

Abstract
A biosimilar medicine is a successor to a reference (‘originator’/’original-brand’) biologic medicine brought to market once the patent and exclusive marketing rights for the reference have expired. Biosimilar natalizumab (PB006 [biosim-NTZ]; developed by Polpharma Biologics S.A. and marketed globally as Tyruko®; Sandoz) has been developed as a successor to reference natalizumab (Tysabri® [ref-NTZ]; Biogen) and is the first US Food and Drug Administration (FDA)-approved and European Medicines Agency (EMA)-approved biosimilar in neurology. As per the FDA and EMA indications for ref-NTZ, biosim-NTZ is approved to treat relapsing forms of multiple sclerosis (USA, EU) and Crohn’s disease (USA only). Approval of biosim-NTZ was based on the ‘totality of evidence’, a comprehensive body of data collected during the development process, demonstrating similarity to its reference medicine. The foundational step of demonstrating structural and functional similarity between biosim-NTZ and ref-NTZ confirmed identical primary and indistinguishable higher order structures, as well as matching binding affinity to α4β1/α4β7 integrins. Following the confirmation of matching structure and function, pharmacokinetic/pharmacodynamic similarity of biosim-NTZ to ref-NTZ in healthy subjects was demonstrated, with no clinically meaningful differences identified in safety and immunogenicity. A comparative, double-blind, randomized study (Antelope) was also conducted in patients with relapsing-remitting multiple sclerosis and demonstrated matching efficacy, safety, and immunogenicity with no clinically meaningful differences between biosim-NTZ and ref-NTZ. This review presents the totality of evidence that confirmed the biosimilarity of biosimilar natalizumab to its reference medicine, which supported its approval by the FDA and the EMA. [Graphical plain language summary available].
Supplementary Information
The online version contains supplementary material available at 10.1007/s40259-024-00671-4.
Key Points
| Biosimilar natalizumab, PB006, was assessed for biosimilarity to reference natalizumab against the totality of evidence, and is the first biosimilar medicine developed and approved for the treatment of patients with relapsing forms of multiple sclerosis. |
| The totality of evidence is a comprehensive package of analytical, functional, pharmacokinetic/pharmacodynamic, and clinical data required for biosimilar approval, demonstrating that a proposed biosimilar matches its reference medicine in terms of quality, efficacy, safety, and immunogenicity. |
| The confirmation of biosimilarity via the totality of evidence establishes a biosimilar to be as safe and effective as its reference medicine, therefore physicians and patients can expect the same clinical outcome. |
Introduction
Multiple sclerosis (MS) is a chronic immune-mediated inflammatory disease that can lead to severe physical and/or cognitive disability [1, 2]. In relapsing-remitting MS (RRMS), individuals experience disease relapses with new or worsening neurological symptoms separated by periods of stability [3]. The MS treatment landscape has changed significantly in recent years, particularly with the introduction of high-efficacy disease-modifying therapies, which include biologic medicines such as monoclonal antibodies [4, 5]. However, the high cost associated with biologic medicines still represents a major limitation for wide access to these treatments [4, 6].
Biosimilar medicines are successors to already approved ‘reference’ biologic medicines [7, 8], and can be brought to market once the market protection period for the reference medicine expires [7]. According to US and European consensus guidelines, biosimilar medicines are considered as effective and safe as the reference medicine when approved within highly regulated areas, confirming that physicians and patients can expect the same clinical outcome when receiving treatment with a biosimilar [4, 8, 9]. Safe and effective switching between reference and biosimilar medicines, and between biosimilar medicines, is also supported by a large body of evidence accumulated from years of clinical practice [10–13]. As seen in these therapy areas, including oncology, metabolic diseases, gastroenterology, and rheumatology [14–17], the anticipated introduction of biosimilar medicines as a less costly biologic treatment option for the first time in MS is expected to increase access to biologics [18, 19]. Increased access would in turn offer both clinical and economic benefits to patients with MS, healthcare systems, and payers, ultimately driving sustainability of care [6, 15, 18, 20].
Biosimilarity to an approved reference medicine is demonstrated via the totality of evidence (ToE) gathered in a comprehensive development process [7, 9, 21, 22]. Unlike the development process for a novel biologic medicine, which must demonstrate the efficacy and safety of the investigational treatment de novo, biosimilarity is based on a comparison of physicochemical and clinical data between the proposed biosimilar medicine and its existing reference medicine to exclude any clinically meaningful differences [7, 9, 21–23]. Extensive physicochemical, structural, and functional characterization using state-of-the-art analytical technologies is applied to demonstrate that the proposed biosimilar matches its reference medicine in terms of structure, function, purity, and other quality attributes (QAs); this characterization is the cornerstone of the biosimilar comparability exercise [9, 22–25]. The pharmacokinetic/pharmacodynamic (PK/PD) profile of the proposed biosimilar medicine versus the reference medicine is also assessed as part of the ToE package. When considered appropriate, additional comparative clinical studies are conducted to generate comparative clinical efficacy and safety data, to reinforce the demonstration of no clinically meaningful differences between the proposed biosimilar and the reference medicine [7, 9, 26] using, as described by the European Medicines Agency (EMA), “sensitive endpoints in a population where product-related differences in clinical performance can be detected” [7]. To demonstrate this process, ToE is often depicted as a pyramid (Fig. 1), where each layer corresponds to the essential evidence required for each step in biosimilar development, and also indicates the weight of that evidence within the overall data package; analytical characterization forms the base of the biosimilarity pyramid, upon which the comparative pharmacokinetic/pharmacodynamic, safety, and efficacy components rely, building toward the top of the pyramid.
Fig. 1.

The totality of evidence data package for the development and approval of biosimilar medicines compared to reference medicines. In vivo preclinical studies are not a requirement from the European Medicines Agency and US Food and Drug Administration for the approval of biosimilar medicines when extensive analytical and functional characterization has already demonstrated the proposed biosimilar and reference medicines to be highly similar [9, 22, 38]. PD pharmacodynamics, PK pharmacokinetics
Biosimilar natalizumab (PB006 [biosim-NTZ]; developed by Polpharma Biologics S.A. and marketed globally as Tyruko®; Sandoz) has been developed as a biosimilar to reference natalizumab (Tysabri® [ref-NTZ]; Biogen, Cambridge, MA, USA) [27–31]. Biosim-NTZ received approval from the US Food and Drug Administration (FDA) in August 2023 and from the EMA in September 2023 [27, 28]. Natalizumab is a highly effective, recombinant, humanized, α4-integrin antibody derived from a murine monoclonal antibody to the human α4β1/α4β7 integrins [5, 27, 29–33]. In the EU, ref-NTZ and biosim-NTZ are approved as monotherapy in adults with highly active RRMS [27, 29]. In the USA, ref-NTZ and biosim-NTZ are approved as monotherapy in adults for the treatment of relapsing forms of MS, to include clinically isolated syndrome, relapsing-remitting disease, and active secondary progressive disease, as well as for the treatment of moderate-to-severe Crohn’s disease with evidence of inflammation, specifically in patients with an inadequate response to conventional therapies and tumor necrosis factor-alpha inhibitors [30, 31].
Natalizumab binds to the α4 subunit of α4β1 (also known as very late antigen 4) and α4β7 integrins expressed on the surface of all leukocytes, except neutrophils [27, 29–31]. It inhibits the α4-integrin-mediated adhesion of leukocytes to their counter-receptor(s) including vascular cell adhesion molecule-1 (VCAM-1), which is expressed on the activated vascular endothelium, and mucosal addressin cell adhesion molecule-1 (MAdCAM-1), present on vascular endothelial cells of the gastrointestinal tract [27, 29–32, 34]. Disruption of the α4β1/VCAM-1 and α4β7/MAdCAM-1 interactions prevents transmigration of leukocytes across the endothelium into inflamed parenchymal tissue, inhibiting further progression of the inflammatory demyelination process in the central nervous system or inflammation of the gastrointestinal tract [27, 29–32, 34].
The development program for biosim-NTZ generated a comprehensive ToE data package comprising extensive comparative analytical and functional characterization [35, 36] alongside four clinical studies, namely: a pilot PK/PD study with EU-approved ref-NTZ (EU-ref-NTZ) in healthy subjects; a supportive safety study with biosim-NTZ in healthy subjects; a pivotal PK/PD study comparing biosim-NTZ to US-licensed ref-NTZ (US-ref-NTZ) and EU-ref-NTZ in healthy subjects; and a pivotal comparative clinical study with biosim-NTZ versus EU-ref-NTZ in patients with RRMS [36].
Biosim-NTZ marks the first biosimilar approved by the FDA and EMA in relapsing forms of MS [28, 37]. This article presents the individual components of the ToE confirming the biosimilarity of biosim-NTZ to ref-NTZ. A graphical plain language summary is available in the Electronic Supplementary Material (ESM).
Comparative Analytical and Functional Characterization
Analytical and functional characterization serves to demonstrate that the active component of a biosimilar medicine is structurally and functionally similar to the active component of the reference medicine, and the biosimilar would thus be expected to behave in the same way as the reference medicine [7, 22, 38]. As noted above, analytical and functional characterization constitutes the foundation and the largest body of evidence of the ToE [22], accordingly represented as the ‘base’ of the biosimilarity development pyramid (Fig. 1).
Methods and assessment criteria of analytical similarity are based on characterization of the reference medicine and its critical quality attributes (CQAs) [39]. QAs are measurable physicochemical characteristics of a biologic medicine that determine its principal properties, and can vary owing to either inherent or manufacturing variability [40]. In biosimilar development, it is therefore fundamental to identify which QAs are CQAs (e.g., oxidation, glycosylation, and binding to Fcγ receptors); that is, have a direct impact on the medicine’s pharmacokinetics, safety, efficacy, and immunogenicity and must be demonstrated to closely match those of the reference medicine, laying within prespecified thresholds [21, 22, 38, 39, 41, 42]. QAs are categorized into very high, high, moderate, low, or very low criticality. All QAs ranked with very high, high, or moderate criticality are considered as CQAs [43].
The biosim-NTZ comparative analytical and functional characterization comprised multiple investigations [35, 36, 44], where the structure and function of biosim-NTZ was compared with that of US-ref-NTZ and EU-ref-NTZ, using quality ranges calculated based on standard deviations from the range of all tested US-ref-NTZ batches (Table 1). The criticality of QAs was assessed based on a risk ranking approach using a preliminary hazards analysis, as per the International Conference on Harmonization (ICH) guidelines (ICH Q9) [36, 45]. Standard deviation multipliers were applied according to risk: 2.5 for ‘high’ risk attributes (those directly related to the mechanism of action), 3 for ‘moderate’ risk attributes (product variants not directly related to the mechanism of action or those attributes directly related to the mechanism of action in which orthogonal methods were included in the comparative analytical assessment), and 4 for ‘low’ risk attributes [35]. For QAs with ‘low’ and ‘very low’ criticality, the data were compared descriptively (a ‘low-risk’ category was also applied for QAs that are not amenable to a statistical evaluation, e.g., if a certain attribute can only be assessed qualitatively) [35].
Table 1.
Overview of the key critical quality attributes assessed as part of the comparative analytical and functional characterization performed in the biosim-NTZ development program [35, 36]
| Characteristic analyzed | Methodology | Outcome: biosim-NTZ vs ref-NTZ |
|---|---|---|
| Amino acid sequence | LC-MS; peptide mapping ultraviolet | Identical amino acid sequence |
| Higher order structures (secondary and tertiary) | Fluorescence emission spectroscopy; Fourier-transform infrared spectroscopy; ultraviolet circular dichroism | Comparable higher order structures |
| Fab-arm exchange | FRET; capillary electrophoresis sodium dodecyl sulphate | Comparable Fab-arm exchange rates |
| Charge variant distribution | CEX | Consistent charge variant distribution, within quality ranges for the acidic species |
| Natalizumab dimers | Size-exclusion chromatography; sedimentation velocity-analytical ultracentrifugation | Comparable levels of natalizumab dimers |
| High-molecular-weight impurities | Size-exclusion chromatography; sedimentation velocity-analytical ultracentrifugation | Comparable |
|
Antibody fragments (low-molecular-weight impurities) |
Capillary electrophoresis sodium dodecyl sulphate; size-exclusion chromatography; sedimentation velocity-analytical ultracentrifugation | Comparable |
| N-glycosylation site occupancy | Capillary electrophoresis sodium dodecyl sulfate; LC-MS | Slightly higher N-glycosylation site occupancy in biosim-NTZ, with differences justified |
| Glycosylation profile | Hydrophilic interaction liquid chromatography | Comparable glycosylation profiles with all major glycosylation peaks present |
| Methionine oxidation | LC-MS | Slightly higher level of methionine oxidation in biosim-NTZ, with differences justified |
| Binding to FcRn | Surface plasmon resonance | Comparable FcRn binding |
| Binding to α4β1 | ELISA; surface plasmon resonance | Comparable α4β1-integrin binding |
| Binding to α4β7 | ELISA; surface plasmon resonance | Comparable α4β7-integrin binding |
| Inhibition of interaction between VCAM-1 and α4β1 |
ELISA; cell-based assay; flow cytometry |
Comparable inhibition of interaction of α4β1-integrin with its cognate receptor VCAM-1 |
| Inhibition of interaction between MAdCAM-1 and α4β7 | ELISA | Comparable inhibition of interaction of α4β7-integrin with its cognate receptor MAdCAM-1 |
Biosim-NTZ biosimilar natalizumab, CEX cation exchange chromatography, ELISA enzyme-linked immunosorbent assay, FcRn neonatal Fc receptor, FRET Förster resonance energy transfer, LC-MS liquid chromatography-mass spectrometry, MAdCAM-1 mucosal vascular addressin cell adhesion molecule 1, ref-NTZ reference natalizumab, VCAM-1 vascular cell adhesion molecule 1
Structural Characterization, Product-Related Variants, and Fab-arm Exchange
The first aspect of the analytical assessment was a comparison of the physicochemical, biophysical, and in vitro functional properties of natalizumab. This included structural characterization, assessment of product-related variants, and studies investigating the Fab-arm exchange under physiologically relevant conditions.
Extensive characterization demonstrated matching outcomes between biosim-NTZ and US-ref-NTZ and EU-ref-NTZ in terms of structural characteristics (Table 1). Peptide mapping followed by liquid chromatography with tandem mass spectrometry confirmed the primary amino acid sequence of biosim-NTZ was identical to US-ref-NTZ and EU-ref-NTZ [35, 36, 44].
The results from Fourier-transform infrared spectroscopy, near and far ultraviolet circular dichroism, fluorescence emission spectroscopy, Förster resonance energy transfer (FRET), and X-ray crystallography demonstrated that the higher order structures of biosim-NTZ, US-ref-NTZ, and EU-ref-NTZ also matched (Table 1) [35, 36, 44]. Comparison of batches of biosim-NTZ and US-ref-NTZ showed that the levels of natalizumab dimers in biosim-NTZ were low and similar to that of US-ref-NTZ, as determined by size-exclusion chromatography [35, 36]. High-molecular-weight impurities were not found in biosim-NTZ or US-ref-NTZ. The purity of biosim-NTZ in terms of the amount of antibody fragments (low-molecular-weight impurities), as determined by capillary electrophoresis under non-reducing and reducing conditions, were within range of US-ref-NTZ [35, 36].
Natalizumab is an immunoglobulin G4 (IgG4) monoclonal antibody that does not mediate Fc-associated activities owing to the low affinity of IgG4 towards Fcγ receptors [46, 47]. Assessments confirmed the lack of Fc-associated effector function for both biosim-NTZ and ref-NTZ (data not shown) [35, 36].
Natalizumab, as an IgG4 monoclonal antibody, is known to undergo a heavy-light chain recombination and therefore a Fab-arm exchange [46]. The Fab-arm exchange takes place when the Fab-arm fragment of a therapeutic antibody exchanges with the Fab-arm fragment of an endogenous antibody, resulting in the formation of a bispecific IgG4 antibody [48]. The kinetics of the Fab-arm exchange between biosim-NTZ and US-ref-NTZ and EU-ref-NTZ was evaluated in real time using Förster resonance energy transfer. The results demonstrated that similar Fab-arm exchange rates were observed for biosim-NTZ, US-ref-NTZ, and EU-ref-NTZ (Supplementary Fig. 1 in the ESM) [35, 36, 44].
Forced Degradation and Stability Studies
The molecules’ performances were then compared under different conditions, including a comparative forced degradation and stability study to ensure biosim-NTZ and ref-NTZ were comparable under long-term (5 ± 3 °C, inverted), accelerated (25 ± 2 °C/60 ± 5% relative humidity, inverted), and stress (40 ± 2 °C/75 ± 5% relative humidity, inverted) conditions through 6 months [35, 36].
Biosim-NTZ, US-ref-NTZ, and EU-ref-NTZ responded in a similar way to applied stress conditions, namely thermal stress (50 °C for 28 days), oxidative stress (5% 2-amidinopropane dihydrochloride at 20 °C for 48 h), light stress (526 W/m2 ultraviolet-A light-hour + 1200 klux-hour visible light), freeze-thaw stress (five cycles of −80 °C for 22 h followed by 20 °C for 2 h), pH stress at acidic pH (pH 4.0 at 30 °C for 21 days), and mechanical stress such as agitation (750 rpm at 20 °C for 24 h) [35, 36, 44].
Comparative stability studies revealed minor differences between biosim-NTZ and ref-NTZ for stress conditions with no expected clinically meaningful impact, while no significant differences were observed under long-term and accelerated storage conditions up to the 6-month timepoint, confirming similarity between biosim-NTZ and both US-ref-NTZ and EU-ref-NTZ [35, 36, 44].
Functional Assessments
The potency of biosim-NTZ was assessed through the detection of any differences in the molecule’s interactions with its known targets and used as a sensitive tool for confirmation of similarity regarding the mechanism of action of biosim-NTZ and ref-NTZ on a functional level [35, 36]. Binding affinity to α4β1 and α4β7 integrins for biosim-NTZ versus US-ref-NTZ and EU-ref-NTZ was assessed via an indirect enzyme-linked immunosorbent assay (ELISA) [Table 1]. The results of the indirect ELISA demonstrated that the binding affinity of biosim-NTZ was within the range of US-ref-NTZ and EU-ref-NTZ [35, 36].
The ability of biosim-NTZ to block the interaction of α4β1 integrin and α4β7 integrin with its cognate receptors VCAM-1 and MAdCAM-1 was also tested by means of a competitive ELISA. Biosim-NTZ, US-ref-NTZ, and EU-ref-NTZ were comparable in terms of blocking the interactions of α4β1 integrin with VCAM-1 and α4β7 integrin with MAdCAM-1 (Fig. 2a, b). The α4β1 integrin and α4β7 integrin indirect ELISA together with the VCAM-1 and MAdCAM-1 competitive ELISAs demonstrated equivalent potency of biosim-NTZ to US-ref-NTZ and EU-ref-NTZ within the predefined quality range [35, 36, 44].
Fig. 2.
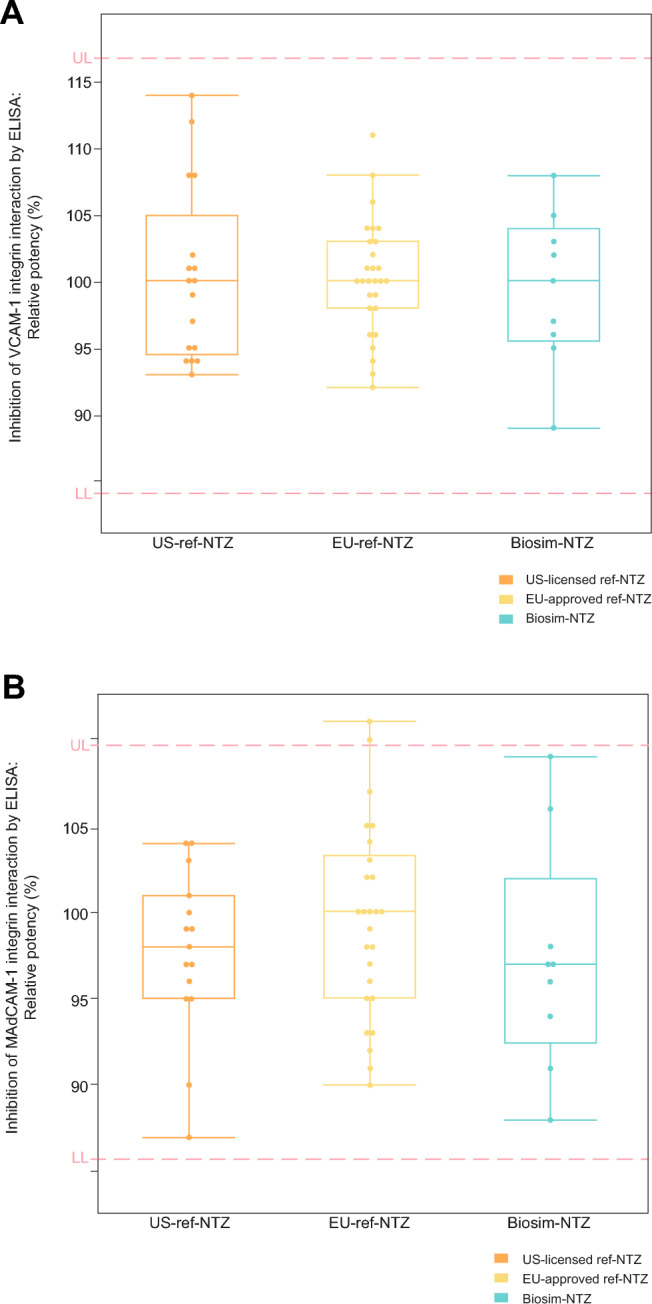
Potency of natalizumab samples by competitive enzyme-linked immunosorbent assay (ELISA). A Inhibition of interaction between vascular cell adhesion molecule 1 (VCAM-1) and α4β1; B Inhibition of interaction between mucosal vascular addressin cell adhesion molecule 1 (MAdCAM-1) and α4β7 integrin. Biosim-NTZ biosimilar natalizumab, ref-NTZ reference natalizumab. The boxes indicate quartiles and the horizontal line in each box represents the median value; the whiskers show the data distribution; the circles represent the individual batches of US-ref-NTZ, EU-ref-NTZ, and biosim-NTZ
Clinical Studies
Data from clinical studies are employed to confirm matching pharmacokinetics/pharmacodynamics, efficacy, safety, and immunogenicity between a proposed biosimilar medicine and its reference medicine [7, 9, 22]. An overview of the clinical studies performed for biosim-NTZ is provided in Table 2.
Table 2.
Overview of clinical studies performed [36]
| Study | Study population | N | Reference origin | Dose | PK | PD | Efficacy | Safety | Immunogenicity |
|---|---|---|---|---|---|---|---|---|---|
| Pilot PB006-01-01 PK/PD studya | Healthy subjects | 36 | EU |
Single 1-mg/kg OR 3-mg/kg OR 6-mg/kg IV infusion of EU-ref-NTZa |
X | X | – | X | – |
| PB006-01-02 safety study | Healthy subjects | 10 | – |
Single 300-mg IV infusion of biosim-NTZb |
– | – | – | X | X |
|
Pivotal PK/PD study EudraCT: 2019-003874-15 |
Healthy subjects | 453 | USA and EU |
Single 3-mg/kg IV infusion of biosim-NTZ OR EU-ref-NTZ OR US ref-NTZ |
X | X | – | X | X |
|
Pivotal Antelope clinical study |
Patients with RRMS | 264 | EU | 300-mg IV infusions of biosim-NTZ OR EU-ref-NTZ, every 4 weeks for a total of 12 infusions | X | – | X | X | X |
Biosim-NTZ, biosimilar natalizumab, EU European Union, IV intravenous, PD pharmacodynamic, PK pharmacokinetic, ref-NTZ reference natalizumab, RRMS relapsing-remitting multiple sclerosis, US USA
X is used to indicate that a particular parameter was measured for the relevant study
aThe pilot PB006-01-01 PK/PD study was conducted with EU-ref-NTZ only
bThe PB006-01-02 safety study was conducted with biosim-NTZ only
Pivotal PK/PD Study
A PK/PD study provides evidential weight that a proposed biosimilar, for which similarity has been confirmed on the analytical and functional level, will behave the same way within the human body [7, 22] (Fig. 1). As a step intended to inform the study design, an initial pilot PK/PD study was conducted using EU-ref-NTZ to confirm the sensitive dose for inclusion in the pivotal PK/PD study (Table 2). Three different doses (1, 3, and 6 mg/kg) of EU-ref-NTZ were assessed in 36 healthy subjects, with the 3-mg/kg dose found to be in the ascending dose–response curve for all PD endpoints, and thus selected for the subsequent pivotal PK/PD study.
Following the identification of the sensitive dose from the pilot study, a randomized, double-blind, three-arm, single-dose, parallel-group, pivotal PK/PD study was conducted in healthy subjects to demonstrate similarity in the pharmacokinetics and pharmacodynamics between biosim-NTZ and ref-NTZ. A total of 453 healthy subjects aged 18–65 years, with a body mass index of 18.5–30.0 kg/m2 received a single dose of 3 mg/kg of biosim-NTZ, US-ref-NTZ, or EU-ref-NTZ in a 1:1:1 ratio, before an 85-day follow-up. The full study design has been reported elsewhere [49].
Biosim-NTZ demonstrated similar serum concentrations for total natalizumab to both US-ref-NTZ and EU-ref-NTZ throughout the entire evaluation period (Fig. 3) [49]. Pharmacokinetic similarity was demonstrated between biosim-NTZ, US-ref-NTZ, and EU-ref-NTZ for all comparisons with the 90% (FDA and EMA) confidence intervals (CIs) for the primary PK endpoint, i.e., area under the curve from time of dosing to infinity within the pre-specified similarity margin of 0.8–1.25 [49].
Fig. 3.
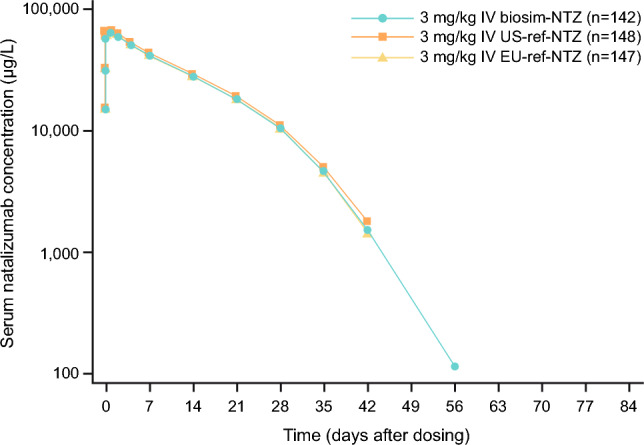
Semi-logarithmic plot of mean total serum concentration over time for biosimilar natalizumab (biosim-NTZ) versus USA (US)-licensed and European Union (EU)-approved reference natalizumab (ref-NTZ) [pharmacokinetic set]. IV intravenous. Wessels et al. [49]. Reprinted by permission of Informa UK Limited, trading as Taylor & Francis Group, https://www.tandfonline.com
Pharmacodynamic similarity was also demonstrated for the primary PD endpoints, namely the area under the effect time curve from time of dosing to 12 weeks for baseline-adjusted CD19+ and α4-integrin percentage receptor saturation were demonstrated, with the 90% CI (FDA) and 95% CI (EMA) residing within the 0.80–1.25 similarity margin (Fig. 4a, b) [49].
Fig. 4.
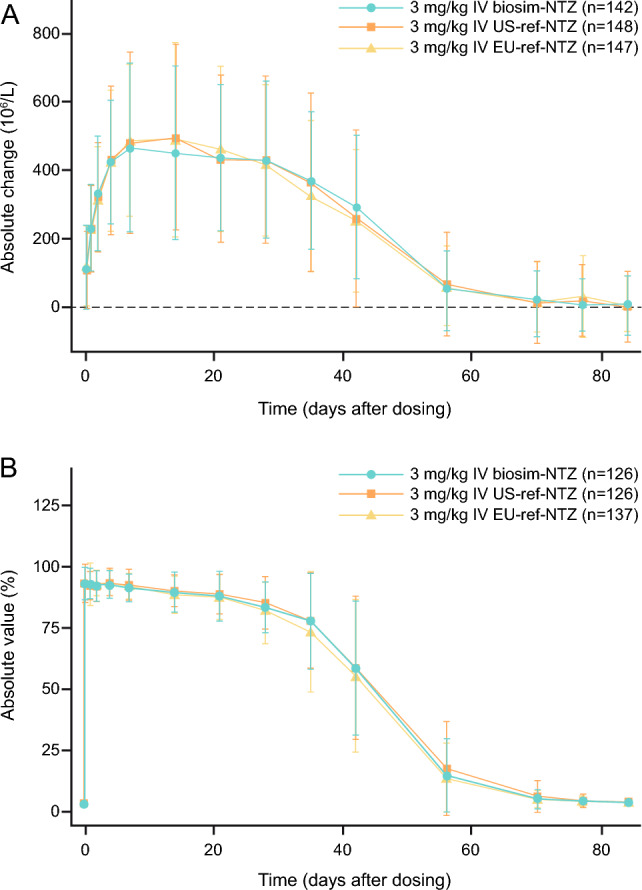
Primary pharmacodynamic endpoints (engagement set). A Change from baseline in mean (SD) CD19+ cell counts over time for biosimilar natalizumab (biosim-NTZ) versus USA (US)-reference natalizumab (ref-NTZ) and European Union (EU)-ref-NTZ. B Change from baseline in mean (SD) α4-integrin percentage receptor saturation (RS) over time for biosim-NTZ versus US-ref-NTZ and EU-ref-NTZ. IV intravenous, PD pharmacodynamic. Wessels et al. [49]. Reprinted by permission of Informa UK Limited, trading as Taylor & Francis Group, https://www.tandfonline.com
Additional PD endpoints further supported PD similarity between biosim-NTZ and ref-NTZ, exhibiting a comparable decrease in mean absolute values for soluble VCAM and MAdCAM across treatment groups [49]. Fab-arm exchange kinetics were also investigated in the pivotal PK/PD study. No difference in Fab-arm exchange was observed between biosim-NTZ and ref-NTZ, corroborating the results from the analytical characterization (Supplementary Tables 1 and 2 of the ESM).
A similar incidence and type of treatment-emergent adverse event (TEAEs) was reported across all treatment groups [49].
Immunogenicity was evaluated throughout the study with highly sensitive and drug-tolerant bioanalytical assays, which were used to assess anti-natalizumab antibodies (anti-drug antibodies [ADAs]) and neutralizing antibodies (NAbs) in the treatment groups (see Chamberlain et al. [Submitted for publication] for further details). These evaluations demonstrated similarity in the incidence and dynamics of treatment-emergent ADAs between biosim-NTZ and US-ref-NTZ and EU-ref-NTZ. In all dosing groups, the majority of subjects tested positive for ADAs at least once during the study period [Chamberlain et al. Submitted for publication]. Furthermore, similarity in the incidence of NAbs and magnitude of the immune response in terms of the ADA titer was shown across all groups. Finally, no treatment-related differences were observed with regard to the impact of ADAs or NAbs on PK or PD measures [49, Chamberlain et al. Submitted for publication]. Overall, the pivotal PK/PD study confirmed PK/PD similarity between biosim-NTZ and both US-ref-NTZ and EU-ref-NTZ in healthy subjects, building upon the foundation of evidence showing matching structure and function of biosim-NTZ compared to ref-NTZ [49].
Pivotal Comparative Antelope Study
In order to demonstrate the biosimilarity of biosim-NTZ to ref-NTZ in a clinical population of interest, a comparative clinical study was conducted in patients with RRMS [50]. Antelope was a pivotal multicenter, double-blind, active-controlled, randomized, parallel-group study conducted between October 2019 and March 2021, in 48 centers across seven countries [50]. The objective of the study was to demonstrate similarity in efficacy, safety, and immunogenicity, and exclude any clinically meaningful differences between biosim-NTZ and ref-NTZ in patients with RRMS [50]. Use of EU-ref-NTZ only in the Antelope study was supported by the results of the comparative analytical and functional characterization and PK/PD study, demonstrating equivalence between US-ref-NTZ and EU-ref-NTZ (see Sects. 2 and 3.1) [49]. Eligible patients, aged 18–60 years, had one or more documented relapse within the previous year and either one or more gadolinium-enhancing T1-weighted or nine or more T2-weighted brain lesions, a Kurtzke Expanded Disability Status Scale (EDSS) score of 0–5.0 (inclusive), and a John Cunningham virus index score of ≤1.5 at screening [50]. The inclusion criteria were less stringent in this study than in the summary of product characteristics (SmPC) of EU-ref-NTZ; however, the eligibility criteria resemble those used in the pivotal phase III studies for the reference medicine. The selected study population was therefore regarded as sufficiently sensitive for the comparative efficacy assessment [36]. Randomized patients received intravenous (IV) infusions of biosim-NTZ or EU-ref-NTZ at a dose of 300 mg every 4 weeks for 12 visits post-baseline visit, in line with the dosing regimen from the SmPC of EU-ref-NTZ (Table 3) [36]. At week 24, a subset of patients was re-randomized from EU-ref-NTZ to biosim-NTZ for assessment of immunogenicity. The full study design has been reported elsewhere [50].
Table 3.
Key characteristics of patients enrolled in the Antelope study
| Characteristic | Biosim-NTZ (n = 131) | EU-ref-NTZ/biosim-NTZ switch (n = 30)a | EU-ref-NTZ (n = 133) |
|---|---|---|---|
| Baseline characteristics | |||
| Age, years, mean (SD) | 36.8 (9.1) | 35.9 (8.29) | 36.6 (9.7) |
| Presence of > 15 T2 lesions, n (%) | 127 (96.9) | 25 (83.30) | 128 (96.2) |
| Positive JCV status (< 1.5), n (%) | 51 (38.9) | 10 (33.30) | 55 (41.4) |
| Baseline EDSS score, mean (SD) | 3.4 (1.1) | 3.1 (1.12) | 3.2 (1.2) |
| Study patients, n | |||
| Randomized | 132 | – | 133 |
| Withdrew consent | 1 | – | – |
| Received treatment | 131 | – | 133 |
| Completed 24-week treatment period | 122 | – | 125 |
| Re-randomized at week 24: | – | – | 125 |
| To remain on ref-NTZ | – | – | 95 |
| To switch to biosim-NTZ | – | 30 | – |
| Completed 48-week treatment period | 117 | 29 | 93 |
Biosim-NTZ biosimilar natalizumab, EDSS Expanded Disability Status Scale, EU European Union, JCV John Cunningham virus, ref-NTZ reference natalizumab, SD standard deviation
aBaseline characteristics for the biosim-NTZ and ref-NTZ group were obtained at week 0. At week 24, baseline characteristics were obtained for the ref-NTZ/biosim-NTZ switch group
Hemmer et al. [50]. Reprinted by permission of JAMA Network
As demonstrated by the study results, no clinically meaningful differences in efficacy were observed between patients treated with biosim-NTZ and EU-ref-NTZ [50]. For the primary efficacy endpoint of the cumulative number of new active lesions, biosim-NTZ demonstrated similarity to EU-ref-NTZ at week 24 (Fig. 5) in the per-protocol population, with 95% CI (−0.61 to 0.94) within the pre-specified margin of ± 2.1 lesions [50]. The margin was derived to preserve at least 50% of the effect size (50% of the lower 95% CI of the treatment effect), based on the most suitable available publication (Miller et al.) that provided data on placebo and ref-NTZ lesions in patients with RRMS [51]. The primary endpoint results were confirmed in the sensitivity analysis in the full population analysis set [50]. All secondary magnetic resonance imaging endpoints further confirmed the similarity between biosim-NTZ and EU-ref-NTZ over 48 weeks [50]. The secondary clinical endpoints, namely annualized relapse rate and mean change from baseline in EDSS score, were similar between all treatment groups in the full analysis set population over 48 weeks [50]. Switching treatment from EU-ref-NTZ to biosim-NTZ at 24 weeks was also not associated with any impact on clinical efficacy over the subsequent 24 weeks [50].
Fig. 5.
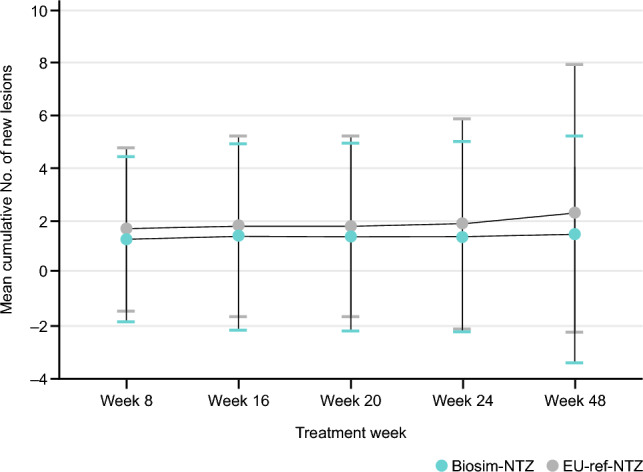
Cumulative number of new active lesions over 48 weeks for biosimilar natalizumab (biosim-NTZ) versus reference natalizumab (ref-NTZ) groups by primary randomization in the Antelope study. Hemmer et al. [50]. Reprinted by permission of JAMA Network
No clinically meaningful difference in the safety profile of biosim-NTZ and EU-ref-NTZ was observed, with a similar incidence of TEAEs across all treatment groups (Supplementary Table 3 of the ESM) [50]. Among all treatment groups, the most reported TEAEs were infections and infestations (biosim-NTZ, n = 39 [29.8%]; EU-ref-NTZ, n = 34 [33.0%]; EU-ref-NTZ/biosim-NTZ switch, n = 15 [50.0%]) and nervous system disorders (biosim-NTZ, n = 33 [25.2%]; EU-ref-NTZ, n = 24 [23.3%]; EU-ref-NTZ/biosim-NTZ switch, n = 8 [26.7%]) [50]. Few adverse events of special interest were reported, with similar proportions across all treatment groups up to week 48 [50]. No fatal or Grade 4 TEAEs under the Common Terminology Criteria for Adverse Events were reported; the majority of reported TEAEs were Grade 1 (Supplementary Table 3 of the ESM) [50]. Eight patients in the biosim-NTZ, three patients in the EU-ref-NTZ group, and one patient in the EU-ref-NTZ/biosim-NTZ switch group experienced TEAEs that led to discontinuation, all except one were at least possibly related to the study drug (one patient in the biosim-NTZ group discontinued because of coronavirus disease 2019 infection) [Supplementary Table 3 of the ESM] [50]. The most common TEAEs leading to discontinuation were pruritus and urticaria, which are known common adverse drug reactions to natalizumab [27, 29–31, 50]. As patients with MS who are treated with natalizumab have a tendency to show an increased white blood cell count and higher lymphocyte count, white blood cell and lymphocyte levels were evaluated during the Antelope study [52]. One case of increased white blood cell count and increased lymphocyte count was reported in the ref-NTZ group and no cases were reported in patients treated with biosim-NTZ, demonstrating a comparable physiological effect between the two treatment arms. No progressive multifocal leukoencephalopathy cases occurred, neither during the treatment period (48 weeks) nor in the progressive multifocal leukoencephalopathy follow-up visit (24 weeks ± 2 weeks after the last study drug infusion) [50].
The immunogenicity profile of biosim-NTZ, including ADA/NAb responses and scale of the impact on relevant clinical parameters, was indistinguishable from that of EU-ref-NTZ during administration to patients with RRMS over 48 weeks, as assessed via the validated high-sensitivity ADA assay [Chamberlain et al. Submitted for publication]. The incidence of ADAs and NAbs at the 24-week and 48-week treatment timepoints was similar for both groups, within the range of 74–79% and 67–69%, respectively, at week 48 (Supplementary Table 3 of the ESM). Additionally, immunogenicity was not impacted during the study period by treatment switching at week 24 and was therefore not associated with an enhanced anti-natalizumab humoral immune response (i.e., ADAs and NAbs) in any of the 30 patients [50].
Building upon the matching analytical and functional characterization and together with confirmation of PK/PD similarity, results of the Antelope study confirmed no clinically meaningful differences in the efficacy, safety, and immunogenicity in patients with RRMS treated with biosim-NTZ or EU-ref-NTZ. Furthermore, no differences were observed in patients who switched from EU-ref-NTZ to biosim-NTZ treatment during the Antelope study for any of the endpoints investigated [50].
Discussion
With biosim-NTZ marking the approval of the first biosimilar medicine in neurology, it is important that the ‘totality of evidence’ concept and its corresponding data are well understood by treating physicians and patients, to aid understanding of how biosimilar medicines are developed and approved (Fig. 6) [7, 9, 28].
Fig. 6.

Sequential demonstration of biosimilar natalizumab (biosim-NTZ) biosimilarity to reference natalizumab (ref-NTZ) via analytical/functional and clinical data. ARR annualized relapse rate, AUC0-inf area under the curve from time of dosing to infinity, CUAL cumulative number of active lesions, EDSS Expanded Disability Status Scale, MAdCAM-1 mucosal vascular addressin cell adhesion molecule 1, PD pharmacodynamic, PK pharmacokinetic, VCAM-1 vascular cell adhesion molecule 1
The clinical development program of biosim-NTZ was extensive. Analytical and functional characterization using state-of-the-art techniques revealed matching physicochemical and functional profiles of biosim-NTZ to ref-NTZ [35, 36]. Both target binding and its immediate pharmacological effect (binding to VCAM-1 and MAdCAM-1) were analyzed in functional assays as well as in healthy subjects [35, 36, 49]. The analytical data on α4ß1 and α4ß7 binding, as well as the functional data on blocking the interactions with the respective blockade of the interaction with the respective ligands VCAM-1 and MAdCAM-1, using highly sensitive assays, were similar [35, 36]. In addition, a clinical study was conducted in patients with RRMS confirming similar efficacy of biosim-NTZ and its reference medicine [50]. These results, together with the profound knowledge of the mechanism of action of natalizumab, resulted in no residual uncertainty regarding the similar efficacy of biosim-NTZ and its reference medicine, following the completion of the analytical/functional comparability exercise.
Furthermore, the FDA and EMA require stringent head-to-head clinical immunogenicity testing of a biosimilar to its reference medicine within a biosimilar development program, using highly sensitive assays [7, 22]. Persistent antibody positivity has been described as relevant during natalizumab treatment owing to its association with reduced treatment effectiveness and increased infusion-related reactions, including hypersensitivity reactions [53–56]. No clinically meaningful differences were detected in the immunogenicity profile of biosim-NTZ compared to US-/EU-ref-NTZ in either the pivotal PK/PD or Antelope studies [49, 50]. Both biosim-NTZ and ref-NTZ reported higher incidences of ADAs in the current studies than reported in clinical studies of ref-NTZ, which can be attributed to the higher sensitivity of this biosimilar development assay used compared with those previously utilized in studies of ref-NTZ (sensitivity of 3.88 ng/mL for the screening assay vs 500 ng/mL), as discussed in Chamberlain et al. [Chamberlain et al. Submitted for publication] [30, 57, 58]. In future developments of biosimilar candidates with similar characteristics as biosim-NTZ, the experience from the biosim-NTZ development program could lead to waiving the efficacy/safety study as laid out by the EMA and World Health Organization [24, 25], facilitating biosimilar development and ultimately increasing access to biosimilars.
Conclusions
The ToE for biosimilar natalizumab presented herein supported its regulatory approval by demonstrating the analytical, functional, and clinical similarity, in terms of efficacy, safety, and immunogenicity, to its reference medicine. Because of the demonstrated similarity, all data collected for ref-NTZ, namely the clinical efficacy, safety, and risk-benefit profile, should equally apply to biosim-NTZ, signifying that patients and prescribers can expect the same treatment effect. Approval of biosimilar natalizumab by the FDA and EMA marks the first biosimilar medicine for the treatment of relapsing forms of MS in the USA and EU [28, 37], representing a potential shift to more affordable and uniform access to high-efficacy biologic treatment in MS.
Supplementary Information
Below is the link to the electronic supplementary material.
Below is the link to the electronic supplementary material.
Acknowledgments
Medical writing assistance during the preparation of this article was provided by Costanza Martelli and Joanne Smith (Syneos Health), and was supported financially by Hexal AG (a Sandoz company).
Declarations
Funding
The investigations and studies included herein were funded/sponsored by Polpharma Biologics S.A. The open access fee for this paper was sponsored by Hexal AG (a Sandoz company). The sponsor (Polpharma Biologics S.A.) had a role in the design and conduct of the studies presented herein. The sponsor and authors had a role in the interpretation of the data and statistical analysis, preparation, review, approval of the manuscript, and decision to submit the manuscript for publication.
Conflicts of Interest/Competing Interests
Krzysztof Selmaj, Karsten Roth, Josef Höfler, Klaus Vitzithum, Rafał Derlacz, Oliver von Richter, Cyrill Hornuss, Johann Poetzl, Barry Singer, and Laura Jacobs. Krzysztof Selmaj has received honoraria for speaking, consulting, and serving on advisory boards for Merck, Novartis, Roche, Biogen, Celgene, and TG Therapeutics. Karsten Roth has received personal compensation for serving as an employee of Polpharma Biologics S.A. Klaus Vitzithum has received personal compensation for serving as an employee of Polpharma Biologics S.A. Josef Höfler has received personal compensation for serving as an employee of Staburo GmbH. Rafał Derlacz has received personal compensation for serving as an employee of Polpharma Biologics S.A. Oliver von Richter has received personal compensation for serving as an employee of Hexal AG (a Sandoz company). Cyrill Hornuss has received personal compensation for serving as an employee of Hexal AG (a Sandoz company). Johann Poetzl has received personal compensation for serving as an employee of Hexal AG (a Sandoz company). Laura Jacobs has received personal compensation for serving as an employee of Hexal AG (a Sandoz company). Barry Singer has received research grant support from AbbVie, Biogen, Bristol Myers Squibb, Greenwich Biosciences, Novartis, Sanofi, and TG Therapeutics, and consulting and/or speaking fees from Alexion, Biogen, Bristol Myers Squibb, Cigna, Cycle, EMD Serono, Genentech, Horizon, Janssen, Novartis, Octave Bioscience, Roche, Sanofi, Sandoz, and TG Therapeutics.
Ethics Approval
All procedures performed in studies involving human participants were in accordance with the International Council for Harmonization Good Clinical Practice, the Declaration of Helsinki, and relevant ethics committee or regulatory agency procedures.
Consent to Participate
Patients or their legally authorized representatives signed statements of informed consent before enrollment and reconsented during the trials.
Consent for Publication
Not applicable.
Availability of Data and Material
The data sets generated and/or analyzed during the analytical and functional characterization study are available from the corresponding author upon reasonable request. Data sharing for the PK/PD and Antelope studies is not applicable to this article as no data sets were generated or analyzed during the current review.
Code Availability
Not applicable.
Authors’ Contributions
All authors were in compliance with the ICMJE criteria. All authors contributed to the concept and design of the studies and the acquisition, analysis, or interpretation of the data. JH performed the statistical analysis of the data. All authors critically reviewed and approved the final version of the manuscript.
References
- 1.Ghasemi N, Razavi S, Nikzad E. Multiple sclerosis: pathogenesis, symptoms, diagnoses and cell-based therapy. Cell J. 2017;19:1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.McGinley MP, Goldschmidt CH, Rae-Grant AD. Diagnosis and treatment of multiple sclerosis: a review. JAMA. 2021;325:765–79. [DOI] [PubMed] [Google Scholar]
- 3.Cunill V, Massot M, Clemente A, et al. Relapsing-remitting multiple sclerosis is characterized by a T follicular cell pro-inflammatory shift, reverted by dimethyl fumarate treatment. Front Immunol. 2018;9:1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Brownlee WJ, Wolf C, Hartung HP, et al. Use of follow-on disease-modifying treatments for multiple sclerosis: consensus recommendations. Mult Scler. 2022;28:2177–89. [DOI] [PubMed] [Google Scholar]
- 5.Voge NV, Alvarez E. Monoclonal antibodies in multiple sclerosis: present and future. Biomedicines. 2019;7:20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.McCamish M, Yoon W, McKay J. Biosimilars: biologics that meet patients’ needs and healthcare economics. Am J Manag Care. 2016;22:S439–42. [PubMed] [Google Scholar]
- 7.EMA. Biosimilars in the EU: information guide for healthcare professionals. 2019. Available from: https://www.ema.europa.eu/en/documents/leaflet/biosimilars-eu-information-guide-healthcare-professionals_en.pdf. Accessed 3 Mar 2023.
- 8.US FDA. Biosimilar and interchangeable biologics: more treatment choices. 2023. Available from: https://www.fda.gov/consumers/consumer-updates/biosimilar-and-interchangeable-biologics-more-treatment-choices. Accessed 3 Oct 2023.
- 9.US FDA. Biosimilar development process. 2023. Available from: https://www.fda.gov/files/drugs/published/Biosimilar-Development-Process.pdf. Accessed 2 Oct 2023.
- 10.Herndon TM, Ausin C, Brahme NN, et al. Safety outcomes when switching between biosimilars and reference biologics: a systematic review and meta-analysis. PLoS ONE. 2023;18: e0292231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mysler E, Azevedo VF, Danese S, et al. Biosimilar-to-biosimilar switching: what is the rationale and current experience? Drugs. 2021;81:1859–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cohen HP, Hachaichi S, Bodenmueller W, Kvien TK, Danese S, Blauvelt A. Switching from one biosimilar to another biosimilar of the same reference biologic: a systematic review of studies. BioDrugs. 2022;36:625–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Allocati E, Godman B, Gobbi M, Garattini S, Banzi R. Switching among biosimilars: a review of clinical evidence. Front Pharmacol. 2022;13:1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gascón P, Tesch H, Verpoort K, et al. Clinical experience with Zarzio® in Europe: what have we learned? Support Care Cancer. 2013;21:2925–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Greenberg B, Giovannoni G. A place for biosimilars in the changing multiple sclerosis treatment landscape. Mult Scler Relat Disord. 2023;77:1–13. [DOI] [PubMed] [Google Scholar]
- 16.Müskens WD, Van Dartel SAAR, Riel PLCMV, Adang EMM. Does etanercept biosimilar prescription in a rheumatology center bend the medication cost curve? J Rheumatol. 2021;48:1803–9. [DOI] [PubMed] [Google Scholar]
- 17.Bhat S, Altajar S, Shankar D, et al. Process and clinical outcomes of a biosimilar adoption program with infliximab-Dyyb. J Manag Care Spec Pharm. 2020;26:410–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tinsley SM, Grande C, Olson K, Plato L, Jacobs I. Potential of biosimilars to increase access to biologics: considerations for advanced practice providers in oncology. J Adv Pract Oncol. 2018;9:699–716. [PMC free article] [PubMed] [Google Scholar]
- 19.Rivera VM. Biosimilar drugs for multiple sclerosis: an unmet international need or a regulatory risk? Neurol Ther. 2019;8:177–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.IMS. Delivering on the potential of biosimilar medicines. 2016. Available from: https://www.medicinesforeurope.com/wp-content/uploads/2016/03/IMS-Institute-Biosimilar-Report-March-2016-FINAL.pdf. Accessed 3 Mar 2023.
- 21.Markus R, Liu J, Ramchandani M, Landa D, Born T, Kaur P. Developing the totality of evidence for biosimilars: regulatory considerations and building confidence for the healthcare community. BioDrugs. 2017;31:175–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.US FDA. Scientific considerations in demonstrating biosimilarity to a reference product. 2015. Available from: https://www.fda.gov/media/82647/download. Accessed 3 Mar 2023.
- 23.Holzmann J, Balser S, Windisch J. Totality of the evidence at work: the first U.S. biosimilar. Expert Opin Biol Ther. 2016;16:137–42. [DOI] [PubMed] [Google Scholar]
- 24.WHO. Guidelines on evaluation of biosimilars. 2022. Available from: https://cdn.who.int/media/docs/default-source/biologicals/annex-3---who-guidelines-on-evaluation-of-biosimilars---sj-ik-5-may-2022.pdf?sfvrsn=9b2fa6d2_1&download=true. Accessed 10 May 2024.
- 25.EMA. Concept paper for the development of a reflection paper on a tailored clinical approach in biosimilar development. November 24, 2023. Available from: https://www.ema.europa.eu/en/documents/other/concept-paper-development-reflection-paper-tailored-clinical-approach-biosimilar-development_en.pdf. Accessed 10 May 2024.
- 26.Kurki P, Kang HN, Ekman N, Knezevic I, Weise M, Wolff-Holz E. Regulatory evaluation of biosimilars: refinement of principles based on the scientific evidence and clinical experience. BioDrugs. 2022;36:359–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sandoz GmbH. Tyruko® (natalizumab): summary of product characteristics. 2023. Available from: https://www.ema.europa.eu/documents/product-information/tyruko-epar-product-information_en.pdf. Accessed 2 Oct 2023.
- 28.US FDA. FDA approves first biosimilar to treat multiple sclerosis. 2023. Available from: https://www.fda.gov/news-events/press-announcements/fda-approves-first-biosimilar-treat-multiple-sclerosis. Accessed 30 Aug 2023.
- 29.Biogen Netherlands B.V. Tysabri® (natalizumab): summary of product characteristics. 2022. Available from: https://www.ema.europa.eu/documents/product-information/tysabri-epar-product-information_en.pdf. Accessed 6 Apr 2023.
- 30.Biogen Inc. Tysabri® (natalizumab): prescribing information. December 2023. Available from: https://www.tysabri.com/content/dam/commercial/tysabri/pat/en_us/pdf/tysabri_prescribing_information.pdf. Accessed 2 Oct 2023.
- 31.Sandoz Inc. Tyruko® (natalizumab-sztn): prescribing information. 2023. Available from: https://www.accessdata.fda.gov/drugsatfda_docs/label/2023/761322s000lbl.pdf. Accessed 27 Oct 2023.
- 32.Rudick RA, Sandrock A. Natalizumab: α4-integrin antagonist selective adhesion molecule inhibitors for MS. Expert Rev Neurother. 2004;4:571–80. [DOI] [PubMed] [Google Scholar]
- 33.Morrow SA, Clift F, Devonshire V, et al. Use of natalizumab in persons with multiple sclerosis: 2022 update. Mult Scler Relat Disord. 2022;65: 103995. [DOI] [PubMed] [Google Scholar]
- 34.Guagnozzi D, Caprilli R. Natalizumab in the treatment of Crohn’s disease. Biologics. 2008;2:275–84. [PMC free article] [PubMed] [Google Scholar]
- 35.US FDA. Tyruko product quality review. 2023. Available from: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2023/761322Orig1s000ChemR.pdf. Accessed 9 May 2024.
- 36.EMA. Tyruko assessment report. July 20, 2023. Available from: https://www.ema.europa.eu/en/documents/assessment-report/tyruko-epar-public-assessment-report_en.pdf. Accessed 9 May 2024.
- 37.GaBI. EC approval of natalizumab, aflibercept and tocilizumab biosimilars. November 2023. Available from: https://gabionline.net/biosimilars/news/ec-approval-of-natalizumab-aflibercept-andtocilizumab-biosimilars. Accessed 31 Jan 2024.
- 38.EMA. Guideline on similar biological medicinal products containing biotechnology-derived proteins as active substance: non-clinical and clinical issues. 2014. Available from: https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-similar-biological-medicinal-products-containing-biotechnology-derived-proteins-active_en-2.pdf. Accessed 3 Mar 2023.
- 39.Vulto AG, Jaquez OA. The process defines the product: what really matters in biosimilar design and production? Rheumatology. 2017;56:iv14-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mascarenhas-Melo F, Diaz M, Gonçalves MBS, et al. An overview of biosimilars: development, quality, regulatory issues, and management in healthcare. Pharmaceuticals. 2024;17:1–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Guillen E, Ekman N, Barry S, Weise M, Wolff-Holz E. A data driven approach to support tailored clinical programs for biosimilar monoclonal antibodies. Clin Pharmacol Ther. 2023;113:108–23. [DOI] [PubMed] [Google Scholar]
- 42.Markus R, McBride HJ, Ramchandani M, et al. A review of the totality of evidence supporting the development of the first adalimumab biosimilar ABP 501. Adv Ther. 2019;36:1833–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Vandekerckhove K, Seidl A, Gutka H, et al. Rational selection, criticality assessment, and tiering of quality attributes and test methods for analytical similarity evaluation of biosimilars. AAPS J. 2018;20:1–9. [DOI] [PubMed] [Google Scholar]
- 44.Selmaj K, Roth K, Höfler J, et al. The totality of evidence for proposed biosimilar natalizumab PB006 confirms biosimilarity to its reference medicine. In: Poster DMT62. Consortium of Multiple Sclerosis Centers; 2023. Accessed 3 may 2023.
- 45.EMA. ICH guideline Q9 on quality risk management. September 2015. Available from: https://www.ema.europa.eu/en/documents/scientific-guideline/international-conference-harmonisation-technical-requirements-registration-pharmaceuticals-human-use-ich-guideline-q9-quality-risk-management-step-5-first-version_en.pdf. Accessed 14 May 2024.
- 46.Labrijn AF, Buijsse AO, van den Bremer ETJ, et al. Therapeutic IgG4 antibodies engage in Fab-arm exchange with endogenous human IgG4 in vivo. Nat Biotechnol. 2009;27:767–71. [DOI] [PubMed] [Google Scholar]
- 47.Vidarsson G, Dekkers G, Rispens T. IgG subclasses and allotypes: from structure to effector functions. Front Immunol. 2014;5:520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.van der Neut KM, Schuurman J, Losen M, et al. Anti-inflammatory activity of human IgG4 antibodies by dynamic Fab arm exchange. Science. 1979;2007(317):1554–7. [DOI] [PubMed] [Google Scholar]
- 49.Wessels H, von Richter O, Velinova M, et al. Pharmacokinetic and pharmacodynamic similarity of biosimilar natalizumab (PB006) to its reference medicine: a randomized controlled trial. Expert Opin Biol Ther. 2023;23:1287–97. [DOI] [PubMed] [Google Scholar]
- 50.Hemmer B, Wiendl H, Roth K, et al. Efficacy and safety of proposed biosimilar natalizumab (PB006) in patients with relapsing-remitting multiple sclerosis: the Antelope phase 3 randomized clinical trial. JAMA Neurol. 2023;80:298–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Miller DH, Khan OA, Sheremata WA, et al. A controlled trial of natalizumab for relapsing multiple sclerosis. N Engl J Med. 2003;348:15–23. [DOI] [PubMed] [Google Scholar]
- 52.Akaishi T, Misu T, Fujihara K, et al. White blood cell count profiles in multiple sclerosis during attacks before the initiation of acute and chronic treatments. Sci Rep. 2021;11:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Vennegoor A, Rispens T, Strijbis EM, et al. Clinical relevance of serum natalizumab concentration and anti-natalizumab antibodies in multiple sclerosis. Mult Scler. 2013;19:593–600. [DOI] [PubMed] [Google Scholar]
- 54.Calabresi PA, Giovannoni G, Confavreux C, et al. The incidence and significance of anti-natalizumab antibodies: results from AFFIRM and SENTINEL. Neurology. 2007;69:1391–403. [DOI] [PubMed] [Google Scholar]
- 55.Subramanyam M. Case study: immunogenicity of natalizumab. In: . Immunogenicity of biopharmaceuticals. Ed: Weert Mv and Møller EH. New York (NY): Springer; 2008, p. 173–87.
- 56.Ciano-Petersen NL, Aliaga-Gaspar P, Hurtado-Guerrero I, et al. Natalizumab-immunogenicity evaluation in patients with infusion related events or disease exacerbations. Front Immunol. 2023;14:1242508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Polman CH, O’Connor PW, Havrdova E, et al. A randomized, placebo-controlled trial of natalizumab for relapsing multiple sclerosis. N Engl J Med. 2006;354:899–910. [DOI] [PubMed] [Google Scholar]
- 58.CHMP. Tysabri assessment report. 2021. Available from: https://www.ema.europa.eu/en/documents/variation-report/tysabri-h-c-603-x-0116-epar-assessment-report-extension_en.pdf. Accessed 3 Oct 2023.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.