

Abstract
Microtubules have spatiotemporally complex posttranslational modification patterns. Tubulin tyrosine ligase-like (TTLL) enzymes introduce the most prevalent modifications on α-tubulin and β-tubulin. How TTLLs specialize for specific substrate recognition and ultimately modification-pattern generation is largely unknown. TTLL6, a glutamylase implicated in ciliopathies, preferentially modifies tubulin α-tails in microtubules. Cryo-electron microscopy, kinetic analysis and single-molecule biochemistry reveal an unprecedented quadrivalent recognition that ensures simultaneous readout of microtubule geometry and posttranslational modification status. By binding to a β-tubulin subunit, TTLL6 modifies the α-tail of the longitudinally adjacent tubulin dimer. Spanning two tubulin dimers along and across protofilaments (PFs) ensures fidelity of recognition of both the α-tail and the microtubule. Moreover, TTLL6 reads out and is stimulated by glutamylation of the β-tail of the laterally adjacent tubulin dimer, mediating crosstalk between α-tail and β-tail. This positive feedback loop can generate localized microtubule glutamylation patterns. Our work uncovers general principles that generate tubulin chemical and topographic complexity.
Microtubules (MTs) are essential noncovalent polymers assembled from αβ-tubulin heterodimers. They build complex cellular structures such as spindles, axonemes and axons to execute diverse cellular processes ranging from cell division and cellular motility to intracellular transport. MTs perform these functions through the recruitment of myriad cellular effectors that organize them, regulate their assembly and disassembly and recruit cargoes for directed transport. Key to MT functional diversity are chemically varied and abundant posttranslational modifications that give spatiotemporally specialized identities to cellular MTs, that is, the tubulin code1. This code regulates both the intrinsic properties of the polymer and the recruitment of cellular effectors such as MT-associated proteins (MAPs) and molecular motors1. Tubulin modifications regulate the recruitment of proteins at the ends of growing and depolymerizing MTs2-4, control interactions between MTs and intermediate filaments5-9, regulate severing enzymes10-13, kinesin motility14-16 and the loading of dynein cargoes17,18.
Glutamylation, the reversible addition of variable numbers of glutamates to genetically encoded internal glutamates in the intrinsically disordered α-tubulin and β-tubulin tails, is the most phylogenetically widespread tubulin modification, conserved from ciliates to humans. Glutamylation is catalyzed by members of the tubulin tyrosine ligases-like (TTLLs) family. Humans have nine TTLL glutamylase genes19 (reviewed in ref. 20). Glutamylation is especially abundant in geometrically complex, stable MT arrays found in terminally differentiated cells such as neurons, as well as the axonemes of cilia and flagella1,21. Loss of TTLL glutamylases leads to defects in axonal transport with an impact on synaptic transmission22,23, and disruption of glutamylation homeostasis causes neurodegeneration24-27. TTLL glutamylases are also critical for the biogenesis28,29 and motility of cilia and flagella28,30,31, and by targeting anterograde intraflagellar transport, glutamylation regulates ciliary localization of signaling complexes in the hedgehog pathway32,33. Several studies have identified regulators that recruit or activate TTLLs in cilia or flagella. For example, TTLL9 interacts with the flagella-associated protein (FAP234), whose loss decreases glutamylation34. TTLL6 requires CEP41, a protein mutated in Joubert syndrome, for ciliary localization35. Cilia and spindle-associated protein, which stimulates TTLL5 and more broadly other TTLLs36, is highly expressed in the brain37. Consistent with its importance in cilia and neuronal function, aberrant glutamylation leads to male infertility28,38 as well as respiratory31 and neurodevelopmental disorders24-27,35.
TTLL glutamylases specialize in modifying either α-tubulin or β-tubulin tails, as well as initiating or elongating glutamate chains from internal, genetically encoded glutamates in the tubulin tails19,39-42. Glutamylation patterns are stereotyped in cells, and the length and position of the glutamate chains differentially regulate interactions with molecular motors and MAPs1. Glutamylation on α-tubulin and β-tubulin regulates MT-severing enzymes spastin and katanin12,13, with α-tubulin glutamylation being exclusively stimulatory and β-tail glutamylation both stimulatory and inhibitory depending on the length and position of the glutamate chain12. Glutamylation increases the binding of tau, MAP1A, MAP1B and MAP2 to MTs43-45 as a function of glutamate chain length, and also regulates the motility of kinesins15,16 and inner-arm dynein motors that power the beating of cilia and flagella46,47. Thus, understanding the substrate specificities and enzymatic properties of TTLL glutamylases is essential to understanding how glutamylation topography is established.
TTLL6 generates long polyglutamate chains on α-tubulin tails. It is important for the biogenesis and motility of cilia29. Its mislocalization from cilia is associated with Joubert syndrome35, and its activity is critical for cancer cell mitosis and survival48. While recent structural and biophysical studies elucidated active site signatures of TTLL enzymes that specialize them for chain initiation or elongation41 and revealed how the TTLL7 glutamylase recognizes β-tubulin tails39, how TTLLs evolved specifically to modify α-tubulin or β-tubulin, and to discriminate between free tubulin versus MTs remains poorly understood. The interplay between the many members of the TTLL family is also unknown. Using cryo-electron microscopy (cryo-EM) coupled with enzyme kinetic analysis and single-molecule fluorescence, we now show how TTLL6 preferentially recognizes MTs and elongates glutamate chains on α-tubulin tails. TTLL6 modifies α-tubulin tails in trans by binding both along and across PFs with two cationic α-helical structural elements. A PF is assembled through the head-to-tail arrangement of αβ-tubulin dimers. Variable numbers of PFs, primarily 13 in humans, associate laterally to form the MT. This recognition in trans by TTLL6 is unprecedented among tubulin modification enzymes and ensures modification of the tubulin dimer only after polymerization. Single-molecule fluorescence experiments show that one of these cationic elements that is unique to TTLL elongases (and absent in initiases) increases the residence time of TTLL6 on MTs to allow efficient glutamate chain elongation. TTLL6 uses this same cationic element to also read the glutamylation status of the β-tail on the laterally adjacent tubulin dimer, leading to a dramatic increase in recruitment of TTLL6 to the MT and increased α-tail glutamylation upon β-tail glutamylation by TTLL7. Our analysis shows that this recognition strategy is shared by other α-tail glutamyl elongases in the TTLL family.
Our work provides a molecular framework for understanding substrate recognition by TTLL enzymes by shedding light on differences in MT recognition between α-tubulin versus β-tubulin and chain initiating versus elongating TTLL glutamylases and reveals that TTLL enzymes read the glutamylation status of adjacent tubulin dimers, thus establishing a spatial positive feedback loop for MT glutamylation and mediating crosstalk between α-tubulin and β-tubulin tails on the MT. This mechanistic understanding of MT recognition principles by TTLL glutamylases will also provide a molecular reference for analyzing the recognition strategies of nontubulin substrates such as nucleosome assembly protein 1, retinitis pigmentosa GTPase regulator and cyclic GMP–AMP synthase, whose glutamylation is important for proper deposition and dynamics of linker histone H1M on chromatin49, photoreceptor function50 and DNA binding regulation51, respectively.
Results
TTLL6 bridges PFs to modify α-tails in trans
To elucidate the structural basis for TTLL6 substrate recognition and understand how it specifically glutamylates the α-tubulin tail, we determined the structure of Mus musculus TTLL6 (residues 51–502) in complex with the MT using cryo-EM (Fig. 1, Extended Data Fig. 1a-i and Supplementary Table 1). TTLL6 (51–502) is active and displays strong specificity for elongating glutamate chains on α-tubulin41. We used unmodified human MTs52 (Methods) polymerized with the nonhydrolyzable guanosine-5′-triphosphate (GTP) analog guanylyl (α,β)-methylene diphosphonate (GMPCPP), as these yielded the most regular decoration (Extended Data Fig. 1b). The majority of MT reconstructions use porcine brain tubulin, which is highly heterogenous and contains a mixture of isoforms as well as chemically diverse post-translational modifications1,53. Our previous work showed that the use of unmodified tubulin can increase the resolution of an MT complex structure39. In our C1 EM reconstruction (Extended Data Fig. 1d), TTLL6 was easily identifiable as a volume spanning two adjacent αβ-tubulin heterodimers (Extended Data Fig. 1d). To improve the EM reconstruction, individual PFs were refined using the ‘PF refinement’ approach, which accounts for structural variations in the MT lattice54 (Extended Data Fig. 1e). The procedure was modified to refine two adjacent PFs because one TTLL6 helix was found spanning adjacent PFs (Fig. 1a). The reconstruction showed substantial improvement for the MT, with local resolution estimates ranging from ~3 Å to ~4 Å (Extended Data Fig. 1f). The density corresponding to TTLL6 was poorly defined, with the exception of a single helix that spans adjacent PFs, indicating that the majority of TTLL6 is flexibly attached to the MT or adopts a range of conformations (Fig. 1a and Extended Data Fig. 1f). To improve the TTLL6 cryo-EM density, we performed focused classification on TTLL6, excluding the MT signal (Methods; Extended Data Fig. 1h). This yielded a map with a nominal resolution of 7.2 Å and local resolution ranging from 5 Å to 14 Å (Extended Data Fig. 1h,i) into which the TTLL6 crystal structure (Protein Data Bank (PDB) 6VZU; ref. 41) could be confidently docked (Methods; Fig. 1b and Supplementary Table 2) with the catalytic core of the enzyme comprising the N-, central- and C-domains spanning the αβ-tubulin longitudinal interdimer interface (Fig. 1b-d) and making limited contacts with the MT through helices α2, α4, α6 and α9 (Fig. 1c,d) consistent with the flexible linkage of the enzyme with the MT.
Fig. 1 ∣. Cryo-EM structure of TTLL6 bound to the MT.
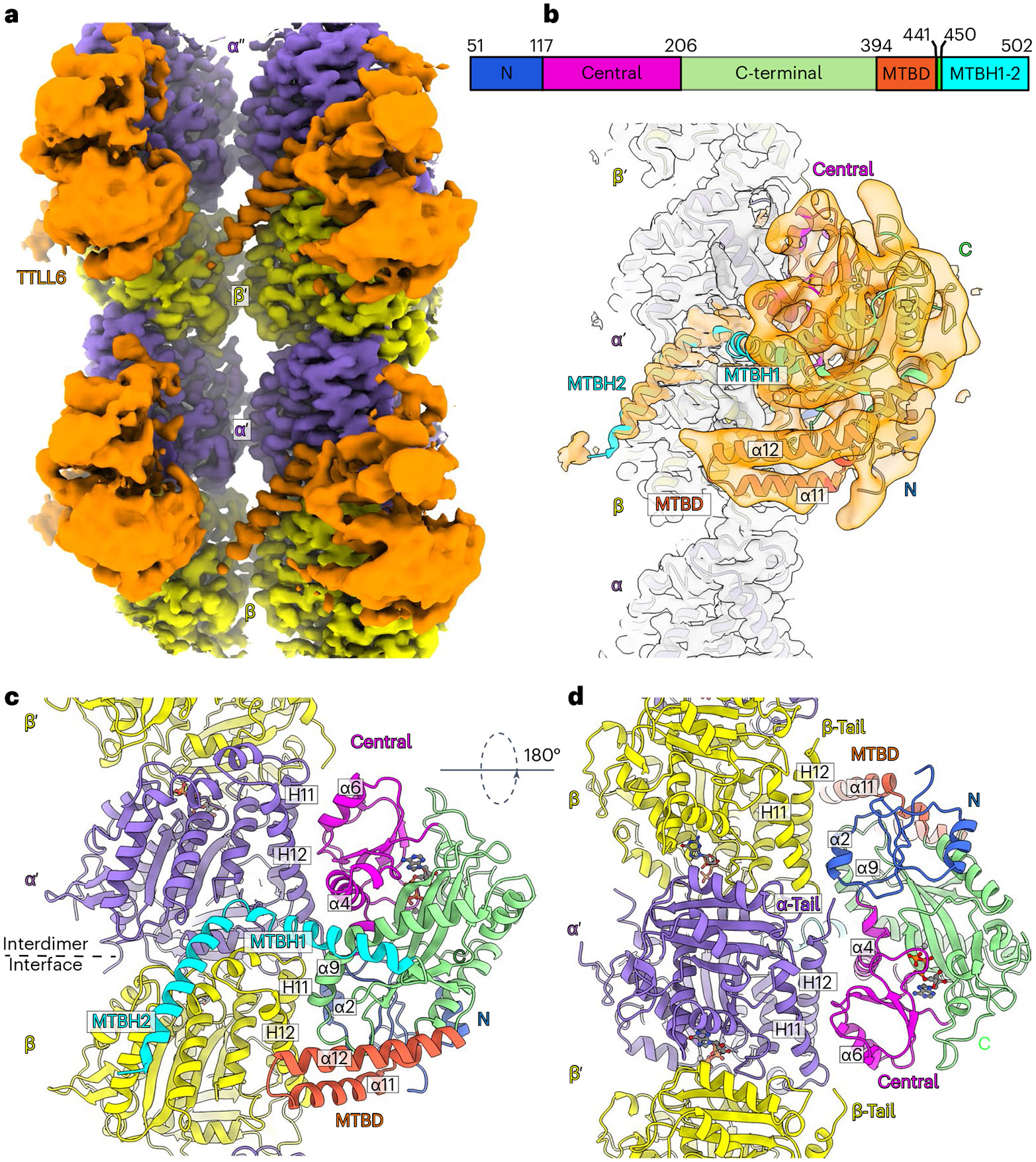
a, Cryo-EM reconstruction showing TTLL6 bound to two PFs in the MT; TTLL6, gold; α-tubulin, purple; β-tubulin, yellow. b, TTLL6 model (X-ray structure PDB 6VZU; ref. 41) fits into the cryo-EM map (Methods). TTLL6 N-, central-, C-, MTBD- and MTBH1-2 domains are blue, magenta, green, orange and cyan, respectively. AlphaFold55 was used to model MTBH1 residues 462–478 (not resolved in PDB 6VZU; ref. 41), as the corresponding cryo-EM density was too noisy for de novo building. c, Model of TTLL6 bound to the MT, color-coded as in b. d, Model of TTLL6 as in c, rotated by 180°. Helices in TTLL6 are denoted as α, and helices in tubulin are denoted as H. The interdimer interface (between α′ and β) is indicated by a dashed line.
The cryo-EM map revealed two prominent structural elements that have substantial interactions with the MT. The first is the cationic MT-binding domain (MTBD; residues 394–440), comprising helices α11 and α12 (Fig. 1b,c) that extend from the ligase core (Fig. 1c,d and Supplementary Fig. 1). The second is the arginine- and lysine-rich helix α14 (MTBH2) that spans the anionic interprotofilament groove and connects to the MTBD and the TTLL core through helix α13 (MTBH1; Fig. 1c). The MTBD was previously identified in the structure of the β-tubulin-specific glutamylase TTLL7 in complex with the MT39, and sequence analysis indicated that it is common to all autonomous TTLL glutamylases39. Both MTBH1 and MTBH2 are highly conserved in TTLL6 sequences from ciliates to humans (Supplementary Fig. 1). MTBH2 is the most well-defined TTLL6 feature in the cryo-EM map, indicating that it is well-ordered on the MT (Fig. 1a,b and Extended Data Fig. 1f,g). Notably, the orientation of MTBH2 relative to the TTLL6 core is dramatically different than that in TTLL6 X-ray structures41 (Extended Data Fig. 2a), indicating that this structural element is intrinsically flexible and stabilized by its interaction with the MT. MTBH1 intersects perpendicularly with the α–β interdimer interface. The MTBH1 C-terminus, which contacts the MT, is disordered in the TTLL6 crystal structures41 (Extended Data Fig. 2a), indicative of its flexibility in the absence of the MT substrate. This section of the MTBH1 (residues 461–474) was modeled using AlphaFold55 (Methods). MTBH1-2 (460–502) in isolation is mostly helical by circular dichroism (CD) (Extended Data Fig. 2b) and binds MTs with an apparent dissociation constant Kd of 3.7 μM (Extended Data Fig. 2c), indicating that it is an autonomous MT-binding element. MTBH1-2 does not form a stable complex with unpolymerized tubulin by analytical gel filtration, even at concentrations as high as 20 μM (Extended Data Fig. 2d), consistent with our cryo-EM structure that shows MTBH1-2 makes MT-specific contacts, spanning the interdimer interface and adjacent PFs. Interestingly, analysis of the alignment parameters derived from the PF refinement of TTLL6-bound MTs versus undecorated MTs (PDB 8V2I; ref. 56) revealed compaction and regularization of the MT wall (Methods; Extended Data Fig. 3). Lateral interactions (between adjacent PFs) are plastic in MTs57,58. While the undecorated MT lattice shows a bimodal wall angle distribution, with two peaks separated by more than 7° (major peak ~23.9° and minor peak ~31.0°), TTLL6-bound MTs show close to a Gaussian distribution (peak ~25.4°; Extended Data Fig. 3a,b), indicating that the MTBH1-2 regularizes the lattice, likely due to its binding in the interprotofilament groove. TTLL6 also compacts the MT longitudinal interface, as evidenced by the change in the dimer repeat distance from 84.22 ± 0.08 Å to 83.61 ± 0.06 Å (P = 0.0083 by one-tailed t test; Extended Data Fig. 3c,d,e; Methods). The addition of excess MTBH1-2 to taxol-stabilized or GMPCPP MTs (either brain MTs or unmodified human MTs) destabilized them, leading to the formation of long sheets (Extended Data Fig. 4a-d). We speculate that the stoichiometric binding of MTBH1-2 induces lateral contacts incompatible with a closed MT structure. Consistent with this, we could not obtain regular decoration and a cryo-EM reconstruction in conditions where TTLL6 was stoichiometric to tubulin, and we did observe subtle variability in helical parameters along the MT lattice under these conditions, requiring the advanced image analyses described in Methods.
Thus, our cryo-EM reconstruction shows that TTLL6 uses two structural elements to specifically modify the α-tubulin tail within the MT context. The first of these, MTBD, shared with all autonomous glutamylases39, positions the enzyme core on the β-tubulin subunit (Fig. 2a). The second, a new cationic helical domain MTBH1-2 (Fig. 2b), bridges adjacent tubulin dimers within one PF and spans across the interprotofilament groove. Together, these two structural elements, MTBD and MTBH1-2, bring a conserved, positively charged groove in the TTLL6 active site in proximity of the α-tail of the longitudinally adjacent tubulin dimer within the same PF (Extended Data Fig. 5a), resulting in selective glutamylation of α-tubulin tails in MTs and not free tubulin or isolated PFs. Mutation of residues in this groove severely impairs glutamylation of both the MT substrate and α-tubulin tail peptides in isolation (Extended Data Fig. 5b,c). Further regioselectivity arises from the specialization of the active site of the enzyme positioned by these two structural elements because TTLL6 prefers branch-containing glutamates on α-tubulin versus β-tubulin tail sequences41. Overall, this recognition paradigm results in selective elongation of glutamate chains on α-tubulin tails on MTs. Consistent with this multipronged MT recognition, MTBD deletion reduces the apparent Michaelis constant KM ~2.2-fold and the catalytic rate kcat ~3.1-fold, MTBH1-2 deletion decreases the apparent KM ~3.5-fold and the kca ~4-fold, while the deletion of both these structural elements decreases the apparent KM ~6.3-fold and the kcat ~14-fold (Fig. 2c).
Fig. 2 ∣. TTLL6 bridges along and across PFs to modify α-tubulin tails in trans.
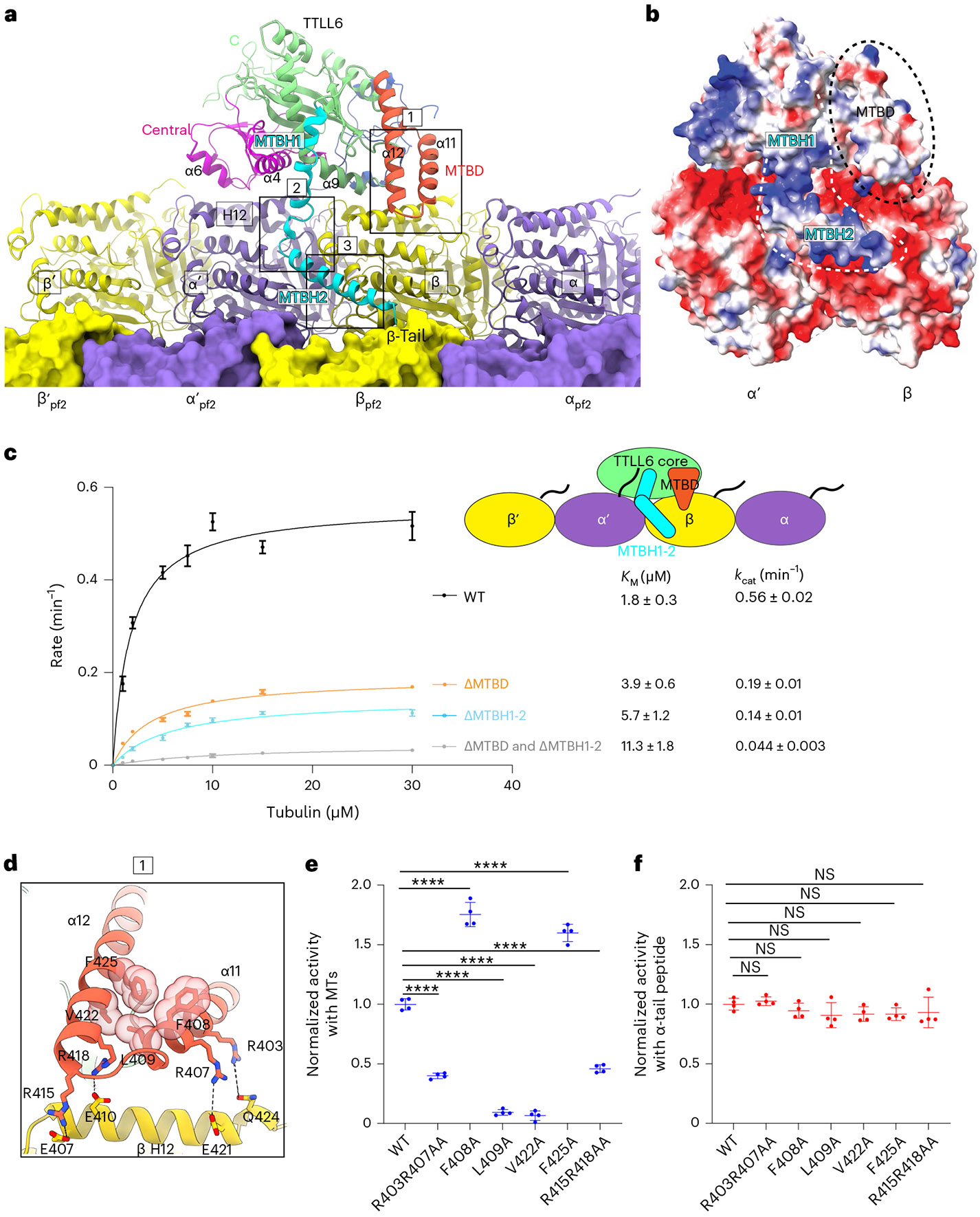
a, Ribbon representation of TTLL6 making interdimer and interprotofilament interactions within the MT. TTLL6 N-, central-, C-, MTBD- and MTBH1-2 domains are blue, magenta, green, orange and cyan, respectively. The tubulin subunits of the adjacent PF are shown in surface rendering. Boxed areas highlight interactions made by MTBD (box 1) and MTBH1-2 (boxes 2 and 3) with the MT. b, Surface representation of TTLL6 in the same orientation as a color-coded for electrostatic potential (red, negative; blue, positive; ranging from −10 KBT to 10 KBT). KB, Boltzmann constant; T, temperature. Electrostatic potential was calculated using Poisson Boltzmann Solver77. MTBD and MTBH1-2 are outlined with black discontinuous lines. c, Michaelis–Menten kinetics of TTLL6, TTLL6ΔMTBD, TTLL6ΔMTBH1-2 and TTLL6ΔMTBDΔMTBH1-2 with taxol-stabilized MTs. Apparent KM and kcat are listed on the right. Error bars represent s.e.m. (n = 4 independent experiments). d, Boxed area 1 in a magnified to show interactions between MTBD helices α11 and α12 and helix H12 of β-tubulin, and hydrophobic interactions between α11 and α12 in the MTBD. e,f, Normalized glutamylation activity of structure-guided TTLL6 mutants with taxol-stabilized MTs (e) and an α1B(-Y) peptide (f). Error bars, s.e.m. n = 4 independent experiments; ****P < 0.0001 and NS as determined by one-way ANOVA with Tukey’s post hoc test. ANOVA, analysis of variance; NS, not significant.
MTBD positions the TTLL6 core by interacting with β-tubulin H12
The MTBD anchors TTLL6 to the MT through contacts with the anionic helix H12 of β-tubulin (Fig. 2d). Conserved lysines and arginines constitute ~25% of the MTBD (Supplementary Fig. 1). The C-terminal region of α11 and the N-terminal region of α12, both proximal to the β-tubulin body, are particularly enriched in basic residues. Invariant R403 and conserved R407 in α11 are within hydrogen bonding distance from E421 in helix H12 of β-tubulin and Q424, respectively (Fig. 2d). Mutation of R403 and R407 to alanine reduces MT glutamylation to ~55% of the wild-type (WT) enzyme (Fig. 2e). Consistent with their role in MT and not α-tail recognition, these mutations have no effect on TTLL6 activity with isolated α-tubulin tail peptides (Fig. 2f). Invariant R415 and R418 in α12 are within hydrogen bonding distance from the negatively charged E407 in helix H12 of β-tubulin and E410, respectively (Fig. 2d). Mutation of R415 and R418 to alanine decreases MT glutamylation by ~47%, without having an effect on activity with α-tail peptides (Fig. 2e,f). The two antiparallel helices α11 and α12 that form the MTBD are held together by conserved hydrophobic interactions, as revealed by the TTLL6 crystal structure41. Invariant F408A of α11 packs against invariant F425 of α12 and invariant Leu409 of α11 against invariant Val422 of α12 (Fig. 2d). Mutating Leu409 or Val422 to alanine decreases TTLL6 activity to less than 10% of the WT without affecting the activity with isolated α-tails (Fig. 2e,f), consistent with the importance of these stabilizing interactions in the MTBD for MT recognition. Surprisingly, mutation of F408 or F425 to alanine increased TTLL6 activity by ~70% and 59%, respectively, without impacting the activity with α-tail peptides (Fig. 2e,f), suggesting that a loosening of the packing of the MTBD helices at this position allows more efficient accommodation of interactions with the MT.
Three cationic clusters recognize distinct interfaces
The MTBH1-2 is shaped like an L, with its elbow wedged between the β–α′ protomers at the interdimer interface (Fig. 2a,b). Cationic residues in MTBH1-2 make extensive electrostatic interactions with the anionic interdimer interface (Fig. 3a,b). The following three clusters of conserved basic residues are critical at this interface: cluster 1 (R474, R476 and R481), cluster 2 (R488, K490, K493 and K494) and cluster 3 (K498, K499, R501 and K502; Supplementary Fig. 1). Cluster 1 interacts with the anionic surface of the α-tubulin and β-tubulin protomers at the interdimer groove (Fig. 3a,b). R474 interacts with invariant D404 and E405 of β-tubulin, and R476 forms a salt bridge with invariant E196 of α′-tubulin, respectively. Mutation of cluster 1 arginines to alanines increases the apparent KM by 2.5-fold, indicative of decreased substrate binding, and decreases the kcat by 2.6-fold (Fig. 3c). Cluster 2 interacts with the negatively charged helices H3 and H11 of β-tubulin, also at the interdimer interface (Fig. 3b). Specifically, R488 is proximal to T399 in β-tubulin helix H12, while K490 and K494 make a network of salt bridges with invariant E108 and E111 in β-tubulin H3 (Fig. 3b). Mutation of cluster 2 positively charged residues to alanines increases the apparent KM by 3.2-fold and decreases the kcat 3.3-fold compared to WT (Fig. 3c). Cluster 3 residues (K498, K499, R501 and K502) extend into the interprotofilament groove and continue the network of salt bridges with acidic residues in helix H3 of β-tubulin D114 and D118 (Fig. 3b). Mutation of cluster 3 residues to alanines increases the apparent KM by 1.9-fold and decreases the kcat by 1.3-fold (Fig. 3c).
Fig. 3 ∣. Three MTBH1-2 cationic clusters recognize different MT interfaces.
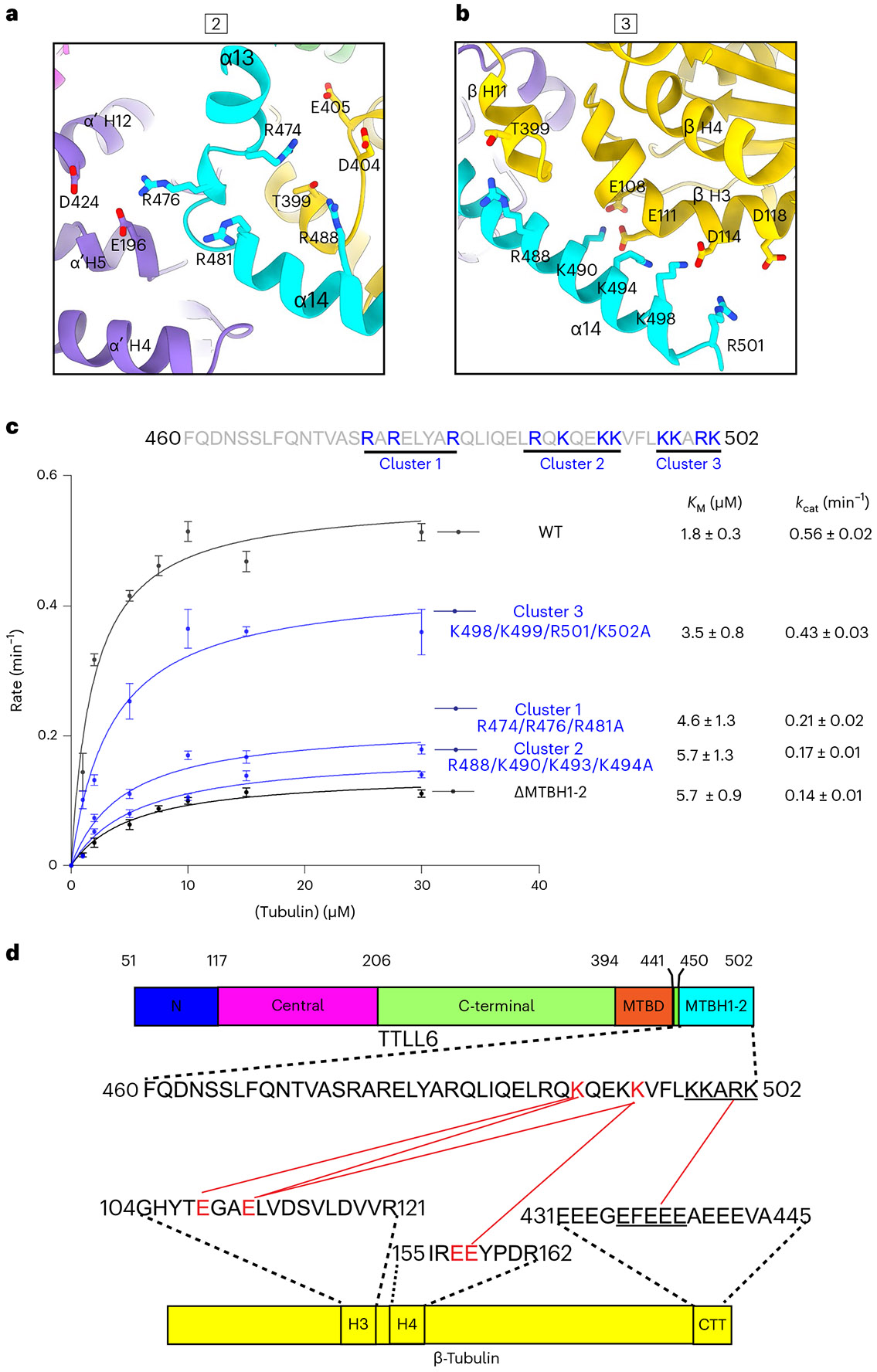
a, Boxed area 2 in Fig. 2a magnified to show interactions made by MTBH1 with the interdimer interface. b, Boxed area 3 in Fig. 2a magnified to show interactions made by MTBH2 with the β-tubulin subunit at the interdimer interface. c, Top, MTBH1-2 sequence with the three clusters of basic residues highlighted. Bottom, Michaelis–Menten kinetics with taxol-stabilized MTs for TTLL6 mutants in the basic residue clusters. Error bars, s.e.m.; n = 4 independent experiments; apparent KM and kcat values listed on the right. d, Domain schematic of TTLL6 showing the location of cross-linked sites between MTBH1-2 and β-tubulin identified from tandem mass spectrometry analysis (Methods). Red lines indicate cross-linked residues.
The network of interactions between the three MTBH1-2 cationic clusters and acidic residues at the interdimer interface is also supported by chemical cross-linking coupled with mass spectrometry (XL–MS; Fig. 3d). We used a carbodiimide-based zero-length cross-linking reagent, 1-ethyl-3-(-3-dimethylaminopropyl) carbodiimide (EDC), that cross-links carboxylic acids and primary amines (Methods) and thus cross-links salt bridges formed by lysines and glutamates. Consistent with our cryo-EM structure and size-exclusion chromatography, MTBH1-2 cross-links only with MTs and not with unpolymerized tubulin (Extended Data Fig. 6a-c). Cross-linked products were visible 30 min after starting the reaction and increased with time. Tandem mass spectrometry of cross-linked products identified five tubulin-MTBH1-2 cross-links (Fig. 3d and Extended Data Fig. 6d-h), consistent with the highly stringent nature of the cross-linking reagent. All of the cross-links were between residues identified in our structure at the interface between MTBH1-2 and β-tubulin (Fig. 3a,b,d). Specifically, K490 of cluster 2 is cross-linked with E108 and E111 in β-tubulin H3, and K494 of cluster 2 is also cross-linked with E111 in β-tubulin H3 (Fig. 3d).
MTBH1-2 interacts with β-tail on adjacent PF
The C-terminus of MTBH2 reaches across the interprotofilament groove and is in close proximity to the disordered β-tubulin tail of the laterally adjacent tubulin dimer (on the neighboring PF; Fig. 2a and Extended Data Fig. 7a). Indeed, our XL–MS analysis revealed an interaction between cluster 3 residues at the MTBH2 C-terminus and glutamates in the β-tail (Fig. 3d and Extended Data Fig. 6h). The side chains for these residues are not well-defined as for the rest of the MTBH2 (Extended Data Fig. 1g), and this interaction is not visible in our cryo-EM structure, indicating a high degree of flexibility and/or transient nature.
To understand the functional significance of this interaction, we used total internal reflection (TIRF) microscopy to measure the association of Atto488-labeled TTLL6 with unmodified WT MTs and MTs missing their β-tubulin tails (βΔ-tails) after controlled subtilisin treatment (Fig. 4a,b and Extended Data Fig. 7b,c). Consistent with our structure and cross-linking results, β-tail loss significantly reduces TTLL6 binding to MTs (Fig. 4a-c). This interaction is mediated primarily through the MTBH1-2 because in its absence TTLL6 no longer shows a strong preference for binding to MTs with intact β-tubulin tails. While TTLL6 recruitment to βΔ-tail MTs is decreased ~18-fold when compared to MTs with intact β-tails (Fig. 4d,e), TTLL6 missing the MTBH1-2 showed only a 40% reduction (Fig. 4f,g), indicating that MTBH1-2 is primarily responsible for binding to the β-tail. Consistent with this, liquid chromatography–mass spectrometry (LC–MS) shows that TTLL6 does not add glutamates to α-tubulin tails on MTs that miss their β-tails, but modifies WT MTs robustly under these conditions (Extended Data Fig. 8a-d).
Fig. 4 ∣. MTBH1-2 interacts with the β-tail of the laterally adjacent tubulin dimer and discriminates between the interdimer and intradimer interface.
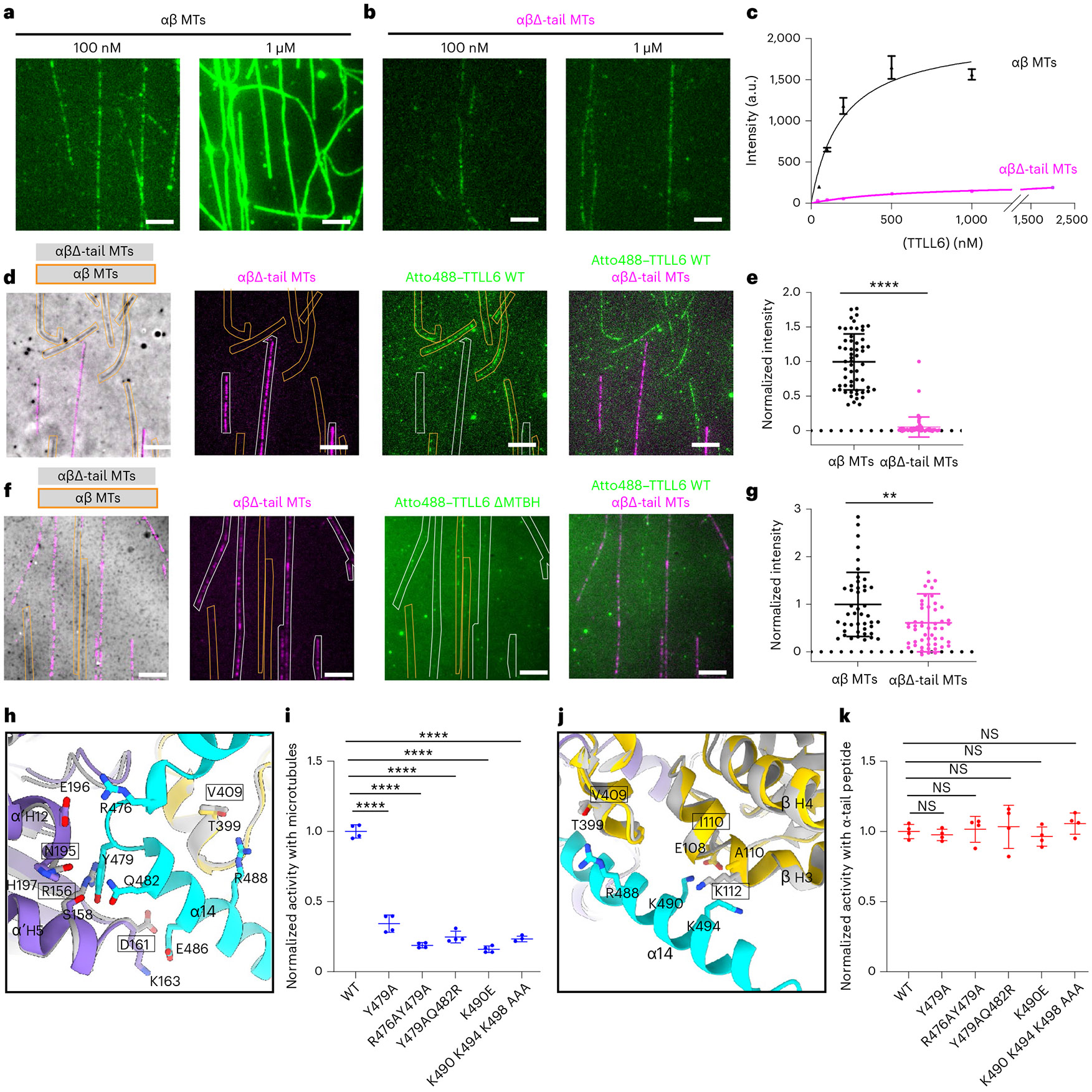
a,b, Representative images from one of three independent experiments showing Atto488–TTLL6 association with intact WT (a) and subtilisin-treated MTs missing their β-tubulin tails, αβΔ-tails (b). Scale bar, 5 μm. c, Titration curves for TTLL6 binding to WT (black) and αβΔ-tail (magenta) MTs. n = 113, 25, 46, 57 and 100 intact WT MTs for 50 nM, 100 nM, 200 nM, 500 nM and 1,000 nM TTLL6, respectively, from three independent experiments. n = 96, 37, 55, 77, 65 and 84 αβΔ-tail MTs for 50 nM, 100 nM, 200 nM, 500 nM, 1,000 nM and 2,000 nM TTLL6, respectively, from three independent experiments. Apparent Kd for WT MTs is ~210 ± 38 nM. Error bars, s.e.m. d, Representative images from 64 and 47 MTs analyzed for WT and αβΔ-tail, respectively, from two independent experiments showing Atto488–TTLL6 recruitment to WT and αβΔ-tail MTs. Scale bar, 5 μm. WT and αβΔ-tail-647-Hilyte MTs are outlined in orange and white, respectively. e, Normalized measurements of Atto488–TTLL6 recruitment to WT and αβΔ-tail MTs. n = 64 and 47 MTs for WT and αβΔ-tail, respectively, from two independent experiments. Data are presented as mean ± s.d. ****P < 0.0001 by two-tailed Mann–Whitney test. f, Representative images from 51 and 44 MTs analyzed for WT and αβΔ-tail, respectively, from two independent experiments showing Atto488–TTLL6ΔMTBH1-2 recruitment to WT and αβΔ-tail MTs. Scale bar, 5 μm. WT unlabeled and αβΔ-tail-647-Hilyte labeled MTs are outlined in orange and white, respectively. g, Normalized measurements of Atto488–TTLL6ΔMTBH1-2 recruitment to WT and αβΔ-tail MTs; n = 51 and 44 MTs for WT and αβΔ-tail, respectively, from two independent experiments. Data are presented as mean ± s.d. **P < 0.01 by two-tailed Mann–Whitney test. h, Ribbon representation highlighting key interactions made by the N-terminus of MTBH2 (cyan) with α’-tubulin and β-tubulin (purple and yellow, respectively) at the interdimer interface of the complex structure. Residues important for interactions with the MTBH1-2 are shown in stick representation in purple and yellow. Modeled are superpositions of β-tubulin (gray) on α′-tubulin of the structure, and α-tubulin (gray) on β-tubulin of the structure. Residues in the modeled intradimer interface are in gray stick representation, and their identities are in boxes. i, Normalized glutamylation activity of structure-guided TTLL6 mutants in the MTBH1-2 with taxol-stabilized MTs. Error bars, s.e.m.; n = 4 independent experiments; ****P < 0.0001 and NS as determined by one-way ANOVA with Tukey’s post hoc test. j, Ribbon representation highlighting key interactions made by the C-terminus of MTBH2 (cyan) with the β-tubulin (yellow) at the interdimer interface. Colors and labels as in h. k, Normalized glutamylation activity of structure-guided TTLL6 mutants in the MTBH1-2 with α1B(-Y) peptide. Error bars, s.e.m.; n = 4 independent experiments; NS as determined by one-way ANOVA with Tukey’s post hoc test.
Consistent with the binding mode of MTBH1-2 along the interdimer interface and across PFs, we find that MTBH1-2 alone can promote tubulin polymerization. While 10 μM porcine brain tubulin did not polymerize as assessed by interference reflection microscopy (IRM), addition of 2.5 or 5 μM MTBH elicited robust polymerization after ~60 s (Extended Data Fig. 9a-c). Mutation of cluster 2 residues K490 and K494 involved in recognizing the interdimer interface dramatically decreased MTBH1-2-induced tubulin polymerization (Extended Data Fig. 9c,d). We were able to observe rare nucleation events only at high concentrations for this mutant (25 μM; Extended Data Fig. 9e). These rare nucleation events were completely abolished upon mutation of additional residues in cluster 3, K498 and K502 (Extended Data Fig. 9f). We note that it is not clear whether the polymeric structures we observe in IRM are MTs or open sheets. The latter would be compatible with the higher flexibility we observed for these polymers as well as our EM data showing the destabilizing effects of the MTBH1-2 on MTs and the promotion of sheet formation (Extended Data Fig. 4).
Interdimer versus intradimer interface recognition by MTBH1-2
The ability of TTLL6 MTBH1-2 to distinguish between the highly homologous α-tubulin and β-tubulin protomers is key to establishing the correct register for the enzyme to modify the α-tubulin and not the β-tubulin tail. Several critical interactions between TTLL6 and residues at the interdimer interface would be weakened or lead to steric clashes at the intradimer interface. Y479 in the loop connecting MTBH1 and MTBH2 interacts with α′-tubulin S158 (α′ denotes the α-tubulin belonging to the neighboring tubulin dimer on the same PF) and α′-tubulin H197 (Fig. 4h). Mutation of Y479 to alanine reduces TTLL6 activity to ~35% of the WT (Fig. 4i). The corresponding residues in β-tubulin are N195 (instead of α-tubulin H197) and R156 (instead of α-tubulin S158; Fig. 4h). Thus, at the intradimer interface, Y479 would lose its aromatic interaction and, notably, clash with the long, bulky side chain of R156. Nearby residues, R476 and Q482, also make interactions with α′-tubulin at the interdimer interface but do not have a significant effect because the TTLL6 double mutants R476A/Y479A and Y479A/Q482R have activities similar to the Y479A single point mutant (Fig. 4i). Invariant K490 in cluster 2 makes a salt bridge with invariant β-tubulin E108 and cross-links with it (Fig. 4j and Extended Data Fig. 6d,f). Consistent with this, mutation of K490 to glutamate reduces TTLL6 activity to less than 20% of the WT (Fig. 4i). E108 is replaced by isoleucine in α-tubulin (I110), and thus TTLL6 would lose this important electrostatic interaction at the intradimer interface (Fig. 4j). Furthermore, A110 of β-tubulin, which faces the positively charged lysines in cluster 2, would be replaced by K112 in α-tubulin, resulting in strong electrostatic repulsion at the intradimer interface (Fig. 4j). Mutation of lysines in cluster 2 reduces TTLL6 activity to ~20% of WT (Fig. 4i). None of these interactions that establish interdimer versus intradimer recognition have an effect on the activity of the enzyme with isolated tubulin peptides (Fig. 4k), consistent with their importance only in MT recognition. Thus, multiple electrostatic and hydrophobic interactions anchor the MTBH1-2 specifically to the interdimer, and not the intradimer, interface.
MTBH1-2 increases substrate residence time to aid catalysis
While MTBD is found in all autonomous TTLL glutamylases39, our sequence and structural analysis shows that MTBH1-2 is absent in enzymes with preferential initiase activity, TTLL4 and TTLL5 (Supplementary Fig. 2). In TTLL7, a β-tubulin-specific glutamylase that preferentially generates short glutamate chains19,39, the helix that corresponds to MTBH2, is folded against the TTLL catalytic core39, away from the MT surface (Fig. 5a-d), and the residues important in TTLL6 for interactions at the interdimer groove are not conserved. Specifically, R481 of cluster 1 in TTLL6 MTBH1-2 is replaced by leucine in TTLL7, while R488 and K490 of cluster 2 are replaced by glutamate and aspartate, respectively (Fig. 5a). TIRF imaging of single Atto488–TTLL6 molecules on brain MTs (Fig. 5e,f and Extended Data Fig. 10a-c) showed that they are static, with minimal to no diffusion on the MT. The average residence time (τ) for TTLL6 on GMPCPP or taxol-stabilized brain MTs is 32.9 s and 37.6 s, respectively (Fig. 5e,f and Extended Data Fig. 10d). In the absence of the MTBH1-2, TTLL6 residence time with the MT decreases ~26-fold to ~1.28 s (Fig. 5g,h), indicating that the MTBH1-2 has a profound effect on the TTLL6 koff rate. Strikingly, the residence time of TTLL6ΔMTBH1-2 is very similar to that measured for TTLL7 (τ~0.87 s; ref. 39), indicating that the extended conformation of the MTBH2 in TTLL6 anchors the enzyme on the MT surface for prolonged residence times that are on the order of the turnover rate of the enzyme (Fig. 5i), ensuring that most binding events result in successful glutamate ligation.
Fig. 5 ∣. MTBH1-2 is an elongase-specific module that controls TTLL6 residence time on the MT.
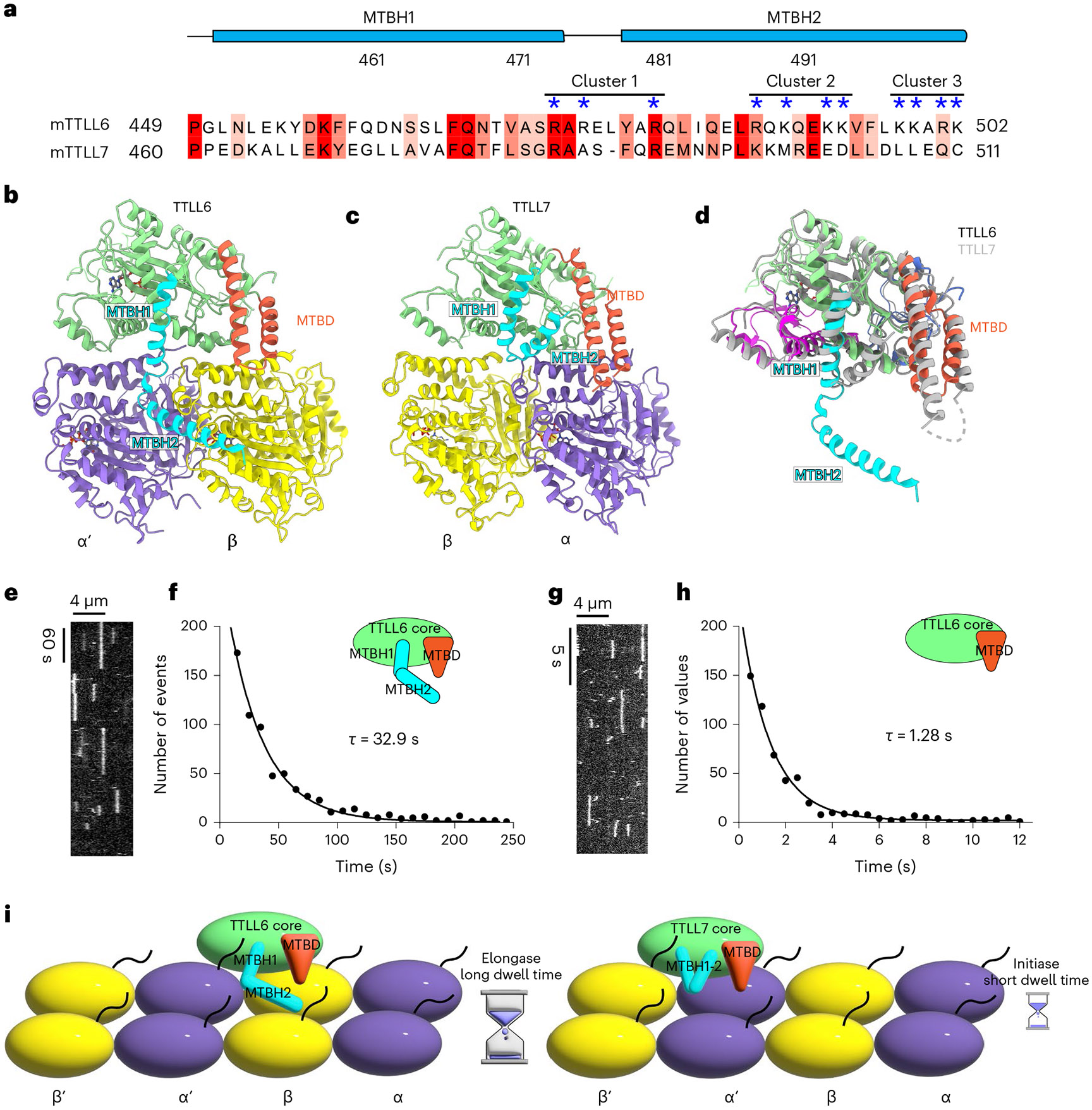
a, Sequence alignment of mouse TTLL6 and TTLL7 MTBH1-2. Residues are colored according to conservation; residues in the cationic clusters important for interaction with the MT are highlighted by blue asterisks. b,c, Ribbon representation of TTLL6 (b) and TTLL7 (c) bound at the interdimer and intradimer interface, respectively. The N-, central-, C-, MTBD- and MTBH1-2 domains are shown in blue, magenta, green, orange and cyan, respectively. α-Tubulin and β-tubulin subunits are shown in purple and yellow, respectively. d, Ribbon representation of superimposed TTLL6 (colored as in b) and TTLL7 (gray) showing the extended conformation of the MTBH1-2 in TTLL6. e, Representative kymograph from one of six independent experiments showing single Atto488–TTLL6 molecules on GMPCPP-stabilized brain MTs (Methods). f, Distribution of MT residence times of Atto488–TTLL6; the mean residence time (τ) was obtained by fitting an exponential curve to the histogram and correcting for photobleaching (R2 = 0.98, n = 951 binding events from six independent experiments). g, Representative kymograph from one of seven independent experiments showing single Atto488–TTLL6ΔMTBH1-2 molecules on GMPCPP-stabilized brain MTs. h, Distribution of MT residence times of Atto488–TTLL6 MTBH1-2. The mean residence time (τ) was obtained by fitting an exponential curve to the histogram and correcting for photobleaching (R2 = 0.95, n = 574 binding events from seven independent experiments). i, Schematic representation showing the interdimer and intradimer interface recognition by TTLL6 and TTLL7, respectively. The extended conformation of the TTLL6 MTBH1-2 domain increases MT residence time for efficient glutamate chain elongation.
TTLL6 reads β-tail glutamylation on adjacent tubulin
Given the important contribution of the β-tail to TTLL6-MT recognition and the strong cationic character of the MTBH1-2, we investigated whether β-tail glutamylation affects TTLL6-MT recruitment. We generated unmodified human MTs and MTs glutamylated primarily on their β-tubulin tails by incubating them with TTLL7, a β-tubulin tail-specific glutamylase39,59,60 (Supplementary Fig. 3), and tested the effect on TTLL6-MT binding using TIRF microscopy (Fig. 6). We found that β-tubulin glutamylation (<nE> ~4.9) increased recruitment of TTLL6 to the MT ~12-fold (Fig. 6a,b). β-Tail glutamylation had no significant effect on the recruitment of a TTLL6 mutant lacking MTBH1-2 (Fig. 6c,d), indicating that this structural element is responsible for sensing glutamylation on the β-tail. Consistent with the increased recruitment to the MT, we find that glutamylation of β-tubulin (<nE> ~3.6) by TTLL7 significantly increases TTLL6 activity as evidenced by the increase in signal from polyglutamate chains on α-tubulin (Fig. 6e-h). Thus, TTLL6 reads the glutamylation status of the β-tail on the tubulin dimer in the neighboring PF (Fig. 2a) through its MTBH1-2 domain and preferentially binds and modifies MTs that are glutamylated.
Fig. 6 ∣. β-Tail glutamylation by TTLL7 stimulates TTLL6.
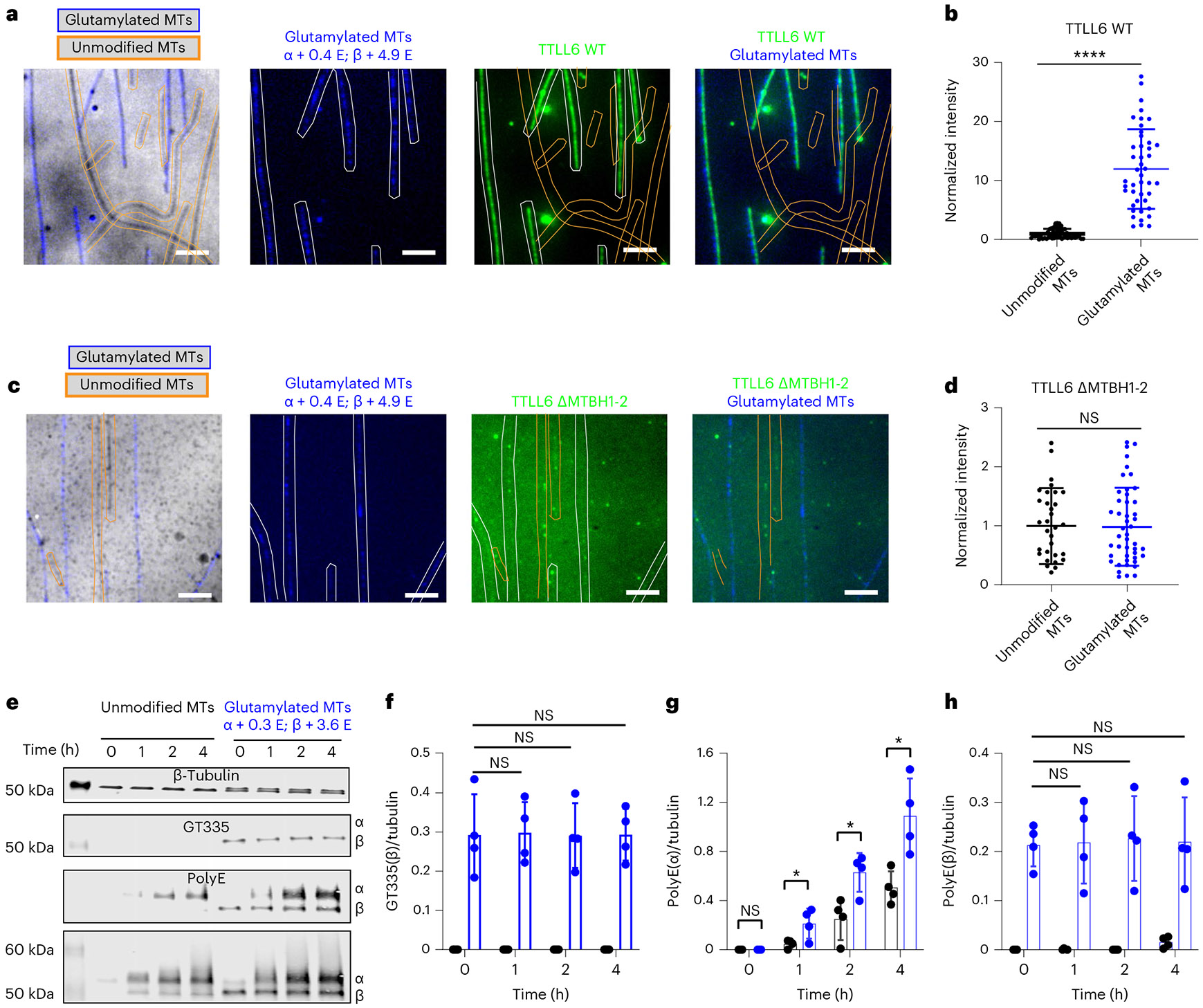
a, Representative images from 44 and 56 MTs analyzed for unmodified and glutamylated MTs, respectively, from two independent experiments showing Atto488–TTLL6 recruitment to unmodified and glutamylated MTs. Unmodified and glutamylated MTs are outlined in orange and blue, respectively. Average number of glutamates added to α-tubulin and β-tubulin is indicated as α + <nE>, β + <nE>, scale bar, 5 μm. For LC–MS spectra of MTs used in assays, see Supplementary Fig. 3. b, Normalized measurements of Atto488–TTLL6 recruitment to unmodified and glutamylated MTs; n = 44 and 56 MTs, respectively, from two independent experiments. Data are presented as mean ± s.d. ****P < 0.0001 as determined by two-tailed Mann–Whitney test. c, Representative images from 42 and 47 MTs analyzed for unmodified and glutamylated MTs, respectively, from two independent experiments showing Atto488–TTLL6ΔMTBH1-2 recruitment to unmodified and glutamylated MTs. Unmodified and glutamylated MTs are outlined in orange and blue, respectively. Average number of glutamates added to α-tubulin and β-tubulin is indicated as α + <nE>, β + <nE>; scale bar, 5 μm. d, Normalized measurements of Atto488–TTLL6ΔMTBH1-2 recruitment to unmodified and glutamylated MTs; n = 42 and 47 MTs, respectively, from two independent experiments. Data are presented as mean ± s.d.; NS by two-tailed Mann–Whitney test. e, Representative western blot images from one of four independent experiments showing TTLL6-mediated glutamylation of MTs glutamylated on their β-tails by TTLL7. Polyglutamylation was monitored on α-tubulin and β-tubulin using an antibodys that specifically detects polyglutamate chains (polyE); branched glutamates were monitored using the GT335 antibody (Methods). f–h, Quantification of monoglutamylation on β-tubulin (f), polyglutamylation on α-tubulin (g) and polyglutamylation on β-tubulin (h) as a function of reaction time. Modifications detected with antibodies are listed in e. n = 4 independent experiments. Error bars, s.d. *P < 0.05 and NS as determined by two-tailed t test.
Discussion
MT spatiotemporal modification patterns are dictated by the substrate specificity and catalytic rates of tubulin modification enzymes. Our work shows that TTLL6 uses two noncatalytic domains, MTBD and MTBH1-2, to recognize both longitudinally and laterally adjacent tubulin dimers. This ensures recognition of the MT, and not tubulin, by virtue of its unique polymer geometry, and positions the enzyme’s catalytic domain preferentially to modify α-tails (Fig. 2a). MT recognition by TTLL6 involves the following four interfaces: the MTBD interface with the β-tubulin body, the catalytic domain with the α-tubulin tail, the MTBH1-2 with the interdimer interface and the MTBH1-2 with the β-tubulin tail of the neighboring tubulin dimer. This complex and unprecedented quadrivalent recognition strategy ensures simultaneous readout of MT geometry and modification status of the tubulin tails. TTLL glutamylases constitute the largest subfamily of tubulin modification enzymes and share a common catalytic domain. Structural comparison between TTLL6 and TTLL7 (ref. 39), a glutamylase specialized in initiating short glutamate chains on β-tails59, suggests a general strategy for α-tail versus β-tail modification within the TTLL glutamylase family—the MTBD, found in autonomous TTLL glutamylases, sits on the protomer whose tail the enzyme does not modify (Fig. 5i). That is, in α-tail-specific glutamylases, the MTBD binds to β-tubulin, while in β-tail-specific glutamylases, it binds to α-tubulin. Thus, α-tail versus β-tail recognition is achieved by moving the register of the MTBD on the MT PF by one tubulin protomer (Fig. 5i). For α-tubulin glutamylases, the active site recognizes the tail in trans, while for β-tubulin glutamylases, it recognizes it in cis.
TTLL6 uses the cationic helical element MTBH1-2 for extensive interactions with the interdimer groove and with the intrinsically disordered β-tubulin tail of the laterally adjacent tubulin, thereby stably anchoring itself onto the MT for efficient elongation of long glutamate chains on the α-tail (Fig. 5i). The interdimer groove between PFs is also used for stable anchoring by other MT effectors such as the spindle and kinetochore-associated (Ska) complex61 and its yeast ortholog, Dam1 complex62, doublecortin63 and end-binding proteins64,65. This suggests a potential regulatory interplay between MT effectors and MT-modifying enzymes that bind at this interface.
Structure-based sequence alignments show that the MTBH1-2 and the positively charged residues critical for its interactions with the interdimer interface are also conserved in the α-tubulin glutamyl elongase TTLL13 (Extended Data Fig. 10e), suggesting a shared MT recognition strategy. Interestingly, the MTBH1-2 is expanded into a larger α-helical domain in TTLL11, also an elongase. In contrast to TTLL6, the MTBH1-2 has a closed conformation in TTLL7, folding against the enzyme core. Both sequence analysis and modeling using Alphfold55 show that an element equivalent to the MTBH1-2 is not found in glutamyl initiases TTLL4 and TTLL5. Consistent with its importance in MT anchoring, MTBH1-2 deletion in TTLL6 decreases the MT residence time of the enzyme by ~26-fold and brings it closer to that reported for TTLL7 (ref. 39). We speculate that TTLL6 and TTLL13, both glutamyl elongases, use the MTBH1-2 to bind longer to the MT for efficient glutamate chain elongation, while TTLL7, which lacks the additional anchoring interactions provided by MTBH1-2, evolved to spend less time on each tubulin dimer and thus scan the MT for unmodified tubulin tails from which it can initiate a new glutamate chain.
We show that the glutamylation status of the β-tail is sensed by MTBH1-2 and increases recruitment of TTLL6 to MTs to provide spatial positive feedback (Fig. 6). This modification-dependent recruitment can generate localized modification patterns, long documented in vivo66 and can also underlie polarity establishment through self-reinforcing accumulation of glutamylation that in turn recruits effectors such as MAPs. In vivo crosstalk between α-tubulin and β-tubulin glutamylation was previously documented in protists. In Tetrahymena, mutation of modification sites on the β-tail decreased glutamylation and glycylation on α-tails67. Our study provides direct biochemical evidence for crosstalk between α-tubulin and β-tubulin glutamylation and a mechanistic basis for these early observations. Interestingly, TTLL6 has higher activity on MTs with detyrosinated α-tails41, and a recent study shows that TTLL6 glutamylation of the α-tail increases recruitment of the α-tubulin detyrosination enzyme VASH1 to the MT68, creating a positive feedback loop between these two chemically distinct modifications. Crosstalk among histone modifications, both on the same histone tail and between different histone tails, is well documented69,70. Thus, just like the histone code, crosstalk among tubulin modification enzymes can create complex, combinatorial modification patterns with functional outputs that are distinct from those of the individual modifications, as recently shown for the MT-severing enzyme katanin12. This crosstalk between modifications also provides a possible explanation for the frequent codysregulation of tubulin modifications in various pathological conditions such as neurodegeneration23,71-73 and cancers74-76. Future work will be aimed at understanding this emerging grammar of the tubulin code.
Methods
Protein expression and purification
M. musculus TTLL6 (51–502) was expressed and purified as described previously41. Briefly, TTLL6 was expressed in Escherichia coli Arctic Express DE3 as an N-terminal glutathione S-transferase (GST) fusion and purified by GST affinity chromatography. A construct containing the first 50 residues was prone to degradation and not suitable for biophysical and structural studies. The protein was further purified on a Heparin column followed by size-exclusion chromatography using a Superdex 75 column (Cytiva Lifesciences) in a buffer solution containing 50 mM HEPES (pH 7.0), 10 mM MgCl2, 400 mM NaCl, 1 mM dithiothreitol (DTT) and 100 μM ATP. ΔMTBD, ΔMTBH1-2 and all point mutants were cloned using the kinase, ligase and DpnI enzyme mix (New England Biolabs) and purified using the same chromatography steps as the WT construct. All TTLL6 constructs used in this study eluted as a single peak from a Superdex 75 gel-filtration column (Cytiva Lifesciences).
Cryo-EM sample preparation
Unmodified tubulin from a human embryonic kidney (tsA201) cell line was purified as described previously52. GMPCPP-bound MTs were prepared as follows. A frozen aliquot of unmodified tubulin was rapidly thawed in a 37 °C water bath, and aggregates were removed by ultracentrifugation at 436,000g for 10 min at 4 °C. Tubulin was diluted to 2.5 mg ml−1 in 1× BRB80 buffer (80 mM PIPES, 1 mM MgCl2 and 1 mM ethylene glycol-bis(β-aminoethyl ether)-N,N,N′,N′-tetraacetic acid (EGTA)) and 1 mM DTT and incubated in the presence of 1 mM GMPCPP (Jena Bioscience) for 5 min on ice, and then polymerized at 37 °C for 2–4 h. MTs were pelleted by centrifugation at 126,000g for 10 min at 30 °C. Supernatant was discarded, and the MT pellet was washed with warm BRB80 buffer and then spun again at 126,000g for 10 min at 30 °C. The final MT pellet was resuspended in warm BRB80 buffer supplemented with 1 mM DTT. The concentration of polymerized MTs was calculated by mixing a small aliquot (1 μl) of MTs with 1 μl of 100 mM CaCl2, measuring absorbance at 280 nm on a Nanodrop and using an extinction coefficient of 115,000 M−1 cm−1. TTLL6 at 12.3 mg ml−1 (230 μM) was rapidly thawed and diluted to 20 μM in EM buffer (40 mM HEPES (pH 7.0), 100 mM KCl, 20 mM MgCl2, 4 mM DTT and 20 μM ATP).
For grid preparation, MTs were diluted to 0.1 mg ml−1 in EM buffer, and TTLL6 was diluted in EM buffer to a 1:0.75 molar ratio of polymerized tubulin to TTLL6 (excess of tubulin). TTLL6 decoration of MTs was heterogeneous, with many MTs remaining undecorated. A large array of TTLL6 stoichiometries was explored, as excess TTLL6 led to highly irregular MT binding that was not suitable for structural determination. In total, 4 μl of MTs were applied to glow discharged 1.2/1.3 UltrAuFoil Holey Gold Films grids (Quantifoil) and allowed to absorb for 30 s before 4 μl of TTLL6 was added and incubated for an additional 30 s on the grids. The sample was blotted with Whatman grade 5 blotting paper on both sides of the grid for 3 s with a blot offset of −1 using a Vitrobot Mark IV (Thermo Fisher Scientific) with a chamber set to 30 °C at 100% humidity and subsequently plunged into liquid ethane.
EM data and image processing
Cryo-EM data were collected on a Talos Arctica (Thermo Fisher Scientific) transmission EM operating at 200 keV, and micrographs were acquired using a K2 Summit (Gatan) direct electron detector, operating in electron counting mode applying a total electron exposure of 42 e− Å−2 as a dose-fractionated movie during a 10 s exposure. The Leginon data collection software78 was used to collect 2,771 movies at ×36,000 nominal magnification (1.15 Å per pixel) with a nominal defocus range of −1.0 to −2.0 μm (Supplementary Table 1).
Frames were aligned and summed within the Appion processing environment79 (Extended Data Fig. 1a). The contrast transfer function (CTF) was estimated using GCTF80. Movie sums with Thon rings beyond 5 Å were selected for further image processing. Due to the highly heterogeneous TTLL6 decoration of MTs (Extended Data Fig. 1b), MTs were manually selected. In total, 59,044 overlapping segments with a box size of 568 pixels were extracted every 82 Å and down-sampled four times to a pixel size of 4.6 Å per pixel. Three-dimensional (3D) reconstruction of the TTLL6-bound MT was carried out using the previously described method81 (Extended Data Fig. 1c). In brief, segment averages were generated for each MT and were subjected to supervised classification against 11–16 PF synthetic MT references, low-pass filtered to 15 Å. The class assignment was the following: 11 PF, 0.3%; 12 PF, 2%; 13 PF, 39%; 14 PF, 48%; 15 PF, 4% and 16 PF, 6%. Fourteen PF MT segments were taken for further processing. Rotation angles and translations were reset to zero, and Psi and Tilt angles were set to priors, followed by one round of refinement against 14 PF synthetic reference. For each MT, the most common rotation angle was derived and assigned to all segments in that MT, followed by 3D refinement against 14 PF synthetic reference with local sampling of Euler angles. Next, X/Y shifts were smoothed to remove mistranslations along MTs, followed by refinement. Four times down-sampled particles were recentered and re-extracted, and segment averages were generated for each MT. Seam location along each MT was assigned by performing a supervised 3D classification without alignment against 28 synthetic references with all possible seam positions. Each MT was then assigned to a common class, followed by adjustments of angles and translations along the helical axis. Unbinned MT segments were re-extracted and recentered. Three-dimensional refinement with and without imposing symmetry was performed. Quality of the resulting 3.8 Å symmetrized map (Extended Data Fig. 1i) was further improved by performing PF refinement54. In short, signal was subtracted from all but a single PF in each MT segment. The process was repeated for each PF, resulting in 14 times more particles. The PF particles were refined as single particles. Although the nominal resolution of the map improved by 0.2 Å (3.6 Å), the quality of the map significantly improved as the method accounts for distortions in the MT lattice. These distortions were quantified by using the refined rotation angles between adjacent PFs (Extended Data Fig. 3a,b). The reconstructions revealed that the TTLL6 MTBH2 domain binds between PFs. Therefore, the PF refinement procedure was performed with particles containing signals for two adjacent PFs (Extended Data Fig. 1e). Due to the heterogeneous decoration of TTLL6 on the MT, the cryo-EM density corresponding to TTLL6 was poor, with the exception of the MTBH2 helix, which was resolved to 3.6 Å (Extended Data Fig. 1f). To improve TTLL6 definition, the signal from one central copy of TTLL6 (including MTBH2) on one of two PFs was left with the rest of the signal subtracted. The resultant particles were subjected to supervised 3D classification without alignment (τ fudge = 40; Extended Data Fig. 1h). One of two 3D classes, containing ~40% of particles, was further refined in 3D. This approach resulted in a better-defined TTLL6 with a nominal resolution of 7.2 Å although the local resolution varied from 5 Å to 14 Å (Extended Data Fig. 1h). Further attempts to improve the definition of the globular part of TTLL6 by subtracting from each particle the signal corresponding to MTBH2 and leaving the rest of the TTLL6 signal were not successful. We also performed refinement with particles containing TTLL6 and helices H11 and H12 of α/β-tubulin (sites of TTLL6 contact), but we saw a slight deterioration in the nominal resolution and the quality of the map, consistent with a highly heterogeneous conformation of the TTLL6 core on the MT. Local resolution was estimated using MonoRes82 and RELION’s local resolution function83 (Extended Data Fig. 1f,h). Both maps, the two-PF MT-TTLL6 and the focused TTLL6 maps, were postprocessed. The two-PF map was modified with DeepEMhancer84, and the focused TTLL6 map was sharpened in RELION with B-factor −18 Å2 using the postprocessing procedure. The MTBH2 density within the focused TTLL6 map was removed using the Volume Eraser tool in UCSF Chimera85, and the two postprocessed maps were merged using the combined focused maps tool in PHENIX86. Figures (Figs. 1, 2a,b,d, 3a,b, 4h,j and 5b-d, and Extended Data Figs. 1, 2a, 5a and 7a) were made in UCSF Chimera85 and UCSF ChimeraX87.
Atomic model fitting
The cryo-EM-derived structure of a human unmodified MT (PDB 5N5N; ref. 53) was used as an initial model for the MT, and the crystal structure of TTLL6 (PDB 6VZU)41 was used to obtain an initial model of TTLL6 bound to the MT. The cryo-EM map region corresponding to the globular core of TTLL6 was poorly resolved (Fig. 1a and Extended Data Fig. 1d,f). Consequently, iterative rounds of structure refinement and rebuilding were applied to the MT and MTBH2 (residues 479–503), corresponding to the most resolved parts in the 2pf cryo-EM map with the TTLL6 core (residues 57–478) fitted rigidly in the composite map (Supplementary Table 1). Specifically, the TTLL6 globular core (residues 57–388) from the crystal structure of TTLL6 (PDB 6VZU)41 was rigidly docked into the corresponding cryo-EM density of the composite map using the UCSF Chimera ‘Fit in Map’ tool85. MTBD residues 389–439 were fit separately as a rigid body in Coot. These steps were followed by the manual correction of clashes at the TTLL6-MT interface in Coot88 installed within SBGrid89, followed by geometry optimization. Secondary structure prediction and circular dichroism analysis of the c-MTBH1-2 (residues 460–503) showed that this region is predominantly α-helical, consistent with the observed density. AlphaFold55 was used to predict the structure for MTBH1 residues 462–478 (not resolved in PDB 6VZU; ref. 41), as the corresponding cryo-EM density was not of sufficient resolution to be used to build de novo the structure in this region. Side chain registry within MTBH1-2 (residues 479–503) was assigned based on cryo-EM densities for positively charged and aromatic side chains. The accuracy of the MTBH1-2 build was further verified by tandem mass spectrometry analyses of the cross-linked MTBH1-2 (residues 460–503) and tubulin. A model containing the MT and the MTBH2, the most resolved part of TTLL6, was iteratively refined against the two half maps of the two-PF map using PHENIX as installed within SBGrid86,89 with manual adjustments of residues in Coot. The quality of the model was validated using the Phenix Comprehensive Validation tool86, including calculations of EMRinger90 and Rama-Z91 scores (Supplementary Table 2).
Dimer repeat distance analysis
To calculate the dimer repeat distances, each of the two cryo-EM datasets (undecorated and TTLL6-decorated) was split into three subsets and independently reconstructed using the MiRP procedure81 to obtain C1 and symmetrized reconstructions. For each reconstruction, the helical symmetry parameters such as rise and rotation per subunit were refined using the Relion helical reconstruction procedure92, and the refined rise per subunit was multiplied by 14 (PF number in a reconstruction) and divided by 1.5 (three-start helix). Similar axial dimer repeat values were obtained by independently fitting two tubulin dimers longitudinally into a C1 reconstruction and then calculating the average distance between all Cα atoms in UCSF Chimera85.
PF angle distribution analysis
PF angle distributions were obtained as described previously54. The angle distributions were fit either to two Gaussian functions for the undecorated MTs (R2 = 0.9848 for nonseam and 0.9848 for seam) or to one Gaussian (R2 = 0.9943 for nonseam and 0.9939 for seam) for the TTLL6-decorated MTs. Fitting the TTLL6-decorated MT angle distributions to two Gaussians results in two major peaks less than 2° apart and not in a significantly better overall fit (R2 = 0.9990), indicating that TTLL6 binding regularizes the lattice.
Circular dichroism spectroscopy
TTLL6 MTBH1-2 was prepared for CD spectroscopy measurements in a buffer solution containing 20 mM sodium phosphate (pH 7.0). Far UV CD measurements were performed with a Jasco J-715 spectropolarimeter (Jasco) at room temperature. CD spectra were recorded between 190 and 260 nm using a 0.02 cm path length. All measurements were corrected by background subtraction, and the spectra presented are the average of ten scans. Data were analyzed to determine the percentage of secondary structures using the K2D2 webserver (https://cbdm-01.zdv.uni-mainz.de/~andrade/k2d2/)93.
MT sedimentation assays
Sedimentation assays were performed in BRB80 buffer (80 mM PIPES (pH 6.8), 1 mM MgCl2 and 1 mM EGTA) and 1 mM DTT. TTLL6 MTBH1-2 was added to different concentrations of taxol-stabilized porcine brain MTs in a total reaction volume of 100 μl. The mixture was incubated for 20 min and then centrifuged at 100,000g for 10 min at 30 °C. The supernatant was collected, and the pellets were resolubilized in 100 μl 1× BRB80 buffer. Equivalent volumes of the supernatant and pellet fractions were analyzed by SDS–PAGE. Images of the Coomassie blue-stained gels were analyzed using the Image Studio Lite software package from LI-COR, and the relative amounts of protein in the supernatant and pellet fractions were quantified. The fraction of the protein bound to the MTs was plotted as a function of MT concentration. The apparent Kd values were obtained by fitting the data to a one-site binding parameter in GraphPad Prism.
Gel-filtration analysis of MTBH1-2:tubulin complex formation
Binding of MTBH1-2 to soluble tubulin was analyzed by size-exclusion chromatography on a Superdex-200 analytical gel-filtration column (GE HealthCare). The column was equilibrated in BRB80 buffer (80 mM PIPES (pH 6.8), 1 mM MgCl2 and 1 mM EGTA) and 1 mM DTT. In total, 10 μM of porcine brain tubulin (Cytoskeleton) was incubated with 10 μM of MTBH1-2 on ice for 10 min and then injected into the column at a flow rate of 0.5 ml min−1.
Glutamylation assays with MTs and tubulin peptides
Glutamylation activity of WT TTLL6 and its mutants was determined by quantifying the incorporation of 3H-labeled glutamate into taxol-stabilized brain MTs. Brain tubulin (Cytoskeleton, T240) was polymerized at 30 μM in 10% dimethyl sulfoxide (DMSO) and 1 mM GTP for 2 h at 37 °C. Taxol at 20 μM concentration was added to stabilize the MTs. Glutamylation reactions were performed in 20 mM HEPES (pH 7.0), 10 mM KCl, 1 mM DTT, 1 mM ATP, 20 μM Taxol, 95 μM 2H-labeled glutamate and 5 μM 3H-labeled glutamate (49.6 Ci per mmol). Enzyme to a final concentration of 1 μM was added to initiate the reaction at 30 °C. At different time points, aliquots were removed, and the reactions were terminated by the addition of 20 mM EDTA. The reaction mix was then spotted on the Amersham Hybond-N+ membrane (GE Lifesciences). Tubulin binds to the membrane, while the unincorporated radioactivity was washed away with phosphate buffer containing 25 mM KCl. The total glutamate transferred to tubulin was quantified by measuring the total radioactivity in the reaction mix with a scintillation counter. Initial rates were calculated by fitting the linear range of the curve in GraphPad Prism 6.1. Reactions were performed in duplicate on two different days.
Peptide activity assays of TTLL6 and structure-guided mutants were performed with the α1B-Y peptide at a 1:10 enzyme-to-substrate ratio (1 μM enzyme and 25 μM peptide) in a buffer solution containing 20 mM HEPES (pH 7.0), 50 mM KCl, 10 mM MgCl2, 5 mM TCEP, 1 mM ATP and 1 mM l-glutamic acid. Reactions were initiated by the addition of the enzyme, and samples were collected at 0, 2, 4, 6, 8, 18 and 24 h. Reactions were quenched by adding an equal volume of 20% acetonitrile with 0.1% trifluoroacetic acid (TFA) and separated on a Zorbax 300SB C18 column (Agilent) using a 0–70% acetonitrile gradient in 0.05% TFA at a flow rate of 0.2 ml min−1. The column was connected to a 6224 ESI-TOF LC–MS (Agilent), and data were analyzed using the Mass Hunter Workstation platform as described previously39,59. The decrease in the m/z intensity of the unmodified peptide was monitored as a function of time. The enzyme that elutes separately from the peptides served as an internal loading standard. All reactions were performed in duplicate on two different days.
Transmission EM of MTBH1-2 with MTs
Unmodified tubulin was purified from tsA201 cells using a tumor overexpression gene affinity column as described previously52. To prepare taxol-stabilized MTs, either 100 μM of porcine brain tubulin (Cytoskeleton) or 30 μM of unmodified tubulin were polymerized in 80 mM PIPES (pH 6.8), 1 mM MgCl2, 1 mM EGTA, 10% DMSO and 1 mM GTP for 1 h at 37 °C followed by the slow addition of taxol to a final concentration of 20 μM. The reaction mix was incubated at 37 °C for 1–2 h. Polymerized MTs were then pelleted and separated from nonpolymerized tubulin by passage through a 60% glycerol cushion (1× BRB80, 60% (vol/vol) glycerol and 20 μM taxol) at 126,000g for 15 min at 37 °C. The pellet containing polymerized MTs was resuspended in 1× BRB80 buffer supplemented with 20 μM taxol and 1 mM GTP.
GMPCPP MTs were prepared by polymerizing either 20 μM of porcine brain tubulin (Cytoskeleton) or 25 μM of unmodified tubulin in 1 mM GMPCPP in 1× BRB80 buffer (80 mM PIPES (pH 6.8), 1 mM MgCl2 and 1 mM EGTA). The mixture was first incubated on ice for 5 min and then in a water bath for 1 h at 37 °C. Polymerized MTs were pelleted down by centrifuging at 126,000g for 5 min at 37 °C. The pellet was washed with BRB80 at 37 °C and then resuspended in an ice-cold BRB80 buffer. MTs were depolymerized by leaving the solution in ice for 30 min and periodically mixing it up and down. GMPCPP was added to the reaction mix to a final concentration of 1 mM and kept on ice for 10 min. The mix was then transferred to 37 °C for 2–4 h. Nonpolymerized tubulin was removed by centrifugation as described above. The pellet was washed and resuspended in a warm BRB80 buffer. In total, 4 μl of MTs at 1 μM concentration were deposited on an EM grid (GF-1.2/1.3-3Au-45nm-50; Electron Microscopy Sciences) held within the environmental chamber of a Leica EM GP2 (Leica Microsystems) and incubated for 30 s at 30 °C and 90% humidity, followed by the addition of 4 μl of MTBH1-2 at 5 μM and additional incubation for 30 s. The grids were blotted for 5 s with Whatman 1 blotting paper and plunge-frozen in liquid ethane cooled by liquid nitrogen. Cryo-EM data were collected with EPU software (Thermo Fisher Scientific) on a Glacios electron microscope equipped with a Falcon 4 camera at ×92,000 magnification corresponding to a pixel size of 1.5 Å and −1.5 μm defocus. The exposure rate was 10.09 e− per pixel per second with a cumulative electron dose of 23.12− Å−2. Light microscopy images were processed and figures (Figs. 4a,b,d,f,5e,g and 6a,c, and Extended Data Figs. 4 and 9b-f) were made using ImageJ94.
Cross-linking
Taxol-stabilized MTs were generated as described above. EDC to a final concentration of 1 mM was first added to 10 μM of taxol-stabilized MTs, followed by the addition of sulfo-N-hydroxysuccinimide (sulfo-NHS) to a final concentration of 2 mM. Sulfo-NHS was included in the reaction to increase the efficiency of coupling reactions. After incubating the reaction mix for 30 min at 30 °C, 10 μM of TTLL6 MTBH1-2 was added to the reaction mix for cross-linking. A parallel reaction was also performed by first activating TTLL6 MTBH1-2 by EDC and sulfo-NHS, followed by the addition of taxol-stabilized MTs. Aliquots were collected at different time points, and the resulting cross-linked products were analyzed on an SDS–PAGE.
EDC and NHS cross-linking experiments were also performed with TTLL6 MTBH1-2 and unpolymerized tubulin. Here EDC to a final concentration of 1 mM was first added to 10 μM of either TTLL6 MTBH or soluble porcine brain tubulin, followed by the addition of sulfo-NHS to a final concentration of 2 mM. All samples were kept on ice throughout the experiment. After incubating the reaction mix for 30 min, 10 μM of tubulin was added to the reaction mix containing TTLL6 MTBH1-2, and TTLL6 MTBH1-2 was added to the reaction mix containing soluble tubulin. Aliquots were analyzed on an SDS–PAGE gel.
Tandem mass spectrometry
In-gel samples (~10 μg per sample) were reduced using 10 mM tris(2-carboxyethyl)phosphine hydrochloride at room temperature for 1 h and alkylated with 10 mM N-ethylmaleimide (NEM) for 10 min. Half of each sample was digested with trypsin (Promega), and the other half was digested with AspN (Sequencing Grade, Roche). Both digestions were performed with an enzyme:sample ratio of 1:20 (wt/wt) at 37 °C for 18 h. Digested samples were desalted using an Oasis HLB μElution plate (Waters). The desalted and dried sample was resuspended in 5 μl loading buffer, and 1 μl of the sample was injected into an Ultimate 3000 HPLC (Thermo Fisher Scientific) during each liquid chromatography tandem mass spectrometry (LC–MS/MS) run. Peptides were separated on an ES802A column. The composition of the mobile phase A (MPA) was 98% H2O, 1.9% ACN and 0.1% formic acid, and that of MPB was 98% ACN, 1.9% H2O and 0.1% formic acid. MPB was increased from 3% to 24% in 54 min for peptide separation. LC–MS/MS data were acquired in data-dependent mode. The MS1 scans were performed in orbitrap with a resolution of 120 K, a mass range of 400–1,500 m/z and an AGC target of 2 × 105. The quadrupole isolation window is 1.6 m/z. The precursor ion intensity threshold to trigger the MS/MS scan was set at 1 × 104. MS2 scans were conducted in an ion trap. Peptides were fragmented with higher-energy collision dissociation (HCD) and electron-transfer/higher-energy collision dissociation (EThcD) methods. The collision energy was fixed at 30% for the HCD method and 20% for the EThcD method. MS1 scan was performed every 3 s. We collected the maximum number of MS2 scans within the 3-s cycle.
Sequest HT in Proteome Discoverer 2.4 software was used for database search. Raw data were searched against the Sprot Human database. For data acquired from trypsin digests, up to four missed cleavages were allowed; for those of AspN digests, up to ten missed cleavages were allowed. NEM on cysteines was set as a static modification. MTBH1-2 fragments generated by trypsin and AspN digestions were included as dynamic modifications. Mass tolerances for MS1 and MS2 scans were set to 10 ppm and 0.6 Da, respectively. All possible cross-linked peptides matched by the database search were manually curated to find real cross-linked products.
Tubulin polymerization assay with MTBH1-2
Flow chambers were made using silanized cover glass and double-stick tape as described previously95. In total, 10 μM of porcine brain tubulin was introduced into the chambers in the absence or presence of different concentrations of TTLL6 MTBH1-2 and its mutants. The final imaging buffer was BRB80 (80 mM PIPES (pH 6.8), 1 mM MgCl2 and 1 mM EGTA) supplemented with 50 mM KCl, 1 mM GTP, 0.1% methylcellulose 4,000 cP, 1% pluronic F-127 and 0.1 mg ml−1 casein. Before imaging, the flow chambers were warmed to 30 °C using an objective heater (Bioptechs). A Nikon Eclipse Ti-E equipped with a Hamamatsu ORCA Flash 4.0 Vc sCMOS camera was used to acquire the IRM images every 5 s for 20 min. Polymer length was extracted using the CurveTrace plugin v.0.3.5 for ImageJ (https://github.com/ekatrukha/CurveTrace). The following detection parameters were used: line width (1.8 pixels), minimum number of points in line (10) and maximum width search (20.0 pixels). The program was specified to split line at junctions. The quantified results were then imported into Prism 6.1 (GraphPad) for visualization.
Generation of MTs missing β-tubulin tails
To selectively remove β-tubulin tails from MTs, taxol-stabilized MTs containing 97% unmodified tubulin, 1% biotin brain tubulin and 2% 647-Hilyte labeled tubulin were treated with subtilisin at a 1:200 (subtilisin:tubulin) mass ratio at room temperature39. The reaction was terminated at 1.5 h by the addition of 5 mM phenylmethylsulfonyl fluoride. MTs were recovered through a glycerol cushion. Mass spectrometry, performed as described in ref. 52, showed the β-tubulin tail completely removed and the α-tail intact (Extended Data Fig. 7b,c). Intact, unlabeled control MTs containing 99% unmodified tubulin and 1% biotinylated tubulin were prepared the same way but without the addition of subtilisin. These MTs were used in TIRF-based assays.
LC–MS analyses of TTLL6-modified MTs
Taxol-stabilized WT and subtilisin-treated MTs (αβ-Δtail) were generated as described above. The proteolytic removal of the β-tubulin tail while the α-tail remained intact was confirmed by LC–MS. Both WT and αβ-Δtail MTs were incubated with M. musculus TTLL6 at an enzyme:MT ratio of 1:10. Reactions were performed in 20 mM HEPES (pH 7.0), 50 mM KCl, 10 mM MgCl2, 5 mM TCEP, 1 mM ATP and 1 mM l-glutamic acid. Reactions were initiated by the addition of TTLL6, and samples were collected at 0 and 2 h. Reactions were quenched by adding an equal volume of 20% acetonitrile with 0.1% TFA and separated on a Zorbax 300SB C18 column (Agilent) using a 0–70% acetonitrile gradient in 0.05% TFA at a flow rate of 0.2 ml min−1. The column was connected to a 6224 ESI-TOF LC–MS (Agilent), and data were analyzed using the Mass Hunter Workstation platform.
Generation of MTs glutamylated on β-tubulin
TTLL7 was purified as previously described39. Taxol-stabilized MTs containing 97% unmodified tubulin, 1% biotin brain tubulin and 2% 647-Hilyte tubulin were treated with Xenopus tropicalis TTLL7 at a molar ratio of 1:10 (TTLL7:tubulin) for 1 h. TTLL7 was removed by adding 300 mM KCl and centrifugation through a 60% glycerol cushion in BRB80 buffer. The number of glutamates added to α-tubulin and β-tubulin was determined by LC–MS as described in ref. 13. The mass spectra display the characteristic distribution of masses with peaks separated by +129 Da corresponding to every glutamate added (Supplementary Fig. 3). Unmodified control MTs containing 99% unmodified tubulin and 1% biotinylated tubulin were prepared the exact same way but without the addition of TTLL7.
Immunoblot assays
Taxol-stabilized TTLL7 glutamylated MTs were generated as described above. The number of glutamates added to β-tubulin was determined by LC–MS as described previously13. The mass spectra displayed the characteristic distribution of masses with peaks separated by +129 Da corresponding to every glutamate added. Both unmodified and glutamylated MTs were incubated with M. musculus TTLL6 at an enzyme:tubulin molar ratio of 1:50. Reactions were carried out in 20 mM HEPES (pH 7.0), 50 mM KCl, 10 mM MgCl2, 5 mM TCEP, 1 mM ATP and 1 mM l-glutamic acid. Reactions were initiated by the addition of TTLL6, and samples were collected at 0, 1, 2 and 4 h. Reactions were quenched with EDTA to a final concentration of 20 mM and then mixed with SDS loading buffer. α-Tubulin and β-tubulin were separated on an SDS–PAGE gel, as described previously96, and transferred to a nitrocellulose membrane. The membrane was treated with 5% nonfat dry milk blocking solution for 1 h at room temperature and subsequently incubated with 1:2,000 dilution of rabbit polyclonal antipolyglutamate chain (polyE) antibody (Adipogen, IN105) and 1:2,000 dilution of mouse monoclonal anti-β-tubulin antibody (Abcam, ab101019). IRdye680 goat anti-mouse and IRdye800 goat anti-rabbit secondary antibodies (1:10,000 dilution) were used (LI-COR Biosciences). The bound antibodies were stripped using Revitablot western blot stripping buffer (Rockland, MB-085-0050). The membrane was then blocked again with 5% nonfat dry milk blocking solution for 1 h at room temperature and probed for monoglutamylation using 1:2,000 dilution of mouse monoclonal antibody GT335 (Adipogen, AG-20B-0020). IRdye680 goat anti-mouse secondary antibody (1:10,000 dilution) was used (LI-COR Biosciences). Densitometric analyses and quantifications were performed with Image Studio Lite (LI-COR Biosciences) and plotted using GraphPad Prism 6.1.
Fluorescent labeling
A synthesized fluorescent peptide, NH2-(Atto488)-LPETGGGG (Bio-Synthesis), was ligated to the N-terminus of TTLL6 using sortase97. Specifically, recombinant TTLL6 was cleaved off its GST tag using tobacco etch virus protease, leaving two glycines on its N-terminus, and then subsequently purified on a Heparin column followed by size-exclusion chromatography. A total of 50 μM of either recombinantly purified TTLL6 or TTLL6ΔMTBH1-2 and 1 mM of the fluorescent peptide were incubated with 100 μM of sortase enzyme for 1 h at room temperature in a buffer solution containing 50 mM Tris (pH 7.0) and 150 mM NaCl. The labeled protein was then purified from the unreacted peptide on a Superose 6 10/300 GL size-exclusion column (GE HealthCare). The labeling efficiency was ~64% for TTLL6 and ~35% for TTLL6ΔMTBH1-2.
TIRF-based microscopy binding assays
To measure the binding of TTLL6 to intact and subtilisin-treated (αβ-Δtail) MTs as a function of TTLL6 concentration, flow chambers were made using silanized cover glass and double-stick tape as described previously95. WT unmodified MTs or αβ-Δtail MTs, prepared as described above, were immobilized on a glass surface using neutravidin (Life Technologies, A-2666). Atto488-labeled TTLL6 at varying concentrations ranging from 0 to 1 μM for WT MTs and 0–2 μM for αβ-Δtail MTs was perfused into the chamber in assay buffer (80 mM PIPES (pH 6.9), 1 mM MgCl2, 1 mM EGTA, 50 mM KCl, 100 μM ATP and 1 mM glutamate) supplemented with 1% F-127 pluronic acid, 20 μM Taxol and oxygen scavengers prepared as previously described95 to remove free oxygen from the solution. An inverted TIRF microscope (Nikon Ti-E with TIRF attachment) was used for image acquisition. The excitation light was provided by a 640 nm Coherent CUBE for Hylite-647 and a 488 nm Coherent CUBE for Atto488, both operating at 20 mW before being coupled to the TIRF attachment via a fiber optic. The excitation light was focused at the back focal plane of a ×100, 1.49 numerical aperture objective (Nikon CFI Apo TIRF 100×). The excitation light was split from the emission light using a 405/488/532/635 quad-band filter (Semrock DiO3-405/488/532/635) and directed to the sample. The emitted light was collected by the same objective and split using an Andor TuCAM equipped with a dichroic mirror (Semrock FF640-FDi02). Two EMCCD cameras (Andor iXON3-897) equipped with either an FF01-550/88 (Semrock) or a BLP01-647R (Semrock) were used to image the 488 and 640 channels, respectively. Images of unlabeled MTs were obtained using IRM with a red LED and imaged on the 640 channel for simultaneous imaging with the 488 channel3. The 488 nm channel was used to image the Atto488-labeled TTLL6 at 100 ms exposure. Multiple fields of view on multiple chambers were imaged for each concentration of Atto488–TTLL6 and TTLL6ΔMTBH1-2. Background-corrected line scan intensities were measured using Fiji136 (ref. 94) and normalized to MT length. Fluorescent intensity was plotted as a function of TTLL6 concentration, and the apparent Kd was obtained by fitting the data to a one-site binding parameter in Prism 6.1 (GraphPad).
To compare TTLL6 binding to WT and βΔ-tail MTs, both unlabeled WT MTs (not fluorescently labeled) and subtilisin-treated αβΔ-tail MTs (containing 2% 647-Hylite labeled tubulin) were immobilized in the same chamber with 0.02 mg ml−1 neutravidin (Thermo Fisher Scientific). Either Atto488-labeled TTLL6 (50 nM) or Atto488-labeled TTLL6ΔMTBH1-2 (250 nM) in 20 mM HEPES (pH 7.0), 50 mM KCl, 10 mM MgCl2 1 mM glutamate, 100 μM ATP, 2 μM taxol, 1% pluronic F-127 and oxygen scavengers (7.5 U μl−1 catalase, 0.15 U μl−1 glucose oxidase and 20 mM glucose) was perfused into the chamber. The chamber was left to equilibrate for 5 min before imaging. Images of the 488 nm and 647 nm excitation channels were acquired at an exposure of 100 ms as described above. MT images were obtained using IRM on an ORCA Flash 4.0 V2 sCMOS camera (Hamamatsu) before acquiring fluorescent images. Multiple fields of view were imaged. Background-corrected line scan intensities were measured using FIJI94, and intensities were normalized to MT length.
To compare TTLL6 binding to unmodified and TTLL7 glutamylated MTs, both unmodified MTs (not fluorescently labeled) and TTLL7 modified glutamylated MTs (containing 2% 647-Hylite labeled tubulin) were immobilized in the same chamber with 0.02 mg ml−1 neutravidin (Thermo Fisher Scientific). Either Atto488–TTLL6 (50 nM) or Atto488–TTLL6ΔMTBH1-2 (250 nM) in 20 mM HEPES pH (7.0), 50 mM KCl, 10 mM MgCl2 1 mM glutamate, 100 μM ATP, 2 μM taxol, 1% pluronic F-127 and oxygen scavengers (7.5 U μl−1 catalase, 0.15 U μl−1 glucose oxidase and 20 mM glucose) was perfused into the chamber. The chamber was left to equilibrate for 5 min before imaging. Images of the 488 nm and 647 nm excitation channels were acquired at an exposure of 100 ms as described above. MT images were obtained using IRM on an ORCA Flash 4.0 V2 sCMOS camera (Hamamatsu) before acquiring fluorescent images. Multiple fields of view on multiple channels were imaged. Background-corrected line scan intensities were measured using FIJI94, and intensities were normalized to MT length.
Single-molecule dwell time analysis
For the dwell time analysis, GMPCPP- or taxol-stabilized MTs consisting of 94% porcine brain tubulin, 5% Hylite-647-labeled tubulin (Cytoskeleton TL590 M) and 1% biotin-labeled tubulin (Cytoskeleton T238P) were prepared as described above. MTs were immobilized on a glass surface using neutravidin (Life Technologies, A-2666). Atto488-labeled TTLL6 (10 nM) was perfused in assay buffer (80 mM PIPES (pH 6.9), 1 mM MgCl2, 1 mM EGTA, 50 mM KCl, 100 μM ATP and 1 mM glutamate) supplemented with 1% F-127 pluronic acid, 20 μM Taxol (in case of taxol-stabilized MTs) and oxygen scavengers (7.5 U μl−1 catalase, 0.15 U μl−1 glucose oxidase and 20 mM glucose) prepared as described previously95. The 640 nm and 488 nm channels were used to image MTs and Atto488–TTLL6, respectively. Images were collected at an exposure of 50 ms for either 1 frame per second in the case of Atto488–TTLL6 or at a frame rate of 20 frames per second in the case of Atto488–TTLL6ΔMTBH1-2. For data analysis, kymographs were generated from the movie stack for each MT using ImageJ. TTLL6 molecules appeared as vertical lines on the kymographs (Fig. 5e,g and Extended Data Fig. 10d). The length of these lines was measured and recorded as dwell times. The results were imported into GraphPad Prism 6.1, and regression analyses were performed to get the mean dwell times. Images of Atto488–TTLL6 bound to a coverslip with no MTs were used to calculate the bleaching time of Atto488 molecules, and the dwell times were corrected for the effects of bleaching (Extended Data Fig. 10c).
Extended Data
Extended Data Fig. 1 ∣. Cryo-EM image processing.
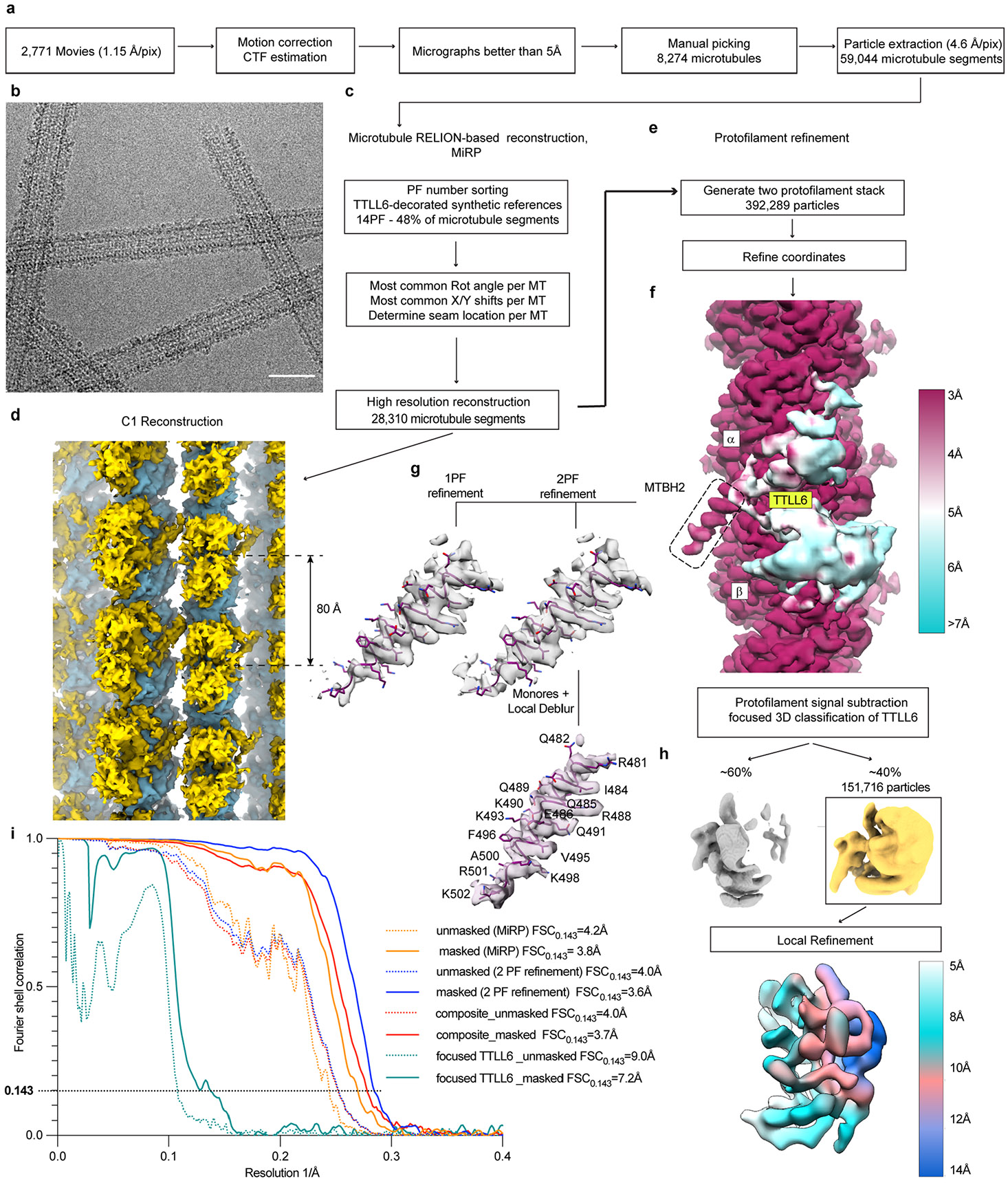
a. Initial image processing was performed in RELION83. In total, 2,771 movies were motion corrected and summed98; high-quality micrographs were selected based on CTF estimation80 and visual inspection for manual picking of microtubule segments. b. Representative cryo-EM micrograph from one of 2,771 movies of microtubules decorated with TTLL6. Decoration is highly heterogeneous. Scale bar, 25 nm. c. Initial reconstruction of microtubule decorated with TTLL6 using MiRP81. Microtubule segments were sorted based on protofilament number; most common rotation angle, x/y shifts and seam positions were assigned to each microtubule segment. d. C1 reconstruction of the TTLL6 bound microtubule. TTLL6, gold, binds every 80 Å, highlighted by the double arrow, along the microtubule lattice, blue. e. Protofilament refinement54 improved map quality for the microtubule and MTBH1-2 in TTLL6. For each microtubule segment all but the signal from two adjacent protofilaments was substracted to generate a two protofilament stack, followed by refinement of particle coordinates. f. Local resolution estimates show the map reaching 3 Å resolution in the microtubule and MTBH1-2 regions and poorly resolved density for the rest of TTLL6. MTBH1-2 is highlighted by a dashed rectangle. Local resolution was determined via Monores82 followed by LocalDeblur99. g. Two protofilament, 2PF, refinement protocol improved definition of the C-terminus of MTBH2 in comparison to a protocol where each particle contained signal for just one protofilament, 1PF. h. Focused classification of TTLL6 improved its definition. Refinement of the minor class yielded a TTLL6 map with local resolution estimates ranging from ~5 Å to 14 Å. i Fourier shell correlation (FSC) curves for each microtubule reconstruction depict their nominal resolution. FSC estimates were calculated at 0.143 criterion. Resolution for the microtubule lattice improved from 3.8 Å, MiRP protocol, to 3.6 Å, two protofilament refinement protocol.
Extended Data Fig. 2 ∣. MTBH1-2 is highly flexible and binds only to microtubules and not soluble tubulin.
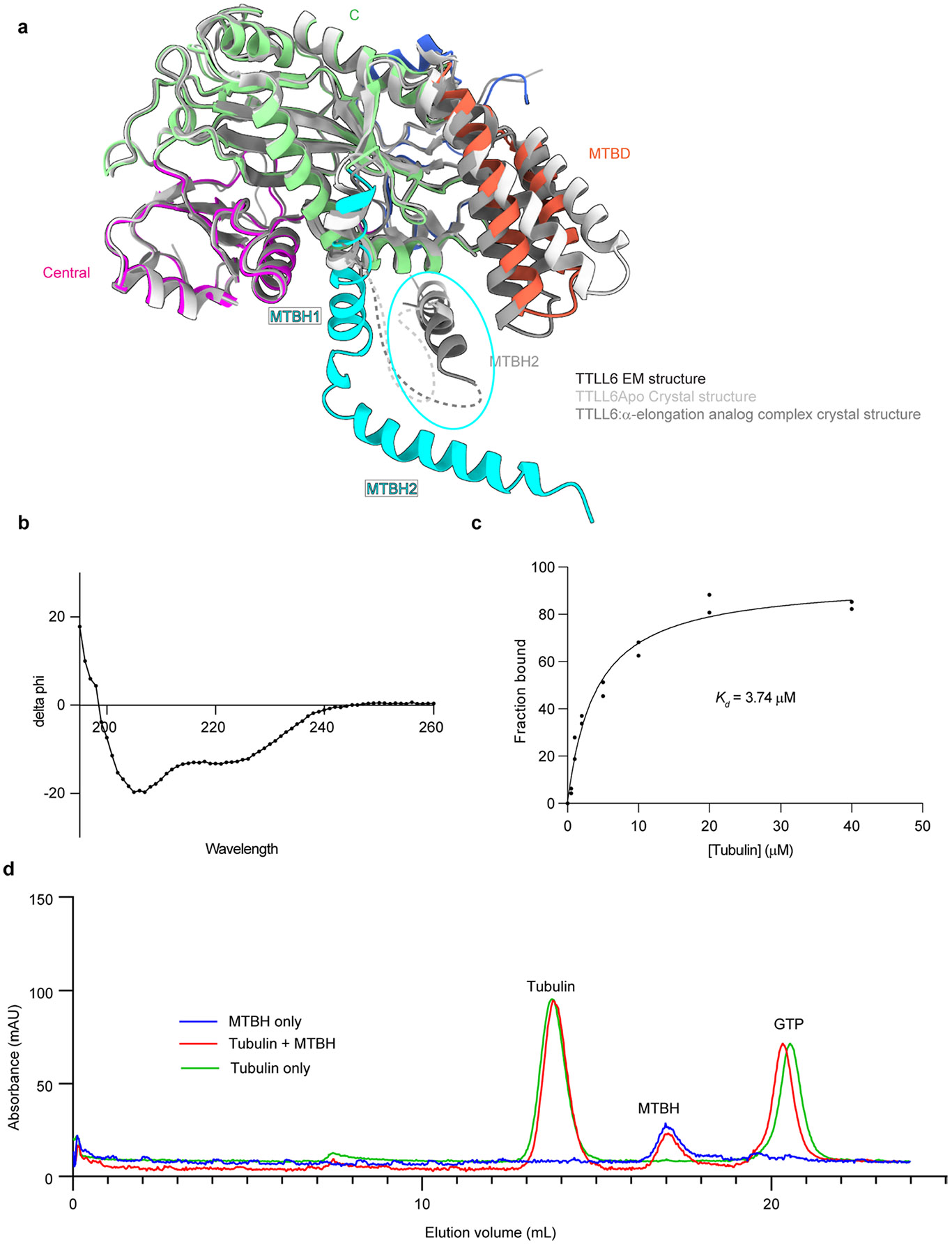
a. Ribbon representation of TTLL6 in the conformation it adopts in complex with the microtubule (domains colored as in Fig. 1) superposed on the X-ray crystal structures of TTLL6 bound to ATP (light gray, PDB 6VZT; ref. 41) or bound to a di-Glu elongation analog (dark gray, PDB 6VZU; ref. 41). A part of the MTBH1-2 was not resolved in the X-ray structures. The unresolved regions are shown as dotted lines. The MTBH1-2 in the TTLL6 X-ray structures, highlighted by a cyan ellipse, is in a different orientation than that in the cryo-EM structure in complex with the microtubule. b. Circular dichroism spectra of recombinantly expressed and purified TTLL6 MTBH1-2 show its secondary structure is predominantly α-helical. c. Binding of TTLL6 MTBH1-2 to taxol-stabilized microtubules assembled from porcine brain tubulin. Kd ~3.7 μM. Error bars, S.E.M (n = 2). d. Gel filtration analysis of recombinant TTLL6 MTBH1-2 and soluble αβ-tubulin mixture showing that the two proteins elute separately on a Superdex-200 analytical gel-filtration column (Methods).
Extended Data Fig. 3 ∣. TTLL6 binding compacts the microtubule lattice and regularizes the microtubule wall.
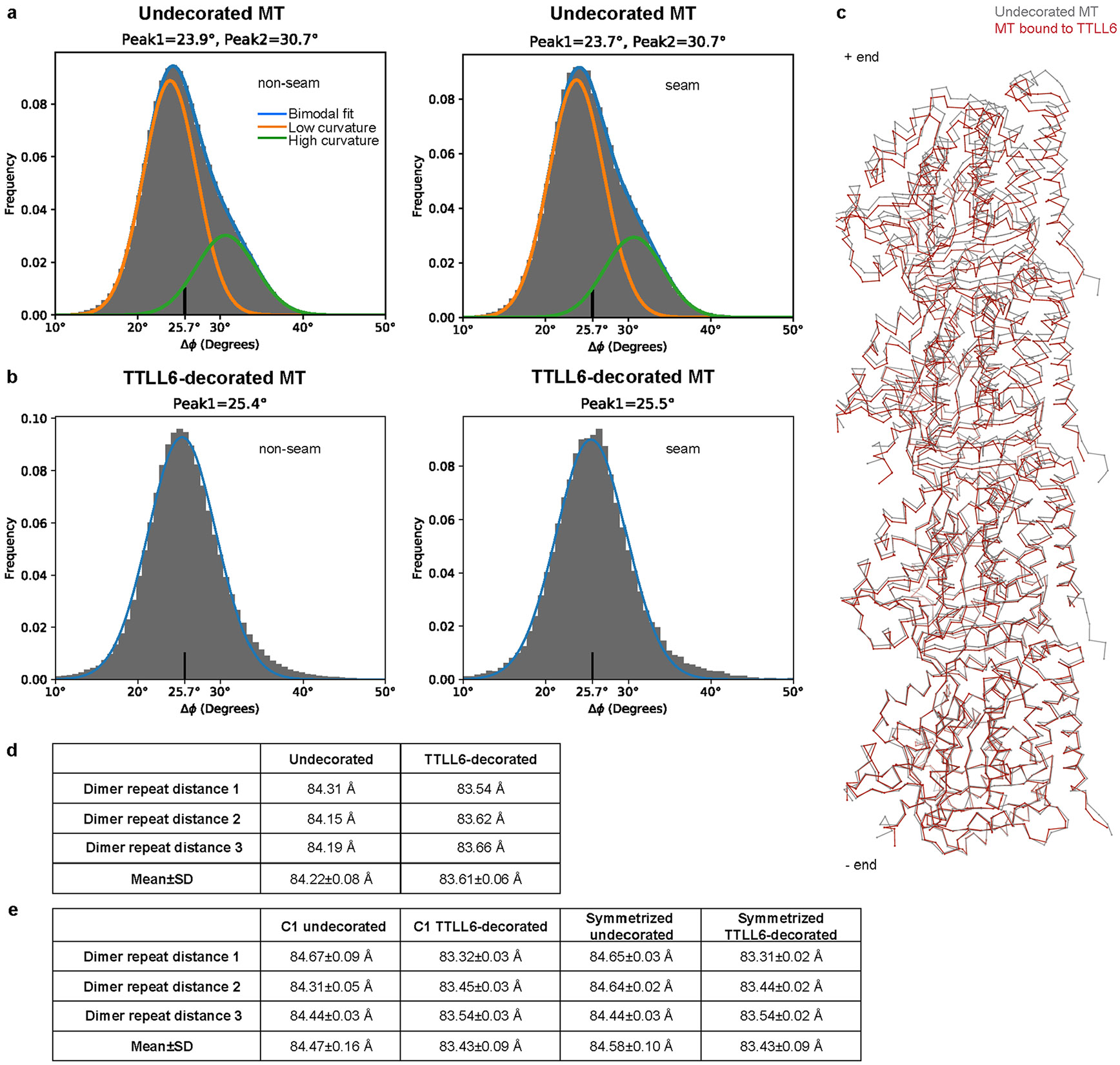
a. Histogram of rotation angles (φ angle) between adjacent protofilaments of undecorated microtubules shows two peaks for non-seam 23.9°, major, and 30.7°, minor; for seam 23.7°, major, 30.7°, minor. These numbers deviate from a symmetric protofilament geometry, φ = 360°/14 protofilaments = 25.7°. b. Histogram of rotation angles (φ angle) between adjacent protofilaments of TTLL6-decorated microtubules shows one peak for non-seam at 25.4° and for the seam at 25.5°, These numbers are close to a symmetric protofilament geometry, φ = 360°/14 protofilaments = 25.7°. c. Atomic models of undecorated, gray, and TTLL6 decorated, red, protofilaments aligned at the minus end on the α1-tubulin subunit show TTLL6-induced structural changes in the microtubule lattice. d. Tubulin dimer repeat distances for undecorated and TTLL6-decorated microtubules obtained in Relion92 by refining the helical rise for 3 independent reconstructions obtained by splitting the data into three datasets, each containing one-third of the data (Methods). e. Tubulin dimer repeat distances obtained from fitting atomic models in 3 independent C1 and symmetrized undecorated and TTLL6-decorated microtubule reconstructions each obtained from one-third of the data (Methods).
Extended Data Fig. 4 ∣. MTBH1-2 destabilizes microtubules.
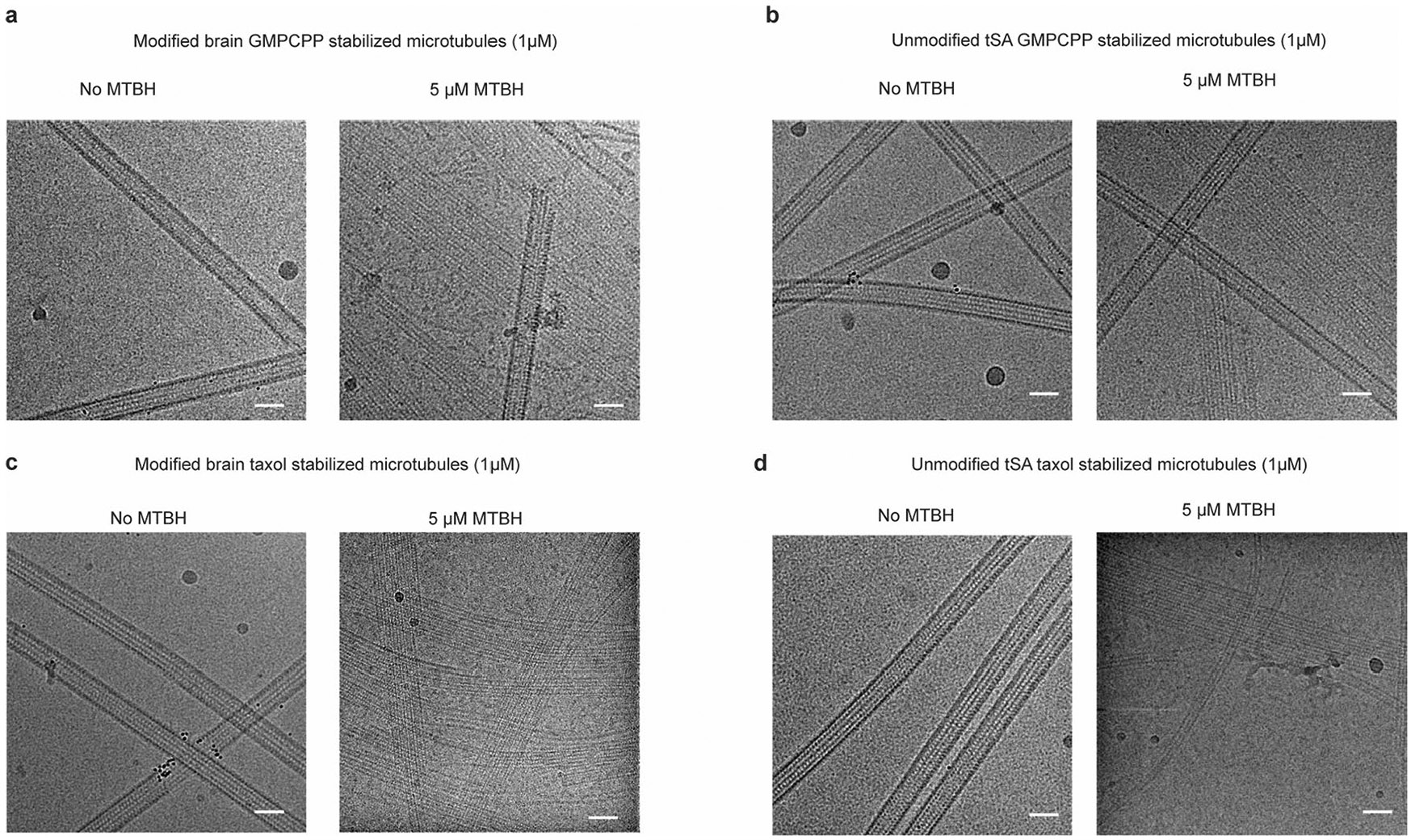
a–d. Representative cryo-EM images from one of two independent experiments showing microtubules alone or in the presence of excess MTBH1-2. MTBH1-2 at 5 μM with 1 μM GMPCPP-stabilized microtubules polymerized from porcine brain tubulin (a), 1 μM GMPCPP-stabilized microtubules polymerized from unmodified human tubulin affinity purified from tSA201 cells52 (b), 1 μM taxol-stabilized microtubules polymerized from porcine brain tubulin (c) and taxol-stabilized microtubules polymerized from unmodified human tubulin (d). Scale bar, 30 nm.
Extended Data Fig. 5 ∣. MTBD and MTBH1-2 optimally position the TTLL6 core to interact with the negatively charged α-tubulin tail through a groove lined with positively charged residues critical for activity.
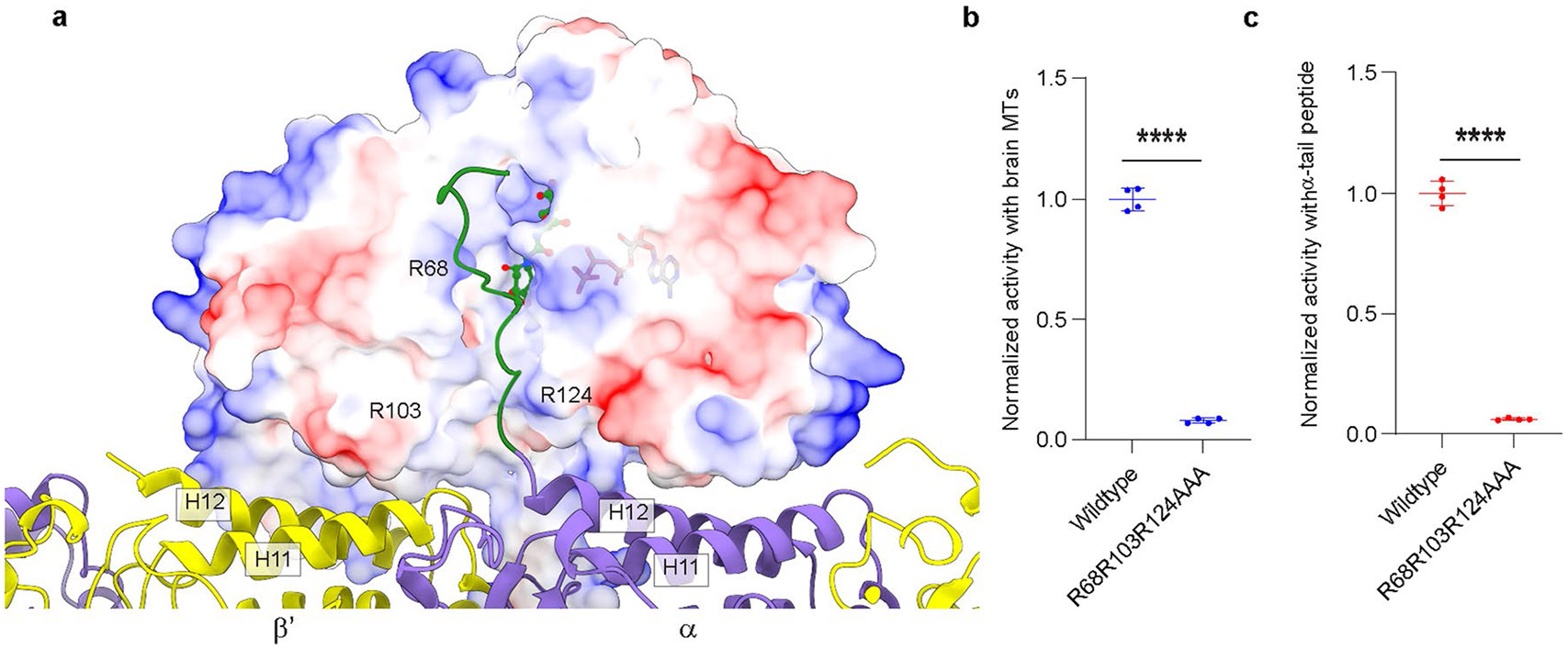
a. View of TTLL6 in complex with the microtubule showing the proximity of the C-terminal tail of α-tubulin to the active site of TTLL6. TTLL6 surface is color-coded for electrostatic potential. The electrostatic potential was calculated using the Poisson Boltzmann Solver77. The α-tubulin tail, shown in green, for which no structural data are available was modeled to maximize side chain interactions without violating stereochemical constraints. ATP and the di-glutamate in the active site are shown in ball-and-stick. The di-glutamate position is based on the X-ray crystal structure of TTLL6 in complex with the α-elongation analog (PDB 6VZU; ref. 41). A groove lined with positively charged residues leads to the active site of TTLL6. We hypothesize that these cationic residues anchor electronegative side chains in the α-tubulin tail, while the hydrophobic groove interacts with the tubulin tail backbone. b,c. Normalized glutamylation activity of structure-guided TTLL6 mutants in the proposed α-tubulin tail binding groove with taxol-stabilized microtubules (b) and isolated α1B(-Y) peptide (c). Error bars, S.E.M. (n = 4). ****p < 0.0001 as determined by two-tailed t-test.
Extended Data Fig. 6 ∣. Identification of TTLL6 MTBH1-2-microtubule interaction sites using cross-linking coupled with tandem mass spectrometry.
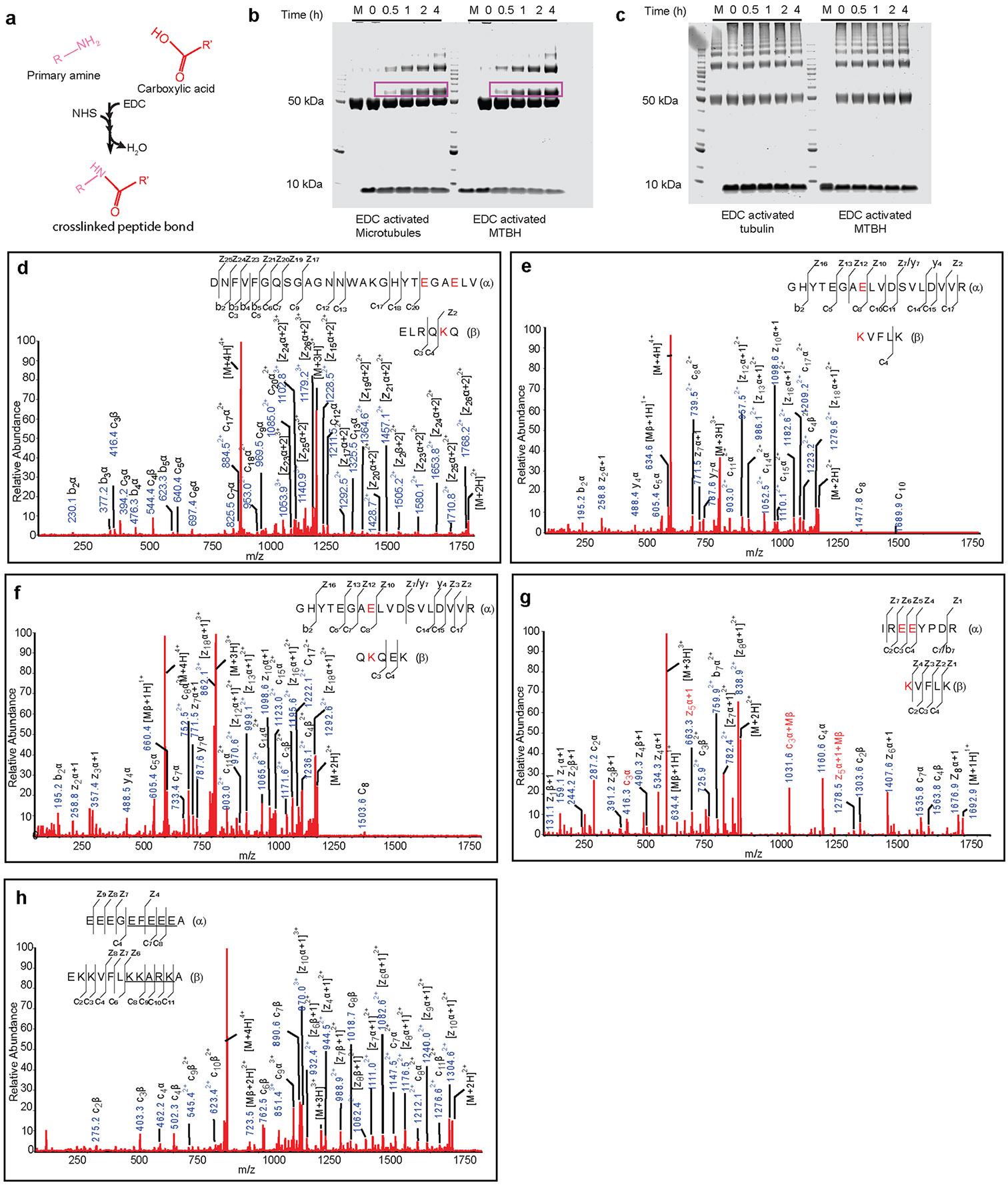
a. Chemical reaction showing the cross-linking of primary amine and carboxylic acid in the presence of 1-ethyl-3-(-3-dimethylaminopropyl) carbodiimide hydrochloride (EDC) and sulfo N-hydroxysuccinimide (sulfo-NHS). b,c. Representative SDS-PAGE gels from one of two independent experiments showing the cross-linking of MTBH1-2 to microtubules (b) but not to soluble tubulin (c). The cross-linked products are outlined with a magenta rectangle. M, mock. d–h. MS/MS sequencing of the cross-linked products. Peptides were fragmented using EThcD decision tree method, so majority of the MS/MS fragments are c- and z-ions, with a small portion of b- and y-ions. The cross-linked product contains 2 peptides. The sequences of the peptides are shown in the spectrum and labeled as (α) or (β) peptides. When a MS/MS fragment contains cross-linked (α) and (β) peptides, it is labeled as normal c- or z-ion. When a fragment is from (α) or (β) peptides alone, α or β is then included in the labeling of that ion, such as [z12α+1] or c3β. The cross-link sites are highlighted in red. Possible cross-linked residues are underlined when it is not possible to locate the precise site.
Extended Data Fig. 7 ∣. Distance between MTBH2 and β-tubulin tails on the microtubule and mass spectra showing subtilisin and subtilisin mediated proteolytic removal of β-tubulin tails from unmodified human microtubules.
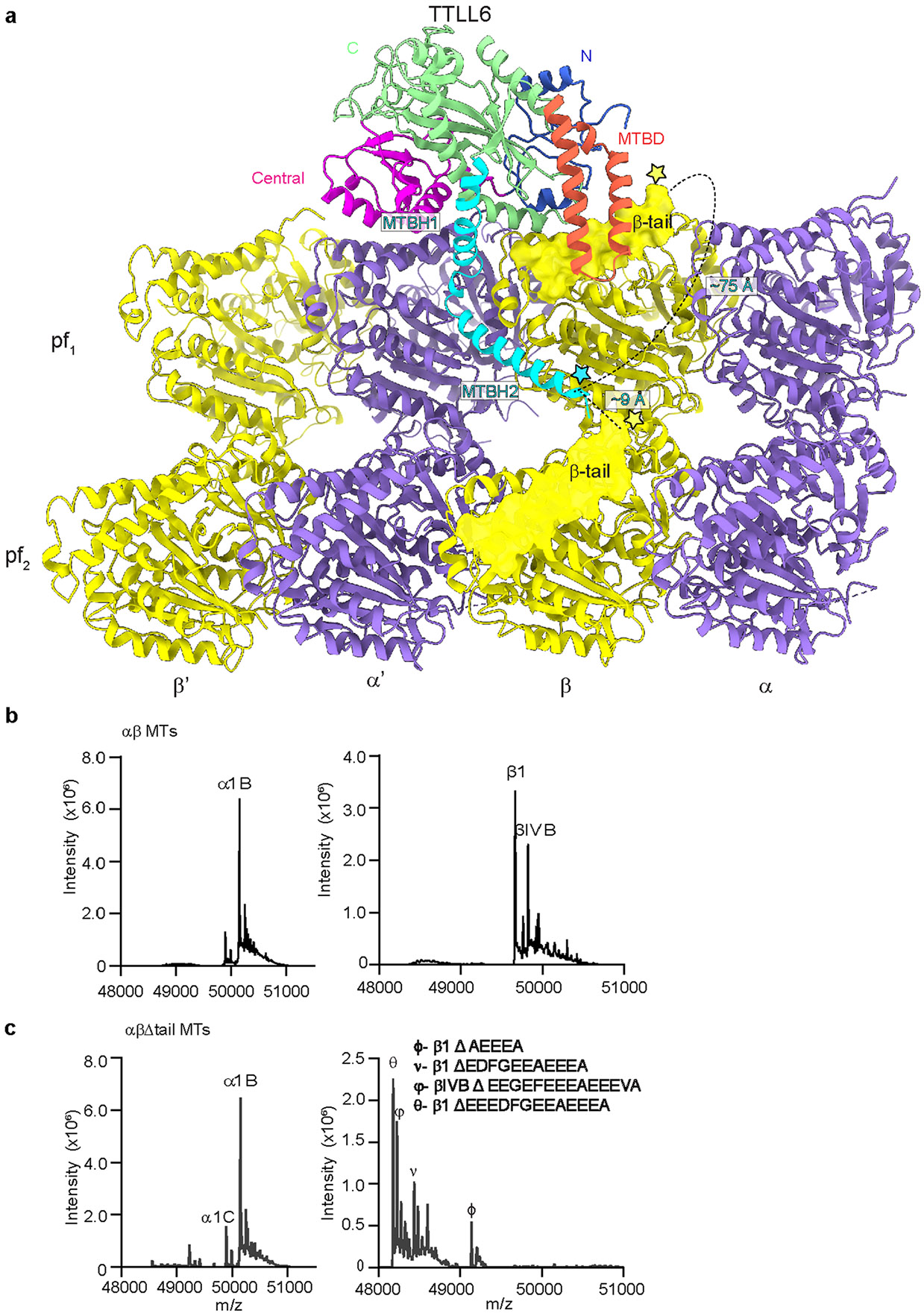
a. Schematic showing the distance between the start of the β-tubulin tail (yellow star) and the MTBH2 (cyan star). The disposition of the β-tubulin tail (its length and position as it emerges from the tubulin body) is such that the MTBH2 is able to interact only with the β-tail from the lateral tubulin dimer because the most C-terminal glutamate that cross-links to the MTBH2 lysine cannot reach closer than ~30 Å from the C-terminus of MTBH2, even when we assume that the β-tail adopts a completely extended random coil conformation (3.8 Å Cα to Cα distance) which would give it the greatest span (the distance between the start of the β-tail and the MTBH2 on the same tubulin dimer is ~75 Å). Even the most C-terminal glutamate residue in the β-tail of the same dimer on which TTLL6 sits cannot reach closer than ~18 Å from MTBH2. In contrast, the Cα of the terminal Lys residue of TTLL6 MTBH2 is positioned ~9 Å away from the last β-tubulin residue visible in our structure (D427, and the start of the flexible β-tubulin tail) that belongs to the laterally adjacent β-tubulin. b,c. LC-MS spectra of wild-type (b) and subtilisin (c) treated unmodified human microtubules used in Fig. 4 (Methods). The tubulin isotypes and the different β-tubulin species after subtilisin treatment are indicated.
Extended Data Fig. 8 ∣. Microtubules missing their β-tubulin tails are poor substrates for TTLL6.
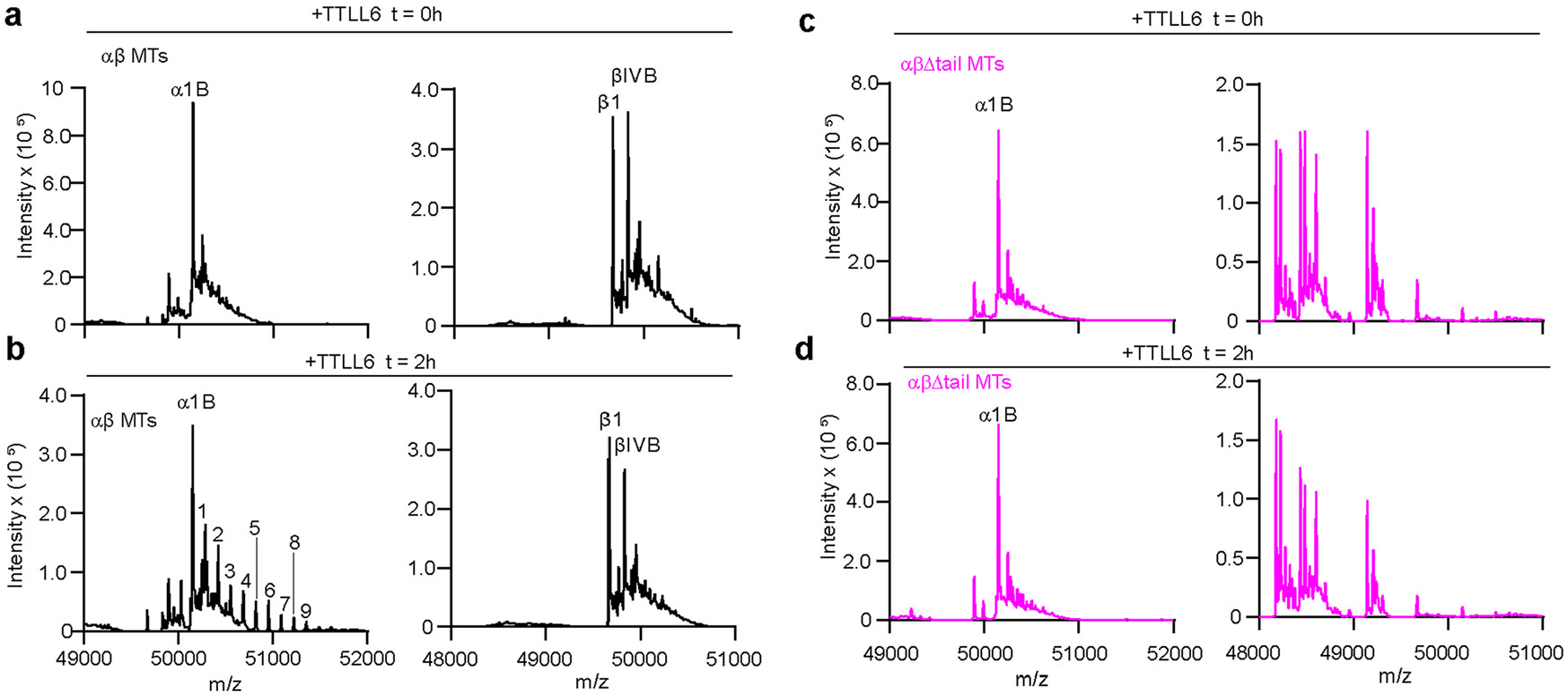
a,b. LC-MS spectra of intact wild-type microtubules at times 0 h (a) and 2 h (b) after incubation with TTLL6. Tubulin isotypes and glutamate numbers are indicated on top. c,d. LC-MS spectra of αβΔ-tail microtubules at times 0 h (c) and 2 h (d) after incubation with TTLL6 (Methods). Spectra labeled as in a.
Extended Data Fig. 9 ∣. MTBH1-2 promotes tubulin polymerization and mutation of residues in the three cationic clusters impairs this activity.
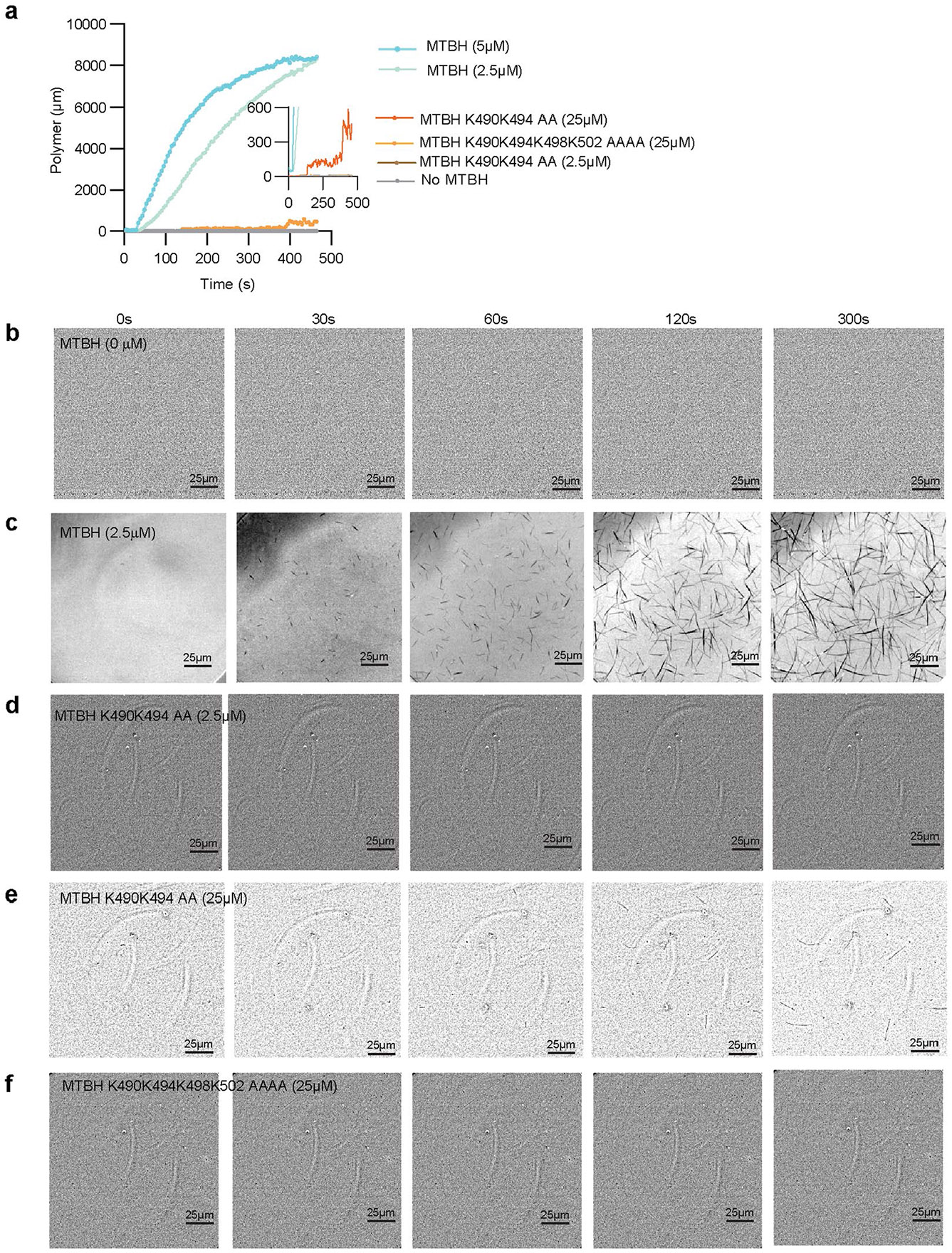
a. Quantification of tubulin polymerization over time in the absence or presence of MTBH1-2 and the structure-guided MTBH1-2 mutants. b–f. Representative interference reflection microscopy images from one of two independent experiments of reactions containing 10 μM porcine tubulin without MTBH1-2 (b) or with 2.5 μM MTBH1-2 (c), 2.5 μM MTBH1-2 (K490A/K494A) (d), 25 μM MTBH1-2 (K490A/K494A) (e) and 25 μM MTBH1-2 (K490A/K494A/K498A/K502A) (f). Scale bar, 25 μm.
Extended Data Fig. 10 ∣. Single molecule characterization of Atto488-TTLL6 interaction with the microtubule.
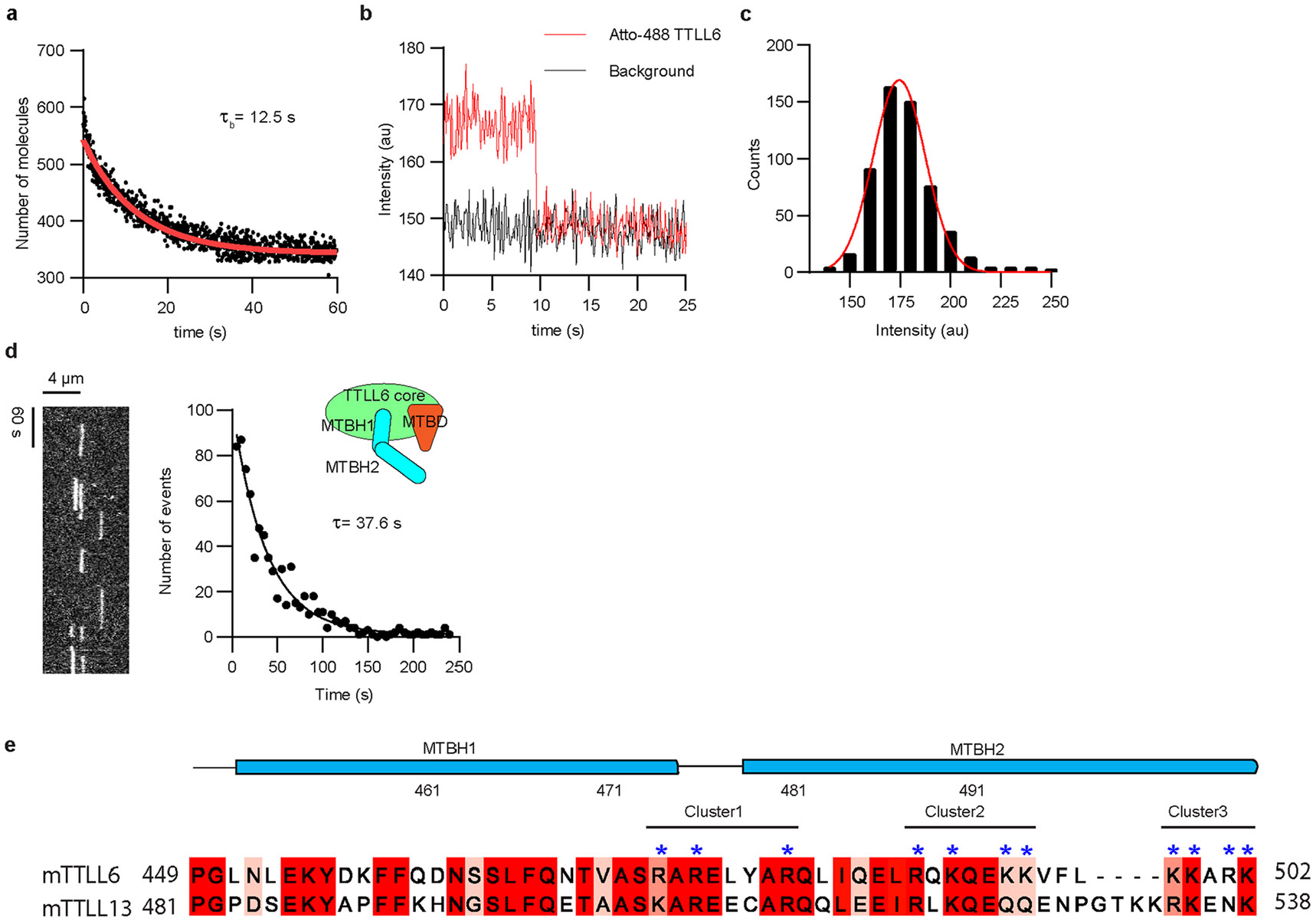
a. Distribution of surface-bound single Atto488 TTLL6 over time. The number of surface-bound molecules decays exponentially as the fluorescence is bleached. An exponential decay fit (red line) yields a bleaching time of 12.5 s. b. One-step photobleaching trace of immobilized Atto488-TTLL6 consistent with the molecule existing as a monomer over the exposure time (50 ms). c. Initial fluorescence intensity distribution of Atto488-TTLL6 molecules immobilized on glass showing a monodisperse profile. d. Representative kymograph of single Atto488-TTLL6 on taxol-stabilized brain microtubules (left). Distribution of residence times of Atto488-TTLL6 with taxol-stabilized brain microtubules. The mean residence time (τ) was obtained by fitting an exponential curve to the histogram and correcting for photobleaching (R2 = 0.95, n = 791 binding events from 6 independent experiments). e. Sequence alignment of the MTBH1-2 from Mus musculus TTLL6 and TTLL13. Positively charged basic residues in the three clusters are marked with blue asterisks on top.
Supplementary Material
Acknowledgements
We thank D.-Y. Lee from the Biochemistry Core (National Heart, Lung and Blood Institute (NHLBI)) for mass spectrometry help, J. Spector (National Institute of Neurological Disorders and Stroke (NINDS)) for microscopy help, D. Hoover (National Institutes of Health (NIH)) for software installation help, G.E. Debs (Yale University) and C.V. Sindelar (Yale University) for advice with PF refinement and modifying the procedure to perform two-PF refinement. Image processing was performed on the Biowulf cluster maintained by the High Performing Computation group at the NIH. G.C.L. (Scripps Research) is supported by NIH NS095892. A.R.-M. is supported by the intramural programs of NINDS and NHLBI.
Footnotes
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Extended data is available for this paper at https://doi.org/10.1038/s41589-024-01599-0.
Supplementary information The online version contains supplementary material available at https://doi.org/10.1038/s41589-024-01599-0.
Competing interests
The authors declare no competing interests.
Online content
Any methods, additional references, Nature Portfolio reporting summaries, source data, extended data, supplementary information, acknowledgements, peer review information; details of author contributions and competing interests; and statements of data and code availability are available at https://doi.org/10.1038/s41589-024-01599-0.
Data availability
The TTLL6MTBH12:MT model has been deposited under accession PDB 8T42; the PDB model of TTLL6:MT from the composite map has been deposited under accession PDB 8U3Z. Maps were deposited at Electron Microscopy Data Bank with accession EMD-42884, B-factor sharpened map, EMD-41018 DeepEmhanced map, EMD-41090, composite map, all with accompanying raw half maps. The map of TTLL6 obtained by focused classification has accession EMD-41022. Motion-corrected micrographs are found under EMPIAR-11798. All plasmids and cell lines used in this study will be shared by the lead contact upon request. This paper does not use any original code. Source data are provided with this paper.
References
- 1.Roll-Mecak A. The tubulin code in microtubule dynamics and information encoding. Dev. Cell 54, 7–20 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bieling P. et al. CLIP-170 tracks growing microtubule ends by dynamically recognizing composite EB1/tubulin-binding sites. J. Cell Biol 183, 1223–1233 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chen J. et al. α-Tubulin tail modifications regulate microtubule stability through selective effector recruitment, not changes in intrinsic polymer dynamics. Dev. Cell 56, 2016–2028 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hotta T. et al. EML2-S constitutes a new class of proteins that recognizes and regulates the dynamics of tyrosinated microtubules. Curr. Biol 32, 3898–3910 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gundersen GG & Bulinski JC Selective stabilization of microtubules oriented toward the direction of cell migration. Proc. Natl Acad. Sci. USA 85, 5946–5950 (1988). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gurland G & Gundersen GG Stable, detyrosinated microtubules function to localize vimentin intermediate filaments in fibroblasts. J. Cell Biol 131, 1275–1290 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kerr JP et al. Detyrosinated microtubules modulate mechanotransduction in heart and skeletal muscle. Nat. Commun 6, 8526 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Palazzo AF, Eng CH, Schlaepfer DD, Marcantonio EE & Gundersen GG Localized stabilization of microtubules by integrin- and FAK-facilitated ρ signaling. Science 303, 836–839 (2004). [DOI] [PubMed] [Google Scholar]
- 9.Robison P. et al. Detyrosinated microtubules buckle and bear load in contracting cardiomyocytes. Science 352, aaf0659 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lacroix B. et al. Tubulin polyglutamylation stimulates spastin-mediated microtubule severing. J. Cell Biol 189, 945–954 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sharma N. et al. Katanin regulates dynamics of microtubules and biogenesis of motile cilia. J. Cell Biol 178, 1065–1079 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Szczesna E. et al. Combinatorial and antagonistic effects of tubulin glutamylation and glycylation on katanin microtubule severing. Dev. Cell 57, 2497–2513 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Valenstein ML & Roll-Mecak A Graded control of microtubule severing by tubulin glutamylation. Cell 164, 911–921 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Barisic M. et al. Mitosis. Microtubule detyrosination guides chromosomes during mitosis. Science 348, 799–803 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lessard DV et al. Polyglutamylation of tubulin’s C-terminal tail controls pausing and motility of kinesin-3 family member KIF1A. J. Biol. Chem 294, 6353–6363 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sirajuddin M, Rice LM & Vale RD Regulation of microtubule motors by tubulin isotypes and post-translational modifications. Nat. Cell Biol 16, 335–344 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.McKenney RJ, Huynh W, Vale RD & Sirajuddin M Tyrosination of α-tubulin controls the initiation of processive dynein-dynactin motility. EMBO J. 35, 1175–1185 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nirschl JJ, Magiera MM, Lazarus JE, Janke C & Holzbaur EL α-Tubulin tyrosination and CLIP-170 phosphorylation regulate the initiation of dynein-driven transport in neurons. Cell Rep. 14, 2637–2652 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.van Dijk J. et al. A targeted multienzyme mechanism for selective microtubule polyglutamylation. Mol. Cell 26, 437–448 (2007). [DOI] [PubMed] [Google Scholar]
- 20.Garnham CP & Roll-Mecak A The chemical complexity of cellular microtubules: tubulin post-translational modification enzymes and their roles in tuning microtubule functions. Cytoskeleton (Hoboken) 69, 442–463 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gundersen GG, Khawaja S & Bulinski JC Generation of a stable, posttranslationally modified microtubule array is an early event in myogenic differentiation. J. Cell Biol 109, 2275–2288 (1989). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bodakuntla S. et al. Tubulin polyglutamylation is a general traffic control mechanism in hippocampal neurons. J. Cell Sci 133, jcs241802 (2020). [DOI] [PubMed] [Google Scholar]
- 23.Magiera MM, Singh P, Gadadhar S & Janke C Tubulin posttranslational modifications and emerging links to human disease. Cell 173, 1323–1327 (2018). [DOI] [PubMed] [Google Scholar]
- 24.Karakaya M. et al. Biallelic variant in AGTPBP1 causes infantile lower motor neuron degeneration and cerebellar atrophy. Am. J. Med. Genet. A 179, 1580–1584 (2019). [DOI] [PubMed] [Google Scholar]
- 25.Maddirevula S. et al. Autozygome and high throughput confirmation of disease genes candidacy. Genet. Med 21, 736–742 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shashi V. et al. Loss of tubulin deglutamylase CCP1 causes infantile-onset neurodegeneration. EMBO J. 37, e100540 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sheffer R. et al. Biallelic variants in AGTPBP1, involved in tubulin deglutamylation, are associated with cerebellar degeneration and motor neuropathy. Eur. J. Hum. Genet 27, 1419–1426 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Konno A. et al. TTLL9−/− mice sperm flagella show shortening of doublet 7, reduction of doublet 5 polyglutamylation and a stall in beating. J. Cell Sci 129, 2757–2766 (2016). [DOI] [PubMed] [Google Scholar]
- 29.Pathak N, Austin CA & Drummond IA Tubulin tyrosine ligase-like genes TTLL3 and TTLL6 maintain zebrafish cilia structure and motility. J. Biol. Chem 286, 11685–11695 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bosch Grau M. et al. Tubulin glycylases and glutamylases have distinct functions in stabilization and motility of ependymal cilia. J. Cell Biol 202, 441–451 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ikegami K, Sato S, Nakamura K, Ostrowski LE & Setou M Tubulin polyglutamylation is essential for airway ciliary function through the regulation of beating asymmetry. Proc. Natl Acad. Sci. USA 107, 10490–10495 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.He K. et al. Axoneme polyglutamylation regulated by Joubert syndrome protein ARL13B controls ciliary targeting of signaling molecules. Nat. Commun 9, 3310 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hong SR et al. Spatiotemporal manipulation of ciliary glutamylation reveals its roles in intraciliary trafficking and Hedgehog signaling. Nat. Commun 9, 1732 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kubo T. et al. A conserved flagella-associated protein in Chlamydomonas, FAP234, is essential for axonemal localization of tubulin polyglutamylase TTLL9. Mol. Biol. Cell 25, 107–117 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lee JE et al. CEP41 is mutated in Joubert syndrome and is required for tubulin glutamylation at the cilium. Nat. Genet 44, 193–199 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bompard G. et al. CSAP acts as a regulator of TTLL-mediated microtubule glutamylation. Cell Rep. 25, 2866–2877 (2018). [DOI] [PubMed] [Google Scholar]
- 37.Backer CB, Gutzman JH, Pearson CG & Cheeseman IM CSAP localizes to polyglutamylated microtubules and promotes proper cilia function and zebrafish development. Mol. Biol. Cell 23, 2122–2130 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mullen RJ, Eicher EM & Sidman RL Purkinje cell degeneration, a new neurological mutation in the mouse. Proc. Natl Acad. Sci. USA 73, 208–212 (1976). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Garnham CP et al. Multivalent microtubule recognition by tubulin tyrosine ligase-like family glutamylases. Cell 161, 1112–1123 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Janke C. et al. Tubulin polyglutamylase enzymes are members of the TTL domain protein family. Science 308, 1758–1762 (2005). [DOI] [PubMed] [Google Scholar]
- 41.Mahalingan KK et al. Structural basis for polyglutamate chain initiation and elongation by TTLL family enzymes. Nat. Struct. Mol. Biol 27, 802–813 (2020). [DOI] [PubMed] [Google Scholar]
- 42.Mukai M. et al. Recombinant mammalian tubulin polyglutamylase TTLL7 performs both initiation and elongation of polyglutamylation on β-tubulin through a random sequential pathway. Biochemistry 48, 1084–1093 (2009). [DOI] [PubMed] [Google Scholar]
- 43.Bonnet C. et al. Differential binding regulation of microtubule-associated proteins MAP1A, MAP1B, and MAP2 by tubulin polyglutamylation. J. Biol. Chem 276, 12839–12848 (2001). [DOI] [PubMed] [Google Scholar]
- 44.Boucher D, Larcher JC, Gros F & Denoulet P Polyglutamylation of tubulin as a progressive regulator of in vitro interactions between the microtubule-associated protein τ and tubulin. Biochemistry 33, 12471–12477 (1994). [DOI] [PubMed] [Google Scholar]
- 45.Genova M. et al. Tubulin polyglutamylation differentially regulates microtubule-interacting proteins. EMBO J. 42, e112101 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kubo T, Yanagisawa HA, Yagi T, Hirono M & Kamiya R Tubulin polyglutamylation regulates axonemal motility by modulating activities of inner-arm dyneins. Curr. Biol 20, 441–445 (2010). [DOI] [PubMed] [Google Scholar]
- 47.Suryavanshi S. et al. Tubulin glutamylation regulates ciliary motility by altering inner dynein arm activity. Curr. Biol 20, 435–440 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhong Y. et al. XBP1 variant 1 promotes mitosis of cancer cells involving upregulation of the polyglutamylase TTLL6. Hum. Mol. Genet 31, 2639–2654 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Miller KE & Heald R Glutamylation of Nap1 modulates histone H1 dynamics and chromosome condensation in Xenopus. J. Cell Biol 209, 211–220 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sun X. et al. Loss of RPGR glutamylation underlies the pathogenic mechanism of retinal dystrophy caused by TTLL5 mutations. Proc. Natl Acad. Sci. USA 113, E2925–E2934 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Xia P. et al. Glutamylation of the DNA sensor cGAS regulates its binding and synthase activity in antiviral immunity. Nat. Immunol 17, 369–378 (2016). [DOI] [PubMed] [Google Scholar]
- 52.Vemu A, Garnham CP, Lee DY & Roll-Mecak A Generation of differentially modified microtubules using in vitro enzymatic approaches. Methods Enzymol. 540, 149–166 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Vemu A, Atherton J, Spector JO, Moores CA & Roll-Mecak A Tubulin isoform composition tunes microtubule dynamics. Mol. Biol. Cell 28, 3564–3572 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Debs GE et al. Dynamic and asymmetric fluctuations in the microtubule wall captured by high-resolution cryoelectron microscopy. Proc. Natl Acad. Sci. USA 117, 16976–16984 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Jumper J. et al. Highly accurate protein structure prediction with AlphaFold. Nature 596, 583–589 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zehr EA & Roll-Mecak A Cryo-EM structures of human α1B/βI+βIVb microtubules shed light on isoform specific assembly. Preprint at bioRxiv 10.1101/2023.12.01.569594 (2023). [DOI] [Google Scholar]
- 57.Kellogg EH et al. Insights into the distinct mechanisms of action of taxane and non-taxane microtubule stabilizers from cryo-EM structures. J. Mol. Biol 429, 633–646 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Sui H & Downing KH Structural basis of interprotofilament interaction and lateral deformation of microtubules. Structure 18, 1022–1031 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Garnham CP, Yu I, Li Y & Roll-Mecak A Crystal structure of tubulin tyrosine ligase-like 3 reveals essential architectural elements unique to tubulin monoglycylases. Proc. Natl Acad. Sci. USA 114, 6545–6550 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ikegami K. et al. TTLL7 is a mammalian β-tubulin polyglutamylase required for growth of MAP2-positive neurites. J. Biol. Chem 281, 30707–30716 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Abad MA et al. Structural basis for microtubule recognition by the human kinetochore Ska complex. Nat. Commun 5, 2964 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Legal T, Zou J, Sochaj A, Rappsilber J & Welburn JP Molecular architecture of the Dam1 complex-microtubule interaction. Open Biol. 6, 150237 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Manka SW & Moores CA The role of tubulin-tubulin lattice contacts in the mechanism of microtubule dynamic instability. Nat. Struct. Mol. Biol 25, 607–615 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Maurer SP, Fourniol FJ, Bohner G, Moores CA & Surrey T EBs recognize a nucleotide-dependent structural cap at growing microtubule ends. Cell 149, 371–382 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Zhang R, Alushin GM, Brown A & Nogales E Mechanistic origin of microtubule dynamic instability and its modulation by EB proteins. Cell 162, 849–859 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Roll-Mecak A. How cells exploit tubulin diversity to build functional cellular microtubule mosaics. Curr. Opin. Cell Biol 56, 102–108 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Redeker V. et al. Mutations of tubulin glycylation sites reveal cross-talk between the C termini of α- and β-tubulin and affect the ciliary matrix in Tetrahymena. J. Biol. Chem 280, 596–606 (2005). [DOI] [PubMed] [Google Scholar]
- 68.Ebberink E. et al. Tubulin engineering by semisynthesis reveals that polyglutamylation directs detyrosination. Nat. Chem 15, 1179–1187 (2023). [DOI] [PubMed] [Google Scholar]
- 69.Latham JA & Dent SY Cross-regulation of histone modifications. Nat. Struct. Mol. Biol 14, 1017–1024 (2007). [DOI] [PubMed] [Google Scholar]
- 70.Suganuma T & Workman JL Crosstalk among histone modifications. Cell 135, 604–607 (2008). [DOI] [PubMed] [Google Scholar]
- 71.Magiera MM et al. Excessive tubulin polyglutamylation causes neurodegeneration and perturbs neuronal transport. EMBO J. 37, e100440 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Zempel H. et al. Amyloid-β oligomers induce synaptic damage via τ-dependent microtubule severing by TTLL6 and spastin. EMBO J. 32, 2920–2937 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Zhang F. et al. Posttranslational modifications of α-tubulin in Alzheimer disease. Transl. Neurodegener 4, 9 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Gadau SD Morphological and quantitative analysis on α-tubulin modifications in glioblastoma cells. Neurosci. Lett 687, 111–118 (2018). [DOI] [PubMed] [Google Scholar]
- 75.Mialhe A et al. Tubulin detyrosination is a frequent occurrence in breast cancers of poor prognosis. Cancer Res. 61, 5024–5027 (2001). [PubMed] [Google Scholar]
- 76.Soucek K. et al. Normal and prostate cancer cells display distinct molecular profiles of α-tubulin posttranslational modifications. Prostate 66, 954–965 (2006). [DOI] [PubMed] [Google Scholar]
- 77.Jurrus E. et al. Improvements to the APBS biomolecular solvation software suite. Protein Sci. 27, 112–128 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Suloway C. et al. Automated molecular microscopy: the new Leginon system. J. Struct. Biol 151, 41–60 (2005). [DOI] [PubMed] [Google Scholar]
- 79.Lander GC et al. Appion: an integrated, database-driven pipeline to facilitate EM image processing. J. Struct. Biol 166, 95–102 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Zhang K. GCTF: real-time CTF determination and correction. J. Struct. Biol 193, 1–12 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Cook AD, Manka SW, Wang S, Moores CA & Atherton J A microtubule RELION-based pipeline for cryo-EM image processing. J. Struct. Biol 209, 107402 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Vilas JL et al. MonoRes: automatic and accurate estimation of local resolution for electron microscopy maps. Structure 26, 337–344 (2018). [DOI] [PubMed] [Google Scholar]
- 83.Scheres SH RELION: implementation of a Bayesian approach to cryo-EM structure determination. J. Struct. Biol 180, 519–530 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Sanchez-Garcia R. et al. DeepEMhancer: a deep learning solution for cryo-EM volume post-processing. Commun. Biol 4, 874 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Pettersen EF et al. UCSF Chimera—a visualization system for exploratory research and analysis. J. Comput. Chem 25, 1605–1612 (2004). [DOI] [PubMed] [Google Scholar]
- 86.Afonine PV et al. Real-space refinement in PHENIX for cryo-EM and crystallography. Acta Crystallogr. D Struct. Biol 74, 531–544 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Pettersen EF et al. UCSF ChimeraX: structure visualization for researchers, educators, and developers. Protein Sci. 30, 70–82 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Emsley P & Cowtan K Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr 60, 2126–2132 (2004). [DOI] [PubMed] [Google Scholar]
- 89.Morin A. et al. Collaboration gets the most out of software. eLife 2, e01456 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Barad BA et al. EMRinger: side chain-directed model and map validation for 3D cryo-electron microscopy. Nat. Methods 12, 943–946 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Sobolev OV et al. A global Ramachandran score identifies protein structures with unlikely stereochemistry. Structure 28, 1249–1258 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.He S & Scheres SHW Helical reconstruction in RELION. J. Struct. Biol 198, 163–176 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Perez-Iratxeta C & Andrade-Navarro MA K2D2: estimation of protein secondary structure from circular dichroism spectra. BMC Struct. Biol 8, 25 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Schneider CA, Rasband WS & Eliceiri KW NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 9, 671–675 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Ziolkowska NE & Roll-Mecak A In vitro microtubule severing assays. Methods Mol. Biol 1046, 323–334 (2013). [DOI] [PubMed] [Google Scholar]
- 96.Banerjee A, Bovenzi FA & Bane SL High-resolution separation of tubulin monomers on polyacrylamide minigels. Anal. Biochem 402, 194–196 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Theile CS et al. Site-specific N-terminal labeling of proteins using sortase-mediated reactions. Nat. Protoc 8, 1800–1807 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Zheng SQ et al. MotionCor2: anisotropic correction of beam-induced motion for improved cryo-electron microscopy. Nat. Methods 14, 331–332 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Ramirez-Aportela E. et al. Automatic local resolution-based sharpening of cryo-EM maps. Bioinformatics 36, 765–772 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The TTLL6MTBH12:MT model has been deposited under accession PDB 8T42; the PDB model of TTLL6:MT from the composite map has been deposited under accession PDB 8U3Z. Maps were deposited at Electron Microscopy Data Bank with accession EMD-42884, B-factor sharpened map, EMD-41018 DeepEmhanced map, EMD-41090, composite map, all with accompanying raw half maps. The map of TTLL6 obtained by focused classification has accession EMD-41022. Motion-corrected micrographs are found under EMPIAR-11798. All plasmids and cell lines used in this study will be shared by the lead contact upon request. This paper does not use any original code. Source data are provided with this paper.