

Abstract
Background
Diabetic cardiomyopathy (DCM) is a serious complication in patients with type 2 diabetes mellitus, and its mechanisms are complex and poorly understood. Despite growing evidence suggesting that ferroptosis plays a significant role in cardiovascular disease, it has been less extensively studied in DCM. Fibroblast growth factor 21 (FGF21), whose mechanism of action is closely related to ferroptosis, is widely utilized in studies focused on the prevention and treatment of glucolipid metabolism-related diseases and cardiovascular diseases.
Objective
To confirm the significant role of ferroptosis in DCM and to investigate whether FGF21 improves DCM by inhibiting ferroptosis and elucidating its specific molecular mechanisms.
Methods
The animal DCM models were established through high-fat feeding combined with streptozotocin injection in C57BL/6J mice or by db/db mice, and the diabetic cardiomyocyte injury model was created using high glucose and high fat (HG/HF) culture of primary cardiomyocytes. Intervention modeling of FGF21 were performed by injecting adeno-associated virus 9-FGF21 in mice and transfecting FGF21 siRNA or overexpression plasmid in primary cardiomyocytes.
Results
The findings indicated that ferroptosis was exacerbated and played a significant role in DCM. The overexpression of FGF21 inhibited ferroptosis and improved cardiac injury and function, whereas the knockdown of FGF21 aggravated ferroptosis and cardiac injury and function in DCM. Furthermore, we discovered that FGF21 inhibited ferroptosis in DCM by directly acting on ferritin and prolonging its half-life. Specifically, FGF21 binded to the heavy and light chains of ferritin, thereby reducing its excessive degradation in the proteasome and lysosomal-autophagy pathways in DCM. Additionally, activating transcription factor 4 (ATF4) served as the upstream regulator of FGF21 in DCM.
Conclusions
The ATF4-FGF21-ferritin axis mediates the protective effects in DCM through the ferroptosis pathway and represents a potential therapeutic target for DCM.
Supplementary Information
The online version contains supplementary material available at 10.1186/s12933-024-02469-8.
Keywords: Diabetic cardiomyopathy, Ferroptosis, FGF21, Ferritin, ATF4, Type 2 diabetes mellitus
Introduction
The global prevalence of diabetes mellitus (DM) is escalating, with the International Diabetes Federation estimating a staggering diabetic population of approximately 529 million in 2021 and predicting that 1.31 billion individuals will have DM by 2050 [1]. Among individuals with DM, cardiovascular complications represent the leading cause of mortality [2]. Diabetic cardiomyopathy (DCM) is a serious and poor prognostic complication of DM [3], characterized by associated vasculopathy, myocardial injury, myocardial fibrosis, and cardiac diastolic and systolic dysfunction, which are significant contributors to heart failure, malignant arrhythmias, and sudden death in diabetic patients [4]. In type 2 diabetes mellitus (T2DM), the accumulation of glucose metabolites and lipids induces cardiac glycotoxicity and lipotoxicity while increasing oxidative stress through excess reactive oxygen species (ROS). Since these factors are critical pathomechanisms of DCM, the inhibition of harmful lipid metabolites and their peroxidation is essential for preventing cardiomyocyte death and protecting cardiac function [4].
Ferroptosis arises from imbalances in cellular metabolism and redox homeostasis, and it is implicated in various disease-related signaling pathways [5, 6]. Excess ROS and polyunsaturated fatty acids are necessary for ferroptosis, triggering unrestricted lipid peroxidation in the presence of excess Fe2+, which leads to the production of phospholipid hydroperoxides—the executors of ferroptosis—thereby causing cell death [7]. While current studies suggest that ferroptosis plays a role in DM and related complications, the existing research is inadequate, and many aspects remain to be clarified.
Fibroblast growth factor 21 (FGF21) is a secreted protein belonging to the fibroblast growth factor family and plays a crucial role in regulating body metabolism across various organs and tissues. Recent studies have underscored the potential of FGF21 as an effective target for the prevention and treatment of metabolism-related diseases, including hepatic lipid and glucolipid metabolism, as well as cardiovascular diseases [8, 9]. The multifaceted impact of FGF21 positions it as a promising avenue for addressing a wide range of metabolic and cardiovascular challenges.
Given the high degree of consistency among the pathophysiologic changes in DCM and ferroptosis, as well as the mechanism of action of FGF21, this prompted us to consider whether ferroptosis plays a role in DCM and whether FGF21 could ameliorate DCM by inhibiting the ferroptosis pathway. In this study, we modeled DCM and utilized mice injected with adeno-associated virus serotype 9 (AAV9)-FGF21, as well as primary cardiomyocytes transfected with FGF21 overexpression (OE) plasmids or siRNA (SI), to explore these speculations. Furthermore, our study illuminated the specific molecular mechanisms through which FGF21 exerted its effects.
Methods
Mice
Adult male wild-type (WT) C57BL/6J mice (8 weeks old) from Beijing Viton Lever Laboratory Animal Technology Co., Ltd. (Charles River Laboratories, Beijing, China) were utilized, and adult male WT C57BL/6J mice (9 weeks old) and db/db mice with C57BLKS/J as background (9 weeks old) from Cyagen Biosciences (Suzhou) Inc. were utilized. Mice were housed in a facility with a 12-hour light/12-hour dark cycle, maintained at 23 ± 3 °C, and with a humidity range of 30–70%. All experiments adhered to the Guidelines for the Care and Use of Laboratory Animals at Jinan University and received approval from the Ethics Committee for Animal Experiments at Jinan University School of Medicine (IACUC-20220512-06 and IACUC-20231214-08).
Mouse DCM model and treatment
For T2DM mice derived from WT mice, following one week of acclimatization, the mice were fed a high-fat diet (HFD) comprising 60% fat, 20% carbohydrate, and 20% protein for a duration of 12 weeks. Subsequently, low-dose streptozotocin (STZ) (Sigma-Aldrich, St. Louis, MO, USA) injections (50 mg/kg body weight, intraperitoneally) were administered for 5 consecutive days to induce the T2DM model, and fasting blood glucose was tested after 2 weeks. For db/db mice, mice were fed with standard chow (STC) comprising 10% fat, 70% carbohydrate, and 20% protein. Mice exhibiting diabetic fasting blood glucose levels ≥ 11.1 mM for two consecutive days were categorized as T2DM mice [10]. Upon confirmation of successful modeling, diabetic mice were stratified into two groups: one receiving injections of an AAV9 viral vector carrying green fluorescent protein (GFP, AAV9-GFP) (Wz Biosciences Inc, Shandong, China) in the tail vein as a model control group, and the other receiving an AAV9 viral vector carrying FGF21 (AAV9-FGF21) as an intervention model group. Mice of the same age and sex maintained on a standard diet comprising 10% fat, 70% carbohydrate, and 20% protein were also divided into two groups as control groups and injected with AAV9-GFP or AAV9-FGF21. All mice were euthanized 12 weeks post-virus injection. Mice were anesthetized with Avertin, and intraperitoneal venous blood was drawn. Subsequently, press down on the back of the mice with left hand, clamp the right armpit of the mice with thumb, clamp the left forelimb with index and middle fingers, and use scissors to cut the head of the mice vertically at the neck with right hand, causing the mice to die due to marrowbrain rupture and massive bleeding, and then their hearts were removed.
Transmission electron microscopy (TEM)
Cardiac samples (1 mm × 1 mm × 1 mm) were immersed in electron microscope fixative. Following post-fixation, embedding, cutting, and mounting at the electron microscope core facility (Servicebio, Wuhan, Hubei, China), ultrathin sections were photographed using a Hitachi H-7800 transmission electron microscope (Hitachi, Tokyo, Japan).
Echocardiography
Mice were anesthetized via respiratory inhalation of isoflurane (Ruipu, Tianjin, China) and subjected to M-mode echocardiography using a Vevo2100/3100 imaging system (VisualSonics, Toronto, Canada). M-mode tracings were acquired from the left ventricle via a short-axis view at mid-nipple, allowing for assessments of ejection fraction and fractional shortening. Cardiac diastolic function was measured in apical four-chamber cardiac views and evaluated by the early to late diastolic transmitral flow velocity (E/A). Each value was averaged across at least three consecutive cardiac cycles.
Hematoxylin-Eosin (HE) staining
Standard HE staining was performed using an HE staining kit (G1005, Servicebio, Wuhan, China) according to the manufacturer’s protocol. Specifically, cardiac paraffin sections were dewaxed to water, subjected to hematoxylin staining and eosin staining sequentially, and finally dehydrated and sealed. Imaging was performed using TissueGnostics Strata FAXS P-S (TissueGnostics, Austria).
Masson staining
Standard Masson staining was performed using a Masson’s trichrome staining kit (G1006, Servicebio, Wuhan, China) according to the manufacturer’s protocol. Specifically, cardiac paraffin sections were dewaxed to water, placed in potassium dichromate overnight, and sequentially stained with hematoxylin, lachrymose red acid magenta, phosphomolybdic acid, and aniline blue, and finally dehydrated and sealed. Imaging was performed using TissueGnostics Strata FAXS P-S.
Prussian blue and diaminobenzidine staining
Tissue sections underwent sequential treatments in xylene and ethanol, followed by water washes. They were stained with a mixture of potassium ferrous hydride and hydrochloric acid, then with a diaminobenzidine solution, monitoring color development under a microscope. After rinsing, sections were counterstained with hematoxylin, differentiated, and returned to blue. Following dehydration and clearing in xylene, sections were mounted. Imaging was performed using TissueGnostics Strata FAXS P-S.
Immunohistochemical staining
Tissues were fixed in cold 4% paraformaldehyde and embedded in paraffin blocks. Following deparaffinization, dehydration, antigen repair, and sealing, tissue sections were incubated with antibodies at 4 °C overnight. Subsequently, all tissue sections underwent incubation with biotinylated goat anti-rabbit IgG for 20 min at room temperature, followed by incubation with streptavidin-horseradish peroxidase for 30 min. Finally, the tissues were stained using diaminobenzidine-H2O2 and hematoxylin. Imaging was performed using TissueGnostics Strata FAXS P-S.
Isolation and culture of neonatal mouse cardiomyocytes
To isolate and culture neonatal mouse cardiomyocytes, hearts from 1- to 3-day-old C57BL/6J mice (purchased from Southern Medical University Laboratory Animal Centre) were initially washed in phosphate-buffered saline (PBS) (Gibco, Carlsbad, CA, USA). Subsequently, the hearts were immersed in a mixture of trypsin (Gibco, Carlsbad, CA, USA) and PBS, placed on a shaker at 4 °C for 8 to 12 h. Following this, the hearts were incubated with complete medium containing 10% fetal bovine serum (Vivacell VC, Biological Industries, Israel) and 1% penicillin and streptomycin (Gibco, Carlsbad, CA, USA) in Dulbecco’s modified Eagle’s medium (DMEM) (Gibco, Carlsbad, CA, USA) to neutralize trypsin; the liquid was then aspirated. The hearts were shaken at 37 °C for 10 min with DMEM spiked with 8.4% collagenase type II (Gibco, Carlsbad, CA, USA), and the cell suspension was then aspirated into a 50 ml centrifuge tube. The collected cell suspension in the centrifuge tube underwent centrifugation (1000 rpm, 5 min), and the supernatant was discarded. The precipitated cells were resuspended in complete medium and spread in petri dishes. After incubating in a humidified 5% CO2 environment for 1.5 h at 37 °C, dissociated cardiomyocytes were collected and added to the corresponding well plates and cultured for 48 h before being utilized for subsequent experiments.
In vitro model of diabetic cardiomyocyte injuries
For the in vitro model of diabetic cardiomyocyte injuries, cardiomyocytes were incubated for 48 h in complete medium with high glucose (HG; 33 mM) along with high fat (0.2 mM palmitic acid, PA), and cardiomyocytes cultured with low glucose (LG; 5.5 mM) were used as controls.
Construction of AAV9 vectors for overexpression or knockdown of FGF21
Coding DNA sequences of mouse FGF21 were amplified by polymerase chain reaction from FGF21 plasmid using the forward primer 5′- CGCAAATGGGCGGTAGGCGTG′ and the reverse primer 5′- TCGCCGGACACGCTGAACT′ and were then inserted into the plasmid to construct the AAV9-FGF21 overexpression vector.
Transfection of cardiomyocytes with plasmid or siRNA
Transfection of cardiomyocytes with plasmids (Wz Biosciences Inc, Shandong, China) and siRNA (Beijing Tsingke Biotech Co., Ltd.) was performed using Lipofectamine 2000 reagent (Invitrogen, Carlsbad, USA) and X-treme gene siRNA transfection reagent (Roche, Basel, Switzerland), respectively. The sequences of siRNAs for mouse FGF21 were: 5′-GUGUCAAAGCCUCUAGGUU-3′ and 5′-AACCUAGAGGCUUUGACAC-3′; for mouse FTH1: 5′-GACACGGUGAUGAGAGCUA-3′ and 5′-UAGCUCUCAUCACCGUGUC-3′; for mouse FTL: 5′-GUCUCCUCGAGUUUCAGAA-3′ and 5′-UUCUGAAACUCGAGGAGAC-3′.
Mitochondrial membrane potential (MMP)
JC1 dye (Thermo Fisher Scientific Inc, T3168, Waltham, MA) was utilized to detect MMP in cardiomyocytes. JC1 accumulates in mitochondria in a potential-dependent manner, causing a shift in fluorescence emission from green (~ 529 nm) to red (~ 590 nm). The detection wavelengths were 488 nm (J-monomers) and 532 nm (J-aggregates). Cardiomyocytes were stained with JC1 dye (2 µM) and incubated for 30 min at 37 °C, 5% CO2. Observation was conducted using a laser scanning confocal microscope (LSCM) (Leica, STELLARIS 8, Weizler, Germany), and fluorescence intensity was measured by the instrument’s own software. Mitochondrial depolarization was indicated by a decrease in the red/green fluorescence intensity ratio (ΔΨm).
Lipid ROS
Lipid ROS levels were measured using the lipid peroxidation sensor BODIPY™ 581/591 C11 (Thermo Fisher Scientific Inc, D3861, Waltham, MA), with excitation/emission maxima of 581/591 nm in a reduced state, which shift to 488/510 nm upon oxidation. Cardiomyocytes were incubated with BODIPY™ 581/591 C11 (5 µM) for 30 min at 37 °C, 5% CO2. Changes in the green to red fluorescence ratio indicated lipid peroxidation. LSCM was used for observation, and fluorescence intensity was measured by the instrument’s own software.
Intracellular Fe2+
FerroOrange (Dojindo, F374, Kyushu Island, Japan), with excitation/emission wavelengths of 532/580 nm, was employed to measure intracellular Fe2+ content following the manufacturer’s instructions. Cardiomyocytes were incubated with FerroOrange (1 µM) for 30 min at 37 °C, 5% CO2. LSCM was used for observation, and fluorescence intensity was measured by the instrument’s own software.
Co-immunoprecipitation (Co-IP) and identification of interacting proteins
Lysates from primary cardiomyocytes underwent Co-IP using an IP antibody and A/G magnetic beads (MedChemExpress, HY-K0202, Monmouth Junction, NJ, USA), as specified in the instructions, followed by resolution using sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and analysis by immunoblotting. The gel at the target position was cut and sent to Novogene Technology Co. (Beijing, China) for protein profiling by liquid chromatography-tandem mass spectrometry. The following antibodies were used in Co-IP: FGF21 (ab171941) from Abcam (Cambridge, MA, USA), FTH1 (sc-376594)/FTL (sc-74513) from Santa Cruz Biotechnology (Dallas, TX, USA).
Immunofluorescence
Isolated cardiomyocytes were adhered to sticky slides and cultured for 48 h before being fixed in 4% paraformaldehyde. After rinsing with PBS, cardiomyocytes were treated with 0.5% Triton X-100 for 8 min, followed by blocking with 10% normal goat serum for 1 h at room temperature. Subsequently, the slides were covered with FGF21 (1:500) and incubated at 4 °C overnight. After rinsing with PBS, a fluorescein 488 antibody (1:500) was added and incubated for 1 h at room temperature. After rinsing with PBS, FTH1 (sc-376594, 1:500)/FTL (sc-74513, 1:500) from Santa Cruz Biotechnology (Dallas, TX, USA) were introduced to the slides and incubated overnight at 4 °C. After rinsing with PBS, nuclei were stained with 4,6-diamidino-2-phenylindole (DAPI) (Sigma-Aldrich, D9542, St. Louis, MO, USA). Mounting Medium, Antifading (Bei jing Solarbio Science & Technology Co., Ltd.) was used to reduce fluorescence quenching and immunofluorescence was examined using LSCM.
Quantitative polymerase chain reaction (qPCR)
Total RNA from cardiomyocytes or tissues was extracted using Trizol reagent. After DNase treatment, RNA was reverse-transcribed using the ReverTra Ace qPCR RT Kit (TOYOBO, Osaka, Japan). qPCR was performed with PerfectStart Green qPCR SuperMix (TransGen Biotech, Beijing, China). Relative gene expression was quantified based on Ct values, normalized against β-actin as an internal control. Primer sequences are available in Table S1.
Protein extraction and western blotting (WB)
Total protein samples from cardiomyocytes and tissues were extracted using RIPA lysis buffer (Beyotime, P0013B, Shanghai, China), supplemented with phenylmethylsulfonyl fluoride (Beyotime, ST506, Shanghai, China). Proteins were separated by SDS-PAGE and subsequently transferred onto polyvinylidene fluoride membranes. The membranes were blocked in Tris-buffered saline with 5% milk and incubated overnight with primary antibodies at 4 °C, with β-actin and β-tubulin serving as internal controls. Following washing with Tris-buffered saline containing Tween, membranes were incubated with horseradish peroxidase-conjugated secondary antibodies (anti-rabbit (7074) and anti-mouse (7076), both at 1:2000, Cell Signaling Technology, Boston, USA) for 1 h at room temperature. The membranes were then scanned and analyzed using an appropriate developer. The following antibodies were employed in WB: β-actin (BM5422, 1:5000) and β-tubulin (A05397-1, 1:5000) from Boster (Wuhan, China); FGF21 (ab171941, 1:1000) from Abcam (Cambridge, MA, USA); FTH1 (AB32180, 1:1000) and FTL (AB32425, 1:1000) from Absci (Oregon, USA); GPX4 (67763-1-Ig, 1:1000) from Proteintech (Wuhan, China); and ATF4 (sc-390063, 1:100) from Santa Cruz Biotechnology (Dallas, TX, USA).
Cell counting Kit-8 (CCK8), malondialdehyde (MDA), and lactate dehydrogenase (LDH) assays
The CCK8 (Life-iLab, AC11L054, Shanghai, China), MDA (Nanjing Jiancheng Bioengineering Institute, A003-4-1/A003-1-2, Nanjing, China), and LDH assay kits (Nanjing Jiancheng Bioengineering Institute, A020-2-2, Nanjing, China) were utilized to assess cell viability, MDA levels, and LDH levels, respectively, following the manufacturers’ instructions.
Other reagents and antibodies
The following reagents were utilized: Bafilomycin A1 (BafA1) (Sigma-Aldrich, B1793, St. Louis, MO, USA), MG132 (MedChemExpress, HY-13259, Monmouth Junction, NJ, USA), 4-hydroxynonenal (4-HNE) (Invitrogen, MA5-27570, Carlsbad, CA, USA), Ferrostatin-1 (Fer-1) (MedChemExpress, HY-100579, Monmouth Junction, NJ, USA), and Artesunate (Art) (MedChemExpress, HY-N0193, Monmouth Junction, NJ, USA).
Statistical analysis
Data are presented as mean ± SEM. Student’s t-test was employed for comparisons between two groups. For comparisons involving more than two groups, one-way ANOVA followed by Tukey’s post hoc analysis was applied for normally distributed variables. For non-normally distributed variables, a Kruskal-Wallis test with Dunn’s multiple comparison post hoc analysis was utilized. All statistical analyses were conducted using GraphPad Prism version 8.0 software (San Diego, CA, USA). Differences with a p-value < 0.05 were considered statistically significant.
Result
Ferroptosis played an important role in pathogenesis of DCM
Ferroptosis was aggravated in DCM mice
Firstly, we confirmed that ferroptosis was aggravated in DCM. T2DM mice have been shown to trigger DCM [11]. In our study, mice treated with HFD and STZ developed fasting hyperglycemia (Fig. S1A), and exhibited cardiac dysfunction including a reduction in early to late diastolic transmitral flow velocity (E/A) (Fig. S1B), ejection fraction (EF) and fractional shortening (FS) (Fig. S1C), as well as myocardial tissue damage and fibrosis observed via hematoxylin and eosin (HE) and Masson’s trichrome staining (Fig. S1D), confirming that DCM modeling was successful. In contrast to wild-type (WT) mice, the cardiac tissues of DCM mice exhibited diminished protein levels of ferroptosis-specific suppressor markers, including glutathione peroxidase 4 (GPX4) and ferritin heavy chain 1 (FTH1) (Fig. 1A), as well as elevated levels of ferroptosis markers such as malondialdehyde (MDA) (Fig. 1B) and prostaglandin endoperoxide synthase 2 (Ptgs2) mRNA (Fig. 1C). Furthermore, transmission electron microscopy (TEM) examination of cardiomyocytes in DCM mice revealed ruptured or shrunken mitochondria, mitochondrial membranes with increased density, and reduced or even absent cristae (Fig. 1D), indicating the occurrence of ferroptosis. These findings collectively confirmed that ferroptosis was aggravated in DCM model.
Fig. 1.

Ferroptosis was aggravated in DCM. A-C, Representative immunoblot images and statistical results of GPX4 and FTH1 (A), levels of MDA (B) and Ptgs2 mRNA (C) in cardiac tissue of DCM mice. D, Representative electron microscopic images of morphology of mitochondria in cardiac tissue of mice with DCM. Scale bar: 1–2 μm. E-I, The levels of GPX4 (E), and FTH1 (F), cellular activity (G), MDA (H) and Ptgs2 mRNA (I) development in primary cardiomyocytes treated with various concentrations of HG (15, 33, 50 mM) and PA (0.2 mM) for 48 h. J-N, The levels of GPX4 (J), FTH1 (K), cellular activity (L), MDA (M), and Ptgs2 mRNA (N) development in primary cardiomyocytes treated with HG (33 mM) and PA (0.2 mM) for different times. *P < 0.05
High glucose and high fat (HG/HF) aggravated ferroptosis in primary cardiomyocytes
HG/HF are key pathogenic factors in T2DM that contribute to the development of DCM via multiple signaling pathways. We established an in vitro model of diabetic heart injuries using HG/HF to investigate ferroptosis in DCM. To determine the appropriate stimulus concentration for ferroptosis in the DCM model, we used high glucose (15–50 mM) and palmitic acid (0.2 mM) to culture primary cardiomyocytes for 48 h. Results revealed that cardiomyocytes exposed to HG (33/50 mM) and palmitic acid (0.2 mM) exhibited significant changes in ferroptosis markers compared to the LG group, including reduced levels of GPX4 (Fig. 1E), FTH1 (Fig. 1F), and cellular activity (Fig. 1G), as well as increased levels of MDA (Fig. 1H) and Ptgs2 mRNA (Fig. 1I). To determine the appropriate time for ferroptosis in the model of diabetic heart injuries, we used HG/HF to culture primary cardiomyocytes for 24 to 72 h. The results showed that protein levels of GPX4 (Fig. 1J), FTH1 (Fig. 1K), and cellular activity (Fig. 1L) decreased significantly, while the levels of MDA (Fig. 1M) and Ptgs2 mRNA (Fig. 1N) increased significantly in cardiomyocytes stimulation at 48 h. Therefore, we selected 33 mM HG and 0.2 mM PA stimulation for 48 h to induce ferroptosis of cardiomyocytes.
The above results further confirmed that ferroptosis was aggravated in DCM and described suitable glycolipid stimulation conditions in the model of diabetic heart injuries.
FGF21 regulated DCM via ferroptosis pathway
FGF21 has been extensively studied in the context of glycolipid metabolism and cardiovascular diseases, and ferroptosis also plays a significant role in these conditions. Considering the mechanisms underlying the close connection among T2DM, ferroptosis, and FGF21, we sought to explore whether FGF21 could alleviate DCM by inhibiting ferroptosis. First, we investigated changes in the expression of FGF21 in DCM. Results revealed significantly elevated mRNA and protein levels of FGF21 (Fig. S2A-D) in DCM, suggesting that FGF21 may regulate DCM to some extent. Next, we examined the role of FGF21 in ferroptosis pathway in DCM using intervention models in cardiac tissues and primary cardiomyocytes.
FGF21 overexpression improved cardiac injury and function in DCM mice via inhibiting ferroptosis
To explore the cardioprotective potential of FGF21 through the ferroptosis pathway, we constructed an FGF21 therapy model in mouse heart. We injected viruses overexpressing FGF21 and corresponding negative control viruses via the tail vein into HFD/STZ-inducedT2DM or WT mice to establish the DCM therapy and corresponding control model (Fig. 2A). FGF21 overexpression significantly increased the mRNA and protein levels of FGF21 (Fig. S2E-F) in cardiac tissues, while inhibiting ferroptosis and improving cardiac injury and function in DCM mice. Specifically, FGF21 overexpression prevented heart enlargement (Fig. 2B) and improved the heart-to-body weight ratio (Fig. 2C), and enhanced cardiac function (Fig. 2D), including elevations in EF, FS, and E/A (Fig. 2E) in DCM mice. TEM images revealed that FGF21 overexpression alleviated mitochondrial damage characteristic in ferroptosis in cardiac tissues of DCM (Fig. 2F). Histologically, FGF21 overexpression improved hypertrophy (as observed via HE staining), fibrosis (as observed via Masson’s trichrome staining), and the accumulation of Fe3+ (as observed via prussian blue staining) and 4-HNE (as observed via immunohistochemistry) in the cardiac tissues of DCM mice (Fig. 2G). Additionally, FGF21 overexpression increased GPX4 protein levels (Fig. 2H), decreased the levels of MDA (Fig. 2I) and Ptgs2 mRNA (Fig. 2J) in the cardiac tissues of DCM mice, and reduced plasma levels of lactate dehydrogenase (LDH) (Fig. 2K). These findings collectively suggested that FGF21 ameliorated cardiac injury and function by inhibiting ferroptosis in the hearts of DCM mice.
Fig. 2.

Overexpression of FGF21 mitigated cardiac injury in mouse DCM via ferroptosis pathway. A, Diagram showing experimental design for the study of overexpression of FGF21 in DCM mice (By MedPeer.). B, Representative images of hearts from each group. C, Heart/body weight ratio. D-E, Representative echocardiography images (D), and statistical results of EF, FS and E/A (E). F, Representative TEM images of morphology of mitochondria in cardiac tissue of mice with DCM. Scale bar: 1–2 μm. G, HE staining, Masson trichrome staining, prussian blue Fe3+ staining (enhance with DAB) and 4-HNE staining of mice hearts from each group. Scale bar: 50 μm. H, Representative immunoblot images and statistical results of GPX4. I-J, Levels of MDA (I) and Ptgs2 mRNA (J) in cardiac tissue. K, Levels of LDH in plasma of mice. *P < 0.05
FGF21 knockdown exacerbated cardiac injury and function in DCM mice via aggravating ferroptosis
Next, we constructed an FGF21 knockdown model in mouse heart. We injected short hairpin RNA (shRNA) for FGF21 and corresponding negative control viruses via the tail vein into db/db or WT mice to establish the DCM intervention and corresponding control model (Fig. 3A). FGF21 knockdown significantly decreased the mRNA and protein levels of FGF21 (Fig. S2G-H) in cardiac tissues, while exacerbating ferroptosis and cardiac injury and function in DCM mice. Specifically, FGF21 knockdown further increased the volume of heart (Fig. 3B) and heart weight/tibial length (Fig. 3C), and exacerbated cardiac function (Fig. 3D), including reduction in EF, FS, and E/A (Fig. 3E) in DCM mice. TEM images revealed that FGF21 knockdown further exacerbated mitochondrial damage characteristic in ferroptosis in cardiac tissues of DCM (Fig. 3F). Histologically, FGF21 knockdown exacerbated hypertrophy and inflammation, fibrosis, and the accumulation of Fe3+ and 4-HNE in the cardiac tissues of DCM mice (Fig. 3G). Additionally, FGF21 knockdown further decreased GPX4 protein levels (Fig. 3H), further increased the levels of MDA (Fig. 3I) and Ptgs2 mRNA (Fig. 3J) in the cardiac tissues of DCM mice, and incrreased plasma levels of LDH (Fig. 3K). These findings collectively suggested that FGF21 knockdown exacerbated cardiac injury and function by aggravating ferroptosis in the hearts of DCM mice.
Fig. 3.

Knockdown of FGF21 exacerbated cardiac injury in DCM mice via ferroptosis pathway. A, Diagram showing experimental design for the study of knockdown of FGF21 in DCM mice (By MedPeer.). B, Representative images of hearts from each group. C, Heart weight/tibial length. D-E, Representative echocardiography images (D), and statistical results of EF, FS and E/A (E). F, Representative TEM images of morphology of mitochondria in cardiac tissue of mice with DCM. Scale bar: 1–2 μm. G, HE staining, Masson trichrome staining, prussian blue Fe3+ staining (enhance with DAB) and 4-HNE staining of mice hearts from each group. Scale bar: 50 μm. H, Representative immunoblot images and statistical results of GPX4. I-J, Levels of MDA (I) and Ptgs2 mRNA (J) in cardiac tissue. K, Levels of LDH in plasma of mice. *P < 0.05
FGF21 overexpression inhibited HG/HF-induced ferroptosis in primary cardiomyocytes
To explore whether FGF21 could mitigate HG/HF-induced ferroptosis in the in vitro diabetic heart injury model, we transfected plasmids for FGF21 overexpression into primary cardiomyocytes for 48 h, subsequently modeling the in vitro diabetic heart injury model (Fig. 4A). Transfection of FGF21-OE elevated both mRNA and protein levels of FGF21 (Fig. S3A-B). Furthermore, FGF21-OE increased GPX4 protein levels (Fig. 4B) and cellular activity (Fig. 4C), and decreased MDA levels (Fig. 4D) and Ptgs2 mRNA (Fig. 4E) in HG/HF-stimulated cardiomyocytes. Additionally, fluorescence staining demonstrated that HG/HF stimulation led to a decreased mitochondrial membrane potential (MMP) - a decrease in JC1 aggregates and an increase in JC1 monomers, increased lipid ROS - a decrease in the reduced state and an increase in the oxidized state of BODIPY™ 581/591 C11 and elevated intracellular free Fe2+ in cardiomyocytes, however, all these adverse conditions were alleviated by FGF21 overexpression (Fig. 4F, S3C-E). To further demonstrate that FGF21 acted through the ferroptosis pathway, we overexpressed FGF21 in HG/HF-stimulated cardiomyocytes concurrently with the ferroptosis inducer artesunate (Art). Art resulted in a further reduction in cell activity (Fig. 4G) and an increase in the levels of MDA (Fig. 4H) and Ptgs2 mRNA (Fig. 4I) in HG/HF-stimulated cardiomyocytes, which were reversed by FGF21 overexpression (Fig. 4G-I). These findings indicated that FGF21 overexpression mitigated further damage induced by Art in HG/HF-stimulated cardiomyocytes. These results collectively suggested that FGF21 overexpression inhibited HG/HF-induced ferroptosis in the in vitro diabetic heart injury model.
Fig. 4.

(By MedPeer.)
FGF21 knockdown exacerbated HG/HF-induced ferroptosis in primary cardiomyocytes
Next, we investigated whether FGF21 knockdown would exacerbate HG/HF-induced ferroptosis in cardiomyocytes. We transfected FGF21-SI into primary cardiomyocytes for 48 h, subsequently modeling the in vitro diabetic heart injury model (Fig. 5A). Transfection of FGF21-SI reduced both mRNA and protein levels of FGF21 (Fig. S4A-B) in cardiomyocytes. Furthermore, FGF21-SI further decreased GPX4 protein levels (Fig. 5B) and cellular activity (Fig. 5C), increased MDA levels (Fig. 5D) and Ptgs2 mRNA (Fig. 5E) in HG/HF-stimulated cardiomyocytes. Additionally, fluorescence staining demonstrated that HG/HF stimulation led to a decreased mitochondrial membrane potential (MMP), increased lipid ROS, and elevated intracellular free Fe2+ in cardiomyocytes, all these adverse conditions were further exacerbated by FGF21 knockdown (Fig. 5F, S4C-E). To further demonstrate that FGF21 acted through the ferroptosis pathway, we knocked down FGF21 in HG/HF-stimulated cardiomyocytes concurrently with the ferroptosis inhibitor ferrostatin-1 (Fer-1). Fer-1 restored cell activity (Fig. 5G) and decreased the levels of MDA (Fig. 5H) and Ptgs2 mRNA (Fig. 5I) in HG/HF-stimulated cardiomyocytes, whereas FGF21 knockdown reversed the improvements induced by Fer-1. These results collectively suggest that FGF21 knockdown exacerbates HG/HF-induced ferroptosis in primary cardiomyocytes.
Fig. 5.

Knockdown of FGF21 aggravated HG/HF-induced ferroptosis in primary cardiomyocytes. A, Diagram showing experimental design for the study of knockdown of FGF21 in the in vitro model of diabetic heart injuries (By MedPeer.). B, Representative immunoblot images and statistical results of GPX4. C-E, Revels of cellular activity (C), MDA (D) and Ptgs2 mRNA (E). F, Representative LSCM images of mitochondria membrane potential, lipid ROS and intracellular Fe2+ were measured using JC1 dye, BODIPY™ 581/591 C11 and FerroOrange respectively in primary cardiomyocytes (per image represents more than 15 cells). Scale bar: 50 μm. G-I, Fer-1 (final concentration of 10 µM) was added to pre-treatment at the last 12 h of transfection, subsequently co-treated of primary cardiomyocytes with HG/HF for 48 h, levels of cellular activity (G), MDA (H) and Ptgs2 mRNA (I) were measured. *P < 0.05
FGF21 exerted inhibitory effect on ferroptosis in DCM by directly targeting ferritin
Protein interactions between FGF21 and ferritin
Considering that FGF21 was a secreted protein, we utilized co-immunoprecipitation (Co-IP) to analyze cardiomyocyte lysates, followed by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) to elucidate how FGF21 functions in ferroptosis in DCM. Following Komas blue staining, specific bands were excised from the gel for the identification of proteins interacting with endogenous FGF21 via liquid chromatography-tandem mass spectrometry, which revealed that FGF21 binds to cell growth and death-associated proteins, including FTH1 and FTL (Fig. 6A-B). FTH1 and FTL are two subunits of ferritin, the hallmark protective protein in ferroptosis, which exert distinct effects on the oxidation of Fe2+ and play critical roles in this process. To validate these interactions, we performed protein co-localization analysis using immunofluorescence techniques in conjunction with laser scanning confocal microscopy, and conducted Co-IP followed by western blotting (WB). The results of immunofluorescence demonstrated significant co-localization between FGF21 and FTH1 or FTL (Fig. 6C, S5A-B), and the interactions were further corroborated by immunoprecipitating endogenous FTH1 and FTL proteins from cardiomyocyte lysates using FGF21 antibody, followed by immunoprecipitation of endogenous FGF21 proteins using FTH1 and FTL antibodies (Fig. 6D). Additionally, we utilized an online resource to predict protein binding sites between FGF21 and FTH1 or FTL (Fig. 6E). Collectively, these results support the interaction between FGF21 and FTH1 or FTL, elucidating FGF21’s mechanism in inhibiting ferroptosis.
Fig. 6.

Protein interactions between FGF21 and ferritin. A-B, Mass spectrometry analysis of proteins interacting with FGF21. Cell lysates were immunoprecipitated with Protein A/G MagBeads and then subjected to immunoblotting, GO analysis results (A), KEGG analysis results (B). C, Representative images of LSCM protein co-localization between FGF21 and FTH1 or FTL. D. Representative immunoblot images of CO-IP confirmed FGF21 interaction with FTH1 or FTL. E, Results of prediction of protein binding sites [12] between FGF21 and ferritin (orange red represents FGF21, purple represents FTH1, and green represents FTL).
FTH1 and FTL were the downstream factors of FGF21 that exerted the inhibitory effect on ferroptosis in DCM
FGF21 regulated changes in protein levels of FTH1 and FTL in DCM
Initially, we assessed the alterations in FTH1 and FTL in DCM models. Previous findings demonstrated a reduction in FTH1 protein levels in cardiac tissues of DCM mice (Fig. 1A) and HG/HF-stimulated primary cardiomyocytes (Fig. 1E, J). Concurrently, our current results revealed diminished FTL protein levels in cardiac tissues of DCM mice (Fig. S6A) and HG/HF-stimulated primary cardiomyocytes (Fig. S6B), thus implicating ferritin’s participation in DCM to some extent. Subsequently, to determine whether FTH1 and FTL serve as downstream mediators of FGF21 in DCM, we performed knockdown or overexpression of FGF21, and assessed the impact of variations in FGF21 expression on FTH1 and FTL in the in vitro model of diabetic cardiac injuries. Results indicated that FGF21 overexpression mitigated the reduction of FTH1 and FTL protein expression levels in cardiac tissues of DCM mice (Fig. 7A) and primary cardiomyocytes under HG/HF (Fig. 7B), whereas knockdown of FGF21 further decreased FTH1 and FTL protein expression in cardiac tissues of DCM mice (Fig. 7C) and primary cardiomyocytes exposed to HG/HF (Fig. 7D).
Fig. 7.

FGF21 regulated changes in protein levels of FTH1 and FTL in DCM. A-B, Representative immunoblot images and statistical results of FTH1 and FTL in cardiac tissues of DCM mice (A) and primary cardiomyocytes under HG/HF (B) with FGF21 overexpression. C-D, Representative immunoblot images and statistical results of FTH1 and FTL in cardiac tissue of DCM mice (C) and primary cardiomyocytes under HG/HF (D) with FGF21 knockdown. *P < 0.05
FTH1-SI and FTL-SI reversed the inhibition of HG/HF-induced ferroptosis by FGF21 overexpression in primary cardiomyocytes
After confirming that FTH1-SI and FTL-SI could reduce the mRNA and protein levels of FTH1 (Fig. S6C-D) and FTL (Fig. S6E-F) in primary cardiomyocytes, we co-transfected cardiomyocytes with FGF21-OE, FTH1-SI and /or FTL-SI. We observed that the transfection of FTH1-SI and/or FTL-SI weakened the protective effect of FGF21 overexpression on cardiomyocytes stimulated by HG/HF via the ferroptosis pathway. Specifically, the transfection of FTH1-SI and/or FTL-SI reversed cell viability (Fig. 8A), GPX4 levels (Fig. 8B), MDA levels (Fig. 8C), and Ptgs2 mRNA levels (Fig. 8D) that were rescued by the overexpression of FGF21. Furthermore, cell fluorescence staining indicated that the benefits of overexpressed FGF21 on MMP, lipid ROS, and intracellular free Fe2+ were reversed by FTH1-SI and/or FTL-SI in cardiomyocytes under HG/HF (Fig. 8E, S6G-I).
Fig. 8.

FTH1 and FTL were the downstream factors of FGF21 that exerted the inhibitory effect on ferroptosis in DCM. A-E, Transfection of FGF21-plasmid and FTH1-SI or/and FTL-SI for 48 h and HG/HF incubation for the following 48 h, levels of cellular activity (A), representative immunoblot images and statistical results of GPX4 (B), MDA (C), Ptgs2 mRNA (D), and representative LSCM images of mitochondria membrane potential, lipid ROS and intracellular Fe2+ were measured using JC1 dye, BODIPY™ 581/591 C11 and FerroOrange respectively (E) in primary cardiomyocytes (per image represents more than 15 cells). Scale bar: 50 μm. F-J, Transfection of FGF21-plasmid and FTH1-siRNA or/and FTL-siRNA for 48 h and HG/HF incubation for the following 48 h, levels of cellular activity (F), representative immunoblot images and statistical results of GPX4 (G), MDA (H) and Ptgs2 mRNA (I), and representative LSCM images of mitochondria membrane potential, lipid ROS and intracellular Fe2+ were measured using JC1 dye, BODIPY™ 581/591 C11 and FerroOrange respectively (J) in primary cardiomyocytes (per image represents more than 15 cells). Scale bar: 50 μm. *P < 0.05
FTH1-OE and FTL-OE reversed the promotion of HG/HF-induced ferroptosis by FGF21 knockdown in primary cardiomyocytes
After confirming that FTH1-OE and FTL-OE could reduce the mRNA and protein levels of FTH1 (Fig. S6J-K) and FTL (Fig. S6L-M) in primary cardiomyocytes, we co-transfected cardiomyocytes with FGF21-SI, FTH1-OE and /or FTL-OE. We observed that the transfection of FTH1-OE and/or FTL-OE reversed the detrimental effects of FGF21 knockdown on cardiomyocytes stimulated by HG/HF via the ferroptosis pathway. Specifically, the transfection of FTH1-OE and/or FTL-OE improved cell viability (Fig. 8F), GPX4 levels (Fig. 8G), MDA levels (Fig. 8H), and Ptgs2 mRNA levels (Fig. 8I) that were aggravated by the knockdown of FGF21. Furthermore, cell fluorescence staining indicated that the disadvantages of FGF21 knockdown on MMP, lipid ROS, and intracellular free Fe2+ were reversed by FTH1-OE and/or FTL-OE in cardiomyocytes under HG/HF (Fig. 8J, S6N-P).
These results collectively suggested that FTH1 and FTL function as downstream factors in FGF21 inhibition of ferroptosis to ameliorate DCM.
FGF21 prolongs the half-life of ferritin by inhibiting ferritin degradation in proteasome and autophagy-lysosome pathways through protein interactions
FGF21 mainly affected the protein level of ferritin rather than the transcriptional level
To elucidate the specific mechanism of FGF21’s action on FTH1 and FTL, we investigated the effects of FGF21 overexpression and knockdown on the mRNA and protein levels of FTH1 and FTL. Consistent with our hypothesis, FGF21 overexpression in WT mice and primary cardiomyocytes significantly mainly elevated the protein levels of FTH1 (Fig. S7A-B) and FTL (Fig. S7C-D), while having minimal effects on the mRNA levels of FTH1 (Fig. S7E-F) and FTL (Fig. S7G-H), as evidenced by the differential changes observed in protein levels relative to mRNA levels. Similarly, FGF21 knockdown in WT mice or primary cardiomyocytes mainly significantly decreased the protein levels of FTH1 (Fig. S7I-J) and FTL (Fig. S7K-L), while exerting minimal effects on the mRNA levels of FTH1 (Fig. S7M-N) and FTL (Fig. S7O-P). These findings suggested that FGF21 primarily acts on FTH1 and FTL through protein interactions rather than transcription-related mechanisms.
FGF21 prolongs the half-life of ferritin by inhibiting ferritin degradation in proteasome and autophagy-lysosome pathways
Next, we assessed the impact of FGF21 on the half-lives of FTH1 and FTL proteins. Forty-eight hours after the transfection of FGF21-OE, primary cardiomyocytes were subjected to HG/HF treatment and varying durations of cycloheximide (CHX). CHX is an antifungal antibiotic that inhibits protein synthesis in eukaryotic cells by blocking protein translation processes. The results indicated that CHX treatment for over 6 h significantly inhibited the synthesis of FTH1 and FTL proteins in HG/HF-stimulated cardiomyocytes, whereas FGF21 overexpression notably delayed their degradation, thereby extending the half-life of ferritin (Fig. 9A). To further investigate FGF21’s interaction with ferritin, we examined FGF21’s role in inhibiting ferritin degradation. Previous studies have demonstrated that ferritin is degraded via proteasomal and lysosomal-autophagy pathways [13]. We knocked down FGF21 in HG/HF-stimulated cardiomyocytes and treated them with either the proteasome inhibitor MG132 or the lysosomal-autophagy inhibitor Baflomycin A1 (BafA1). The results indicated that, compared with the control, both MG132 and BafA1 rescued the reduction in protein levels of FTH1 (Fig. 9B) and FTL (Fig. 9C) in cardiomyocytes resulting from HG/HF stimulation and/or FGF21 knockdown. These results suggested that ferritin was degraded via both proteasomal and lysosomal-autophagy pathways in HG/HF-stimulated cardiomyocytes, and that FGF21 inhibited ferritin degradation through both pathways.
Fig. 9.

FGF21 prolongs the half-life of ferritin by inhibiting ferritin degradation in proteasome and autophagy-lysosome pathways. A, After overexpression of FGF21 for 48 h, cardiomyocytes were stimulated with HG/HF for 48 h while different durations of CHX (final concentration of 20 µg/ml) were administered, and the protein levels of FTH1 and FTL in cardiomyocytes were measured by WB. B-C, Transfection of FGF21-siRNA for 48 h, BafA1 (final concentration of 100 nM) and MG132 (final concentration of 5 µM) was added to pre-treatment at the last 2 h and 0.5 h of transfection respectively, followed by 48 h incubation with HG/HF, representative immunoblot images and statistical results of FTH1 (B) and FTL (C) in primary cardiomyocytes. *P < 0.05
Activating transcription factor 4 (ATF4) regulated FGF21 transcription in DCM
Our study revealed a marked increase in FGF21 expression in DCM (Fig. S2A-D), which is attributed to the accumulation of lipid peroxidation and reactive oxygen species (ROS) resulting from HG/HF conditions, thereby fostering an organism’s stress-protective response. Previous studies have linked FGF21 gene expression to the stimulation of ATF4 [14], and we confirmed this mechanism in our in vitro model of diabetic heart injuries. We observed increased mRNA and protein levels of ATF4 in cardiac tissues of DCM (Fig. S8A-B) and HG/HF-stimulated cardiomyocytes (Fig. S8C-D) compared to controls. Furthermore, ATF4 overexpression increased the mRNA and protein levels of both ATF4 and FGF21 (Fig. S8E-G) in primary cardiomyocytes, indicating that ATF4 functions as an activator promoting FGF21 transcription in DCM. Therefore, ATF4 served as an upstream regulator of FGF21 in DCM.
Discussion
As an important complication of T2DM, the mechanisms and effective treatments for DCM remain inadequately explored, highlighting an urgent need for further investigation. Ferroptosis, an iron-dependent and modifiable mode of cell death, results from the accumulation of lipid peroxidation and redox imbalance [15]. Ferroptosis has emerged as a novel and regulatable mode of cell death involved in various diseases, garnering significant research attention due to its importance in organismal growth and potential therapeutic implications. However, the study of its role in diabetes is still in its infancy [16–18]. The pathogenesis of DCM is linked to the dysregulation of redox homeostasis caused by oxidative stress and lipid accumulation under high glucose/high fat (HG/HF) conditions [19], and increased iron levels in cardiomyocytes have been observed in DCM [20, 21], DCM and ferroptosis are mechanistically intertwined, therefore it is hypothesized that ferroptosis plays an important role in DCM (Fig. 10).
Fig. 10.
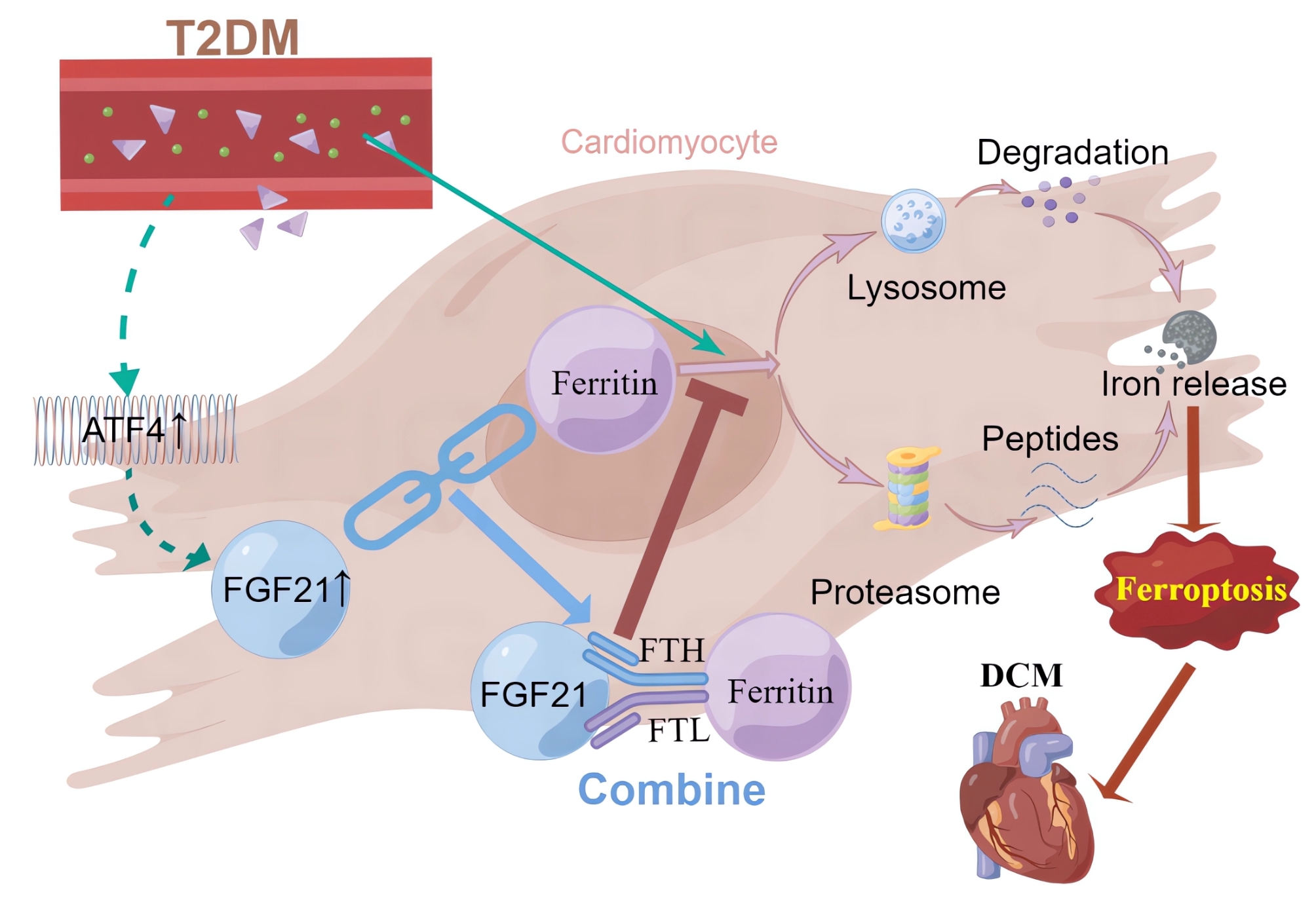
Schematic representation of the mechanism by which FGF21 improves DCM by inhibiting ferroptosis via ferritin pathway (By Figdraw.)
Our study identified the activation of ferroptosis in DCM, evidenced by reduced levels of cellular activity, FTH1 and GPX4, significantly elevated levels of MDA and Ptgs2 mRNA. Additionally, TEM of cardiac tissues from DCM mice further revealed characteristic mitochondrial changes indicative of ferroptosis [5]. These findings provide compelling evidence for the critical role of ferroptosis in DCM.
Previous studies have shown that FGF21 alleviates several cardiovascular diseases [9]. Notably, FGF21 has been found to inhibit iron overload-induced hepatic ferroptosis by promoting the ubiquitination of heme oxygenase-1 and enhancing NRF2 activity, thereby mitigating liver injury and fibrosis [22]. Although FGF21 levels often increase compensatorily under stress, endogenous FGF21 is typically deficient and has a short half-life, necessitating exogenous supplementation to achieve therapeutic effects [23, 24], which is similar to that of B-type natriuretic peptide in heart failure. Similarly, our study demonstrated that overexpression of FGF21 inhibited ferroptosis and improved myocardial injury and cardiac function in DCM models. Conversely, FGF21 knockdown exacerbated ferroptosis, myocardial injury, and cardiac dysfunction, confirming FGF21’s crucial role in inhibiting ferroptosis in DCM. This is the first study to report that FGF21 exerts therapeutic effects in DCM through the inhibition of ferroptosis.
Initially, we assumed that FGF21 inhibited ferroptosis primarily by improving lipid metabolism and reducing oxidative stress. However, Co-IP analysis revealed that FGF21 may bind to both FTH1 and FTL. These two subunits of ferritin form a spherical-shell structure that serves as the predominant intracellular iron-storage protein and plays a vital role in ferroptosis [25]. Typically, excess iron within the cell is sequestered by ferritin, which maintains iron homeostasis and mitigates oxidative stress induced by the Fenton reaction, thus reducing ferroptosis [26]. Research indicates that ferritin has been found to have an important role in cardiovascular diseases, and the expression of both FTH and FTL decreases during ferroptosis, impairing the ability to sequester Fe2+, leading to cardiovascular disease [26–29]. Therefore, we explored the interactions between FGF21 and both FTH1 and FTL. Our findings from confocal fluorescence co-localization analysis, Co-IP, protein binding site prediction, and WB confirmed that FGF21 interacts with FTH and FTL. Subsequently, we established that FTH1 and FTL are downstream factors through which FGF21 exerted its inhibitory effect on ferroptosis in DCM. We further elucidated the specific mechanism by which FGF21 acted on ferritin, demonstrating that FGF21 primarily interacted with ferritin through protein interactions rather than transcriptional regulation. Specifically, FGF21 prolonged the half-life of ferritin by inhibiting its excessive degradation through proteasomal and lysosomal-autophagy pathways in DCM.
Activation of ATF4, a crucial response element in the regulation of oxidative stress-related genes, positions FGF21 as a key regulator of cellular responses to oxidative stress, linking it to genes such as NRF2, TBP-2, UCP3, SOD2, ERK, and p38 [14]. However, whether the stress-induced elevation of FGF21 in DCM is also regulated by ATF4 remains to be established. To test this hypothesis, we first confirmed the upregulation of ATF4 mRNA and protein levels in DCM. Following this, we observed an increase in FGF21 expression upon ATF4 overexpression in primary cardiomyocytes, thereby establishing that ATF4 promotes FGF21 transcription and mediates its stress-induced increase during DCM. Our results confirmed that ATF4 was an upstream regulator of FGF21 in the context of DCM.
Conclusion
In conclusion, our study underscores the significance of ferroptosis in DCM and elucidates the role and mechanism of FGF21 in ameliorating DCM through the inhibition of ferroptosis. Specifically, we propose a novel molecular mechanism whereby FGF21 inhibits ferroptosis in DCM by binding to FTH1 and FTL, preventing their excessive degradation via the autophagy-lysosomal and proteasomal pathways, thereby prolonging ferritin’s half-life and upregulating its expression. Our findings provide new insights into the pathogenesis and treatment of DCM and offer robust evidence for the therapeutic potential of FGF21 in T2DM, particularly concerning DCM.
Supplementary Information
Acknowledgements
We would like to thank the Diabetes Complications Consortium (www.diacomp.org) for providing the methodology to establish a mouse model of T2DM and the protein binding site prediction function provided free of charge by The Yang Zhang Lab (https://zhanggroup.org/).
Author contributions
Conception or design of the study: Lihong Wang, Xiaofang Zhang, and Ruxin Wang; data collection: Ruxin Wang, Haowen Ye, Yongting Zhao, Jiaxin Wang, Ying Wang, and Han Liu; data analysis and interpretation: Ruxin Wang, Haowen Ye, Meixin Yu, Liangyan Wu and Xian Yang; drafting the article: Ruxin Wang, Caixia Ma, and Yun Wen; critical revision of the article: Lihong Wang, Xiaofang Zhang, and Ruxin Wang. All authors approved the final version of the manuscript.
Funding
This study was supported by National Natural Science Foundation of China (Grant Number: 82200417), Science and Technology Projects in Guangzhou (Grant Number: 2023A04J1280), Talent Introduction Funding Project of The First Affiliated Hospital of Jinan University (Grant Number: 808026), and Basic and Applied Basic Research Foundation of Guangdong (Grant Number: 2023A1515110934). The funders had no role in the study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Data availability
No datasets were generated or analysed during the current study.
Declarations
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Ruxin Wang, Xiaofang Zhang, Haowen Ye have contributed equally to this work and share first authorship.
References
- 1.Global, regional, and national burden of diabetes from,. to 2021, with projections of prevalence to 2050: a systematic analysis for the Global Burden of Disease Study 2021. Lancet. 1990;2023(402):203–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sattar N, McMurray J, Borén J, et al. Twenty years of cardiovascular complications and risk factors in patients with type 2 diabetes: a nationwide Swedish cohort study. Circulation. 2023;147:1872–86. [DOI] [PubMed] [Google Scholar]
- 3.Jia G, Hill MA, Sowers JR. Diabetic cardiomyopathy: an update of mechanisms contributing to this clinical entity. Circ Res. 2018;122:624–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tan Y, Zhang Z, Zheng C, Wintergerst KA, Keller BB, Cai L. Mechanisms of diabetic cardiomyopathy and potential therapeutic strategies: preclinical and clinical evidence. Nat Rev Cardiol. 2020;17:585–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dixon SJ, Lemberg KM, Lamprecht MR, et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell. 2012;149:1060–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jiang X, Stockwell BR, Conrad M. Ferroptosis: mechanisms, biology and role in disease. Nat Rev Mol Cell Biol. 2021;22:266–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wang R, Ye H, Wei J, Wang Y, Wang L, Zhang X. Research progress of ferroptosis and its association with diabetes mellitus and related complications. Chin J Diabetes Mellitus. 2022;14:1326–31. [Google Scholar]
- 8.Geng L, Lam KSL, Xu A. The therapeutic potential of FGF21 in metabolic diseases: from bench to clinic. Nat Rev Endocrinol. 2020;16:654–67. [DOI] [PubMed] [Google Scholar]
- 9.Zhang Y, Liu D, Long XX, Fang QC, Jia WP, Li HT. The role of FGF21 in the pathogenesis of cardiovascular disease. Chin Med J (Engl). 2021;134:2931–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Diabetic Complications Consortium. https://www.diacomp.org/. Accessed 10 November, 2022.
- 11.Sun Y, Zhou S, Guo H, et al. Protective effects of sulforaphane on type 2 diabetes-induced cardiomyopathy via AMPK-mediated activation of lipid metabolic pathways and NRF2 function. Metabolism. 2020;102: 154002. [DOI] [PubMed] [Google Scholar]
- 12.The Yang Zhang Lab. https://zhanggroup.org/. Accessed May 10, 2024.
- 13.Zhang N, Yu X, Xie J, Xu H. New insights into the role of ferritin in iron homeostasis and neurodegenerative diseases. Mol Neurobiol. 2021;58:2812–23. [DOI] [PubMed] [Google Scholar]
- 14.Gómez-Sámano M, Grajales-Gómez M, Zuarth-Vázquez JM, et al. Fibroblast growth factor 21 and its novel association with oxidative stress. Redox Biol. 2017;11:335–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Liang D, Minikes AM, Jiang X. Ferroptosis at the intersection of lipid metabolism and cellular signaling. Mol Cell. 2022;82:2215–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ni T, Huang X, Pan S, Lu Z. Inhibition of the long non-coding RNA ZFAS1 attenuates ferroptosis by sponging miR-150-5p and activates CCND2 against diabetic cardiomyopathy. J Cell Mol Med. 2021;25:9995–10007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang N, Ma H, Li J, et al. HSF1 functions as a key defender against palmitic acid-induced ferroptosis in cardiomyocytes. J Mol Cell Cardiol. 2021;150:65–76. [DOI] [PubMed] [Google Scholar]
- 18.Wang X, Chen X, Zhou W, et al. Ferroptosis is essential for diabetic cardiomyopathy and is prevented by sulforaphane via AMPK/NRF2 pathways. Acta Pharm Sin B. 2022;12:708–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Meng Z, Liang H, Zhao J, et al. HMOX1 upregulation promotes ferroptosis in diabetic atherosclerosis. Life sci. 2021;284: 119935. [DOI] [PubMed] [Google Scholar]
- 20.Farhangkhoee H, Khan ZA, Mukherjee S, et al. Heme oxygenase in diabetes-induced oxidative stress in the heart. J Mol Cell Cardiol. 2003;35:1439–48. [DOI] [PubMed] [Google Scholar]
- 21.Fernández-Real JM, McClain D, Manco M. Mechanisms linking glucose homeostasis and iron metabolism toward the onset and progression of type 2 diabetes. Diabetes Care. 2015;38:2169–76. [DOI] [PubMed] [Google Scholar]
- 22.Wu A, Feng B, Yu J, et al. Fibroblast growth factor 21 attenuates iron overload-induced liver injury and fibrosis by inhibiting ferroptosis. Redox Biol. 2021;46: 102131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kharitonenkov A, Wroblewski VJ, Koester A, et al. The metabolic state of diabetic monkeys is regulated by fibroblast growth factor-21. Endocrinology. 2007;148:774–81. [DOI] [PubMed] [Google Scholar]
- 24.Hecht R, Li YS, Sun J, et al. Rationale-based engineering of a potent long-acting FGF21 analog for the treatment of type 2 diabetes. PLoS ONE. 2012;7: e49345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lee NK, Cho S, Kim IS. Ferritin - a multifaceted protein scaffold for biotherapeutics. Exp Mol Med. 2022;54:1652–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fang X, Cai Z, Wang H, et al. Loss of cardiac ferritin H facilitates cardiomyopathy via SLC7A11-mediated ferroptosis. Circ Res. 2020;127:486–501. [DOI] [PubMed] [Google Scholar]
- 27.Shesh BP, Connor JR. A novel view of ferritin in cancer. BBA-Rev Cancer. 2023;1878: 188917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.López-Castro JD, Delgado JJ, Perez-Omil JA, et al. A new approach to the ferritin iron core growth: influence of the H/L ratio on the core shape. Dalton T. 2012;41:1320–4. [DOI] [PubMed] [Google Scholar]
- 29.Hu J, Sha X, Li Y, et al. Multifaceted applications of ferritin nanocages in delivering metal ions, bioactive compounds, and enzymes: a comprehensive review. J Agr Food Chem. 2023;71:19903–19. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
No datasets were generated or analysed during the current study.