

Abstract
Delta helicase is a 5′ to 3′ DNA helicase that partially co-purifies with DNA polymerase delta (pol delta) from fetal bovine thymus tissue. We describe the resolution of delta helicase from pol delta on heparin–agarose chromatography and its purification to apparent homogeneity by affinity purification on single-stranded DNA–cellulose chromatography, unique-sequence RNA–agarose chromatography, and ceramic hydroxyapatite chromatography. Delta helicase isolated from fetal bovine thymus had an apparent Mr of 115 kDa in SDS–PAGE, and photo-crosslinked to [α-32P]ATP. Tandem mass spectrometry peptide mass data derived from the bovine polypeptide matched to human UPF1 (HUPF1), a 5′ to 3′ RNA and DNA helicase, and a requisite component of the mRNA surveillance complex. Antisera against HUPF1 cross-reacted with delta helicase on western analysis, and delta helicase activity was immunoinactivated by pre-incubation with antibodies to HUPF1, suggesting that delta helicase is the bovine homolog of HUPF1. Immunoprecipitation experiments demonstrated that HUPF1 interacts with the 66-kDa third subunit of pol delta in vivo.
INTRODUCTION
Helicases, an important class of enzymes involved in numerous nucleic acid transactions, are partly defined by the presence of seven conserved amino acid sequence domains found in two major helicase superfamilies (1–4). Helicases are nucleic acid-dependent NTPases that unwind duplex nucleic acids by translocating unidirectionally along one strand, either 5′ to 3′ or 3′ to 5′, thus displacing the other strand (1–3). It is widely believed that hydrolysis of the nucleoside triphosphate fuels the helicase translocation, by driving the energetically unfavorable disruption of the stable hydrogen bonds between duplex nucleic acid strands (2–6). Many of the known helicases have been shown to be active as oligomers, either hexamers or dimers, whereas some appear to be active as monomers (3,4). Interest in this class of enzymes has been heightened by the ever-growing list of human diseases attributed to defects in genes encoding DNA helicases, e.g. Cockayne’s syndrome (7), xeroderma pigemtosum (7), Bloom’s syndrome (8), Werner’s syndrome (9) and Rothmund–Thompson syndrome (10). All of these diseases are characterized by genomic instability and predisposition to various forms of cancer (7,11,12).
Faithful and timely replication of the genome is essential for cell viability. This has been shown to require DNA helicase(s) to open replication origins as well as to unwind duplex DNA at replication forks in several model systems (13). DnaB, a hexameric 5′ to 3′ DNA helicase, is the replicative helicase in Escherichia coli (14,15). The identity of the replicative helicase(s) in eukaryotes is still unknown, however, it may prove to be the mini-chromosome maintenance (MCM2-7) proteins that have recently been shown to be required for replication fork progression (16) as well as for initiation of DNA replication (17).
RNA helicase activities are required for many RNA processes including transcription, translation initiation and splicing, as well as for transcript stability (18). Several helicases have been reported to have little or no substrate specificity, and can thus utilize duplex RNA or DNA as substrates, e.g. nucleolin (19), nuclear DNA helicase II (20), SV-40 large T-antigen (2), eIF-4A (2) and yeast Upf1 (21), an ATP-dependant 5′ to 3′ helicase required for mRNA surveillance or nonsense-mediated decay (NMD). The NMD pathway ensures that unproductive mRNAs are not translated by selectively degrading mRNAs containing premature termination codons (22,23). Yeast NMD requires several genes, including up-frameshift 1 (UPF1), UPF2 and UPF3 (24). The NMD pathway and the requisite helicases involved are conserved across the eukaryotes from yeast to humans (25). The human homolog of yeast Upf1 is the human UPF1 (HUPF1) helicase (26). HUPF1 has been reported to have an ATP-dependent 5′ to 3′ DNA helicase activity, and ATPase activity dependent on RNA or DNA substrates (27). At least in the yeast model, NMD requires an appropriately positioned downstream element (DSE) (28). Further, it has been reported that yeast DSEs must be bound by a heteronuclear ribonuclear protein (hnRNP), Hrp1/Nab4, which interacts with Upf1 to facilitate NMD (29). All of the DSEs studied in yeast thus far have contained an invariant core nucleotide sequence, GAUG (30).
In this report we describe the purification of delta helicase, an ATP-dependent DNA helicase that co-purifies with DNA polymerase delta (pol delta) through a number of chromatographic columns, but separates from pol delta on heparin–agarose chromatography. The helicase has been shown to have a 5′ to 3′ directionality and a strong preference for a fork-like DNA substrate (31). Delta helicase was thought to be a candidate for a replicative DNA helicase because of its substrate preference and its association with pol delta (31). In an effort to identify the cDNA encoding delta helicase, the enzyme was purified to homogeneity from fetal bovine thymus and analyzed by tandem mass spectrometry. Surprisingly, the resulting peptide mass data derived from the bovine polypeptide matched HUPF1. In addition, western analyses, immunoinactivation studies and comparisons of biochemical characteristics indicated that delta helicase is the bovine homolog of HUPF1.
MATERIALS AND METHODS
Materials
[3H]dTTP was obtained from ICN. Both [α-32P]ATP and [γ-32P]ATP were from New England Nuclear. dA2500 and dT12–18 were from Midland Certified Reagents (Midland, TX). Monoclonal antibody to human hnRNP L (4D11) was a kind gift from Dr G. Dreyfuss, University of Pennsylvania School of Medicine (Philadelphia, PA). Polyclonal antipeptide rabbit antibody to the 66-kDa subunit of human pol delta and the p66-pET19b plasmid were generous gifts from Dr P. Hughes (CNRS, Villejuif, France). Polyclonal chicken antibody to the 50-kDa subunit of human pol delta was prepared by Alpha Diagnostic International (San Antonio, TX). Polyclonal rabbit antibody to the N-terminal one-third of the 125-kDa subunit of human pol delta, expressed as a glutathione-S-transferase (GST)-fusion protein, was prepared as previously described (32). Polyclonal antipeptide rabbit antibody to HUPF1 was prepared as previously described (26). Polyclonal antiserum to the N-terminal 266 residues of HUPF1, expressed as a GST-fusion protein, was a kind gift from Dr L. E. Maquat, Roswell Park Cancer Institute (Buffalo, NY). Monoclonal antibody to cMyc (9E10) was a generous gift from Dr Jin-Qiu Zhou, Princeton University (Princeton, NJ). Horseradish peroxidase (HRP)-conjugated anti-cMyc (9E10) monoclonal antibody was from Roche Molecular Biochemicals (Indianapolis, IN). HRP-conjugated rabbit anti-mouse immunoglobulin (IgG), HRP-conjugated mouse anti-rabbit IgG and HRP-conjugated donkey anti-chicken IgY were from Jackson ImmunoResearch Laboratories (West Grove, PA). Partially purified delta helicase was isolated from fetal bovine thymus with pol delta through step 5 of the pol delta purification procedure (33). Pol delta and the proliferating cell nuclear antigen (PCNA) were prepared as described previously (33). Heparin II-S agarose and single-stranded DNA–cellulose (ssDNAc) chromatographic matrices were obtained from Sigma-Aldrich (St Louis, MO). The ceramic hydroxyapatite (CHA) column (Bio Scale CHT5-I HA) was obtained from Bio-Rad Laboratories (Hercules, CA).
Chromatography buffers
Buffer A: 50 mM Tris–HCl (pH 7.8), 20% glycerol, 0.5 mM EDTA, 0.1 mM EGTA, 1 mM dithiothreitol (DTT) and 0.2 mM phenylmethylsulfonyl fluoride (PMSF). Buffer B: 50 mM Tris–HCl (pH 7.8), 1 mM DTT, 0.2 mM PMSF, 0.5 mM EDTA, 0.1 mM EGTA, 10% glycerol, 0.01% NP-40, 0.2 µg/ml aprotinin, 0.2 µg/ml leupeptins, 0.1 µg/ml pepstatin A, 0.1 µg/ml antipain. Buffer C: the same as Buffer B except that glycerol was 20%. Buffer D: 20 mM potassium phosphate (KPi) (pH 7.0), 10% glycerol, 0.01% NP-40 and 20 mM KCl. FPLC buffers were filtered through a 0.22 µm filter before use.
Helicase assay
Helicase activity was assayed as previously described (31). Briefly, reaction mixtures (25 µl) contained 80 mM KPi (pH 6.5), 2.5 µg/ml bovine serum albumin (BSA), 3 mM MgCl2, 0.8 mM ATP-MgCl2, 0.5 mM DTT, 1–3 fmol of 5′-[32P]-labeled substrate (9 µCi/pmol), and helicase. Reaction mixtures were incubated at 37°C for 60–120 min, stopped with load/run buffer, loaded onto 8% non-denaturing PAGE and resolved with cooling at 12°C in a Hoeffer SE600 apparatus. Helicase substrate was prepared as described (31). It consists of a 5′-[32P]-50mer partially annealed to M13mp19 (+) DNA. The 5′-[32P]-50mer was designed such that only the 5′ 30 nt are complimentary to M13mp19 (+) DNA and the remaining 3′ 20 nt are non-complimentary, resulting in the formation of a fork-like substrate. Helicase activity was measured by the displacement of the 5′-[32P]-50mer from the partial duplex. Reaction products were visualized and quantified with a PhosphorImager and ImageQuant (Molecular Dynamics). One unit of helicase activity is defined as 15% displacement of 5′-[32P]-50mer/30 min.
Polymerase assay
DNA polymerase activity was assayed with poly (dA)/oligo (dT) (10:1 ratio) as the template/primer in the presence and absence of PCNA as described (32).
Preparation of TK119/RNA–Sepharose
RNA was synthesized with a RIBOMAX™ Large Scale RNA Production System (Promega) according to the manufacturer’s instructions, utilizing a pGEM plasmid (pGEM/TK119) encoding a 119-nt sequence from the herpes simplex virus thymidine kinase (HSV-TK) gene that was kindly provided by Dr L. E. Mertz, University of Wisconsin (Madison, WT). The RNA was isolated and coupled to CNBr-activated Sepharose 4B (Pharmacia) according to manufacturer’s protocols.
UV photo-crosslinking
Photo-crosslinking of purified delta helicase with [α-32P]ATP was performed essentially as described (34), with some modification. The final reaction volume (30 µl) contained 66 mM KPi (pH 7.0), 4 mM MgCl2, 5% glycerol, 1 mM DTT, 40 µM [α-32P]ATP (0.6 µCi/pmol) and ∼0.4 µg delta helicase. Reaction mixtures were pre-incubated on ice for 5 min, and then irradiated with a 15 W/254 nm UV lamp at a distance of 3 cm for 10 min. Reaction products were resolved on SDS–PAGE (4% stacking gel, 7.5% separating gel) and visualized by autoradiography. Protein bands were detected with a Colloidal Blue Staining Kit (Novex) according to the manufacturer’s protocol.
Tandem mass spectrometry peptide analysis
Purified delta helicase was dialyzed against water and dried by vacuum. The dried sample was dissolved in 1× SDS–PAGE sample buffer and resolved on SDS–PAGE (4% stacking gel, 10% separating gel). The 115-kDa peptide band was excised, and the gel slice was equilibrated in 50% CNCH3. The polypeptide was digested with trypsin, and the tryptic peptides were resolved on microcapillary reverse-phase HPLC and subjected to nano-electrospray tandem mass spectrometry (µLC/MS/MS) analysis on a Finnagan LCQ quadrapole ion trap mass spectrometer at the Harvard Microchemistry Facility (Cambridge, MA). The resulting tryptic peptide mass data were matched against a database containing the predicted tryptic peptide masses for all of the known human genes.
Microsequencing
Bovine hnRNP L was either directly resolved on SDS–PAGE (4% stacking gel, 7.5% separating gel) or first subjected to cyanogen bromine (CNBr) digestion prior to SDS–PAGE and then electroblotted onto PVDF membranes. The electroblotted bands were excised and subjected to microsequencing by the Protein Core Facility, University of Miami School of Medicine (Miami, FL) or at the ICBR Protein Chemistry Core Facility, University of Florida (Gainesville, FL).
Immunoinactivation of helicase activity
Delta helicase reaction mixtures were pre-incubated on ice for 1 h with the indicated antibodies, then incubated at 37°C for an additional hour. Products were resolved on non-denaturing 8% PAGE and quantified as described above for helicase assays. Prior to use, antibodies were purified on protein A–Sepharose and the IgG concentration determined by the Bradford method with ovalbumin as standard.
Immunoblotting
Protein samples were resolved by SDS–PAGE (4% stacking gel, 7.5 or 10% separating gel) and electroblotted onto nitrocellulose membranes. Membranes were blocked for 1 h with blocking buffer (1× PBS, pH 7.4, 0.1% Tween-20, 5% non-fat milk), and incubated overnight with the indicated primary antibody. Membranes were washed with blocking buffer for 1 h, then incubated with either HRP-conjugated rabbit anti-mouse IgG, HRP-conjugated mouse anti-rabbit IgG, or HRP-conjugated donkey anti-chicken IgY for 1 h. Membranes were washed with wash buffer (1× PBS, pH 7.4, 0.1% Tween-20) four times for 15 min each, then developed with Super Signal West Pico Chemiluminesent Substrate (Pierce) and visualized by fluorography. Protein samples from transfected HeLa cell lysates were treated as described above except that membranes were incubated with HRP-conjugated primary antibody, HRP-conjugated anti-cMyc (9E10) monoclonal antibody, at 1:1000 dilution.
Construction of cMyc tagged expression vectors
All PCR amplifications were carried out utilizing PCR High Fidelity Super Mix (Invitrogen). All primers contained KpnI or NotI restriction sites (underlined). The coding sequence for the 66-kDa subunit of human pol delta was PCR amplified using a pET19b plasmid containing the KIAA0039 coding sequence as template (35), and the following primers: forward primer, 5′-TTTGGTACCATGGCGGACCTTTATCTG; reverse primer, 5′-TTTGCGGCCGCCTAGATGAACTTACATAG. The coding sequence for the 50-kDa subunit of human pol delta was PCR amplified using the plasmid pET16b-p50 (36) as template and the following primers: forward primer, 5′-TTTGGTACCATGTTTTCTGAGCAGGCTGCC; reverse primer, 5′-TTTGCGGCCGCTCAGGGGCCCAGCCC. The coding sequence for β-lactamase was PCR amplified using the plasmid pcDNA3.1 (Invitrogen) containing the β-lactamase gene (bla) as template and the following primers: forward primer, 5′-TTTGGTACCATGAGTATTCAACATTTC; reverse primer, 5′-TTTGCGGCCGCTTACCAATGCTTAATCAGTG. The coding sequence for β-galactosidase was PCR amplified using a genomic template derived from E.coli DB3.1 (Invitrogen) containing the β-galactosidase gene (lacZ) and the following primers: forward primer, 5′-TTTGGTACCATGACCATGATTACGGATTCACTG; reverse primer, 5′-TTTGCGGCCGCTTATTTTTGACACCAGACCAACTGG. The PCR products and the pCMV-Myc vector plasmid were digested with KpnI and NotI (New England Biolabs), purified on a 1% agarose gel and recovered using a Gel Extraction Kit (Qiagen).
The HUPF1 coding sequence (26) was obtained directly from a pGEM-HUPF1 construct after digestion with XhoI (New England Biolabs), purified on a 1% agarose gel, and recovered using a Gel Extraction Kit (Qiagen). The pCMV-Myc vector plasmid was also digested with XhoI, then dephosphorylated twice with calf intestinal alkaline phosphatase (Invitrogen).
The PCR-amplified cDNAs were ligated into the pCMV-Myc vector digested with KpnI and NotI, and the HUPF1 cDNA was ligated into the dephosphorylated, XhoI-digested pCMV-Myc vector using T4 Ligase (New England Biolabs). Ligation reactions were used to transform E.coli DH-5α cells, and positive clones were used to prepare large quantities of plasmid DNA for transfection with a QIAFilter Plasmid Maxi Kit (Qiagen). Plasmids used for transfections were first confirmed by DNA sequence analysis at the Vector Core Facility (Sylvester Comprehensive Cancer Center, Miami, FL).
Transfection of HeLa cells
Transfections were performed utilizing LIPOFECTIN and PLUS reagents (Invitrogen) according to the manufacturer’s protocol for HeLa cells. Briefly, HeLa cells were grown to 60–80% confluence in 60 mm tissue culture plates in DMEM with 10% fetal bovine serum (FBS) and 0.1 mM non-essential amino acids (NEAA). LIPOFECTIN and PLUS reagents were preincubated with 2 µg of plasmid DNA, then added to tissue culture dishes with freshly replaced DMEM containing only 0.1 mM NEAA. Transfections were incubated for 3 h at 37°C in 5% CO2 before adding DMEM with 0.1 mM NEAA and 15% FBS, to a final FBS concentration of 10%. Cells were incubated at 37°C in 5% CO2 for 40 h before harvesting.
Co-immunoprecipitation
Transfected HeLa cells were washed twice with cold PBS, lysed by adding 0.6 ml lysis buffer [10 mM Tris–HCl (pH 7.4), 10 mM EDTA, 1% NP-40, 0.4% deoxycholic acid, 8 µg/ml aprotinin, 10 µg/ml leupeptins, 10 µg/ml antipain, 2 µg/ml pepstatin A], incubated on ice for 15 min and the lysates were centrifuged at 12 000 g for 2 min. The co-immunoprecipitation reactions were assembled on ice in the following order: 0.25 ml of cleared lysate supernatant, 0.25 ml of NET gel buffer [0.1% NP-40, 50 mM Tris–HCl (pH 7.4), 1 mM EDTA, 150 mM NaCl, 0.25% gelatin, 0.2% NaN3], 5 µg anti-cMyc monoclonal antibody (9E10). Reactions were incubated on ice for 2 h then 30 µl of Gamma Bind Plus Sepharose beads (Amersham Pharmacia Biotech) were added and mixed for 1 h. The immunocomplexes were precipitated by centrifugation at 700 g for 5 min, washed with 0.5 ml of a mixture of equal parts lysis buffer and NET gel buffer for 5 min and the washing was repeated four times. The washed pellets were resuspended in 2× SDS–PAGE sample buffer, and heated at 95°C for 4 min. The heated samples were centrifuged at 10 000 g for 90 s and the supernatants were recovered for SDS–PAGE.
RESULTS
Separation of delta helicase from pol delta on heparin–agarose chromatography and further purification on ssDNAc chromatography
The series of chromatographic steps utilized to co-purify pol delta and delta helicase were as previously described (33), and the series was, in order: DEAE–cellulose, phenyl–Sepharose, phosphocellulose and hydroxyapatite. The starting material for the purification of delta helicase was a hydroxyapatite-purified pol delta sample, i.e. Step 5 enzyme (33), of a pol delta preparation from 1.5 kg of fetal bovine thymus. The Step 5 enzyme was loaded onto a heparin–agarose column (2.3 × 5.6 cm) in Buffer A containing 75 mM KCl, washed with the same buffer and eluted with a 600 ml gradient from 75 to 235 mM KCl in Buffer A. Fractions were assayed for pol delta activity in the presence and absence of PCNA and for delta helicase activity in the presence and absence of ATP (Fig. 1). ATP-dependent helicase activity eluted between 105 and 175 mM KCl, with a broad peak of activity centered at ∼125 mM KCl, while PCNA-dependent pol delta activity eluted between 175 and 235 mM KCl, with a sharp activity peak at 200 mM.
Figure 1.

Elution profiles of pol delta and delta helicase activities on heparin–agarose chromatography. DNA polymerase activity was assayed using poly(dA)/oligo(dT) as template/primer in the presence (closed squares) and absence (open squares) of PCNA, as described in Materials and Methods, and is reported as pmol dTMP incorporated/15 min. Helicase activity was assayed in the presence (closed circles) and absence (open circles) of ATP using a fork-like substrate constructed from single-stranded M13 (+) DNA and a partially annealed 5′-[32P]-50mer, as described in Materials and Methods, and is reported as integrated volume of displaced 50mer. The KCl concentration of the fractions (open triangles) is also indicated (mM).
The helicase-containing fractions from three heparin–agarose columns (∼4.5 kg of fetal bovine thymus total starting material) were pooled, dialyzed against Buffer B containing 100 mM KCl and loaded onto a pre-equilibrated HR 10/10 FPLC ssDNAc column (1 × 8.6 cm). After washing with Buffer B containing 250 mM KCl, then with 500 mM KCl, the column was eluted stepwise with 1 M (fractions 59–71) and 2 M (fractions 87–97) KCl in Buffer B (Fig. 2). Aliquots of eluted fractions were dialyzed against Buffer B and assayed for helicase activity in the presence (Fig. 2A) and absence (Fig. 2B) of ATP. Most of the helicase activity eluted at 1 and 2 M KCl, whereas most of the contaminating nuclease activity eluted at 250 and 500 mM KCl (data not shown). Nuclease activity also eluted at 1 M KCl, as evidenced by the ATP-independent disappearance of substrate and lack of appearance of [32P]-50mer (Fig. 2B, fractions 62–63).
Figure 2.

Affinity purification of delta helicase on FPLC ssDNAc chromatography. Helicase activity was assayed in the presence (A) and absence (B) of ATP as described in Materials and Methods. Depicted in (A) and (B) are phosphorimages of helicase assays resolved on non-denaturing 8% PAGE gels. The migration positions of the fork-like helicase substrate (Substrate) and the displaced 50mer (Product) are indicated to the left. The letters above the lanes are: H, substrate heated at 90°C for 10 min as a positive control; U, unheated substrate as a negative control; BC, before column sample; FT, flow through sample after loading. The numbers above the lanes indicate the column fraction numbers. (C) Aliquots of the ssDNAc column fractions were resolved by SDS–PAGE and polypeptide bands were detected by silver staining, as described in Materials and Methods. (D) Aliquots of the ssDNAc column fractions were resolved by SDS–PAGE and western analysis using a monoclonal antibody against the human hnRNP L protein were performed as described in Materials and Methods. For (C and D), the letters above the lanes are: BC, before column sample; M, molecular weight markers with the known kDa position indicated to the left. The numbers above the lanes indicate the column fraction numbers. For (A)–(D), fractions 59–71 eluted with 1 M KCl, and fractions 87–97 eluted with 2 M KCl.
Aliquots of the ssDNAc fractions were subjected to SDS–PAGE and silver stained (Fig. 2C). Polypeptides of the Mr previously reported for delta helicase, i.e. 52–57 kDa (31), were present in the fractions that eluted at 1 M KCl. Sequence analysis of these polypeptides, as well as CNBr fragments, suggested that the polypeptides were highly homologous (63–76% identity) to human hnRNP L (data not shown). Although the reported Mr for human hnRNP L ranges from 64 to 68 kDa (37), the polypeptides isolated here ranged from 57 to 65 kDa, possibly the result of partial degradation. Western analysis of the ssDNAc fractions with antibody to human hnRNP L showed that a major component of the fractions eluting at 1 M KCl was bovine hnRNP L (Fig. 2D). A higher ratio of helicase activity to hnRNP L was detected in the fractions eluting at 2 M KCl (Fig. 2A and D, fractions 87–97), which were also relatively free of nuclease activity (Fig. 2B, fractions 87–97). The major polypeptides in these fractions had an Mr of 110–115 kDa (Fig. 2C, fractions 87–97).
Resolution of delta helicase and hnRNP L on RNA-affinity chromatography
It was previously reported that hnRNP L binds specifically to a 119-nt sequence present in HSV-TK mRNA (38). Consequently, we used this sequence to prepare an RNA–Sepharose column, TK119/RNA–Sepharose, to specifically remove hnRNP L, as described in Materials and Methods. Helicase fractions that eluted from the ssDNAc column at 2 M KCl were dialyzed against Buffer C and loaded onto a pre-equilibrated FPLC TK119/RNA–Sepharose column (1 × 10.8 cm), washed with equilibration buffer containing 75 mM KCl (Fig. 3, fractions 4–20) followed by stepwise elution with Buffer C containing 150 (Fig. 3, fractions 28–40), 225, 300, 400 and 500 mM KCl (data not shown). Essentially all of the helicase activity passed through the TK119/RNA–Sepharose column in the flow through and in the washing buffer (Fig. 3A, FT and fractions 4–12). SDS–PAGE of the TK119/RNA fractions, followed by silver staining (Fig. 3B) detected polypeptides of 95, 112 and 115 kDa that coincided with the helicase activity (Fig. 3B, FT and fractions 4–12). Western analysis with a monoclonal antibody against hnRNP L (Fig. 3C) indicated that hnRNP L eluted at higher KCl concentrations than the helicase activity (Fig. 3C, fractions 12–40). The flow-through fraction and fractions 1–8, which were essentially free of hnRNP L, were pooled and further purified by hydroxyapatite chromatography.
Figure 3.
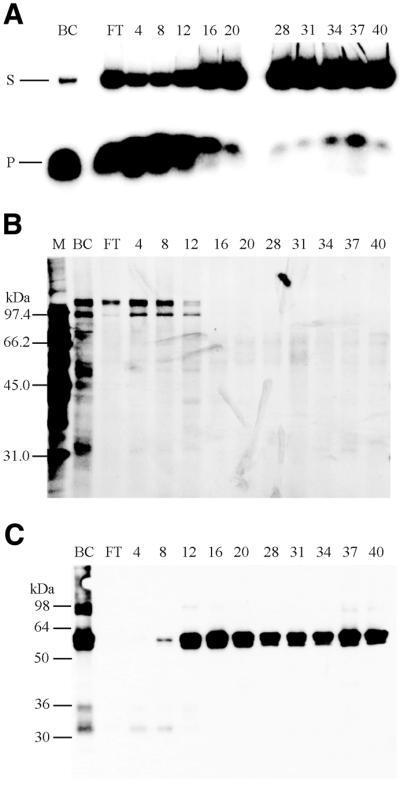
Resolution of delta helicase and hnRNP L on FPLC TK119/RNA–Sepharose chromatography. (A) Helicase activity was assayed in the presence of ATP, reaction products were resolved on 8% non-denaturing PAGE gels, and products were detected by phosphorimaging as described in Materials and Methods. The migration positions of the helicase substrate (S) and the displaced 50mer reaction product (P) are indicated to the left. The letters above the lanes are: BC, before column sample; FT, flow through sample after loading. The numbers above the lanes indicate the fraction numbers. (B) Aliquots of the TK119/RNA–Sepharose column fractions were resolved by SDS–PAGE and polypeptide bands were detected by silver staining, as described in Materials and Methods. (C) Aliquots of the TK119/RNA–Sepharose column fractions were resolved by 10% SDS–PAGE gels and western analysis was carried out as described in Materials and Methods, using a monoclonal antibody against the human hnRNP L protein. For (B) and (C), the letters above the lanes are: BC, before column sample; FT, flow through sample after loading; M, molecular weight markers with the known kDa position indicated to the left. The numbers above the lanes indicate the fraction numbers. For (A)–(C), fractions 4–20 eluted with 75 mM KCl, and fractions 24–40 eluted with 150 mM KCl.
Further purification of delta helicase on CHA FPLC chromatography
The pooled helicase fractions from TK119/RNA–Sepharose chromatography were dialyzed against Buffer D and loaded onto a CHA FPLC column (1 × 6.4 cm) pre-equilibrated with Buffer D. After washing with Buffer D, a 150 ml linear gradient from 20 to 500 mM KPi was developed. A peak of helicase activity eluted sharply at ∼150 mM KPi (Fig. 4A, fractions 50–60). Two polypeptide bands with an apparent Mr of 112 and 115 kDa coincided with the helicase activity (Fig. 4B, fractions 50–60). A fraction containing only a 115-kDa polypeptide, which eluted in the earliest fractions of the helicase activity peak (Fig. 4A and B, fraction 50), was subjected to tandem mass spectrometry analysis.
Figure 4.

Purification of delta helicase on CHA FPLC chromatography. (A) Helicase activity was assayed in the presence of ATP and reaction products were resolved on 8% non-denaturing PAGE gels and detected by phosphorimaging as described in Materials and Methods. The migration positions of the fork-like helicase substrate (Substrate) and the displaced 50mer reaction product (Product) are indicated to the left. The letters above the lanes are: H, substrate heated at 90°C for 10 min as a positive control; U, unheated substrate as a negative control; BC, before column sample; F1 and F2, flow through samples after loading. The numbers above the lanes indicate the fraction numbers. (B) Aliquots of the column fractions were resolved by 10% SDS–PAGE gels and polypeptide bands were detected by silver staining, as described in Materials and Methods. The numbers above the lanes indicate the fraction numbers. M, molecular weight markers with the known kDa position indicated to the left.
UV photo-crosslinking with [α-32P]ATP
A purified sample of delta helicase from CHA FPLC chromatography containing both the 112- and 115-kDa polypeptides (Fig. 4B, fraction 55) was concentrated ∼50-fold with a Centricon-30 spin column. The concentrated sample and a BSA control were UV photo-crosslinked to [α-32P]ATP, as detailed in Materials and Methods, and reaction products were resolved by SDS–PAGE. As shown in Figure 5A, 112- and 115-kDa polypeptides crosslinked with [α-32P]ATP were detected by autoradiography, but the BSA negative control did not crosslink. The gel was stained using Colloidal Blue Staining Kit (Fig. 5B). Both the BSA and helicase 112- and 115-kDa bands were detected. Interestingly, the two [α-32P]ATP crosslinked products were generated when the total ATP concentration was at the Km for delta helicase (40 µM) (31), but not when the total ATP concentration was only 3 µM (data not shown).
Figure 5.
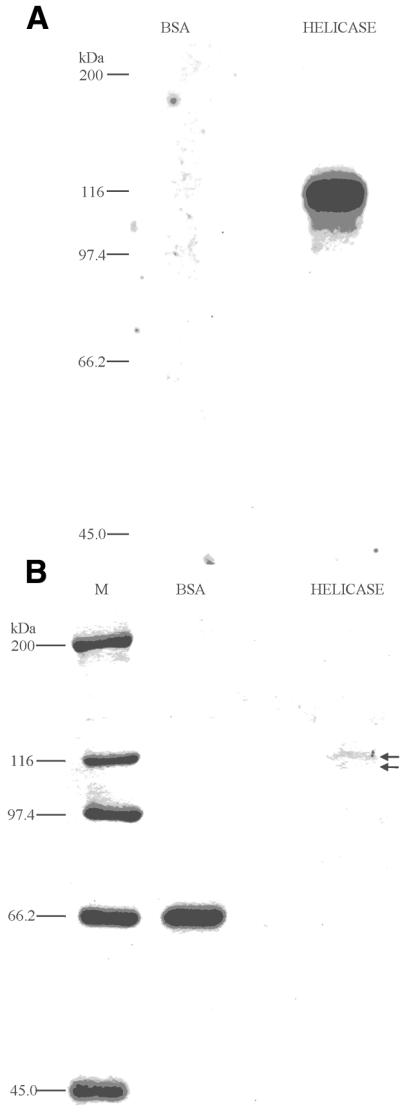
Purified delta helicase UV photo-crosslinks with [α-32P]ATP. Purified delta helicase (Fig. 4, fraction 55) was concentrated ∼50-fold and samples were photo affinity-labeled with [α-32P]ATP by irradiating with UV, as described in Materials and Methods. (A) Photo-crosslinked reaction products were resolved on 10% SDS–PAGE gels and [32P]-labeled products were visualized by autoradiography. (B) Polypeptides were visualized by staining with a Colloidal Blue Staining Kit. The arrows indicate the positions of the 112- and 115-kDa polypeptides. For (A) and (B), the labels above the lanes are: M, molecular weight markers with the known kDa positions indicated to the left; Helicase, concentrated homogeneous delta helicase sample (∼0.4 µg); BSA, bovine serum albumin (3 µg).
Identification of delta helicase as the bovine homolog of HUPF1
The 115-kDa polypeptide from a delta helicase preparation (Fig. 4A, fraction 50) was digested with trypsin and the tryptic peptides were subjected to µLC/MS/MS analysis. The resulting peptide mass data matched the reported sequence of HUPF1 (Fig. 6). The bovine peptide mass data that were consistent with HUPF1(Fig. 6, red letters) encompassed 36.3% of the HUPF1 protein sequence and included both putative zinc finger motifs (Fig. 6, ZnFr1 and ZnFr2) and five of the seven helicase domains (Fig. 6, motifs Ia, Ib, IV, V, VI). Several of the residues known to be invariant in the Superfamily I helicases (Fig. 6, bold letters) are also included in the bovine peptide sequence (Fig. 6, red letters). Moreover, some of the invariant Upf1 homolog zinc finger motif residues that are conserved in yeast and humans (Fig. 6, blue letters) overlap the bovine peptide sequence data (Fig. 6, purple letters). HUPF1 has a 51% overall identity with yeast Upf1, but the central regions containing the zinc fingers and all seven helicase motifs are 60% identical (74% similar), and the regions that consist of the helicase motifs only are 63% identical (85% similar) between yeast Upf1 and HUPF1 (26).
Figure 6.

Tandem mass spectrometry data from delta helicase and sequence of HUPF1. The complete 1129 amino acid sequence deduced from the human HUPF1 cDNA is given. Tryptic peptides of the purified 115-kDa delta helicase polypeptide were subjected to µLC/MS/MS analysis. The tryptic peptide mass data derived from delta helicase matched with the predicted masses for the HUPF1 regions that are indicated in red letters. Two unique zinc finger motifs (ZnFr1 and ZnFr2) and the seven classic helicase domains (Ia, Ib, II, III, IV, V, VI) are underlined. Putative zinc finger motif cysteine (C) and histidine (H) residues are in a larger font size. Residues within the putative zinc finger motifs that are conserved in mammalian, C.elegans, S.cerevisiae and S.pombe Upf1 homologs are indicated by blue letters, and those conserved residues that overlap with the delta helicase sequence are indicated by purple letters. Residues conserved in the Superfamily I helicase domains are in bold.
Upf1 and HUPF1 are 5′ to 3′ RNA helicases required for nonsense-mediated mRNA decay (21,27). Both human and yeast enzymes unwind DNA substrates in vitro, and have essentially identical physical and enzymatic properties. The published biochemical properties of bovine delta helicase (31), yeast Upf1 (21) and HUPF1 (27) are compared in Table 1. Of the properties examined the three proteins differed only in their pH optima. However, it should be noted that both yeast Upf1 and HUPF1 were reported to have very broad pH optima, and remain quite active (at least 50% maximum activity) from pH 4.0 to 6.5 for HUPF1 (27) and from pH 6.5 to 9.5 for yeast Upf1 (21). These findings further support the hypothesis that delta helicase is the bovine homolog of HUPF1.
Table 1. Biochemical properties of delta helicase, Upf1 and HUPF1.
| Delta helicase | Upf1 | HUPF1 | |
|---|---|---|---|
| Directionality |
5′ to 3′ |
5′ to 3′ |
5′ to 3′ |
| Helicase activity |
DNA (RNA?) |
DNA/RNA |
DNA (RNA?) |
|
Km for ATP/dATP (µM) |
40 |
37 |
Not tested |
| Optimum [Mg2+] (mM) |
3 |
2 |
Not tested |
| NTP requirement |
ATP, dATP |
ATP, dATP |
ATP |
| Optimum pH |
6.5 |
8.0 |
5.0 |
| [KCl] inhibition (mM) |
200 |
200 |
Not tested |
| Molecular weight (kDa) | 115 | 109 | 130 |
Immunoinactivation of helicase activity
In order to further test the hypothesis that delta helicase is indeed the bovine homolog of HUPF1, as well as to rule out the possibility that helicases other than bovine UPF1 might co-purify with pol delta, immunoinactivation studies were carried out using antipeptide antibodies to HUPF1 and step 6 delta helicase (following heparin–agarose chromatography) as the source of helicase activity. The heparin–agarose purified delta helicase sample was used because it was the first purification fraction assayed for ATP-dependent helicase activity, and would therefore have to contain any other detectable helicase(s) that might be present. The control antibodies were pre-immune antibodies, antibodies against the N-terminal one-third of the 125-kDa subunit of pol delta, and antipeptide antibodies against the 66-kDa subunit of pol delta. As shown in Figure 7, the pre-immune and anti-p125 antibodies had no appreciable effect on the helicase activity. However, the addition of antibodies against HUPF1 inhibited helicase activity, and complete inhibition was seen at 22 µg of anti-HUPF1 antibody. Interestingly, anti-p66 antibodies had a stimulatory effect on the helicase activity, i.e. up to 50%.
Figure 7.

Immunoinactivation of delta helicase. Immunoinactivation reactions were performed and quantified as described in Materials and Methods and the results represented in the histogram. Briefly, helicase reactions were preincubated with one of four antibodies tested, in three different quantities (either 5.5, 11 or 22 µg of IgG). Reaction products were resolved on 8% PAGE, then detected by phosphorimaging and quantified with ImageQuant (Molecular Dynamics). The results were entered in an Excel (Microsoft) spreadsheet, the percent of the total substrate consumed was calculated, and then converted into a histogram. The abbreviations under the histogram columns are: PreImm, pre-immune antibodies; HUPF1, antipeptide antibody against HUPF1; p125, antibody against an N-terminal pol delta p125 subunit–GST fusion protein; p66, antipeptide antibody against the pol delta p66 subunit.
Co-immunoprecipitation of p66 and HUPF1
In order to determine whether bovine UPF1 physically interacts with pol delta, co-immunoprecipitation experiments were carried out using Step 5 delta helicase and antibodies to HUPF1 and the 125-, 50- and 66-kDa subunits of pol delta. No interaction was detected between bovine UPF1 and either p125 or p50, but a weak interaction was detected between bovine Upf1 and p66 (data not shown). In order to determine whether p66 and HUPF1 interact in vivo, HeLa cells were transfected with expression vectors encoding either cMyc-tagged p66, cMyc-tagged p50, cMyc-tagged HUPF1, cMyc-tagged β-lactamase or cMyc-tagged β-galactosidase, with the latter two used as negative controls. The expressed cMyc-tagged proteins were immunoprecipitated from transfected HeLa cell lysates with a monoclonal antibody against the cMyc epitope and co-immunoprecipitated proteins identified by western analysis as detailed in Materials and Methods. As shown in Figure 8, endogenous p66 co-immunoprecipitated with cMyc-tagged HUPF1, as detected by western analysis for p66 (Fig. 8A, HUPF1 lane). The reciprocal interaction between HUPF1 and p66 was demonstrated by the co-immunoprecipitation of endogenous HUPF1 with cMyc-tagged p66, as detected by western analysis for HUPF1 (Fig. 8B, p66 lane). cMyc-tagged p50, β-lactamase or β-galactosidase did not co-immunoprecipitate endogenous p66 (Fig. 8A, p50, lacZ and bla lanes) or endogenous HUPF1 (Fig. 8B, p50, lacZ and bla lanes). Furthermore, even though the expression levels of cMyc-tagged p66 were three to five times lower than the negative controls, as determined by quantification of western analysis for cMyc in transfected HeLa cell lysates (Fig. 8D), the cMyc-tagged p66 still co-immunoprecipitated endogenous HUPF1, whereas the other three cMyc-tagged proteins did not (Fig. 8B). Similar results were obtained with expression vectors encoding HA-tagged proteins (data not shown). These results suggest that HUPF1 may interact with the third subunit of pol delta in vivo.
Figure 8.
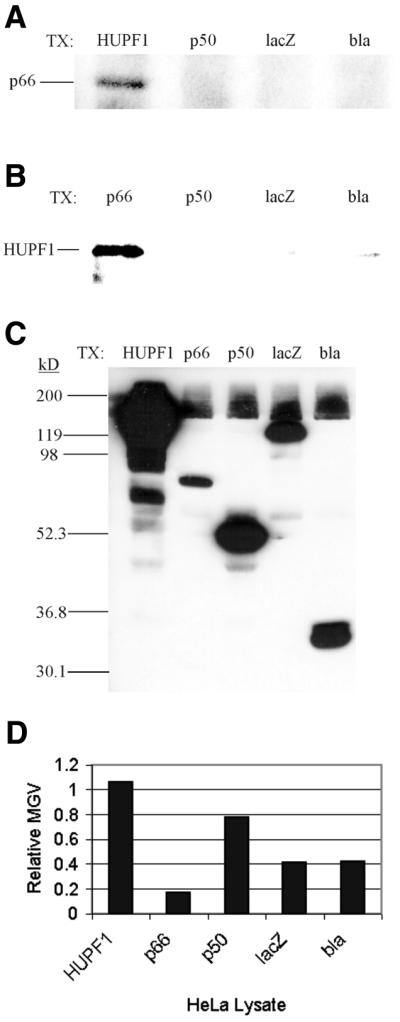
Co-immunoprecipitation of pol delta p66 and HUPF1. Immunoprecipitation complexes were collected from HeLa cells transfected with one of five pCMV-cMyc expression vectors. The complexes were resolved on 10% SDS–PAGE and western analyses were carried out, followed as described in Materials and Methods. (A) Western analysis for endogenous p66 with antipeptide antiserum against the pol delta p66 subunit. (B) Western analysis for endogenous HUPF1 with affinity purified antipeptide antibodies against HUPF1. (C) Western analysis for cMyc with a monoclonal antibody against cMyc (9E10) conjugated to HRP. For (A)–(C) the abbreviations to the right of TX: and above the lanes indicate the pCMV-cMyc expression construct that the HeLa cells were transfected with, as described in Materials and Methods: HUPF1, pCMV-Myc/HUPF1; p66, pCMV-Myc/p66; p50, pCMV-Myc/p50; lacZ, pCMV-Myc/β-galactosidase; bla, pCMV-Myc/β-lactamase. The known positions of p66, HUPF1 and molecular weight markers are indicated to the left. (D) The transfected HeLa cell lysates were resolved by 10% SDS–PAGE and cMyc tagged proteins were detected by western analyses with a monoclonal antibody against cMyc (9E10) conjugated to HRP, followed by fluorography, as described in Materials and Methods. The detected bands were quantified with ImageJ (NCBI) and the measured mean gray values are depicted in an Excel (Microsoft) histogram. The transfected HeLa cell lysates are indicated below the columns, and the mean gray values are indicated on the vertical axis.
Bovine UPF1 co-elutes with the 66-kDa subunit of pol delta but not the 125- and 50-kDa subunits on heparin–agarose chromatography
The behavior of bovine UPF1, the subunits of bovine pol delta and bovine hnRNP L on heparin–agarose chromatography was determined by western analyses of the fractions shown in Figure 1. As shown in Figure 9, polypeptides immunoreactive with antipeptide antibodies to HUPF1 (Fig. 9A) eluted in a broad peak from fraction 40 to 110, consistent with the elution profile of delta helicase activity (Fig. 1). The 125- (Fig. 9B) and 50-kDa (Fig. 9E) subunits of pol delta eluted in fractions 110–150, consistent with the pol delta activity in Figure 1. However, the third subunit of pol delta, p66 (Fig. 9D), separated from the 125- and 50-kDa subunits of the enzyme and eluted in fractions 40–60, coincident with hnRNP L (Fig. 9C) in fractions 40–70, and overlapping the elution profile of bovine UPF1.
Figure 9.

Western analyses of proteins resolved on heparin–agarose chromatography. Aliquots of the heparin–agarose fractions depicted in Figure 1 were subjected to 10% SDS–PAGE, followed by western analyses using (A) antipeptide antibodies against HUPF1, (B) antibodies against pol delta p125, (C) a monoclonal antibody against hnRNP L, (D) antipeptide antibodies against p66, and (E) antiserum against p50. For (A)–(E), the letters above the lanes are: BC, before column sample; L, flow through sample after loading; W, wash sample. The numbers above the lanes indicate the fraction number. The known positions of the molecular weight markers are indicated to the left.
DISCUSSION
We have isolated a 115-kDa polypeptide from fetal bovine thymus tissue that is responsible for the helicase activity originally reported as delta helicase, a 5′ to 3′ DNA helicase that partially co-purifies with pol delta (31). The µLC/MS/MS tryptic peptide mass data derived from the isolated 115-kDa helicase polypeptide matched with HUPF1 (26), a 5′ to 3′ RNA and DNA helicase required for mRNA surveillance (24). The bovine tryptic peptide mass data that matched to HUPF1 encompass more than one-third (36.3%) of the total HUPF1 sequence, and these consistent regions include five of the seven conserved helicase domains and both of the unique putative zinc finger motifs. The identification by µLC/MS/MS peptide analysis of delta helicase as the bovine UPF1 polypeptide was supported by comparison of the physical and enzymatic properties of bovine delta helicase with those of both human and yeast Upf1 (Table 1), by immunoinactivation studies, and by western analyses. In order to corroborate that delta helicase is the bovine homolog of HUPF1, and to demonstrate the presence of bovine UPF1 at all steps of the delta helicase purification, antiserum to the N-terminal 266 residues of HUPF1 was used in western analyses of pooled samples from the delta helicase purification. The data clearly indicate that bovine UPF1 was present in all purification samples (data not shown).
Human and yeast Upf1 are members of the Superfamily I group of helicases. Both the human and yeast enzymes contain two unique putative zinc finger motifs near their N-terminal ends. These zinc fingers are unique because they have unusually long intervening stretches of amino acids between the coordinating cysteine/histidine residues. One of these two motifs contains an extra pair of conserved cysteine residues known as a zinc finger/knuckle motif, found in proteins with dual roles as both RNA and DNA binding proteins (39). Both of the zinc finger motifs are found in yeast Upf1 and HUPF1, as well as in the Caenorhabditis elegans homolog, SMG-2 (40), whereas they are not found in any of the other Superfamily I helicases (26). It is of interest that bacteriophage T7 gene 4 primase/helicase contains a similarly placed zinc finger (41) and the HSV-1 helicase-primase has a single, unusually large zinc finger at the C-terminal end of the UL52 subunit (42).
The significance of the physical interaction of the third subunit of pol delta with HUPF1 is not presently clear, even though we have demonstrated an in vivo interaction between p66 and HUPF1. An interaction between pol delta and HUPF1 might imply a role for HUPF1 in DNA replication or repair, a role for p66 in mRNA surveillance, or a possible sequestering function for one enzyme or the other, such as that seen with the WRN protein recruiting pol delta to the nucleolus (43). In fact, it was previously reported that the Saccharomyces cerevisiae homolog of mammalian p66, Pol32, was required for the functional interaction of pol delta with WRN (44). Pol32 was also found to interact both genetically and physically with a DNA repair helicase, Srs2 (45). Additionally, a helicase involved in Schizosarccharomyces pombe DNA replication, Dna2, was reported to interact both genetically and physically with the S.pombe p66 homolog, Cdc27 (46). Our western analyses indicate that p66 co-elutes with HUPF1 on heparin–agarose chromatography, and our co-immunoprecipitation data strongly suggest an interaction between p66 and HUPF1. However, the possibility of mammalian Upf1 having a physiologically relevant interaction with pol delta is complicated by the difference in the subcellular locations of these two proteins. The nuclear processes that pol delta is known to be involved in, DNA replication and DNA repair, are segregated from the cytoplasmic process that Upf1 has been shown to be required for, i.e. mRNA surveillance. It has been reported that yeast Upf1 is associated with polysomes in the cytoplasm (47), and the subcellular location of the mammalian enzyme is also reported to be cytoplasmic by immunofluorescence (26,48) and western blot analyses (26,49). However, it is possible that a small amount of HUPF1 could be present in the nucleus. HUPF1 has a putative bipartite nuclear localization signal (50), and it was suggested that nonsense-mediated decay occurs both in the cytoplasm and in association with the nucleus in mammalian cells. Other evidence that the translation machinery functions within the mammalian nucleus includes a report that the presence of a premature termination codon, which would be recognized by translation machinery, can influence co-transcriptional events within the nucleus of mouse cells (51), and a recent report that 10–15% of cellular translation is coupled to transcription within the nucleus of mammalian cells (52). These studies suggest the possibility that mammalian Upf1 is, at least in part, localized in the nucleus and is therefore able to interact with mammalian p66. It has recently been shown that murine Upf1 is required for the cellular viability of embryonic stem cells (53). Considering that Upf1 is non-essential in yeast (54), the absolute requirement for murine UPF1 may indicate another critical function for mammalian UPF1 in addition to its function in NMD. In a recent study in which co-localization of p66 with PCNA and the catalytic subunit of pol delta was examined in the nucleus of S-phase HeLa cells, it was found that the majority of p66 did not co-localize with these proteins at replication foci, nor did all replication foci contain p66 (55). The third subunit of pol delta has now been identified in fission yeast (Cdc27), budding yeast (Pol32) and mammalian cells (p66). Cdc27, the product of an essential gene in S.pombe, has been shown to co-purify with the catalytic and second subunits of the enzyme and to be required for interaction of pol delta with its processivity factor, PCNA (56). POL32, on the other hand, is not an essential gene in S.cerevisiae, and pol delta can be isolated from budding yeast as either a heterodimer or heterotrimer (57). Although the third subunit (Pol32) is not required for interaction of PCNA with pol delta in S.cerevisiae, it stabilizes the interaction (58). In mammals, it has been reported that the third subunit of pol delta (p66) is absolutely required for interaction of the enzyme with PCNA (55,59). However, in the present study we have demonstrated that calf thymus p66 is readily separated from the core pol delta heterodimer (p125/p50) by chromatography on heparin–agarose, although it co-eluted with the core polymerase during earlier purification steps. Furthermore, complete removal of p66 from the heterodimeric pol delta had no effect on its ability to functionally interact with PCNA. These findings raise the question as to whether p66 is an integral component of mammalian pol delta.
The 57–65-kDa proteins that co-eluted with helicase activity on ssDNAc chromatography were identified as bovine hnRNP L by sequencing of the full-length polypeptides and CNBr generated fragments. This identification was confirmed by western analysis with a monoclonal antibody raised against human hnRNP L. Numerous purification strategies were unsuccessful at resolving this major contaminant from bovine UPF1, including chromatography on cationic exchange resins, anionic exchange resins, hydroxyapatite, ssDNAc, polyU–agarose and ATP–agarose, as well as gel filtration (data not shown). The only method which was found to be successful in resolving hnRNP L and bovine UPF1 was sequence-specific RNA-affinity chromatography which selectively retained hnRNP L. The RNA sequence utilized to construct this chromatography matrix was reported to interact with hnRNP L specifically (38). A member of the yeast hnRNP family, Hrp1/Nab4, has been reported to physically interact with Upf1, and to be required for binding to a DSE for NMD to occur (29). Furthermore, the invariant DSE core sequence was found to be GAUG (30), the first 4 nt in the 119-nt RNA sequence that binds hnRNP L (38). Our data further suggest that hnRNP L and HUPF1 may physically interact, specifically the co-elution of HUPF1 and hnRNP L on both heparin–agarose and ssDNAc affinity chromatography. These three pieces of circumstantial evidence, taken together, suggest that mammalian hnRNP L may be performing an analogous function to Hrp1/Nab4, and this would suggest that the mechanism of nonsense-mediated mRNA decay is highly conserved between yeast and mammalian cells. Consistent with this hypothesis, it has been reported that some of the genes involved in NMD are conserved between yeast (22,24,60), C.elegans (40) and mammals (22,24,60).
Acknowledgments
ACKNOWLEDGEMENTS
We want to thank Gunter Kraus for his advice on cloning and Min You for his help with computer graphics. This work was supported by a grant from the National Institutes of Health (DK26206) to A.G.S.
REFERENCES
- 1.Hall M.C. and Matson,S.W. (1999) Helicase motifs: the engine that powers DNA unwinding. Mol. Microbiol., 34, 867–877. [DOI] [PubMed] [Google Scholar]
- 2.Matson S.W. and Kaiser-Rogers,K.A. (1990) DNA helicases. Annu. Rev. Biochem., 59, 289–329. [DOI] [PubMed] [Google Scholar]
- 3.Lohman T.M. and Bjornson,K.P. (1996) Mechanisms of helicase-catalyzed DNA unwinding. Annu. Rev. Biochem., 65, 169–214. [DOI] [PubMed] [Google Scholar]
- 4.Gorbalenya A.E. and Koonin,E.V. (1993) Helicases: amino acid sequence comparisons and structure function relationships. Curr. Biol., 3, 419–429. [Google Scholar]
- 5.Lohman T.M. (1992) Escherichia coli DNA helicases: mechanisms of DNA unwinding. Mol. Microbiol., 6, 5–14. [DOI] [PubMed] [Google Scholar]
- 6.Caruthers J.M., Johnson,E.R. and McKay,D.B. (2000) Crystal structure of yeast initiation factor 4A, a DEAD-box helicase. Proc. Natl Acad. Sci. USA, 97, 13080–13085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Weeda G., van Ham,R.C., Vermeulen,W., Bootsma,D., var der Eb,A.J. and Hoeijmakers,J.H. (1990) A presumed DNA helicase encoded by ERCC-3 is involved in the human repair disorders xeroderma pigmentosum and Cockayne’s syndrome. Cell, 62, 777–791. [DOI] [PubMed] [Google Scholar]
- 8.Passarge E. (1995) A DNA helicase in full Bloom. Nature Genet., 11, 356–357. [DOI] [PubMed] [Google Scholar]
- 9.Yu C.-E., Oshima,J., Fu,Y.-H., Wijsman,E.M., Hisama,F., Alisch,R., Mathews,S., Nakura,J., Miki,T., Ouais,S., Martin,G.M., Mulligan,J. and Schellenberg,G.D. (1996) Positional cloning of the Werner’s syndrome gene. Science, 272, 258–262. [DOI] [PubMed] [Google Scholar]
- 10.Kitao S., Shimamoto,A., Goto,M., Miller,R.W., Smithson,W.A., Lindor,N.M. and Furuichi,Y. (1999) Mutation in RECQL4 cause a subset of cases of Rothmund–Thomson syndrome. Nature Genet., 22, 82–84. [DOI] [PubMed] [Google Scholar]
- 11.Mohaghegh P. and Hickson,I.D. (2001) DNA helicase deficiencies associated with cancer predisposition and premature aging disorders. Hum. Mol. Genet., 10, 741–746. [DOI] [PubMed] [Google Scholar]
- 12.Yamagata K., Kato,J.-I., Shimamoto,A., Goto,M., Furuichi,Y. and Ideka,H. (1998) Bloom’s and Werner’s syndrome genes suppress hyperrecombination in yeast sgs1 mutant: implication for genomic instability in human diseases. Proc. Natl Acad. Sci. USA, 95, 8733–8738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.So A.G. and Downey,K.M. (1992) Eukaryotic DNA replication. Crit. Rev. Biochem. Mol. Biol., 27, 129–155. [DOI] [PubMed] [Google Scholar]
- 14.LeBowitz J.H. and McMacken,R. (1986) The Escherichia coli dnaB replication protein is a DNA helicase. J. Biol. Chem., 261, 4738–4748. [PubMed] [Google Scholar]
- 15.Kornberg A. and Baker,T.A. (1992) DNA Replication, 2nd Edn. W.H. Freeman and Company, San Francisco, CA.
- 16.Labib K., Tercero,J.A. and Diffley,J.F.X. (2000) Uninterrupted MCM2-7 function required for DNA replication fork progression. Science, 288, 1643–1647. [DOI] [PubMed] [Google Scholar]
- 17.Lee D.G. and Bell,S.P. (2000) ATPase switches controlling DNA replication initiation. Curr. Opin. Cell Biol., 12, 280–285. [DOI] [PubMed] [Google Scholar]
- 18.Eison A. and Lucchessi,J.C. (1998) Unraveling the role of helicases in transcription. Bioessays, 20, 634–641. [DOI] [PubMed] [Google Scholar]
- 19.Tuteja N., Huang,N.W., Skopac,D., Teteja,R., Hrvatic,S., Zhang,J., Pongor,S., Joseph,G., Faucher,C., Amalric,F. and Falaschi,A. (1995) Human DNA helicase IV is nucleolin, an RNA helicase modulated by phosphorylation. Gene, 160, 143–148. [DOI] [PubMed] [Google Scholar]
- 20.Zhang S., Maacke,H. and Grosse,F. (1995) Molecular cloning of the gene encoding nuclear DNA helicase II. J. Biol. Chem., 270, 16422–16427. [DOI] [PubMed] [Google Scholar]
- 21.Czaplinski K., Weng,Y., Hagan,K.W. and Peltz,S.W. (1995) Purification and characterization of the Upf1 protein: a factor involved in translation and mRNA degradation. RNA, 1, 610–623. [PMC free article] [PubMed] [Google Scholar]
- 22.Culbertson M.R. (1999) RNA surveillance. Unforeseen consequences for gene expression, inherited genetic disorders and cancer. Trends Genet., 15, 74–80. [DOI] [PubMed] [Google Scholar]
- 23.Lykke-Anderson J. (2001) mRNA quality control: marking the message for life or death. Curr. Biol., 11, R88–R91. [DOI] [PubMed] [Google Scholar]
- 24.Czaplinski K., Ruiz-Echevarria,M.J., Gonzalez,C.I. and Peltz,S.W. (1999) Should we kill the messenger? The role of the surveillance complex in translation and mRNA turnover. Bioessays, 21, 685–696. [DOI] [PubMed] [Google Scholar]
- 25.Perlick H.A., Medghalchi,S.M., Spencer,F.A., Kendzior,R.J.,Jr and Dietz,H.C. (1996) Mammalian orthologues of a yeast regulator of nonsense transcript stability. Proc. Natl Acad. Sci. USA, 93, 10928–10932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Applequist S.E., Selg,M., Raman,C. and Jack,H.-M. (1997) Cloning and characterization of HUPF1, a human homologue of the Saccharomyces cerevisiae nonsense mRNA-reducing UPF1 protein. Nucleic Acids Res., 25, 814–821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bhattacharya A., Czaplinski,K., Trifillis,P., He,F., Jacobson,A. and Peltz,S.W. (2000) Characterization of the biochemical properties of the human Upf1 gene product that is involved in nonsense-mediated mRNA decay. RNA, 6, 1226–1235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ruiz-Echevarria M.J., Czaplinski,K. and Peltz,S.W. (1996) Making sense of nonsense in yeast. Trends Biochem. Sci., 21, 433–438. [DOI] [PubMed] [Google Scholar]
- 29.Gonzalez C.I., Ruiz-Echevarria,M.J., Vasudevan,S., Henry,M.F. and Peltz,S.W. (2000) The yeast hnRNP-like protein Hrp1/Nab4 marks a transcript for nonsense-mediated mRNA decay. Cell, 5, 489–499. [DOI] [PubMed] [Google Scholar]
- 30.Zhang S., Ruiz-Echevarria,M.J., Quan,Y. and Peltz,S.W. (1995) Identification and characterization of a sequence motif involved in nonsense-mediated mRNA decay. Mol. Cell. Biol., 15, 2231–2244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Li X., Tan,C.-K., So,A.G. and Downey,K.M. (1992) Purification and characterization of delta helicase from fetal calf thymus. Biochemistry, 31, 3507–3513. [DOI] [PubMed] [Google Scholar]
- 32.Zhou J.-Q., He,H., Tan,C.-K., Downey,K.M. and So,A.G. (1997) The small subunit is required for functional interaction of DNA polymerase delta with the proliferating cell nuclear antigen. Nucleic Acids Res., 25, 1094–1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Downey K.M. and So,A.G. (1995) Purification of mammalian DNA polymerases: DNA polymerase delta. Methods Enzymol., 262, 84–92. [DOI] [PubMed] [Google Scholar]
- 34.Foiani M., Linder,A.J., Hartmann,G.R., Lucchini,G. and Plevani,P. (1989) Affinity labeling of the active center and ribonucleoside triphosphate binding site of the yeast DNA primase. J. Biol. Chem., 264, 2189–2194. [PubMed] [Google Scholar]
- 35.Hughes P., Tratner,I., Ducoux,M., Piard,K. and Baldacci,G. (1999) Isolation and identification of the third subunit of mammalian DNA polymerase delta by PCNA-affinity chromatography of mouse FM3A cell extracts. Nucleic Acids Res., 27, 2108–2114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhang J., Tan,C.-K., McMullen,B., Downey,K.M. and So,A.G. (1995) Cloning of the cDNAs for the small subunits of bovine and human DNA polymerase delta and chromosomal localization of the gene (POLD2). Genomics, 29, 179–186. [DOI] [PubMed] [Google Scholar]
- 37.Dreyfuss G., Matunis,M., Pinol-Roma,S. and Burd,C.G. (1993) HnRNP proteins and the biogenesis of mRNA. Annu. Rev. Biochem., 62, 289–321. [DOI] [PubMed] [Google Scholar]
- 38.Liu X. and Mertz,J.E. (1995) HnRNP L binds a cis-acting RNA sequence element that enables intron-independent gene expression. Genes Dev., 9, 1766–1780. [DOI] [PubMed] [Google Scholar]
- 39.Burd C.G. and Dreyfuss,G. (1994) Conserved structures and diversity of functions of RNA-binding proteins. Science, 265, 615–621. [DOI] [PubMed] [Google Scholar]
- 40.Page M.F., Carr,B., Anders,K.R., Grimson,A. and Anderson,P. (1999) SMG-2 is a phosphorylated protein required for mRNA surveillance in Caenorhabditis elegans and related to Upf1p of yeast. Mol. Cell. Biol., 19, 5943–5951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bernstein J.A. and Richardson,C.C. (1988) A 7-kDa region of the bacteriophage T7 gene 4 protein is required for primase but not for helicase activity. Proc. Natl Acad. Sci. USA, 85, 396–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Biswas N. and Weller,S.K. (1999) A mutation in the C-terminal putative Zn2+ finger motif of UL52 severely affects the biochemical activities of the HSV-1 helicase primase subcomplex. J. Biol. Chem., 274, 8068–8076. [DOI] [PubMed] [Google Scholar]
- 43.Szekely A.M., Chen,Y.-H., Zhang,C., Oshima,J. and Weissman,S.M. (2000) Werner protein recruits DNA polymerase delta to the nucleolus. Proc. Natl Acad. Sci. USA, 97, 11365–11370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kamath-Loeb A.S., Johansson,E., Burgers,P.M.J. and Loeb,L.A. (2000) Functional interaction between the Werner’s syndrome protein and DNA polymerase delta. Proc. Natl Acad. Sci. USA, 97, 4603–4608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Huang M.E., de Calignon,A., Nicolas,A. and Galibert,F. (2000) POL32, a subunit of Saccharomyces cerivisiae DNA polymerase delta, defines a link between DNA replication and the mutagenic bypass repair pathway. Curr. Genet., 38, 178–187. [DOI] [PubMed] [Google Scholar]
- 46.Kang H.Y., Choi,E., Bae,S.H., Lee,K.H., Gim,B.S., Kim,H.D., Park,C., MacNeil,S.A. and Seo,Y.S. (2000) Genetic analyses of Schizosarccharmyces pombe dna2+ reveal that Dna2 plays an essential role in Okazaki fragment metabolism. Genetics, 155, 1055–1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Atkin A.L., Altamura,N., Leeds,P. and Culbertson,M.R. (1995) The majority of the yeast Upf1 co-localizes with polysomes in the cytoplasm. Mol. Biol. Cell, 6, 611–625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lykke-Anderson J., Shu,M.-D. and Steitz,J.A. (2000) Human Upf proteins target an mRNA for nonsense-mediated decay when bound downstream of a termination codon. Cell, 103, 1121–1131. [DOI] [PubMed] [Google Scholar]
- 49.Pal M., Ishigaki,Y., Nagy,E. and Maquat,L. (2001) Evidence that phosphorylation of human Upf1 protein varies with intracellular location and is mediated by a wortmannin-sensitive and rapamycin-sensitive PI 3-kinase-related kinase signaling pathway. RNA, 7, 5–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sun X., Perlick,H.A., Dietz,H.C. and Maquat,L.E. (1998) A mutated human homologue to yeast Upf1 protein has a dominant negative effect on the decay of nonsense-containing mRNA in mammalian cells. Proc. Natl Acad. Sci. USA, 95, 10009–10014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Muhlemann O., Mock-Casagrande,C.S., Wang,J., Li,S., Custodio,N., Carmo-Fonseca,M., Wilkinson,M.F. and Moore,M.J. (2001) Precursor RNAs harboring nonsense codons accumulate near the site of transcription. Mol. Cell, 8, 33–44. [DOI] [PubMed] [Google Scholar]
- 52.Iborra J.F., Jackson,D.A. and Cook,P.R. (2001) Coupled transcription and translation within the nuclei of mammalian cells. Science, 293, 1139–1142. [DOI] [PubMed] [Google Scholar]
- 53.Medghalchi S.M., Frischmeyer,P.A., Mendell,J.T., Kelly,A.G., Lawler,A.M. and Dietz,H.C. (2001) Rent1, a trans-effector of nonsense-mediated mRNA decay, is essential for mammalian embryonic viability. Hum. Mol. Genet., 10, 99–105. [DOI] [PubMed] [Google Scholar]
- 54.He F., Peltz,S.W., Donahue,J.L., Rosbash,M. and Jacobson,A. (1993) Stabilization and ribosome association of unspliced pre-mRNAs in a yeast upf1– mutant. Proc. Natl Acad. Sci. USA, 90, 7034–7038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ducoux M., Urbach,S., Baldacci,G., Hubscher,U., Koundrioukoff,S., Christensen,J. and Hughes,P. (2001) Mediation of proliferating cell nuclear antigen PCNA-dependent DNA replication through a conserved p21(Cip1)-like PCNA-binding motif present in the third subunit of human DNA polymerase delta. J. Biol. Chem. 276, 49258–49266. [DOI] [PubMed] [Google Scholar]
- 56.Zou S., Gibbs,E., Kelman,Z., Wang,T.S.-F., O’Donnell,M., MacNeill,S. and Hurwitz,J. (1997) DNA polymerase delta isolated from Schizosaccharomyces pombe contains five subunits. Proc. Natl Acad. Sci. USA, 94, 11244–11249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Gerik K.J., Li,X., Pautz,A. and Burgers,P.M.J. (1998) Characterization of the two small subunits of Saccharomyces cerevisiae DNA polymerase δ. J. Biol. Chem., 273, 19747–19755. [DOI] [PubMed] [Google Scholar]
- 58.Burgers P.M.J. and Gerik,K.J. (1998) Structure and processivity of two forms of Saccharomyces cerevisiae DNA polymerase δ. J. Biol. Chem., 273, 19756–19762. [DOI] [PubMed] [Google Scholar]
- 59.Shikata K., Ohta,S., Yamada,K., Obuse,C., Yoshikawa,H. and Tsurimoto,T. (2001) The human homologue of fission yeast Cdc27, p66, is a component of active human DNA polymerase δ. J. Biochem., 129, 699–708. [DOI] [PubMed] [Google Scholar]
- 60.Serin G., Gersappe,A., Black,J.D., Aronoff,R. and Maquat,L.E. (2001) Identification and characterization of human orthologues to Saccharomyces cerevisiae Upf2 protein and Upf3 protein. Mol. Cell. Biol., 21, 209–223. [DOI] [PMC free article] [PubMed] [Google Scholar]