


Abstract
The long-standing suspicion that Epstein–Barr virus nuclear antigen 5 (EBNA5) is involved in transcription regulation was recently confirmed by the observation by several groups that EBNA5 cooperates with EBNA2 in activation of the LMP1 promoter. In attempts to elucidate the molecular basis for the EBNA5-mediated enhancement of EBNA2 transactivation, we obtained evidence of an additional function of EBNA5: at high but still biologically relevant levels, EBNA5 acted as a repressor of gene expression by interfering with the processing of pre-mRNA. Transient transfections with reporter plasmids revealed that EBNA5 repressed reporter mRNA and protein expression in the cytoplasm, but did not lower the steady-state level of reporter RNA in the total cellular RNA fraction. We have excluded that repression occurred as a consequence of cell death induced by EBNA5. Using the RNase protection assay with a probe comprising the pre-mRNA cleavage and polyadenylation site, EBNA5 was found to inhibit 3′-end cleavage and polyadenylation of pre-mRNAs from the reporter plasmids investigated. The effect of inhibitory levels of EBNA5 on chromosomal genes was examined in transient transfections by expression profiling using a cDNA microarray panel containing 588 genes. The results showed that EBNA5 could also inhibit the expression of chromosomal genes and did it in a discriminatory manner. This is consistent with the notion that a regulatory mechanism exists in the cell that confers specificity to the selection by EBNA5 of target genes for repression.
INTRODUCTION
Epstein–Barr virus (EBV) is a ubiquitous herpesvirus in humans that can infect, establish latency and induce proliferation in B lymphocytes. EBV infects 90% of the world’s population and is associated with a wide range of benign and malignant diseases, including infectious mononucleosis, Burkitt’s lymphoma (BL), nasopharyngeal carcinoma, Hodgkin’s lymphoma, oral hairy leukoplakia and post-transplantation lymphoproliferative disorders (reviewed in 1). The 172 kb EBV genome contains more than 80 genes that encode structural and regulatory proteins. Six of these have been shown to be required for the process that leads to efficient differentiation of EBV-infected resting B cells into proliferating B lymphoblasts (immortalisation), including the nuclear proteins Epstein–Barr virus nuclear antigen (EBNA) 1, EBNA2, EBNA3 (also designated EBNA3A), EBNA5 (also designated EBNA-LP) and EBNA6 (also designated EBNA3C), and the membrane protein LMP1 (reviewed in 2). The objective of the present investigation was to further define the role of EBNA2 and EBNA5 as regulators of gene expression.
In the EBV-infected lymphoblastoid cell all of the EBNA genes have been implicated in the control of gene expression. They belong to the same transcription unit and the different mRNAs are generated by alternative splicing of a large primary transcript. In the early stages of infection transcription is initiated from the Wp promoter, but within 24–48 h there is a switch to the use of the upstream Cp promoter. The EBNA2- and EBNA5-encoding sequences are promoter proximal in the EBNA transcription unit and the protein products appear within 6 h after infection (3,4). EBNA2 mRNA encompasses an untranslated leader region consisting of repeating W1 and W2 exons and single copies of the Y1 and Y2 exons, which is spliced to an exon that contains the EBNA2-encoding open reading frame (ORF) (BYRF1). EBNA5 mRNA is formed by alternative splicing between a promoter-proximal exon and the first exon of the repeat. This splice creates a translation initiation codon and an EBNA5-encoding ORF composed of the repeating W1 and W2 and the unique Y1 and Y2 exons. This ORF is translated into a protein containing multiple copies of a 66 amino acid repeat domain and a 45 amino acid unique domain. In the infected B cell EBNA5 occurs as a series of polypeptides of different size (5), which is presumably due mainly to exon skipping during assembly of leader exons from the primary transcripts resulting in mRNAs carrying different numbers of W1/W2 exon pairs.
Many aspects of the function of EBNA2 have been clarified. EBNA2 is a transcriptional activator that regulates the activity of viral (EBNA Cp, LMP1, LMP2A and LMP2B) and cellular (CD21, CD23, c-fgr and BLR2) promoters (reviewed in 2). EBNA2 does not bind to DNA directly but is recruited to the target promoters by interaction with DNA-binding transcription factors like RBP-Jκ (6–8), PU.1 (9–11), POU domain proteins (9) and ATF-2–c-jun (12) or the SNF5/Ini1 component of the human SNF–SWI chromatin remodelling complex (13). The precise mechanism by which EBNA2 regulates the activity of responsive promoters is, however, still not clarified. Much less is known about the biochemical function of EBNA5 and its role in the immortalisation process. The level of EBNA5 during infection rises to high levels during the first 3–4 days of infection and then decreases to the levels found in established lymphoblastoid cell lines (LCLs) (14). EBNA5 antigens appear very early and are first diffusely distributed throughout the nucleoplasm. After a few days the EBNA5 immunostaining condenses into discrete foci which coincide with nuclear bodies known as ND10 domains or PML oncogenic domains (PODs) (14–16). Evidence is accumulating suggesting that PODs are involved in transcription regulation, cell cycle regulation and regulation of cell death. At a later stage of infection and in established cell lines EBNA5 is almost exclusively located at the POD sites. Mutations in EBV strains that prevent expression of the Y1 and Y2 exon reduce the efficiency of immortalisation at least 10-fold and the transformed cells grow slowly and require the presence of a fibroblast feeder layer (17,18). Some evidence exists which suggests that EBNA5 is involved in regulation of the cell cycle. The phosphorylation status of EBNA5 changes during the cell cycle as shown by an increased number of phosphorylated serine residues in EBNA5 during the G2 phase, reaching a maximum of ∼50% at the G2/M boundary (19). Moreover, cyclin D2 expression was up-regulated in resting B lymphocytes in response to co-transfection with EBNA2 and EBNA5 expression vectors, making the cells leave the G0 phase and enter the cell cycle (20). It should be noted, however, that cyclin D2 expression cannot be induced in a constitutively EBNA5-expressing lymphoblastoid cell line by EBNA2 in the absence of de novo protein synthesis, suggesting that cyclin D2 expression is a secondary event driven by the primary viral and cellular EBNA2 targets (21). Further support for the role of EBNA5 in transcriptional regulation is lent by recent data showing that EBNA5 cooperates with EBNA2 to activate expression of LMP1 (8,22).
The molecular basis for the EBNA5-mediated enhancement of EBNA2 transactivation of promoter activity is still not fully understood. Here we have begun to examine the role of EBNA5 in the regulation of gene expression using transient transfection of lymphoid cell lines with reporter plasmids. During the course of the study evidence appeared indicating that EBNA5, in addition to being a positive transcription factor, at high but biologically relevant concentrations also acted as a repressor of gene expression. We have excluded that repression occurred as a consequence of cell death induced by EBNA5. We show that EBNA5 interferes with RNA processing by inhibiting 3′-end cleavage and polyadenylation of pre-mRNAs from reporter plasmids.
MATERIALS AND METHODS
Cell lines, cell culture and antibodies
DG75 is an EBV genome-negative BL cell line (23). The Rael line is derived from an endemic BL and displays a BL type I phenotype in which EBNA1 is the only detectable viral protein (24). CBC-Rael is an LCL obtained by infection of cord blood cells with the Rael virus strain (25). The IB4 LCL was derived by transformation of human placental lymphocytes with EBV strain B95-8 (26). The WW1 LCL was established by in vitro infection with the EBV isolate QIMR-Wil of normal B cells from the same patient (27). P3HR-1 is a BL line of type II phenotype and carries a transformation-defective virus strain with a deletion that removes parts of the EBNA2 and EBNA5 genes (28). Raji is a BL line exhibiting a type II phenotype (29) and Namalwa (30) is a BL line of type III phenotype. The cell lines were routinely cultured in RPMI 1640 medium (Gibco, Paisley, UK) supplemented with 10% (v/v) heat-inactivated fetal calf serum (Gibco), penicillin and streptomycin.
EBNA2 was detected with the monoclonal anti-EBNA2 antibody PE2 purchased from Dako (Glostrup, Denmark) and EBNA5 was detected with the monoclonal anti-EBNA5 antibody JF186 (31), obtained from Dr M. Masucci (MTC, Karolinska Institute, Stockholm, Sweden). In all immunoblots the primary antibody was detected by a secondary alkaline phosphatase (AP)-conjugated antibody against the IgH chain of the primary antibody used (mouse or rabbit). The EBNA proteins were visualised on the blots with a nitroblue tetrazolium/5-bromo-4-chloro-indolylphosphate (BCIP/NBT) colourimetric AP reaction (Promega, Madison, WI).
Plasmids
The EBNA5 expression vector p3.1W-LP was obtained from Paul Farrell (20). The EBNA5-encoding reading frame in this plasmid is derived from the IB4 cell line and contains seven copies of the W1 and W2 exons and one copy each of the Y1 and Y2 exons. The p3.1W-LP plasmid was cleaved with XbaI and the staggered ends were filled in by T4 DNA polymerase (Boehringer-Mannheim, Mannheim, Germany). The fragment containing the EBNA5 reading frame was excised by cleavage with EcoRI and inserted into the pCI vector (Promega), which had been opened with XhoI, blunted with T4 DNA polymerase and cleaved with EcoRI. The resulting plasmid, pCI-EBNA5, contains the EBNA5 reading frame under the control of the CMV promoter. pcDNA6-EBNA5 is an expression vector for EBNA5. The EBNA5-encoding region was excised from the pCI-EBNA5 plasmid with NheI and EcoRI and ligated into NheI + EcoRI-opened pcDNA6/V5-His A vector (Invitrogen). The EBNA2 expression vector pEΔA6 has been described previously (32).
A series of 5′-deleted Wp-containing fragments was transferred to pgCAT (33) from a corresponding deletion series, pBΔWCAT-1–pBΔWCAT-5, described previously (34). The AvaI–AvaI Wp fragment was excised from pBΔWCAT-1 and cloned in pCATGem using HindIII linkers. The resulting plasmid was designated pgBΔW(–557)CAT. The BglII–AvaI, NarI–AvaI and ApaI–AvaI segments of Wp together with the CAT gene were excised as single fragments from pBΔWCAT-2, -3 and -5, repectively, by cleavage with BamHI and SalI and inserted between the BamHI and SalI sites in pGEM-3Zf(+) (Promega). The resulting constructs were designated pgBΔW(–439)CAT, pgBΔW(–299)CAT and pgBΔW(–135)CAT, respectively. The 3′-end of all the inserted Wp fragments was at nucleotide +76 relative to the transcription initiation site while the position of the 5′-ends differed as indicated by the designation of the plasmids. The pgBΔC(–1024)CAT reporter plasmid was made by cloning the C promoter region and CAT gene of pBΔC3CAT (35) as a continuous SalI–BamHI fragment in the pGEM-3Zf(+) vector. The pgLRS(–54)CAT, pgLRS(–634)CAT, pgSVECAT (33) and pgTKCAT reporter plasmids have been described earlier (9). The pCMVLuc reporter plasmid carries the luciferase reporter gene. It was created by excising the CMV promoter region from the pCI vector (Promega) with BglII and NheI, blunting the ends with the Klenow DNA polymerase fragment and inserting the promoter fragment into SmaI-cleaved pGL3-Basic vector (Promega).
The pBΔW(–557)globin (36), pGlobinT and pSVEglobin (35) plasmids were used for S1 endonuclease protection assays. For the construction of pGlobinT, a fragment containing the rabbit globin gene promoter and the globin-encoding sequence was excised from pSXb+ (37) by cleavage with BglII and inserted into the same pBR322-based vector as pSVEglobin. The CD2 expression vector pE300CY6, which contains a truncated version of the rat CD2 surface antigen under control of the CMV promoter, was a kind gift of Dr E. Lundgren (University of Umeå, Sweden). The TS24 CAT reporter plasmid was described previously. This plasmid contains upstream regulatory sequences of the human 4-hydroxyfenylpyruvate dioxygenase gene (HPD, nucleotides –327 to +13 relative to the transcription initiation site) (38).
A plasmid for the generation of riboprobes to detect luciferase RNA was constructed by inserting a fragment encompassing a part of the LMP1 promoter and the first 200 bp of the luciferase gene into pGEM-3Zf(+). The pLRS(–106)Luc plasmid was cleaved with Csp45I and the staggered ends were filled in with Klenow fragment (Boehringer Mannheim). The fragment containing the LRS sequence and a part of the luciferase reading frame was excised by cleavage with SacI and inserted into the pGEM-3Zf(+) vector (Promega), which had been opened with SacI and SmaI. The resulting plasmid was designated pgProbeLRS(–106)ΔLuc. A plasmid for the generation of riboprobes to detect cleaved and non-cleaved luciferase primary transcripts was constructed by PCR amplification of the SV40 late polyadenylation signal from the pGL3-Control plasmid (Promega) with the primers 5′-TTTTTGGATCCTTCGAGCAGACATGATAAGATACA-3′ (sense) and 5′-TTTTTGGATCCAACCTCCCACACCTCCCCCTGAAC-3′ (antisense). The PCR fragment was cleaved with BamHI (introduced in the PCR primers) and inserted into the BamHI-cleaved pGEM-3Zf(+) vector (Promega). The resulting plasmid was designated pgProbePolyA.
All constructs made were verified by dideoxy sequencing using ABI 373 or ABI Prism 310 automated sequencers (Applied Biosystem, Foster City, CA) and standard protocols. Reagents for cloning were from Boehringer Mannheim unless otherwise stated.
DNA transfections and reporter gene assays
The day before transfection, the cells were diluted to a density of 5 × 105 cells/ml medium. Transfections were performed with 5 × 106 or 107 cells in 250 µl medium using electroporation with a Bio-Rad Genepulser and 4 mm gap cuvettes (Bio-Rad, Hercules, CA) at 260 V and 960 µF for all cell lines except CBC-Rael, where 250 V and 500 µF were used. Appropriate molar amounts of empty plasmid DNA (pCI, pSV2gpt or pcDNA6/V5-HisA) were added to compensate for variations in the amount of expression vector. The transfected cells were harvested after 3 days and aliquots of cell lysates were assayed for reporter gene activity. The chloramphenicol acetyltransferase (CAT) assay has been described previously (34). Luciferase activity in transfected cells was determined with the Luciferase Assay System (Promega) using a TD 20/20 luminometer (Turner Designs Instruments, Sunnyvale, CA).
RNA purification and analysis
Total cellular RNA was isolated by acid guanidinium thiocyanate/phenol/chloroform extraction essentially as described by Chomczynski and Sacchi (39). Cytoplasmic RNA was prepared by the sucrose cushion method of Favaloro et al. (40) with 10 mM vanadylribonucleoside complexes as RNase inhibitors, treated with RQ1 DNase (Promega) and stored at –70°C. Poly(A)+ RNA was isolated using magnetic oligo(dT)25-conjugated beads following the protocol supplied by the manufacturer (Dynal, Norway). The poly(A)+ and poly(A)– RNA fractions were extracted with chloroform/phenol and ethanol precipitated. RNA was analysed by S1 endonuclease or RNase protection analysis as described previously (34,41). Protected fragments were resolved by electrophoresis in 4% polyacrylamide gels and visualised by phosphorimaging (Molecular Dynamics).
Differential expression of genes in cells transfected with the pCI-EBNA5 expression vector and the empty pCI vector, respectively, was analysed using Human Atlas cDNA array membranes (7740-1) purchased from Clontech. Portions of 5 × 106 DG75 cells were transfected with 540 fmol pCI or pCI-EBNA5 DNA together with 5 µg pE300CY6 DNA in 250 µl medium as described above. Aliquots of 108 cells of each transfection were pooled and the CD2-expressing cells were selected with anti-rCD2 antibody-coated magnetic beads. Total RNA was prepared and the poly(A)+ fraction isolated using oligo(dT)-conjugated beads as described above. Aliquots of 1 µg were used as template for the synthesis of 32P-labelled cDNA probes in a reverse transcriptase-catalysed reaction with [α-32P]dATP as described by the manufacturer of the DNA array membranes. The membranes were incubated for 16 h at 68°C in hybridisation solution containing 106 c.p.m./ml 32P-labelled cDNA in roller bottles and were then washed extensively. The composition of the hybridisation and washing solutions was as described by the manufacturer (Clontech). [32P]cDNA hybridised to the membrane was quantified by phosphorimaging (Molecular Dynamics) with the settings 88 µm resolution and 800 V PMT voltage and ImageQuant software (Molecular Dynamics).
Cell sorting, [3H]thymidine incorporation and FACS analysis
In experiments requiring the selection of transfected cells, 10 µg rat CD2 expression vector pE300CY6 was included in the transfection mixture. Sorting of CD2-expressing cells was performed the day after transfection using anti-rCD2 antibodies as described (42). The CD2-positive cells were transferred in portions of 90 000 cells/well to a 96-well plate and diluted to a density of 5 × 105 cells/ml with culture medium. For the determination of thymidine incorporation, 1 µCi [3H]methylthymidine (DuPont NEN, Boston, MA) was added after 24 h, the incubation was continued for 24 h and the cells were harvested, washed once with PBS (180 mM NaCl, 3.6 mM KCl, 11 mM Na2HPO4 and 2 mM KH2PO4) and suspended in 200 µl 0.25 M Tris–HCl, pH 8.0. The cells were broken by three repeated cycles of freezing in liquid nitrogen and thawing at 37°C. DNA was collected by filtration through a glass microfibre filter (Whatman International, Kent, UK) using a vacuum manifold. The filters were washed, dried in air and the radioactivity was determined in a scintillation counter. For determination of the relative level of EBNA5, the cells were harvested on the same day and in the same way as for the determination of incorporation of [3H]thymidine with the additional step that insoluble matter was removed by centrifugation. Aliquots were applied to Hybond-C Extra membranes (Amersham) using a vacuum manifold. EBNA5 was detected with the monoclonal antibody JF186 and visualised with a BCIP/NBT colourimetric AP reaction (Promega, Madison, WI).
For FACS analysis 105 cells were pelleted by centrifugation and washed twice with PBS. Aliquots of the cells were incubated with Annexin V–FITC, with propidium iodide and with a mixture of Annexin V–FITC and propidium iodide, respectively. The fractions of early apoptotic/Annexin V–FITC stained cells or necrotic/propidium iodide stained cells were analysed by cell sorting of 5000 cells with a FACScan apparatus using CellQuest software for the evaluation (Becton Dickinson, San Jose, CA).
RESULTS
EBNA5 modulates the activity of reporter plasmids carrying viral or cellular promoters
It has been reported that EBNA5 cooperates with EBNA2 in activation of the LMP1 promoter in Eli, Akata and BJAB cells, and EBNA5-responsive sequences in the promoter region have been identified (8,22). In order to relate these observations to our previous work and increase our knowledge about the mechanism of the co-activator function of EBNA5, we initiated a study of the effect of EBNA5 on LMP1 promoter activity using transient transfection experiments. Increasing amounts of the EBNA5 expression vector pCI-EBNA5 or the empty pCI vector were transfected into EBV-negative DG75 cells together with the EBNA2 expression vector pEΔA6 and CAT reporter plasmids. The latter plasmids contained the complete –634 to +40 LMP1 gene regulatory sequence (LRS) or the constitutively active –54 to +40 part of LRS. Somewhat unexpectedly, the EBNA2-induced activity of pgLRS(–634)CAT decreased in a manner that correlated with the levels of EBNA5 in the cells (Fig. 1). The pCI vector without insert had no significant effect on promoter activity (data not shown). Notably, the expression of EBNA2 decreased with increasing EBNA5 levels in parallel with the decrease in activity of the reporter plasmids. This would be compatible with the notion that the effect of EBNA5 on the reporter plasmids was indirect via a repression of expression of EBNA2. However, the pCI-EBNA5 plasmid also repressed the EBNA2-independent basal activity of the pgLRS(–634)CAT and pgLRS(–54)CAT plasmids in the absence of the EBNA2 expression vector (Fig. 1), which speaks against this interpretation. Thus, our results were compatible with the hypothesis that under the conditions of the transient transfection experiments EBNA5 acts as a repressor of LMP1 promoter activity in DG75 cells and that the target sequence for EBNA5 is located in the –54/+40 part of LRS. It should be noted that EBNA2 did not seem to modulate the function of EBNA5.
Figure 1.

Effect of EBNA5 on constitutive and EBNA2-induced expression of LRS-containing reporter plasmids. (A) The EBV-negative B cell line DG75 (5 × 106 cells) was transfected with the pgLRS(–634)CAT reporter plasmid together with the EBNA2 expression vector pEΔA6 (main panel) or with an equivalent amount of the empty vector pSV2gpt (inset) and increasing amounts of the EBNA5 expression vector pCI-EBNA5. The CAT activity is given as relative chloramphenicol acetylation expressed as a percentage of the activity obtained with pgLRS(–634)CAT in the presence of EBNA2 and in the absence of EBNA5. The 100% value corresponds to acetylation of 260% of the substrate in the assay (measured value × dilution factor). The values shown are the mean of five independent experiments. Error bars indicate standard errors of the mean (SEM). Variations in the amount of expression vector were compensated for by the addition of appropriate molar amounts of pCI or pSV2gpt DNA to the transfection mixtures. (B) The pgLRS(–54)CAT reporter plasmid was transfected together with increasing amounts of the EBNA5 expression vector pCI-EBNA5 into 5 × 106 DG75 cells (constitutive expression). The CAT activity is given as relative chloramphenicol acetylation expressed as a percentage of the activity obtained with pgLRS(–54)CAT in the absence of EBNA5. The 100% value corresponds to acetylation of 110% of the substrate in the assay (measured value × dilution factor). The mean values ± SEM of five independent experiments are presented. Variations in the amount of expression vectors were compensated for by addition of the appropriate molar amounts of pCI to the transfection mixtures.
The effect of EBNA5 on the EBV Wp promoter was investigated in DG75 cells using reporter plasmids that carried a 5′ deletion series of the Wp regulatory region (Fig. 2). Large amounts of the EBNA5 expression vector repressed the activity of all reporter plasmids, including the one with the shortest segment (–135 to +76) of the promoter regulatory sequence. At low levels of EBNA5 a moderate stimulation of the activity of the Wp promoter containing reporter constructs was observed (Fig. 2). The stimulatory effect was most evident for the reporter with the longest Wp insert (–557/+76), which was activated ∼2-fold over basal activity at the EBNA5 concentration corresponding to 27 fmol pCI-EBNA5 DNA in the transfection mixture. The effect of EBNA5 on C promoter-containing reporter constructs was also investigated in DG75 cells. Low levels of pCI-EBNA5 DNA activated a plasmid containing a long C promoter fragment [pCp(–1024)CAT] 2–3-fold while high concentrations of EBNA5 repressed the activity (Fig. 2B).
Figure 2.

Effect of EBNA5 on the expression of reporter genes in plasmids containing the EBV W and C promoters. (A) The pgBΔW(–557)CAT (diamonds), pgBΔW(–439)CAT (filled squares), pgBΔW(–299)CAT (unfilled squares) and pgBΔW(–135)CAT (filled circles) reporter plasmids, respectively, were transfected together with increasing amounts of pCI-EBNA5 in 5 × 106 DG75 cells. The CAT activity is given as relative chloramphenicol acetylation expressed as a percentage of the activity obtained with pgBΔW(–557)CAT in the absence of EBNA5. The 100% value corresponds to acetylation of 150% of the substrate in the assay (measured value × dilution factor). The mean values ± SEM of three independent experiments are presented. The interval between 0 and 54 fmol added pCI-EBNA5 is shown as an expanded inset. (B) The pgBΔC(–1024)CAT plasmid was transfected together with increasing amounts of pCI-EBNA5 in 5 × 106 DG75 cells. The inset shows the activity of pgBΔC(–1024)CAT at low pCI-EBNA5 concentrations. The CAT activity is given as relative chloramphenicol acetylation expressed as a percentage of the activity obtained with the respective reporter plasmid in the absence of EBNA5. The 100% value corresponds to acetylation of 150% of the substrate in the assay (measured value × dilution factor). The mean values ± SEM of three independent experiments are presented.
To corroborate our preliminary conclusion that the observed inhibitory effect of EBNA5 on the activity of the reporter plasmids occurred at the RNA level, we performed S1 nuclease analysis of reporter gene expression (Fig. 3). Co-transfection of the pgBΔW(–557)globin plasmid with pCI-EBNA5 DNA into DG75 cells showed that small amounts of the EBNA5 expression vector increased and large amounts decreased the level of the protected globin RNA fragment in the cytoplasm (Fig. 3A). Similarly, EBNA5 decreased the amount of globin RNA in the cytoplasm expressed from reporter plasmids that contained the β-globin gene under the control of the β-globin promoter or the SV40 early promoter (Fig. 3B).
Figure 3.

Effect of EBNA5 on the level of cytoplasmic RNA transcribed from reporter plasmids carrying the Wp, SV40 early and rabbit β-globin promoters, respectively. The pgBΔW(–557)Globin, pSVE40Globin and pbGlobinT reporter plasmids were co-transfected with increasing amounts of pCI-EBNA5 in 5 × 106 DG75 cells. Cytoplasmic RNA was prepared and 40 µg aliquots were analysed by the S1 nuclease protection assay. The expected length of a β-globin mRNA protected fragment is 210 bp. Fragments obtained by MspI cleavage of pBR322 DNA were used as size markers and the length in bp of the appropriate marker fragments is indicated on the left side of each figure. The experiments were repeated at least twice for each reporter construct. (A) PAGE of 32P-labelled fragments protected by RNA from cells transfected with increasing amounts of pCI-EBNA5 and 10 µg pgBΔW(–557)Globin. (B) PAGE of 32P-labelled fragments protected by RNA from cells transfected with 0 or 540 fmol pCI-EBNA5 and 10 µg pSVE40Globin (lanes 2 and 3) or pbGlobinT (lanes 4 and 5). The SV40 virus early promoter/enhancer drives transcription of the β-globin gene in the pSVE40Globin construct and the rabbit β-globin promoter drives transcription of the β-globin gene in the pbGlobinT plasmid.
To verify that the repressor effect was mediated by the EBNA5 protein and did not arise as a secondary phenomenon from plasmid DNA or transcribed RNA, we generated two deletion mutants of the EBNA5 expression vector. One of these contained one copy of the W exon-encoding part (W1 and W2) and no Y sequence, the other only the Y exon-encoding part (Y1 and Y2). Neither of the mutated vectors displayed any repressor activity (data not shown). We conclude that repression of the activity of the reporter plasmid was mediated by the translated product of the EBNA5-encoding reading frame in the expression vectors and was not a direct effect of vector DNA or transcribed RNA.
EBNA5 inhibits processing of the primary transcript
The data presented so far suggest that EBNA5 exerts its repressive effect on gene expression at the level of transcription, although we have not discriminated between effects on initiation of transcription, elongation of the primary transcript or mRNA maturation, transport or stability. To determine if EBNA5 interferes with pre-mRNA processing, a luciferase reporter plasmid driven by the SV40 promoter was transfected together with the pCI-EBNA5 expression vector or the empty pCI vector into DG75 cells. Protein extracts and total and cytoplasmic RNA fractions were prepared for determination of the reporter activity at the protein and RNA levels. Assay of luciferase activity in the cell extracts showed that expression of the reporter gene was repressed >20 fold in the presence of EBNA5 (data not shown). Aliquots of the total and cytoplasmic RNA fractions containing similar amounts of RNA were analysed by the RNase protection assay. Since >75% of the total cellular RNA is located in the cytoplasm, a given amount of RNA of either fraction corresponded to roughly the same number of cells (43). As expected, the level of luciferase RNA in the cytoplasmic fraction was considerably lower in cells transfected with the pCI-EBNA5 expression vector than in those transfected with the pCI plasmid (Fig. 4A). However, the level of luciferase RNA in the total RNA fraction was about the same in the pCI-EBNA5-transfected and the pCI-transfected control cells, suggesting that EBNA5-mediated repression of gene expression did not occur at the level of transcription initiation but at some later stage of transcript processing. To assess the effect of EBNA5 on mRNA stability, the half-life of luciferase RNA in the presence and absence of EBNA5 was determined using the transient transfection assay. RNA synthesis was inhibited by the addition of actinomycin D to the cell culture medium 4 and 8 h before cell harvest. Total cellular RNA was prepared and the level of luciferase RNA determined at the two time points using the RNase protection assay. A comparison of the rate of disappearence of luciferase-specific RNA in cells transfected with the pGL3-Control reporter plasmid together with the pCI-EBNA5 expression vector or the empty pCI plasmid revealed that the presence of EBNA5 did not significantly affect the stability of the luciferase RNA (data not shown).
Figure 4.
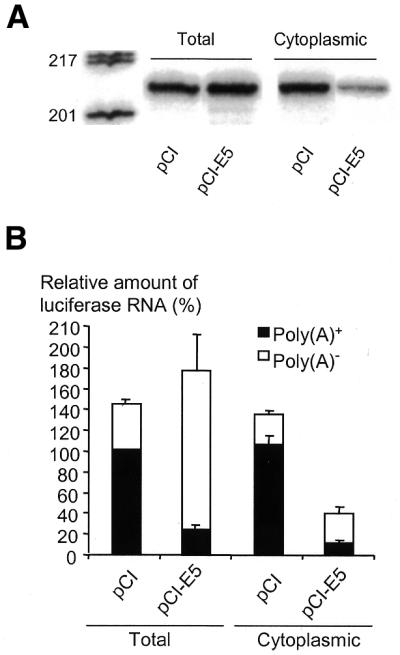
Effects of EBNA5 on the processing of reporter gene pre-mRNA. (A) EBNA5 diminishes the fraction of reporter gene RNA exported from the nucleus. The pGL3-Control (20 µg) reporter plasmid was co-transfected with 1080 fmol either pCI or pCI-EBNA5 (designated pCI-E5 in the figure) into 107 DG75 cells. Cytoplasmic and total RNA fractions were prepared and the level of luciferase RNA determined by RNase protection analysis using a riboprobe that protects the first 200 bp of luciferase RNA transcribed from the reporter gene. The RNA fragments were separated by PAGE under denaturing conditions. The length in bp of the DNA fragments used as size markers is indicated on the left side of the figure. Similar results were obtained in five independent experiments. (B) EBNA5 inhibits polyadenylation and nuclear export of reporter gene RNA. The RNA obtained from the transfections above were separated into poly(A)+ and poly(A)– fractions with oligo(dT)-conjugated magnetic beads. Luciferase RNA was quantified by RNase protection analysis as described above. The relative level of polyadenylated luciferase RNA in total RNA in pCI-transfected cells was defined as 100% and the levels in the other RNA fractions were expressed as a percentage of this value. The means ± SEM of three independent experiments are presented.
Inhibition of polyadenylation during processing of the primary transcript leads to accumulation in the nucleus of RNA lacking the poly(A) tail (44). To assess the possibility that EBNA5 interferes with the polyadenylation process, cytoplasmic and total RNA preparations obtained in transfection experiments similar to those described in the previous section were separated into poly(A)+ and poly(A)– RNA fractions. Luciferase-specific RNA in the fractions was determined using RNase protection analysis and the relative amounts of specific transcripts were quantified with a phosphorimager. The total amount of polyadenylated and non-polyadenylated luciferase RNA in the total RNA preparations from pCI-EBNA5-transfected cells was largely the same as in pCI-transfected cells. The fraction of poly(A)+ luciferase RNA was, however, considerably lower in the cells transfected with pCI-EBNA5 (15%) than in the control cells transfected with pCI (70%) (Fig. 4B). Moreover, in cytoplasmic RNA from EBNA5-expressing cells both the total amount and the polyadenylated fraction of luciferase RNA were smaller than in the cells transfected with the empty vector. We conclude that EBNA5 can interfere with polyadenylation of the primary transcript and thereby reduce the level of mRNA in the cytoplasm.
To test the possibility that EBNA5 decreased the formation of polyadenylated RNA by inhibiting cleavage of the primary transcript, we analysed the relative amounts of non-cleaved and cleaved pre-mRNA in transient transfections using RNase protection analysis. The antisense RNA probe employed encompassed the SV40 late polyadenylation signal sequence for RNA cleavage and polyadenylation in the pGL3-Control luciferase reporter plasmid. DG75 cells were transfected with the luciferase reporter plasmid together with either the pCI-EBNA5 plasmid or the empty pCI plasmid. The RNase protection assays showed that transcripts hybridising to the probe for cleavage were much more abundant in cells also transfected with the CMV promoter-driven pCI-EBNA5 and pCI plasmids than in those transfected with the SV40 promoter-controlled pGL3-Control reporter vector alone (Fig. 5A). This was presumably due to the fact that the CMV promoter is considerably more active than the SV40 promoter in DG75 cells (unpublished results). Moreover, the RNase protection probe did not discriminate between the pre-mRNAs transcribed from these plasmids since both the pGL3-Control reporter plasmid and the pCI-EBNA5 and pCI plasmids contained the SV40-derived cleavage signal sequence. Therefore, the results obtained in this series of experiments mainly reflected the processing of primary transcripts from the EBNA5 expression vector pCI-EBNA5 in the presence of EBNA5 as compared with the presumably normal processing of transcripts from the empty pCI control vector in the absence of EBNA5. It is also worth noting that the total RNA fraction from mock-transfected cells did not produce detectable protected hybrids with the RNase protection probe (lane 5), indicating that the SV40-derived 3′-untranslated region (3′-UTR) sequence containing the polyadenylation signal is not common in cellular genes. The fraction of cleaved SV40-derived 3′-UTR transcripts was ∼7-fold lower in cells expressing EBNA5 than in cells transfected with the pCI control plasmid. The total amount of RNA hybridising with the cleavage probe, i.e. the sum of cleaved and uncleaved RNA, was the same in cells co-transfected with pGL3-Control and pCI DNA and in cells co-transfected with pGL3-Control and pCI-EBNA5 DNA (Fig. 5A, lanes 1 and 2). This is consistent with the notion that EBNA5 does not repress transcription at the level of initiation.
Figure 5.

EBNA5 inhibits cleavage of the 3′-UTR of pre-mRNA. (A) DG75 cells (107) were transfected with pCI, pCI-EBNA5 and pGL3-Control plasmid DNA. The amounts of pCI and pCI-EBNA5 DNA in pmol and pGL3-Control DNA in µg are indicated in the figure. Total RNA was prepared and 40 µg aliquots were analysed for the presence of cleaved and uncleaved primary transcripts by RNase protection analysis using a probe that comprised the SV40 polyadenylation signal. Uncleaved pre-mRNA protected 195 bp and cleaved pre-mRNA 147 bp of the hybridisation probe. The fragments were designated U and C, respectively, and are indicated by arrowheads in the figure. The lanes marked Pr and S contain the undigested probe and the size markers, respectively. Similar results were obtained in three independent experiments. (B) EBNA5 inhibits cleavage of the luciferase reporter pre-mRNA. Aliquots of 107 DG75 cells were transfected with the pcDNA6-EBNA5 expression vector or the empty pcDNA6/V5-His A plasmid and the pCMVLuc (lanes 1–3) or pGL3-Control reporter plasmid (lanes 4–6). The amounts of pcDNA6-EBNA5 and pcDNA6/V5-His A DNA in pmol and pCMVLuc and pGL3-Control DNA in µg are indicated in the figure. Total RNA was prepared and the relative levels of cleaved and uncleaved luciferase RNA determined as above. Similar results were obtained in three independent experiments.
To show that the processing of pre-mRNA synthesised from luciferase reporter plasmids was also inhibited by EBNA5, an EBNA5 expression vector with a different 3′-UTR, pcDNA6-EBNA5, was created. This plasmid contained the CMV promoter, the EBNA5 reading frame and the 3′ pre-mRNA processing signal from the bovine growth hormone gene. DG75 cells were transfected with the EBNA5 expression vector pcDNA6-EBNA5 or the corresponding empty vector pcDNA6/V5-HisA and the pCMVLuc or pGL3-Control DNA reporter plasmids and then analysed for extent of processing of the primary transcripts with the same cleavage riboprobe as in the previous experiments. As the sequence of this probe was derived from the SV40 3′-UTR it will only generate specific fragments with RNA from the pCMVLuc or pGL3-Control reporter plasmids. The fraction of uncleaved reporter pre-mRNA increased with increasing amounts of pcDNA6-EBNA5 DNA (Fig. 5B). The total amount of RNA hybridising with the cleavage probe, i.e. the sum of cleaved and uncleaved RNA, depended on the reporter but was independent of the expression of EBNA5 (Fig. 5B). This corroborates the notion that EBNA5 does not repress transcription at the level of initiation.
Taken together, the results show that EBNA5 specifically inhibits endonucleolytic cleavage of pre-mRNA and the formation of polyadenylated mRNA at cellular concentrations at which EBNA5 represses gene expression. In the context of our model system, transient transfection of DG75 cells with reporter plasmids, EBNA5-induced inhibition of mRNA processing seemed to be independent of promoter and reporter gene sequence.
In order to decide whether pre-mRNA sequences encoded by the reporter gene or sequences encoded by other parts of the plasmid were the targets for the repressor function of EBNA5, a DNA fragment encompassing the luciferace gene including the 3′-UTR was synthesised by nested PCR and purified. The fragment was transfected together with the EBNA5 expression vector into DG75 cells under standard conditions. Total RNA was prepared and analysed for cleavage of pre-mRNA by the RNase protection assay using a probe encompassing the 3′-UTR cleavage site (Fig. 6). Circular reporter plasmid DNA, reporter plasmid DNA where the luciferase gene-containing fragment had been released by restriction enzyme cleavage with BamHI and NheI, and the isolated PCR fragment containing the luciferase gene, all showed essentially the same pattern of inhibition of pre-mRNA cleavage by EBNA5.
Figure 6.

EBNA5-induced inhibition of pre-mRNA processing depends on sequences in the reporter gene transcript. The reporter gene region of pCMVLuc encompassing the promoter (CMV), the luciferase-encoding sequence and the 3′-UTR [the SV40 late poly(A) signal] was amplified by nested PCR. The primers in the first round were 5′-AGTGCAAGTGCAGGTGCCAG-3′ (sense) and 5′-TGACTGGGTTGAAGGCTCTC-3′ (antisense) and in the second round 5′-GAGCTCTTACGCGTGCTAGC-3′ (sense) and 5′-TCTCAAGGGCATCGGTCGAC-3′ (antisense). The primer sequences were derived from pGL3-Basic vector (Promega) regions surrounding the reporter gene. Aliquots of 5 × 106 DG75 cells were co-transfected with circular pCMVLuc DNA (I), pCMVLuc DNA cleaved with BamHI and NheI to release the luciferase gene region (II) and the PCR-amplified DNA fragment of the luciferase gene region (III), respectively, and increasing amounts (0, 340 and 1500 fmol) of pcDNA6-EBNA5 DNA. (A) Total RNA was prepared and analysed for the presence of cleaved and uncleaved luciferase pre-mRNA using a RNase protection assay with a riboprobe comprising the SV40 3′-UTR cleavage signal. The relative levels of cleaved (C) and uncleaved (U) probe fragments were quantified by phosphorimaging. The results of five independent experiments are presented as the means ± SEM of the ratio between the uncleaved and the sum of cleaved and uncleaved fragments. (B) Analysis of the pCMVLuc reporter activity in the transfected cell extracts. The measured values for each set of experiments (I–III) are expressed as a percentage of the value obtained in the absence of EBNA5. The means ± SEM of five independent experiments are presented.
EBNA5 expression in transfected DG75 cells and EBV-transformed B cell lines
EBNA5 expression in EBV-transformed LCLs and BL cell lines vary considerably between different isolates but the level is generally much lower than that found in the early stages of infection (5,31,45). To relate the levels of EBNA5 obtained in DG75 cells in our transfection experiments to the levels observed in EBV-transformed established B lymphoid cell lines under normal growth conditions and after transient transfection, DG75 cells were transfected with increasing amounts of pCI-EBNA5. To avoid a massive background of untransfected cells, it was necessary to isolate the fraction of cells that had taken up plasmid DNA during the transfection procedure. To that end, the transfection mixture was supplemented with the pE300CY6 expression vector for the rat cell surface protein CD2. The transfectants were selected according to cell surface expression of CD2 using magnetic beads coated with anti-rat CD2 antibodies (42). Expression of EBNA5 in the rCD2-expressing cells was compared with that in the IB4 and WW1 LCLs by dot immunoblot analysis using the JF186 anti-EBNA5 antibody (Fig. 7A). The IB4 cell line was chosen because it has been well characterised by several groups with regard to EBNA5 expression (4,5). The average level of EBNA5 in IB4 cells was similar to that in DG75 cells transfected with 14–54 fmol pCI-EBNA5 DNA under standard conditions. These levels of EBNA5 activated or only slightly repressed expression of the reporter plasmid in transfected DG75 cells. Transfection of DG75 cells with 135–540 fmol pCI-EBNA5 DNA resulted in EBNA5 levels considerably higher than the endogenous level in IB4 cells. Immunofluorescence staining with the JF186 anti-EBNA5 antibody showed that EBNA5 was distributed both as discrete nuclear bodies reminiscent of those seen in EBNA5-expressing LCLs and as a diffuse staining of the whole nucleus (data not shown).
Figure 7.
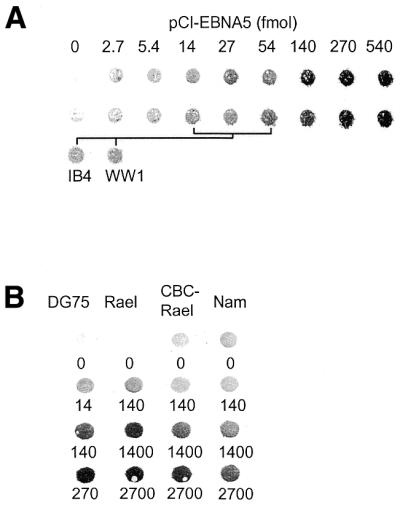
Comparative analysis of vector-induced expression of EBNA5 in DG75 cells and constitutive expression of EBNA5 in EBV-transformed B-lymphoid cell lines. (A) Analysis of the relative level of EBNA5 expression in DG75 cells transfected with different amounts of pCI-EBNA5 DNA and that in IB4 and WW1 LCLs. DG75 cells were transfected with increasing amounts of pCI-EBNA5 DNA together with 5 µg pE300CY6 DNA. The transfected cells were purified on the basis of rCD2 expression using anti-rCD2 antibody-coated magnetic beads. The relative concentration of EBNA5 protein in the cells was determined by dot immunoblot analysis. Cell extract corresponding to 50 000 cells was applied in each dot and EBNA5 expression was detected using the JF186 anti-EBNA5 antibody. The upper two rows in the figure show EBNA5 expression in two independent series of transfections of DG75 cells compared with the endogenous levels of EBNA5 expression in IB4 and WW1 cells. The amounts of pCI-EBNA5 DNA that induced EBNA5 expression levels in the transfected cells corresponding to the endogenous levels in IB4 and WW1 cells are indicated in the figure. (B) Analysis of the relative EBNA5 expression levels in DG75, Rael, CBC-Rael and Namalwa cells transfected with increasing amounts of pCI-EBNA5 DNA together with 5 µg pE300CY6 DNA. The transfected cells were purified on the basis of rCD2 expression and the relative levels of EBNA5 determined as described above. The amount of pCI-EBNA5 DNA per 5 × 106 cells (in fmol) in the transfection is indicated below each dot.
To compare the levels of EBNA5 after transfection in cells of different phenotype, DG75, Rael, CBC-Rael and Namalwa cells were transfected with increasing amounts of pCI-EBNA5 DNA together with the rCD2 expression vector. rCD2-expressing cells were isolated with magnetic beads coated with anti-rat CD2 antibodies and analysed by dot immunoblotting using the JF186 anti-EBNA5 antibody. The expression of EBNA5 was generally about one order of magnitude lower for a given amount of pCI-EBNA5 DNA in the transfected EBV-transformed BL lines and LCLs than in the transfected DG75 cell line (Fig. 7B). The inhibitory effect of EBNA5 on reporter gene expression was investigated in EBV-transformed B lymphoid cell lines of different phenotypes. CAT reporter plasmids containing fragments of the Cp, Wp, LMP1 or SV40 promoter regions [pgBΔC(–1024)CAT, pgBΔW(–559)CAT, pgLRS(–54)CAT and pSVECAT] were transfected together with pCI-EBNA5 into the cell lines indicated in Table 1. All of the reporter plasmids were significantly active in all cell lines and all plasmids were repressed by high levels of EBNA5 (Table 1). Thus, the intracellular level of EBNA5 protein required for repression of the reporter plasmids was approximately the same in the EBV-positive BL type I (Rael) and type III (Namalwa) cell lines as in the EBV-negative DG75 cell line.
Table 1. EBNA5 repression of viral promoter-containing reporter plasmids in EBV-positive cell lines.
| Cell line | Plasmid | Relative CAT activitya (%) | ||
|---|---|---|---|---|
| 0b | 540b | 2700b | ||
| Rael | pgBΔC(–1024)CAT | 100 (48) | 22 ± 4.8 | 3.6 ± 1.0 |
| pgBΔW(–559)CAT | 100 (61) | 16 ± 3.1 | 7.1 ± 1.6 | |
| pgLRS(–54)CAT | 100 (48) | 7.0 ± 1.8 | 3.5 ± 0.5 | |
| pSVE40CAT | 100 (180) | 8.6 ± 1.2 | 4.1 ± 0.1 | |
| P3HR-1 | pgBΔC(–1024)CAT | 100 (5.2) | 24 ± 2.4 | 4.5 ± 1.6 |
| pgBΔW(–559)CAT | 100 (17) | 31 ± 3.0 | 7.6 ± 2.4 | |
| pgLRS(–54)CAT | 100 (17) | 8.4 ± 1.6 | 3.2 ± 1.5 | |
| pSVE40CAT | 100 (130) | 15 ± 5.0 | 6.0 ± 2.0 | |
| Raji | pgBΔC(–1024)CAT | 100 (31) | 21 ± 4.6 | 1.8 ± 0.7 |
| pgBΔW(–559)CAT | 100 (33) | 20 ± 11 | 1.8 ± 0.6 | |
| pgLRS(–54)CAT | 100 (32) | 4.6 ± 1.1 | 5.6 ± 3.8 | |
| pSVE40CAT | 100 (480) | 22 ± 5.0 | 3.1 ± 1.0 | |
| Namalwa | pgBΔC(–1024)CAT | 100 (6.0) | 37 ± 0.9 | 6.4 ± 1.3 |
| pgBΔW(–559)CAT | 100 (7.0) | 28 ± 1.9 | 5.6 ± 1.5 | |
| pgLRS(–54)CAT | 100 (7.0) | 15 ± 2.2 | 5.2 ± 0.5 | |
| pSVE40CAT | 100 (44) | 14 ± 1.7 | 7.3 ± 0.5 | |
aThe activities were calculated as a percentage of the activity in cells transfected with the empty pCI vector. The mean CAT activity in the absence of EBNA5 measured as percentage acetylation under standard assay conditions is denoted within parentheses (measured value × dilution factor). Means ± SEM of three independent experiments are presented.
bThe amount of pCI-EBNA5 DNA per 5 × 106 cells (in (fmol) in the transfections.
Inhibitory levels of EBNA5 are not cytotoxic in transient transfections
The observed negative effect of EBNA5 might conceivably be explained by a non-specific cytotoxic effect of the protein leading to the induction of cell death and mRNA and protein degradation. In a first series of experiments to investigate possible cytotoxic effects of EBNA5 on the transfected cell, two markers for cell viability, [3H]thymidine incorporation and Trypan Blue exclusion, were employed. The EBNA5 expression vector pCI-EBNA5 and the pSVECAT reporter plasmid were co-transfected with the CD2 expression vector pE300CY6 into DG75 cells and the transfected cells selected with regard to cell surface expression of CD2 with magnetic beads coated with anti-rat CD2 antibodies (42). Expression of EBNA5 did not change the total number of cells recovered after selection or the fraction of CD2-positive cells as determined by FACS analysis (data not shown). The level of EBNA5 protein in the cells correlated positively with the amount of pCI-EBNA5 DNA added to the transfection mixture (Fig. 8A). The rate of [3H]thymidine incorporation and the uptake of Trypan Blue by the cells were not significantly affected by pCI-EBNA5 at levels up to those corresponding to 540 fmol vector DNA in the transfection mixture (Fig. 8B). In contrast, the activity of the reporter plasmid was repressed by increasing amounts of EBNA5. Inhibition was nearly complete (>90%) at the EBNA5 level corresponding to 540 fmol pCI-EBNA5 DNA in the transfection mixture (Fig. 8A).
Figure 8.

Repressor function of EBNA5 does not correlate with the induction of cell death. DG75 cells were co-transfected with the CD2 expression vector pE300CY6, the pSVECAT reporter plasmid and increasing amounts of the EBNA5 expression vectors pCI-EBNA5 (A) or pcDNA6-EBNA5 (B and C). The transfected cells were selected using anti-CD2 antibody-coated magnetic beads, plated and assayed for EBNA5 expression, CAT reporter gene expression, luciferase reporter gene expression, Trypan Blue exclusion (cell viability), [3H]thymidine incorporation (DNA synthesis rate) and staining with Annexin V–FITC (apoptosis) and propidium iodide (necrosis), respectively, as described in Materials and Methods. The results of three independent experiments are presented as the means ± SEM. (A) Trypan Blue exclusion, [3H]thymidine incorporation and CAT reporter gene expression at different EBNA5 levels. The observed values of each parameter are expressed as a percentage of the value obtained in the absence of EBNA5 expression. Thus, the 100% value corresponds to the [3H]thymidine incorporation rate, the fraction of cells excluding Trypan Blue or the CAT activity in cells transfected with 540 fmol empty pCI plasmid. The means ± SEM of three independent experiments are presented. EBNA5 expression in the transfected cells was determined by dot immunoblot analysis using the anti-EBNA5 antibody JF186. Cell extract corresponding to 90 000 transfected cells was applied in each dot. The amount of pCI-EBNA5 DNA per 5 × 106 cells used in the transfections is indicated in the figure. (B) Frequency of apoptosis and necrosis of EBNA5-expressing cells. Aliquots of transfected and selected cells were incubated with Annexin V–FITC, with propidium iodide and with a mixture of Annexin V–FITC and propidium iodide, respectively. The fractions of Annexin V–FITC (early apoptosis) and propidium iodide (necrosis and late apoptosis) stained cells were determined by cell sorting of 5000 cells with a FACScan apparatus. The means ± SEM of three independent experiments are presented. (C) The activity of the pCMVLuc reporter plasmid in the cells in (B). The means ± SEM of three independent experiments are presented.
In a second series of experiments, EBNA5 toxicity was assessed by monitoring cell death by necrosis by FACScan analysis of propidium iodide stained cells and death by apoptosis by analysis of Annexin V–FITC stained cells. DG75 cells were co-transfected with the pcDNA6-EBNA5 plasmid, the CD2 expression vector pE300CY6 and the pCMVLuc reporter plasmid and CD2-expressing cells were isolated as above. The level of EBNA5 in the cells increased with increasing amounts of pcDNA6-EBNA5 DNA in the transfection (data not shown). The fraction of apoptotic cells and necrotic cells both remained essentially constant over the concentration range of EBNA5 studied (0–3000 fmol pcDNA6-EBNA5 DNA; Fig. 8B). It should be noted that expression of the pcDNA6-EBNA5 vector is approximately five times lower per fmol DNA than that of the pCI-EBNA5 vector (Fig. 8C and data not shown). The inhibition of reporter gene activity was ∼80% at the EBNA5 level corresponding to 1500 fmol in the transfection mixture. Taken together, the results strongly suggest that cell death is not responsible for the inhibition of CAT or luciferase pre-mRNA processing in EBNA5-transfected cells. The transfected DG75 cells almost doubled in number during the 72 h period of incubation, irrespective of whether the cells were transfected with the pCI-EBNA5 plasmid, the pcDNA6-EBNA5 plasmid or the empty control vector (data not shown). The doubling time of growing DG75 cells not subjected to the transfection procedure was ∼24 h.
Inhibition by EBNA5 is gene dependent in the context of the chromosome
Our results above show that EBNA5 is not cytotoxic even at relatively high intracellular levels. Thus, EBNA5 is not an indiscriminant inhibitor of the expression of all cellular genes, and a regulatory mechanism must exist for the selection of target genes. To further establish the existence of such a control mechanism at the cellular level, we determined the effect of EBNA5 expressed at levels resulting in >90% repression of reporter gene activity on the expression of a panel of cellular genes in transient transfection studies using the DNA array technique. Cells that had taken up plasmid DNA were isolated with magnetic beads coated with antibody using their expression of the rat surface CD2 antigen as the selectable marker. Expression of EBNA5 did not significantly affect the yield of cells obtained after transfection. The poly(A)+ RNA fraction was isolated with oligo(dT)-conjugated magnetic beads and used as template for the synthesis of 32P-labelled cDNA probes in a reverse transcriptase-catalysed reaction. The specific radioactivity of the cDNA probes synthesised from the EBNA5-positive and EBNA5-negative cells, respectively, were essentially the same. The probes were incubated under hybridisation conditions with membranes that contained an array of cDNA fragments derived from 588 human genes, 145 of which were expressed in cells transfected with the empty pCI vector (Fig. 9). Cells transfected with the EBNA5 expression vector displayed an expression pattern identical in qualitative but different in quantitative terms. A >2-fold increase or decrease in gene expression in three out of three independent experiments was regarded as significant. Using this criterion, two of the 145 expressed genes were repressed in cells that had been exposed to EBNA5 at levels conferring repression in the reporter plasmid system. None of the genes showed increased expression. Expression of the BNIP3 gene was down-regulated ∼3-fold (relative expression level 0.30 ± 0.04, n = 3) and the CD27L (CD70) gene ∼2-fold (relative expression level 0.46 ± 0.14, n = 3). To confirm this observation, RNA from the transfected cells was analysed with the RNase protection technique using probes specific for actin, glyceraldehyde 3-phosphate dehydrogenase (GAPDH) and BNIP3 mRNA, respectively. The results of three independent experiments showed a significant down-regulation of BNIP3 expression (relative expression level 0.37 ± 0.02, n = 3) in the presence of EBNA5 whereas expression of the GAPDH and actin genes remained unchanged (relative expression levels 0.78 ± 0.03, n = 3, and 0.92 ± 0.01, n = 3, respectively).
Figure 9.
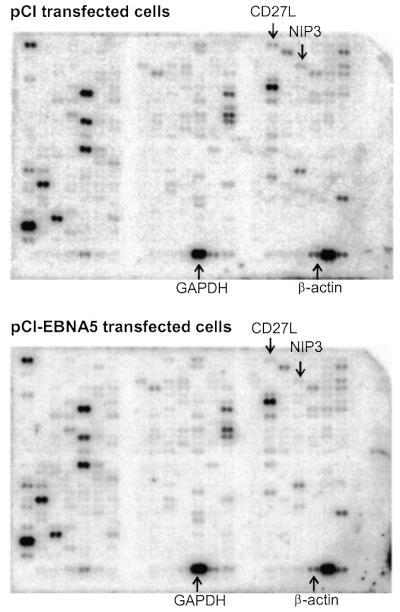
Effect of EBNA5 on the expression of cellular genes on the chromosomes. DG75 cells were transfected with the pE300CY6 plasmid and pCI-EBNA5 or pCI. The transfected cells were purified on the basis of rCD2 expression and the poly(A)+ RNA fraction isolated and used as template for the preparation of 32P-labelled cDNA. Human Atlas DNA microarray membranes (Clontech) containing cDNA fragments representing 588 human genes were incubated under hybridisation conditions with probes derived from pCI-transfected cells (upper) and pCI-EBNA5-transfected cells (lower), respectively. The figure shows the representative results of one of three independent experiments. The positions corresponding to the BNIP3, CD27L, GAPDH and β-actin cDNAs are indicated. The expression level of each gene as reflected by the amount of 32P hybridised to the membrane was quantified by phosphorimaging. The influence of EBNA5 on expression of the genes represented on the membrane was calculated as the ratio between the hybridisation signal obtained with the probe derived from cells transfected with the EBNA5 expression vector and the probe from cells transfected with the control plasmid.
DISCUSSION
It has been suggested that the role of EBNA5 in EBV-mediated primary B lymphocyte growth transformation resides in its ability to enhance transcriptional activation by EBNA2. In the present paper we present evidence of another property of EBNA5 compatible with a role in the early stages of EBV infection and cell transformation, namely as a repressor of the expression of certain viral and host cell genes. The precise molecular mechanism of EBNA5-induced repression remains, however, to be established. We have excluded that the repressive effect of EBNA5 only reflects the induction of cell death and subsequent arrest of gene expression. The data presented are consistent with the notion that as a repressor EBNA5 acts not at the level of transcription initiation but on RNA processing by inhibiting 3′-end cleavage and polyadenylation of certain cellular and viral pre-mRNAs. As a consequence, nuclear export of functional mRNA to the cytoplasm is repressed. Post-transcriptional 3′-end processing of cellular pre-mRNAs occurs in two tightly coupled steps: endonucleolytic cleavage of the primary transcripts and poly(A) addition to the upstream product. The reaction requires the presence of at least two sequences in the pre-mRNA: a conserved AAUAAA hexamer located 10–30 nt upstream of the cleavage site and a variable G/U-rich element downstream of the cleavage site. A large complex of factors is required to reconstitute the complete polyadenylation reaction in vitro, and most of these factors consist of several subunits. The cleavage and polyadenylation specificity factor (CPSF) binds to the AAUAAA hexamer and plays a key role in coordinating both the cleavage and poly(A) addition reactions. It can also form a stable complex with the transcription factor TFIID and is involved in linking polyadenylation to transcription. In addition to CPSF, cleavage requires the cleavage stimulation factor, which binds to the G/U-rich element, and cleavage factors I and II. After cleavage, poly(A) polymerase catalyses the synthesis of long poly(A) tails in a processive reaction that requires a poly(A)-binding protein (PABII). As far as is known, the poly(A) tails of EBV mRNAs are generated by the cellular 3′-end processing machinery without contributions by virally encoded proteins. Obviously, our results do not allow conclusions regarding the precise target for the repressive action of EBNA5 among the components of the basal polyadenylation machinery. Recent data indicate that the major steps in pre-mRNA processing, capping, splicing, 3′-end cleavage and polyadenylation, are tightly coupled to transcription (46). This is achieved by recruitment of the capping enzyme, splicing factors, and the 3′-processing factors to the unique C-terminal domain of RNA polymerase II, forming a ‘transcriptosome’ (47,48). It represents an efficient system for delivering processing factors to the nascent pre-mRNA as and when they are needed. Furthermore, it has been proposed that RNA polymerases associate into transient complexes or ‘transcription factories’ which would act to increase the local concentration of processing factors at sites of active transcription. This gives rise to visible structures (‘speckles’), with components in dynamic equilibrium with the surrounding nucleoplasm. Thus, it is clear that eukaryotic nuclei are not homogeneous with regard to the distribution of factors but specific mechanisms exist which serve to increase local concentrations of factors at discrete locations where they are needed. It has, for example, been shown that a specific subset of SR proteins, a family of proteins involved in splicing, are concentrated at a subset of sites of active transcription (49). One might speculate that EBNA5 is at least temporarily enriched at sites in the vicinity of selected genes to reach concentrations where its function as an inhibitor of pre-mRNA cleavage and polyadenylation dominates. In this connection it is interesting to note that EBNA5 immunofluorescence in the transiently transfected DG75 cells in our study was distributed both as discrete nuclear bodies (speckles) reminiscent of those seen in EBNA5-expressing LCLs and as a diffuse staining of the whole nucleus (data not shown). The localisation of EBNA5 in B lymphocytes during infection has been characterised in some detail (15,16). EBNA5 antigens appear very early (hours) after infection and are first distributed throughout the nucleoplasm. After a few days the EBNA5 immunostaining condenses into nuclear bodies designated PODs or ND10 domains.
The molecular basis for the difference in sensitivity of various genes to inhibition by EBNA5 also remains to be delineated. In particular, the question of why the specificity of the inhibitory function of EBNA5 with regard to genes in a chromosomal context is transformed into apparent promiscuity when the target gene is located extrachromosomally in an episomal reporter plasmid has to be explored. Conceptually, multiple factors might contribute to the regulation of pre-mRNA processing, including the coding sequence and intron organisation of the gene itself, the sequence of the 3′-UTR and its polyadenylation signals, and local variations in the abundance of specific components of the basal polyadenylation machinery in the nucleus. The processing of pre-mRNA synthesised from a linear DNA fragment, which encompassed the reporter gene including the 3′-UTR, was inhibited by EBNA5 in the same manner as pre-mRNA derived from the same sequence in a circular plasmid (Fig. 6). This strongly suggests that EBNA5-induced repression is mediated by sequence elements in the gene region. We have not performed a systematic investigation of the correlation between the inhibitory effect of EBNA5 and the presence of introns in the primary transcript of the reporter gene. However, the majority of the reporter constructs employed in the present study contain at least one intron at different locations in the transcription unit (5′ of, 3′ of or within the coding sequence). Two of the constructs do not contain any splice sites. Expression of the reporter gene in all constructs was, however, down-regulated by high levels of EBNA5. Thus, the intron–exon structure of the target genes does not seem to determine their response to inhibition by EBNA5. Furthermore, although the majority of the constructs contain the SV40 late or early signals for pre-mRNA cleavage and polyadenylation, one plasmid contains the EBV EBNA2 signal sequence and another the bovine growth hormone signal sequence. Still, all plasmids were sensitive to inhibition by EBNA5. It is clearly not possible to dismiss the sequence of the 3′-UTR as one of the decisive factors for the response of the gene to EBNA5 inhibition on the basis of these few observations. We presently favour a hypothetical model for the function of EBNA5 that includes enrichment of EBNA5 to levels consistent with the inhibitory effect at specific loci in the nucleus at an early stage of infection. EBNA5 might interact with a signal sequence in the 3′-UTR of discrete pre-mRNAs or form a complex with a distinct component of the polyadenylation apparatus in a manner that leads to inhibition of endonucleolytic cleavage of the pre-mRNA molecule.
The various EBNA-encoding mRNAs in EBV-transformed B lymphoma cells are formed by alternative polyadenylation and splicing of a large primary transcript initiated at one of two promoters (2). Thus, transcription initiation cannot account for the differentiated expression levels of the different EBNAs. The large transcripts contain several polyadenylation signals: EBNA2 and EBNA5 have a common polyadenylation signal but each of EBNA1, EBNA3, EBNA4 and EBNA 6 have a unique polyadenylation signal. The differential use of latent infection polyadenylation sites probably determines upstream splicing and thereby regulates synthesis of the six EBNA mRNAs from the same promoter. The molecular details of this process are not known. The results presented in this communication suggest that EBNA5 plays a role in pre-mRNA processing by, in a concentration-dependent manner, inhibiting 3′-UTR cleavage and polyadenylation. Thus, it is conceivable that changes in the levels of EBNA5 play a role in the regulated use of alternative polyadenylation sites.
EBNA5 expression in EBV-transformed LCLs varies considerably between different isolates but the level is generally much lower than that found in the early stages of infection (5,31,45). In the present study, DG75 cells transfected with 54 fmol pCI-EBNA5 DNA under our standard conditions were shown to express the EBNA5 protein at a level similar to the endogenous level in IB4 cells. Comparative determinations of EBNA5 protein levels in the transfected cells showed that the concentration range generated by the different amounts of EBNA5 expression vector encompassed the EBNA5 levels in EBV-transformed LCLs and in freshly infected B lymphocytes. Thus, the repressor effect of EBNA5 in the transfected cells was exerted at concentrations that are biologically relevant. The high level of EBNA5 in the early stages of infection is compatible with the hypothesis that repression of certain viral and cellular genes by EBNA5 might be an important step early in EBV infection and initiation of cell transformation.
The effect of inhibitory levels of EBNA5 on the expression of cellular genes in a chromosomal context was analysed in transient transfection experiments with an EBNA5 expression vector and using DNA microarray technology. Under these circumstances, only 2 of the 145 expressed genes in the panel of 588 genes were repressed. This shows that EBNA5 can also inhibit expression of genes in a chromosomal environment and does it in a discriminatory manner. Thus, a regulatory mechanism must exist in the cell which confers specificity to the selection of target genes for EBNA5. One of the repressed genes coded for the BNIP3 protein. Notably, this protein has pro-apoptotic activity and interacts with anti-apoptotic proteins, including BCL-2, adenovirus E1B and the EBV-encoded BCL-2 homologue BHRF1 (50). BNIP3 binding of BCL-2 inhibits its anti-apoptotic function. It appears that BCL-2, whose expression is up-regulated by LMP1, plays an important role in protecting latency type III cells from apoptosis induced by growth factor withdrawal (51,52). Conceivably, down-regulation of BNIP3 by EBNA5 promotes the survival of EBV-infected cells.
Acknowledgments
ACKNOWLEDGEMENTS
We thank Dr Maria Masucci for the anti-EBNA5 JF186 antibody, Dr Paul J. Farrell for the p3.1W-LP plasmid and Dr Erik Lundgren for the CD2 expression vector pE300CY6. This investigation was supported by grants from the Swedish Research Council (project 5667), the Swedish Cancer Society and the Sahlgrenska University Hospital.
REFERENCES
- 1.Rickinson A.B. and Kieff,E. (2001) Epstein–Barr virus. In Knipe,D.M. and Howley,P.M. (eds), Fields Virology, 4th Edn. Lipincott Williams & Wilkins, Philadelphia, PA, pp. 2575–2628.
- 2.Kieff E. and Rickinson,A.B. (2001) Epstein–Barr virus and its replication. In Knipe,D.M. and Howley,P.M. (eds), Fields Virology, 4th Edn. Lipincott Williams & Wilkins, Philadelphia, PA, pp. 2511–2573.
- 3.Dillner J. and Kallin,B. (1988) The Epstein-Barr virus proteins. Adv. Cancer Res., 50, 95–158. [DOI] [PubMed] [Google Scholar]
- 4.Alfieri C., Birkenbach,M. and Kieff,E. (1991) Early events in Epstein-Barr virus infection of human B lymphocytes. Virology, 181, 595–608. [DOI] [PubMed] [Google Scholar]
- 5.Finke J., Rowe,M., Kallin,B., Ernberg,I., Rosén,A., Dillner,J. and Klein,G. (1987) Monoclonal and polyclonal antibodies against Epstein-Barr virus nuclear antigen 5 (EBNA-5) detect multiple protein species in Burkitt’s lymphoblastoid cell lines. J. Virol., 61, 3870–3878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Henkel T., Ling,P.D., Hayward,S.D. and Peterson,M.G. (1994) Mediation of Epstein-Barr virus EBNA2 transactivation by recombination signal-binding protein J kappa. Science, 265, 92–95. [DOI] [PubMed] [Google Scholar]
- 7.Zimber-Strobl U., Strobl,L.J., Meitinger,C., Hinrichs,R., Sakai,T., Furukawa,T., Honjo,T. and Bornkamm,G.W. (1994) Epstein-Barr virus nuclear antigen 2 exerts its transactivating function through interaction with recombination signal binding protein RBP-J kappa, the homologue of Drosophila Suppressor of Hairless. EMBO J., 13, 4973–4982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Harada S. and Kieff,E. (1997) Epstein-Barr nuclear protein LP stimulates EBNA-2 acidic domain mediated transcriptional activation. J. Virol., 71, 6611–6618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sjoblom A., Jansson,A., Yang,W., Lain,S., Nilsson,T. and Rymo,L. (1995) PU box-binding transcription factors and a POU domain protein cooperate in the Epstein-Barr virus (EBV) nuclear antigen 2-induced transactivation of the EBV latent membrane protein 1 promoter. J. Gen. Virol., 76, 2679–2692. [DOI] [PubMed] [Google Scholar]
- 10.Johannsen E., Koh,E., Mosialos,G., Tong,X., Kieff,E. and Grossman,S.R. (1995) Epstein-Barr virus nuclear protein 2 transactivation of the latent membrane protein 1 promoter is mediated by J kappa and PU.1. J. Virol., 69, 253–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Laux G., Adam,B., Strobl,L.J. and Moreau-Gachelin,F. (1994) The Spi-1/PU.1 and Spi-B ets family transcription factors and the recombination signal binding protein RBP-Jκ interact with an Epstein-Barr virus nuclear antigen 2 responsive cis-element. EMBO J., 13, 5624–5632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sjoblom A., Yang,W., Palmqvist,L., Jansson,A. and Rymo,L. (1998) An ATF/CRE element mediates both EBNA2-dependent and EBNA2-independent activation of the Epstein-Barr virus LMP1 gene promoter. J. Virol., 72, 1365–1376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wu D.Y., Kalpana,G.V., Goff,S.P. and Schubach,W.H. (1996) Epstein-Barr virus nuclear protein 2 (EBNA2) binds to a component of the human SNF-SWI complex, hSNF/Ini1. J. Virol., 70, 6020–6028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Szekely L., Jiang,W.-Q., Pokrovskaja,K., Wiman,K.G., Klein,G. and Ringertz,N. (1995) Reversible nucleolar translocation of Epstein-Barr virus encoded EBNA5 and hsp70 proteins after exposure to heat shock or cell density congestion. J. Gen. Virol., 76, 2423–2432. [DOI] [PubMed] [Google Scholar]
- 15.Szekely L., Pokrovskaja,K., Jiang,W.-Q., de The,H., Ringertz,N. and Klein,G. (1996) The Epstein-Barr virus-encoded nuclear antigen EBNA-5 accumulates in PML-containing bodies. J. Virol., 70, 2562–2568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Szekely L., Pokrovskaja,K., Jiang,W.-Q., Selivanova,G., Löwbeer,M., Ringertz,N., Wiman,K.G. and Klein,G. (1995) Resting B-cells, EBV-infected B-blasts and established lymphoblastoid cell lines differ in their Rb, p53 and EBNA-5 expression patterns. Oncogene, 10, 1869–1874. [PubMed] [Google Scholar]
- 17.Hammerschmidt W. and Sugden,B. (1989) Genetic analysis of immortalizing functions of Epstein-Barr virus in human B lymphocytes. Nature, 340, 393–397. [DOI] [PubMed] [Google Scholar]
- 18.Mannick J.B., Cohen,J.I., Birkenbach,M., Marchini,A. and Kieff,E. (1991) The Epstein-Barr virus nuclear protein encoded by the leader of the EBNA RNAs is important in B-lymphocyte transformation. J. Virol., 65, 6826–6837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kitay M.K. and Rowe,D.T. (1996) Cell cycle stage-specific phosphorylation of the Epstein-Barr virus immortalization protein EBNA-LP. J. Virol., 70, 7885–7893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sinclair A.J., Ignacio,P., Peters,G. and Farrell,P. (1994) EBNA-2 and EBNA-LP cooperate to cause G0 to G1 transition during immortalization of resting human B lymphocytes by Epstein-Barr virus. EMBO J., 13, 3321–3328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kaiser C., Laux,G., Eick,D., Jochner,N., Bornkamm,G.W. and Kempkes,B. (1999) The proto-oncogene c-myc is a direct target gene of Epstein-Barr virus nuclear antigen 2. J. Virol., 73, 4481–4484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nitsche F., Bell,A. and Rickinson,A. (1997) Epstein-Barr leader protein enhances EBNA-2 mediated transactivation of latent membrane protein 1 expression: a role for the W1W2 repeat domain. J. Virol., 71, 6619–6627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ben Bassat H., Goldblum,N., Mitrani,S., Goldblum,T., Yoffey,J.M., Cohen,M.M., Bentwich,Z., Ramot,B., Klein,E. and Klein,G. (1977) Establishment in continuous culture of a new type of lymphocyte from a “Burkitt like” malignant lymphoma (line d.G.-75). Int. J. Cancer, 19, 27–33. [DOI] [PubMed] [Google Scholar]
- 24.Klein G., Dombos,L. and Gothoskar,B. (1972) Sensitivity of Epstein-Barr virus (EBV) producer and non-producer human lymphoblastoid cell lines to superinfection with EB-virus. Int. J. Cancer, 10, 44–57. [DOI] [PubMed] [Google Scholar]
- 25.Ernberg I., Falk,K., Minarovits,J., Busson,P., Tursz,T., Masucci,M.G. and Klein,G. (1989) The role of methylation in the phenotype-dependent modulation of Epstein-Barr nuclear antigen 2 and latent membrane protein genes in cells latently infected with Epstein-Barr virus. J. Gen. Virol., 70, 2989–3002. [Erratum (1990) J. Gen. Virol., 71, 499] [DOI] [PubMed] [Google Scholar]
- 26.King W., Thomas Powell,A.L., Raab Traub,N., Hawke,M. and Kieff,E. (1980) Epstein-Barr virus RNA. V. Viral RNA in a restringently infected, growth-transformed cell line. J. Virol., 36, 506–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rooney C.M., Gregory,C.D., Rowe,M., Finerty,S., Edwards,C., Rupani,H. and Rickinson,A.B. (1986) Endemic Burkitt’s lymphoma: phenotypic analysis of tumor biopsy cells and of derived tumor cell lines. J. Natl Cancer Inst., 77, 681–687. [DOI] [PubMed] [Google Scholar]
- 28.Hinuma Y. and Grace,J.T.,Jr (1967) Cloning of immunoglobulin-producing human leukemic and lymphoma cells in long-term cultures. Proc. Soc. Exp. Biol. Med., 124, 107–111. [DOI] [PubMed] [Google Scholar]
- 29.Epstein M.A., Achong,B.G., Barr,Y.M., Zajac,B., Henle,G. and Henle,W. (1966) Morphological and virological investigations on cultured Burkitt tumor lymphoblasts (strain Raji). J. Natl Cancer Inst., 37, 547–559. [PubMed] [Google Scholar]
- 30.Altiok E., Minarovits,J., Hu,L.F., Contreras Brodin,B., Klein,G. and Ernberg,I. (1992) Host-cell-phenotype-dependent control of the BCR2/BWR1 promoter complex regulates the expression of Epstein-Barr virus nuclear antigens 2–6. Proc. Natl Acad. Sci. USA, 89, 905–909. [Erratum (1992) Proc. Natl Acad. Sci. USA, 89, 6225] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dillner J., Kallin,B., Alexander,H., Ernberg,I., Uno,M., Ono,Y., Klein,G. and Lerner,R.A. (1986) An Epstein-Barr virus (EBV)-determined nuclear antigen (EBNA5) partly encoded by the transformation-associated Bam WYH region of EBV DNA: preferential expression in lymphoblastoid cell lines. Proc. Natl Acad. Sci. USA, 83, 6641–6645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ricksten A., Svensson,C., Welinder,C. and Rymo,L. (1987) Identification of sequences in Epstein-Barr virus DNA required for the expression of the second Epstein-Barr virus-determined nuclear antigen in COS-1 cells. J. Gen. Virol., 68, 2407–2418. [DOI] [PubMed] [Google Scholar]
- 33.Fahraeus R., Jansson,A., Sjoblom,A., Nilsson,T., Klein,G. and Rymo,L. (1993) Cell phenotype-dependent control of Epstein-Barr virus latent membrane protein 1 gene regulatory sequences. Virology, 195, 71–80. [DOI] [PubMed] [Google Scholar]
- 34.Ricksten A., Olsson,A., Andersson,T. and Rymo,L. (1988) The 5′ flanking region of the gene for the Epstein-Barr virus-encoded nuclear antigen 2 contains a cell type specific cis-acting regulatory element that activates transcription in transfected B-cells. Nucleic Acids Res., 16, 8391–8410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nilsson T., Sjoblom,A., Masucci,M.G. and Rymo,L. (1993) Viral and cellular factors influence the activity of the Epstein-Barr virus BCR2 and BWR1 promoters in cells of different phenotype. Virology, 193, 774–785. [DOI] [PubMed] [Google Scholar]
- 36.Jansson A., Masucci,M. and Rymo,L. (1992) Methylation of discrete sites within the enhancer region regulates the activity of the Epstein-Barr virus BamHI W promoter in Burkitt lymphoma lines. J. Virol., 66, 62–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Banerji J., Rusconi,S. and Schaffner,W. (1981) Expression of a beta-globin gene is enhanced by remote SV40 DNA sequences. Cell, 27, 299–308. [DOI] [PubMed] [Google Scholar]
- 38.Ruetschi U., Rymo,L. and Lindstedt,S. (1997) Human 4-hydroxyphenylpyruvate dioxygenase gene (HPD). Genomics, 44, 292–299. [DOI] [PubMed] [Google Scholar]
- 39.Chomczynski P. and Sacchi,N. (1987) Single-step method of RNA isolation by acid thiocyanate-phenol-chloroform extraction. Anal. Biochem., 162, 156–159. [DOI] [PubMed] [Google Scholar]
- 40.Favaloro J., Treisman,R. and Kamen,R. (1980) Transcription maps of polyoma virus-specific RNA: analysis by two-dimensional nuclease S1 gel mapping. Methods Enzymol., 65, 718–749. [DOI] [PubMed] [Google Scholar]
- 41.Zetterberg H., Stenglein,M., Jansson,A., Ricksten,A. and Rymo,L. (1999) Relative levels of EBNA1 gene transcripts from the C/W, F and Q promoters in Epstein-Barr virus-transformed lymphoid cells in latent and lytic stages of infection. J. Gen. Virol., 80, 457–466. [DOI] [PubMed] [Google Scholar]
- 42.Pilon M., Gullberg,M. and Lundgren,E. (1991) Transient expression of the CD2 cell surface antigen as a sortable marker to monitor high frequency transfection of human primary B cells. J. Immunol., 146, 1047–1051. [PubMed] [Google Scholar]
- 43.Brandhorst B. and McConkey,E. (1974) Stability of nuclear RNA in mammalian cells. J. Mol. Biol., 85, 451–463. [DOI] [PubMed] [Google Scholar]
- 44.Huang Y. and Carmichael,G.C. (1996) Role of polyadenylation in nucleocytoplasmic transport of mRNA. Mol. Cell. Biol., 16, 1534–1542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wang F., Petti,L., Braun,D., Seung,S. and Kieff,E. (1987) A bicistronic Epstein-Barr virus mRNA encodes two nuclear proteins in latently infected, growth transformed lymphocytes. J. Virol., 61, 945–954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lewis J.D. and Tollervey,D. (2000) Like attracts like: getting RNA processing together in the nucleus. Science, 288, 1385–1389. [DOI] [PubMed] [Google Scholar]
- 47.Gall J.G., Bellini,M., Wu,Z. and Murphy,C. (1999) Assembly of the nuclear transcription and processing machinery: Cajal bodies (coiled bodies) and transcriptosomes. Mol. Biol. Cell, 10, 4385–4402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Corden J.L. and Patturajan,M. (1997) A CTD function linking transcription to splicing. Trends Biochem Sci., 22, 413–416. [DOI] [PubMed] [Google Scholar]
- 49.Neugebauer K.M. and Roth,M.B. (1997) Distribution of pre-mRNA splicing factors at sites of RNA polymerase II transcription. Genes Dev., 11, 1148–1159. [DOI] [PubMed] [Google Scholar]
- 50.Yasuda M., Theodorakis,P., Subramanian,T. and Chinnadurai,G. (1998) Adenovirus E1B-19K/BCL-2 interacting protein BNIP3 contains a BH3 domain and a mitochondrial targeting sequence. J. Biol. Chem., 273, 12415–12421. [DOI] [PubMed] [Google Scholar]
- 51.Henderson S., Rowe,M., Gregory,C., Croom-Carter,D., Wang,F., Longnecker,R., Kieff,E. and Rickinson,A. (1991) Induction of bcl-2 expression by Epstein-Barr virus latent membrane protein 1 protects infected B cells from programmed cell death. Cell, 65, 1107–1115. [DOI] [PubMed] [Google Scholar]
- 52.Finke J., Fritzen,R., Ternes,P., Trivedi,P., Bross,K.J., Lange,W., Mertelsmann,R. and Dolken,G. (1992) Expression of bcl-2 in Burkitt’s lymphoma cell lines: induction by latent Epstein-Barr virus genes. Blood, 80, 459–469. [PubMed] [Google Scholar]