that 125I-CMG901 and 125I-CM311 were mainly distributed in the vascular system, followed by highly perfused organs (lung, ovaries, liver, adrenal glands, kidneys, and spleen) and stomach (Figure 4D; Figure S5I). A lower level of radioactivity was also observed in the heart, pancreas, testicle, bone, fat, and muscle. No gender difference was observed in distribution patterns. The concentrations of 125I-CMG901 and 125I-CM311 in the brain were low, indicating that 125I-CMG901 and 125I-CM311 cannot easily penetrate the blood-brain barrier. Plasma stability results showed CMG901 was stable in human and cynomolgus monkey plasma as evidenced by no more than 1% of the maximum theoretical amount of MMAE from CMG901 at 37°C over 21 days (Figure 4E). CMG901 was less stable in rat plasma, generating almost 6% of the maximum theoretical amount of MMAE from CMG901.
Toxicity of CMG901 in cynomolgus monkeys and rats
A 16-week non-GLP study was conducted in cynomolgus monkeys to evaluate the safety of CMG901 and CM311. CMG901 doses of 3, 8, and 12 mg/kg were administered q3w for five times (on day 1, 22, 43, 64, and 85), with CM311 dosed at 12 mg/kg for comparison. Higher doses of CM311 at 30 and 100/200 mg/kg were also tested. Clinical signs, reduction in body weight, and hematology changes associated with reduced hematopoiesis, including reduced white blood cells (WBCs) and red blood cells (RBCs), were observed at a dose of 12 mg/kg CMG901. Clinical chemistry parameters, including creatine kinase, lactate dehydrogenase, alanine transaminase (ALT), and aspartate aminotransferase, were occasionally elevated at CMG901 doses ≥8 mg/kg and CM311 dose of 12 mg/kg. Changes in immunophenotype were observed in all CMG901-treated groups and CM311 doses of ≥30 mg/kg. Pathological changes in cecum and lymph nodes at 8 mg/kg and in lungs at 12 mg/kg were observed in the CMG901-treated group, while colon and cecum showed pathological changes at CM311 dose of 100/200 mg/kg.
In a 10-week study in cynomolgus monkeys, CMG901 doses of 1, 3, and 6 mg/kg were administered q3w for four times (on day 1, 22, 43, and 64). CM311 and MMAE were dosed at 6 and 0.06 mg/kg, respectively, corresponding to their amount in 6 and 3 mg/kg CMG901, respectively. Decreases in red cell mass, including RBCs, hemoglobin, hematocrit, and mean corpuscular hemoglobin, associated with a decreased reticulocyte count (#RET) and reticulocyte percentage (RET%), were noted at doses of 6 mg/kg CMG901 and 0.06 mg/kg MMAE. Decreases in total leukocytes, lymphocytes, and monocytes were also observed in both groups. Neutrophils were decreased initially on day 7 (Table S4) but showed an increase on day 21 (Table S5) and 70 (Table S6). Microscopic examination revealed that hematology changes were associated with decreased hematopoietic cells in the bone marrow (CMG901 ≥ 3 mg/kg; MMAE 0.06 mg/kg), lymphocytes necrosis, and increased thymus mitosis (CMG901 6 mg/kg; MMAE 0.06 mg/kg). All changes were found to be reversible. The highest non-severely toxic dose (HNSTD) of CMG901 in cynomolgus monkeys in this study was determined to be 6 mg/kg.
In a 6-week dose-ranging finding (DRF) study conducted in rats (q3w × 2 IV), treatment-related adverse effects were observed at all doses of CMG901 (7.5, 15, and 30/20 mg/kg administered on day 1 and 22). Mortality occurred at the highest tested dose of 30 mg/kg, resulting in a dose reduction to 20 mg/kg for the second dose. Adverse clinical signs and reduced body weight gain were observed at 30/20 mg/kg CMG901. Doses of CMG901 ≥ 15 mg/kg resulted in a significant decrease in RBC mass and associated changes, with a decrease in WBCs observed at the 30/20 mg/kg dose. Additionally, doses of CMG901 ≥ 15 mg/kg q3w × 2 IV resulted in increased ALT and total bile acid levels, along with a decreased albumin to globulin (A/G) ratio. No treatment-related effects were observed with CM311 administered at doses of 30/20 mg/kg, except for decreased activity in the animals.
In a 10-week repeat-dose toxicity study with an 8-week recovery period in rats, CMG901 was administered at doses of 2.5, 5, or 10 mg/kg, CM311 at 10 mg/kg, and MMAE at 0.2 mg/kg. The test articles were administered q3w (on day 1, 22, 43, and 64) for 10 weeks. Decreased activity was observed at 10 mg/kg in both CMG901 and CM311 groups, while a reduction in body weight was only observed with MMAE. Similar to the DRF study, a reduction in red cell mass and associated changes were observed at 10 mg/kg CMG901 and 0.2 mg/kg MMAE. Reduction in leukocytes was also present in both groups, with more pronounced reduction of neutrophils and eosinophils in the MMAE-treated group. In contrast, CMG901 treatment showed only reduction in eosinophils. These changes were associated with the myelosuppressive and cytotoxic effect of MMAE, resulting in decreased hematopoietic cells in the bone marrow, extramedullary hematopoiesis in the spleen, and reduced lymphocytes in the thymic cortex. Microscopic changes in mammary gland and degenerative changes in kidney of males at a 10 mg/kg CMG901 dose were attributed to MMAE and CM311, respectively. A reduction in testes and epididymides weight and size at doses ≥5 mg/kg and microscopic changes in these organs at doses ≥2.5 mg/kg were observed in CMG901 groups. All changes except those in testes and epididymides were reversible at the end of the recovery period (Table S7). A dose of 10 mg/kg was identified as the HNSTD in rats.
Discussion
There are 1.1 million newly diagnosed gastric cancer cases and 0.77 million deaths worldwide annually. For pancreatic cancer, there are approximately 495,773 new cases and 466,003 deaths worldwide.16 Systemic chemotherapy remains the mainstay for the treatment of advanced gastric and pancreatic cancer. However, targeted therapy and immunotherapy have revolutionized the treatment of various cancers in the past decade. HER2-targeted therapies and PD-1-targeted immunotherapies show responses in specific population in patients with gastric cancer,17 but about 80%–90% of patients with gastric and pancreatic cancer respond poorly to PD-(L)1 antibody treatment18,19 and HER2-targeted therapies are currently approved for only a small (<20%) segment of gastric cancer patients. The five-year survival rates of gastric and pancreatic cancers are a mere 35.1% and 7.2%, respectively, with the standard-of-care treatments in China.20,21
CMG901 is a CLDN18.2-specific targeted cancer therapy with a highly potent MMAE payload attached to a Claudin18.2 antibody. The ADC effectively kills tumor cells through three mechanisms: (1) release of the cytotoxic payload (MMAE) after internalization into tumor cells and lysosomal trafficking, (2) the induction of ADCC and CDC effects of the immune system, and (3) bystander killing activity of the released MMAE warhead. Compared to a zolbetuximab analog,3 CMG901’s unconjugated antibody specifically binds to Claudin18.2 with higher affinity and leads to more potent cell killing by ADCC and CDC, in vitro. Furthermore, upon internalization of CMG901 by tumor cells, MMAE is subsequently released inside of tumor cells. The payload is highly cytotoxic and has the capability of bystander killing of nearby Claudin18.2-negative tumor cells. In animal models of gastric and pancreatic cancers, CMG901 exhibited much stronger antitumor activity in comparison to unconjugated CLDN18.2 antibody, CM311, or zolbetuximab analog. Furthermore, CMG901 has shown favorable tolerability and safety in our preclinical animal studies. In comparison to the combination of monoclonal antibodies and chemotherapies, ADCs are thought to deliver cytotoxic agents specifically to tumor cells, thus minimizing systemic toxicity in normal tissues and increasing therapeutic index.1 In repeated-dose toxicity studies, the HNSTD of CMG901 was determined as 6 mg/kg and 10 mg/kg in cynomolgus monkeys and rats, respectively. These dose levels are much higher than the lowest efficacious dose (0.3 mg/kg) determined in in vivo animal efficacy studies. The major findings are hematological changes associated with myelosuppressive and cytotoxic effects of MMAE,22 which are recoverable. This demonstrates that CMG901 can potentially improve antitumor efficacy while minimizing toxicity.
Claudin18.2 is highly expressed in various solid tumors, including gastric, pancreatic cancer, and esophageal adenocarcinoma, while its expression is limited in normal gastric mucosa. The overall positivity rate of Claudin18.2 in gastric and esophageal adenocarcinoma ranges from 21% to 95%, with moderate to high expression (≥2+) having a positive rate of 31%–86%.23,24,25 In our study, using a Claudin18.2-specific antibody, we identified a positive rate (≥2+ ≥20%) of 39% in a gastric cancer tissue array from a commercial source. With 43-14A, an immunohistochemical antibody used as a companion diagnostic for a Claudin18.2-targeting therapy, positive rates were reported as 44.8% (≥2+ ≥50%), 49% (≥2+ ≥40%), and 38.5% (≥2+ ≥75%).3,26,27 Kubota et al. reported that Claudin18.2-positive (≥2+ ≥75%) was identified as 24.0% with almost equal distribution in the four molecular subtypes or combined positive score subgroups.28 It can be concluded that different combinations of detecting antibody and varying cutoff values for interpretation yielded divergent results. In contrast to the reported homogeneous expression pattern of Claudin18.2 in gastric cancer,29 we found its expression to be significantly heterogeneous, evidenced by diverse staining intensities with varying ratios. Therefore, Claudin18.2 expression needs to be interpreted with caution.
Certain chemotherapies can induce immunogenic cell death in tumor cells, releasing antigens that stimulate the immune system.30 Microtubule inhibitors, including vinblastine and auristatin analogs dolastatin 10, MMAE, and MMAF, have been reported to act as potent activators of dendritic cell (DC) maturation and antigen response.30,31 Accordingly, ADCs that release cytotoxic drugs can also enhance host immunity. Trastuzumab deruxtecan, consisting of trastuzumab and the DNA topoisomerase I inhibitor DXd, increases DC and CD8+ T cell tumor infiltration, upregulates tumor cell surface programmed death-ligand 1 (PD-L1) and major histocompatibility complex class I molecules, and effectively inhibits tumor growth when combined with anti-PD-1.32 Rovalpituzumab tesirine, an ADC drug consisting of a monoclonal antibody targeting DLL3 and the DNA-damaging pyrrolobenzodiazepine, can increase Ki67+/Granzyme B+/CD8+ T cells in tumors when used in combination with PD-1 inhibitors.33 Brentuximab vedotin, an ADC drug consisting of targeting CD30 antibody and MMAE, can also increase tumor-specific immune responses by activating patient DCs, T cells, and B cells.34 Preclinical studies have shown enhanced antitumor activity when zolbetuximab is combined with anti-PD-1, and a phase 2 clinical trial (NCT03505320) is currently investigating the combined antitumor activity of zolbetuximab and PD-1 inhibition.35 In our study, CMG901, in combination with anti-PD-1, significantly suppressed tumor growth compared with CMG901 monotherapy. Therefore, combining ADCs with immune therapeutic strategies, such as immune checkpoint inhibitors PD-(L)1, can potentially overcome drug resistance and can achieve long-term benefits.
The safety and efficacy of CMG901 is currently under exploration in patients with advanced solid tumors, including gastric cancer, GEJ adenocarcinoma, and pancreatic cancer, as part of a phase 3 trial (NCT06346392). In 2022, the Food and Drug Administration granted CMG901 the fast-track designation and orphan drug designation and later received breakthrough therapy designation by the Center for Drug Evaluation in China for relapsed/refractory metastatic GEJ cancer. In summary, CMG901 has a well-defined mechanism of action and a favorable safety profile, positioning it as a potential therapeutic option for patients with advanced solid tumors.
Limitations of the study
The main limitation of this study lies in the simultaneous evaluation of both ADC (direct cytotoxicity) and ADCC activities of CMG901 in animal models. Significant differences exist in the effective dosage between ADC and unconjugated monoclonal antibodies acting through ADCC and CDC effects, making simultaneous testing of both ADC activity and ADCC and CDC activity challenging. Additionally, the combination of CMG901 with anti-PD-1 showed synergistical TGI compared with CMG901 monotherapy. Compared to CMG901 monotherapy, the combination increased the ratio of intra-tumoral CD8+ T cells and NK cells. However, the detailed mechanisms underlying these effects warrant further investigation.
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Bo Chen (knybochen@keymedbio.com).
Materials availability
All unique/stable reagents generated in this study will be made available upon request to the lead contact.
Data and code availability
-
•
All data reported in this paper will be shared by the lead contact upon request.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
Acknowledgments
The authors would like to thank the staff of AstraZeneca for the critical review of the manuscript. The authors would also like to thank Dr. Qing Chu for reviewing the manuscript and Mr. Xiuqiang Sun for assisting with the production of the figures. The study was supported by Keymed Biosciences (Chengdu) Limited.
Author contributions
G.X.: conceptualization, formal analysis, supervision, investigation, methodology, and project administration. W.L.: conceptualization, data curation, formal analysis, supervision, validation, investigation, methodology, project administration, writing – original draft, and writing – review and editing. Y.W.: conceptualization, investigation, project administration, and visualization. X.W.: conceptualization, investigation, project administration, and writing – review and editing. F.L.: conceptualization, investigation, project administration, and writing – review and editing. Y.H.: investigation, formal analysis, methodology, project administration, and writing – review and editing. L.Z.: investigation, data curation, and formal analysis. Q.S.: investigation, validation, and methodology. Z.L.: investigation and methodology. C.W.: conceptualization, resource, formal analysis, investigation, methodology, and writing – review and editing. R.X.: conceptualization, resources, supervision, investigation, project administration, and writing – review and editing. B.C.: conceptualization, resources, supervision, funding acquisition, investigation, project administration, and writing – review and editing.
Declaration of interests
G.X., W.L., Y.W., Y.H., L.Z., Q.S., C.W., and B.C. are employees of Keymed Biosciences (Chengdu) Limited. The patents disclosed included WO2022078523A1 and its patent family.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Zolbetuximab | This paper | N/A |
| anti-PD-1 | This paper | N/A |
| CM311 | This paper | N/A |
| CMG901 (ADC) | Shanghai Miracogen Inc. | N/A |
| Anti-KLH hIgG1 | This paper | N/A |
| Anti-KLH-hIgG1-ADC | Shanghai Miracogen Inc. | N/A |
| Claudin18.2 antibody clones (8-1 v8) | This paper | N/A |
| Claudin18.2 antibody clones (EPR19202-244) | This paper | N/A |
| Claudin18.2 antibody clones (43-14A) | This paper | N/A |
| Human Lamp-1/CD107a antibody | R&D | MAB4800 |
| AF488-Fab’2 Goat anti-mIgG(H + L) | Jackson | 115-546-146 |
| AF594 -Fab’2 Goat anti-hIgG(H + L) | Jackson | 109-586-088 |
| Alexa Fluor 647-conjugated AffiniPure F(ab’)2 fragment Goat anti-Human IgG, Fcγ Fragment specific | Jackson ImmunoResearch | 109-606-170 |
| ChromPure Mouse IgG,whole molecule | Jackson ImmunoResearch | 015-000-003 |
| Biological samples | ||
| Peripheral blood mononuclear cells (PBMC) | AllCells Shanghai | LP181218 |
| Human Plasma | Shanghai Miracogen Inc. | N/A |
| Monkey Plasma | Shanghai InnoStar Bio Ltd. | N/A |
| Rat Plasma | Shanghai InnoStar Bio-Tech Co., Ltd. | N/A |
| Organoids | Sun Yat-sen University Cancer Center/ZOS biotechnology | N/A |
| Human normal and cancer tissue for microarray | Jingqin path | N/A |
| Frozen tissues of human, cynomolgus monkey and rat for TCR assay | Shanghai Institute of Materia Medica, Chinese Academy of Sciences. | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| ProLong™ Gold Antifade Mountant with DAPI | Thermo | P36941 |
| L-Glutamine | Sigma | G7531-100ML |
| PerfectStart® Green qPCR SuperMix | TRANS, China | AQ602-22 |
| Lipofectamine™ RNAiMAX Transfection Reagent | Thermo | 13778150 |
| Fetal bovine serum (FBS) | Excell | FSP500 |
| Bovine calf serum (BCS) | Hyclone | SH30626.03 |
| Bovine serum albumin (BSA) | Sangon Biotech | A500023-0100 |
| Triton X-100 | BBI Life Sciences | 9002-93-1 |
| 4% Paraformaldehyde fixative solution | Beyotime | P0099-500 mL |
| Trypsin solution in EDTA (0.25%) | Shanghai Basalmedia | S310KJ |
| TRIzol reagent | Tiangen, China | DP424 |
| PBS (pH7.4) | Gibco | C10010500BT |
| RPMI 1640 | Gibco | C11875500BT |
| IMDM basal medium | Gibco | C12440500BT |
| DMEM | Gibco | C11995500BT |
| Puromycin | Gibco | A1113803 |
| Collagenase | Sigma-Aldrich | C9407 |
| adDMEM/F12 | Life Technologies | 12634–034 |
| Propidium iodide (PI) | Sigma-Aldrich | P4170 |
| Cultrex reduced growth factor basement membrane extract (BME) type 2 | R&D Systems | 3536-005-02 |
| B27 supplement | Life Technologies | 17504–044 |
| Penicillin-streptomycin | Life Technologies | 15140–122 |
| N-acetyl-L-cysteine | Sigma-Aldrich | A9165 |
| Nicotinamide | Sigma-Aldrich | N0636 |
| Human EGF | PeproTech | AF-100-15 |
| Human FGF10 | PeproTech | 100–26 |
| Prostaglandin E2 | Tocris Bioscience | 2296 |
| Gastrin | R&D Systems | 3006 |
| Human complement | QUIDEL | A113 |
| HEPES | Life Technologies | 15630–056 |
| Critical commercial assays | ||
| CCK-8 kit | DOJINDO | CK04 |
| CytoTox-ONE Homogeneous Membrane Integrity Assay kit | Promega | G7891 |
| pHrodo™ Antibody Labeling Kits | Thermo | P35355 |
| cDNA reverse transcription kit | Thermo | 18080051 |
| Experimental models: Cell lines | ||
| KATOIII-hCLDN18.2 | This paper | N/A |
| HEK293-hCLDN18.2 | This paper | N/A |
| HEK293-hCLDN18.1 | This paper | N/A |
| 3T3-hCLDN18.2 | This paper | N/A |
| HEK293-cynoCLDN18.2 | This paper | N/A |
| HEK293-ratCLDN18.2 | This paper | N/A |
| HEK293-hCLDN18.1 | This paper | N/A |
| MC38-hCLDN18.2 | This paper | N/A |
| MC38 | a gift from Biocytogen Pharmaceuticals (Beijing) Co., Ltd | N/A |
| KATOIII | ATCC | HTB-103 |
| Experimental models: Organisms/strains | ||
| Pancreatic cancer patient-derived xenograft (PDX) model (PAN#026) | GenenDesign | N/A |
| Gastric cancer patient-derived xenograft (PDX) model STO#025 | GenenDesign | N/A |
| Cynomolgus monkeys | Guangxi Fangcheng Gang Spring Biological Technology Development Corporation Ltd | N/A |
| C57BL/6 mice | Vital River | N/A |
| Rats | Vital River | N/A |
| Balb/c mice | Vital River | N/A |
| B-NDG | Biocytogen | M/A |
| Hu-HSC-NPG mice | Vital River | N/A |
| Oligonucleotides | ||
| siRNA sequences, primers for RT-qPCR and sgRNA sequences | This paper | See Table S8 for details |
| Recombinant DNA | ||
| Human Claudin18.2 plasmid | This paper | N/A |
| pSPgRNA plasmid | Addgene | 47108 |
| Software and algorithms | ||
| GraphPad Prism 5 | GraphPad Software | https://www.graphpad.com/scientific-software/prism/ |
| WinNonlin 6.4 software | WinNonlin 6.4 software | https://www.certara.com/software/phoenix-winnonlin/ |
Experimental model and study participant details
Cell lines
Each cell line was cultured in its specific medium and grown at 37°C on a 5% CO2 air incubator. Cell lines including KATOIII, NIH/3T3 (3T3), BxPC-3, CFPAC-1 and HEK293 were purchased from the American Type Culture Collection. BxPC-3 and CFPAC-1were cultured in RPMI-1640 with 10% fetal bovine serum (FBS). HEK293 were maintained in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% FBS and 2 mM L-Glutamine. KATOIII were maintained in IMEM supplemented with 10% FBS. 3T3 were cultured in DMEM with 20% FBS and 2 mM L-Glutamine. MC38 cell line was a gift from Biocytogen Pharmaceuticals (Beijing) Co., Ltd, and cultured in DMEM with 10% FBS, 1 mM non-essential amino acid (NEAA), 1 mM Sodium Pyruvate, 10 mM HEPES, 2 mM L-Glutamine (HyClone). The constructed Claudin18.2 plasmid was transfected into different cells by electro-transfection or lentiviral construct to generate stable cell lines, including KATOIII-hCLDN18.2 (human Claudin18.2-expressing KATOIII cells), HEK293-hCLDN18.2 (human Claudin18.2-expressing HEK293 cells), 3T3-hCLDN18.2 (human Claudin18.2-expressing 3T3 cells), HEK293-cynoCLDN18.2 (cynomolgus monkey Claudin18.2-expressing HEK293 cells), HEK293-ratCLDN18.2 (rat Claudin18.2-expressing HEK293 cells), and HEK293-hCLDN18.1 (human Claudin18.1-experssing HEK293 cells). HEK293-CLDN18.2 and HEK293-hCLDN18.1 were cultured in DMEM with 10% FBS and 2 μg/ml puromycin. KATOIII-CLDN18.2 cells were cultured in IMEM with 10%FBS and 1 μg/ml puromycin.
Organoid culture
Organoids culture was performed as described previously.36 Briefly, pancreatic cancer tissues taken from biopsy were vigorously shaken in 1 mg/mL collagenase (Sigma-Aldrich; C9407) at 37°C for 30–60 min. Cell suspension was diluted with basic medium (advanced DMEM/F12, supplemented with 1× GlutaMAX, penicillin-streptomycin and 10 mM HEPES) and strained by 70 μm filter, followed by 300 g centrifuging. The resulting pellet was resuspended in Cultrex reduced growth factor basement membrane extract (BME) type 2. After the BME had solidified (15–30 min at 37°C), organoids were maintained in culture medium (basic medium with 50% Wnt3a-conditioned medium, 1×B27 supplement, 1.25 mM N-acetyl-L-cysteine, 10 mM nicotinamide, 50 ng/mL human EGF, 500 nM A83-01, 100 ng/mL human FGF10, 1 μM prostaglandin E2, 10 nM gastrin, 4% RSPO, and Noggin) at 37°C with 5% CO2. Culture medium was refreshed every 2–3 days, and organoids were passaged every 6–7 days.
Mouse models
For pancreatic cancer patient-derived organoid xenograft (PC-PDOX) model, female B-NDG mice (aged 6–8 weeks) and female Hu-HSC-NPG mice (aged 16–18 weeks) were obtained from Biocytogen Pharmaceuticals (Beijing) Co., Ltd. and Beijing Vitalstar Biotechnology Co., Ltd., respectively. All animals were housed in in standard conditions, in a 12/12 h dark/light cycle, at 20°C–26°C, with free access to food and water. All animal experimental procedures were approved by the Institutional Animal Care and Use Committee of Biocytogen Pharmaceuticals (Beijing) Co., Ltd. (Experimental animals use license number: SYXK (Jing) 2020-0020).
For the efficacy of CMG901 in patient-derived xenograft (PDX) model, female Balb/c nude mice (aged 6–8 weeks) were obtained from Vital River and used for tumor fragment implantation. Animals were fed with normal nude mice diet and housed in SPF animal facility at GenenDesign in accordance with the Guide for Care and Use of Laboratory Animals and regulations of the Institutional Animal Care and Use Committee.
For the combination of CMG901 and anti-PD-1 in PDX model, female Hu-HSC-NPG mice were obtained from Beijing Vital River Laboratory Animal Technology Co., Ltd. and housed in SPF animal facility at GenenDesign with free access to food and water in accordance with the Guide for Care and Use of Laboratory Animals and regulations of the Institutional Animal Care and Use Committee.
For the combination of CMG901 and anti-PD-1 in cell line-derived xenograft studies, Female C57BL/6 mice (aged 6–8 weeks) were obtained from Beijing Vital River Laboratory Animal Technology Co., Ltd. and group-housed in standard conditions, in a 12/12 h dark/light cycle, at 20°C–26°C, with free access to food and water. All animal experimental procedures were approved by the Institutional Animal Care and Use Committee of WestChina-Frontier PharmaTech Co., Ltd.
Rat models
For acute toxicity of CMG901 in rats, female and male SD rats (aged 6–8 weeks) were obtained from Beijing Vital River Laboratory Animal Technology Co., Ltd. and group-housed in standard conditions, in a 12/12 h dark/light cycle, at 20°C–26°C, with free access to food and water. All animal experimental procedures were approved by the Institutional Animal Care and Use Committee of Shanghai InnoStar Bio-Tech Co., Ltd. (InnoStar) (Experimental animals use license number: SYXK (Hu) 2019-0009).
In tissue distribution and excretion study of 125I-CMG901 and 125I-CM311 after single intravenous administration in rats, female and male rats (7–11 weeks) were obtained from Zhejiang Vital River Laboratory Animal Technology Co., Ltd. and housed in standard conditions, in a 12/12 h dark/light cycle, at 18°C–26°C, with free access to food and water. The protocol, amendment and procedures concerning the care and use of animals in the study were reviewed and approved by the Institutional Animal Care and Use Committee (IACUC) of InnoStar Bio-tech Haimen Co., Ltd. (InnoStarHM) (Experimental animals use license number: SYXK (Su) 2018-0034).
In dose range finding toxicity study of CMG901 in rats, female and male SD rats (aged 6–9 weeks) were obtained from Beijing Vital River Laboratory Animal Technology Co., Ltd. and group-housed in standard conditions, in a 12/12 h dark/light cycle, at 20°C–26°C, with free access to food and water. All animal experimental procedures were approved by the Institutional Animal Care and Use Committee of Shanghai InnoStar Bio-Tech Co., Ltd. (InnoStar) (Experimental animals use license number: SYXK (Hu) 2014-0009, SYXK (Hu) 2019-0009).
In 10-week repeat-dose toxicity study of CMG901 in rats with an 8-week recovery period, female and male SD rats (aged 7–9 weeks) were obtained from Zhejiang Vital River Laboratory Animal Technology Co., Ltd. and housed in standard conditions, in a 12/12 h dark/light cycle, at 20°C–33°C, with free access to food and water. All animal experimental procedures were approved by the Institutional Animal Care and Use Committee of Shanghai InnoStar Bio-Tech Co., Ltd. (InnoStar) (Experimental animals use license number: SYXK (Hu) 2019-0009).
Non-human primate studies
For acute toxicity of CMG901 in cynomolgus monkeys, female and male cynomolgus monkeys (aged 2.5–3 years) were obtained from Guangxi Guidong Quadrumana Development & Laboratory CO., Ltd. and group-housed by sex in standard conditions, in a 12/12 h dark/light cycle, at 18°C–26°C, with free access to food and water. All animal experimental procedures were approved by the Institutional Animal Care and Use Committee of Shanghai InnoStar Bio-Tech Co., Ltd. (InnoStar) (Experimental animals use license number: SYXK (Hu) 2019-0009).
In preliminary toxicological study of CMG901 in cynomolgus monkeys, female and male cynomolgus monkeys (aged 3–5 years) were obtained from Guangxi Fangcheng Gang Spring Biological Technology Development Corporation Ltd. and housed in standard conditions, in a 12/12 h dark/light cycle, at 18°C–26°C, with free access to food and water. All animal experimental procedures were approved by the Institutional Animal Care and Use Committee of Shanghai HkeyBio Technology Co., Ltd. (Experimental animals use license number: SYXK (Gui) 2018-0004).
In 10-week repeat-dose toxicity study of CMG901 in cynomolgus monkeys with an 8-week recovery period, female and male Cynomolgus monkeys (aged 3–4 years) were obtained from Guangxi Guidong Quadrumana Development & Laboratory CO., Ltd. and group-housed by sex in standard conditions, in a 12/12 h dark/light cycle, at 18°C–28°C, with free access to food and water. All animal experimental procedures were approved by the Institutional Animal Care and Use Committee of Shanghai InnoStar Bio-Tech Co., Ltd. (InnoStar) (Experimental animals use license number: SYXK (Hu) 2019-0009).
Method details
Claudin18.2 expression analysis of TCGA and GTEx data
Transcript expression data for cancer patients and normal tissues were downloaded from University of California Santa Cruz (UCSC) Xena website (TcgaTargetGtex_rsem_isoform_tpm, accessed on 29 July 2022) and Gene-Tissue Expression (GTEx), respectively.37,38 To differentiate the expression of Claudin18.2 from Claudin18.1, the specific isoform transcript annotation information was retrieved from The Ensembl.39 Positive was defined as the transcript expression levels in patients at 2-fold higher than that in the corresponding normal tissues.
Antibody and ADC
The humanized antibody CM311 targeting the extracellular loop 1 (ECL-1) of human Claudin18.2 was generated using the hybridoma technology. Briefly, female Balb/c mice were immunized with human Claudin18.2 plasmid using gene gun for 4 cycles. Mice were sacrificed after final boost with 3T3-hCLDN18.2 (human Claudin18.2-expressing 3T3 cells), and the splenocyte and lymph node were harvested and fused with sp2/0 myeloma. Hybridoma was screened to identify antibodies specifically binding to Claudin18.2 using flow cytometry. Antibody sequences including both heavy chain and light chain were sequenced from corresponding hybridoma and assembled into the chimera. Fc-mediated antibody-dependent cellular cytotoxicity (ADCC) and complement-dependent cytotoxicity (CDC) assays were then used to evaluate all chimeras, and the selected ones were further screened for cellular internalization property. Final candidate antibody was humanized and expressed in CHO host cells, referring to CM311. Synthesis and quality control of the antibody-drug conjugate CMG901 was performed according to the internal protocol by Shanghai Miracogen Inc, and drug-to-antibody ratio (DAR) was set as approximately 3.8.
Zolbetuximab analog was recombinantly expressed in CHO host cells according to the disclosed sequence (www.imgt.org).
CLDN18.1 detection using reverse transcription PCR (RT-PCR)
Primers KNB14 and KNB15 were designed to amplify the region between the second transmembrane domain and the C-terminus of Claudin18.1 and Claudin18.2, yielding a characteristic 504 bp band. Furthermore, the Claudin18.2 N-terminal specific primer KNB16 was designed to be used with the C-terminal specific primer KNB15, to amplify the full-length Claudin18.2 fragment, producing a typical 780 bp band. KATOIII cells, which express Claudin18.2, was used as a positive control. Total RNA from cells was extracted using TRNzol and transcribed it into cDNA using Super Script III reverse transcriptase. Use this cDNA as a template to amplify DNA products containing the target gene, followed by agarose gel electrophoresis to verify if the band sizes match theoretical expectations.
pHrodo-based internalization assay
KATOIII-hCLDN18.2 and HEK293-hCLDN18.2 cells were seeded at the density of 1×105 cells/well in a 96-well plate and cultured overnight. CMG901 or Anti-KLH-hIgG1 were labeled with pHrodo Deep Red tetrafluorophenyl (TFP) ester as per the manufacturer’s instruction and diluted to 10 μg/mL in culture medium. The labeled antibodies were added to the cells at 100 μL/well (33 nM), followed by a 1 h incubation on ice. After washing, cells were incubated at 37°C with 5% CO2 in culture medium for 1 h, 2 h, 4 h, 6 h, 24 h. Subsequently, cells were stained with propidium iodide (PI) for 5 min on ice, washed with 1% BSA, resuspended in 50 μL/well PBS, and analyzed for mean fluorescence intensity (MFI) using flow cytometry.
Affinity of CMG901 to Fcγ receptors, FcRn receptors, and complement C1q
CM5 chip was coated with anti-His-Tag antibody by amino conjugation, and his-tagged recombinant proteins of FcγRI, FcγRIIAH131, FcγRIIBC, FcγRIIIAV158, or FcγRIIIAF158 were allowed to flow through the chip, and were captured onto sensor chip surface through interaction between the His-tag and the anti-His-tag antibody, then CMG901 or CM311 solutions at different concentrations was flew through the sensor chip. CM5 chip was also coated with FcRn protein by amino conjugation, then, CMG901 or CM311 solutions at different concentrations flowed through the sensor chip. Finally, CMG901 or CM311 was flowed through Protein L chip which specifically binds to, Fab part of the antibody, while Fc segment of the antibody was fully exposed to the solution, then human C1q protein solutions at different concentrations were flew through the sensor chip. Binding affinity of the antibody to each receptor was measured and fitted using Langmuir 1:1 kinetics model or steady state model by using Biacore T200 Evaluation software (version 3.1) to calculate binding affinity of CMG901 and CM311 to Fcγ receptors, FcRn, and C1q.
Binding of CMG901 on Claudin18.2-expressing cell lines
CMG901, CM311, and Anti-KLH hIgG1, and Anti-KLH hIgG1 ADC were diluted with 1% bovine serum albumin (BSA) to 90 μg/mL (600 nM), and then diluted serially 3-fold into 10 dilutions in duplicate. HEK293-CLDN18.1, HEK293-CLDN18.2, and KATOIII-CLDN18.2 cells were digested with trypsin EDTA solution (0.25%), spun down the cells at 4°C, 300 g for 5 min and harvested the cells. Resuspended cells with blocking buffer to 1×107 cells/mL, placed on ice for 30 min. After blocking, adjusted cell density to 5×105 cells/mL with flow cytometry buffer, then dispensed the cell suspension into a 96-well plate at 100 μL/well, followed by centrifuged to harvest cells, resuspended the cells with 50 μL/well of test articles diluent accordingly and incubated on ice for 45 min. After incubation wash the cells 2 times with 200 μL of wash buffer. Resuspended the cells with prepared secondary antibody diluent at 50 μL/well, then incubated on ice for 45 min. Washed the cells 3 times and resuspended the cells with 60 μL/well of propidium iodide (PI) staining buffer and recorded mean fluorescence intensity (MFI) using flow cytometry. The drug concentration-MFI curve was obtained by 4-parameter logistic model fitting, and half maximum effective concentration (EC50) of test articles binding to cells was calculated.
ADC cytotoxicity assays
KATOIII-hCLDN18.2 (human Claudin18.2-expressing KATOIII cells) and HEK293-hCLDN18.2 (human Claudin18.2-expressing HEK293 cells) as target cells and HEK293-hCLDN18.1 (human Claudin18.1-expressing HEK293 cells) as negative control cells were incubated with different concentrations of CMG901, CM311, Anti-KLH hIgG1, or Anti-KLH hIgG1 ADC (control ADC), respectively. Cell viability was measured using cell counting kit-8 (CCK8). Diluted CMG901, CM311, Anti-KLH hIgG1, and Anti-KLH hIgG1 ADC to a final concentration of 20 μg/mL (133 nM) with culture medium into 9 dilutions with a dilution factor of 4. KATOIII-CLDN18.2, HEK293-CLDN18.2, and HEK293-CLDN18.1 were cultured at 37°C. After digestion with trypsin EDTA solution (0.25%), centrifuged the cells at 300 g 4°C for 10 min, then discarded the supernatant and harvested the cells. Washed the cells twice with 10 mL of PBS and resuspended with culture medium to 5×103 cells/mL, then added 100 μL/well into a 96-well culture plate in triplet and incubated at 37°C with 5% CO2 for 18–20 h. 100 μL of test articles at different concentrations were added into each well, and incubated at 37°C with 5% CO2 for 72 h. Spun down cells and discarded the supernatant, 200 μL/well of the CCK-8 chromogen solution was added into each well, and incubated at 37°C with 5% CO2 for 2 h. Added culture medium into the target cell well as the control well, added CCK-8 chromogen solution into wells as blank well, assayed in the same way, and then documented the results.
The data were processed using GraphPad Prism by 4-parameter logistic regression to calculate EC50 of test articles. Sample concentration (abscissa) was plotted against relative viability (%) (ordinate). Relative viability (%) was calculated using the following formula:
Test well: containing cell suspension, sample, and chromogen solution; Control well: containing cell suspension, RPMI 1640 culture medium, and chromogen solution; Blank well: CCK-8 chromogen solution.
ADCC assays
KATOIII-hCLDN18.2 and HEK293-hCLDN18.2 as target cells and peripheral blood mononuclear cells (PBMC) as effector cells were incubated with diluted concentrations of CMG901, CM311, Anti-KLH hIgG1, or Anti-KLH hIgG1 ADC, respectively. Lactate dehydrogenase (LDH) released was measured using CytoTox-ONE Homogeneous Membrane Integrity Assay kit to calculate the viability of the target cells. Diluted CMG901, CM311, Anti-KLH hIgG1 and Anti-KLH hIgG1 ADC with reaction buffer (RPMI 1640 medium supplement with 2% FBS) to 30 μg/mL (200 nM), then serially diluted 10-fold for 6 dilutions, duplicate for each dilution. After digested with trypsin EDTA solution (0.25%), KATOIII-CLDN18.2, HEK293-CLDN18.2, and HEK293-CLDN18.1 cells were collected by centrifugation at 4°C, 300 g for 10 min; washed once with PBS and suspended with reaction buffer at 1×106 cells/mL; then dispensed the cell suspension into a 96-well U-bottom plate, 50 μL per well in doublets. Thawed PBMCs and suspended with reaction buffer at 1×107 cells/mL, 50 μL of test articles and 50 μL of PBMC effector cells were added into the wells, and incubated at 37°C with 5% CO2 for 4 h. Release of lactate dehydrogenase (LDH) in the supernatant was assayed with CytoTox-ONE Homogeneous Membrane Integrity Assay kit (Promega). Wells for spontaneous release were prepared by addition of 100 μL reaction buffer into target cells; Wells for maximum release were prepared by addition of 100 μL lysis solution (including in assay kit) into target cells, then measured LDH release as other test wells and recorded results.
The data were processed using GraphPad Prism by 4-parameter logistic regression to calculate EC50 of the sample. Sample concentration (abscissa) was plotted against lysis (%) (ordinate). Lysis (%) was calculated using the following formula:
Test well: containing targeted cells, effector cells, and sample; Spontaneous target cell release well: containing target cells suspension only; Maximum target cell release well: containing target cell and lysis solution.
CDC assays
KATOIII-hCLDN18.2 and HEK293-hCLDN18.2 as target cells mixed with 30% normal human serum complement were incubated with diluted CMG901, CM311, Anti-KLH hIgG1, or Anti-KLH hIgG1 ADC, respectively, and CCK8 was used to detect the cell viability. After digested with trypsin EDTA solution (0.25%), KATOIII-CLDN18.2, HEK293-CLDN18.2, and HEK293-CLDN18.1 cells were suspended with RPMI 1640 culture medium at density of 1.2×106 cells/mL; each cell suspension was then seeded onto a 96-well plate, 50 μL per well in doublet. CMG901, CM311, Anti-KLH hIgG1, and Anti-KLH hIgG1 ADC were diluted with medium to 30 μg/mL (200 nM); then diluted serially into 6 dilutions. Added 50 μL of 30% complement cocktail and 50 μL of test articles at different concentrations into each well, then incubated the plate at 37°C in 5% CO2 for 2 h. Added 50 μL of 40% CCK-8 cocktail into each well and further incubated for 2 h. Taken out the plates and vortexed for 10 s, and Optical density (OD) at 450 nm wavelength was measured using plate reader.
The data were processed using GraphPad Prism by 4-parameter logistic regression to result in EC50 of the sample. Sample concentration (abscissa) was plotted against lysis (%) (ordinate). Lysis (%) was calculated using the following formula:
Test well: containing cell suspension, CCK-8, complement, and test article; Reference well: containing cell suspension, CCK-8, and complement; Blank well: RPMI 1640 culture medium, CCK-8, and complement.
Bystander killing effect of CMG901 on cell lines
Claudin18.2-positive KATOIII-hCLDN18.2 and Claudin18.2-negative KATOIII cells were incubated with 1 μg/ml of CMG901 for 5 days, and live cell number was determined, and ratio of Claudin18.2-positive and Claudin18.2-negative cells was analyzed by flow cytometry.
Bystander killing effect of CMG901 on organoids
Bystander killing activity of CMG901 on organoids was performed in Sun Yat-sen University Cancer Center. 10 μg/ml CMG901 and Anti-KLH hIgG1 ADC were added into organoids culture plate and organoids were harvested and dissociated into single cells after 6 days treatment. Live cell number and ratio of Claudin18.2-positive and Claudin18.2-negative cells were determined by a cell counter and a flow cytometry, respectively.
Claudin18.2 knockout in organoids
Organoids culture and knockout was performed by ZOS Biotechnology. Organoids were engineered using CRISPR/Cas9 system. Single-guide RNAs were designed using an online web-tool (www.atum.bio/eCommerce/cas9/input) and cloned into the pSPgRNA plasmid as described by Ran et al.40 Human pancreatic cancer patient-derived organoids were transfected by electroporation, as described previously.41 Briefly, organoids were converted to single cells using TrypLe, washed twice with basic medium and resuspended in 130 μL of Opti-MEM containing the DNA mixture. Electroporation was performed using ECM2001. After 20 min recovery in pancreatic cancer organoids medium with additional Y-27632 (final concentration, 10 μM), single cells were recovered and washed with basic medium, and plated in 100 μL of BME suspension into one well of a 12-well plate (three droplets per well). Cells were cultured in complete PC medium with additional Y-27632 (final concentration, 10 μM) until small organoids appeared, then were shifted to regular PC medium. When organoids were co-transfected with the hygromycin-piggyBac two-plasmid system, selection was started when organoids of small size formed. Hygromycin B Gold was retained for roughly 12 days. Single surviving organoids were picked, converted to small fragments/single cells by TrypLe, and plated into a single BME droplet well of a 24-well plate and expanded into clonal lines.
In vitro ADC cytotoxicity, ADCC, and CDC effects for Claudin18.2 WT/KO organoids
Human pancreatic cancer organoids were harvested and trypsinized into single cells by trypLe. For ADC cytotoxicity assay and CDC effect evaluation, cells were seeded in 2 μL with BME in 96-well cell culture plate in a concentration of 2x103 cells per well for 2-day culture. For ADC cytotoxicity assay, test articles were diluted and added into organoids culture medium for 5 days, and cell viability was measured by CTG assay. For CDC assay, normal human serum was used as source of complement and natural antibodies. Organoids were incubated with diluted antibody CM311 and 15% normal human serum for 6 h, followed by cell viability measurement. For ADCC assay, 1x104 organoids were harvested and gently seeded into each well of 96-well ultra-low attachment plated for 2 h, and human PBMC was added into each well with the 1:100 target-effector cell ratio. After 6 h of incubation, LDH release assay was performed using a CytoTox 96Non-Radioactive Cytotoxicity Assay Kit.
Intracellular internalization, lysosomal localization, and internalization efficiency analysis of CMG901
KATOIII-CLDN18.2 cells were centrifuged at 300 g for 5 min and then washed once with 5 mL PBS. The cells were resuspended at density of 2.5×105 cells/mL with complete medium (IMDM supplement with 10% FBS), and seeded 800 μL/well into a 4-well 20 mm glass-bottom plate and incubated at 37°C with 5% CO2 for 48 h. The cells were washed twice with PBS and placed on ice. Diluted CMG901, CM311, Anti-KLH hIgG1 and Anti-KLH hIgG1 ADC with buffer (PBS supplemental with 4% bovine calf serum) to 10 μg/mL. Dispensed diluted test articles into a plate at 250 μL/well, and incubated on ice for 45 min. Spun down and resuspended the cells with 500 μL of culture medium, incubated at 37°C with 5% CO2 for indicated time. Collect cell samples at 0 h, 0.5 h, 1.0 h, 2.0 h, 4.0 h, and 6.0 h, and washed twice with cold PBS, then added 250 μL/well of 4% paraformaldehyde solution to fix the cells at room temperature (RT) for 20 min. The cells were then washed twice with PBS. Added 250 μL/well of 5% Triton X-100 solution to permeabilize cell membrane for 5 min. Washed the cells twice with PBS, and then Added 250 μL/well of 3% BSA/PBS solution to block the cells, and placed the plate on ice for 30 min. After spun down the cells, AF594-Fab’2 Goat anti-hIgG (H + L) antibody was added at 150 μL/well, incubated the cells at RT for 45 min and then washed twice with PBS. 8 μg/mL Human LAMP-1/CD107a antibody was prepared with 4% BCS/PBS and added at 150 μL/well and incubated at RT for 45 min, then washed twice with PBS. AF488-Fab’2 Goat anti-mIgG (H + L) (1:300) was added at 150 μL/well, incubated the cells at RT for 45 min, then washed twice with PBS. After spinning down the cells, one drop of anti-fade solution (ProLong Gold Antifade Mountant with DAPI) was added into each well, and the plate was covered with a cover slide and kept in dark at RT for 24 h before photographed with a confocal microscope or stored in dark at 4°C. Images were acquired and analyzed using THUNDER imaging systems (Leica).
6×105 cells/mL were seeded onto 24-well plates at 500 μL/well, and incubated at 37°C with 5% CO2. After 48 h culture, added 500 μL/well of 4% BCS/PBS to wash the cells twice, placed the culture plate on ice. The prepared samples were added to plates at a volume of 250 μL/well, then the plates were incubated on ice for 45 min. Discarded the supernatant and washed the cells twice with 500 μL/well of 4% BCS/PBS. Added 500 μL/well of 37°C pre-incubated complete medium into the wells, and cells were then further cultured for 0 h, 0.5 h, 1.0 h, 2.0 h, 4.0 h, and 6.0 h in the incubator at 37°C with 5% CO2. After removal of IMDM complete medium, 500 μL/well of cold 4% BCS/PBS was added to wash the cells twice. AF647-conjugated Goat Anti-Human IgG (Fcγ Fragment specific) diluted at a ratio of 1:300 was added at 150 μL/well, and put on ice for 45 min in dark. Cells were washed twice with 500 μL/well of cold PBS. Added 250 μL/well trypsin-EDTA solution to digest the cells at RT for about 7 min, and then added 500 μL/well of cold PBS to stop the digestion. The cell suspension was collected and centrifuged at 300 g for 5 min, and then washed with cold PBS once, resuspended with cold PI solution at volume of 200 μL/well, and then analyzed by flow cytometry. MFI of AF647 in PI-negative cell population was analyzed, and internalization percentage was calculated.
Cell proliferation
To evaluate the effects of Claudin18.2 on cancer cells proliferation, BxPC-3 and CFPAC-1 cells (60000 cells/well) were seeded into 24-well plates. Cell proliferation ability was analyzed 0, 24, 48 h post-transfection of Claudin18.2 siRNA using a Cell Counting Kit-8 (CCK-8) Assay Kit. The OD value of each well was measured at 450 nm under microplate reader.
Knockdown of Claudin18.2
To knockdown of Claudin18.2, BxPC-3 and CFPAC-1 cells were transfected with Claudin18.2 or NC siRNA using Lipofectamine RNAiMAX Transfection Reagent with a sequence listed in key resources table.
qRT-PCR for Claudin18.2 and Claudin18.1
The mRNA levels of Claudin18.2 and Claudin18.1 were quantified by real-time PCR (qRT-PCR) using SYBR Green and normalized to GAPDH mRNA levels. The sequences of primers used were listed in key resources table. The value of 2−ΔΔCt was used to evaluate relative gene expression.
The efficacy of CMG901 in patient-derived xenograft (PDX) model
Gastric cancer STO#025, STO#580, and pancreatic cancer PAN#026 patient derived xenograft (PDX) model with different Claudin 18 mRNA expression were established at GenenDesign. The detected CLDN18 RPKM values for STO#25, STO#580 and PAN#026 were 721.43, 217.07 and 731.178, respectively. Tumor fragments of about 15–30 mm3 in size were prepared for implantation subcutaneously into female Balb/c nude mice. Mice were anesthetized with isoflurane throughout the implantation procedure. Tumor fragments were placed into the trochar needle and implanted into a subcutaneous pocket in the right flank. Mice with tumor volumes ranging from 143 to 269 mm3 in STO#025, 150 to 249 mm3 in STO#580, or 150 to 259 mm3 in PAN#026 were randomly assigned into the vehicle, CM311 (3 mg/kg), Anti-KLH hIgG1 ADC (3 mg/kg), and CMG901 (0.3, 1, and 3 mg/kg), and test articles were administered intravenously once every three weeks (q3w). Tumors were measured twice a week in two dimensions with calipers. Tumor volume was calculated using following formula: tumor volume = (length x width2) × 0.5. Tumor growth inhibition at end of treatment was assessed by calculating the ΔT/ΔC% according to following formulas: when ΔT/ΔC% (for mean values >0): [(Tumor volume in drug group at end of treatment - Tumor volume in drug group at start of treatment)/(Tumor volume in vehicle group at end of treatment - Tumor volume in vehicle group at start of treatment)] × 100%]; when ΔT/ΔC% (for mean value) ≤ 0, i.e., regression: [(Tumor volume in drug group at end of treatment - Tumor volume in drug group at start of treatment)/Tumor volume in drug group at start of treatment] × 100%. When ΔT/ΔC % (for mean values) > 0, TGI% = (1-ΔT/ΔC) × 100%.
Combination of CMG901 and anti-PD-1 in tumor growth inhibition
For the combination of CMG901 and anti-PD-1 in PDX model, tumor fragments of about 15–30 mm3 from STO#025 were implanted subcutaneously into right flanks of female Hu-HSC-NPG mice. When tumor volume reached near 158–264 mm3, mice were randomly assigned into vehicle, anti-PD-1 (10 mg/kg), CMG901 (0.5 mg/kg), the combination of CMG901 (0.5 mg/kg) and anti-PD-1 (10 mg/kg). CMG901 was administered intravenously once every week (qw, 3 doses) and anti-PD-1 was given intravenously once every three days (q3d, 10 doses).
For the combination of CMG901 and anti-PD-1 in cell line-derived xenograft studies, 2 × 106 MC38-hCLDN18.2 cells (human Claudin18.2-expressing MC38 cells) were inoculated subcutaneously into female C57BL/6 mice. When average tumor volume reached 150–200 mm3, mice were grouped and treated either with vehicle control, CMG901 (0.1, 0.3, and 1.0 mg/kg), anti-PD-1 (0.1, 0.3, and 1.0 mg/kg), or the combination of CMG901 (0.3, 1.0 mg/kg) and anti-PD-1 (0.1, 0.3 mg/kg). Tumor volume was measured to evaluate the tumor growth inhibition of the combination.
Pancreatic cancer patient-derived organoid xenograft (PC-PDOX) model
The organoid expansion and transplantation experiments were conducted by ZOS Biotechnology. Briefly, human tissue-derived pancreatic cancer organoids were cultured according to the protocol described by Driehuis et al.36 To this end, a strain of pancreatic cancer organoids was expended and transplanted into B-NDG mice for passage generations. Following expansion, the organoids were dissociated from matrigel using TrypLE Express, harvested, and quantified. Subsequently, approximately 1.5 × 105 cells in a 200 μL matrigel-organoid suspension were subcutaneously injected into B-NDG mice (Biocytogen) for passaging. After two rounds of passaging in the pancreatic cancer patient-derived organoid xenograft (PC-PDOX) model, tumor tissues were excised, and sectioned into small pieces approximately 2 mm in each dimension and transplanted into Hu-HSC-NPG mice (Vitalstar). Tumor growth was monitored using a vernier caliper. When tumor volumes reached ∼150–200 mm3, each group of mice were given intravenous 1 mg/kg of Anti-KLH MMAE and CMG901 (administered once a week for 2 doses), 40 mg/kg of CM311 and Zolbetuximab (administered twice a week for 4 doses), respectively. Mice were euthanized at the end of the study. PC-PDOX tumors were formalin-fixed, paraffin-embedded, and subjected to hematoxylin and eosin (HE) staining and immunohistochemistry (IHC). All procedures were performed in compliance with animal welfare regulations.
Tissue distribution and excretion study of 125I-CMG901 and 125I-CM311 after single intravenous administration in rats
48 rats (Group 1–8) received a single intravenous dose of 125I-CMG901 or 125I-CM311. 24 rats (3/sex/radiolabeled test articles) were sacrificed at 24 h, 72 h, 168 h, and 336 h post-dose. Tissue and body fluid samples were collected and processed using the trichloroacetic acid (TCA) precipitation method. Total and precipitation radioactivity were measured by a γ counter to calculate exposure levels.
8 rats (Group 9) received 125I-CMG901 (1 mg/kg) for bile excretion analysis, and 6 rats (Group 10) for feces/urine excretion. Radioactivity was measured using a γ counter at different time points. Serum/urine samples from 1 animal/group (Groups 1–8) were prepared and separated by HPLC and analyzed using a γ counter for drug metabolism.
Single-dose toxicity study of CMG901 in rats
The acute toxicity of CMG901 was also assessed in female and male rats (aged 6–8 weeks) after a single intravenous injection at doses 0, 12, and 24 mg/kg. The index included morbidity and mortality, clinical observations, body weights, and gross and microscopic pathology.
Dose range finding toxicity study of CMG901 in rats
A 4-week dose range finding (DRF) toxicity study of CMG901 in female and male SD rats was conducted to evaluate toxicity and for supporting the design and selection of dose levels for the GLP repeat-dose toxicity study in rats. CMG901 doses of 7.5, 15 and 30/20 mg/kg were employed in the DRF study, based on available data from an in vivo efficacy study in tumor mice models while CM311 dose was kept at 30/20 mg/kg for comparison. Parameters monitored included mortality/moribundity, clinical observations, body weight, food consumption, hematology/coagulation/clinical chemistry, bone marrow smears, and gross pathology.
10-week repeat-dose toxicity study of CMG901 in rats with an 8-week recovery period
The GLP-compliant study was conducted to assess the toxicity, immunogenicity, and TK profiles of CMG901, when administered to female and male SD rats via intravenous infusion (q3w, on day 1, 22, 43 and 64) for 10 weeks and to assess the reversibility of toxic effects after an 8-week recovery period. CMG901 doses of 2.5, 5 and 10 mg/kg were administered in the 10-week pivotal study, the doses of CM311 and MMAE were 10 mg/kg and 0.2 mg/kg, respectively, corresponding to the amount at CMG901 dose of 10 mg/kg. Reversibility of effects was evaluated after an 8-week treatment free period. Parameters monitored during in-life phase included morbidity and mortality, clinical observations, body weights, food consumption and ophthalmology. Hematology, coagulation, clinical chemistry, cardiac troponin I, immune function, and urinalysis were assessed for all surviving animals in the main study once prior to the terminal and recovery necropsies. All surviving animals in the main study and recovery animals were subjected to necropsy on Day 71 and Day 127, respectively. Organ weights were recorded at scheduled necropsy and bone marrow smears from femur were prepared for examination. Histopathology evaluation was performed for gross lesions and tissues collected at necropsy. Tissues collected from control and CMG901 high dose, and CM311 and small molecule (MMAE) groups were subjected to histopathology evaluation. Tissues/organs showing test articles-related lesions were also microscopically examined for animals at low and mid dose groups.
Pharmacokinetic (PK)/Toxicokinetic (TK) in rats
Pharmacokinetic (PK)/Toxicokinetic (TK) studies were accompanied by 10-week repeated-dose toxicity study with an 8-week recovery period in conjunction with the Good Laboratory Practice (GLP) compliant. The serum concentrations of CMG901, total antibody (TAb), CM311 were measured by the validated ELISA analytical methods. The lower limit of quantitation (LLOQ) of CMG901, TAb, and CM311, were 50, 50, and 50 ng/mL in rats. The serum concentrations of MMAE were measured by the validated LC-MS/MS method. The LLOQ of MMAE in rats were 20 pg/mL. Plasma stability of CMG901 was tested in vitro with normal rat plasma. The tissue distribution and excretion of CMG901 were evaluated in rats.
Single-dose toxicity study of CMG901 in cynomolgus monkeys
In single-dose toxicity study, CMG901 was assessed in cynomolgus monkeys after intravenous infusion at doses of 0, 8, and 12 mg/kg. The parameters included morbidity and mortality, clinical observations, body weights, food consumption, body temperature, electrocardiograms examination, clinical pathology (hematology, coagulation, clinical chemistry) and gross pathology.
Preliminary toxicological study of CMG901 in cynomolgus monkeys
The DRF (16-week) study in female and male cynomolgus monkeys was performed using CMG901 at doses of 3, 8 and 12 mg/kg and CM311 at 12 mg/kg for comparison (administered q3w×5; on Day 1, 22, 43, 64 and 85). Higher doses of CM311 30 mg/kg (qw×4; on Day 1, 8, 15 and 22) and 100/200 mg/kg (100 mg/kg: qw×4 followed by 200 mg/kg: qw×2) were also tested. Parameters included clinical signs (including injection site observation), body weight, feed consumption, clinical pathological examinations (hematology, serum chemistry and blood coagulation), immunophenotyping and ADA analysis. At the end of the study, the animals were necropsied for toxicity evaluation, and stomach (containing the superior end of gastric cardia and the proximal end of duodenum), lungs, spleen, liver, kidneys, heart, and any tissue with lesions found in dissection were collected for histopathological examinations. Samples for clinical pathology, immunophenotyping and ADA were collected. ADA was analyzed using an exploratory ECL assay based on meso scale discovery (MSD) platform. Only screening analysis and confirmatory analysis were performed in this study and antibody titers were not measured.
10-Week repeat-dose toxicity study of CMG901 in cynomolgus monkeys with an 8-week recovery period
The pivotal 10-week female and male Cynomolgus monkey study was performed at CMG901 doses of 1, 3 and 6 mg/kg based on the results of the DRF study; the CM311 dose was similar to CMG901 high dose of 6 mg/kg while the MMAE dose was kept at 0.06 mg/kg which is equal amount conjugated in 3 mg/kg CMG901. The test articles were administered q3w for 4 times (on Day 1, 22, 43 and 64) by intravenous infusion. Reversibility of toxicity was evaluated after an 8-week treatment free period. Parameters monitored included mortality, clinical observations, dose site irritation observations, body weights, food consumption, ophthalmology, hematology, clinical chemistry, cardiac troponin I, immune function, urinalysis, occult blood in feces, bone marrow smear, organ weights and gross and microscopic pathology. Organ weights were recorded at scheduled necropsy, and bone marrow smears from ribs were prepared for examination. Histopathology evaluation was performed for gross lesions and tissues collected at necropsy. Tissues collected from control and CMG901 high dose, CM311, and MMAE dose groups were initially subjected to histopathology evaluation. Tissues/organs showing test articles-related lesions were also microscopically examined for animals in low and middle dose groups.
Pharmacokinetic (PK)/Toxicokinetic (TK) in cynomolgus monkeys
Pharmacokinetic (PK)/Toxicokinetic (TK) studies were accompanied by 10-week repeated-dose toxicity study with an 8-week recovery period in conjunction with the Good Laboratory Practice (GLP) compliant. The serum concentrations of CMG901, total antibody (TAb), CM311 were measured by the validated ELISA analytical methods. The lower limit of quantitation (LLOQ) of CMG901, TAb, and CM311, were 100, 100, and 100 ng/mL in cynomolgus monkeys. The serum concentrations of MMAE were measured by the validated LC-MS/MS method. The LLOQ of MMAE in cynomolgus monkeys were 50 pg/mL. Plasma stability of CMG901 was tested in vitro with normal human and monkey plasma.
In vitro plasma stability
Plasma stability of CMG901 at a concentration of 250 μg/mL was evaluated after incubation with normal human, cynomolgus monkey and rat plasma. The released MMAE in each sample was expressed as a percentage of the maximum theoretical amount of MMAE.
Tissue cross reactivity in human, cynomolgus monkey and rat tissues
Tissue cross reactivity (TCR) was completed by Shanghai Institute of Materia Medica, Chinese Academy of Sciences. Briefly, CMG901 and monoclonal antibody CM311 were biotinylated, and streptavidin-peroxidase (SP) immunohistochemistry method was used. Gastric cancer PDX tissue was used as positive control tissue, while lymphoma PDX tissue was used as negative control tissue. Human IgG1 was used as isotype control. TCR with normal frozen tissues of human, cynomolgus monkey and rat were performed with biotinylated CMG901, CM311 and isotype control antibody at the concentrations of 5 and 20 μg/mL.
Quantification of CMG901, total antibody (TAb), and CM311 in serum
For quantification of CMG901 in cynomolgus monkey or rat serum, microplates were pre-coated with anti-MMAE antibody. After blocking, diluted standards, quality controls, matrix blank and test samples were added into the plates, respectively. Subsequently, horseradish peroxidase (HRP)-labeled goat anti-human-Fab antibody was added to bind CMG901 in cynomolgus monkey serum or biotinylated CM311aIAm (a mouse specific antibody against CM311) was added to allow binding of CMG901 followed by addition of Streptavidin (SA) Poly-HRP in rat serum. Finally, TMB was added to the microplates to allow color development, and the absorbance at 450/650 nm was measured on a microplate reader. The standard curves were fitted with a 4-parameter fitting model and concentrations of test samples were calculated.
For quantification of TAb (CMG901 and unconjugated antibody) in cynomolgus monkey or rat serum, microplates were pre-coated with CM311aIAm. After blocking, standards, quality controls, matrix blank, and test samples were added into the plates, respectively. After incubation, TAb in the samples was detected by HRP-labeled goat anti-human-Fc antibody for cynomolgus monkey serum or HRP-labeled goat anti-human-Fab antibody for rat serum. Next steps were same as the quantification of CMG901 in serum.
The same procedures were carried out as above for the quantification of CM311 in cynomolgus monkey or rat serum with the exception that CM311 was used to replace CMG901 in preparing standards and quality controls.
Quantification of MMAE in serum
High-performance liquid chromatography-tandem mass spectrometry (HPLC-MS/MS) method was developed to quantify the serum MMAE. Briefly, the test samples along with the blank matrix and the quality control samples were removed from the low temperature refrigerators and thawed. 50 μL of each sample was added into a 96-well plate and mixed with 20 μL of acetonitrile. The mixture was then passed through a solid phase extraction (SPE) column (Oasis MCX, 30 mg). After washing the column with 0.2% formic acid, the sample was eluted with 2% ammonium hydroxide, dried, and finally re-dissolved in 0.1% formic acid for analysis by LC-MS/MS.
Detection of anti-CMG901 antibodies (ADA) in serum
This assay employed a bridging electrochemiluminescence (ECL) method to detect ADA in the monkey serum. Briefly, the method contains three steps: 1) screening assay, which identifies initial positive or negative samples, 2) confirmation assay, which assesses specificity of the positive screen samples, and 3) titration assay, which estimates the titer level of the ADA for the confirmed positive samples. The assay contained an acid dissociation step of the samples followed by a bridging ECL readout. Acidified samples were added to a mixture which contained Tris base, biotin-labeled CMG901, and Sulfo-TAG-labeled CMG901, and were allowed to form complex with ADA. The labeled drug-ADA complexes are transferred to a streptavidin plate where they were captured. The plate was washed to remove any non-specific binders, and read buffer was added, and the plate was read on an MSD Sector Imager.
Immunohistochemistry (IHC)
To detect CLDN18.2 expression on stable cells, HEK293-CLDN18.2 cells coated on coverslips were fixed with 4% formaldehyde solution and incubated with different antibodies at indicated concentrations at 4°C overnight, followed by incubation with HRP-conjugated secondary antibody at room temperature for 1 h. Chromogenic 3,3′-diaminobenzidine (DAB) substrate was added to visualize the expression of the target proteins.
To detect the expression of CLDN18.2 in human normal and cancer tissue microarray (Jingqin path) and in animal tissue, tissue slides were probed with primary antibodies at indicated concentrations overnight at 4°C, followed by incubation with HRP-conjugated secondary antibody at room temperature for 1 h. Chromogenic 3,3′-diaminobenzidine (DAB) substrate was added to visualize the expression of the target proteins.
Quantification and statistical analysis
Statistical analysis in pharmacokinetic parameters calculation
The primary pharmacokinetic parameters, such as Tmax (Peak time), Cmax (Peak concentration), AUCinf (Area under the concentration-time curve extrapolated from the beginning of administration to infinity time), AUC0-t (Area under the concentration-time curve from the beginning of administration to the last time), Cl (Apparent clearance), MRT (Mean residence time), Vss (Steady-state apparent distribution volume), and Vz (Apparent distribution volume), were calculated by WinNonlin 6.4 software using non-compartmental analysis (NCA). If the concentrations of more than half samples were below LLOQ at the same time point, the data was excluded from the calculation and statistical analysis for average. Dose proportionality was evaluated by linear regression of the exposure parameters and dose in the power model, based on power exponent β calculation. The acceptable criteria for β was 0.8–1.25 with 90% confidence interval.42
Statistical analysis in animal studies
Computer program GraphPad Prism 5 was used for statistical calculation. Tumor volume data were analyzed by Two Way Analysis of Variance (two-Way ANOVA) followed by Bonferroni post-hoc multiple pairwise comparisons (ρ ≤ 0.05).
Published: September 3, 2024
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.xcrm.2024.101710.
Contributor Information
Ruihua Xu, Email: xurh@sysucc.org.cn.
Bo Chen, Email: knybochen@keymedbio.com.
Supplemental information
References
- 1.Coats S., Williams M., Kebble B., Dixit R., Tseng L., Yao N.S., Tice D.A., Soria J.C. Antibody-Drug Conjugates: Future Directions in Clinical and Translational Strategies to Improve the Therapeutic Index. Clin. Cancer Res. 2019;25:5441–5448. doi: 10.1158/1078-0432.CCR-19-0272. [DOI] [PubMed] [Google Scholar]
- 2.Sahin U., Koslowski M., Dhaene K., Usener D., Brandenburg G., Seitz G., Huber C., Türeci O. Claudin-18 splice variant 2 is a pan-cancer target suitable for therapeutic antibody development. Clin. Cancer Res. 2008;14:7624–7634. doi: 10.1158/1078-0432.CCR-08-1547. [DOI] [PubMed] [Google Scholar]
- 3.Sahin U., Türeci Ö., Manikhas G., Lordick F., Rusyn A., Vynnychenko I., Dudov A., Bazin I., Bondarenko I., Melichar B., et al. FAST: a randomised phase II study of zolbetuximab (IMAB362) plus EOX versus EOX alone for first-line treatment of advanced CLDN18.2-positive gastric and gastro-oesophageal adenocarcinoma. Ann. Oncol. 2021;32:609–619. doi: 10.1016/j.annonc.2021.02.005. [DOI] [PubMed] [Google Scholar]
- 4.Hong J.Y., An J.Y., Lee J., Park S.H., Park J.O., Park Y.S., Lim H.Y., Kim K.M., Kang W.K., Kim S.T. Claudin 18.2 expression in various tumor types and its role as a potential target in advanced gastric cancer. Transl. Cancer Res. 2020;9:3367–3374. doi: 10.21037/tcr-19-1876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tureci, Mitnacht-Kraus R., Woll S., Yamada T., Sahin U. Characterization of zolbetuximab in pancreatic cancer models. OncoImmunology. 2019;8:e1523096. doi: 10.1080/2162402X.2018.1523096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cao W., Xing H., Li Y., Tian W., Song Y., Jiang Z., Yu J. Claudin18.2 is a novel molecular biomarker for tumor-targeted immunotherapy. Biomark. Res. 2022;10:38. doi: 10.1186/s40364-022-00385-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhang J., Dong R., Shen L. Evaluation and reflection on claudin 18.2 targeting therapy in advanced gastric cancer. Chin. J. Cancer Res. 2020;32:263–270. doi: 10.21147/j.issn.1000-9604.2020.02.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shitara K., Lordick F., Bang Y.J., Enzinger P., Ilson D., Shah M.A., Van Cutsem E., Xu R.H., Aprile G., Xu J., et al. Zolbetuximab plus mFOLFOX6 in patients with CLDN18.2-positive, HER2-negative, untreated, locally advanced unresectable or metastatic gastric or gastro-oesophageal junction adenocarcinoma (SPOTLIGHT): a multicentre, randomised, double-blind, phase 3 trial. Lancet. 2023;401:1655–1668. doi: 10.1016/S0140-6736(23)00620-7. [DOI] [PubMed] [Google Scholar]
- 9.Xu R.-h., Shitara K., Ajani J.A., Bang Y.-J., Enzinger P.C., Ilson D.H., Lordick F., Van Cutsem E., Gallego Plazas J., Huang J., et al. Zolbetuximab + CAPOX in 1L claudin-18.2+ (CLDN18.2+)/HER2− locally advanced (LA) or metastatic gastric or gastroesophageal junction (mG/GEJ) adenocarcinoma: Primary phase 3 results from GLOW. J. Clin. Oncol. 2023;41:405736. doi: 10.1200/JCO.2023.41.36_suppl.405736. [DOI] [Google Scholar]
- 10.Qi C., Gong J., Li J., Liu D., Qin Y., Ge S., Zhang M., Peng Z., Zhou J., Cao Y., et al. Claudin18.2-specific CAR T cells in gastrointestinal cancers: phase 1 trial interim results. Nat. Med. 2022;28:1189–1198. doi: 10.1038/s41591-022-01800-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fu Z., Li S., Han S., Shi C., Zhang Y. Antibody drug conjugate: the "biological missile" for targeted cancer therapy. Signal Transduct. Target. Ther. 2022;7:93. doi: 10.1038/s41392-022-00947-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kellner C., Otte A., Cappuzzello E., Klausz K., Peipp M. Modulating Cytotoxic Effector Functions by Fc Engineering to Improve Cancer Therapy. Transfus. Med. Hemother. 2017;44:327–336. doi: 10.1159/000479980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tsuchikama K., An Z. Antibody-drug conjugates: recent advances in conjugation and linker chemistries. Protein Cell. 2018;9:33–46. doi: 10.1007/s13238-016-0323-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Staudacher A.H., Brown M.P. Antibody drug conjugates and bystander killing: is antigen-dependent internalisation required? Br. J. Cancer. 2017;117:1736–1742. doi: 10.1038/bjc.2017.367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Muller P., Martin K., Theurich S., Schreiner J., Savic S., Terszowski G., Lardinois D., Heinzelmann-Schwarz V.A., Schlaak M., Kvasnicka H.M., et al. Microtubule-depolymerizing agents used in antibody-drug conjugates induce antitumor immunity by stimulation of dendritic cells. Cancer Immunol. Res. 2014;2:741–755. doi: 10.1158/2326-6066.CIR-13-0198. [DOI] [PubMed] [Google Scholar]
- 16.Sung H., Ferlay J., Siegel R.L., Laversanne M., Soerjomataram I., Jemal A., Bray F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA. Cancer J. Clin. 2021;71:209–249. doi: 10.3322/caac.21660. [DOI] [PubMed] [Google Scholar]
- 17.Shitara K., Bang Y.J., Iwasa S., Sugimoto N., Ryu M.H., Sakai D., Chung H.C., Kawakami H., Yabusaki H., Lee J., et al. Trastuzumab Deruxtecan in Previously Treated HER2-Positive Gastric Cancer. N. Engl. J. Med. 2020;382:2419–2430. doi: 10.1056/NEJMoa2004413. [DOI] [PubMed] [Google Scholar]
- 18.Zhao B., Zhao H., Zhao J. Efficacy of PD-1/PD-L1 blockade monotherapy in clinical trials. Ther. Adv. Med. Oncol. 2020;12 doi: 10.1177/1758835920937612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jeffrey Chi R.P., Rehman H., Goyal S., Saif M.W. Recent advances in immunotherapy for pancreatic cancer. J Cancer Metastasis Treat. 2020;6:43. [Google Scholar]
- 20.Hu J.X., Zhao C.F., Chen W.B., Liu Q.C., Li Q.W., Lin Y.Y., Gao F. Pancreatic cancer: A review of epidemiology, trend, and risk factors. World J. Gastroenterol. 2021;27:4298–4321. doi: 10.3748/wjg.v27.i27.4298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yan X., Lei L., Li H., Cao M., Yang F., He S., Zhang S., Teng Y., Li Q., Xia C., Chen W. Stomach cancer burden in China: Epidemiology and prevention. Chin. J. Cancer Res. 2023;35:81–91. doi: 10.21147/j.issn.1000-9604.2023.02.01. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Donaghy H. Effects of antibody, drug and linker on the preclinical and clinical toxicities of antibody-drug conjugates. mAbs. 2016;8:659–671. doi: 10.1080/19420862.2016.1156829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pellino A., Brignola S., Riello E., Niero M., Murgioni S., Guido M., Nappo F., Businello G., Sbaraglia M., Bergamo F., et al. Association of CLDN18 Protein Expression with Clinicopathological Features and Prognosis in Advanced Gastric and Gastroesophageal Junction Adenocarcinomas. J. Pers. Med. 2021;11:1095. doi: 10.3390/jpm11111095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Xu B., Liu F., Liu Q., Shi T., Wang Z., Wu N., Xu X., Li L., Fan X., Yu L., et al. Highly expressed Claudin18.2 as a potential therapeutic target in advanced gastric signet-ring cell carcinoma (SRCC) J. Gastrointest. Oncol. 2020;11:1431–1439. doi: 10.21037/jgo-20-344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Weng W., Zhang M., Ni S., Tan C., Xu M., Wang X., Sun H., Wang L., Huang D., Sheng W. Decreased expression of claudin-18.2 in alpha-fetoprotein-producing gastric cancer compared to conventional gastric cancer. J. Gastrointest. Oncol. 2022;13:1035–1045. doi: 10.21037/jgo-22-462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tureci O., Sahin U., Schulze-Bergkamen H., Zvirbule Z., Lordick F., Koeberle D., Thuss-Patience P., Ettrich T., Arnold D., Bassermann F., et al. A multicentre, phase IIa study of zolbetuximab as a single agent in patients with recurrent or refractory advanced adenocarcinoma of the stomach or lower oesophagus: the MONO study. Ann. Oncol. 2019;30:1487–1495. doi: 10.1093/annonc/mdz199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Data on file. Northbrook, Ill. Astellas Pharma Inc. 2023.
- 28.Kubota Y., Kawazoe A., Mishima S., Nakamura Y., Kotani D., Kuboki Y., Bando H., Kojima T., Doi T., Yoshino T., et al. Comprehensive clinical and molecular characterization of claudin 18.2 expression in advanced gastric or gastroesophageal junction cancer. ESMO Open. 2023;8:100762. doi: 10.1016/j.esmoop.2022.100762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jiang H., Shi Z., Wang P., Wang C., Yang L., Du G., Zhang H., Shi B., Jia J., Li Q., et al. Claudin18.2-Specific Chimeric Antigen Receptor Engineered T Cells for the Treatment of Gastric Cancer. J. Natl. Cancer Inst. 2019;111:409–418. doi: 10.1093/jnci/djy134. [DOI] [PubMed] [Google Scholar]
- 30.Montes de Oca R., Alavi A.S., Vitali N., Bhattacharya S., Blackwell C., Patel K., Seestaller-Wehr L., Kaczynski H., Shi H., Dobrzynski E., et al. Belantamab Mafodotin (GSK2857916) Drives Immunogenic Cell Death and Immune-mediated Antitumor Responses In Vivo. Mol. Cancer Ther. 2021;20:1941–1955. doi: 10.1158/1535-7163.MCT-21-0035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kroemer G., Galluzzi L., Kepp O., Zitvogel L. Immunogenic cell death in cancer therapy. Annu. Rev. Immunol. 2013;31:51–72. doi: 10.1146/annurev-immunol-032712-100008. [DOI] [PubMed] [Google Scholar]
- 32.Iwata T.N., Ishii C., Ishida S., Ogitani Y., Wada T., Agatsuma T. A HER2-Targeting Antibody-Drug Conjugate, Trastuzumab Deruxtecan (DS-8201a), Enhances Antitumor Immunity in a Mouse Model. Mol. Cancer Ther. 2018;17:1494–1503. doi: 10.1158/1535-7163.MCT-17-0749. [DOI] [PubMed] [Google Scholar]
- 33.Vitorino P., Chuang C.H., Iannello A., Zhao X., Anderson W., Ferrando R., Zhang Z., Madhavan S., Karsunky H., Saunders L.R. Rova-T enhances the anti-tumor activity of anti-PD1 in a murine model of small cell lung cancer with endogenous Dll3 expression. Transl. Oncol. 2021;14:100883. doi: 10.1016/j.tranon.2020.100883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Huang L., Wang R., Xie K., Zhang J., Tao F., Pi C., Feng Y., Gu H., Fang J. A HER2 target antibody drug conjugate combined with anti-PD-(L)1 treatment eliminates hHER2+ tumors in hPD-1 transgenic mouse model and contributes immune memory formation. Breast Cancer Res. Treat. 2022;191:51–61. doi: 10.1007/s10549-021-06384-4. [DOI] [PubMed] [Google Scholar]
- 35.Samuel J., Klempner J.A.A., Al-Batran S.-E., Bang Y.-J., Catenacci D.V.T., Enzinger P.C. Phase II study of zolbetuximab plus pembrolizumab in claudin 18.2: Positive locally advanced or metastatic gastric or gastroesophageal junction adenocarcinoma (G/GEJ)—ILUSTRO Cohort 3. J. Clin. Oncol. 2021;39 [Google Scholar]
- 36.Driehuis E., van Hoeck A., Moore K., Kolders S., Francies H.E., Gulersonmez M.C., Stigter E.C.A., Burgering B., Geurts V., Gracanin A., et al. Pancreatic cancer organoids recapitulate disease and allow personalized drug screening. Proc. Natl. Acad. Sci. USA. 2019;116:26580–26590. doi: 10.1073/pnas.1911273116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.GTEx Consortium The Genotype-Tissue Expression (GTEx) project. Nat. Genet. 2013;45:580–585. doi: 10.1038/ng.2653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Goldman M.J., Craft B., Hastie M., Repečka K., McDade F., Kamath A., Banerjee A., Luo Y., Rogers D., Brooks A.N., et al. Visualizing and interpreting cancer genomics data via the Xena platform. Nat. Biotechnol. 2020;38:675–678. doi: 10.1038/s41587-020-0546-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Howe K.L., Achuthan P., Allen J., Allen J., Alvarez-Jarreta J., Amode M.R., Armean I.M., Azov A.G., Bennett R., Bhai J., et al. Ensembl 2021. Nucleic Acids Res. 2021;49:D884–D891. doi: 10.1093/nar/gkaa942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ran F.A., Hsu P.D., Wright J., Agarwala V., Scott D.A., Zhang F. Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 2013;8:2281–2308. doi: 10.1038/nprot.2013.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hendriks D., Artegiani B., Hu H., Chuva de Sousa Lopes S., Clevers H. Establishment of human fetal hepatocyte organoids and CRISPR-Cas9-based gene knockin and knockout in organoid cultures from human liver. Nat. Protoc. 2021;16:182–217. doi: 10.1038/s41596-020-00411-2. [DOI] [PubMed] [Google Scholar]
- 42.Smith B.P., Vandenhende F.R., DeSante K.A., Farid N.A., Welch P.A., Callaghan J.T., Forgue S.T. Confidence interval criteria for assessment of dose proportionality. Pharm. Res. (N. Y.) 2000;17:1278–1283. doi: 10.1023/a:1026451721686. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
All data reported in this paper will be shared by the lead contact upon request.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.