

SUMMARY
In this high-throughput proteomic study of autosomal dominant Alzheimer’s disease (ADAD), we sought to identify early biomarkers in cerebrospinal fluid (CSF) for disease monitoring and treatment strategies. We examined CSF proteins in 286 mutation carriers (MCs) and 177 non-carriers (NCs). The developed multi-layer regression model distinguished proteins with different pseudo-trajectories between these groups. We validated our findings with independent ADAD as well as sporadic AD datasets and employed machine learning to develop and validate predictive models. Our study identified 137 proteins with distinct trajectories between MCs and NCs, including eight that changed before traditional AD biomarkers. These proteins are grouped into three stages: early stage (stress response, glutamate metabolism, neuron mitochondrial damage), middle stage (neuronal death, apoptosis), and late presymptomatic stage (microglial changes, cell communication). The predictive model revealed a six-protein subset that more effectively differentiated MCs from NCs, compared with conventional biomarkers.
Graphical abstract
In brief
Examination of CSF proteins in mutation carriers and non-carriers for autosomal dominant Alzheimer’s disease uncovers early proteomic changes and generates a robust ADAD prediction model. The findings highlight the similarities and differences between ADAD and sporadic Alzheimer’s disease, which can lead to personalized medicine for mutation carriers in ADAD genes.
INTRODUCTION
Autosomal dominant Alzheimer’s disease (ADAD) accounts for approximately 1% of all AD cases.1 ADAD is characterized by the presence of autosomal dominant mutations in the amyloid precursor protein (APP), presenilin-1 (PSEN1), or presenilin-2 (PSEN2) genes.1,2 This rare form of AD has been instrumental in elucidating critical pathological mechanisms and the temporal progression of brain changes associated with AD.3 Thus, a comprehensive study of the cerebrospinal fluid (CSF) proteome changes in this form of the disease can help advance our understanding of its pathophysiology and contribute to the identification of previously unreported biomarkers and potential therapeutic strategies.
The Dominantly Inherited Alzheimer Network (DIAN) observational study is a worldwide effort to study individuals with ADAD mutations. It involves longitudinal assessments including imaging, cognitive evaluations, and fluid collection (CSF and plasma).4 Traditional CSF AD biomarkers, such as β-amyloid 42 (Aβ42), total Tau (Tau), and phosphorylated tau181 (pTau), have demonstrated their analytical validity in ADAD as well as sporadic Alzheimer’s disease (sAD).5 CSF biomarkers change earlier in the disease course than amyloid or Tau in positron emission tomography (PET) imaging6 with the added benefits of being more cost-effective, faster, and easily applicable in clinical settings.7 Although proteome-based biomarkers in the plasma for ADAD and sAD are being identified, CSF, which is in direct contact with the brain, is relatively unaffected by proteins from other organs and is ideal for detection of brain-related changes.8
ADAD exhibits several similarities to sAD, including phenotype, clinical progression, and neuropathology.1,9,10 However, it is essential to note that the etiology and onset mechanisms of these two AD forms follow distinct patterns.11 Particularly in ADAD, mutation carriers (MCs) from the same family generally exhibit a similar age at symptom onset (AAO), and therefore, it is possible to calculate the “estimated year of onset (EYO)” for each family member by extrapolating from the known AAO in individuals who share the same mutation.12 For this, EYOs are determined by subtracting the age at study assessment minus the mean mutation AAO associated with their specific mutation.12 A recent CSF proteomic study in 14 carriers of ADAD mutations reported 56 proteins associated with mutation status.13 In another study that included 22 MC and 20 non-carrier (NC) individuals, 66 CSF proteins associated with ADAD were identified, some of which were also dysregulated in sAD.14 However, the predictive value of those proteins was not explored. In another recent study, researchers analyzed 59 CSF proteins, selected based on prior brain studies, in 286 MCs and 184 NCs samples of which 33 were associated significantly with ADAD after correcting for EYO.15
There has been a significant increase in the volume of AD proteomic research. Proteomic studies have been conducted on a small scale but have been able to capture some actual biological substrates and go beyond the traditional neuropathological features like amyloid plaques, neurofibrillary tangle, and cerebral amyloid angiopathy,16–22 leading to a better biological understanding of the disease as well as revealing potential new therapeutic targets. When evaluating individual studies, including the previously published ADAD studies, they shared some common limitations such as limited sample size and a small number of investigated proteins. Previous studies were focused on identifying proteins associated with mutation status, not disease onset. Although some were corrected by the EYO,15 EYO information has neither been fully leveraged to identify proteins that show different trajectories between MCs and NCs nor to determine the time in which protein changes occur in relation to the clinical onset. Overall, these limitations highlight the need for a large-scale and high-throughput study to identify early proteomic changes in the ADAD.
RESULTS
Study design
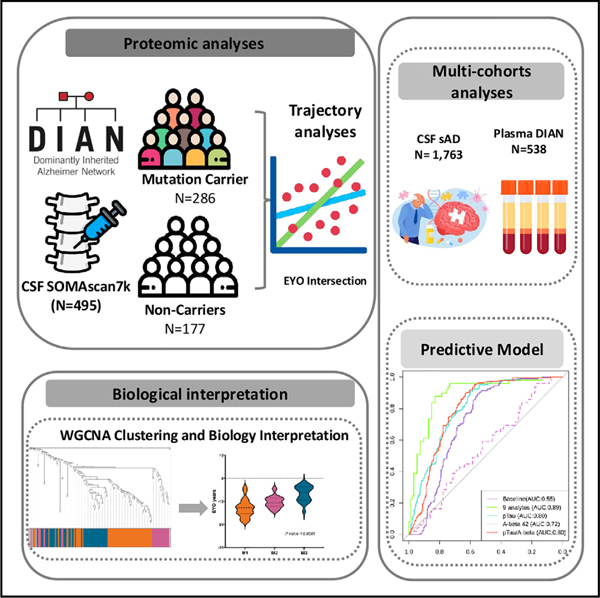
The primary goal of this study is to identify proteins that present early changes in ADAD MCs, compared with NCs, in CSF and plasma. We aimed to identify the earliest biomarkers of ADAD, with the potential to provide valuable insights into the presymptomatic phases of ADAD and to enable early intervention strategies. To do this, we used a unique approach that leverages the estimated AAO to assess pseudo-trajectories (using cross-sectional data to simulate longitudinal data) and identified proteins with significant differences between MCs and NCs. We subsequently determined the time when those pseudo-trajectories become divergent in relation to the EYO.
We generated and leveraged large-scale proteomics from CSF (6,163 proteins) and plasma (6,022 proteins) (Figure 1) from samples with and without autosomal dominant AD mutations from the DIAN study (Table 1). MCs include symptomatic and presymptomatic DIAN participants with pathogenic variants in one of three ADAD genes (APP, PSEN1, or PSEN2).23 Presymptomatic participants are cognitively normal MCs. The matched NCs were sourced from the families of the MCs. Participants in this control group share similar genetics backgrounds and environmental influences, further enhancing the power of our study to detect reliable proteomic alterations.
Figure 1. Study overview.
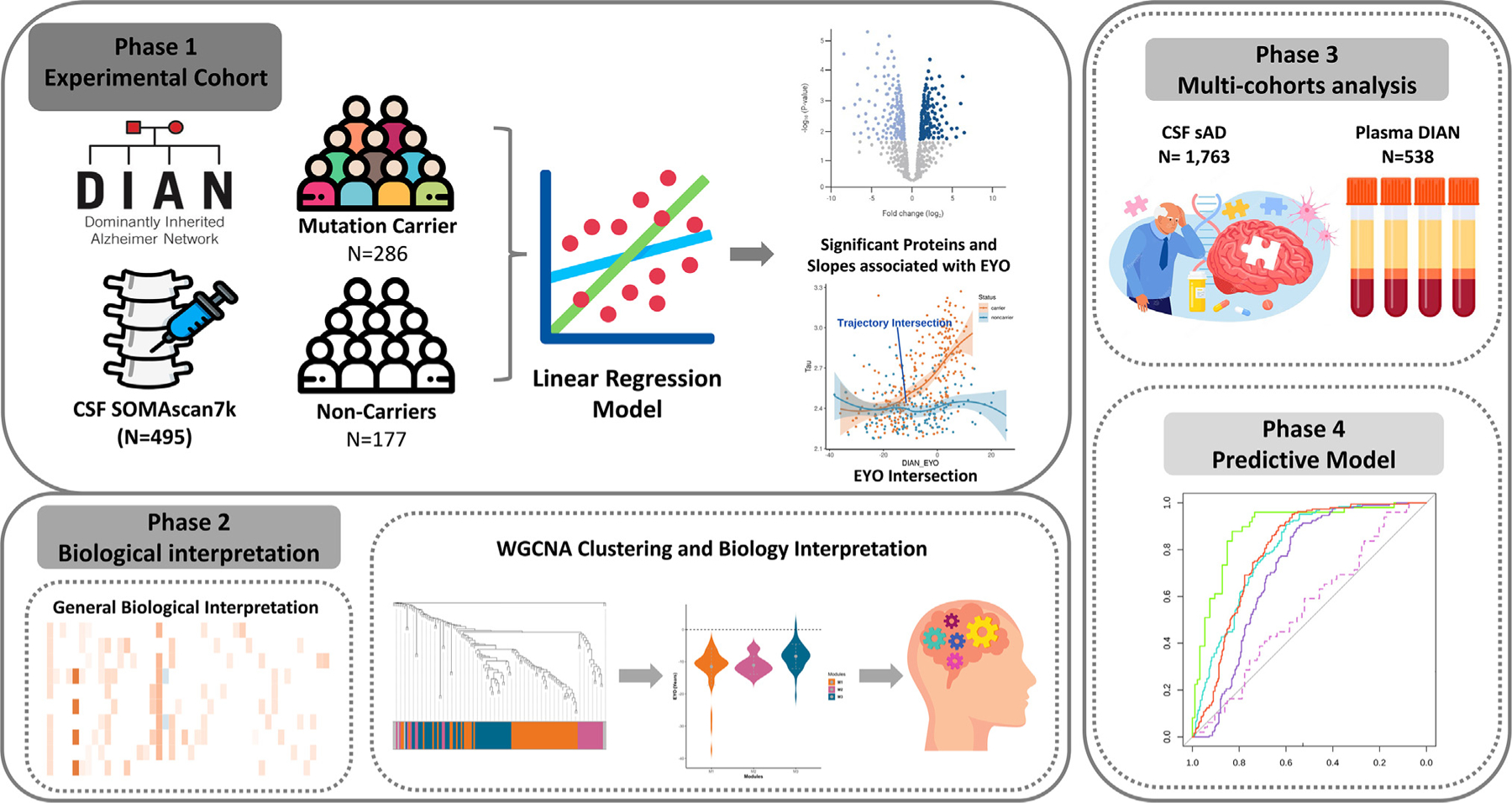
A total of 6,163 proteins were measured in CSF sample from 286 mutation carriers (MCs) and 177 non-carriers (NCs). Differential pseudo-trajectory analyses were performed between MCs and NCs. Trajectory intersections were calculated for significant pseudo-trajectory proteins. Biological functions were identified by protein co-expression network analysis and pathway enrichment. A total of 1,763 sAD CSF samples and 538 DIAN plasma samples were analyzed to validate the approach and contextualize the findings. Several publicly available external proteomic datasets were used to validate our findings as well. Finally, the LASSO model was used to select significant trajectory proteins and to create predictive models for ADAD.
Table 1.
Demographics of participants of CSF and plasma in DIAN
| Descriptive | CSF |
Plasma |
||||
|---|---|---|---|---|---|---|
| MCs | NCs | p value | MCs | NCs | p value | |
| Sample size | 286 | 177 | NT | 325 | 213 | NT |
| PSEN1 (% of MCs) | 212 (74) | 0 | 240 (74) | 0 | ||
| PSEN2 (% of MCs) | 23 (8) | 0 | 25 (8) | 0 | ||
| APP (% of MCs) | 51 (18) | 0 | 60 (18) | 0 | ||
| Affected carrier (% of MCs) | 102 (36) | 0 | 7.58 × 10−103 | 127 (39) | 0 | 2.77 × 10−116 |
| Presymptomatic carrier (% of MCs) | 184 (64) | 0 | 198 (61) | 0 | ||
| AD non-carrier (% of NCs) | 0 | 6 (4) | 0 | 12 (6) | ||
| Control non-carrier (% of NCs) | 0 | 171 (96) | 0 | 201 (93) | ||
| Age mean (SD), years | 40.0 (10.4) | 39.6 (11.6) | 0.63 | 40.4 (10.6) | 40.7 (11.6) | 0.70 |
| Female Sex n (%) | 159 (55) | 109 (59) | 0.36 | 178 (55) | 127 (59) | 0.38 |
| EYO (SD), years | −6.6 (10.4) | −7.8 (12.5) | 0.39 | −6.3 (11.06) | −7.9 (11.7) | 0.12 |
| APOE ε4+ N (%), at least one allele | 83 (29) | 61 (33) | 0.30 | 97 (30) | 71 (33) | 0.46 |
Chi-squared test used to test subtypes differences between MCs and NCs; not tested (NT), because of all the NCs without any mutation, chi-squared test does not apply to compare the subtypes of mutations between MCs and NCs.SD, standard deviation; MCs, mutation carriers; NCs, non-carriers.
To identify robust proteins associated with ADAD, we divided the samples into discovery and replication datasets through random sampling, while maintaining the proportion of MCs (Table S1). In the discovery stage, we identified the proteins associated with pseudo-trajectories after false discovery rate (FDR) correction. The significant proteins were then tested in the replication dataset (Figure 2A). Of these proteins tested, only those that passed FDR in replication were considered as study-wide significant. Subsequently, we pinpointed when these significant protein changes start, providing invaluable insights into the disease progression. We then conducted analysis based on mutation status as a phenotype, comparing MCs and NCs to further validate these findings (Figure 2B).
Figure 2. Significant pseudo-trajectory proteins and significant proteins associated with ADAD mutation status in CSF.
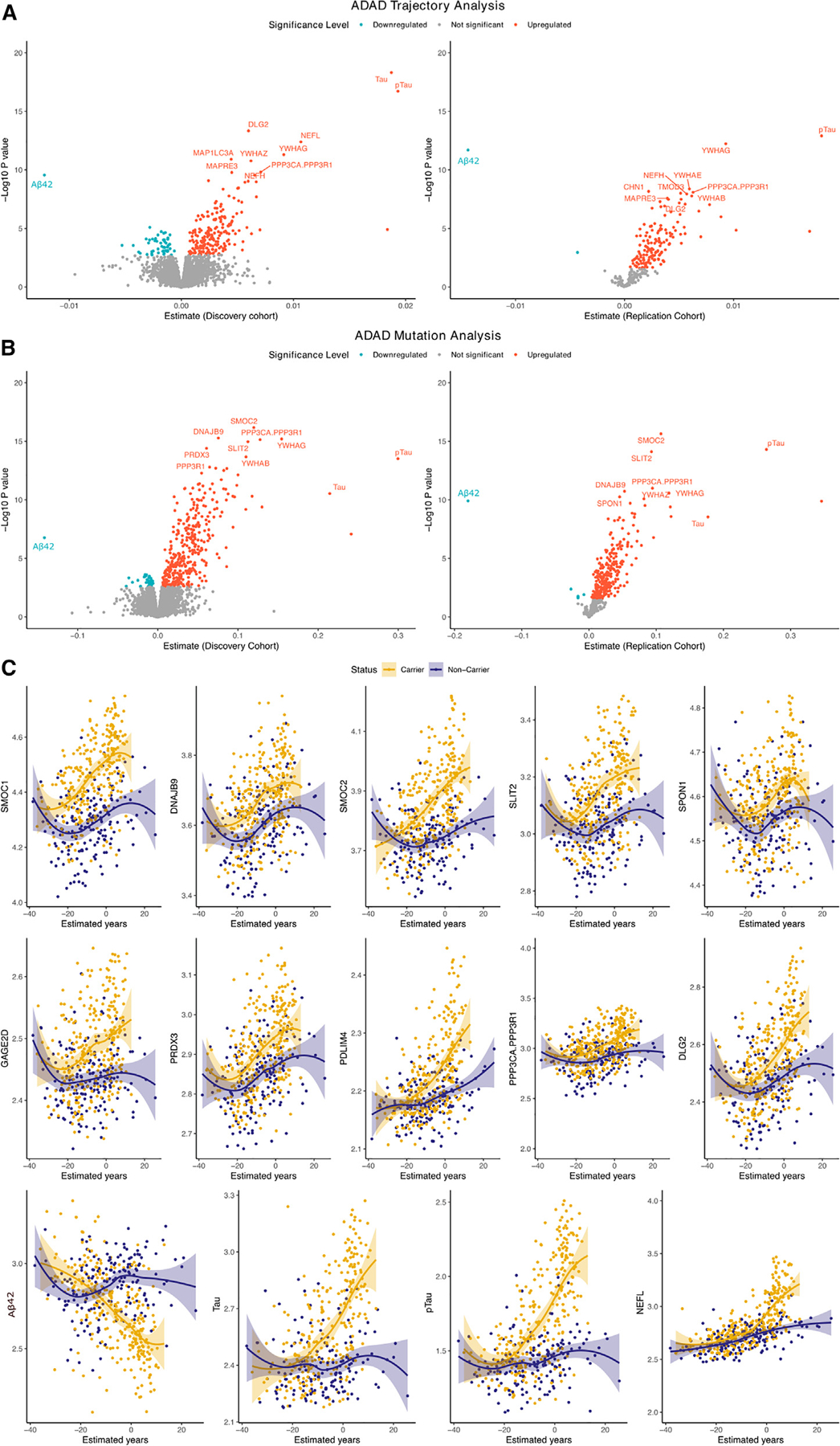
(A and B) Volcano plots displaying the estimate change (x axis) against −log10 statistical differences (y axis) for all tested proteins for pseudo-trajectory analyses between MCs and NCs (A) and ADAD mutation status only (B). The red dots show the significantly upregulated proteins, and the blue dots show the significantly downregulated proteins after multiple test correction (FDR p value < 0.05).
(C) Twelve significant pseudo-trajectory proteins that changed earlier than Tau, pTau, and Aβ42.
We applied network and pathway enrichment analyses to create clusters of proteins that share functional relationships. Pathway enrichment analysis for these groups of proteins was applied to identify specific biological pathways dysregulated in ADAD. We utilized the same analytical pipeline in plasma to determine whether plasma could provide information consistent with that obtained from CSF. We investigated whether the proteins associated with ADAD were also linked to late-onset sAD. Finally, we leveraged those dysregulated proteins to create Aβ- and pTau-independent predictive models. We applied a machine learning approach to identify the most relevant proteins that could serve as promising biomarkers for ADAD. The overarching goal was to pinpoint potential biomarkers that could aid in monitoring disease progression, assessing treatment effectiveness, and designing well-informed therapeutic strategies.
Significantly different pseudo-trajectory protein identification in CSF samples
We analyzed 6,163 unique proteins (7,008 SomaLogic aptamers), which passed quality control (QC), from 286 MCs and 177 NCs. In the discovery stage (143 MCs and 88 NCs), we identified 247 proteins (259 aptamers) that exhibited significantly different pseudo-trajectories between MCs vs. NCs (Figure 2A; Table S1). In the replication stage, only those 247 proteins (259 aptamers) were tested and 137 proteins (145 protein aptamers) replicated at FDR < 0.05 and in the same direction of effects between discovery and replication (Figure 2A; Table S1). Aβ, pTau, and Tau were included for comparison as they are known and well-validated AD biomarkers (Figure 2C).24,25
The top 30 hits included proteins reported to be associated with AD, such as neurofilament (NEFH, NEFL),2,14,26 calcineurin complex (PPP3CA, PPP3R1),27 and 14–3-3 proteins (14–3-3 beta, gamma, zeta).11,13 Other proteins significant in our analyses were not reported in previous studies and included those for extracellular matrix binding (SMOC2, SLIT2), negative regulation of the protein metabolic process (PEBP1, GPI, PTPA, CRKL, PIN1), cytoskeletal protein binding (PDLIM4, PDCD5, STMN2, TMOD3, or MAPRE3), cytosol function (CHN1, DLG2),28 and for protein binding (TCEAL5, GARS1).28,29 There was only one protein that displayed downregulation in ADAD MCs: NPTX2 (p = 1.34 × 10−6). Other reported AD markers, such as TREM2 or YKL-40, did not pass multiple test correction (Figure 3). Additional analyses were done for the 137 identified proteins stratifying the individuals by the gene in which the mutation is located (i.e., individuals with mutation in PSEN1 only and PSEN2 only) (Figure S1). All the proteins that were found significant in the main analyses were also found to be significant in PSEN1 carriers. Despite only being 51 APP carriers, 85 of the 148 analytes were also significant in this group. In PSEN2 carriers, 11 total analytes were nominally significant, of which 6 were common in all three types of MCs. The correlation of effect sizes between these mutations’ specific analyses was very high when compared with our main analysis with very similar EYO estimation (Figure S1).
Figure 3. Validation and replication of significant pseudo-trajectory proteins.
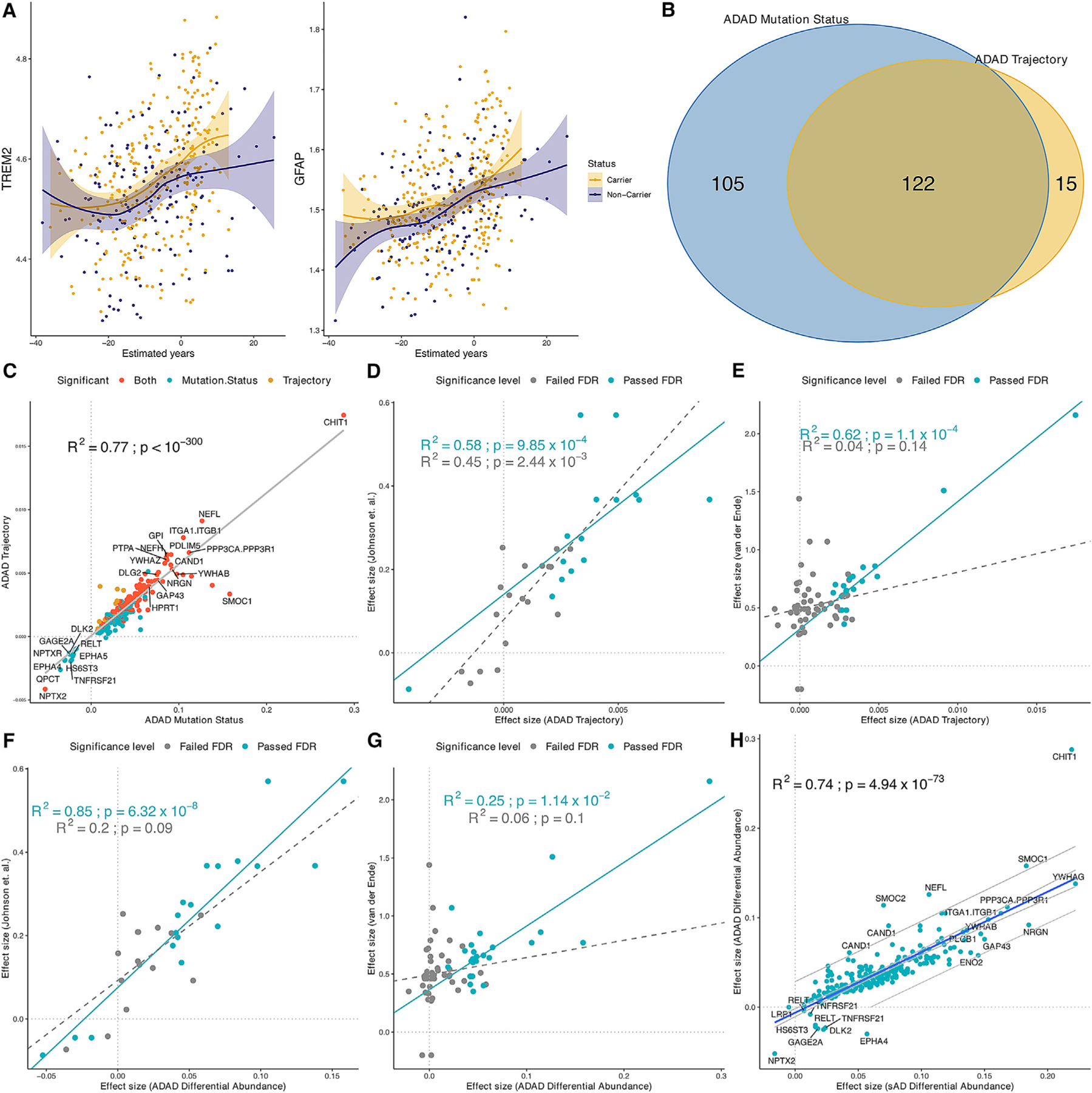
(A) The pseudo-trajectory curve for TREM2 and GFAP, but these two proteins did not pass the FDR threshold between MCs vs. NCs. The yellow curve indicates the MCs’ proteins change with EYO, and purple curve indicates the NCs’ proteins change with EYO.
(B) The overlapped significant proteins from trajectory analysis and ADAD mutation status analysis of MCs vs. NCs at FDR threshold.
(C–H) Scatterplot of the significant trajectory proteins replicated in (C) CSF ADAD mutation status analysis; (D) significant pseudo-trajectory proteins replicated in Johnson et al.15 finding; (E) significant trajectory proteins replicated in Van der Ende et al.14 finding; (F) significant proteins associated with mutation status replicated in Johnson et al. finding; (G) significant proteins associated with mutation status replicated in Van der Ende et al. finding; (H) significant proteins associated with mutation status replicated in significant proteins associated with sAD. Gray line represent 95% confidence interval.
To determine the reliability of our pseudo-trajectory analysis, we conducted further analysis based on mutation status as a phenotype, comparing MCs and NCs (Figure 2B; Table S2). We identified 227 proteins (240 aptamers) associated with mutation status (Figure 3B). Among the 227 significant proteins, 14 proteins were downregulated in MCs and the remaining 239 proteins were upregulated. We found that all, but 16, proteins associated with the pseudo-trajectory (n = 137) were also significant in this analysis (Figure 3B). The top upregulated proteins associated with mutation status included chitotriosidase (CHIT1) (p = 7.97 × 10−17), NEFL (p = 2.66 × 10−18), YWHAG (p = 2.72 × 10−26), ITGA1/ITGB1 (p = 1.18 × 10−13), and PPP3CA/PPP3R1 (p = 1.10 × 10−26). Downregulated proteins included NPTX2 (p = 1.61 × 10−7), EPHA4 (p = 5.22 × 10−6), or RELT (p = 2.27 × 10−6) (Table S2). We found strong correlation between the effect sizes from these two analyses (R2 = 0.77, p = <1.00 × 10−300) (Figure 3C), indicating that both methods captured the same overall findings, even though mutation status analysis seems to provide more statistical power, as 105 additional proteins were found in this analysis (Figure 3B).
We then compared our results with two other recent ADAD proteomic studies,14,15 to replicate our findings, and determined potential previously unreported findings. Johnson et al.15 employed a mass spectrometry (MS) approach to measure 59 proteins in 286 MCs and 184 NCs from DIAN.15 As this study analyzed the same cohort, this represents a technical replication (Figure 3D; Table S3). Among the proteins identified in this study, 12 were present in Johnson et al., and all of them were also associated in this study (hypergeometric p = 2.15 × 10−14; fold enrichment: 20.76). Van de Ende et al.14 used Olink-based proteomics to measure 808 proteins in 22 MCs and 20 age- and sex-matched controls. This cohort is independent from the samples included in this study, representing an external replication. Of the 19 significant proteins that were present on the Olink panel, 16 of which passed FDR, two showed nominally significant associations (GFAP and SFRP1; hypergeometric p = 3.24 × 10−13; fold change: 32.13; Figure 3E; Table S3). When we integrated the data from both studies, out of the 137 pseudo-trajectory proteins, 26 unique proteins were present in at least one of these studies, and all of them exhibit nominal associations in one of these studies and consistent direction of association (Table S3), supporting the robustness of our findings. Similar results were found when comparing the 227 associated with mutation status (Figures 3F and 3G).
Comparison of significantly altered proteins in ADAD and sAD
We then determined if the proteins associated with ADAD mutation status exhibit an association with sAD. This analysis aimed to provide additional validation for the proteins we initially identified in ADAD and to identify potential differences and commonalities with sAD. To investigate this, we generated and analyzed proteomic data from the CSF samples from 1,763 individuals diagnosed with sAD (A+T+: 848, A−T−: 915) from multiple cohorts (Table S4). Among the identified 137 significant pseudo-trajectory proteins, all of them were also significant after Bonferroni correction in sAD with consistent direction of effect.
When looking at the 227 proteins associated with mutation status in DIAN, we found a very high correlation in the effect sizes between sAD and ADAD mutation status analyses (R2 = 0.74, p = 4.94 × 10−73; Figure 3H). There were seven proteins that were associated with sAD, also after multiple test correction, but in the opposite direction (Figure 3H; Table S4): DLK2, EPHA4, GAGE2A, TNFRSF21, HS6ST3, RELT, and LRP1. DLK2 is implicated in neurite outgrowth,30 synaptic plasticity,31 and neuroprotection32,33 and has been associated with anxiety and depression.34 EPHA4 has been reported to be involved in the progression of AD due to synaptic dysfunction and is enriched in neurons. GAGE2A has been widely studied in its immune system function in cancer, but its role in neurological disease remains unclear. However, the other two proteins have been shown to play a role in the blood-brain barrier (BBB). RELT, a member of the tumor necrosis factor (TNF) receptor superfamily, has been shown to contribute to the acquisition and development of barrier properties in the BBB. Both TNFRSF21 and RELT (a.k.a TNFRSF19) are downstream targets of the Wnt/beta-catenin signaling pathway in BBB endothelial cells. When the TNFRSF21/TNFRSF19 signaling is dysregulated, it can result in the breakdown of the BBB’s endothelial layer. It is worth noting that the Wnt/beta-catenin signaling pathway is essential for central nervous system (CNS) angiogenesis but not for the development of peripheral vasculature.35,36 HS6ST3 and GAGE2A are both implicated in immune response and inflammation. EPHA4 and LRP1 are involved in synaptic structure and intracellular trafficking.37 These findings highlight how the differential regulation of distinct proteins and alterations in endothelial cells, the BBB, inflammation, and the endolysosomal pathways result in different disease outcomes (ADAD and sAD).
To investigate which proteins showed a difference in strength in their association with sAD or ADAD, we identified proteins with effect sizes outside of the 95% confidence interval (CI) for the regression for all the 227 proteins associated with ADAD. There were five proteins (CHIT1, SMOC1, SMOC2, NEFL, and CAND1) that showed significantly higher effect size in ADAD, compared with that in sAD (Figure 3H; Table S4). The protein with the highest difference in ADAD was CHIT1 (p = 7.97 × 10−17, β = 0.29). CHIT1, a putative marker of microglial activation,38 has already been shown to be elevated in the CSF and peripheral blood of AD patients.39 Another protein with higher effect size in ADAD is CAND1 (p = 1.39 × 10−14, β = 0.09), which has been involved in modulating the ubiquitin-proteasome pathway (UPP).40 This pathway is pivotal in removing proteins, including the clearance of misfolded proteins, and plays a crucial role in controlling various cellular functions. CAND1 directly interacts with Cullins in their unneddylated form, regulating the assembly of Cullin-RING ligase (CRL) complexes. When Cullins are neddylated, CAND1 dissociates, allowing Cullins to form active CRLs, which are essential for targeting specific substrates for ubiquitination. Two other proteins with higher effect size in ADAD are SMOC1 and NEFL, which have already been proposed as AD biomarkers in previous studies.14,41,42 The only protein that exhibited a significantly lower effect size in ADAD was NPTX2, which is downregulated in both ADAD and sAD. NPTX2, known as a glutamate receptor, found in established synapses, has been implicated in non-apoptotic cell death of dopaminergic nerve cells (Figure 3H).43
In summary, this analysis revealed a significant degree of overlap between ADAD and sAD, while also validating the MC proteomic analyses in sAD, and highlighted several proteins and potential mechanisms that are different between sAD and ADAD.
Earliest CSF proteomic changes
Owing to the study design, which leverages mutation status and EYO, we estimated when proteins levels start changing in MCs, compared with NCs, in relationship to the EYO. We found that of the 137 identified proteins, 124 began to change before the EYO, with approximately 93% of these changes occurring within the range of −20 to −3 years (Figure 2C). PPIL1 was the only protein displaying changes one year after the estimated onset (change +1.9 years in relationship to EYO; Table S1). Established biomarkers changed between 11 and 17 years before onset: pTau (−17 years), Tau (−12 years), and Aβ42 (−11 years). Among the 137 proteins significant in the pseudo-trajectory analyses, 12 initiated changes earlier than established biomarkers, such as SMOC1 (−31 years), followed by DNAJB9 (−26 years) and SMOC2 (−20 years) (Figure 2A). Several proteins began to change around −18 years (SLIT2 and SPON1) and −17 years (PRDX3, GAGE2D, PDLIM4, PPP3CA, PPP3R1, and DLG2). Compared with the 33 significant proteins associated with EYO in Johnson et al.,15 we identified 116 additional proteins with significant trajectory changes.
Plasma-specific analyses find low overlap with CSF
We applied an identical model to perform pseudo-trajectory and mutation status analyses between 325 MCs and 213 NCs from ADAD plasma samples. In the pseudo-trajectory analysis, out of 6,022 proteins, three proteins were significant after FDR correction (FDR p < 0.05; Figure S2A; Table S5). Complexin-2 (CPLX2, p = 0.049) and syntaxin-1A (STX1A, p = 0.049) were upregulated, while vesicle amine transport 1 (VAT1, p = 0.049) was downregulated in MCs. In examining the time to symptom onset, CPLX2’s pseudo-trajectory began changing approximately seven years before symptom onset, whereas alterations in STX1A’s started around four years before symptom onset (Figure S2B).
In the MCs vs. NCs analyses, nine proteins were associated with mutation status after FDR (Figure S2C; Table S5), including CPLX2 and STX1A. Additional proteins identified in this analysis include MAD1L1, SMOC1, PPP4R3A, CPLX1, and CAST that were upregulated, while two proteins (SSBP1 and VOPP1) were downregulated. SMOC1 was the only protein identified in both plasma and CSF specimens (Figure S2D). Overall, we identified a limited number of significant proteins associated with ADAD in plasma, compared with CSF (Figures S2E and S2F).
CSF-brain comparison identified complex protein dynamics across tissues
Next, we analyzed the overlap in the proteins associated with ADAD in brain and CSF. To do this, we leveraged data from a recent study that included proteomic data (SomaLogic 1.3K) from 24 ADAD carriers and 25 control parietal brain samples and identified 92 associated with ADAD.11 A total of 89, of those 92 proteins, passed QC in this study, and 14 were nominally associated in CSF (fold enrichment: 6.3; p = 4.88 × 10−8). Out of the 137 proteins showing significant trajectory in CSF, 20 proteins, all with positive direction in CSF, were also present in the brain dataset. Among these 20 proteins, 11 proteins showed a nominal association with ADAD (p < 0.05; Figure S3; Table S6). Five proteins (SMOC1, osteopontin, 14–3-3E, SUMO3, and UFC114–3-3E) were also elevated in ADAD individuals (compared with healthy controls). The remaining six proteins (CAMK2B, EPHA3, 14–3-3 protein zeta/delta, IF4G2, PHI, and 14–3-3 protein gamma) had the opposite direction in brain, showing a lower abundance in ADAD individuals (compared with controls). This could represent complex proteomic dynamics across tissues, as in the case of Aβ, or a false positive in the brain studies driven by the limited sample size in this tissue.
CSF-dysregulated proteins capture pathways implicated in neuronal death and inflammation
We analyzed if the proteins identified in our analyses were reported to be implicated in other diseases by performing gene-disease network enrichment analysis. Of these 45 disease terms, 16 belong to neurodegenerative disease traits, including “familial Alzheimer disease (FAD),” which was one of the top hits (FDR p = 3.29 × 10−4; Figure S4A; Table S7); “Creutzfeldt-Jakob disease” (FDR p =5 × 10−5); “frontotemporal dementia” (FDR p = 9.65 × 10−4); and “senile plaques” (FDR p = 7.20 × 10−4). The association with these disease terms are led by a common core of proteins known to be implicated in neurodegeneration in general (SPP1, GFAP, NRGN, NEFL, ACHE, YWHAZ, or PIN1; Table S7). Some of these proteins are also key connectors in various cellular functions such as postsynaptic signaling (NRGN, NEFL, GAP43, PIN1), neuron projection (STMN2, ENO1, GPI), and cell junction (PPIA, YWHAZ, ACHE),28 suggesting that they may be common contributors across these diseases (Figure S4B). In addition, 18 of the 45 diseases belong to cancer or immune-related traits, including several disease terms for neuroblastomas, carcinomas, fibrosarcomas, or gliomas. The association with these diseases is led by known proteins implicated in immune response and inflammation or expressed in glial cells: ITGB1, SPP1, TNFRSF1B, or GFAP.
We also conducted pathway enrichment analyses of these 137 proteins to gain insights into the biological functions and the biological processes leading to disease. There were 60 significantly enriched pathways belonging to 13 major categories (Table S7). Major overrepresented pathway categories were “signal transduction” (17 pathways), followed by “neuronal system” (15 path-ways) and “disease” (7 pathways) (Figure S4C). Signal transduction (FDR p = 7.80 × 10−4, logFC (log fold change)= 1.96) included 13 proteins: cholinergic synapse proteins (ACHE, CAMK2B, PLCB1, and PRKCG), glutamatergic synapse proteins (GLUL and HOMER1), signaling proteins (DLG2 and DLG4), cell communication proteins (LIN7A, LIN7B, and NRGN), proteins implicated in anatomical structure development (PDLIM5), and known AD biomarkers (NEFL).44 Specific pathways within the “neuronal system” category includes several pathways related to NMDA receptors (NMDARs) (Table S7), and includes NEFL, DLG4, DLG2, and CAMK2B. Another significant pathway was “activation of BAD and translocation to mitochondria” (FDR p = 2.63 × 10−9) and belongs to the super-pathway “programmed cell death” (FDR p = 2.08 × 10−5, logFC = 3.69; Figure S4D). This super-pathway is connected with “autophagy” (FDR p = 1.78 × 10−2, logFC = 2.28; Figure S4D; Table S7) through the 14–3-3 proteins (14–3-3 beta, epsilon, gamma, zeta), which are likely capturing early neuronal death and autophagy events in ADAD.
Co-expression network analysis identified modules that capture the chronological progression in ADAD
Our analyses suggest that there are many pathways dysregulated in ADAD (Table S7; Figure S4). To determine if specific pathways are dysregulated at different disease stages, we performed weighted gene co-expression network analyses (WGCNA) and then pathway analyses in each module. We first performed WGCNA in the 6,163 proteins/7,005 aptamers that passed QC, identifying 18 modules that collectively included a total of 4,909 proteins (Figure S5). The remaining 2,096 proteins were not assigned into any of the 18 modules and therefore grouped into the M0 (gray) module. The M1 (cyan) module contained 129 (137 aptamers) out of the 137 (145 aptamers) proteins associated with different pseudo-directories in the MCs. Pathway analysis within M1 module revealed multiple pathways that are dysregulated at different stages of the disease onset. They covered cellular response to stimuli (in very early stages), metabolism and immune pathways (in the middle stages), and autophagy (near or right after the onset) (Table S8). In particular, programmed cell death (activation of BAD and translocation to mitochondria, FDR = 1.38 × 10−5) included calcineurin (PPP3CA/PPP3R1, EYO = −16.04; PPP3R1, EYO = −12.67) along with 14–3-3 family proteins (YWHAG, EYO = −14.61; YWHAB, EYO = −13.86; YWHAE, EYO = −12.91; YWHAZ, EYO = −11.81) that had changes in early stages (Table S8). Other pathways involved similarly in early stages included FOXO-mediated transcription (FDR = 9.95 × 10−3; median EYO = −11.17) that regulates cell survival, growth, differentiation, and metabolism in response to environmental changes such as growth factor deprivation, starvation, and oxidative stress. Several pathways, such as innate immune system and cytokine signaling pathways (median EYO = −9.95), dysregulation of signal transduction (median EYO = −9.87), negative regulation of MET activity (EYO = −7.27), and signaling by EGFR and netrin-1 signaling (median EYO = −6.94) were observed and included proteins that changed roughly between 11 and 6 years before symptom onset (subsequently after Aβ42 changes). Finally, multiple signaling transduction pathways (regulation of FZD by ubiquitination, EYO = −5.78; nuclear signaling by ERBB4, median EYO = −5.03; FLT3 signaling in disease, EYO = −3.85) along with hemostasis pathways (median EYO = −3.85) were observed near the onset of disease.
As most of the associated proteins were included in the same co-expression network module (M1), we further performed additional clustering focusing only on those 137 identified proteins (145 aptamers). We identified four modules: MEgray (n = 6), MEchocolate (M1, n = 64), MEhotpink (M2, n = 17), and MEdeepskyblue (M3, n = 50) (Figures 4 and S5; Table S9). The MEgray module comprised proteins not clustered into the other three modules, and it was therefore not included for further analyses. We found that the nodes presented different mean EYOs, with M1 being enriched in proteins with the earliest changes (mean EYO: −11.14 ± 4.78 years), followed by M2 (mean EYO: −11.07 ± 2.7 years) and M3 (mean EYO: −8.32 ± 3.80 years) (Table S9). The difference between the mean EYO for M1 and M2 was not statistically significant; however, significant differences were observed when comparing the mean EYOs of M1 and M3 (p = 0.001) as well as those of M2 and M3 (p = 0.005).
Figure 4. Co-expression network analysis of significant pseudo-trajectory proteins and pathway enrichment for each module.
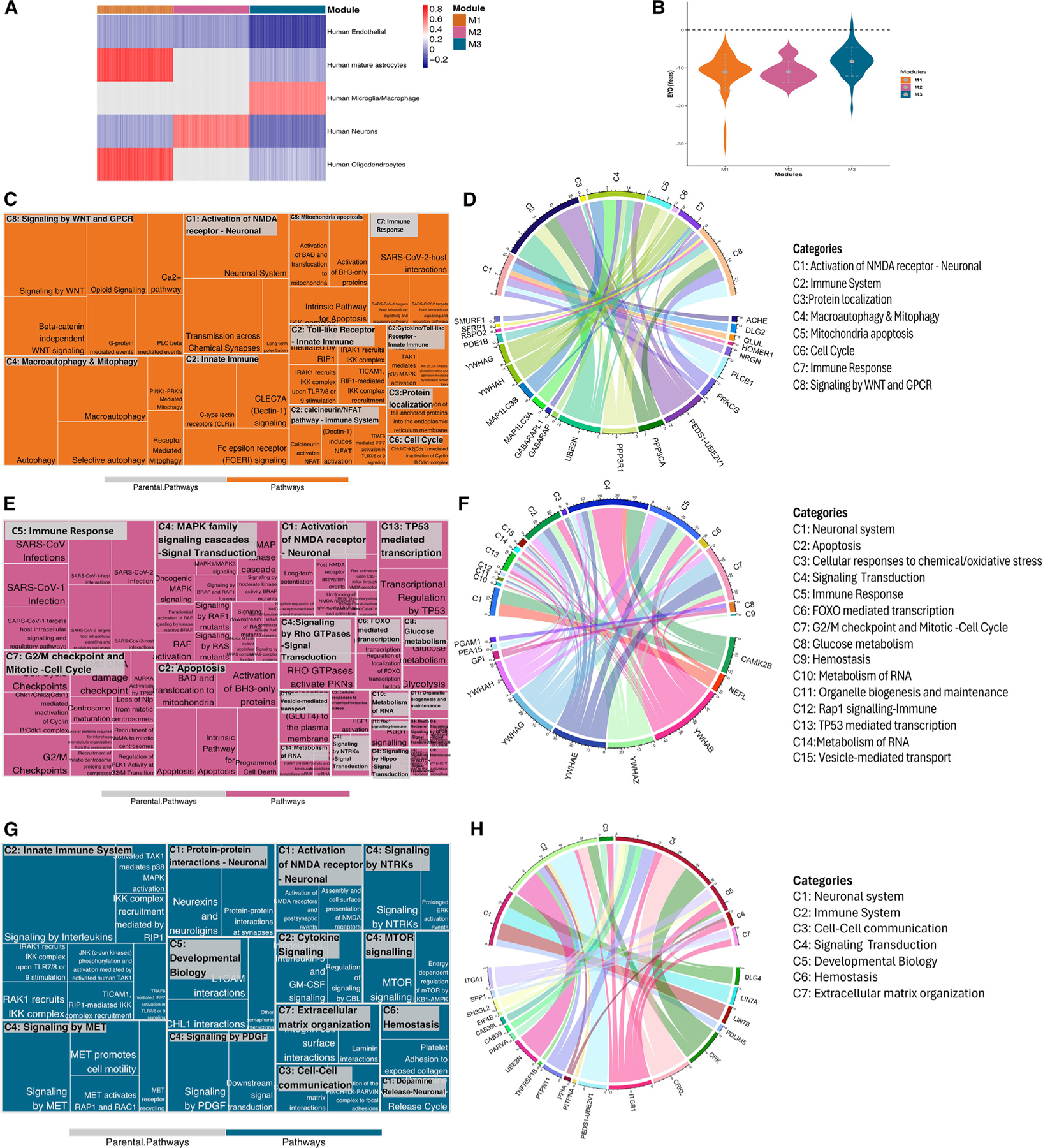
(A) Heatmap showing GSVA scores for each cell type across modules. Higher GSVA score is shown in red, highlighting an enrichment for that cell type within the module, whereas lower GSVA scores, represented in blue, show depletion. Gray color indicates that the module did not have any genes/proteins associated with the cell type.
(B) EYO comparison for functional identified proteins from Reactome pathway analysis.
(C–H) Reactome pathway analysis for each module. Treemap (C, E, and G) represents the significantly enriched pathways with summarized categories (C#, such as C1); chord diagram (D, F, and H) shows the enriched proteins in categorized pathways. The colored patterns labeling proteins represent the different cell types, and the colors are consistent with bar colors in cell-type enrichment (A).
See also Figures S4 and S5.
The M1 module, which showed the earliest proteomic changes, was mainly enriched in astrocyte and oligodendrocyte proteins and depleted in neuronal proteins (Figure 4A; Table S8). Protein-protein interaction revealed enrichment in calcineurin complex (p = 2.41 × 10−2, FC = 2.00), autophagosome membrane (p = 7.00 × 10−3, FC = 1.37), postsynaptic density (p = 1.34 × 10−2, FC = 0.59), and glutamatergic synapse (p = 1.34 × 10−2, FC = 0.80) pathways (Figure S6A; Table S10). Of the 64 proteins present in M1, 22 proteins displayed significant enrichment in 34 different pathways. These 24 pathways belong to 8 super-pathways (the super-pathways are denoted as C1–C8; Table S10). The top pathway categories include NMDAR synapse signaling (C1: p = 1.23 × 10−2), calcineurin/NFAT pathway (C2: p = 1.53 × 10−2), protein localization (C3: p = 5.18 × 10−3), macroautophagy and mitophagy (C4: p = 1.82 × 10−2), cell cycle (C6: p = 1.71 × 10−2), and several immune-related pathways (p = 1.91 × 10−2) (Figures 4C–4D; Table S10). In these pathways, 7 out of 24 proteins were present in tier 1 or tier 2 of the druggable genome database, such as proteins involved in the NFAT pathway (C1: ACHE, PRKCG, GLUL), calcineurin (C2: PPP3CA/PPP3R1), and signaling (SFRP1) (Table S9). The mitochondrial apoptosis pathway (C5) was driven by the 14–3-3 gamma (p = 6.04 × 10−8, EYO −14.65 years), 14–3-3 eta (p = 7.00 × 10−5, EYO −10.65 years), and PPP3R1 (p = 4.19 × 10−18, EYO −16.03 years), which were some of the proteins with the lowest EYO. The identified microtubule-associated proteins (MAP1LC3A, MAP1LC3B) enriched in macroautophagy and mitophagy (C4; Figure 4C) and the neuronal proteins (Figure 4C; Table S10) suggest that early neuron pathology changes may involve microtubule dysfunctions. Several proteins in this module have been reported to be implicated in AD pathogenesis, such as ACHE, HOMER1,45 and calcineurin (PPP3R1/PPP3CA).27 Other proteins such as NRGN are known and validated AD biomarkers and part of this module.46 In summary, this module seems to capture very early neuronal dysfunction, due to the presence of ADAD mutations.
Proteins in M2 were found to be enriched mainly in neuronal cells and depleted of endothelial cells (Figure 4A), which included proteins that change trajectories after those in M1, suggesting the capture of later processes. Of the 17 proteins in M2, 10 were enriched in biological pathways (Tables S9 and S10). Pathway analyses revealed many processes related to apoptosis (C2: p = 9.27 × 10−11; Figure 4E; Table S10), death receptor signaling (C4: p = 7.83 × 10−3), and pathways related to signaling (C4) and very early immune response (C5: p = 7.96 × 10−11). Protein-protein interaction analysis revealed that 14–3-3 proteins (beta, epsilon, eta, gamma, zeta) interact with CAMK2B and neurofilament proteins (NEFH, NEFL) within the postsynaptic intermediate filament cytoskeleton (Figure S6B). Unlike the other two modules, M2 is significantly marked by its involvement in inflammation signaling transduction pathways and early immune responses. The eight identified proteins (the five 14–3-3 proteins, CAMK2B, NEFL, and PEA15) participated in complex biological pathway interaction (Figure 4E). The networks include mitogen-activated protein kinase (MAPK) family signaling cascades and signaling transduction pathways involving G protein-coupled receptors (GPCR), Hippo, Notch, neurotrophic tropomyosin receptor kinases (NTRKs), and Rho GTPases (C4: p = 6.38 × 10−7), as well as cell injury/death pathways, such as apoptosis (C2: p = 9.27 × 10−11), and Rap1 signaling (C12: p = 4.78 × 10−4). The M3 module, which is closer to symptom onset, was enriched in microglial cell types and linked to the pathways involving the innate immune system (C2: p = 1.31 × 10−2) and cell-cell communication (C3: p = 3.22 × 10−3). Proteins enriched in microglia, such as CAB39/CAB39L, CRK/CRKL, EIF4B, and SPP1, were found to interact within signaling pathways of mammalian target of rapamycin (mTOR), MET,47 NTRKs,48 and platelet-derived growth factor (PDGF),49 which are known to be involved in neurodegenerative disorders (Figures 4G and 4H). Additionally, ubiquitin proteins like UBE2N, UBE2V1, and UBE2V2, enriched in C2 (innate immune system), were found to interact with the integrin alpha1-beta1 complex (ITGA1, ITGB1), involving intermediates like PPIA and CRK. This complex further interacts with the MPP7-DLG1-LIN7 complex (LIN7A, LIN7B) through DLG4 (Figure S6C). Pathways related to cell-cell communication (C3: p = 3.2 × 10−3), developmental biology (C5: p = 8.69 × 10−3), and extra-cellular matrix organization (C7: p = 3.25 × 10−2) appeared to reflect an effort to regain functionality by rebuilding and establishing new connections to maintain communication. PARVA, part of the cell-cell communication pathway, which is involved in reorganizing the actin cytoskeleton and cell polarity, was identified for the first time in the context of ADAD.50,51 In summary, by performing network and subsequent pathway analyses, we were able to provide a better overview of the pathways that are disrupted in ADAD at different stages of the disease.
Predictive models of ADAD
Next, we determined if the proteins identified in this study could predict mutation status (MCs vs. NCs; independently of the clinical status). Using machine learning approaches, we identified the minimum number of proteins that maximizes predictive power. We applied 50 iterations of random sampling to split the CSF DIAN cohort into unique training (60%) and testing (40%) datasets each time (Table S11). We then comprehensively selected highly informative proteins (see STAR Methods) to develop a proteomic signature of six proteins (GFAP, NPTX2, PEA15, SMOC1, SMOC2, and TNFRSF1B) that was able to predict mutation status (Table S11). The signature included previously identified CSF AD-associated proteins, such as SMOC111,15 or NPTX2, among others.52 This model showed a strong predictive power for classifying ADAD mutation status, with an area under the curve (AUC) of 0.911 (Figure 5A; Table S11). The AUC of this proteomic signature was significantly better than those for pTau181 (AUC: 0.755), Aβ42 (AUC: 0.709), and pTau/Aβ42 (AUC: 0.783) (Table S11). The negative predictive value (NPV) and positive predictive value (PPV) were 0.876 and 0.777, respectively, showing better performance than the classical AD biomarkers when differentiating between MCs and NCs.
Figure 5. Predictive models for ADAD.
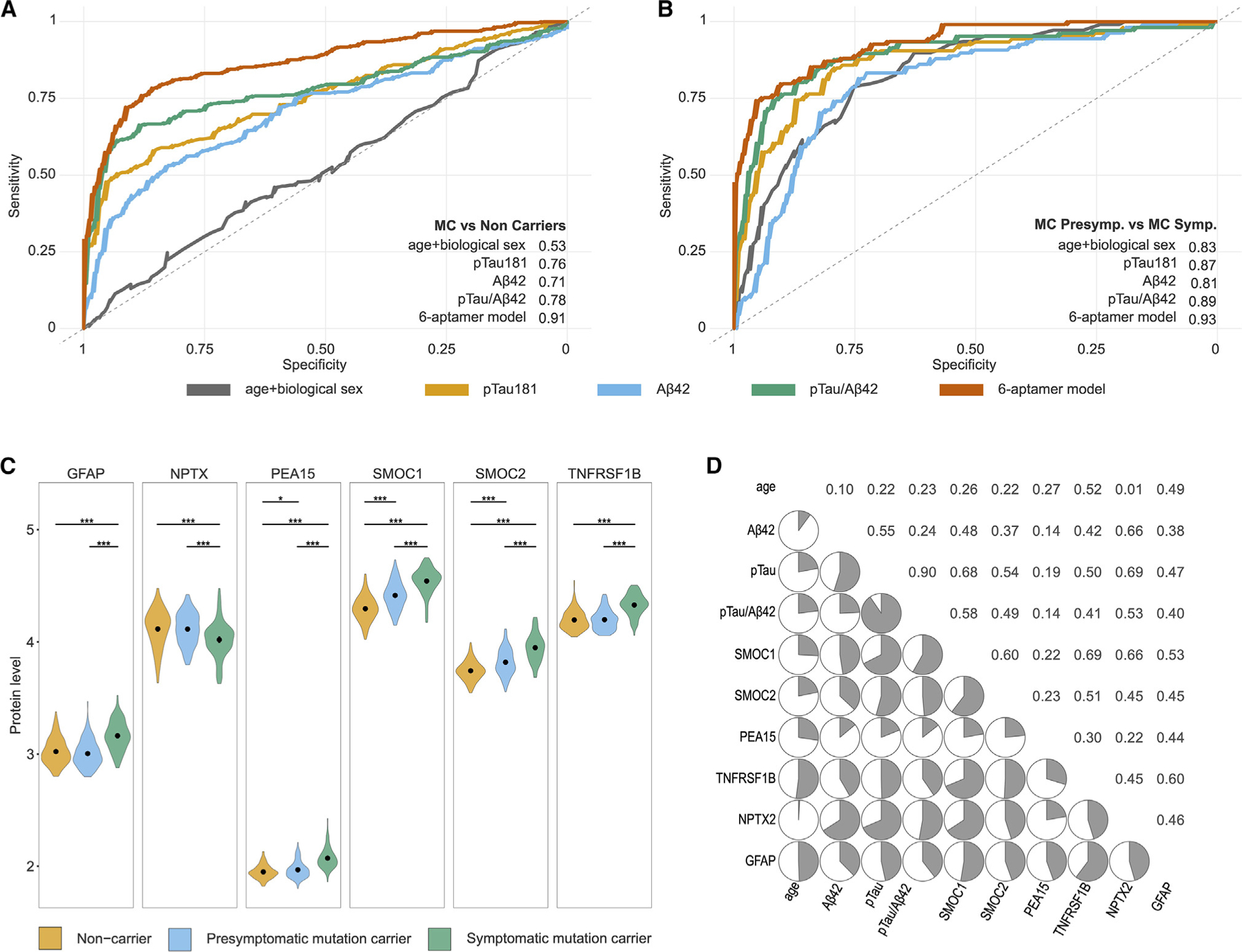
(A and B) The ROC and AUC corresponding to the final protein signature fitted into a generalized linear model (GLM) model, compared with the predictive power of age and sex; β-amyloid 42, pTau 181, and the pTau181/Abeta42 ratio in predicting mutation carriers compared with non-carriers (A); and symptomatic mutation carriers compared with non-symptomatic mutation carriers (B).
(C) Violin plots comparing the levels of the six proteins included in the predictive model in non-carriers, non-symptomatic carriers, and symptomatic carriers. Gray dots with extended lines in each violin plot represent the median ± SD. * represents level of p value significance. ***p < 0.001; **p < 0.01; *p < 0.05; ns = not significant.
(D) Correlation matrix corresponding to the Spearman correlations for the six proteins included in the predictive models, age, β-amyloid 42, pTau 181, and the pTau181/ABeta42 ratio in non-carriers.
Further, we categorized MCs into subgroups based on their clinical status at CSF draw date. Within the MCs, there were 186 individuals without clinical symptoms (presymptomatic) and 105 symptomatic individuals. We tested if this model could also distinguish the presymptomatic from the symptomatic individuals. We used the same proteomic signature (six proteins). The six-protein predictive model showed an AUC = 0.930 (Figure 5B) to differentiate MCs affected vs. the non-affected. The NPV (0.847) and the sensitivity (0.915; Table S11) were similar to those for pTau181/Aβ42 (NPV 0.825 and sensitivity 0.902), suggesting that the six-protein (Figure 5C) signature might be capturing very early disease changes.
To validate this predictive model, we used two different approaches. In the first one, we performed an orthogonal validation by generating Olink HT1 data for the samples from which SomaLogic data were available. Of the six proteins included in the initial predictive model (SMOC1, SMOC2, GFAP, NPTX2, TNFRSF1B, and PEA15), all but PEA15 were present. The predictive model with the five Olink proteins showed an AUC of 0.837 for mutation status (SomaLogic AUC = 0.91; Table S11; Figure S7) and an AUC of 0.879 when comparing presymptomatic vs. symptomatic (SomaLogic = 0.930). Then, we performed a second validation using a non-overlapping subset of DIAN CSF samples that we assessed using the Nulisa CNS panel (Table S11). Three proteins were included in this panel (SMOC1, GFAP, and NPTX2), which overlap with those included in the predictive model. Even with only three proteins, the predictive model for mutation status showed a high predictive power for both mutation status (AUC = 0.781) and clinical status (AUC = 0.876) (Table S11; Figure S7). These two platforms (Olink and Alamar) are antibody-based approaches and constitute an orthogonal validation of the original findings and predictive model.
DISCUSSION
In this study, we conducted an extensive analysis of CSF and plasma proteomics in ADAD (Figure 1). Our goal was to uncover early proteomic changes, understand the underlying biology, and to develop effective predictive models for early diagnosis. We measured 6,163 proteins in 463 CSF samples and 6,022 proteins in 538 plasma samples from the DIAN study. We identified 137 proteins with a significant difference in pseudo-trajectory analysis and 227 significant proteins associated with ADAD mutations status. These pseudo-trajectory changes could be traced back to 30 years before symptom onset (Figure 2; Table S1). For example, SMOC1 undergoes alterations as early as 15 years prior to established AD biomarkers like pTau, Tau, and Aβ42. Some of the top findings include known AD biomarkers such as NEFL and NRGN. However, most of the proteins identified in our analyses were not reported before, such as PPP3R1, which has been associated with disease progression and pTau levels in sAD27 or in GAGE2D that also showed protein changes 19 years before clinical onset.
We leveraged two previous studies14,15 to replicate our findings from the ADAD trajectory analysis. These studies, by either using a different proteomic platform or different samples, increase confidence in our findings and indicate that SomaLogic results can be replicated with other technologies and independent datasets. We also found that most of the proteins associated with different trajectories in MCs vs. NCs were also associated with sAD (Table S4). These results further support our findings and indicate a large overlap between ADAD and sAD. However, we identified seven proteins (EPHA4, DLK2, GAGE2A, TNFRSF21, HS6ST3, RELT, and LRP1) that exhibited opposite directions between ADAD and sAD. One potential explanation of this opposite effect could be due to the dynamic changes of these proteins in relation to disease status. In the presymptomatic phase of ADAD, some metabolic and TNF receptors (DLK2, GAGE2A, TNFRSF21)31,53 are observed downregulated, possibly as a compensatory response to early pathological changes. However, as ADAD progresses to the symptomatic phase, these markers might increase, indicative of deteriorating neuronal function and synaptic integrity. This hypothesis aligns with observations about heightened activation of EPHA4 signaling, which directly impacts motor neurons and leads to their degeneration.54 Additionally, we found five proteins (CHIT1, SMOC1, NEFL, SMOC2, and CAND1) with a higher effect size in ADAD. These findings suggest that there are distinct and diverse biological processes that are dysregulated in ADAD.
Of the 137 proteins associated with ADAD in this study, 111 were previously unreported and represent proteins from which changes start at−22 to −15 years before onset, such as DNAJB9 (EYO = −26.09 years), GAGE2D (EYO = −16.60 years), and PPP3R1 (EYO = −16.03 years). Network and pathway enrichment analyses for the associated proteins unveiled a significant enrichment within three distinct modules, each linked to different stages of the disease (Figure 6). In the earliest stage, the M1 module that captures early neuron pathology displayed changes in mitochondrial damage (GABARAP, GABARAPL1, MAP1LC3A, MAP1LC3B, PPP3R1) and NMDAR synapse signaling (ACHE, PLCB1, NRGN, GLUL, DLG2, PRKCG, HOMER1). The M1 module was depleted on neuronal proteins that might be caused by the cellular stress affecting their mitochondria and microtubules, leading to programmed cell death in neurons. As time progresses and the disease trajectory transitions to the M2 signaling transduction stage, these stimuli begin. Many studies have shown evidence of excitotoxicity of glutamatergic neurotransmission through postsynaptic NMDAR on the neurons.55–57 Other studies suggested that various cellular stressors, such as oxidative stress and endoplasmic reticulum stress, could dysregulate MAPK pathways.58,59 These findings show that oxidative stress or disruptions in glutamate metabolism, which initiate in the early and middle stages of ADAD, may promote the disease progression. Subsequently, under the regulation of cytokine, innate immune, and mTOR signaling pathways, microglia and macrophages become involved in an autophagy process that extends to other neurons and their microenvironments, as captured in the M3 module. This module also captures proteins and biological pathways that are involved in repairing and restoring damaged neurons, axons, dendrites, and synapses. This was mainly recapitulated in the M3 stage where dysregulation of ubiquitin-conjugating enzymes (UBE2N, PEDS1-UBE2V1) was observed. Non-degradative ubiquitin signaling is essential for homeostatic mechanisms crucial to neuronal function and survival,60 and it plays a role in inflammatory processes,60 thus supporting our hypothesis regarding self-rescue mechanisms at play during this stage.
Figure 6. Multiple biological process trajectories’ summary.
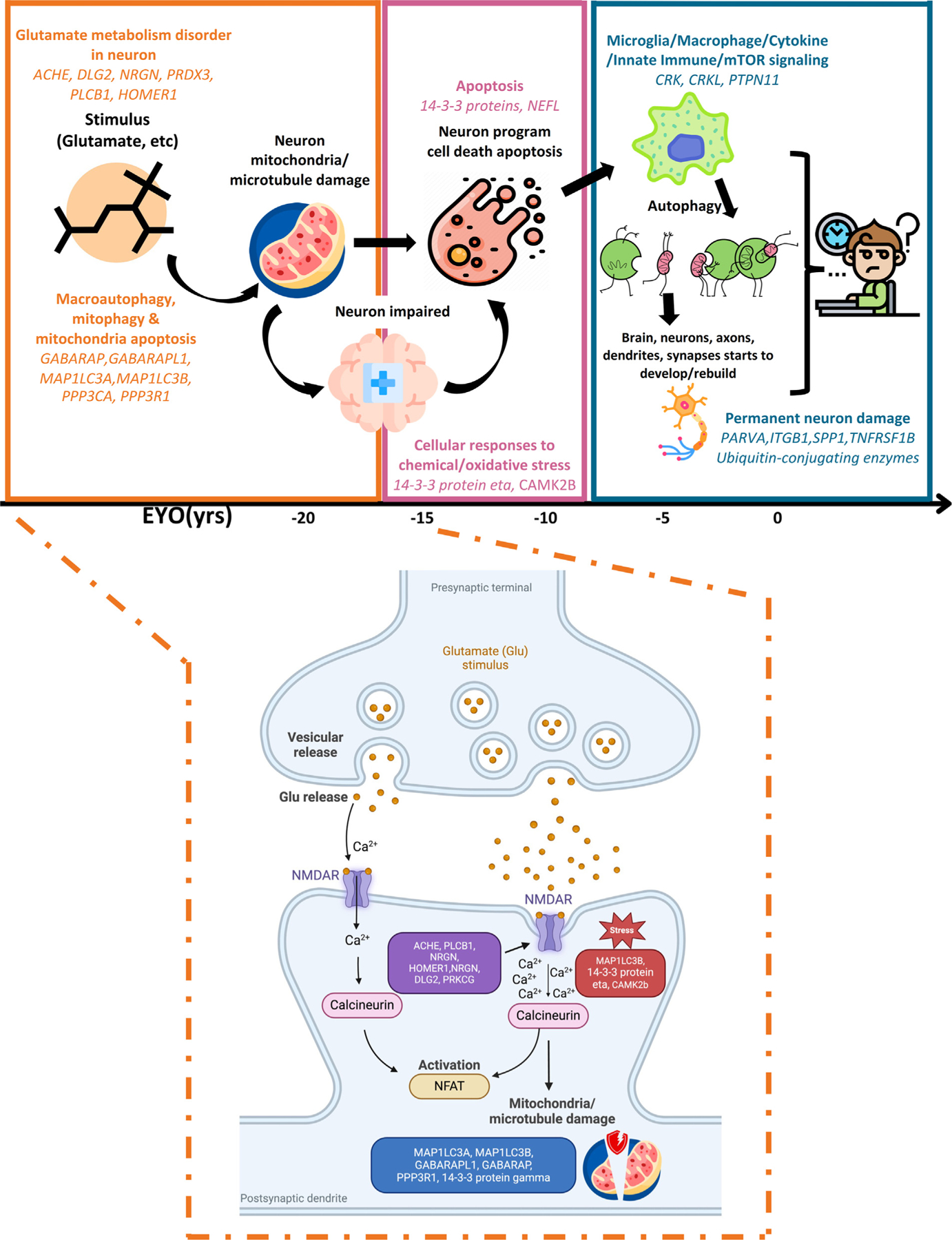
Highlighted pathology process and enriched significant trajectory proteins in each module by chronological order and highlighted biological process of early stage of ADAD (M1). It includes normal state and early ADAD disease stage. x axis represents the EYO in years.
The proteins identified in this study were used to create a robust ADAD predictive model. We applied a least absolute shrinkage and selection operator (LASSO) model to select a robust subset of significant pseudo-trajectory proteins. The predictive model based on six proteins accurately distinguished MCs from NCs, with an AUC of 0.91 in the testing set, which is better than Aβ42, pTau, and pTau/Aβ42 ratio. As some of the MCs were not symptomatic at the time of the lumbar puncture, we also analyzed if our model could distinguish between symptomatic and presymptomatic individuals carrying mutations. This model showed a similar performance (AUC: 0.95; PPV: 0.86; NPV: 0.84) when compared with pTau/Aβ42 (AUC: 0.93; PPV: 0.86; NPV: 0.82; Figure 5B; Table S11). In the last decades, many studies have provided evidence for the predictive values of CSF Tau, pTau, or pTau/Aβ42 for cognitive decline.61–63 Our model showed similar NPV, PPV, and sensitivity to distinguish MCs with clinical symptoms from those without clinical symptoms, compared with existing biomarkers, but it has several advantages: (1) it is highly sensitive to detect early protein changes, effectively distinguishing between early and late stages of ADAD; (2) the six proteins identified in our model can serve as secondary or confirmatory tests following initial screening with the known biomarkers that have high overall accuracy; and (3) as new therapies are targeting Aβ or Tau, there is a need to develop additional predictive models that do not rely on these proteins. We further validated these predictive models using orthogonal and antibody-based platforms (Olink and Alamar). The predictive power (AUC) for the Alamar and Olink is slightly lower than the original AUC, but this could be explained because neither of the two platforms included the six proteins. In addition, in several instances, the 95% CI intervals between the original model and Olink or Alamar overlap, indicating that these models are comparable.
Limitations of the study
Despite being the largest proteomic study in ADAD and having sAD as comparison group, this study has several limitations. First, our primary research cohort was the DIAN study, which despite being uniquely the largest ADAD cohort and extensive in terms of proteomic screening, it lacked a comparable cohort for systematic validation of our findings. Second, even though we were able to leverage the EYO by approximating cross-sectional data to longitudinal changes (pseudo-trajectories), follow-up studies using true longitudinal measure of proteomic changes may be needed to validate these results. However, conducting such studies may pose various practical challenges as our analyses cover a lifespan of 60 years (EYO from −40 to +20). Third, very few significant proteins were found in plasma in the same subjects and using the same methodology. The limited number of plasma findings in our study could be due to the lack of sensitivity of the platform used in measuring brain-related proteins in plasma. Thus, additional studies with orthogonal proteomic approaches are needed for plasma biomarkers. Fourth, the Somascan platform has additional limitations besides those already discussed. Owing to its targeted nature, only the proteins included in its panel are tested. Thus, it does not represent a true unbiased screening. In this study, we were able to find supportive evidence for all of our significant proteins in other platforms, but it is known that some SomaLogic aptamers do not correlate well with Olink, ELISA, or MS.64,65 However, this lack of replication is not unique to the SomaLogic platform but has been reported across the field. This can be attributed to one of three scenarios: (1) one of the platforms is accurately measuring the reported proteins, but the other is not; (2) both platforms failed to accurately quantify the protein; or (3) both are reporting accurate measures but are targeting different proteoforms. In the absence of validated information on exact proteoforms being measured across platforms, head-to-head comparisons between these cannot be made. Fifth, additional analyses by gene or even specific mutation with a larger sample size will be needed to understand the role of each of these mutations. Sixth, even though we used two different antibody-based platforms to validate the predictive model, neither of them included all six proteins selected for the initial model, which limits the assessment of the real predictive power of this model. To translate this model from the bench to the bedside, specific multiplex panels that include all six proteins will need to be developed to further validate the model and to determine the specific weights and cutoffs for the new platform. As the field of biomarkers in neurodegenerative diseases is moving from single biomarkers to multi-proteomic panels to capture different aspects of these diseases, there is a need to implement and use technologies that can multiplex multiple assays, and those that only measure one or two different gene-coding proteins are unlikely to be informative in the future.
In summary, our study leverages the DIAN participants and represents a comprehensive proteomic analysis of ADAD to date. Our methodology employs extensive protein panels, covering a broad range of biological processes. The detection of numerous dysregulated proteins through a robust approach, exhibiting altered patterns early in the disease and maintaining statistical significance even after rigorous multiple test corrections, highlights profound changes in the CSF proteome in ADAD. This reinforces the value of CSF proteomics in investigating the disease’s pathophysiology. Additionally, our study draws parallels between the CSF proteomes of ADAD and sAD, indicating the notable similarities but also identifying differences that can lead to personalized medicine for those carrying any mutation in ADAD genes. Ultimately, our results lay the groundwork for creating predictive models and identifying potential therapeutic targets, enhancing our understanding of ADAD and fostering the development of more effective future treatments.
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources should be directed to and will be fulfilled by the lead contact, Carlos Cruchaga (cruchagac@wustl.edu).
Materials availability
This study did not generate new materials. However, CSF and plasma proteomics data in DIAN was generated for this study (see data and code availability below).
Data and code availability
The individual-level data from DIAN cannot be publicly shared. Due the rarity of this disease it makes the data identifiable. This has been corroborated with the IRB and confirmed with the NIH, thus the data cannot be shared in public repositories. However, data are available to approved investigators via data request through https://dian.wustl.edu/our-research/for-investigators/diantu-investigator-resources/dian-tu-biospecimen-request-form/. The proteomics and individual-level genetic data obtained from the ADNI cohort can be requested through ADNI’s website (adni.loni.usc.edu) after access has been approved. The proteomics and individual-level genetic data obtained from the Knight-ADRC can be requested through the NIAGADS website (ng00130). The proteomics and individual-level genetic data obtained from Fundació ACE Alzheimer Center Barcelona can be requested through Fundacio ACE’s website (www.fundacioace.com). The summary results generated from this manuscript is available in Tables S1, S2, S3, S4, S5, S6, S7, S8, S9, S10, and S11. In addition, they are available to the scientific community through a public web browser https://proteomics.wustl.edu/csf/.
All our analyses used open-source software listed under key resources table. This paper does not report codes used.
KEY RESOURCES TABLE.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
CONSORTIA
The members of Dominantly Inherited Alzheimer Network are James M. Noble, Gregory S. Day, Neill R. Graff-Radford, Jonathan Voglein, Ricardo Allegri, Patricio Chrem Mendez, Ezequiel Surace, Sarah B. Berman, Snezana Ikonomovic, Neelesh Nadkarni, Francisco Lopera, Laura Ramirez, David Aguillon, Yudy Leon, Claudia Ramos, Diana Alzate, Ana Baena, Natalia Londono, Sonia Moreno Mathias Jucker, Christoph Laske, Elke Kuder-Buletta, Susanne Graber-Sultan, Oliver Preische, Anna Hofmann, Takeshi Ikeuchi, Kensaku Kasuga, Yoshiki Niimi, Kenji Ishii, Michio Senda, Raquel Sanchez-Valle, Pedro Rosa-Neto, Nick Fox, Dave Cash, Jae-Hong Lee, Jee Hoon Roh, Meghan Riddle, William Menard, Courtney Bodge, Mustafa Surti, Leonel Tadao Takada, Martin Farlow, Jasmeer P. Chhatwal, V.J. Sanchez-Gonzalez, Maribel Orozco-Barajas, Alison Goate, Alan Renton, Bianca Esposito, Celeste M. Karch, Jacob Marsh, Carlos Cruchaga, Victoria Fernandez, Brian A. Gordon, Anne M. Fagan, Gina Jerome, Elizabeth Herries, Jorge Llibre-Guerra, Allan I. Levey, Erik C.B. Johnson, Nicholas T. Seyfried, Peter R. Schofield, William Brooks, Jacob Bechara, Randall J. Bateman, Eric McDade, Jason Hassenstab, Richard J. Perrin, Erin Franklin, Tammie L.S. Benzinger, Allison Chen, Charles Chen, Shaney Flores, Nelly Friedrichsen, Nancy Hantler, Russ Hornbeck, Steve Jarman, Sarah Keefe, Deborah Koudelis, Parinaz Massoumzadeh, Austin McCullough, Nicole McKay, Joyce Nicklaus, Christine Pulizos, Qing Wang, Sheetal Mishall, Edita Sabaredzovic, Emily Deng, Madison Candela, Hunter Smith, Diana Hobbs, Jalen Scott, Johannes Levin, Chengjie Xiong, Peter Wang, Xiong Xu, Yan Li, Emily Gremminger, Yinjiao Ma, Ryan Bui, Ruijin Lu, Ralph Martins, Ana Luisa Sosa Ortiz, Alisha Daniels, Laura Courtney, Hiroshi Mori, Charlene Supnet-Bell, Jinbin Xu, and John Ringman.
STAR★METHODS
EXPERIMENTAL MODEL AND STUDY PARTICIPANT DETAILS
ADAD and sporadic AD cohorts
DIAN is a long-term observational study that employs standardized clinical and cognitive assessments (Clinical Dementia Rating®(CDR)®), neuropsychological testing, imaging modalities (magnetic resonance imaging (MRI), PIB-PET, and 18F-FDG),73 and collects biological fluids (blood and CSF). The main goal of DIAN cohort is to detect alterations in individuals who possess known gene mutations that cause AD. Sporadic AD individuals, as part of validation cohorts, were obtained from four different sites: Charles F. and Joanne Knight Alzheimer Disease Research Center (Knight-ADRC), Alzheimer’s Disease Neuroimaging Initiative (ADNI), Fundació ACE Alzheimer Center Barcelona, and Barcelona-1), totaling 1,763 sample. The phenotypes information of DIAN participants, sporadic AD participants and the approval of institutional review board were described in supplementary methods.
ADAD cohort
We applied the Somascan® 7K panel to analyze the CSF proteome from 286 MCs, 177 NCs, and the plasma proteome from 325 MCs and 213 NCs from DIAN participants. In total, there were 448 participants that have both CSF and plasma proteomics data.
CSF samples were distributed as follows: 286 MCs [212 (74%) PSEN1, 23 (8%) PSEN2, and 51 (18%) APP]. Of these, 102 (36%) were symptomatic MCs, and 184 (64%) were presymptomatic MCs. The 177 NCs in the study were recruited from family members associated with MCs. The mean age of MCs was 40.0 years; 55% were females, with a mean EYO of −6.6 ±10.4 years (yrs), and 29% of MCs carried at least one APOE ε4 allele. The mean age of NCs was 39.6 years, 59% were females, the mean EYO was −7.8 ± 12.5 yrs, and 33% of NCs presented at least one APOE ε4 allele. Age, sex, EYO and APOE ε4 allele did not show any significant difference between MCs and NCs in CSF (Table 1).
Plasma was available for 325 MCs [240 (74%) PSEN1, 25 (8%) PSEN2, and 60 (18%) APP]. Of those, 127 (39%) were asymptomatic MCs, and 198 (61%) were presymptomatic MCs. The mean age of MCs was 40.4 years; with a mean EYO of −6.3 ±11.06 yrs, 30% carried at least one APOE ε4 allele, and 55% were females. There were 213 NCs with a mean age of 40.7 years; 59% were females, with the mean of −7.9 ± 11.7 yrs, and 33% of NCs carried at least one APOE ε4 allele. There was no significant difference observed in the distribution of sex, age at draw, EYO, and frequency of APOE ε4 carriers as well in the individual with plasma samples and at the specific blood draw used in this study.
Sporadic late-onset Alzheimer’s disease proteomics data cohorts
In this study, we utilized the cerebrospinal fluid (CSF) proteomics data from four different cohorts, with a total of 1,763 samples, including Knight Alzheimer’s Disease Research Center (Knight ADRC), the Alzheimer’s Disease Neuroimaging Initiative (ADNI), Ace Alzheimer Center Barcelona (FACE), Barcelona-1 (Table S4).
Knight ADRC
The Knight ADRC at Washington University School of Medicine has been recruiting and longitudinally assessing community-dwelling adults older than 45 years old since 1979. The Memory and Aging Project (MAP) at the Knight ADRC collects biofluids, and conducts annual clinical assessments, neuropsychological testing, neuroimaging studies, and autopsies of brain samples. Eligible participants may be asymptomatic or have mild dementia at the time of enrollment. All participants are required to participate in core study procedures, including annual longitudinal clinical assessments, neuropsychological testing, neuroimaging, and biofluid biomarker studies. Annual cognitive assessments of the participants were conducted by experienced clinicians. Multi-tissue data from brain, CSF, and plasma has been utilized for generating multi-omics data encompassing genetics, epigenomics, transcriptomics, proteomics, metabolomics, and lipidomics with the aim of identifying new risk and protective variants for dementia as well as novel potential drug targets.
ADNI
CSF proteomics data used in the preparation of this manuscript was obtained from the ADNI database (https://adni.loni.usc.edu/). Launched in 2003, ADNI represents a public-private partnership led by Principal Investigator Michael W. Weiner, MD. The primary objective of ADNI study has been to investigate whether the combination of serial magnetic resonance imaging (MRI), positron emission tomography (PET), other biological markers, and clinical and neuropsychological assessments can effectively measure the progression of mild cognitive impairment (MCI) and early AD.
FACE
We also acquired CSF samples from FACE, a private non-profit organization dedicated to AD research. Established in 1995 and based in Barcelona, FACE has diagnosed over 30,000 patients, collected 20,000 blood samples, 1,831 cerebrospinal fluid samples, and analyzed nearly 13,000 genetic samples.74,75 Additionally, it has been involved in nearly 150 clinical trials during its existence. Additional information about the FACE cohort is available at their website (www.fundacioace.com/en).
Barcelona-1
For this study, CSF samples were also sourced from Barcelona-1, a study led by the University Hospital Mutua de Terrassa in Terrassa, Spain. Barcelona-1 is a longitudinal study comprising approximately 300 individuals. Only those individuals, diagnosed with MCI or more severe conditions, underwent PET scans and CSF collection and follow-up analyses were conducted to monitor disease progression. The study encompassed individuals with diagnoses of subjective memory complaints (SMC), MCI, AD dementia (ADD), and non-AD dementias (non-ADD).
METHOD DETAILS
Clinical assessment and EYO in ADAD
Dementia was evaluated using the CDR®, with clinical assessors blinded to participants’ mutation status. The EYO for a participant was computed during each assessment, factoring in their age at the visit and the anticipated period of dementia symptom onset specific to their mutation.12 This anticipated onset age for a given mutation was established by averaging reported symptom onset ages among individuals sharing the same mutation. When the mutation-specific expected age of symptom onset was unavailable, the EYO was derived from the age of cognitive decline onset in the participant’s parents or family members, as determined through a thorough semi-structured interview using comprehensive historical data. It is crucial to note that the EYO calculation procedure remained consistent for both MCs and NCs. In the present study, we leveraged the DIAN-EYO, which enhances EYO accuracy by integrating an individual’s actual decline age into the EYO determination process rather than solely relying on the mean mutation or parental/familial age at onset.12 For simplification, EYO stands for DIAN-EYO.12 Mutation status was determined using a PCR-based amplification of the relevant exon, followed by Sanger sequencing.2
CSF proteomic data collection, processing, and quality control
The CSF proteomic data was profiled using the Somascan® 7K Platform, an aptamer-based technology superior to ELISA and MS in handling dynamic range and missing values. CSF samples, collected post-fasting via lumbar puncture, were uniformly processed and stored at −80 °C. This study included 495 DIAN and 1,763 sAD samples, assessed using 7,584 aptamers on the Somascan® Platform. The samples were sent together to SomaLogic to minimize batch effects and randomly distributed across plates. Protein levels were measured and normalized to account for plate variability and technical variation, as described previously.64,76 To ensure data quality, individual-level quality control (QC) was carried out to detect and exclude outlier aptamers and samples, as described previously.64,76 Overall at the end of QC, in DIAN and sAD cohorts, there were 845 and 866 aptamers targeting more than one protein, respectively; and in total, we included 6,163 unique proteins in the analysis. Also, 463 DIAN samples and 1763 sAD samples were available. Additionally, levels of CSF Aβ42, Tau, and pTau were measured using Lumipulse® G Assays.
Plasma proteomics data collection, processing, and QC
Blood samples were collected at the time of the visit, immediately centrifuged, and the plasma stored at −80°C. Clinical status (case-control) was determined by the CDR® at the time of draw. We measured 7,584 aptamers in 580 plasma samples from DIAN participants using SomaLogic’s Somascan® Platform. Initial data normalization and QC was performed as described above. Ultimately, 542 samples and 6,022 aptamers targeting 6,022 unique proteins were kept for downstream analysis. In the subsequent description, we will only talk about unique proteins for both CSF and plasma, instead of protein aptamers.
ATN classification
We grouped the sAD cohorts based on the ATN classification (where A+T+ and A−T− serve as proxies for sAD and controls).77 However, the lack of consensus in universal biomarker cutoffs is a major caveat as biomarker levels, and subsequently their cutoffs for dichotomization, can be influenced by the technique of measurement. Thus, we utilized Gaussian mixture models to dichotomize quantitative Aβ42 and pTau measures into positive and negative groups and applied separately for each sAD cohort.78,79 Individuals with low CSF Aβ42 and high pTau levels were classified as amyloid/tau positive (A+T+). Individuals with high Aβ42 and low pTau levels were defined as controls (A−T−). Detail description of the dichotomization and cut-off determination for each cohort can be found elsewhere.79
In the case of the Knight ADRC cohort, measurements for both Aβ42 and pTau were conducted using the LumiPulse® G platform. For Aβ42, samples with values below 630 were classified as Aβ42-positive (A+). As for pTau, a cutoff of z-score = 0.61 was determined, which corresponded to a raw value of 62.9. Samples with values above 62.9 were considered pTau-positive (T+). In the ADNI cohort, Aβ42 measurements were conducted using Innotest by Fujirebio, while pTau measurements were performed using Elecsys by F. Hoffmann-La Roche Ltd in Switzerland. A z-score cutoff of 0.616, corresponding to a raw value of 196 pg/mL, and 0.197, corresponding to 27.8 pg/ml, were determined as cutoff for Aβ42 and pTau respectively. Similarly in FACE and Barcelona-1 cohort, both Aβ42 and pTau measurements were conducted using Innotest (Fujirebio). For Aβ42, a z-score cutoff of 0.468 was established, which corresponded to a raw Aβ42 value of 856 pg/mL. Samples with values below 856 were categorized as Aβ42-positive (A+). For pTau, a z-score cutoff of −0.018 was identified, corresponding to a raw value of 67. Samples with values greater than 67 were classified as pTau-positive (T+). In Barcelona-1, a z-score cutoff of 1.04, corresponding to a raw Aβ42 value of 1325 pg/mL, was determined whereas a z-score cutoff of −0.163 was identified for pTau, corresponding to a raw value of 58.
QUANTIFICATION AND STATISTICAL ANALYSIS
Cohorts demographics
We processed demographic characteristics using GraphPad Prism 9. To compare cohorts characteristics between MCs’ and NCs’ groups we used unpaired Student’s t tests and Chi-squared tests for continuous and binary variables, respectively. Continuous variables were reported as mean ± standard deviation. p values of <0.05 after the false discovery rate correction method was considered significant.
Differential pseudo-trajectory analysis
To study how alterations in protein levels changes over time, we employed a multi-layer regression model to infer the pseudo-temporal trajectory from the cross-sectional proteomic data, called pseudo-trajectory analysis. This model constructed a more comprehensive framework, capable of capturing proteins with significant pseudo-trajectory differences between MCs and NCs. In the pseudo-trajectory calculation model, we incorporated the first two surrogate variables (SVs) to correct for unmeasured heterogeneity. These SVs were generated by applying the sva() function within the R (version 4.1.3) package sva (version 3.42.0). False Discovery Rate (FDR) correction (FDR p < 0.05) was used to define significance. Pseudo-trajectory calculations were performed in three steps: (1) we used a linear regression, lm() function from the stats package, to build the model with log 10-transformed proteins as the dependent variable, EYO as the independent variable, sex, and the first two SV as covariates for MCs and NCs, separately (Formula 1); (2) we compared the coefficient-estimates (b) of EYOs between MCs and NCs (Formula 2); and (3) we calculated when the pseudo-trajectories significantly deviate from each other in EYO by Student’s t test (Formula 3, 4).
| Formula 1: |
| Formula 2: |
| Formula 3: |
| Formula 4: |
Pseudo-trajectory intersections in ADAD
For each protein that displayed substantial differences, we calculated the pseudo-trajectory intersection point between MCs and NCs. This was done to calculate the exact moment when the protein levels between these two groups started to diverge significantly. A two-step process was employed to derive these pseudo-trajectory intersections for each protein. In the initial step, we utilized the predict() function to generate the predicted 95% confidence intervals (CI) for MCs and NCs independently (Formula 5). In the second step, we extracted the lower bound and upper bound values from the predicted 95% CIs of both MCs and NCs, assembling them into a new matrix. We established another linear regression model from this new matrix by selecting either the predicted lower bound or upper bound values for significant protein based on each protein’s specific β. If the β for a particular protein were > 0, indicating that its protein level was higher in MCs than NCs, we opted for the lower bound from the MCs matrix and the upper bound from the NCs matrix. Conversely, if the β < 0, indicating that the protein level was higher in NCs than MCs, we selected the upper bound from the MCs matrix and the lower bound from the NCs matrix. Following this selection, we extracted the two βs from the new linear regression models (Formula 6).
| Formula 5: |
| Formula 6: |
Then we obtained the two coefficients from the new linear regression models (Formula 6), and applied solve() function to calculate the pseudo-trajectory intersections for each protein.
ADAD protein change and sAD protein change analysis in ADAD and sAD
Protein level changes between mutation status groups (MCs vs. NCs) in the DIAN cohort and between ATN status in sporadic late-onset AD cohorts (A+T+ vs. A−T−) were identified using the linear regression model. In these models, the log 10-transformed protein levels served as the dependent variable, mutation or ATN and the group status as the independent variable, and age at CSF draw, sex, and the first two SVs included as covariates (Formula 7).
| Formula 7: |
Proteomic comparison across studies
To compare the analyses and results across cohorts and analyses, we evaluated the consistency of CSF ADAD results with other studies by comparing the effect sizes for each protein. These comparisons included (1) another two publicly available CSF ADAD studies,14,15 (2) the analyses for CSF in sporadic AD and (3) DIAN plasma ADAD results. We obtained correlation (R2) and significance (p value) by performing correlation tests using Spearman’s correlation test from the cor.test() function in R to test effect sizes of studies. The prediction line and regression line’s 95% confidence interval were utilized to assess the effect size of significant proteins between ADAD and sAD.
Predictive models
To identify the minimum and robust set of informative proteins to build a model to predict ADAD mutations status, we used the least absolute shrinkage and selection operator (LASSO – L1 regularization) regression. For those aptamers targeting the same protein, only the most significant one was included in the LASSO input, leaving the input matrix with 132 aptamer measurements. The full dataset was divided in 60% training 40% testing randomly 50 times keeping the original proportion of carriers and noncarriers for each split. Minimum lambda was calculated at each iteration using cross validation prior to training and testing in each iteration using the glmnet function in the caret R package version 6.0. A final summary model was calculated using the full population. Predictive models performances were assessed by plotting the receiver operator characteristic (ROC) curves using pROC R package version 1.18.2.80
To further optimize the model and minimize the number of proteins included, we evaluated the predictive power of RIDGE (L2 regularization) based models. We computed 50 models, each of them based on the 50 random iterations. In brief, we computed how many times each of the 132 aptamers are selected by LASSO as informative and create RIDGE models in an incremental manner. In other words, the first model contains those proteins that were deemed informative by LASSO 50 times, the second model those that were selected by LASSO 50 and 49 times, and subsequentially until all aptamers that were included at least one time were included. Finally, we picked the model with the best performance and the minimum number of aptamer and fitted a final GLM model. All assessments were made with the complete population. We further interrogated the selected protein signature in the prediction of symptomatic vs non-symptomatic mutation carriers by re-fitting a new GLM model for the selected proteins.
Pathway analyses
We preformed pathway enrichment analysis, weighted gene correlation network analysis, and cell type enrichment analysis to interpret the function of significant trajectory proteins. We used the ClusterProfiler R package version 4.8.068 for pathway enrichment analysis to identify the biological functions of proteins, utilizing default parameters and an FDR P-value <0.05 for significant pathway identification. The enriched pathways were categorized based on the 2022 version Reactome database’s81 "Event Hierarchy" and analyzed for disease associations using the DisGeNET database.70 Additionally, we employed the WGCNA R package version 1.72.1 to create a network of protein co-expression modules, using hierarchical clustering and dynamic tree cutting for module identification.71 For cell type enrichment analysis, we focused on specific cell markers, particularly in human astrocytes, neurons, oligodendrocytes, microglia/macrophages, and endothelial cells, to evaluate protein specificity. We matched proteins to gene symbols and assessed cell-type specificity, determining enrichment based on expression data across these cell types. We described the package versions, used functions, parameters, and thresholds for the significance cut-off in supplementary methods. In addition, protein-pro-ein interaction (PPI) networks were obtained using STRING.
Cell type enrichment
Cell type enrichment was performed using single sample using the GSVA package (v1.48) in R. The reference database used for the analysis was obtained from the study conducted by Zhang et al.44 This included markers specific to human astrocytes, neurons, oligodendrocytes, microglia/macrophages, and endothelial cells. In each of the three modules identified by our WGCNA analysis, we used the “ssGSEA” algorithm to calculate the GSVA scores for each cell type within each sample based on the expression levels of proteins included within the module. Protein levels were log10 transformed and missing values were imputed using random sampling prior to the analysis.
Co-expression network analyses
We utilized the blockwiseModules function from the WGCNA package with the following parameters: power=3, corType=“ bicor”, networkType=“ signed”, deepSplit=4, and mergeCutHeight=0.07. After adjusting for age, sex, and plate ID for proteomic samples, the soft threshold power was determined as 3 by considering both the scale-free topology model fit (R2= 0.87) and mean connectivity. To measure similarity more robustly, we used bicor method, which is based on medians and less influenced by outliers compared to Pearson or Spearman correlation coefficient. A signed network was considered to account for both positive and negative correlations. The topological overlap matrix (TOM) was calculated to determine the interconnection within the network structure. Hierarchical clustering analysis was then conducted based on the calculated 1-TOM values to classify the proteins into distinct modules. The final modules were defined by grouping and rearranging similar groups.
ADDITIONAL RESOURCES
Data visualization
Data visualization plots were mainly generated by ggplot2 R package version 3.4.2.82 Specifically, the results of the differential proteins analysis in the form of significantly up- and down-regulated proteins were visualized as a volcano plot using the geom_point() function. Pseudo-trajectory curve and trajectory intersections were visualized by stat_smooth() function with ‘loess’ method.
Web browser for navigating results
We created a web portal (https://proteomics.wustl.edu/csf/) with Shiny R package to facilitate both exploration of our analysis and further investigation into individual proteins across MC and NC. The browser provides three tabs. The first tab provides a brief explanation of the web portal, and a description of the datasets used. The second tab (Abundance distribution) displays a table including proteomic abundance levels on each analyte that passed our QC process, along with its effect and p value for each comparison presented here. The table allows the user to select a protein, which displays the distribution of the selected protein levels between MC and NC. The third tab (Volcano plot) displays the volcano plots for each comparison.
Supplementary Material
Highlights.
137 CSF proteins’ trajectories differed between ADAD mutation carriers and non-carriers
The identified proteins started changing between 15 and 20 years before onset
The identified proteins are involved in neuronal death and immune pathways
Six proteins showed high predictive power for mutation and cognitive status
ACKNOWLEDGMENTS
We thank all the participants and their families as well as the many involved institutions and their staff.
This work was supported by grants from the National Institutes of Health (R01AG044546 [C.C.], RF1AG053303 [C.C.], RF1AG058501 [C.C.], U01AG058922 [C.C.], RF1AG074007 [Y.J.S.], and RF1AG083744 [R.T.]), the Chan Zuckerberg Initiative (C.Z.I.), and the Alzheimer’s Association Zenith Fellows Award (ZEN-22–848604, awarded to C.C.). Anonymous Foundation and access to equipment were made possible by the Hope Center for Neurological Disorders, the NeuroGenomics and Informatics Center (NGI: https://neurogenomics.wustl.edu/), and the Departments of Neurology and Psychiatry at Washington University School of Medicine.
DIAN resources: data collection and sharing for this project was supported by The Dominantly Inherited Alzheimer Network (DIAN, U19AG032438) funded by the National Institute on Aging (NIA) and the Alzheimer’s Association (SG-20–690363-DIAN).
FACE resources: the Genome Research @ Ace Alzheimer Center Barcelona project (GR@ACE) is supported by Grifols SA, Fundación bancaria “La Caixa,” Ace Alzheimer Center Barcelona, and CIBERNED.
Proteomic data generation was supported by NIH: RF1AG044546.
Footnotes
DECLARATION OF INTERESTS
C.C. has received research support from GSK and EISAI. C.C. is a member of the advisory board of Circular Genomics and owns stocks in these companies. C.C. is on the advisory board of ADmit.
J.L. reports speaker fees from Bayer Vital, Biogen, EISAI, TEVA, Zambon, Merck, and Roche; consulting fees from Axon Neuroscience, EISAI, and Biogen; author fees from Thieme medical publishers and W. Kohlhammer GmbH medical publishers; and is inventor in a patent “Oral Phenylbutyrate for Treatment of Human 4-Repeat Tauopathies” (EP 23 156 122.6). He receives compensation for serving as chief medical officer for MODAG GmbH and is a beneficiary of the phantom share program of MODAG GmbH.
E.M. reports research support received from NIA (U01AG059798), Anonymous Foundation, GHR, Alzheimer Association, Eli Lilly Eisai, and Hoffmann-La Roche and paid consulting for Eli Lilly, Alector, Alzamend, Sanofi, AstraZeneca, Hoffmann-La Roche, Grifols, and Merck.
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.cell.2024.08.049.
Supplemental figures
Figure S1. Gene-specific analyses, related to STAR Methods (A) Upset plot showing number of significant analytes shared between different mutation carriers, as identified by pseudo-trajectory analysis at p < 0.05 threshold. Scatterplot showing correlation of slope difference and EYO between joint analysis and APP carriers (B and C); between joint analysis and PSEN1 carriers (D and E); between joint analysis and PSEN2 carriers (F and G).
Figure S2. The significant findings of trajectory and ADAD mutation status analyses from MCs vs. NCs in plasma, related to STAR Methods (A and B) Volcano plots displaying the estimate change (x axis) against −log10 statistical differences (y axis) for all tested proteins. The red dots show the upregulated significant proteins, and the blue dots show the downregulated significant proteins at FDR-corrected threshold. (A) The significant trajectory proteins from MCs and NCs; (B) the significant proteins from ADAD mutation status analysis. (C–F) (C) CPLX2 and STX1A showed the significant trajectory in plasma; (D) identify the overlapped significant proteins between CSF and plasma trajectory and ADAD mutation status analysis. The UpSet intersection diagram shows the overlapped proteins in the analysis. (E) The effect size correlations of significant trajectory proteins between CSF and plasma. (F) The effect size correlations of significant proteins between CSF and plasma ADAD mutation analysis.
Figure S3. Effect size comparison between the proteins identified in brain and CSF, related to STAR Methods (A) The effect size of the proteins associated with ADAD in brain was compared with the effect size of those in CSF. (B) The effect size of the proteins with significant trajectory in ADAD CSF was compared with the effect size of those in brain.
Figure S4. Pathway enrichment analysis for significant trajectory proteins in CSF, related to Figure 4 (A) Top 20 significantly enriched diseases in DisGeNET enrichment analysis; (B) network map of the top 20 significantly enriched diseases; (C) top 20 significant pathways in Reactome pathway analysis for the significant proteins in trajectory analysis; pathways ordered based on gene ratio; (D) the top 20 significantly enriched Reactome pathways and their associated genes presented as network.
Figure S5. Co-expression network analyses in the full dataset, related to main Figure 4 (A) WGCNA analysis with 6,163 aptamers identified 18 modules. (B) M1 (cyan) module (2,402 analytes; 2,182 proteins), which contained 137 out of 145 analytes with significant trajectory difference between MCs and NCs, showed separation from the remaining 17 modules. (C) Pathways with M1 module spanned pathways that occur before Aβ42 changes (integrin cell surface interactions) and pathways right after the onset (chaperon-mediated autophagy). They included proteins including calcineurin (PPP3CA/PPP3R1), and 14–3-3 family proteins (YWHAG) are shown.
Figure S6. PPI network analysis for each module, related to STAR Methods (A) PPI network for M1 in GO cellular component. (B) PPI network for M2 in STRING network clusters. (C) PPI network for M3 in biological process.
Figure S7. Protein correlations between SomaLogic and orthogonal platforms, related to STAR Methods (A) Olink predictive models. AUC analyses were performed using Olink HT1 data, including the proteins identified using SomaLogic proteomic data. ROC and forest plot for the multi-proteins and single protein for (upper) mutation status and (lower) symptomatic vs. presymptomatic models. AUC and 95% CI for the SomaLogic model are also included as reference. (B) Alamar predictive models. AUC analyses were performed using the Alamar CNS panel data, including the proteins identified using SomaLogic proteomic data. ROC and forest plot for the multi-proteins and single protein for (upper) mutation status and (lower) symptomatic vs. presymptomatic models. AUC and 95% CI for the SomaLogic model are also included as reference.
REFERENCES
- 1.Bateman RJ, Aisen PS, De Strooper B, Fox NC, Lemere CA, Ringman JM, Salloway S, Sperling RA, Windisch M, and Xiong C (2011). Autosomal-dominant Alzheimer’s disease: a review and proposal for the prevention of Alzheimer’s disease. Alzheimers Res. Ther 3, 1. 10.1186/alzrt59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bateman RJ, Xiong C, Benzinger TLS, Fagan AM, Goate A, Fox NC, Marcus DS, Cairns NJ, Xie X, Blazey TM, et al. (2012). Clinical and biomarker changes in dominantly inherited Alzheimer’s disease. N. Engl. J. Med 367, 795–804. 10.1056/NEJMoa1202753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Levitis E, Vogel JW, Funck T, Hachinski V, Gauthier S, Vöglein J, Levin J, Gordon BA, Benzinger T, Iturria-Medina Y, et al. (2022). Differentiating amyloid beta spread in autosomal dominant and sporadic Alzheimer’s disease. Brain Commun 4, fcac085. 10.1093/braincomms/fcac085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tang M, Ryman DC, McDade E, Jasielec MS, Buckles VD, Cairns NJ, Fagan AM, Goate A, Marcus DS, Xiong C, et al. (2016). Neurological manifestations of autosomal dominant familial Alzheimer’s disease: a comparison of the published literature with the Dominantly Inherited Alzheimer Network observational study (DIAN-OBS). Lancet Neurol 15, 1317–1325. 10.1016/S1474-4422(16)30229-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Oboudiyat C, Gefen T, Varelas E, Weintraub S, Rogalski E, Bigio EH, and Mesulam MM (2017). Cerebrospinal fluid markers detect Alzheimer’s disease in nonamnestic dementia. Alzheimers Dement 13, 598–601. 10.1016/j.jalz.2017.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Meyer PF, McSweeney M, Gonneaud J, and Villeneuve S (2019). AD molecular: PET amyloid imaging across the Alzheimer’s disease spectrum: from disease mechanisms to prevention. Prog. Mol. Biol. Transl. Sci 165, 63–106. 10.1016/bs.pmbts.2019.05.001. [DOI] [PubMed] [Google Scholar]
- 7.Graff-Radford J, Yong KXX, Apostolova LG, Bouwman FH, Carrillo M, Dickerson BC, Rabinovici GD, Schott JM, Jones DT, and Murray ME (2021). New insights into atypical Alzheimer’s disease in the era of biomarkers. Lancet Neurol 20, 222–234. 10.1016/S1474-4422(20)30440-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wesenhagen KEJ, Teunissen CE, Visser PJ, and Tijms BM (2020). Cerebrospinal fluid proteomics and biological heterogeneity in Alzheimer’s disease: A literature review. Crit. Rev. Clin. Lab. Sci 57, 86–98. 10.1080/10408363.2019.1670613. [DOI] [PubMed] [Google Scholar]
- 9.Schindler SE, and Fagan AM (2015). Autosomal Dominant Alzheimer Disease: A Unique Resource to Study CSF Biomarker Changes in Preclinical AD. Front. Neurol 6, 142. 10.3389/fneur.2015.00142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lippa CF, Saunders AM, Smith TW, Swearer JM, Drachman DA, Ghetti B, Nee L, Pulaski-Salo D, Dickson D, Robitaille Y, et al. (1996). Familial and sporadic Alzheimer’s disease: neuropathology cannot exclude a final common pathway. Neurology 46, 406–412. 10.1212/wnl.46.2.406. [DOI] [PubMed] [Google Scholar]
- 11.Sung YJ, Yang C, Norton J, Johnson M, Fagan A, Bateman RJ, Perrin RJ, Morris JC, Farlow MR, Chhatwal JP, et al. (2023). Proteomics of brain, CSF, and plasma identifies molecular signatures for distinguishing sporadic and genetic Alzheimer’s disease. Sci. Transl. Med 15, eabq5923. 10.1126/scitranslmed.abq5923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ryman DC, Acosta-Baena N, Aisen PS, Bird T, Danek A, Fox NC, Goate A, Frommelt P, Ghetti B, Langbaum JBS, et al. (2014). Symptom onset in autosomal dominant Alzheimer disease: a systematic review and meta-analysis. Neurology 83, 253–260. 10.1212/WNL.0000000000000596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ringman JM, Schulman H, Becker C, Jones T, Bai Y, Immermann F, Cole G, Sokolow S, Gylys K, Geschwind DH, et al. (2012). Proteomic changes in cerebrospinal fluid of presymptomatic and affected persons carrying familial Alzheimer disease mutations. Arch. Neurol 69, 96–104. 10.1001/archneurol.2011.642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.van der Ende EL, In ‘t Veld SGJG, Hanskamp I, van der Lee S, Dijkstra JIR, Hok-A-Hin YS, Blujdea ER, van Swieten JC, Irwin DJ, Chen-Plotkin A, et al. (2023). CSF proteomics in autosomal dominant Alzheimer’s disease highlights parallels with sporadic disease. Brain 146, 4495–4507. 10.1093/brain/awad213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Johnson ECB, Bian S, Haque RU, Carter EK, Watson CM, Gordon BA, Ping L, Duong DM, Epstein MP, McDade E, et al. (2023). Cerebrospinal fluid proteomics define the natural history of autosomal dominant Alzheimer’s disease. Nat. Med 29, 1979–1988. 10.1038/s41591-023-02476-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Drummond E, Kavanagh T, Pires G, Marta-Ariza M, Kanshin E, Nayak S, Faustin A, Berdah V, Ueberheide B, and Wisniewski T (2022). The amyloid plaque proteome in early onset Alzheimer’s disease and Down syndrome. Acta Neuropathol. Commun 10, 53. 10.1186/s40478-022-01356-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Drummond E, Pires G, MacMurray C, Askenazi M, Nayak S, Bourdon M, Safar J, Ueberheide B, and Wisniewski T (2020). Phosphorylated tau interactome in the human Alzheimer’s disease brain. Brain 143, 2803–2817. 10.1093/brain/awaa223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Drummond E, Nayak S, Faustin A, Pires G, Hickman RA, Askenazi M, Cohen M, Haldiman T, Kim C, Han X, et al. (2017). Proteomic differences in amyloid plaques in rapidly progressive and sporadic Alzheimer’s disease. Acta Neuropathol 133, 933–954. 10.1007/s00401-017-1691-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liao L, Cheng D, Wang J, Duong DM, Losik TG, Gearing M, Rees HD, Lah JJ, Levey AI, and Peng J (2004). Proteomic characterization of postmortem amyloid plaques isolated by laser capture microdissection. J. Biol. Chem 279, 37061–37068. 10.1074/jbc.M403672200. [DOI] [PubMed] [Google Scholar]
- 20.Xiong F, Ge W, and Ma C (2019). Quantitative proteomics reveals distinct composition of amyloid plaques in Alzheimer’s disease. Alzheimers Dement 15, 429–440. 10.1016/j.jalz.2018.10.006. [DOI] [PubMed] [Google Scholar]
- 21.Hondius DC, Koopmans F, Leistner C, Pita-Illobre D, Peferoen-Baert RM, Marbus F, Paliukhovich I, Li KW, Rozemuller AJM, Hoozemans JJM, et al. (2021). The proteome of granulovacuolar degeneration and neurofibrillary tangles in Alzheimer’s disease. Acta Neuropathol 141, 341–358. 10.1007/s00401-020-02261-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zellner A, Müller SA, Lindner B, Beaufort N, Rozemuller AJM, Arzberger T, Gassen NC, Lichtenthaler SF, Kuster B, Haffner C, et al. (2022). Proteomic profiling in cerebral amyloid angiopathy reveals an overlap with CADASIL highlighting accumulation of HTRA1 and its substrates. Acta Neuropathol. Commun 10, 6. 10.1186/s40478-021-01303-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Campion D, Dumanchin C, Hannequin D, Dubois B, Belliard S, Puel M, Thomas-Anterion C, Michon A, Martin C, Charbonnier F, et al. (1999). Early-onset autosomal dominant Alzheimer disease: prevalence, genetic heterogeneity, and mutation spectrum. Am. J. Hum. Genet 65, 664–670. 10.1086/302553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bayart JL, Hanseeuw B, Ivanoiu A, and van Pesch V (2019). Analytical and clinical performances of the automated Lumipulse cerebrospinal fluid Aβ42 and T-Tau assays for Alzheimer’s disease diagnosis. J. Neurol 266, 2304–2311. 10.1007/s00415-019-09418-6. [DOI] [PubMed] [Google Scholar]
- 25.Paciotti S, Sepe FN, Eusebi P, Farotti L, Cataldi S, Gatticchi L, and Parnetti L (2019). Diagnostic performance of a fully automated chemiluminescent enzyme immunoassay for Alzheimer’s disease diagnosis. Clin. Chim. Acta 494, 74–78. 10.1016/j.cca.2019.03.1612. [DOI] [PubMed] [Google Scholar]
- 26.Zetterberg H, Skillbäck T, Mattsson N, Trojanowski JQ, Portelius E, Shaw LM, Weiner MW, and Blennow K; Alzheimer’s Disease Neuroimaging Initiative (2016). Association of Cerebrospinal Fluid Neurofilament Light Concentration With Alzheimer Disease Progression. JAMA Neurol 73, 60–67. 10.1001/jamaneurol.2015.3037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cruchaga C, Kauwe JSK, Mayo K, Spiegel N, Bertelsen S, Nowotny P, Shah AR, Abraham R, Hollingworth P, Harold D, et al. (2010). SNPs associated with cerebrospinal fluid phospho-tau levels influence rate of decline in Alzheimer’s disease. PLoS Genet 6, e1001101. 10.1371/journal.pgen.1001101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gene Ontology Consortium, Aleksander SA, Balhoff J, Carbon S, Cherry JM, Drabkin HJ, Ebert D, Feuermann M, Gaudet P, Harris NL, et al. (2023). The Gene Ontology knowledgebase in 2023. Genetics 224, iyad031. 10.1093/genetics/iyad031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.McMacken G, Whittaker RG, Wake R, Lochmuller H, and Horvath R (2023). Neuromuscular junction involvement in inherited motor neuropathies: genetic heterogeneity and effect of oral salbutamol treatment. J. Neurol 270, 3112–3119. 10.1007/s00415-023-11643-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Snyder JL, Kearns CA, and Appel B (2012). Fbxw7 regulates Notch to control specification of neural precursors for oligodendrocyte fate. Neural Dev 7, 15. 10.1186/1749-8104-7-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Alberi L, Liu S, Wang Y, Badie R, Smith-Hicks C, Wu J, Pierfelice TJ, Abazyan B, Mattson MP, Kuhl D, et al. (2011). Activity-induced Notch signaling in neurons requires Arc/Arg3.1 and is essential for synaptic plasticity in hippocampal networks. Neuron 69, 437–444. 10.1016/j.neuron.2011.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hitoshi S, Seaberg RM, Koscik C, Alexson T, Kusunoki S, Kanazawa I, Tsuji S, and van der Kooy D (2004). Primitive neural stem cells from the mammalian epiblast differentiate to definitive neural stem cells under the control of Notch signaling. Genes Dev 18, 1806–1811. 10.1101/gad.1208404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mizutani K, Yoon K, Dang L, Tokunaga A, and Gaiano N (2007). Differential Notch signalling distinguishes neural stem cells from intermediate progenitors. Nature 449, 351–355. 10.1038/nature06090. [DOI] [PubMed] [Google Scholar]
- 34.Gan YL, Wang CY, He RH, Hsu PC, Yeh HH, Hsieh TH, Lin HC, Cheng MY, Jeng CJ, Huang MC, et al. (2022). FKBP51 mediates resilience to inflammation-induced anxiety through regulation of glutamic acid decarboxylase 65 expression in mouse hippocampus. J. Neuroinflammation 19, 152. 10.1186/s12974-022-02517-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Daneman R, Agalliu D, Zhou L, Kuhnert F, Kuo CJ, and Barres BA (2009). Wnt/beta-catenin signaling is required for CNS, but not non-CNS, angiogenesis. Proc. Natl. Acad. Sci. USA 106, 641–646. 10.1073/pnas.0805165106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Liebner S, Corada M, Bangsow T, Babbage J, Taddei A, Czupalla CJ, Reis M, Felici A, Wolburg H, Fruttiger M, et al. (2008). Wnt/beta-catenin signaling controls development of the blood-brain barrier. J. Cell Biol 183, 409–417. 10.1083/jcb.200806024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Szabo MP, Mishra S, Knupp A, and Young JE (2022). The role of Alzheimer’s disease risk genes in endolysosomal pathways. Neurobiol. Dis 162, 105576. 10.1016/j.nbd.2021.105576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Steinacker P, Verde F, Fang L, Feneberg E, Oeckl P, Roeber S, Anderl-Straub S, Danek A, Diehl-Schmid J, Fassbender K, et al. (2018). Chitotriosidase (CHIT1) is increased in microglia and macrophages in spinal cord of amyotrophic lateral sclerosis and cerebrospinal fluid levels correlate with disease severity and progression. J. Neurol. Neurosurg. Psychiatry 89, 239–247. 10.1136/jnnp-2017-317138. [DOI] [PubMed] [Google Scholar]
- 39.Yu X, Yu W, Wu L, Yang W, and Lü Y (2021). Chitotriosidase attenuates brain inflammation via HDAC3/NF-kB pathway in D-galactose and aluminum-induced rat model with cognitive impairments. Neurosci. Res 172, 73–79. 10.1016/j.neures.2021.05.014. [DOI] [PubMed] [Google Scholar]
- 40.Huang X, Liu X, Li X, Zhang Y, Gao J, Yang Y, Jiang Y, Gao H, Sun C, Xuan L, et al. (2023). Cullin-associated and Neddylation-dissociated protein 1 (CAND1) alleviates NAFLD by reducing ubiquitinated degradation of ACAA2. Nat. Commun 14, 4620. 10.1038/s41467-023-40327-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Roberts JA, Varma VR, Candia J, Tanaka T, Ferrucci L, Bennett DA, and Thambisetty M (2023). Unbiased proteomics and multivariable regularized regression techniques identify SMOC1, NOG, APCS, and NTN1 in an Alzheimer’s disease brain proteomic signature. NPJ Aging 9, 18. 10.1038/s41514-023-00112-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jung Y, and Damoiseaux JS (2024). The potential of blood neurofilament light as a marker of neurodegeneration for Alzheimer’s disease. Brain 147, 12–25. 10.1093/brain/awad267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhou J, Wade SD, Graykowski D, Xiao MF, Zhao B, Giannini LAA, Hanson JE, van Swieten JC, Sheng M, Worley PF, et al. (2023). The neuronal pentraxin Nptx2 regulates complement activity and restrains microglia-mediated synapse loss in neurodegeneration. Sci. Transl. Med 15, eadf0141. 10.1126/scitranslmed.adf0141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhang Y, Sloan SA, Clarke LE, Caneda C, Plaza CA, Blumenthal PD, Vogel H, Steinberg GK, Edwards MSB, Li G, et al. (2016). Purification and Characterization of Progenitor and Mature Human Astrocytes Reveals Transcriptional and Functional Differences with Mouse. Neuron 89, 37–53. 10.1016/j.neuron.2015.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Dube U, Del-Aguila JL, Li Z, Budde JP, Jiang S, Hsu S, Ibanez L, Fernandez MV, Farias F, Norton J, et al. (2019). An atlas of cortical circular RNA expression in Alzheimer disease brains demonstrates clinical and pathological associations. Nat. Neurosci 22, 1903–1912. 10.1038/s41593-019-0501-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mathys H, Davila-Velderrain J, Peng Z, Gao F, Mohammadi S, Young JZ, Menon M, He L, Abdurrob F, Jiang X, et al. (2019). Single-cell transcriptomic analysis of Alzheimer’s disease. Nature 570, 332–337. 10.1038/s41586-019-1195-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Liu H, Luo K, and Luo D (2018). Guanosine monophosphate reductase 1 is a potential therapeutic target for Alzheimer’s disease. Sci. Rep 8, 2759. 10.1038/s41598-018-21256-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kim E, Kim H, Jedrychowski MP, Bakiasi G, Park J, Kruskop J, Choi Y, Kwak SS, Quinti L, Kim DY, et al. (2023). Irisin reduces amyloid-beta by inducing the release of neprilysin from astrocytes following downregulation of ERK-STAT3 signaling. Neuron 111, 3619–3633.e8. 10.1016/j.neuron.2023.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sweeney MD, Ayyadurai S, and Zlokovic BV (2016). Pericytes of the neurovascular unit: key functions and signaling pathways. Nat. Neurosci 19, 771–783. 10.1038/nn.4288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Huang AH, Pan SH, Chang WH, Hong QS, Chen JJW, and Yu SL (2015). PARVA promotes metastasis by modulating ILK signalling pathway in lung adenocarcinoma. PLoS One 10, e0118530. 10.1371/journal.pone.0118530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Patel NR, K C R, Blanks A, Li Y, Prieto MC, and Meadows SM (2022). Endothelial cell polarity and extracellular matrix composition require functional ATP6AP2 during developmental and pathological angiogenesis. JCI Insight 7, e154379. 10.1172/jci.insight.154379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Haque R, Watson CM, Liu J, Carter EK, Duong DM, Lah JJ, Wingo AP, Roberts BR, Johnson ECB, Saykin AJ, et al. (2023). A protein panel in cerebrospinal fluid for diagnostic and predictive assessment of Alzheimer’s disease. Sci. Transl. Med 15, eadg4122. 10.1126/scitranslmed.adg4122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Chen Y, Fu AKY, and Ip NY (2012). Eph receptors at synapses: implications in neurodegenerative diseases. Cell. Signal 24, 606–611. 10.1016/j.cellsig.2011.11.016. [DOI] [PubMed] [Google Scholar]
- 54.Zhao J, Stevens CH, Boyd AW, Ooi L, and Bartlett PF (2021). Role of EphA4 in Mediating Motor Neuron Death in MND. Int. J. Mol. Sci 22, 9430. 10.3390/ijms22179430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wang R, and Reddy PH (2017). Role of Glutamate and NMDA Receptors in Alzheimer’s Disease. J. Alzheimers Dis 57, 1041–1048. 10.3233/JAD-160763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zhang Y, and Bhavnani BR (2006). Glutamate-induced apoptosis in neuronal cells is mediated via caspase-dependent and independent mechanisms involving calpain and caspase-3 proteases as well as apoptosis inducing factor (AIF) and this process is inhibited by equine estrogens. BMC Neurosci 7, 49. 10.1186/1471-2202-7-49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Léveillé F, El Gaamouch F, Gouix E, Lecocq M, Lobner D, Nicole O, and Buisson A (2008). Neuronal viability is controlled by a functional relation between synaptic and extrasynaptic NMDA receptors. FASEB J 22, 4258–4271. 10.1096/fj.08-107268. [DOI] [PubMed] [Google Scholar]
- 58.Steinacker P, Aitken A, and Otto M (2011). 14–3-3 proteins in neurodegeneration. Semin. Cell Dev. Biol 22, 696–704. 10.1016/j.semcdb.2011.08.005. [DOI] [PubMed] [Google Scholar]
- 59.Kim EK, and Choi EJ (2010). Pathological roles of MAPK signaling pathways in human diseases. Biochim. Biophys. Acta 1802, 396–405. 10.1016/j.bbadis.2009.12.009. [DOI] [PubMed] [Google Scholar]
- 60.Schmidt MF, Gan ZY, Komander D, and Dewson G (2021). Ubiquitin signalling in neurodegeneration: mechanisms and therapeutic opportunities. Cell Death Differ 28, 570–590. 10.1038/s41418-020-00706-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Therriault J, Pascoal TA, Lussier FZ, Tissot C, Chamoun M, Bezgin G, Servaes S, Benedet AL, Ashton NJ, Karikari TK, et al. (2022). Biomarker modeling of Alzheimer’s disease using PET-based Braak staging. Nat Aging 2, 526–535. 10.1038/s43587-022-00204-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Roe CM, Fagan AM, Grant EA, Hassenstab J, Moulder KL, Maue Dreyfus D, Sutphen CL, Benzinger TLS, Mintun MA, Holtzman DM, et al. (2013). Amyloid imaging and CSF biomarkers in predicting cognitive impairment up to 7.5 years later. Neurology 80, 1784–1791. 10.1212/WNL.0b013e3182918ca6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Blennow K, Shaw LM, Stomrud E, Mattsson N, Toledo JB, Buck K, Wahl S, Eichenlaub U, Lifke V, Simon M, et al. (2019). Predicting clinical decline and conversion to Alzheimer’s disease or dementia using novel Elecsys Aβ(1–42), pTau and tTau CSF immunoassays. Sci. Rep 9, 19024. 10.1038/s41598-019-54204-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Timsina J, Gomez-Fonseca D, Wang L, Do A, Western D, Alvarez I, Aguilar M, Pastor P, Henson RL, Herries E, et al. (2022). Comparative Analysis of Alzheimer’s Disease Cerebrospinal Fluid Biomarkers Measurement by Multiplex SOMAscan Platform and Immunoassay-Based Approach. J. Alzheimers Dis 89, 193–207. 10.3233/JAD-220399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Dammer EB, Ping L, Duong DM, Modeste ES, Seyfried NT, Lah JJ, Levey AI, and Johnson ECB (2022). Multi-platform proteomic analysis of Alzheimer’s disease cerebrospinal fluid and plasma reveals network biomarkers associated with proteostasis and the matrisome. Alzheimers Res. Ther 14, 174. 10.1186/s13195-022-01113-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Cruchaga C, Ali M, Shen Y, Do A, Wang L, Western D, Liu M, Beric A, Budde J, Gentsch J, et al. (2024). Multi-cohort cerebrospinal fluid proteomics identifies robust molecular signatures for asymptomatic and symptomatic Alzheimer’s disease. Preprint at Research Square. 10.21203/rs.3.rs-3631708/v1. [DOI] [Google Scholar]
- 67.Leek JT, Johnson WE, Parker HS, Jaffe AE, and Storey JD (2012). The sva package for removing batch effects and other unwanted variation in high-throughput experiments. Bioinformatics 28, 882–883. 10.1093/bioinformatics/bts034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Yu G, Wang L-G, Han Y, and He Q-Y (2012). clusterProfiler: an R Package for Comparing Biological Themes Among Gene Clusters. OMICS 16, 284–287. 10.1089/omi.2011.0118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Yu G, and He QY (2016. Feb). ReactomePA: an R/Bioconductor package for reactome pathway analysis and visualization. Mol. Biosyst 12, 477–479. 10.1039/c5mb00663e. [DOI] [PubMed] [Google Scholar]
- 70.Piñero J, Bravo À, Queralt-Rosinach N, Gutiérrez-Sacristán A, Deu-Pons J, Centeno E, García-García J, Sanz F, and Furlong LI (2017). DisGeNET: a comprehensive platform integrating information on human disease-associated genes and variants. Nucleic Acids Res 45, D833–D839. 10.1093/nar/gkw943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Langfelder P, and Horvath S (2008). WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics 9, 559. 10.1186/1471-2105-9-559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Hänzelmann S, Castelo R, and Guinney J (2013-01–16). GSVA: gene set variation analysis for microarray and RNA-Seq data. BMC Bioinformatics 14, 7. 10.1186/1471-2105-14-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Morris JC, Aisen PS, Bateman RJ, Benzinger TLS, Cairns NJ, Fagan AM, Ghetti B, Goate AM, Holtzman DM, Klunk WE, et al. (2012). Developing an international network for Alzheimer research: the Dominantly Inherited Alzheimer Network. Clin. Investig. (Lond) 2, 975–984. 10.4155/cli.12.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Moreno-Grau S, de Rojas I, Hernández I, Quintela I, Montrreal L, Alegret M, Hernández-Olasagarre B, Madrid L, González-Perez A, Maroñas O, et al. (2019). Genome-wide association analysis of dementia and its clinical endophenotypes reveal novel loci associated with Alzheimer’s disease and three causality networks: The GR@ACE project. Alzheimers Dement 15, 1333–1347. 10.1016/j.jalz.2019.06.4950. [DOI] [PubMed] [Google Scholar]
- 75.de Rojas I, Moreno-Grau S, Tesi N, Grenier-Boley B, Andrade V, Jansen IE, Pedersen NL, Stringa N, Zettergren A, Hernández I, et al. (2021). Common variants in Alzheimer’s disease and risk stratification by polygenic risk scores. Nat. Commun 12, 3417. 10.1038/s41467-021-22491-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Yang C, Farias FHG, Ibanez L, Suhy A, Sadler B, Fernandez MV, Wang F, Bradley JL, Eiffert B, Bahena JA, et al. (2021). Genomic atlas of the proteome from brain, CSF and plasma prioritizes proteins implicated in neurological disorders. Nat. Neurosci 24, 1302–1312. 10.1038/s41593-021-00886-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Jack CR Jr., Bennett DA, Blennow K, Carrillo MC, Feldman HH, Frisoni GB, Hampel H, Jagust WJ, Johnson KA, Knopman DS, et al. (2016). A/T/N: An unbiased descriptive classification scheme for Alzheimer disease biomarkers. Neurology 87, 539–547. 10.1212/WNL.0000000000002923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Ali M, Sung YJ, Wang F, Fernández MV, Morris JC, Fagan AM, Blennow K, Zetterberg H, Heslegrave A, Johansson PM, et al. (2022). Leveraging large multi-center cohorts of Alzheimer disease endophenotypes to understand the role of Klotho heterozygosity on disease risk. PLoS One 17, e0267298. 10.1371/journal.pone.0267298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Timsina J, Ali M, Do A, Wang L, Western D, Sung YJ, and Cruchaga C (2024). Harmonization of CSF and imaging biomarkers in Alzheimer’s disease: Need and practical applications for genetics studies and preclinical classification. Neurobiol. Dis 190, 106373. 10.1016/j.nbd.2023.106373. [DOI] [PubMed] [Google Scholar]
- 80.Robin X, Turck N, Hainard A, Tiberti N, Lisacek F, Sanchez JC, and Müller M (2011). pROC: an open-source package for R and S+ to analyze and compare ROC curves. BMC Bioinformatics 12, 77. 10.1186/1471-2105-12-77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Gillespie M, Jassal B, Stephan R, Milacic M, Rothfels K, Senff-Ribeiro A, Griss J, Sevilla C, Matthews L, Gong C, et al. (2022). The reactome pathway knowledgebase 2022. Nucleic Acids Res 50, D687–D692. 10.1093/nar/gkab1028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Villanueva RAM, and Chen ZJ (2019). ggplot2: Elegant Graphics for Data Analysis (Taylor & Francis; ). [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The individual-level data from DIAN cannot be publicly shared. Due the rarity of this disease it makes the data identifiable. This has been corroborated with the IRB and confirmed with the NIH, thus the data cannot be shared in public repositories. However, data are available to approved investigators via data request through https://dian.wustl.edu/our-research/for-investigators/diantu-investigator-resources/dian-tu-biospecimen-request-form/. The proteomics and individual-level genetic data obtained from the ADNI cohort can be requested through ADNI’s website (adni.loni.usc.edu) after access has been approved. The proteomics and individual-level genetic data obtained from the Knight-ADRC can be requested through the NIAGADS website (ng00130). The proteomics and individual-level genetic data obtained from Fundació ACE Alzheimer Center Barcelona can be requested through Fundacio ACE’s website (www.fundacioace.com). The summary results generated from this manuscript is available in Tables S1, S2, S3, S4, S5, S6, S7, S8, S9, S10, and S11. In addition, they are available to the scientific community through a public web browser https://proteomics.wustl.edu/csf/.
All our analyses used open-source software listed under key resources table. This paper does not report codes used.
KEY RESOURCES TABLE.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.