

Abstract
The PIM kinase family, consisting of PIM1, PIM2, and PIM3, is a group of serine/threonine protein kinases crucial for cellular growth, immunoregulation, and oncogenesis. PIM1 kinase is often overexpressed in solid and hematopoietic malignancies, promoting cell survival, proliferation, migration, and senescence by activating key genes. In vitro and in vivo studies have established the oncogenic potential of PIM1 kinases. These kinases have been implicated in tumor progression, metastasis, and resistance to chemotherapy, underscoring their potential as a therapeutic target for cancer therapy. This review delves into the intricate molecular mechanisms through which PIM1 interacts with specific substrates in different tumor tissues, leading to diverse outcomes in various human cancers. Over the past decade, the inhibition of PIM1 in cancers has garnered significant attention as a potential standalone treatment. Various in vitro, in vivo, and early clinical trial data have provided support for this approach to varying extents. Novel compounds that inhibit PIM1 kinase have shown effectiveness and a favorable toxicity profile in preclinical studies. Several of these substances are now being studied in clinical trials due to their promising outcomes. This article provides a thorough examination of the PIM1 kinase pathways and the recent advancements in producing PIM1 kinase inhibitors for the treatment of cancer.
Keywords: Cancer, PIM1 Kinase, NDRG1, RBMY, Dynamin related protein 1, Hu antigen R
Introduction
PIM1 kinase was discovered in 1989 at the proviral insertion site of the Moloney murine leukemia virus. It is identified as a serine/threonine kinase and belongs to the PIM kinase family, which also includes PIM2 and PIM3. All three of these protein kinases are members of the Ca2 + /calmodulin-dependent family. [1, 2]. While PIM1 is located on chromosome 6p21.2, PIM2 and PIM3 are on chromosome X and chromosome 22q13, respectively [3]. Human PIM proteins exhibit significant sequence similarity at the amino acid level. PIM1 and PIM3 share 71% homology, PIM1 and PIM2 have 61% homology, and PIM2 and PIM3 display 44% homology [3]. The transcript of PIM1 messenger RNA (mRNA) is expressed by six exons, accompanied by extensive five ′ and three ′ untranslated regions (UTRs). These regions include a GC-rich area and five instances of AUUA destabilizing motifs, which are generated through alternative splicing (Fig. 1A) [4, 5]. PIM1 generates two protein isoforms, 34 kDa and 44 kDa, because of an alternate translation start occurring at a CUG codon located upstream. The shorter isoform is found in both the cytoplasm and nucleus, while the longer form is localized to the plasma membrane. Both isoforms feature a proline-rich N-terminal motif that binds to the ETΚ-SH3 domain, aiding their recruitment to cell membranes [4]. In contrast, PIM2 produces three isoforms -34 kDa, 37 kDa, and 40 kDa—while PIM3 is known to produce only one protein variant. Despite differences in molecular mass among these isoforms, they all retain serine/threonine kinase activity (Fig. 1A) [5, 6]. The crystallographic structure of PIM1 kinase, displays a conventional bi-lobed kinase architecture. It consists of an N-terminal lobe (residues 37–122) that is largely composed of β-sheets, alongside a C-terminal lobe (residues 126–305) that is mainly formed of α-helices, with a significant cleft in between [7]. This cleft contains several residues that act as the ATP binding site, which are conserved among all three PIM kinases. The connection between the two lobes is facilitated by a distinctive hinge region (residues 123–125), which includes a proline residue with a tertiary amine, a feature unique to PIM kinases. This particular attribute provides an opportunity for the selective inhibition of PIM kinases without impacting other kinases. Crystallographic analyses indicate that PIM2 possesses a conformation that is analogous to that of PIM1 [8]. While the crystal structure of PIM3 kinase has yet to be elucidated, scientists believe it likely resembles the structure of PIM1, given the high level of sequence homology between PIM1 and PIM3. The expression patterns of these kinases differ across tissues: PIM1 is predominantly present in hematopoietic cells, while PIM2 is found in the brain and lymphocytes. PIM3 is expressed in the kidneys, breast, and brain cells [9]. Unlike other proteins, these kinases lack a regulatory domain, resulting in their constant activation and regulation through transcription, translation, and proteasomal degradation [10]. Additionally, microRNAs and hormones influence PIM1 expression post-transcriptionally, in addition to a number of substances that regulate it at the transcriptional level.
Fig. 1.

A PIM kinase family gene structures. The gene structures of the PIM kinase family consist of six codons surrounded by large untranslated regions (UTR). Within each gene, there are alternative start codons (*) that correspond to different mRNA transcripts, resulting in various isoforms. These isoforms contain a serine/threonine kinase domain, with molecular weights ranging from 34–44 kDa. The largest isoform is depicted in detail, highlighting the locations of the kinase domain, hinge regions, and key sites such as K67, H68, P81, P123, and S261 for PIM1; P119, K121, D128, and E171 for PIM2; and Q86V86 and S190 for PIM3. B Transcription factors responsible for PIM1 gene transcription. Growth factors and cytokines found in cancer cells trigger the activation of intracellular tyrosine kinases and receptor-associated kinases, such as Janus Kinase. This leads to the activation of STATs through tyrosine phosphorylation. Once phosphorylated, STAT3s form dimers through phosphotyrosine-SH2 linkage and move to the nucleus. In the nucleus, they promote the transcription of target genes, such as PIM1, resulting in the upregulation of PIM1 mRNA. Additionally, PIM protein levels are increased by hypoxia due to HIF1α. Green and red arrows represent activating and inactivating pathways, respectively
PIM1 is a protein with a relatively short lifespan, typically lasting only 5–15 min in primary cells [11]. However, in specific tumor cells like K562 and BV173 that express the BCR-ABL fusion protein, its half-life can be extended up to 100 min. This prolonged lifespan is thought to be a result of HSP90-mediated protection, which prevents ubiquitylation along with subsequent proteasomal degradation [12, 13]. Moreover, additional researchers have found that the protein phosphatase PP2A can destabilize PIM1. Dephosphorylation of PIM kinases by PP2A facilitates the ubiquitination and subsequent degradation of these proteins via the proteasome [14, 15]. Several research teams have observed elevated levels of PIM proteins when small-molecule PIM inhibitors are employed, suggesting that PIM1 kinase can influence their own degradation process through auto-/transphosphorylation [16, 17]. The stability of PIM proteins is carefully controlled by protein–protein interactions, with chaperone proteins such as HSP90 and HSP70 playing a crucial role in this process. The association of PIM1 with HSP90 prevents its degradation, while HSP70 binds to ubiquitinated PIM1, facilitating its breakdown. These protein interactions are essential for regulating the stability of PIM1 proteins [12, 13]. Treatment with PIM inhibitors lead to an increase in PIM protein levels by preventing its degradation and increasing the half-life of the protein. Collectively, these findings underscore the critical role of upstream kinases and/or phosphatases in the signaling pathways of PIM kinases [14, 15].
The increased expression of PIM1, which is linked to a negative prognosis, has been observed in various cancerous tissues, including leukemia, liver, lung, myeloma, prostate, and breast cancers. However, the exact mechanisms by which PIM1 regulates cancer are not yet fully comprehended. This review explores the molecular mechanisms involved in the recruitment of distinct tissue-specific substrates by PIM1, resulting in diverse outcomes in different types of solid tumor.
Transcription factors responsible for PIM1 gene activation
PIM1 kinase genes are categorized as rapid response genes, which are transiently activated by specific stimuli such as growth factors, interleukins, granulocyte-colony stimulating factor, granulocyte–macrophage colony-stimulating factor, and interferons [1]. These stimuli initiate the JAK/STAT signaling pathway, a critical mechanism for regulating PIM1 gene expression. The JAK/STAT pathway is triggered when cytokines and growth factors connect to their specific receptors. This interaction results in the phosphorylation of cytoplasmic receptor domains by JAK kinases, which in turn provide binding sites for STATs and other signaling proteins (Fig. 1B). JAK phosphorylates STATs, which causes them to dimerize and move into the nucleus. Once in the nucleus, these dimers bind to specific gene promoter regions, such as the ISFR/ GAS sequence found in the PIM1 promoter, to up-regulate PIM1 gene expression [18–20]. Conversely, PIM1 negatively regulates the JAK/STAT pathway by binding to and activating suppressors of cytokine signaling (SOCS) proteins in hematopoietic cells [21].
PIM1 is also involved in tumorigenesis by influencing the transcriptional activities of Myc, cap-dependent protein translation, and cell cycle progression [22–24]. It regulates survival signals primarily through the phosphorylation of BAD and apoptosis signaling kinase 1 (ASK1) [25, 26]. Furthermore, PIM1 expression can be increased through the activation of homeobox protein HOXA9 and nuclear factor-κB (NF-κB) (Fig. 1B) [27–29]. HOXA9 interacts with a distant region of the PIM1 promoter, leading to the upregulation of PIM1 in hematopoietic progenitor cells [27]. NF-κB can rapidly induce PIM1 expression in response to tumor necrosis factor-α (TNF-α) stimuli, and this process is mediated by STAT5 [28]. In addition to phosphorylating RelA/p65, the primary subunit of NF-κB, PIM1 inhibits its degradation through ubiquitin-mediated proteolysis [29]. Phosphorylation of RelA/p65 by PIM1 introduces a novel and vital mechanism for controlling NF-κB function, affecting its stability and activity.
PIM1 functions in cancer
PIM1 kinase acts as a proto-oncogene and is accountable for phosphorylating a broad range of protein substrates that govern key biological functions, including cell cycle advancement, apoptosis, and transcriptional activation [30]. Phosphorylation modulates numerous aspects of protein function, such as enzymatic activity, subcellular localization, stability, and interactions with other proteins. It is also associated with various signaling pathways and disease states. Elevated levels of PIM1 expression have been noted in several kinds of cancers, like breast, prostate, pancreatic, gastric, colorectal, leukemia, lung, and head and neck squamous cell carcinomas [31]. This overexpression is associated with the emergence of resistance to treatment in these malignancies. The pro-tumorigenic effects of PIM1 kinases stem from their capacity to phosphorylate specific serine/threonine motifs, which boosts the activity of target proteins involved in critical cellular processes such as proliferation, survival, cap-dependent translation, metastasis, and tumor development [32].
Role of PIM1 in Breast Cancer
Breast cancer tissues express PIM1 at far higher levels than normal breast epithelium [33–35]. Moreover, PIM1 is excessively expressed in TNBC and is linked to cell survival, tumor growth, and resistance to chemotherapy, indicating its potential as a dependable biomarker for TNBC [35–37]. The expression of PIM1 is contingent upon various cytokines, growth factors, and mitogens. It has been previously reported that estrogen and estrogen receptor alpha-binding regions are the potential enhancers of PIM1 [38]. There are four estradiol-bound ER-α binding sites located far upstream of the PIM1 promoter, which may act as estrogen-regulated enhancers for PIM1 [38]. Estradiol-induced PIM1 overexpression leads to the phosphorylation of key proteins, which in turn suppresses the expression of cell cycle inhibitors (CDKN1A and CDKN2B). This process impedes apoptosis, accelerates cell cycle progression, and enhances the invasiveness of breast cancer tumors. (Fig. 2A) [38]. IL-6 is a cytokine secreted by immune cells, fibroblasts, and cancer cells and has been shown to be a crucial component for the aggressiveness of breast cancer. In their study, Gao et al. showed that PIM1 was activated at the transcriptional level by the IL-6/STAT3 pathway, serving as an essential factor in the induction of EMT and stemness in breast cancer cells stimulated by IL-6 [39]. EMT and stemness were induced through the c-Myc, a target /cofactor for PIM1 [24, 39]. Triple negative breast cancers (TNBCs) exhibit an elevated expression of the Myc oncogene, which collaborates with PIM1 to impede cell proliferation, migration, and apoptosis in TNBCs. Suppression of PIM1 influences the expression of Myc and its transcriptional target MCL1 through various mechanisms involving the phosphorylation of histone-H3 at S10 and the phosphorylation of c-Myc at S62 (Table 1). These phosphorylation events are recognized as crucial for c-MYC-driven transcriptional activation along with the development of oncogenic properties [40]. An additional mechanism that appears vital in promoting Myc-driven tumorigenesis is the survival signaling pathway mediated by PIM1 kinase, which modulates the threshold for apoptosis induction through BAD phosphorylation (Fig. 2A) [24]. The interaction between PIM1 and c-myc influences the expression of SHP2 (PTPN11) and EPHA2, both of which are oncogenic proteins with critical roles in breast cancer. [41]. Tumor-initiating cells and the advancement of breast cancer are linked to the phosphatase SHP2, which is encoded by PTPN11 [40]. Along with PIM1, EPHA2 is associated with the control of SHP2 and belongs to a class of kinases associated with a poor prognosis in ER-negative breast tumors.
Fig. 2.

A PIM1 enhances aggressive oncogenic characteristics in breast cancer through multiple mechanisms. PIM1 expression is triggered by estradiol in breast cancer cells, and this triggering is facilitated by ERα-regulated enhancers situated distally upstream from the gene. Increased PIM1 levels enhanced the proliferation of breast cancer cells, accompanied by decreased expression of cyclin-dependent protein kinase inhibitors CDKN1A and CDKN2B. PIM1 was also transcriptionally activated by the IL-6/STAT3 axis, playing a crucial role in promoting EMT and stemness in breast cancer cells. This process involves phosphorylation of c-Myc, a target/cofactor for PIM1, which contributes to MYC-driven tumorigenesis through phosphorylation of Histone-H3 at S10. PIM1 could enhance the stem cell-like traits of breast cancer cells by promoting the phosphorylation and cytoplasmic localization of RUNX3. The translocation of RUNX3 from the nucleus hindered its function as a tumor suppressor, leading to the acquisition of stem cell traits by breast cancer cells. PIM-mediated phosphorylation inhibits CSL-dependent activity of Notch3 but promotes that of Notch1. Despite the difference, both cases of phosphorylation ultimately lead to increased tumor growth. Green circles show activating phosphorylation events. Green and red arrows represent activating and inactivating pathways, respectively. B PIM1 promotes prostate cancer through the phosphorylation of diverse substrates. PIM1 plays a crucial role in prostate cancer by phosphorylating the AR at S213, affecting AR target gene expression. Additionally, PIM1 targets the AR co-activator, 14–3-3 ζ, at S64, leading to alterations in AR transcriptional activity. PIM1 also phosphorylates the metastasis suppressor protein NDRG1 at S330, resulting in reduced stability, nuclear localization, and interaction with AR. PIM1 enhances NFATC1 transcriptional activity by phosphorylating it at S257 and S335, thereby promoting prostate cancer cell migration, invasion, metastasis, and tumor angiogenesis. Additionally, PIM1 phosphorylates Capza1 at S106 and S126, and Capzb2 at S182, S192, and S226, resulting in enhanced adhesion and migration of human prostate cancer cells
Table 1.
Direct PIM1 substrates in various solid cancers
| No | Substrate | Cancer Types | Phosphorylation sites | Protein types | Function | References |
|---|---|---|---|---|---|---|
| 1 | c-Myc | Breast cancer | S62 | Transcription factor | Cell proliferation | [40] |
| 2 | H3 | Breast cancer | S10 | Histone proteins | Stimulates transcription of specific subset of myc dependent genes | [40] |
| 3 | Notch1 | Breast cancer | S2152 | Transmembrane protein | Notch1 nuclear localization, cell migration, Glycolytic switch and CSL-dependent tumor growth | [42] |
| 4 | Notch3 | Breast cancer | S1673 | Transmembrane protein | Nuclear localization and CSL-independent tumor growth | [43] |
| 5 | AR | Prostate cancer | S213 | Transcription factor | Higher grade tumors, castration-resistant disease, and altered AR transcriptional activity | [44, 45] |
| 6 | 14–3-3 ζ | Prostate cancer | S64 | AR co-activator | Altered AR transcriptional activity | [46] |
| 7 | NDRG-1 | Prostate cancer | S330 | Stress responsive protein | Cell migration and invasion | [47] |
| 8 | NFATC1 | Prostate cancer |
S257 S335 |
Transcription factor | Increased invasion, migration and angiogenesis | [48] |
| 9 | Capza1 | Prostate cancer |
S106 S126 |
Capping protein |
Increase adhesion and migration |
[48] |
| 10 | Capzb2 | Prostate cancer |
S182 S192 S226 |
Capping protein |
Increase adhesion and migration |
[48] |
| 11 | ABI-2 | Prostate cancer | S183 | Wave regulatory complex protein | Increase protein stability, WARC formation, enhanced actin dynamics and tumor invasion | [49] |
| 12 | AKT | HCC | S473 | Kinase | Tumor growth, cell invasion and metastasis | [50] |
| 13 | RBMY | HCC | - |
RNA binding motif protein |
Increased proliferation, invasion and migration |
[51] |
| 14 | eIF4B | Lung cancer | S406 | Eukaryotic translation initiation factor |
Activation of c-MET downstream signaling pathway |
[52] |
| 15 | c-Myc | Ovarian cancer | S62 | Transcription factor |
Increase cell proliferation and glycolysis |
[53] |
Liu and colleagues demonstrated the function of PIM1 kinase in regulating breast cancer stem cells (BrCSCs) and discovered potential new targets for eliminating the BrCSC population. Evidence suggests that PIM1 improves breast cancer cells' stem cell characteristics via increasing human runt-related transcription factor 3 (RUNX3) phosphorylation and cytoplasmic localization. This process leads to the inactivation of RUNX3, as its displacement from the nucleus imparts stem cell-like characteristics to breast cancer cells. Inhibition of PIM1 activity significantly increases the nuclear localization of RUNX3, restores its transcriptional function, and reduces the stem cell-like features in breast cancer cells. (Fig. 2A) [34]. The dysregulation of the Notch signaling pathway, that has a vital function in the homeostasis and development of tissues, can contribute to the progression of tumors. Prior research has shown that the oncogenic PIM1 kinase phosphorylates Notch1 and Notch3 but not Notch2 [42, 43]. Specifically, PIM1 kinase phosphorylates Notch1 on S2152 (equivalent to human NOTCH1 S2162) within the intracellular domain (Table 1). This phosphorylation event enhances the nuclear localization as well as transcriptional activity of Notch1. The interaction between PIM1 and Notch1 proteins promotes tumorigenicity and cell proliferation in a CSL (C promoter–binding factor 1, Suppressor of Hairless, Lag-1) dependent manner [42]. PIM1 kinases are responsible for phosphorylating Notch3 at S1673(equivalent to human NOTCH3 S1672) in the RAM domain, leading to the disruption of its interaction with CSL and subsequent inhibition of the canonical CSL-dependent transcriptional program (Table-1) [43]. Despite this, phosphorylated Notch3 persists in supporting cell survival and tumor proliferation under estrogenic conditions (Fig. 2A) [43]. In summary, PIM kinases are essential in modulating the signaling pathways of different Notch paralogs, thereby influencing breast cancer development through both CSL-dependent and CSL-independent pathways.
PIM1 acts as an oncogene in Prostate Cancer
Among male genitourinary system tumors, prostate cancer ranks high and is the 2nd most prevalent cause of cancer-related mortality. It primarily affects older men [54]. High-grade prostate intraepithelial neoplasia and castration-resistant prostate cancer tissues show elevated levels of PIM1 kinase compared to normal prostatic tissue and benign prostatic hyperplasia [55]. The frequent coexpression of PIM1 kinase and the Myc gene in human prostate cancer significantly enhances c-Myc-driven tumorigenesis in a manner dependent on the kinase activity [56]. In mouse studies conducted both in vitro and in vivo, it has been observed that PIM1 exhibits weak tumorigenic properties. However, when coexpressed with Myc, it synergizes significantly. Previous research suggests that PIM1 could facilitate Myc tumorigenesis by stabilizing the Myc protein or boosting its transcriptional activity [57]. Wang et al. showed that silencing PIM1 led to reduced cellular proliferation, decreased Erk signaling, and diminished cancer malignancy, even though Myc levels did not change. This suggests the main function of PIM1 in maintaining tumorigenicity [57]. As a result, targeting PIM1 could prove to be an effective approach for prostate cancer therapy.
PIM1 plays a significant role in prostate cancer by phosphorylating the androgen receptor (AR) at Serine 213 (S213), which is crucial for AR's function and affects its target gene expression, as detailed in Table 1 [44, 45]. This particular phosphorylation event is connected to more aggressive tumors, castration-resistant forms of the disease, and alterations in AR's transcriptional activity, as depicted in Fig. 2B [44, 45, 58]. Nonetheless, the precise mechanism that determines the phosphorylation of AR at S213 affects its ability to activate transcription remains unclear. Recent research has shown that PIM1 also phosphorylates the AR co-activator 14–3-3 ζ at Serine 64 (S64), which affects AR transcriptional activity in prostate cancer (Table 1) [46]. This phosphorylation by PIM1 is thought to modulate AR’s interaction with 14–3-3 ζ and, subsequently, their chromatin binding, potentially affecting AR’s activity by recruiting additional co-regulators like hnRNPK and TRIM28 [46]. Furthermore, PIM1 interacts with the metastasis suppressor protein N-Myc Downstream-Regulated Gene 1 (NDRG1) at Serine 330 (S330), leading to reduced stability, altered nuclear localization, and diminished interaction with AR (Table 1) [47]. This results in enhanced cell migration and invasion in prostate cancer. Cellular Migration and adhesion are two important characteristics of cancer. PIM1 also phosphorylates three other substrates: NFATC1 [48] and capza1 and capzb2 [59]. Phosphorylation of PIM1 target sites by several other kinases like PKA and GSK3 contributes to the nuclear exit and the transcriptional inactivation of NFATC1. PIM1 does not influence the cellular localization of NFATC1; it directly phosphorylates the NFATC1 at S257 and S335 and increases its transcriptional activity (Table 1) [48]. When PIM1 phosphorylates NFATC1, it promotes prostate cancer cell motility, invasion, metastasis, and angiogenesis (Fig. 2B) [48].
The motility and invasion of cancer cells are attributed to irregularities in normal cellular movements, which are controlled by actin proteins. These actin proteins are regulated by the polymerization and depolymerization of capping proteins. Heterodimers of capping proteins attach to the elongating ends of actin filaments, thereby regulating actin dynamics [59]. It has been discovered that the PIM1 protein phosphorylates Capza1 at positions S106 and S126, as well as Capzb2 at positions S182, S192, and S226 (Table 1). Phosphorylation of these proteins leads to increased adhesion and migration of human prostate cancer cells. Moreover, this modification reduces the protective role of capping proteins at the ends of actin filaments., intensifying the fluctuations in actin dynamics (Fig. 2B) [59]. The PIM1 protein is elevated in response to hypoxia, and its increased expression is essential for the invasion and formation of cellular protrusions under hypoxic conditions [49]. Within the WAVE regulatory complex, ABI-2, a member, has been identified as a fresh substrate of PIM kinase [49]. PIM1 phosphorylates ABI2 at S183, thereby augmenting the activity of the WAVE regulatory complex and enhancing actin dynamics, ultimately propelling tumor cell invasion (Table 1) (Fig. 2B) [49]. These discoveries establish the initial signal transduction pathway that connects hypoxia towards the control of actin cytoskeletal dynamics and unveil PIM1 as a novel target for counteracting hypoxia-induced metastasis.
Role of PIM1 in Hepatocellular carcinoma (HCC)
Primary hepatocellular carcinomas (HCCs) have high levels of PIM1 kinase expression, and this expression is much higher in the extra-hepatic metastatic tissues of HCCs than it is in the original tumors [50]. Under hypoxic conditions, PIM1 expression is upregulated in various HCC cell lines, leading to increased nuclear translocation of the protein and enhanced protein stability. By manipulating cell metabolism, PIM1 has an important role in sustaining HCC growth as well as progression. This process is mediated through the activation of AKT phosphorylation and its downstream targets, which in turn promotes AKT-driven glycolysis. This pathway generates energy that supports the growth and progression of HCC in the hypoxic tumor microenvironment (Fig. 3A) [50]. RBMY (RNA binding motif protein Y-linked), an oncofetal protein, has been suggested as a potential biomarker for poor prognosis in HCC patients. In adult tissues, RBMY is primarily expressed in the testis, where it functions in spermatogenesis by encoding a nuclear protein specific to germ cells. This protein features an N-terminal RNA recognition motif, i.e., RRM, and includes four repeat regions at the C-terminus that are rich in serine, arginine, glycine, and tyrosine residues, collectively known as SRGY boxes [60]. Additionally, it has been observed that PIM1 phosphorylates RBMY, leading to its cytoplasmic accumulation and activation of oncogenic functions (Table 1) [51]. The binding of PIM1 to RBMY led to a reciprocal stabilization and significant relocation of RBMY from the nuclei to the mitochondria. Consequently, EMT was induced by regulating the levels of Snail1 and ZEB1 proteins. After RBMY is phosphorylated by PIM1, PIM1 associates with nuclear RBMY to enhance the transcription of Snail1 and ZEB1. Additionally, PIM1 interacts with cytoplasmic RBMY to promote the nuclear translocation of Snail1. These actions collectively promote the transcription of ZEB1 and vimentin while suppressing E-cadherin expression. Additionally, the PIM1-RBMY interaction facilitates a shift toward mitochondrial fission, which enhances ATP and ROS production, thereby supporting mitochondrial mobility. This mechanism contributes to tumor metastasis and adversely affects the prognosis of HCC patients (Fig. 3A) [51].
Fig. 3.

A PIM1 signaling in Hepatocellular carcinoma. PIM1 enhances AKT-driven glycolysis by phosphorylating AKT at S473, leading to the activation of its downstream targets. This process generates energy and supports the growth and advancement of HCC in the hypoxic tumor microenvironment. PIM1 phosphorylates RBMY, resulting in its cytoplasmic accumulation and activation of oncogenic functions. Upon phosphorylation PIM1 collaborates with nuclear RBMY to facilitate the transcription of Snail1 and ZEB1, and with cytoplasmic RBMY to induce the nuclear translocation of Snail1. These dual pathways synergistically boost the transcription of ZEB1 and vimentin leading to EMT and metastasis. The interaction of PIM1 with RBMY causes a reciprocal stabilization and substantial translocation of RBMY from the nuclei to the mitochondria. This PIM1-RBMY interaction prompts a shift towards mitochondrial fission, resulting in elevated ATP and ROS production for mitochondrial movement, acting as a mechanism to drive tumor metastasis. B Molecular mechanisms of PIM1 regulation in pancreatic Cancer. Under hypoxic conditions the cis-acting AU-rich elements present within a 38-base pair region of the PIM1 mRNA 3’-untranslated region mediate a regulatory interaction with the mRNA stability factor HuR. HuR, primarily expressed in the nucleus of PDAC cells, relocates to the cytoplasm in response to hypoxic stress, resulting in the stabilization and overexpression of PIM1 protein leading to enhance cell survival, DNA repair and development of chemoresistance. IL-6 stimulation triggers autophosphorylation of JAK and subsequently activates STAT3 protein. Phosphorylated STAT3, upon binding to activated JAK proteins, forms dimers that then translocate to the nucleus to control the transcription of crucial genes responsible for cell proliferation and cell cycle advancement. Green circles show activating phosphorylation events. Green and red arrows represent activating and inactivating pathways, respectively
PIM1 acts as an oncogene in Pancreatic Cancer
The PIM1 protein exhibits a marked increase in malignant pancreatic tumors compared to benign, inflammatory, and precancerous conditions, such as the normal pancreas, chronic pancreatitis, and benign intraductal papillary mucinous neoplasms [61]. Xu et al. found that PIM1 levels were markedly higher in pancreatic ductal adenocarcinoma (PDAC) tissues compared to their normal counterparts. Additionally, PIM1 expression was notably elevated in PDAC cell lines relative to the immortalized human pancreatic nestin-expressing epithelial cell line HPNE [62]. PIM1 is a hypoxia-inducible protein that is critical for cell transformation and tumorigenesis [63]. It has a short half-life and is predominantly degraded via the ubiquitin–proteasome pathway. In pancreatic cancer, PIM1 levels are elevated under hypoxic conditions compared to normoxic conditions. This increase occurs because, under normoxic conditions, PIM1 is targeted for degradation by the ubiquitin–proteasome pathway, whereas hypoxia inhibits this degradation process [63]. Under hypoxia, the PIM1 protein is mostly localized within the nucleus, whereas under normoxia, it is found in both the cytoplasm and nucleus. The mechanism by which PIM1 is regulated in the context of tumor hypoxia remains unknown. Evidence indicates that elevated PIM1 expression induced by hypoxia occurs independently of hypoxia-inducible factor 1α (HIF-1α) [63], and there are no known genetic alterations in PIM1 that can explain its abundance in PDAC cells. HuR, or Hu antigen R, is an RNA-binding protein essential for post-transcriptional gene regulation in cancer. Predominantly located in the nucleus, HuR aids in the export of mRNA transcripts to the cytoplasm and then returns to the nucleus. Under stress conditions, HuR moves to the cytoplasm, where it increases the stability of certain prosurvival mRNAs [64, 65]. The cis-acting adenylate-uridylate-rich elements (AREs) are located within a 38-base pair segment of the 3’-untranslated region of PIM1 mRNA (3’-UTR) interact with HuR to regulate mRNA stability under conditions of tumor hypoxia (Fig. 3B). In PDAC cells, HuR is mainly expressed in the nucleus but shifts to the cytoplasm when exposed to hypoxic stress. This translocation leads to the stabilization of the PIM1 mRNA, entailing elevated levels of PIM1 protein. Consequently, this interaction contributes to PIM1-mediated chemoresistance in PDAC cells [66]. Pancreatic cancer is characterized by the deregulation of multiple cellular signaling pathways. One specific target that has been extensively researched in PDAC is the oncoprotein K-Ras, which is found to be mutated in over 90% of PDAC cases [67]. PIM1 kinase, in contrast, is consistently present in pancreatic cell lines and is modulated by K-Ras signaling [62]. Acting downstream of K-Ras signaling, PIM1 kinase may serve as a marker for oncogenic K-Ras activity. Knocking down PIM1 with shRNA results in alterations in its downstream targets, Bad and p27, and leads to reduced anchorage-dependent and -independent growth, diminished invasion through Matrigel, and decreased radioresistance [62]. KRAS hyperactivation activates stromal fibroblasts and draws inflammatory mediators to the tumor microenvironment, boosting IL-6 production and tumor growth and invasion [68]. IL-6 stimulation triggers JAK autophosphorylation and subsequent STAT3 protein activation. Phosphorylated STAT3, upon binding to activated JAK proteins, generates dimers that translocate towards the nucleus and control the transcription of key genes connected to cell proliferation, survival, cell cycle advancement, angiogenesis, and immune suppression (Fig. 3B) [69]. The proto-oncogene PIM1 is a target gene regulated by STAT3-driven transcription, contributing significantly to tumor development and progression. PIM1 over-expression in Panc-1 and MiaPaCa2 cell lines did not change the rates of basal apoptosis, cell cycle, or proliferation. However, previous studies have shown that inhibiting PIM1 kinase through shRNA in MiaPaCa2 cells can induce apoptosis, reduce cell growth in anchorage-dependent and independent conditions, inhibit invasion through matrigel, along with reduce radioresistance [70]. In pancreatic cancer, a feedback loop exists between PIM1 and the EGFR signaling pathway. Activation of the EGFR pathway drives cell proliferation, anti-apoptotic processes, angiogenesis, metastasis, and resistance to both gemcitabine and EGFR-TKIs. Furthermore, it boosts the activity of stem cells across various cancers [71]. PIM1 influences sensitivity to the chemotherapy agent erlotinib and regulates the generation of pancreatic cancer stem cell markers via modulating the EGFR signaling pathway.
PIM1 promotes Lung cancer
Worldwide, lung cancer is the leading killer of cancer patients. There are two main forms of lung cancer based on histology such as small cell lung cancer (SCLC), which represents (15–20%) of cases, and non-small cell lung cancer (NSCLC), which constitutes (80–85%) of cases. SCLC is recognized for its more aggressive nature compared to NSCLC. Warnecke-Eberz et al. noticed that the expression of PIM1 mRNA was downregulated in NSCLC compared to normal tissues [72]. The progression of cancer to the lymph nodes was intimately linked to this downregulation. On the contrary, Jin et al. found that overexpression of PIM1 was linked to poor differentiation, advanced clinical stage, and the presence of lymph node and distant metastasis [73]. Pang et al. demonstrated that NSCLC, PIM1 expression was significantly aligned with tumor size, lymph node metastasis, histological type, and clinical staging [74]. Similarly, Jiang et al. observed that increased nuclear levels of PIM1 were correlated with lymph node metastasis, histological characteristics, and diminished survival in NSCLC patients [75]. Additionally, Cao et al. identified a positive correlation between PIM1 and c-MET expression in lung adenocarcinoma tissues. An important role for PIM1 in regulating c-MET and the signaling pathways leading from it, such as RAS/ERK, was suggested in their model, PI3K/AKT, and STAT3, through the phosphorylation of eIF4B at Serine 406 (S406) [52]. Depletion of PIM1 resulted in reduced cell proliferation, migration, invasion, and colony formation in vitro, as well as diminished tumor growth in vivo. These impacts were partly reversed by the restoration of c-MET expression, suggesting the involvement of the PIM1/c-MET signaling axis (Fig. 4A) [52]. In MET-amplified NSCLC cell lines, the application of a MET inhibitor led to the up-regulation of PIM1, which served as a mechanism for drug resistance. However, the sensitivity of cell lines resistant to MET inhibitors was restored by the application of a PIM inhibitor [76]. PIM1 contributes to the growth of chemoresistance in NSCLC by enhancing mitochondrial fusion. The equilibrium between mitochondrial fusion and fission is crucial for maintaining mitochondrial function, which in turn influences various cellular processes related to tumorigenesis, including metabolic alterations, proliferation, and apoptosis [77]. PIM kinases have been identified as regulators of dynamin-related protein 1 (DRP1), a main actor in mitochondrial fission. Inhibition of PIM results in elevated levels and increased activation of DRP1, leading to mitochondrial fragmentation and, consequently, apoptosis in lung cancer cells (Fig. 4A) [78].
Fig. 4.

A PIM1-mediated signaling pathways in lung cancer. The overexpression of PIM1 in lung adenocarcinoma leads to the phosphorylation of eIF4B at S406. This phosphorylated eIF4B then accelerates c-MET mRNA translation, which in turn enhances the RAS/ERK, PI3K/AKT, and STAT3 pathways in collaboration with oncogenic drivers, resulting in heightened cell proliferation, survival, migration, and invasion. PIM kinases also affect mitochondrial dynamics, ROS production, and the response to chemotherapy in lung cancer. Inhibiting PIM1 significantly raises the protein levels and mitochondrial localization of Drp1, causing marked fragmentation of mitochondria, free radical generation, and apoptosis. B PIM1 promotes colorectal cancer (CRC) via several mechanisms. PIM1 is integrated into a complex regulatory network involving mitogenic and antiapoptotic signals. Inhibiting PIM1 has been found to result in significant changes in oncogenic signal transduction, affecting p21Cip1/WAF1, STAT3, c-jun-N-terminal kinase (JNK), c-Myc, and survivin, as well as the levels of apoptosis-related proteins Puma, Bax, and Bcl-xL. PIM1 facilitates the Warburg effect in CRC cells. When glucose is scarce, AMPK is activated through phosphorylation, leading to the upregulation of PIM1 expression. PIM1 then stimulates the expression of several glycolytic enzymes, such as HK2 and LDHA. Additionally, the Warburg effect is intensified as a compensatory mechanism to enhance the survival and proliferation of CRC cells. The proliferation of CRC is facilitated by TAB3 through the upregulation of PIM1 expression. TAB3 modulates the activity of PIM1 via the STAT3 signaling pathway. From a mechanistic standpoint, TAB3 regulates the expression of PIM1 by stimulating the phosphorylation and activation of STAT3 through the formation of the TAB3-TAK1-STAT3 complex. Therefore, targeting specific inhibitors that affect this TAB3-TAK1-STAT3 complex may hold promise as potential therapeutic agents for CRC. Green circles show activating phosphorylation events. Green and red arrows represent activating and inactivating pathways, respectively
PIM1 acts as an oncogene in Colorectal cancer
Colorectal cancer (CRC) is a major life-threatening cancer where PIM1 is crucial in promoting cell survival and growth. In CRC, PIM1 is notably present in tumors, tumor stroma, and adjacent mucosa, serving as a valuable prognostic indicator for the disease [79, 80]. Weirauch et al. showed that the antitumor effect of PIM1 involved alteration in several downstream proteins that control apoptosis, cell survival, and cell migration [81]. PIM1 knockdown decreased survivin mRNA and protein levels and phosphorylation of STAT3 at T705 and S72. Previous studies have indicated that STAT3 regulates PIM1 and survivin expression. Furthermore, PIM1 knockdown involved a slight reduction in c-Myc levels and a more significant decrease in p53 expression. Interestingly, the suppression of E-cadherin expression was also observed upon PIM1 knockdown. This loss of E-cadherin leads to EMT, which disrupts cell–cell contacts and promotes increased migration and metastasis. Moreover, PIM1 knockdown induced apoptosis and led to the downregulation of the antiapoptotic protein Bcl-xL and the upregulation of the proapoptotic protein Bax. Additionally, the proapoptotic protein Puma, which is directly regulated by p53, was also downregulated upon PIM1 inhibition (Fig. 4B) [81]. Zhang et al. showed that PIM1 acts as an oncogene that is responsive to glucose deprivation in CRC [82]. In the context of glucose deprivation, PIM1 expression was increased in CRC tissues, thereby promoting the Warburg effect as a compensatory mechanism to enhance CRC cell survival and proliferation [82]. The altered expression of PIM1 predominantly influences glucose consumption and lactate production by regulating the genes Hexokinase 2 (HK2) and Lactate Dehydrogenase A (LDHA) [82]. TAK1-Binding Protein 3 (TAB3) is a member of the TAB family and is activated by Transforming Growth Factor-β-Activated Kinase 1 (TAK1), is integral to processes such as autophagy, immune response, and cancer progression [83–85]. In CRC tissues, both TAB3 and PIM1 are aberrantly upregulated, and their expression levels are positively correlated [86]. Furthermore, in vitro and in vivo surveys have shown that TAB3 is enhanced by increasing the expression of PIM1. This regulation of PIM1 by TAB3 is primarily mediated through the STAT3 signaling pathway. The TAB3-TAK1 complex interacts with STAT3, thereby influencing its phosphorylation and activation [86]. Mechanistically, TAB3 promotes PIM1 expression by facilitating the phosphorylation and activation of STAT3 by forming the TAB3-TAK1-STAT3 complex. (Fig. 4B). Any malfunction in TAB3/PIM1 signaling could be a contributing factor to CRC tumorigenesis.
Role of PIM1 in Ovarian Cancer
After breast cancer, ovarian cancer (OC) stands as the second most deadly cancer affecting women on a large scale. PIM1 kinase is upregulated in OC and is crucial in regulating various proteins linked to tumorigenesis [53]. The phosphoinositide 3-kinase (PI3K)/protein kinase B (AKT)/mammalian target of rapamycin (mTOR) signaling pathway is a crucial driver in the progression of OC [87]. PIM kinases are involved in maintaining the activity of the PI3K/AKT/mTOR pathway (Fig. 5A) [88]. They can influence this pathway by modulating PI3K through insulin receptor substrates (IRS) and affecting AKT via reactive oxygen species (ROS) [89]. PIM and AKT intersect in their control of cell growth and translation, phosphorylating numerous shared substrates to regulate mTORC1. These PI3K/AKT/mTOR pathway constituents are essential in ovarian cancer [90]. The interactions between PIM kinases and each component of the PI3K/AKT/mTOR pathway suggest that they might influence OC either directly or through modulation of the PI3K/AKT/mTOR signaling network. Cancer is characterized by various hallmarks, with deregulating cellular metabolism having a significant impact on OC [88]. The Warburg effect, or aerobic glycolysis, is crucial in regulating cellular metabolism in cancer cells. This process involves rapid glucose metabolism into lactate, regardless of oxygen availability, to support cell survival and biomass production. In OC, the process of aerobic glycolysis plays a vital role in the growth of tumors [91]. C-Myc controls aerobic glycolysis through the overexpression of numerous important glycolytic genes like LDHA, GLUT1, and PKM2 [92–94]. Wu et al. have shown the involvement of PIM1 in regulating cell metabolism through glycolysis in ovarian cancer [53]. The phosphorylation of c-Myc at S62, which is downstream of PIM1, serves as a significant controller of OC metabolism (Table 1) [53]. PIM1 could potentially influence metabolic alterations in OC cells through the c-Myc-PGK1 pathway (Fig. 5A).
Fig. 5.

A PIM1 activates AKT signaling in ovarian cancer. PIM kinases are responsible for the activation of mTOR and its upstream effectors, such as AKT. The activation of both PIM1 and AKT contributes to the proliferation of OC cells by inhibiting apoptosis (through the phosphorylation of BAD) and preventing cell cycle arrest by hindering the nuclear translocation of p53 and p27. Additionally, PIM1 plays a crucial role in regulating OC metabolism. It phosphorylates c-Myc at S62 and controls aerobic glycolysis by upregulating several key glycolytic genes, ultimately leading to tumor growth and metastasis. Green and red arrows represent activating and inactivating pathways, respectively. B miR-33a directly targets PIM1 in gastric cancer cells. In gastric cancer tissues, the downregulation of miR‑33a correlated with the upregulation of CDK‑6, CCND1, and PIM1, leading to the suppression of gastric cancer cell growth by inducing G1 phase arrest. C PIM1 signaling in glioblastoma. EGFR is involved in promoting PIM1 expression in GBM cells. The regulatory mechanisms of PIM1 may vary depending on whether EGFR is wild-type or mutant. EGFRwt mainly activates MAP/ERK kinase (MEK) and signal transducer and activator of transcription 3 signaling, whereas EGFRvIII triggers both the MEK and the PI3K-Akt1 cascade. As a result, it is conceivable that PIM1 is more highly expressed in GBM with EGFRvIII due to the combined stimulation of PIM1 expression by MEK/ERK and PI3K-Akt1 pathways. Green and red arrows represent activating and inactivating pathways, respectively
Oncogenic PIM1 in Gastric cancer
PIM1 kinase was found to be overexpressed in gastric cancer as well as connected with tumor grading [95]. In healthy gastric epithelial cells, PIM1 activity was limited to the nucleus. However, abnormal expression and localization were observed in 75.6%, i.e., 68/90 of human gastric cancer tissues, characterized by reduced nuclear expression and/or rise in cytoplasmic expression [96]. The altered expression pattern showed a notable inverse relationship between nuclear and cytoplasmic levels. Immunostaining results indicated stronger expression in gastric carcinoma, with 46% of cases exhibiting high levels, compared to 16% in gastric glands [95]. Clinicopathological analyses revealed that diminished nuclear PIM1 expression was associated with poorer survival outcomes and more extensive tumor invasion, whereas elevated cytoplasmic PIM1 expression was inversely related to lymphovascular invasion [96]. Members of the miR-33 family, particularly miR-33a, are involved in regulating cholesterol metabolism, fatty acid oxidation, and insulin signaling. In the context of gastric cancer, miR-33a functions as an oncogene suppressor. Research conducted by Wang et al. has demonstrated that the expression of miR-33a is reduced in gastric tumors [97]. Increased miR-33a levels lead to the downregulation of PIM1, cyclin D1, and cyclin-dependent kinase (CDK6), the proliferation of stomach cancer cells, and induce cell cycle arrest (Fig. 5B) [97]. Among these three target genes, the interaction between miR-33a and PIM1 significantly impacts gastric cancer [97].
PIM1is an oncogene in Glioblastoma
Glioblastoma multiforme (GBM) represents the primary brain tumor in adults. PIM1 expression in GBM is significantly elevated in comparison to non-malignant brain tissue [98]. This increase in PIM1 kinase activity may provide for the generation and progression of GBM, as PIM1 has a vital role in controlling tumor proliferation, apoptosis, and migration [98]. Research shows that the expression of PIM1 is influenced by the epidermal growth factor receptor (EGFR) [99], which is frequently overexpressed in GBM (Fig. 5C). In LN18 glioblastoma cells, treatment with EGF leads to a significant upregulation of PIM1 mRNA and protein levels, whereas the EGFR kinase inhibitor AG1478 greatly decreased PIM1 expression. Screening patients for EGFR wild-type (EGFRwt) and the deletion mutant EGFRvIII using PCR identified that 18.5% of GBM cases were positive for the EGFRvIII mutation, consistent with previous reports on glioblastoma [100, 101]. Notably, in PIM1, there was a substantial increase in mRNA expression in GBM with the EGFRvIII variant in comparison with those with the EGFRwt variant. This suggests distinct regulatory mechanisms for PIM1 by the wild-type versus mutant EGFR. While EGFRwt mainly activates the MAP/ERK kinase (MEK) pathway along with STAT3, EGFRvIII stimulates both the MEK and the PI3K-Akt1 pathways (Fig. 5C) [102]. Consequently, it is plausible that PIM1 is more upregulated in GBM with EGFRvIII due to the dual induction of PIM1 expression by MEK/ERK and PI3K-AKT1 pathways. These findings propose that EGFR plays a role in stimulating PIM1 expression in GBM cells, suggesting the possibility of combining PIM1 inhibitors with other treatments to increase the effectiveness of EGFR tyrosine kinase inhibitors [100]. Suppression of GBM growth was noted when PIM1 was silenced using U1 adaptor-mediated methods in GBM cells within a subcutaneous xenograft model in mice [103]. Additionally, inhibition of PIM1 increased the sensitivity of GBM cells to apoptosis triggered by ABT-737, which targets BCL2, BCL2-like 1 and BCL2-like 2 proteins [104]. Therefore, the blockade of PIM1 could offer a novel therapeutic approach for GBM. Although substantial research has been conducted on the functional link between PIM1 and GBM in recent years, the specific function of PIM1 in glioblastoma growth and maintenance remains inadequately understood. Inhibition of PIM1 has been found to reduce the expression of stem cell markers CD133 and Nestin in GBM cells (LN-18, U-87 MG) [105]. Conversely, an increase was observed in the expression of CD44 and the astrocytic differentiation marker GFAP. Additionally, it modulates the expression of SPARC (secreted protein acidic and rich in cysteine), which enhances GBM cell invasion while reducing cell proliferation [105]. Moreover, PIM1 expression was found to be enhanced in neurospheres, which serve as a model for GBM stem-like cells. Administering PIM1 inhibitors (LY294002, Quercetagetin, and TCS PIM1) to neurospheres led to reduced cell viability, decreased DNA synthesis rate, heightened caspase three activity, lowered PCNA protein expression, along with diminished neurosphere [105]. These findings suggest that PIM1 influences the behavior of glioblastoma stem cells, and targeting it could potentially eliminate glioblastoma stem-like cells, highlighting PIM1 as a promising target for anti-glioblastoma therapy.
PIM1 is oncogenic in Oral Cancer
Chiang et al. reported a notable increase in both PIM1 mRNA and protein expression levels in oral squamous cell carcinoma (OSCC) in comparison with non-cancerous matched tissue from the tumor periphery [106]. On the other hand, Beier et al. found that the PIM1 protein was overexpressed in 98% of invasive Head and Neck Squamous Cell Carcinoma (HNSCC), while it was absent in non-neoplastic head and neck squamous cell epithelium [107]. PIM1 expression in OSCC is a predictor of metastasis because it stimulates cancer cell motility even in the early stages of cancer development. PIM1 primarily boosts the activity of Rac1, influencing the organization of cytoskeleton filaments. This, in turn, enhances cell motility and metastatic ability [108]. Salivary gland adenoid cystic carcinoma (SACC) is a malignant tumor of the salivary gland, representing about 10% of all epithelial salivary gland tumors, with a 5-year survival rate below 20%. Due to its limited response to radiotherapy and chemotherapy, surgical resection is the most common treatment approach. The role of PIM1 in ACC has not been extensively studied. Zhu et al. found that silencing PIM1 led to decreased cell proliferation, induced apoptosis, caused cell cycle arrest, and reduced cell invasion in SACC-83 and SACC-LM cell lines [109]. RUNX3 is present in both normal salivary glands and ACC and is associated with histopathological growth patterns, T stage, distant metastasis, and patient survival. Zhu et al. employed immunohistochemistry to reveal a significant inverse correlation between PIM1 and RUNX3. Both PIM1 and RUNX3 levels were significantly related to T stage and nerve invasion. Furthermore, higher PIM1 expression was linked to reduced radiosensitivity. Peltola et al. demonstrated that PIM1 expression helps protect HNSCC cells from radiotherapy-induced damage [100].
PIM1 role in other solid cancers
PIM1 Kinase has been identified as an important factor in several other solid tumor types, such as endometrial cancer, melanoma, renal cancer, and papillary thyroid cancer. In endometrial cancer, specifically uterine serous carcinoma (USC), PIM1 is found to be overexpressed and associated with a poor prognosis, driving tumor cell proliferation, migration, and invasion through the phosphorylation of MYC and Wee1 [110]. Inhibiting PIM1 with SGI-1776 has revealed potential therapeutic advantages [110]. PIM kinases have also been shown to be significantly overexpressed in samples and cell lines derived from melanoma patients [111]. PIM1 inhibition, either through knockdown techniques or the application of the clinically available PIM kinase inhibitor demonstrated a reduction in cell proliferation, viability and invasion in preclinical models. Furthermore, cells exhibiting high levels of PIM1 were associated with resistance to BRAF (B-Raf proto-oncogene) and MEK (Mitogen-Activated Protein Kinase) inhibitors. Consequently, the use of PIM inhibitor, SGI-1776, was found to improve treatment outcomes when used in conjunction with these agents [111]. Mahalingam et al. demonstrated that PIM1 kinase levels are elevated in renal cell carcinoma cell lines when compared to normal renal proximal tubule epithelial cells (RPTEC) [112]. PIM1 kinase can phosphorylate the oncogene c-Myc, resulting in its stabilization and increased transcriptional activity. The use of SGI-1776 was shown to lower both phosphorylated and total c-Myc levels, thereby modulating the expression of c-Myc target genes. Moreover, the combination of SGI-1776 with sunitinib led to a further decrease in c-Myc levels, enhancing the cytotoxicity of sunitinib against renal cell carcinoma [112]. Finally, in papillary thyroid carcinoma (PTC), PIM1 was identified as a key factor in the regulation of oxidative stress [113]. The heightened expression of PIM1 in PTCs correlates positively with adverse prognostic indicators, with NOX-4 facilitating the management of ROS and supporting tumor proliferation. It appears that NOX4 may stimulate the expression of PIM1, which subsequently activates an antioxidant response that mitigates the buildup of harmful ROS, thereby sustaining an appropriate oxidative stress environment conducive to tumor advancement [113].
Clinical statuses of PIM1 kinase inhibitors
PIM1 kinase plays a major function in cellular growth, survival, differentiation, senescence, apoptosis, and drug resistance. Its over-expression or dysregulation has been linked to numerous cancers, making it a prime target for drug development. Moreover, its interactions with diverse proteins and involvement in multiple signaling pathways emphasize its significance as a possible target for treatment. Currently, several small molecule inhibitors have been created to target PIM kinase activity specifically. These PIM kinase inhibitors can be classified into three distinct categories according to the nature of their ligand interactions with specific amino acid residues. The first category consists of ATP mimetic inhibitors, where Glu121 is involved in the interaction. The second category encompasses non-ATP mimetic or ATP-competitive inhibitors, which engage with Lys67 located in the hinge region of PIM1 kinase. The third category comprises inhibitors that exhibit interactions characteristic of both ATP mimetic and non-ATP mimetic binding modes. These inhibitors function by binding to the enzyme's catalytic domain, inhibiting its function, and disrupting downstream signaling pathways that could promote cancer growth. While some inhibitors like AZD1208, CX-4945, CXR1002, LY-2835219 and TP3654 are undergoing clinical trials, many others are still in the preclinical research stage. Among the growing array of PIM1 kinase inhibitors, this review focuses on 15 such inhibitors in various solid tumors.
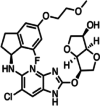
A953864.1 (Compound 14j)
This compound has demonstrated low nanomolar activity against PIM3, PIM1 and PIM2 (0.5 nM, 2 nM, and 3 nM, respectively) and exhibits high selectivity for PIM kinases (Table 2) [114]. It is a highly bioavailable compound that can be administered orally. It has demonstrated good overall efficiency in both enzymatic and cell-based tests, according to several investigations. It has also been discovered to have a lengthy half-life in liver microsomes, low protein binding, and excellent permeability. Compound 14j exhibits a significant level of specificity towards PIM kinases, leading to the suppression of BAD phosphorylation and triggering cell death by reducing the expression of Myc transcriptional target genes in cell lines obtained from Eμ-myc mice [114, 115]. This pan-PIM inhibitor functions by halting the growth of cells in LNCaP cell lines through the inhibition of Bad phosphorylation [31, 116].
Table 2.
PIM1 inhibitors in preclinical and clinical trials
| No | Compound | Structure | Class | PIM Inhibition (IC50) | Clinical status and Clinical trial identification | Cancer | References |
|---|---|---|---|---|---|---|---|
| 1 |
A953864.1 (Compound 14j) |
|
3H-benzo [4,5] thieno [3, 2-d] pyrimidin-4-one |
PIM1: 2 nM PIM2: 3 nM PIM3: 0.5 nM |
Preclinical | Prostate cancer | [31, 114–116] |
| 2 | AUM302 |
|
Triple kinase inhibitor | PIM1: 410 nM | Preclinical | Pancreatic cancer | [117] |
| 3 | AZD1208 |
|
Thiazolidene |
PIM1: 0.4 nM PIM2: 5 nM PIM3: 1.9 nM |
Phase I |
Gastric cancer, Prostate cancer and TNBC |
[40, 118–122] |
| 4 | CX-4945 |
|
Napthyridines |
PIM1: 48 nM PIM2: 186 nM |
Phase I/II |
Breast cancer | [122–125] |
| 5 | CX-6258 |
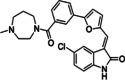
|
7-(4H-1,2,4-triazol-3-yl) benzo[c][2,6] naphthyridines |
PIM1: 5 nM PIM2: 25 nM PIM3: 16 nM |
Preclinical | Solid tumors | [31, 116, 125, 126] |
| 6 | CXR1002 |

|
Ammonium salt of perfluorooctanoic Acid |
PIM1: 40 nM PIM2: 170 nM PIM3: 240 nM |
Phase I | Pancreatic and Ovarian cancer | [31, 116, 126, 127] |
| 7 | DHPCC-9 |
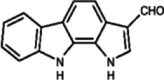
|
Pyrrolo [2,3-α] carbazole |
PIM1: 120 nM PIM2: 510 nM PIM3: 10 nM |
Preclinical |
Breast cancer, Prostate cancer, HNSCC |
[31, 116, 128, 129] |
| 8 | IBL-302 |
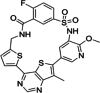
|
- |
PIM1: 22.8 nM PIM2: 7.74 nM PIM3: 5.86 nM |
Preclinical |
Breast cancer, Prostate cancer |
[130, 131] |
| 9 | LY-2835219 |
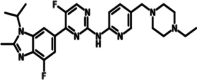
|
- |
PIM1: 50 nM PIM2: 3.4 µM |
Phase I |
NSCLC, Breast cancer, Brain cancer |
[132, 133] |
| 10 | Quinoxaline derivatives |
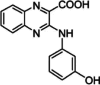
|
Quinoxaline-2-carboxylic acid |
PIM1: 74 nM PIM2: 2100 nM |
Preclinical | Colorectal cancer | [134, 135] |
| 11 | Quercetagetin |
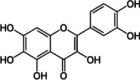
|
Flavonol quercetagetin (3,3′,4′,5,6,7-hydroxyflavone) (plexxicon) |
PIM1: 340 nM PIM2: 3450 nM |
Preclinical | Prostate cancer | [136, 137] |
| 12 | SGI-1776 |
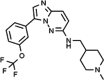
|
Imidazopyridazine |
PIM1: 7 nM PIM2: 363 nM PIM3: 69 nM |
Phase I (failed) |
Prostate cancer | [134, 135, 139, 141, 142, 144, 146, 147] |
| 13 | SMI-4a |
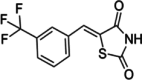
|
Benzylidene-thiazolidene-2, 4- dione |
PIM1: 24 nM PIM2: 100 nM |
Preclinical | Prostate cancer | [138–140] |
| 14 | 7-(4H-1,2,4-triazol-3-yl) benzo[c][2,6]naphthyridine |
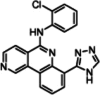
|
Napthyridine |
PIM1: 1 nM PIM2: 6 nM PIM3: 86 nM |
Phase I |
Kidney cancer, Medulloblastoma, Solid cancers |
[122] |
| 15 |
TP-3654 or SGI-9481 |
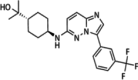
|
Imidazo[1,2-b] pyridazine and pyrazolo[1,5-A] pyrimidine |
PIM1: 5 nM PIM2: 239 nM PIM3: 42 nM |
Phase I |
Ovarian cancer, Colon cancer, NSCLC | [119, 141, 142] |
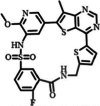
AUM302
AUM302 has been recognized as a powerful inhibitor of cell proliferation and viability across different PDAC cell lines, displaying a lower IC50 value, which signifies its increased effectiveness in reducing cell growth and viability [117]. Treatment with AUM302 has been shown to significantly impact the cell cycle distribution by raising the number of cells in the G0 or G1 along with G2 or M phases while decreasing cells in the S phase, indicating a notable influence on cell cycle dynamics. Moreover, AUM302 treatment has been found to elevate the subG1 population in specific PDAC cell lines such as BxPC-3 and Capan-2, suggesting the induction of apoptosis. Additionally, AUM302 exerts a significant inhibitory effect on the phosphorylation of key proteins involved in the PI3K/AKT/mTOR signaling pathway, leading to reduced levels of c-Myc, a critical transcription factor for cellular metabolism and proliferation. Notably, AUM302 has demonstrated efficacy against gemcitabine-resistant PDAC cells, positioning it as a promising candidate for PDAC therapy due to its diverse effects on cell proliferation, cell cycle progression, apoptosis induction, and inhibition of PI3K/AKT/mTOR signaling (Table 2) [117].
AZD1208
AZD1208 serves as a potent and orally active pan-PIM kinase inhibitor, with IC50 values of 0.4 nM (PIM1), 5 nM (PIM2) and 1.9 nM (PIM3) respectively (Table 2) [118]. This compound is derived from 1,3-thiazolidine-2,4-dione and functions as an ATP-competitive inhibitor. It achieves this by forming hydrogen bonds with Lys67, Asp128, and Glu171 via its thiazolidine framework, while also engaging in hydrophobic interactions with Pro123, Val126, and Leu174 through the phenyl ring [119]. In gastric cancer AZD1208 treatment alone caused marked cell death through induction of autophagy. In addition, the combination of AZD1208 with an Akt inhibitor produced synergistic effects against tumors by affecting the regulation of the DNA damage repair pathway [120]. AZD1208 has been shown to be effective across different models of prostate cancer in both mice and humans. Its mechanism involves the suppression of p53 protein and a combined therapeutic approach that includes radiation-mediated c-Myc suppression [121]. Treatment with AZD1208 impaired the growth of both TNBC cell line and patient-derived xenografts and sensitized them to chemotherapy-induced apoptotic cell death through downregulation of Mcl-1 and Bcl-2 protein [40]. AZD1208 demonstrated its ability to inhibit acute myeloid leukemia (AML) through the modulation of PIM1 and STAT-5 expression [143]. It triggered apoptosis and caused cell cycle arrest in the MOLM-16 cell line by decreasing the phosphorylation of PIM kinase targets such as SE-BP1, Bad, and p70S6K, while simultaneously activating p27 and caspase-3 [143]. While the Phase 1 trial for AML was discontinued, it was successfully completed for advanced solid tumors and malignant lymphoma, demonstrating safety and tolerability (Table 2) [122].
CX-4945
CX-4945 functions as an inhibitor of casein kinase 2 (CK2α) and also targets PIM kinase. It effectively suppresses PIM1 and PIM2 isoforms at very low concentrations in the nanomolar range. The potent anti-proliferative impact was observed in cell lines including MiPaca-2, PC-3, MDAMB231, K562, and MV4:11. Biochemical assays revealed that the IC50 values for PIM1 and PIM2 were 48 nm and 186 nm, respectively (Table 2). This suggests that the in vivo inhibitory effect is a result of inhibiting both the kinases CK2 and PIM [122]. This drug inhibits the phosphorylation of Bad at Serine-112 and exhibits an inhibited cell growth in the MV-4–11 AML cell line [123]. Phase I and II clinical studies for cholangiocarcinoma are now underway, and it is taken orally [124]. The structural analysis of the PIM1-CX-4945 complex indicates that the inhibitor interacts with the protein through the formation of two direct hydrogen bonds [125]. The initial hydrogen bond occurs between the carboxylate group of CX-4945 and the amino group of Lys67 located at the active site of PIM1. The second hydrogen bond is established between the carboxylate group of the inhibitor and the main chain amide of Asp186 situated within the A-loop [125].
CX-6258
CX-6258, a derivative of furanamide, functions as a broad-spectrum inhibitor of PIM kinases (Table 2). It has exhibited potent anti-proliferative effects in cell lines originating from both human solid tumors and hematological malignancies. When administered to the MV4:11 cell line, CCX-6258 decreased the phosphorylation of BAD and 4E-BP1, while FLT3 autophosphorylation remained unchanged. In PC-3 prostate cancer cell lines, CCX-6258, when combined with doxorubicin and paclitaxel, exhibited a synergistic antiproliferative effect. Oral administration of CCX-6258 in MV4:11 and PC-3 tumor xenografts led to a dose-dependent reduction in tumor growth [31, 116, 126]. CX-6258, interacts at the adenine binding site with an unusual oxindole ring. The oxygen within the inhibitor oxindole forms a hydrogen bond with the side chain amide of Lys67 and is further stabilized by carbonyl-π interaction with the sidechain of Phe49. The oxindole secondary amine forms a water mediated contact with the main chain amide of Asp186. Further, the water network anchors the inhibitor at the sidechain of Glu89. The oxindole moiety is additionally stabilized by hydrophobic interactions contributed by the sidechains of Val52, Ala65, Ile104, Leu120, Leu174 and Ile185 [125].
CXR1002
CXR1002 is a perfluorooctanoic acid ammonium salt that serves as a lipid mimic, triggering ER stress and impeding PIM1 kinase activity (Table 2). This compound alters the level of PIM1 kinase in K562 cells, causing the inhibition of Mdm2 phosphorylation via PIM1. Various cell lines, including HT29, PC-3, A549, HepG2, and Panc-1 xenograft models, have exhibited a strong response to CXR1002. Furthermore, CXR1002 has demonstrated a potent synergistic effect when combined with gemcitabine and doxorubicin in hepatic, ovarian, and pancreatic cancer cells [31, 127].
DHPCC-9
The compound DHPCC-9 (1,10-dihydropyrrolo[2,3-a] carbazole-3- carbaldehyde) is a PIM selective inhibitor that inhibits PIM1-dependent survival of cytokine-deprived myeloid cells (IC50 = 4–6 µM). It reduces the intracellular phosphorylation of PIM1's downstream targets. Bad and prevents apoptosis. Additionally, treatment with DHPCC-9 decreases the migration of both squamocellular carcinoma cells (UT-SCC-12A) and prostate cancer cells (PC-3) without impacting PIM1 protein levels or cell viability. Notably, it exhibits efficient inhibition of PIM1 and PIM3 in comparison to PIM2 (Table 2) [31, 116, 128]. Crystallization study have proposed that DHPCC-9 falls into the second category of PIM kinase inhibitors, competing with ATP for the binding site at the conserved Lys67 residues in PIM1 kinase active site [129].
IBL-302
IBL-302 represents a significant advancement in oncology by targeting both PIM and the PI3K/AKT/mTOR signaling pathways. This oral kinase inhibitor simultaneously addresses pan-PIM kinase, pan-PI3K and mTOR, providing a comprehensive treatment approach. Preclinical studies evaluated IBL-302's efficacy in various breast cancer cell lines (SKBR-3, BT-474, and HCC-1954) in vitro and in BALB/c nude mice in vivo. The results demonstrated that IBL-302 effectively suppressed pAKT, pmTOR, and pBAD, showcasing its promising anti-tumor effects across different breast cancer models [130, 131]. As a novel PIM and PI3K/mTOR inhibitor, IBL-302 holds substantial potential as a breast cancer treatment and warrants further exploration in clinical trials (Table 2).
LY-2835219
LY2835219 is an orally available dual PIM1 and CDK4/6 inhibitor. It effectively targets CDK4/6 and PIM1 and stands out for its ability to permeate the blood–brain barrier. The inhibitor is currently in a Phase-1 trial and has exhibited G-1 phase arrest in colon xenografts, as well as in vivo anti-tumor activity (Table 2) [132]. The combination of Gemcitabine has shown impressive efficacy across various tumors, especially when used in conjunction with Temozolomide for targeting orthotopic brain tumor xenografts. This effectiveness is attributed to Gemcitabine's ability to cross the blood–brain barrier, highlighting its potential in treating central nervous system-related tumors [133]. Additionally, the dual inhibition of CDK4/6 and PIM1 has demonstrated success in synergizing with Sunitinib in renal cancer cell lines [133].
Quinoxaline derivatives
A derivative of quinoxaline, quinoxaline-2-carboxylic acid, has demonstrated strong inhibition of PIM1 enzymatic activity at sub-micromolar concentrations (IC50 of 74 nM) and moderate activity against PIM2, with an IC50 of 2.10 µM (Table 2) [134]. A new series of quinoxaline-2-carboxylic acid compounds with dual PIM1/2 inhibitory properties were synthesized through a structure-based design strategy, showing optimized activity on both PIM1 and PIM2 isoforms. Two major compounds, 5c (6-Cl) and 5e (6-Br), were detected, demonstrating submicromolar potency against both PIM1 and PIM2 isoforms, along with a favorable among various mammalian kinases [135]. These compounds were also effective in inhibiting the proliferation of human cell lines MV4-11 (acute myeloid leukemia) and HCT-116 (colorectal carcinoma), which are characterized by elevated levels of PIM1/2 kinases. These promising results suggest that these compounds could be strong candidates for further pharmacomodulation studies.
Quercentagetin
Quercentagetin is a flavonoid that functions as a highly specific ATP-competitive inhibitors of PIM1 kinase (Table 2). In RWPE2 prostate cancer cell lines with overexpressed PIM1, Quercentagetin demonstrated a dose-dependent inhibition of BAD phosphorylation [136]. The inhibitory effect of Quercentagetin is dependent on the levels of PIM1 kinase in other prostate epithelial cell lines. In vascular smooth muscle cells, stimulation with PDGF-BB led to elevated PIM1 mRNA expression, which subsequently caused an increase in protein levels and cell proliferation. This proliferative effect, however, could be effectively inhibited by either Quercentagetin or PIM1-targeted shRNA, indicating a potential therapeutic approach [137].
SGI-1776
SuperGen Inc., USA, identified this pan-PIM 1st generation inhibitor. It was created by enhancing SC-47, an imidazole [1, 2-b] pyridazine derivative, using various in-silico screening techniques [144]. The inhibitor SGI-1776 inhibits all the three PIM kinases and targets Flt3 and Haspin, leading to uncertainty regarding its actual contribution and effectiveness (Table 2). SGI-1776 acts as an ATP competitive inhibitor of the PIM1 kinase, establishing a hydrogen bond with Lys67 located in the kinase active site. Additionally, it also exhibits robust interactions with the residues Phe49 and Asp128/Glu171 [134]. Treatment of CLL cell lines with SGI-1776 decreased phosphorylation and overall protein levels of c-Myc while simultaneously enhancing the expression of the antiapoptotic protein MCL-1, ultimately facilitating apoptosis [145]. Prostate cancer cells triggered apoptosis and G1 phase arrest by reducing phosphorylated p21Clp1/WAF1 and Bad levels, demonstrating enhanced cytotoxic effects when paired with Paclitaxel/Dovetail [146]. For PDAC cells, it lowered phospho-Bad levels and induced alterations in the cell cycle, with more favorable outcomes observed when combined with Gemcitabine [147]. In Multiple Myeloma, PIM kinase expression was diminished by inhibiting protein synthesis pathways such as p70S6K and 4E-BP1 [148]. However, this compound did not progress past Phase 1 clinical trials owing to its toxicity.
SMI-4a
SMI-4a is a potent, specific, cell-penetrating, and ATP-competitive inhibitor of PIM1, showing an IC50 value of 24 nM. In addition, SMI-4a can inhibit PIM2 with an IC50 of 100 nM while not exerting significant inhibition on other serine/threonine or tyrosine kinases (Table 2). Molecular docking study of SMI-4a with PIM1 kinase suggested that the thiazolidine NH group donates a hydrogen bond to the carbonyl oxygen of Glu121, and the trifluorophenyl group makes numerous van der Waals contacts with the hydrophobic cleft [138]. SMI4a halts the G1 phase of the cell cycle in prostate and AML cell lines by blocking Cdk2 and relocating the PIM substrate p27kip1 [139]. The combination of SMI4a and rapamycin synergistically stops mTOR activity, reduces 4E-BP-1 expression via phosphorylation, and suppresses the proliferation of leukemic cells. Co-administration of SMI4a and ABT-737, a Bcl2 antagonist, effectively induced apoptosis in prostate cancer cells in both in vitro and in vivo experiments. Inhibition of PIM1 led to increased transcription of the BH3 protein Noxa by activating the unfolded protein response, which in turn caused eIF-2α phosphorylation and elevated expression of CHOP [140]. This drug is currently undergoing preclinical development for AML with a focus on improving specificity and reducing cytotoxicity.
7-(4H-1,2,4-triazol-3-yl) benzo[c][2,6] naphthyridine
The Triazolo-benzo[c]-2–6-napthyridines represent a class of pan-PIM kinase inhibitors, derived from the structural modification of the CK2 inhibitor CX-4945. These inhibitors were engineered by incorporating triazole or amide (II) groups at the C (7) position, along with 2'-halogenoanilines at the C (5) position. This strategic design led to the development of highly potent inhibitors, targeting both PIM1 and PIM2 isoforms, with several analogs demonstrating efficacy at nanomolar concentrations (Table 2). The compound inhibited BAD phosphorylation at Serine 112 and showed significant antiproliferative effects on the AML cell line MV-4–11 (IC50 < 30 nM). Additionally, it markedly inhibited the proliferation of various other cell lines, such as MDAMB231, K562, and PC-3. Biochemical assays revealed IC50 values of 48 nm for PIM1 and 186 nm for PIM2, indicating that the in vivo inhibitory effect is owing to the dual inhibition of CK2 and PIM [122].
TP-3654 or SGI-9481
TP-3654 is a 2nd generation potent PIM1 kinase inhibitor that is administered orally and has displayed activity against PIM2 and PIM3 while also exhibiting favorable selectivity against other kinases (Table 2). This compound inhibits PIM kinase by forming hydrogen bonds at Lys67 and Asp128 & hydrophobic interaction with Phe49 and Leu174, respectively [119]. This chemical selectively disrupts drug transport mediated by ABCG2, a protein that plays a vital role in drug efflux and multidrug resistance in cancer cells. Clinical trials are underway to examine the potential of TP-3654 in treating advanced solid tumors and myelofibrosis. Studies have shown that TP-3654 effectively reverses multidrug resistance in cancer cells that overexpress ABCG2, making them more responsive to cytotoxic drugs such as topotecan, SN-38, and mitoxantrone. By targeting ABCG2 specifically, TP-3654 can be a valuable addition to combination therapies aimed at enhancing the efficacy of existing anticancer drugs without interfering significantly with other drug transporters like ABCB1 [141, 142].
Conclusion and future prospects
PIM1 kinase is a critical player in cell growth, differentiation, survival, and resistance to chemotherapy. The overexpression of PIM1 proto-oncogene is a common occurrence in different cancer types, leading to unfavorable clinical outcomes. Its interactions with various proteins and involvement in signaling pathways highlight PIM1 as a promising target for anti-cancer research. Research have demonstrated that targeting PIM1 can effectively slow down tumor growth in animal models without causing side effects, suggesting its potential as a therapeutic strategy for certain solid tumors. Despite ongoing clinical trials, no pan-PIM kinase or PIM1-specific inhibitors have been authorised by the FDA. Despite the fact that several pan-PIM inhibitors have undergone clinical trials, their limited selectivity has raised concerns about potential side effects. Nonetheless, the development of new PIM1 inhibitors remains active, with several promising candidates undergoing preclinical evaluation (Table 2).
Data from preclinical research suggests that PIM1 inhibitors are as potent as monotherapies. Nonetheless, their effectiveness is further amplified when combined with chemotherapy or other targeted therapies. Targeting PIM1 alone has proven to be less effective due to the presence of alternative signaling routes that restrict the safe and acceptable dose due to inhibitor toxicity and acquired resistance. A multi-modal strategy for treatment, integrating PIM1 inhibitors with inhibitors of other signaling pathways, such as PI3K/AKT/mTOR, may help overcome these challenges. This strategy could reduce toxicity, enhance patient quality of life, and potentially improve survival outcomes. Within our review, we have highlighted the diverse PIM1 signaling pathways present in different cancer types, all of which hold potential as targets for cancer therapy. This review has explored the various PIM1 signaling pathways across different types of cancer, highlighting their potential as therapeutic targets. Some combination therapies have already shown promising leads to both in vitro and in vivo studies, underscoring the need for clinical trials to evaluate their potential benefits for patients [126, 127, 133, 149]. Moreover, exploring combination therapies that incorporate PIM1 inhibitors with compounds that have the ability to increase the expression of particular tumor suppressor molecules is crucial. By creating and implementing these synergistic treatment approaches, we can unleash the complete potential of PIM1 as a critical role for cancer therapy.
Acknowledgements
The authors would also like to thank Reseapro Scientific Services (P) Ltd., India, for the English editing of the manuscript.
Abbreviations
- PIM
Provirus Integration site for Moloney Leukemia Virus
- mRNA
Messenger RNA
- UTR
Untranslated Regions
- JAK/STAT
Janus Kinase / Signal Transducers and Activators of Transcription
- SOCS
Suppressor of Cytokine Signaling
- MYC
Myelocytomatosis Oncogene
- ASK1
Apoptosis Signaling Kinase 1
- HoxA9
Homeobox Protein
- NF-κB
Nuclear Factor-κB
- TNBC
Triple Negative Breast Cancer
- ERB
ER-α-Binding Region
- BrCSC
Breast Cancer Stem Cells
- RUNX3
Runt-Related Transcription Factor 3
- CSL
C promoter-binding factor 1, Suppressor of Hairless, Lag 1
- AR
Androgen Receptor
- NDRG1
N-Myc Downstream-Regulated Gene 1
- HCC
Hepatocellular Carcinoma
- RBMY
RNA Binding Motif protein Y-linked
- RRM
RNA Recognition Motif
- HPNE
Human Pancreatic Nestin-expressing Epithelial cell line
- HIF-1α
Hypoxia Inducible Factor-1α
- HuR
Hu antigen R
- ARE
Adenylate uridylate-Rich Element
- PDAC
Pancreatic Ductal Adenocarcinoma
- SCLC
Small Cell Lung Cancer
- NSCLC
Non-Small Cell Lung Cancer
- eIF4B
Eukaryotic translation Initiation Factor 4B
- DRP1
Dynamin-Related Protein 1
- CRC
Colorectal Cancer
- HK2
Hexokinase 2
- LDHA
Lactate Dehydrogenase A
- TAK1
Transforming Growth factor-β-activated Kinase 1
- TAB3
Transforming Growth factor-β-activated Kinase 1 Binding protein 3
- OC
Ovarian Cancer
- PI3K
Phosphoinositide 3-Kinase
- mTOR
Mammalian Target of Rapamycin
- IRS
Insulin Receptor Substrates
- ROS
Reactive Oxygen Species
- CDK
Cyclin Dependent Kinase
- GBM
Glioblastoma Multiforme
- EGFR
Epidermal Growth Factor Receptor
- MEK
MAP/ERK Kinase
- GSC
Glioblastoma Stem Cells
- SPARC
Secreted Protein Acidic and Rich in Cysteine
- OSCC
Oral Squamous Cell Carcinoma
- NCMT
Non-Cancerous Match Tissue
- HNSCC
Head and neck Squamous Cell Carcinoma
- SACC
Adenoid Cystic Carcinoma
- RPTEC
Renal Proximal Tubule Epithelial Cells
- PTC
Papillary Thyroid Carcinoma
- AML
Acute Myeloid Leukemia
- CK2
Casein kinase 2
Authors’ contributions
R.C. (writing original draft, creating figures and reviewing), C.K.B. (writing original draft and reviewing), I.P.R. (writing original draft and reviewing), P.D. (writing original draft and reviewing), I.P. (writing original draft and reviewing), S.M. (writing original draft and reviewing) S.R.M. (reviewing and editing) S.P (reviewing and editing) and K.N. (reviewing, editing, creating figures and funding acquisition). All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by the Department of Biotechnology, Government of India Ramalingaswami Re-entry Fellowship (grant number BT/RLF/Re-entry/29/2019).
Data availability
No datasets were generated or analysed during the current study.
Declarations
Ethics approval and consent to participate
Not applicable.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Chandan Kumar Bahadi, Ipsa Pratibimbita Ray, Pragyanshree Dash, Isha Pattanaik and Suman Mishra contributed equally to this work.
References
- 1.Narlik-Grassow M, Blanco-Aparicio C, Carnero A. The PIM family of serine/threonine kinases in cancer. Med Res Rev. 2014;34(1):136–59. 10.1002/med.21284. [DOI] [PubMed] [Google Scholar]
- 2.Bachmann M, Möröy T. The serine/threonine kinase Pim-1. Int J Biochem Cell Biol. 2005;37(4):726–30. 10.1016/j.biocel.2004.11.005. [DOI] [PubMed] [Google Scholar]
- 3.Nawijn MC, Alendar A, Berns A. For better or for worse: the role of Pim oncogenes in tumorigenesis. Nat Rev Cancer. 2011;11(1):23–34. 10.1038/nrc2986. [DOI] [PubMed] [Google Scholar]
- 4.Zhu N, Ramirez LM, Lee RL, Magnuson NS, Bishop GA, Gold MR. CD40 signaling in B cells regulates the expression of the Pim-1 kinase via the NF-κB pathway. J Immunol. 2002;168(2):744–54. 10.4049/jimmunol.168.2.744. [DOI] [PubMed] [Google Scholar]
- 5.Fox CJ, Hammerman PS, Cinalli RM, Master SR, Chodosh LA, Thompson CB. The serine/threonine kinase Pim-2 is a transcriptionally regulated apoptotic inhibitor. Genes & development. 2003;17(15):1841–1854. http://www.genesdev.org/cgi/doi/10.1101/gad.1105003. [DOI] [PMC free article] [PubMed]
- 6.Fujii C, Nakamoto Y, Lu P, Tsuneyama K, Popivanova BK, Kaneko S, Mukaida N. Aberrant expression of serine/threonine kinase Pim-3 in hepatocellular carcinoma development and its role in the proliferation of human hepatoma cell lines. Int J Cancer. 2005;114(2):209–18. 10.1002/ijc.20719. [DOI] [PubMed] [Google Scholar]
- 7.Qian KC, Wang L, Hickey ER, Studts J, Barringer K, Peng C, et al. Structural basis of constitutive activity and a unique nucleotide binding mode of human Pim-1 kinase. J Biol Chem. 2005;280(7):6130–7. 10.1074/jbc.m409123200. [DOI] [PubMed] [Google Scholar]
- 8.Bullock AN, Russo S, Amos A, Pagano N, Bregman H, Debreczeni JE, et al. Crystal structure of the PIM2 kinase in complex with an organoruthenium inhibitor. PloS one. 2009;4(10):e7112. 10.1371/2Fjournal.pone.0007112 [DOI] [PMC free article] [PubMed]
- 9.Blanco-Aparicio C, Carnero A. Pim kinases in cancer: diagnostic, prognostic and treatment opportunities. Biochem Pharmacol. 2013;85(5):629–43. 10.1016/j.bcp.2012.09.018. [DOI] [PubMed] [Google Scholar]
- 10.Julson JR, Marayati R, Beierle EA, Stafman LL. The role of PIM kinases in pediatric solid tumors. Cancers. 2022;14(15):3565. 10.3390/cancers14153565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Amson R, Sigaux F, Przedborski S, Flandrin G, Givol D, Telerman A. The human protooncogene product p33pim is expressed during fetal hematopoiesis and in diverse leukemias. Proc Natl Acad Sci. 1989;86(22):8857–61. 10.1073/pnas.86.22.8857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mizuno K, Shirogane T, Shinohara A, Iwamatsu A, Hibi M, Hirano T. Regulation of pim-1 by hsp90. Biochem Biophys Res Commun. 2001;281(3):663–9. 10.1006/bbrc.2001.4405. [DOI] [PubMed] [Google Scholar]
- 13.Petersen Shay K, Wang Z, Xing PX, McKenzie IF, Magnuson NS. Pim-1 kinase stability is regulated by heat shock proteins and the ubiquitin-proteasome pathway. Mol Cancer Res. 2005;3(3):170–81. 10.1158/1541-7786.MCR-04-0192. [DOI] [PubMed] [Google Scholar]
- 14.Losman JA, Chen XP, Vuong BQ, Fay S, Rothman PB. Protein phosphatase 2A regulates the stability of Pim protein kinases. J Biol Chem. 2003;278(7):4800–5. 10.1074/jbc.M208246200. [DOI] [PubMed] [Google Scholar]
- 15.Ma J, Arnold HK, Lilly MB, Sears RC, Kraft AS. Negative regulation of Pim-1 protein kinase levels by the B56β subunit of PP2A. Oncogene. 2007;26(35):5145–53. 10.1038/sj.onc.1210323. [DOI] [PubMed] [Google Scholar]
- 16.Park YK, Obiang-Obounou BW, Lee KB, Choi JS, Jang BC. AZD1208, a pan-Pim kinase inhibitor, inhibits adipogenesis and induces lipolysis in 3T3-L1 adipocytes. J Cell Mol Med. 2018;22(4):2488–97. 10.1111/jcmm.13559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kreuz S, Holmes KB, Tooze RM, Lefevre PF. Loss of PIM2 enhances the anti-proliferative effect of the pan-PIM kinase inhibitor AZD1208 in non-Hodgkin lymphomas. Mol Cancer. 2015;14:1–4. 10.1186/s12943-015-0477-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yip-Schneider MT, Horie M, Broxmeyer HE. Transcriptional induction of pim-1 protein kinase gene expression by interferon gamma and posttranscriptional effects on costimulation with steel factor. Blood. 1995;85:3494–502. 10.1182/blood.V85.12.3494.bloodjournal85123494. [PubMed] [Google Scholar]
- 19.Matikainen S, Sareneva T, Ronni T, Lehtonen A, Koskinen PJ, Julkunen I. Interferon-α Activates Multiple STAT Proteins and Upregulates Proliferation-Associated IL-2Rα, c-myc, and pim-1 Genes in Human T Cells. Blood, The Journal of the American Society of Hematology. 1999;93(6):1980–91. 10.1182/blood.V93.6.1980.406k20_1980_1991. [PubMed] [Google Scholar]
- 20.Block KM, Hanke NT, Maine EA, Baker AF. IL-6 stimulates STAT3 and Pim-1 kinase in pancreatic cancer cell lines. Pancreas. 2012;41(5):773–7781. 10.1097/MPA.0b013e31823cdd10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Peltola KJ, Paukku K, Aho TL, Ruuska M, Silvennoinen O, Koskinen PJ. Pim-1 kinase inhibits STAT5-dependent transcription via its interactions with SOCS1 and SOCS3. Blood. 2004;103(10):3744–50. 10.1182/blood-2003-09-3126. [DOI] [PubMed] [Google Scholar]
- 22.Shirogane T, Fukada T, Muller JM, Shima DT, Hibi M, Hirano T. Synergistic roles for Pim-1 and c-Myc in STAT3-mediated cell cycle progression and antiapoptosis. Immunity. 1999;11(6):709–19. 10.1016/S1074-7613(00)80145-4. [DOI] [PubMed] [Google Scholar]
- 23.Zhang Y, Wang Z, Li X, Magnuson NS. Pim kinase-dependent inhibition of c-Myc degradation. Oncogene. 2008;35:4809–19. 10.1038/onc.2008.123. [DOI] [PubMed] [Google Scholar]
- 24.Horiuchi D, Camarda R, Zhou AY, Yau C, Momcilovic O, Balakrishnan S, et al. PIM1 kinase inhibition as a targeted therapy against triple-negative breast tumors with elevated MYC expression. Nat Med. 2016;11:1321–9. 10.1038/nm.4213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Macdonald A, Campbell DG, Toth R, McLauchlan H, Hastie CJ, Arthur JS. Pim kinases phosphorylate multiple sites on Bad and promote 14–3–3 binding and dissociation from Bcl-X L. BMC cell biology. 2006:1–4. 10.1186/1471-2121-7-1 [DOI] [PMC free article] [PubMed]
- 26.Gu JJ, Wang Z, Reeves R, Magnuson NS. PIM1 phosphorylates and negatively regulates ASK1-mediated apoptosis. Oncogene. 2009;48:4261–71. 10.1038/onc.2009.276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hu YL, Passegué E, Fong S, Largman C, Lawrence HJ. Evidence that the Pim1 kinase gene is a direct target of HOXA9. Blood, The Journal of the American Society of Hematology. 2007;109(11):4732–8. 10.1182/blood-2006-08-043356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Brault L, Gasser C, Bracher F, Huber K, Knapp S, Schwaller J. PIM serine/threonine kinases in the pathogenesis and therapy of hematologic malignancies and solid cancers. haematologica. 2010;95(6):1004. 10.3324/haematol.2009.017079 [DOI] [PMC free article] [PubMed]
- 29.Nihira K, Ando Y, Yamaguchi T, Kagami Y, Miki Y, Yoshida K. Pim-1 controls NF-κB signalling by stabilizing RelA/p65. Cell Death Differ. 2010;4:689–98. 10.1038/cdd.2009.174. [DOI] [PubMed] [Google Scholar]
- 30.Rout AK, Dehury B, Parida SN, Rout SS, Jena R, Kaushik N, et al. A review on structure-function mechanism and signaling pathway of serine/threonine protein PIM kinases as a therapeutic target. International Journal of Biological Macromolecules. 2024:132030. 10.1016/j.ijbiomac.2024.132030 [DOI] [PubMed]
- 31.Panchal NK, Sabina EP. A serine/threonine protein PIM kinase as a biomarker of cancer and a target for anti-tumor therapy. Life Sci. 2020;255: 117866. 10.1016/j.lfs.2020.117866. [DOI] [PubMed] [Google Scholar]
- 32.Zhang X, Song M, Kundu JK, Lee MH, Liu ZZ. PIM kinase as an executional target in cancer. Journal of Cancer Prevention. 2018;23(3):109. 10.15430/JCP.2018.23.3.109 [DOI] [PMC free article] [PubMed]
- 33.Liu XJ, Xiao QL, Wu X, Jin M, Shan T. Differential expression and clinical characteristics of Pim1 protein in different lesions of breast. Journal of Hainan Medical University. 2019;25(1):40–4. [Google Scholar]
- 34.Liu H, Chen C, Ma D, Li Y, Yin Q, Li Q, et al. Inhibition of PIM1 attenuates the stem cell–like traits of breast cancer cells by promoting RUNX3 nuclear retention. J Cell Mol Med. 2020;11:6308–23. 10.1111/jcmm.15272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chen J, Tang G. PIM-1 kinase: a potential biomarker of triple-negative breast cancer. OncoTargets and therapy. 2019:6267–73. 10.2147/OTT.S212752 [DOI] [PMC free article] [PubMed]
- 36.Zhao W, Qiu R, Li P, Yang J. PIM1: a promising target in patients with triple-negative breast cancer. Med Oncol. 2017;34(8):142. 10.1007/s12032-017-0998-y. [DOI] [PubMed] [Google Scholar]
- 37.Mahata S, Sahoo PK, Pal R, Sarkar S, Mistry T, Ghosh S, et al. PIM1/STAT3 axis: a potential co-targeted therapeutic approach in triple-negative breast cancer. Med Oncol. 2022;39(7):74. 10.1007/s12032-022-01675-2. [DOI] [PubMed] [Google Scholar]
- 38.Malinen M, Jääskeläinen T, Pelkonen M, Heikkinen S, Väisänen S, Kosma VM, et al. Proto-oncogene PIM-1 is a novel estrogen receptor target associating with high grade breast tumors. Mol Cell Endocrinol. 2013;365(2):270–6. 10.1016/j.bcp.2012.10.018. [DOI] [PubMed] [Google Scholar]
- 39.Gao X, Liu X, Lu Y, Wang Y, Cao W, Liu X, et al. PIM1 is responsible for IL-6-induced breast cancer cell EMT and stemness via c-myc activation. Breast Cancer. 2019;26:663–71. 10.1007/s12282-019-00966-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Brasó-Maristany F, Filosto S, Catchpole S, Marlow R, Quist J, Francesch-Domenech E, et al. PIM1 kinase regulates cell death, tumor growth and chemotherapy response in triple-negative breast cancer. Nat Med. 2016;11:1303–13. 10.1038/nm.4198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gazinska P, Grigoriadis A, Brown JP, Millis RR, Mera A, Gillett CE, et al. Comparison of basal-like triple-negative breast cancer defined by morphology, immunohistochemistry and transcriptional profiles. Mod Pathol. 2013;26(7):955–66. 10.1038/modpathol.2012.244. [DOI] [PubMed] [Google Scholar]
- 42.Santio NM, Landor SK, Vahtera L, Ylä-Pelto J, Paloniemi E, Imanishi SY, et al. Phosphorylation of Notch1 by Pim kinases promotes oncogenic signaling in breast and prostate cancer cells. Oncotarget. 2016;7(28):43220. 10.18632/oncotarget.9215 [DOI] [PMC free article] [PubMed]
- 43.Landor SK, Santio NM, Eccleshall WB, Paramonov VM, Gagliani EK, Hall D, et al. PIM-induced phosphorylation of Notch3 promotes breast cancer tumorigenicity in a CSL-independent fashion. Journal of Biological Chemistry. 2021;296. 10.1016/j.jbc.2021.100593 [DOI] [PMC free article] [PubMed]
- 44.Ha S, Iqbal NJ, Mita P, Ruoff R, Gerald WL, Lepor H, et al. Phosphorylation of the androgen receptor by PIM1 in hormone refractory prostate cancer. Oncogene. 2013;32(34):3992–4000. 10.1038/onc.2012.412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Linn DE, Yang X, Xie Y, Alfano A, Deshmukh D, Wang X, et al. Differential regulation of androgen receptor by PIM-1 kinases via phosphorylation-dependent recruitment of distinct ubiquitin E3 ligases. J Biol Chem. 2012;287(27):22959–68. 10.1074/jbc.M111.338350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ruff SE, Vasilyev N, Nudler E, Logan SK, Garabedian MJ. PIM1 phosphorylation of the androgen receptor and 14–3-3 ζ regulates gene transcription in prostate cancer. Communications Biology. 2021;4(1):1221. 10.1038/s42003-021-02723-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ledet RJ, Ruff SE, Wang Y, Nayak S, Schneider JA, Ueberheide B, et al. Identification of PIM1 substrates reveals a role for NDRG1 phosphorylation in prostate cancer cellular migration and invasion. Communications Biology. 2021;4(1):36. 10.1038/s42003-020-01528-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Eerola SK, Santio NM, Rinne S, Kouvonen P, Corthals GL, Scaravilli M, et al. Phosphorylation of NFATC1 at PIM1 target sites is essential for its ability to promote prostate cancer cell migration and invasion. Cell Communication and Signaling. 2019;17:1–6. 10.1186/s12964-019-0463-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Jensen CC, Clements AN, Liou H, Ball LE, Bethard JR, Langlais PR, et al. PIM1 phosphorylates ABI2 to enhance actin dynamics and promote tumor invasion. J Cell Biol. 2023;222(6): e202208136. 10.1083/jcb.202208136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Leung CO, Wong CC, Fan DN, Kai AK, Tung EK, Xu IM, et al. PIM1 regulates glycolysis and promotes tumor progression in hepatocellular carcinoma. Oncotarget. 2015;6(13):10880. 10.18632/oncotarget.3534 [DOI] [PMC free article] [PubMed]
- 51.Chua HH, Chang MH, Chen YH, Tsuei DJ, Jeng YM, Lee PH, et al. PIM1-Induced Cytoplasmic Expression of RBMY Mediates Hepatocellular Carcinoma Metastasis. Cell Mol Gastroenterol Hepatol. 2023;15(1):121–52. 10.1016/j.jcmgh.2022.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Cao L, Wang F, Li S, Wang X, Huang D, Jiang R. PIM1 kinase promotes cell proliferation, metastasis and tumor growth of lung adenocarcinoma by potentiating the c-MET signaling pathway. Cancer Lett. 2019;444:116–26. 10.1016/j.canlet.2018.12.015. [DOI] [PubMed] [Google Scholar]
- 53.Wu Y, Deng Y, Zhu J, Duan Y, Weng W, Wu X. Pim1 promotes cell proliferation and regulates glycolysis via interaction with MYC in ovarian cancer. Onco Targets Ther. 2018;11:6647–56. 10.2147/OTT.S180520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Adamaki M, Zoumpourlis V. Prostate Cancer Biomarkers: From diagnosis to prognosis and precision-guided therapeutics. Pharmacol Ther. 2021;228: 107932. 10.1016/j.pharmthera.2021.107932. [DOI] [PubMed] [Google Scholar]
- 55.Dhanasekaran SM, Barrette TR, Ghosh D, Shah R, Varambally S, Kurachi K, et al. Delineation of prognostic biomarkers in prostate cancer. Nature. 2001;412(6849):822–6. 10.1038/35090585. [DOI] [PubMed] [Google Scholar]
- 56.Wang J, Kim J, Roh M, Franco OE, Hayward SW, Wills ML, et al. Pim1 kinase synergizes with c-MYC to induce advanced prostate carcinoma. Oncogene. 2010;17:2477–87. 10.1038/onc.2010.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wang J, Anderson PD, Luo W, Gius D, Roh M, Abdulkadir SA. Pim1 kinase is required to maintain tumorigenicity in MYC-expressing prostate cancer cells. Oncogene. 2012;31(14):1794–803. 10.1038/onc.2011.371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.McAllister MJ, McCall P, Dickson A, Underwood MA, Andersen D, Holmes E, et al. Androgen receptor phosphorylation at serine 81 and serine 213 in castrate-resistant prostate cancer. Prostate Cancer Prostatic Dis. 2020;23(4):596–606. 10.1038/s41391-020-0235-1. [DOI] [PubMed] [Google Scholar]
- 59.Santio NM, Vainio V, Hoikkala T, Mung KL, Lång M, Vahakoski R, et al. PIM1 accelerates prostate cancer cell motility by phosphorylating actin capping proteins. Cell Communication and Signaling. 2020;18:1–8. 10.1186/s12964-020-00618-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Tsuei DJ, Hsu HC, Lee PH, Jeng YM, Pu YS, Chen CN, et al. RBMY, a male germ cell-specific RNA-binding protein, activated in human liver cancers and transforms rodent fibroblasts. Oncogene. 2004;23(34):5815–22. 10.1371/journal.pone.0026948. [DOI] [PubMed] [Google Scholar]
- 61.Reiser-Erkan C, Erkan M, Pan Z, Bekasi S, Giese NA, Streit S, et al. Hypoxia-inducible proto-oncogene Pim-1 is a prognostic marker in pancreatic ductal adenocarcinoma. Cancer Biol Ther. 2008;7(9):1352–9. 10.4161/cbt.7.9.6418. [DOI] [PubMed] [Google Scholar]
- 62.Xu D, Allsop SA, Witherspoon SM, Snider JL, Yeh JJ, Fiordalisi JJ, et al. The oncogenic kinase Pim-1 is modulated by K-Ras signaling and mediates transformed growth and radioresistance in human pancreatic ductal adenocarcinoma cells. Carcinogenesis. 2011;32(4):488–95. 10.1093/carcin/bgr007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Chen J, Kobayashi M, Darmanin S, Qiao Y, Gully C, Zhao R, et al. Hypoxia-mediated up-regulation of Pim-1 contributes to solid tumor formation. Am J Pathol. 2009;175(1):400–11. 10.2353/ajpath.2009.080972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Burkhart RA, Pineda DM, Chand SN, Romeo C, Londin ER, Karoly ED, et al. HuR is a post-transcriptional regulator of core metabolic enzymes in pancreatic cancer. RNA Biol. 2013;10:1312–23. 10.4161/rna.25274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Lal S, Burkhart RA, Beeharry N, Bhattacharjee V, Londin ER, Cozzitorto JA et al. HuR posttranscriptionally regulates WEE1: implications for the DNA damage response in pancreatic cancer cells. Cancer Res 2014; 74: 1128–1140 10.1158/0008-5472.CAN-13-1915 [DOI] [PMC free article] [PubMed]
- 66.Blanco FF, Jimbo M, Wulfkuhle J, Gallagher I, Deng J, Enyenihi L, et al. The mRNA-binding protein HuR promotes hypoxia-induced chemoresistance through posttranscriptional regulation of the proto-oncogene PIM1 in pancreatic cancer cells. Oncogene. 2016;35(19):2529–41. 10.1158/0008-5472.CAN-13-1915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Malumbres M, Barbacid M. RAS oncogenes: the first 30 years. Nat Rev Cancer. 2003;3(6):459–65. 10.1038/nrc1097. [DOI] [PubMed] [Google Scholar]
- 68.Ancrile B, Lim KH, Counter CM. Oncogenic Ras-induced secretion of IL6 is required for tumorigenesis. Genes Dev. 2007;21(14):1714–9. 10.1101/gad.1549407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Scheller J, Ohnesorge N, Rose-John S. Interleukin-6 trans-signalling in chronic inflammation and cancer. Scand J Immunol. 2006;63(5):321–9. 10.1111/j.1365-3083.2006.01750.x. [DOI] [PubMed] [Google Scholar]
- 70.Chen J, Kobayashi M, Darmanin S, Qiao Y, Gully C, Zhao R, et al. Pim-1 plays a pivotal role in hypoxia-induced chemoresistance. Oncogene. 2009;28(28):2581–92. 10.1038/onc.2009.124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Xu J, Xiong G, Cao Z, Huang H, Wang T, You L, et al. PIM-1 contributes to the malignancy of pancreatic cancer and displays diagnostic and prognostic value. J Exp Clin Cancer Res. 2016;35:1–2. 10.1186/s13046-016-0406-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Warnecke-Eberz U, Bollschweiler E, Drebber U, Pohl A, Baldus SE, Hoelscher AH, et al. Frequent down-regulation of pim-1 mRNA expression in non-small cell lung cancer is associated with lymph node metastases. Oncol Rep. 2008;20(3):619–24. 10.3892/or_00000050. [PubMed] [Google Scholar]
- 73.Jin Y, Tong DY, Chen JN, Feng ZY, Yang JY, Shao CK, Li JP. Overexpression of osteopontin, αvβ3 and Pim-1 associated with prognostically important clinicopathologic variables in non-small cell lung cancer. PLoS ONE. 2012;7(10): e48575. 10.1371/journal.pone.0048575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Pang W, Tian X, Bai F, Han R, Wang J, Shen H, et al. Pim-1 kinase is a target of miR-486-5p and eukaryotic translation initiation factor 4E, and plays a critical role in lung cancer. Mol Cancer. 2014;13:1–5. 10.1186/1476-4598-13-240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Jiang R, Wang X, Jin Z, Li K. Association of nuclear PIM1 expression with lymph node metastasis and poor prognosis in patients with lung adenocarcinoma and squamous cell carcinoma. J Cancer. 2016;7(3):324. 10.7150/jca.13422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Attili I, Bonanno L, Karachaliou N, Bracht JW, Berenguer J, Codony-Servat C, et al. SRC and PIM1 as potential co-targets to overcome resistance in MET deregulated non-small cell lung cancer. Translational Lung Cancer Research. 2020;9(5):1810. 10.21037/tlcr-20-681 [DOI] [PMC free article] [PubMed]
- 77.Westermann B. Mitochondrial fusion and fission in cell life and death. Nat Rev Mol Cell Biol. 2010;11(12):872–84. 10.1038/nrm3013. [DOI] [PubMed] [Google Scholar]
- 78.Chauhan SS, Toth RK, Jensen CC, Casillas AL, Kashatus DF, Warfel NA. PIM kinases alter mitochondrial dynamics and chemosensitivity in lung cancer. Oncogene. 2020;39(12):2597–611. 10.1038/s41388-020-1168-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Peng YH, Li JJ, Xie FW, Chen JF, Yu YH, Ouyang XN, et al. Expmunression of pim-1 in tumors, tumor stroma and tumor-adjacent mucosa co-determines the prognosis of colon cancer patients. PLoS ONE. 2013;8(10): e76693. 10.1371/journal.pone.0076693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Zhou Y, Zhou Y, Liu S, Wang J, Ji R, Yan X. Prognostic and imomodulatory effects of PIM1 in colorectal carcinoma. Journal of BU ON: Official Journal of the Balkan Union of Oncology. 2021;26(2):513–20. [PubMed] [Google Scholar]
- 81.Weirauch U, Beckmann N, Thomas M, Grünweller A, Huber K, Bracher F, et al. Functional role and therapeutic potential of the pim-1 kinase in colon carcinoma. Neoplasia. 2013;15(7):783-IN28. 10.1593/neo.13172 [DOI] [PMC free article] [PubMed]
- 82.Zhang M, Liu T, Sun H, Weng W, Zhang Q, Liu C, et al. Pim1 supports human colorectal cancer growth during glucose deprivation by enhancing the Warburg effect. Can Sci. 2018;109(5):1468–79. 10.1111/cas.13562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Criollo A, Niso-Santano M, Malik SA, Michaud M, Morselli E, Mariño G, et al. Inhibition of autophagy by TAB2 and TAB3. EMBO J. 2011;30(24):4908–20. 10.1038/emboj.2011.413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Hu E, Ding L, Miao H, Liu F, Liu D, Dou H, et al. MiR-30a attenuates immunosuppressive functions of IL-1β-elicited mesenchymal stem cells via targeting TAB3. FEBS Lett. 2015;589(24):3899–907. 10.1016/j.febslet.2015.11.001. [DOI] [PubMed] [Google Scholar]
- 85.Luo C, Yuan R, Chen L, Zhou W, Shen W, Qiu Y, et al. TAB3 upregulates Survivin expression to promote colorectal cancer invasion and metastasis by binding to the TAK1-TRAF6 complex. Oncotarget. 2017;8(63):106565. 10.18632/oncotarget.22497 [DOI] [PMC free article] [PubMed]
- 86.Li Q, Chen L, Luo C, Ge J, Zhu Z, Wang K, et al. TAB3 upregulates PIM1 expression by directly activating the TAK1-STAT3 complex to promote colorectal cancer growth. Exp Cell Res. 2020;391(1): 111975. 10.1016/j.yexcr.2020.111975. [DOI] [PubMed] [Google Scholar]
- 87.Mabuchi S, Kuroda H, Takahashi R, Sasano T. The PI3K/AKT/mTOR pathway as a therapeutic target in ovarian cancer. Gynecol Oncol. 2015;137(1):173–9. 10.1016/j.ygyno.2015.02.003. [DOI] [PubMed] [Google Scholar]
- 88.Rebello RJ, Huglo AV, Furic L. PIM activity in tumours: A key node of therapy resistance. Advances in biological regulation. 2018;67:163–9. 10.1038/s41388-020-1202-y. [DOI] [PubMed] [Google Scholar]
- 89.Song JH, Padi SK, Luevano LA, Minden MD, DeAngelo DJ, Hardiman G, et al. Insulin receptor substrate 1 is a substrate of the Pim protein kinases. Oncotarget. 2016;7(15):20152. 10.18632/oncotarget.7918 [DOI] [PMC free article] [PubMed]
- 90.Aziz AU, Farid S, Qin K, Wang H, Liu B. PIM kinases and their relevance to the PI3K/AKT/mTOR pathway in the regulation of ovarian cancer. Biomolecules. 2018;8(1):7. 10.3390/biom8010007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Suh DH, Kim MK, No JH, Chung HH, Song YS. Metabolic approaches to overcoming chemoresistance in ovarian cancer. Ann N Y Acad Sci. 2011;1229(1):53–60. 10.1111/j.1749-6632.2011.06095.x. [DOI] [PubMed] [Google Scholar]
- 92.Gordan JD, Thompson CB, Simon MC. HIF and c-Myc: sibling rivals for control of cancer cell metabolism and proliferation. Cancer Cell. 2007;12(2):108–13. 10.1016/j.ccr.2007.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Morais M, Dias F, Teixeira AL, Medeiros R. MicroRNAs and altered metabolism of clear cell renal cell carcinoma: Potential role as aerobic glycolysis biomarkers. Biochimica et Biophysica Acta (BBA)-General Subjects. 2017;1861(9):2175–85. 10.1016/j.bbagen.2017.05.028 [DOI] [PubMed]
- 94.Labak CM, Wang PY, Arora R, Guda MR, Asuthkar S, Tsung AJ, et al. Glucose transport: meeting the metabolic demands of cancer, and applications in glioblastoma treatment. Am J Cancer Res. 2016;6(8):1599. [PMC free article] [PubMed] [Google Scholar]
- 95.Warnecke-Eberz U, Bollschweiler E, Drebber U, Metzger R, Baldus SE, Hoelscher AH, et al. Prognostic impact of protein overexpression of the proto-oncogene PIM-1 in gastric cancer. Anticancer Res. 2009;29(11):4451–5. 10.7150/jca.13422. [PubMed] [Google Scholar]
- 96.Yan B, Yau EX, Samanta S, Ong CW, Yong KJ, Ng LK, et al. Clinical and therapeutic relevance of PIM1 kinase in gastric cancer. Gastric Cancer. 2012;15:188–97. 10.1007/s10120-011-0097-2. [DOI] [PubMed] [Google Scholar]
- 97.Wang Y, Zhou X, Shan B, Han J, Wang F, Fan X, et al. Downregulation of microRNA-33a promotes cyclin-dependent kinase 6, cyclin D1 and PIM1 expression and gastric cancer cell proliferation. Mol Med Rep. 2015;12(5):6491–500. 10.3892/mmr.2015.4296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Herzog S, Fink MA, Weitmann K, Friedel C, Hadlich S, Langner S, et al. Pim1 kinase is upregulated in glioblastoma multiforme and mediates tumor cell survival. Neuro Oncol. 2015;17(2):223–42. 10.1093/neuonc/nou216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Peltola K, Hollmen M, Maula SM, Rainio E, Ristamäki R, Luukkaa M, et al. Pim-1 kinase expression predicts radiation response in squamocellular carcinoma of head and neck and is under the control of epidermal growth factor receptor. Neoplasia. 2009;11(7):629-IN1. 10.1593/neo.81038 [DOI] [PMC free article] [PubMed]
- 100.Montano N, Cenci T, Martini M, D’Alessandris QG, Pelacchi F, Ricci-Vitiani L, et al. Expression of EGFRvIII in glioblastoma: prognostic significance revisited. Neoplasia. 2011;13(12):1113-IN6. 10.1593/neo.111338 [DOI] [PMC free article] [PubMed]
- 101.Hatanpaa KJ, Burma S, Zhao D, Habib AA. Epidermal growth factor receptor in glioma: signal transduction, neuropathology, imaging, and radioresistance. Neoplasia. 2010;12(9):675–84. 10.1593/neo.10688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Pines G, Köstler WJ, Yarden Y. Oncogenic mutant forms of EGFR: lessons in signal transduction and targets for cancer therapy. FEBS Lett. 2010;584(12):2699–706. 10.1016/j.febslet.2010.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Weirauch U, Grünweller A, Cuellar L, Hartmann RK, Aigner A. U1 adaptors for the therapeutic knockdown of the oncogene pim-1 kinase in glioblastoma. nucleic acid therapeutics. 2013;23(4):264–72. 10.1089/nat.2012.0407 [DOI] [PMC free article] [PubMed]
- 104.Remy J, Linder B, Weirauch U, Konovalova J, Marschalek R, Aigner A, et al. Inhibition of PIM1 blocks the autophagic flux to sensitize glioblastoma cells to ABT-737-induced apoptosis. Biochimica et Biophysica Acta (BBA)-Molec Cell Res. 2019;1866(2):175–89. 10.1016/j.bbamcr.2018.10.017. [DOI] [PubMed] [Google Scholar]
- 105.Seifert C, Balz E, Herzog S, Korolev A, Gaßmann S, Paland H, et al. PIM1 inhibition affects glioblastoma stem cell behavior and kills glioblastoma stem-like cells. Int J Mol Sci. 2021;22(20):11126. 10.3390/ijms222011126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Chiang WF, Yen CY, Lin CN, Liaw GA, Chiu CT, Hsia YJ, et al. Up-regulation of a serine–threonine kinase proto-oncogene Pim-1 in oral squamous cell carcinoma. Int J Oral Maxillofac Surg. 2006;35(8):740–5. 10.1016/j.ijom.2006.01.027. [DOI] [PubMed] [Google Scholar]
- 107.Beier UH, Weise JB, Laudien M, Sauerwein H, Görögh T. Overexpression of Pim-1 in head and neck squamous cell carcinomas. Int J Oncol. 2007;30(6):1381–7. 10.3892/ijo.30.6.1381. [PubMed] [Google Scholar]
- 108.Tanaka S, Kitamura T, Higashino F, Hida K, Ohiro Y, Ono M, et al. Pim-1 activation of cell motility induces the malignant phenotype of tongue carcinoma. Mol Med Rep. 2009;2(2):313–8. 10.3892/mmr_00000102. [DOI] [PubMed] [Google Scholar]
- 109.Zhu X, Xu JJ, Hu SS, Feng JG, Jiang LH, Hou XX, et al. Pim-1 acts as an oncogene in human salivary gland adenoid cystic carcinoma. J Exp Clin Cancer Res. 2014;33:1–2. 10.1186/s13046-014-0114-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Takeuchi H, Miyamoto T, Fuseya C, Asaka R, Ida K, Ono M, et al. PIM1 is a poor prognostic factor for and potential therapeutic target in serous carcinoma of the endometrium. Int J Gynecol Pathol. 2023;42(3):282–92. 10.1097/PGP.0000000000000882. [DOI] [PubMed] [Google Scholar]
- 111.Shannan B, Watters A, Chen Q, Mollin S, Dörr M, Meggers E, et al. PIM kinases as therapeutic targets against advanced melanoma. Oncotarget. 2016;7(34):54897. 10.18632/oncotarget.10703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Mahalingam D, Espitia CM, Medina EC, Esquivel JA, Kelly KR, Bearss D, et al. Targeting PIM kinase enhances the activity of sunitinib in renal cell carcinoma. Br J Cancer. 2011;105(10):1563–73. 10.1038/bjc.2011.426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Wen QL, Yi HQ, Yang K, Yin CT, Yin WJ, Xiang FY, et al. Role of oncogene PIM-1 in the development and progression of papillary thyroid carcinoma: Involvement of oxidative stress. Mol Cell Endocrinol. 2021;523: 111144. 10.1016/j.mce.2020.111144. [DOI] [PubMed] [Google Scholar]
- 114.Tao ZF, Hasvold LA, Leverson JD, Han EK, Guan R, Johnson EF, et al. Discovery of 3 H-benzo [4, 5] thieno [3, 2-d] pyrimidin-4-ones as potent, highly selective, and orally bioavailable inhibitors of the human protooncogene proviral insertion site in moloney murine leukemia virus (PIM) kinases. J Med Chem. 2009;52(21):6621–36. 10.1021/jm900943h. [DOI] [PubMed] [Google Scholar]
- 115.Forshell LP, Li Y, Forshell TZ, Rudelius M, Nilsson L, Keller U, et al. The direct Myc target Pim3 cooperates with other Pim kinases in supporting viability of Myc-induced B-cell lymphomas. Oncotarget. 2011;2(6):448. 10.18632/oncotarget.283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Rathi A, Kumar D, Hasan GM, Haque MM, Hassan MI. Therapeutic targeting of PIM KINASE signaling in cancer therapy: Structural and clinical prospects. Biochimica et Biophysica Acta (BBA)-General Subjects. 2021;1865(11):129995.10.1016/j.bbagen.2021.129995. [DOI] [PubMed] [Google Scholar]
- 117.Ingle K, LaComb JF, Graves LM, Baines AT, Bialkowska AB. AUM302, a novel triple kinase PIM/PI3K/mTOR inhibitor, is a potent in vitro pancreatic cancer growth inhibitor. PLoS ONE. 2023;18(11): e0294065. 10.1371/journal.pone.0294065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Dakin LA, Block MH, Chen H, Code E, Dowling JE, Feng X, et al. Discovery of novel benzylidene-1, 3-thiazolidine-2, 4-diones as potent and selective inhibitors of the PIM-1, PIM-2, and PIM-3 protein kinases. Bioorg Med Chem Lett. 2012;22(14):4599–604. 10.1016/j.bmcl.2012.05.098. [DOI] [PubMed] [Google Scholar]
- 119.Le BT, Kumarasiri M, Adams JR, Yu M, Milne R, Sykes MJ, et al. Targeting Pim kinases for cancer treatment: opportunities and challenges. Future Med Chem. 2015;7(1):35–53. 10.4155/fmc.14.145. [DOI] [PubMed] [Google Scholar]
- 120.Lee M, Lee KH, Min A, Kim J, Kim S, Jang H, et al. Pan-Pim kinase inhibitor AZD1208 suppresses tumor growth and synergistically interacts with Akt inhibition in gastric cancer cells. Cancer Research and Treatment: Official Journal of Korean Cancer Association. 2019;51(2):451–63. 10.4143/crt.2017.341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Kirschner AN, Wang J, Van Der Meer R, Anderson PD, Franco-Coronel OE, Kushner MH, et al. PIM kinase inhibitor AZD1208 for treatment of MYC-driven prostate cancer. J Nat Canc Inst. 2015;107(2):dju407. 10.1093/jnci/dju407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Pierre F, Regan CF, Chevrel MC, Siddiqui-Jain A, Macalino D, Streiner N, et al. Novel potent dual inhibitors of CK2 and Pim kinases with antiproliferative activity against cancer cells. Bioorg Med Chem Lett. 2012;22(9):3327–31. 10.1016/j.bmcl.2012.02.099. [DOI] [PubMed] [Google Scholar]
- 123.Garcia PD, Langowski JL, Wang Y, Chen M, Castillo J, Fanton C, Ison M, Zavorotinskaya T, Dai Y, Lu J, Niu XH. Pan-PIM kinase inhibition provides a novel therapy for treating hematologic cancers. Clin Cancer Res. 2014;20(7):1834–45. 10.1158/1078-0432.CCR-13-2062. [DOI] [PubMed] [Google Scholar]
- 124.Chon HJ, Bae KJ, Lee Y, Kim J. The casein kinase 2 inhibitor, CX-4945, as an anti-cancer drug in treatment of human hematological malignancies. Front Pharmacol. 2015;6:70. 10.3389/fphar.2015.00070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Bogusz J, Zrubek K, Rembacz KP, Grudnik P, Golik P, Romanowska M, Wladyka B, Dubin G. Structural analysis of PIM1 kinase complexes with ATP-competitive inhibitors. Sci Rep. 2017;7(1):13399. 10.1038/s41598-017-13557-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Haddach M, Michaux J, Schwaebe MK, Pierre F, O’Brien SE, Borsan C, Tran J, Raffaele N, Ravula S, Drygin D, Siddiqui-Jain A. Discovery of CX-6258. A potent, selective, and orally efficacious pan-Pim kinases inhibitor. ACS medicinal chemistry letters. 2012 ;3(2):135–9. 10.1021/ml200259q [DOI] [PMC free article] [PubMed]
- 127.Barnett A, Ding S, Murray C, Chamberlain M, Plummer S, Evans TR, MacPherson I, Bissett D, Elcombe CR, Wolf CR. Anti-tumor activity of CXR1002, a novel anti-cancer clinical phase compound that induces ER stress and inhibits PIM kinases: human tumor xenograft efficacy and in vitro mode of action. EJC Suppl. 2010;8(7):45–6. 10.1093/toxsci/kfy035. [Google Scholar]
- 128.Santio NM, Vahakoski RL, Rainio EM, Sandholm JA, Virtanen SS, Prudhomme M, Anizon F, Moreau P, Koskinen PJ. Pim-selective inhibitor DHPCC-9 reveals Pim kinases as potent stimulators of cancer cell migration and invasion. Mol Cancer. 2010;9:1–3. 10.1186/1476-4598-9-279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Akué-Gédu R, Rossignol E, Azzaro S, Knapp S, Filippakopoulos P, Bullock AN, Bain J, Cohen P, Prudhomme M, Anizon F, Moreau P. Synthesis, kinase inhibitory potencies, and in vitro antiproliferative evaluation of new Pim kinase inhibitors. J Med Chem. 2009Oct 22;52(20):6369–81. [DOI] [PubMed] [Google Scholar]
- 130.Kennedy SP, O’Neill M, Cunningham D, Morris PG, Toomey S, Blanco-Aparicio C, Martinez S, Pastor J, Eustace AJ, Hennessy BT. Preclinical evaluation of a novel triple-acting PIM/PI3K/mTOR inhibitor, IBL-302, in breast cancer. Oncogene. 2020;39(14):3028–40. 10.1038/s41388-020-1202-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Mohlin S, Hansson K, Radke K, Martinez S, Blanco-Apiricio C, Garcia-Ruiz C, Welinder C, Esfandyari J, O’Neill M, Pastor J, von Stedingk K. Anti-tumor effects of PIM/PI 3K/mTOR triple kinase inhibitor IBL-302 in neuroblastoma. EMBO Mole Med. 2019;11(8):e10058. 10.15252/emmm.201810058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Gelbert LM, Cai S, Lin X, Sanchez-Martinez C, Del Prado M, Lallena MJ, et al. Preclinical characterization of the CDK4/6 inhibitor LY2835219: in-vivo cell cycle-dependent/independent anti-tumor activities alone/in combination with gemcitabine. Invest New Drugs. 2014;32:825–37. 10.1007/s10637-014-0120-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Small JS, Holder SL. Targeting PIM1 and CDK4/6 kinases in renal cell carcinoma. Canc Res. 2015;75(15_Supplement):5323. 10.1158/1538-7445.AM2015-5323. [Google Scholar]
- 134.Oyallon B, Brachet-Botineau M, Logé C, Bonnet P, Souab M, Robert T, Ruchaud S, Bach S, Berthelot P, Gouilleux F, Viaud-Massuard MC. Structure-based design of novel quinoxaline-2-carboxylic acids and analogues as Pim-1 inhibitors. Eur J Med Chem. 2018;154:101–9. 10.1016/j.ejmech.2018.04.056. [DOI] [PubMed] [Google Scholar]
- 135.Oyallon B, Brachet-Botineau M, Logé C, Robert T, Bach S, Ibrahim S, Raoul W, Croix C, Berthelot P, Guillon J, Pinaud N. New quinoxaline derivatives as dual Pim-1/2 kinase inhibitors: Design, synthesis and biological evaluation. Molecules. 2021;26(4):867. 10.3390/molecules26040867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Holder S, Zemskova M, Zhang C, Tabrizizad M, Bremer R, Neidigh JW, Lilly MB. Characterization of a potent and selective small-molecule inhibitor of the PIM1 kinase. Mol Cancer Ther. 2007;6(1):163–72. 10.1158/1535-7163.MCT-06-0397. [DOI] [PubMed] [Google Scholar]
- 137.Willert M, Augstein A, Poitz DM, Schmeisser A, Strasser RH, Braun-Dullaeus RC. Transcriptional regulation of Pim-1 kinase in vascular smooth muscle cells and its role for proliferation. Basic Res Cardiol. 2010;105:267–77. 10.1007/s00395-009-0055-x. [DOI] [PubMed] [Google Scholar]
- 138.Beharry Z, Zemskova M, Mahajan S, Zhang F, Ma J, Xia Z, et al. Novel benzylidene-thiazolidine-2, 4-diones inhibit Pim protein kinase activity and induce cell cycle arrest in leukemia and prostate cancer cells. Mol Cancer Ther. 2009;8(6):1473–83. 10.1158/1535-7163.mct-08-1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Bellon M, Lu L, Nicot C. Constitutive activation of Pim1 kinase is a therapeutic target for adult T-cell leukemia. Blood, The Journal of the American Society of Hematology. 2016;127(20):2439–50. 10.1182/blood-2015-11-685032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Arunesh GM, Shanthi E, Krishna MH, Sooriya Kumar J, Viswanadhan VN. Small molecule inhibitors of PIM1 kinase: July 2009 to February 2013 patent update. Expert Opin Ther Pat. 2014;24(1):5–17. 10.1517/13543776.2014.848196. [DOI] [PubMed] [Google Scholar]
- 141.Wu CP, Li YQ, Chi YC, Huang YH, Hung TH, Wu YS. The second-generation PIM kinase inhibitor TP-3654 resensitizes ABCG2-overexpressing multidrug-resistant cancer cells to cytotoxic anticancer drugs. Int J Mol Sci. 2021;22(17):9440. 10.3390/ijms22179440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Foulks JM, Carpenter KJ, Luo B, Xu Y, Senina A, Nix R, Chan A, Clifford A, Wilkes M, Vollmer D, Brenning B. A small-molecule inhibitor of PIM kinases as a potential treatment for urothelial carcinomas. Neoplasia. 2014;16(5):403–12. 10.1016/j.neo.2014.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Keeton EK, McEachern K, Dillman KS, Palakurthi S, Cao Y, Grondine MR, et al. AZD1208, a potent and selective pan-Pim kinase inhibitor, demonstrates efficacy in preclinical models of acute myeloid leukemia. Blood, The Journal of the American Society of Hematology. 2014;123(6):905–13. 10.1182/blood-2013-04-495366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Xu Y, Brenning BG, Kultgen SG, Foulks JM, Clifford A, Lai S, Chan A, Merx S, McCullar MV, Kanner SB, Ho KK. Synthesis and biological evaluation of pyrazolo [1, 5-a] pyrimidine compounds as potent and selective Pim-1 inhibitors. ACS Med Chem Lett. 2015;6(1):63–7. 10.1021/ml500300c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Chen LS, Redkar S, Bearss D, Wierda WG, Gandhi V. Pim kinase inhibitor, SGI-1776, induces apoptosis in chronic lymphocytic leukemia cells. Blood, The Journal of the American Society of Hematology. 2009;114(19):4150–7. 10.1182/blood-2009-03-212852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Mumenthaler SM, Ng PY, Hodge A, Bearss D, Berk G, Kanekal S, Redkar S, Taverna P, Agus DB, Jain A. Pharmacologic inhibition of Pim kinases alters prostate cancer cell growth and resensitizes chemoresistant cells to taxanes. Mol Cancer Ther. 2009;8(10):2882–93. 10.1158/1535-7163.MCT-09-0293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Xu D, Cobb MG, Gavilano L, Witherspoon SM, Williams D, White CD, Taverna P, Bednarski BK, Kim HJ, Baldwin AS, Baines AT. Inhibition of oncogenic Pim-3 kinase modulates transformed growth and chemosensitizes pancreatic cancer cells to gemcitabine. Cancer Biol Ther. 2013;14(6):492–501. 10.4161/cbt.24343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Cervantes-Gomez F, Chen LS, Orlowski RZ, Gandhi V. Biological effects of the Pim kinase inhibitor, SGI-1776, in multiple myeloma. Clin Lymphoma Myeloma Leuk. 2013;13:S317–29. 10.1016/j.clml.2013.05.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Luszczak S, Kumar C, Sathyadevan VK, Simpson BS, Gately KA, Whitaker HC, Heavey S. PIM kinase inhibition: co-targeted therapeutic approaches in prostate cancer. Signal Transduct Target Ther. 2020Jan 31;5(1):7. 10.1038/s41392-020-0109-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
No datasets were generated or analysed during the current study.