

Abstract
Glycogen storage disease (GSD) is the most prevalent inherited disorder of glycogen metabolism for which no causal treatment is available. In recent years, thanks to the improved clinical management, the life expectancy of these patients extended, disclosing previously unidentified adverse conditions in other organs. In this study, we evaluated the clinical bone complications and the cellular responses in 20 patients (aged 14.1 ± 3.4 years) affected by GSD type I. Fragility fractures were reported in 35% of the patients, which were older than unfractured patients. They involved appendicular skeletal segments, while no vertebral deformity was detected. 60% of the patients had a bone mineral density (BMD) “below the expected range for age”, and lumbar spine (LS) BMD Z-scores positively correlated with muscle strength. Circulating mineral and bone markers showed reduction in the older subjects, with no increase in the pubertal age. Significant correlations could not be detected between circulating markers and LS BMD Z-scores, except for sclerostin levels, which also correlated with muscle strength. The osteoclasts differentiated from patients’ peripheral blood mononuclear cells did not show cell-autonomous alterations. However, circulating osteoclast precursors from healthy individuals cultured in the presence of patients’ sera exhibited increased osteoclastogenesis compared to control sera suggesting that GSD type I serum factors could affect osteoclast function in a non-autonomous manner. In contrast, circulating osteoprogenitors were unremarkable.
Supplementary Information
The online version contains supplementary material available at 10.1007/s00223-024-01302-4.
Keywords: Glycogen storage disease, Bone, Bone Mineral Density, Marker of Bone turnover, Osteoclast, Osteoblast
Introduction
Glycogen storage diseases (GSDs) are severe inherited disorders of glycogen metabolism caused by deficiencies in the enzymes involved in the synthesis or breakdown of glycogen [1, 2], characterised by excess or glycogen accumulation in various tissues [3]. Based on the deficient enzyme(s) and the affected organ(s), over 20 different types of GSDs are classified, some of which extremely rare [2]. The overall incidence of GSDs is estimated at 1:20,000 to 1:43,000 live births [2].
Liver and muscles are more commonly and more seriously affected by GSDs [3], with symptoms usually appearing in infancy [4]. In the severe forms, if the diagnosis is delayed, serious or even lethal consequences may develop, including hepatic [5] and renal damage [6], neoplasms [7], and infections [8]. Prognosis and occurrence of complications depend on metabolic control and avoidance of life-threatening hypoglycaemia or lactic acidosis [4].
As in other severe chronic systemic diseases, bone metabolism may be negatively affected by GSDs, with possible long-term alterations in bone mass and strength and increased risk of fractures [8, 9]. GSD-induced bone complications are likely to have a multifactorial origin, related to poor nutrition and fluctuations in glucose control [10, 11], reduced muscular force [12, 13], release of inflammatory cytokines, lactic acidosis, and hypogonadism [8]. Furthermore, liver damage may lead to reduced activation and consequent deficiency of vitamin D, with secondary hyperparathyroidism and an increase in bone turnover [14]. All these alterations negatively impact on the bones, as demonstrated by the link between poor disease control and impaired growth and delayed pubertal spurt [14].
Due to scanty and inconsistent data, the actual relevance of bone problems in GSDs has not been reliably assessed. Very few data are available on the prevalence and severity of bone complications in GSDs while their pathogenesis and risk factors for bone loss and fractures have not been investigated. Very few studies on small cohorts, mostly limited to the evaluation of bone mineral density (BMD) have been published so far [14], suggesting that bone complications, including fragility fractures, are typical mainly of GSD type I [15]. This form of GSD, also known as Von Gierke disease [16], is characterised by a very severe impact on metabolic and glycaemic homeostasis and is subclassified in GSD Ia and GSD Ib [17]. The former depends on deficiency of the glucose-6-phosphatase encoded by the G6PC gene, while the latter shows normal G6Pase activity but insufficiency in the glucose-6-phosphate translocase transporter enzyme encoded by the SLC37A4 gene [18]. Patients manifest hypoglycaemia and metabolic acidosis already at the age of 3–4 months. The management of GSD patients is based on specific diet to maintain normal blood glucose levels, prevent hypoglycaemia, and provide optimal nutrition for growth and development [19]. Extending the lifespan of GSD patients with specialised management makes bone complications more likely.
Learning from the experience with other diseases such as cystic fibrosis and Duchenne’s muscular dystrophy, in which new treatments have significantly extended the survival [20, 21], important bone complications are now more often observed also in patients with GSDs, requiring intervention to reduce the severe impairment of skeletal growth and development, loss of bone mass, bone pain and fractures, which negatively influence the patient’s quality of life and the outcome of the primary disease [9–11]. Therefore, the aim of our work was to investigate the skeletal complications in GSD type 1, which represents the most common type of GSD (about 25% of all cases). To this end, we focussed on the bone alterations in GSD Ia–Ib, performing clinical observations and cellular studies.
Results
Clinical Study
Fractures
At the anamnestic collection at time of enrolment (T0), 5 out of 20 patients (25%) reported previous fractures in hand, second toe, fifth metatarsal radius, wrist, and humerus, respectively. After 24 months (T24), two further patients, who had not reported any fracture events at T0, experienced fracture of the distal phalanx of the 3rd finger and wrist, respectively (Table 1). Therefore, 7 out of 20 patients (35%) suffered fracture events in their lifetime, up to and including T24. All fractures occurred in the absence of trauma and were described by parents and/or patients themselves. Therefore, they were considered fragility fractures given that, similar to those observed in adult osteoporotic patients, they occurred in the absence of a high-energy trauma.
Table 1.
Fractures in GSD type 1 patients
| Time | Sex | Age (Years) | Weight (Kg) | Height (cm) | Tanner stage | Physical exercise | Fractured skeletal segment |
|---|---|---|---|---|---|---|---|
| T0 |
M F M F M |
17 17 18 11 15 |
55.0 58.0 73.8 29.8 81.8 |
174 162 186 120 179 |
5 5 5 1 5 |
Yes Yes Yes No Yes |
Right hand Right toe Left V metatarsal radius Right wrist Right humerus |
| T24 |
F F |
13 13 |
41.1 40.9 |
153 148 |
2/3 3 |
Yes Yes |
Left III finger distal phalanx Left wrist SX |
M: male; F: female
Fractured patients were older than unfractured patients, while their Body Mass Index (BMI) was similar (Table 2). Of note, the fractured subjects showed germline mutations in both GSD type I genes, one in SLC37A4 and 4 in G6PC, whereas the other two patients did not receive a genetic diagnosis. GSD patients were also investigated for vertebral deformities by conventional spine radiography; at T0 and T24, no vertebral morphometric deformity was detected.
Table 2.
Clinical and biochemical features of fractured and unfractured GSD type I patients
| Parameters* | Normal range | Fractured patients | Unfractured patients | p-value |
|---|---|---|---|---|
| F/M | – | 4/3 | 4/9 | |
| Age (years) | – | 14.1 ± 3.4 | 10.1 ± 4.4 | 0.049 |
| BMI (kg/m2) | – | 20.3 ± 3.1 | 18.4 ± 3.3 | 0.227 |
| LS Z-scores | > −2.0 | − 2.10 ± 0.73 | − 2.45 ± 0.96 | 0.438 |
| Muscle strength | – | 15.6 (8.9–29.4) | 9.5 (6.8–14.1) | 0.290 |
| Mineral metabolic markers | ||||
| 25OHD (ng/ml) | > 20 | 39.3 ± 18.4 | 32.5 ± 17.5 | 0.427 |
| S Calcium (mg/dl) | 8.4–10.4 | 10.4 ± 0.6 | 10.4 ± 0.6 | 0.967 |
| S Phosphate (mg/dl) | 3.5–5.7 | 4.3 ± 0.7 | 5.1 ± 0.4 | 0.007 |
| P PTH (pg/ml) | 20–64 | 17.3 ± 8.2 | 20.4 ± 12.4 | 0.566 |
| Bone markers | ||||
| BSAP (µg/L) | 20–62 | 72.8 ± 51.5 | 77.0 ± 37.5 | 0.837 |
| OC (ng/ml) | 20–60 | 48.9 ± 25.2 | 62.6 ± 30.8 | 0.357 |
| P1NP (ng/ml) | 300–1200 | 302.7 ± 210.9 | 348.2 ± 200.0 | 0.639 |
| β-CTX (pg/ml) | 400–2000 | 937.0 ± 527.4 | 1089 ± 543.3 | 0.553 |
| RANKL (pmol/L) | – | 417.6 ± 203.6 | 649.2 ± 553.2 | 0.304 |
| OPG (pmol/L) | – | 3.34 ± 1.65 | 3.86 ± 2.51 | 0.620 |
| RANKL/OPG ratio | – | 151.4 ± 87.9 | 214.0 ± 156.4 | 0.344 |
| DKK1 (pmol/L) | – | 51.6 ± 22.7 | 46.3 ± 20.6 | 0.601 |
| Sclerostin (pmol/L) | – | 26.0 ± 8.8 | 20.0 ± 7.5 | 0.114 |
*F, female; M, male; BMI, body mass index; 25OHD, 25 hydroxyvitamin D; S serum, P plasma, PTH parathormone, BSAP bone-specific alkaline phosphatase, OC osteocalcin, P1NP N-terminal pro-peptide sequence of type I collagen, CTX β-isomerised C-terminal cross-linked telopeptide of type I collagen, RANKL receptor activator of NF-κB ligand, OPG osteoprotegerin, IL-6 interleukin 6, DKK1 Dickkopf-1. Data are the mean ± SD
Bone Mineral Density
BMD was evaluated by dual-energy X-ray absorptiometry (DXA) in the GSD type I patients at T0 and after 12 (T12) and 24 (T24) months. At T0, 12 out of 20 subjects (60%) had lumbar spine (LS) BMD values, corrected for sex, age and height growth, "below the expected range for age", defined as LS Z-score < −2.0 (Table 3). At T24, 16 subjects were re-evaluated by DXA and 9 (56%) were confirmed to present LS BMD values, corrected for sex, age and height growth, “below the expected range for age” (Table 3). One patient at T0, 7 patients at T12 and 4 patients at T24 were unable or unwilling to repeat the DXA scan.
Table 3.
BMD in GSD type I patients
| LS BMD Z-scores | ||||
|---|---|---|---|---|
| Time of evaluation | T0 | T12 | T24 | |
| Patients | 1 | −3.6 | −3.1 | −2.4 |
| 2 | −3.3 | −2.2 | −2.4 | |
| 3* | −2.1 | −1.9 | −1.6 | |
| 4 | −1.6 | −1.7 | −1.6 | |
| 5 | −1.1 | −1.3 | −1.0 | |
| 6 | −2.5 | −2.3 | −3.0 | |
| 7 | −1.7 | −2.5 | −1.5 | |
| 8* | n.a.# | 1.3 | 1.2 | |
| 9 | −1.0 | −1.6 | −0.9 | |
| 10 | −2.1 | −1.8 | −0.9 | |
| 11 | −2.2 | −2.3 | −2.4 | |
| 12 | −3.2 | n.a | −2.9 | |
| 13* | −1.9 | n.a | −2.1 | |
| 14 | −3.0 | n.a | −3.0 | |
| 15 | −1.2 | n.a | n.a | |
| 16 | −4.2 | −4.1 | −4.4 | |
| 17 | −2.5 | n.a | n.a | |
| 18 | −1.8 | n.a | n.a | |
| 19* | −3.3 | −3.0 | −3.7 | |
| 20* | −2.2 | n.a | n.a | |
| Mean ± SD | −2.34 ± 0.89 | −2.04 ± 1.25 | −2.04 ± 1.32 | |
*Patients with fractures; #n.a.: Not available
No significant changes in the mean LS BMD Z-scores could be detected at T12 and T24 compared to T0 (Fig. 1).
Fig. 1.
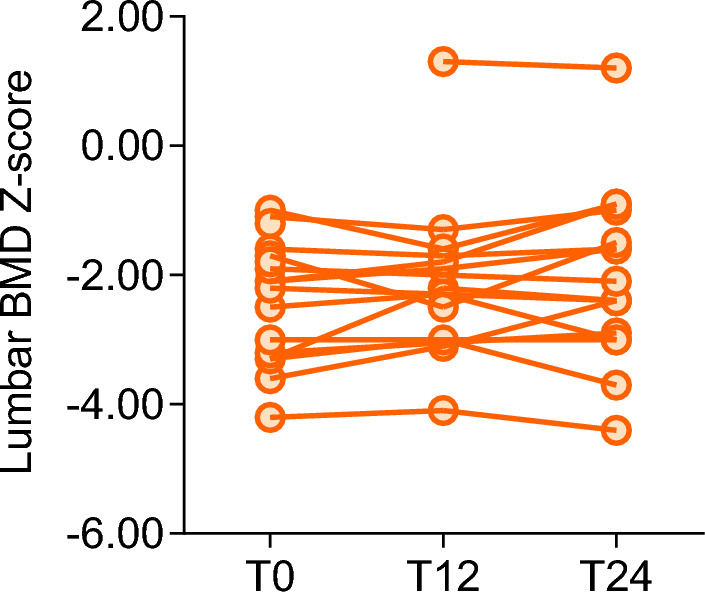
LS BMD Z-scores detected at the indicated time points, expressed as months from the start of the study (T0)
At T0, GSD type I patients with LS BMD Z-scores < −2.0 did not differ from patients with appropriate LS BMD Z-scores in any anthropometric, clinical, genetic, and biochemical parameters. LS BMD Z-scores of fractured patients did not differ from those of unfractured patients (Table 2). Of note, LS BMD Z-scores correlated with muscle strength measured by the hand grip test (Fig. 2).
Fig. 2.

Correlation of patients’ LS BMD Z-scores with muscle strength measured by the hand grip test
Mineral and Bone Turnover Parameters
At T0, 80% of patients (16 out of 20) had vitamin D sufficient levels (> 20 ng/ml). Fourteen of them were supplemented with calcifediol or cholecalciferol. Hyperparathyroidism was absent and circulating calcium and phosphate levels were within normal range in all patients. However, as expected, patients aged 15–19 years had lower, although not significant, mean serum calcium and phosphate levels than younger patients (Fig. 3).
Fig. 3.

Circulating mineral and bone markers. BSAP: bone specific alkaline phosphatase; OC: osteocalcin; CTX: C-telopeptide of type I collagen cross-links; OPG: osteoprotegerin. Statistics: one-way ANOVA
Circulating mineral markers did not correlate with LS BMD, while upper limb muscle strength measured by hand grip test showed a negative correlation with serum calcium (r = −0.573, p = 0.010) and a trend toward negative correlation with serum phosphate levels (r = −0.432, p = 0.065), which is likely related to increasing age.
As far as the bone markers are concerned, circulating BSAP showed a trend of decrease and OC, β-CTX and OPG levels were significantly lower in patients aged 15–19 years than those in younger patients, as observed in the healthy children and adolescents. Indeed, in the present series, the increase of bone markers at puberty could not be detected (age 9–14, Fig. 3). No differences were observed between the two genotypes for the baseline values of the serum markers of bone metabolism (Table 4).
Table 4.
Comparison of bone markers levels at T0 between patients harbouring the G6PC and SLC37A4 germline mutations (n = 12)
| Sex, age and bone markers* | G6PC (17q21) | SLC37A4 (11q23) | p-value |
|---|---|---|---|
| N (F/M) | 8 (6/2) | 4 (0/4) | – |
| Age (years) | 12.4 ± 3.9 | 9.3 ± 5.3 | 0.2719 |
| 25OHD (ng/ml) | 36.9 ± 18.7 | 85.2 ± 77.6 | 0.1120 |
| S calcium (mg/dl) | 10.6 ± 0.5 | 9.9 ± 0.4 | 0.0536 |
| S Phosphate (mg/dl) | 4.6 ± 0.7 | 4.9 ± 0.5 | 0.3869 |
| PTH (pg/ml) | 20.5 ± 10.6 | 25.9 ± 13.4 | 0.4633 |
| BSAP (µg/L) | 78.8 ± 49.5 | 55.4 ± 13.1 | 0.3841 |
| OC (ng/ml) | 51.0 ± 23.2 | 78.4 ± 11.0 | 0.0517 |
| P1NP (ng/ml) | 314.7 ± 198.9 | 441.5 ± 98.3 | 0.2640 |
| β-CTX (pg/ml) | 1055.0 ± 533.6 | 1035.0 ± 231.8 | 0.9453 |
| RANKL (pmol/L) | 429.5 ± 232.4 | 652.3 ± 465.8 | 0.2830 |
| OPG (pmol/L) | 4.3 ± 1.5 | 2.7 ± 1.0 | 0.0918 |
| RANKL/OPG ratio | 125.6 ± 107.6 | 243.0 ± 179.1 | 0.1804 |
| DKK1 (pmol/L) | 54.0 ± 27.6 | 42.4 ± 13.6 | 0.4561 |
| Sclerostin (pmol/L) | 25.3 ± 8.0 | 21.7 ± 5.3 | 0.4396 |
*Abbreviations are shown in Table 2
Of note, serum sclerostin levels positively correlated with LS BMD Z-scores (Fig. 4). Besides, muscle strength of the upper limbs correlated with serum OPG (r = −0.537, p = 0.018) and sclerostin levels (r = 0.464, p = 0.045).
Fig. 4.
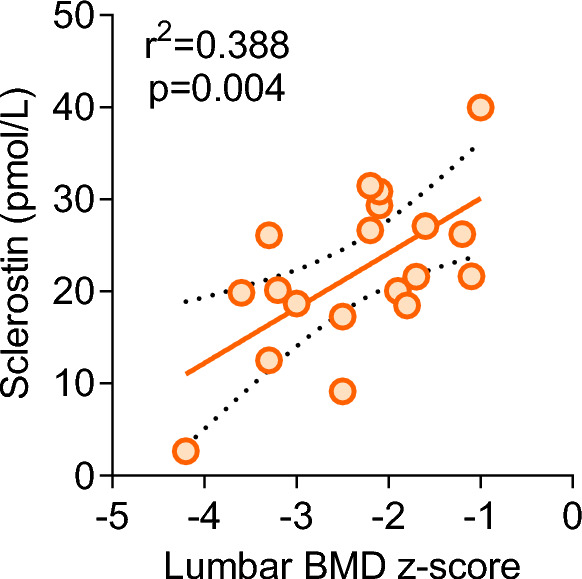
Correlation between serum sclerostin levels and LS BMD Z-scores
Cellular Study
Circulating Osteoclast Precursors
From a cellular point of view, we found no significant differences between patients and healthy controls in the number of osteoclasts differentiated in culture from peripheral blood mononuclear cells (PBMCs), in the number of nuclei per osteoclast and in the osteoclast size (Fig. 5A). Similarly, we found no difference in the release of the bone resorption marker CTX and the osteoclast marker TRAcP between osteoclasts differentiated from patient and control PBMCs, cultured on bone slices (Fig. 5B), suggesting that GSD type I does not cause a cell-autonomous impairment of osteoclastogenesis and bone resorption.
Fig. 5.

A PBMCs isolated from control and GSD type I patients at T0 were differentiated in osteoclasts by treatment with M-CSF and RANKL. After 2 weeks, cultures where fixed and evaluated histochemically for TRAcP activity to measure osteoclast number (left panel), number of nuclei per osteoclast (middle panel) (by co-staining with the nuclear dye DAPI) and osteoclast size (right panel). B CTX and TRAcP released in the culture media by GSD type I and control osteoclasts cultured for three weeks on bone slices. C Osteoclasts from healthy donors differentiated from PBMCs as described in (A) were cultured in the presence of foetal bovine serum (FBS), sera from control subjects or from GSD type I patients, stained histochemically for the expression of TRAcP and counted. Statistics: Student’s t test
In contrast, the cultures of PBMCs from healthy subjects incubated in the presence of GSD sera showed a significant increase of osteoclastogenesis versus standard cultures performed in the presence of foetal bovine serum (FBS) or in the presence of human control sera (Fig. 5C). These results may indicate that circulating factors could contribute to enhanced osteoclastogenesis in GSD type I patients. However, further work is necessary to confirm this hypothesis.
Circulating Alkaline Phosphatase Positive Osteoprogenitors
Finally, we isolated circulating ALP positive (ALP +) OsteoProgenitor (COP) cells (Fig. 6A) from GSD type I patients and healthy donors. This latter group had a mean age higher than the group of patients, given that they were obtained from blood donors (years, healthy donors: 33.33 ± 11.52; GSD patients: 13.71 ± 4.71). GSD patients showed higher percent of ALP + COPs normalised over PBMCs total number compared to controls (Fig. 6B), as expected in growing vs. adult individuals. Furthermore, osteoblasts collected from healthy individuals and cultured in the presence of GSD type I sera showed no changes in the expression of osteoblast genes versus control sera, except for a significant increase of ALP in cultures challenged by sera from GSD type 1 patients (Fig. 6C).
Fig. 6.

A Imaging illustrating isolation of ALP + COPs from serum at T12. B Number of ALP + COPs in controls and GSD type I patient sera. C Osteoblasts from healthy donor were cultured in the presence of pools of sera from healthy individuals (dotted line) and GSD type I patients. RNA was then subjected to real time RT-PCR for the indicated osteoblast genes. Results are expressed as fold changes of GSD type I versus healthy donor sera treated cells, normalised by GAPDH. Number of samples treated with healthy sera pool: 9. SD of gene expression in cultures treated with healthy sera pool before normalisation to 1: RUNX2 = 0.35; COL1A1 = 0.50; ALP = 0.49; OCN = 0.7; RANKL = 0.26; OPG = 0.53. Statistics: Student’s t test
Taken together, these results suggest that the major cellular alteration in GSD type I patients could be ascribed to the osteoclast lineage, with a non-cell autonomous increase of osteoclast generation, possibly induced by circulating factors.
Discussion
GSDs, including GSD type I, are rare diseases for which no orphan drugs nor enzyme replacement therapies are available even off-label, and patients are currently treated with a special dietary management to avoid hypoglycaemia [22].This treatment, associated with appropriate prevention and control of infections, extends the life expectancy but uncover the appearance of multiple organ disease with a wide spectrum of severity, including long-term skeletal problems [9], reduced bone accrual and/or increased bone loss [10, 11, 13, 14]. To expand the knowledge on bone complications in GSD type I, we collected data from 20 GSD Ia–Ib patients, evaluating the changes in BMD and in bone turnover/metabolism parameters over 2 years.
About one-third of GSD type I patients in our population suffered fracture events prior to T0 and at the anamnestic collection at T24. This prevalence may be not different from that reported in the healthy young population, with fractures similarly involving mainly the upper and lower limbs [23–26]. All fractured patients, but one, performed physical activity, either regular or irregular. However, fractures were reported to occur in the absence of high-energy trauma. Furthermore, at T0 60% of patients had LS BMD values measured by DXA “below the expected range for age”, which persisted at T24, while patients experiencing fractures were older than those unfractured. Altogether, these results suggest that fractures could be associated with fragility and increasing burden of the metabolic disease with age, but further studies are necessary to test this hypothesis. Importantly, the high prevalence of low BMD should be considered in case of liver transplantation in GSD patients, a condition associated with increased risk of fragility fractures due to immunosuppressive therapy. Patients should be evaluated for BMD and managed for bone complications starting immediately after the surgery.
GSD patients with fractures and/or low LS BMD did not differ from unfractured non-osteoporotic GSD patients in their anthropometric, genetic, and clinical data nor in their circulating mineral and bone marker levels. Therefore, we failed to identify a specific phenotype associated with an increased risk of low LS BMD. Indeed, in the present series, we detected a positive correlation between LS BMD Z-scores and muscle strength, similarly to what recently reported in young paediatric cancer survivors [27]. Interestingly, a positive correlation emerged between LS BMD Z-scores and muscle strength with circulating sclerostin levels.
Serum sclerostin is secreted locally from bone matrix embedded osteocytes, where it antagonises Wnt/β-catenin signalling in osteoblasts [28]. High levels of sclerostin are observed in postmenopausal osteoporotic women. Therefore, the finding of relatively higher sclerostin levels in GSD type I patients with higher LS BMD is unexpected. Recent studies have highlighted a hypothetical role of sclerostin in myogenesis, modulating the interaction between bone and muscle [29, 30]. Accordingly, GSD type I patients with relatively high circulating levels of sclerostin had higher LS BMD and higher muscle strength.
In addition to the clinical observations, this study provided evidence that bone cells can be altered in GSD type I. In fact, healthy PBMCs showed a non-autonomous increase of osteoclastogenesis when challenged with GSD type I sera, while osteoclast parameters were unaltered in cultures performed with patients’ PBMCs in standard medium supplemented with FBS or with sera from control subjects. This result suggests that no cell-autonomous alterations are induced by GSD type I in the osteoclast lineage. However, dysregulated factors may be present in the bone microenvironment and in sera of GSD type I patients, causing a non-cell autonomous increase of osteoclast function compatible with the low BMD observed in the GSD type I population analysed in this study. In contrast, the circulating ALP + COP population did not appear to be altered in patients, nor to be modified by sera from GSD type I patients, although this observation does not allow to draw any hypothesis on the involvement of the bone-resident osteogenic lineage in the onset of the observed alterations of patients’ BMD. Therefore, further work is necessary to elucidate these aspects, especially because, due the COVID-19 pandemics, our control sera could not be obtained from healthy kids. Therefore, we used control sera from adult healthy blood donors, which represents a limitation of the study.
Another limitation was due to the partial coincidence of the study with the COVID-19 pandemics, which prevented the evaluation of a larger number of patients and caused some withdrawal. More patients should be investigated and long-term analysis should be performed through new analytical algorithms applied to DXA [31–33], for the early identification of GSD subjects at risk of fracture/imminent fracture. Furthermore, further investigations are necessary to shed light on the structural and cellular features altered in GSD. However, despite these limitations, we can propose that low BMD characterises the bone phenotype of GSD type I patients, with mechanisms potentially interconnected with muscle alterations and involving a non-cell autonomous increase of osteoclastogenesis.
Materials and Methods
Ethical Aspects
The study involved standard clinical follow-up procedures for patients already taken care of at the participating Centres, with no experimental drugs used. The clinical protocol and the informed consent were approved by the Ethics Committee of all participating Centres (n° 31/2018 for the University of L’Aquila and n° 2017_12_19_05 for Istituto Auxologico Italiano). The study was conducted in accordance with the Declaration of Helsinki, adhering to the procedures and principles of Good Clinical Practice. The informed consent was signed and dated by the parent(s)/guardian(s) for patients under 18 years of age, or by the patients themselves if ≥ 18 years of age, and by the Centre’s Principal Investigator or another delegated member of the investigation team. In the case of children, oral consent for those between 3 and 10 years, or signed consent for those between 11 and 18 years, were also required. The project was subjected to all the law requirements regarding the insurance of clinical studies, according to the indications of the Coordinating Centre’s Ethics Committee.
Study Design
This project was a 24-month multicentre prospective interventional non-pharmacologic study on 20 young patients (age range 3–20 years) affected by GSD Ia-Ib to: (1) identify and evaluate the different patterns/degrees of bone involvement; (2) investigate the biological mechanisms involved in the pathogenesis of bone alterations and determine their type/severity; (3) identify the specific risk factors for bone loss and bone fragility; (4) identify patients with a higher risk of bone mass loss and fractures.
Study Protocol
Patients were visited at baseline (T0), 12 (T12) and 24 (T24) months, using a Clinical Record Form. Skeletal exams and laboratory tests performed are described in Table 5.
Table 5.
Skeletal exams and laboratory tests
| Clinical Record | Skeletal Exams | Laboratory tests | |||
|---|---|---|---|---|---|
| Serum bone formation biomarkers | Serum bone resorption biomarkers | Serum cytokines | |||
|
-Complete physical examination (auxological parameters, Tanner stage for children/adolescents, menstrual history) |
-DXA scans to evaluate LS BMD |
-Calcium -Phosphate -Creatinine -25-OH Vitamin D -Parathyroid hormone |
-Osteocalcin -Bone specific alkaline phosphatase -Procollagen type I N-t terminal propeptide |
-Type I collagen C- telopeptide |
-Osteoprotegerin -Receptor activator of nuclear factor kappa-B ligand -Dickkopf-related protein 1 (Dkk1), sclerostin |
|
-Evaluation of metabolic control laboratory values |
-BMD Z- scores calculated using a suitable reference sample of healthy subjects |
||||
|
-Current/previous treatments (drugs, dosage and duration) |
-X-rays of lateral d dorsal and LS spine to evaluate vertebral d deformities |
||||
| -Fracture history |
-Wrist X-rays to estimate bone age at T0 |
||||
| -Functional tests for muscle strength | |||||
|
-Questionnaires for pain, well-being and quality of life |
|||||
According to the definition of the Official Positions of the International Society for Clinical Densitometry, a Z-score ≤ − 2.0 was defined as “below the expected range for age,” and a Z-score > − 2.0 was regarded as "within the expected range for age” [34]. According to the consensus report published by the European Society for Paediatric Endocrinology (ESPE) in 2016, 25(OH)D3 levels were defined as normal for > 20 ng/ml, insufficient for 12–20 ng/ml, and deficient for < 12 ng/ml [35].
Study Population
Patients with GSD Ia-Ib were recruited at the Rare Metabolic Diseases Unit, IRCCS Foundation San Gerardo dei Tintori in Monza, Italy, and at the Rare Metabolic Diseases Unit, Institute Giannina Gaslini IRCCS in Genova, Italy. They presented the project to the patients and, full information was provided to those who expressed interested followed by signing the informed consent form. Withdrawal from the study was allowed at any time. Inclusion and exclusion criteria are described in Table 6.
Table 6.
Inclusion and exclusion criteria for patient enrolment
| Inclusion criteria | Exclusion criteria |
|---|---|
|
-Clinical diagnosis of GSD type Ia or type Ib -Age range 3–20 years -Availability to undergo all visits/tests according to the planned time schedule -Signed and dated informed consent form |
-Presence of other chronic systemic diseases -Presence of other conditions affecting bone mass and metabolism (i.e. rickets, hyperparathyroidism, etc.) -Use of oestrogen/testosterone replacement therapy, corticosteroids, or other drugs affecting bone metabolism (current use, programmed use, past use during the previous 6 months) |
Cellular Studies
Reagents
Dulbecco’s modified Minimum Essential Medium (DMEM), penicillin, streptomycin, and trypsin were from Euroclone (Milan, Italy). Foetal bovine serum (FBS) was from GIBCO (Uxbridge, UK). Sterile plastic ware was from Greiner bio-one (Kremsmünster, Austria) or Euroclone (Milan, Italy). TRIzol reagent (cat#15,596,018), primers and reagents for RT-PCR (cat#k1622) were from Invitrogen (Carlsbad, CA, USA). The qRT-PCR assays Luna Universal One Step RT qPCR Kit (#E3006) was from New England Biolab. Bone Slices (DT-1BON1000-96), Carboxy Terminal collagen crosslinks (CTX, cat#AC-06F1) and Tartrate-Resistant Acid Phosphatase (TRAcP, cat#SB-TR103) immunoenzymatic kits were from Immunodiagnostic Systems (IDS®, The Boldons, UK). Macrophage-Colony Stimulating Factor (M-CSF) and Receptor Activator of NK-ĸB Ligand (RANKL) were from Preprotech (London, UK). All other reagents were of the purest grade from Sigma Aldrich Co. (St. Louis, MO, USA).
Osteoclast Preparation from Peripheral Blood Mononuclear Cells (PBMCs)
Human peripheral blood from healthy donors and GSD type I patients was diluted in Hank’s balanced salt solution (1:1), layered over Histopaque 1077 solution, centrifuged at 400 g for 30 min, washed twice with Hank’s solution and further centrifuged at 250 g for 10 min. PBMCs were collected with the buffy coat and resuspended in Dulbecco’s modified Minimum Essential Medium (DMEM) containing 4 mM L-glutamine, 100 U/ml penicillin, 100 μg/ml streptomycin, and 10% FBS; 3 × 106 cells/cm2 were seeded on tissue culture plate and incubated at 37 °C in humidified atmosphere with 5% CO2. After 1 h, cultures were rinsed to remove nonadherent cells and maintained for 7–21 days in medium containing 10% FBS or human serum from GSD patients or healthy donors, in the presence of 50 ng/ml human M-CSF and 30 ng/ml human RANKL. Cultures from healthy donors were also performed in the presence of 50 ng/ml human M-CSF and 0.5 ng/ml human RANKL and 10% GDS serum pool.
TRAcP Activity Assay
Cells were fixed in 4% paraformaldehyde in PBS for 15 min and washed in PBS. TRAcP activity was detected histochemically using Sigma-Aldrich kit (87A-1KT), according to the manufacturer’s instruction.
Bone Resorption Assay
PBMCs were cultured for 3 weeks in the presence of 10% FBS, 25 ng/ml M-CSF and 30 ng/ml RANKL, on bone slices. Then the media were collected and analysed for the release of the bone resorption biomarker CTX and the osteoclast biomarker TRAcP.
Isolation of Circulating Osteo-progenitors (COPs)
Blood samples (10 ml) from healthy donors and GSD type I patients were layered over Histopaque 1077 density gradients. Mononuclear cells were isolated and incubated with anti-ALP-biotin primary antibody (anti-MSCA-1), followed by a second incubation with anti-biotin microbeads conjugated secondary antibody. The marked cells were processed by magnetic activated cell sorting (MACS) using MS columns for smaller single cells. Following the magnetic sorting, ALP + COPs were then counted and their percentage over the total number of PBMCs was computed.
Gene Expression in Osteoblasts from Healthy Subjects Treated with Healthy Donor or GSD Type I Sera
Bone fragments were obtained from healthy subjects who underwent surgery for traumatic fractures. Bone fragments were digested with 1 mg/ml Clostridium histolyticum type IV collagenase and 0.25% trypsin for 20 min at 37 °C in Hank’s buffer solution with gentle agitation. The procedure was repeated twice for 40 min at the same temperature, and cells from the latter digestion were plated and grown to confluence in DMEM supplemented with antibiotics and 10% FBS. Cells were then replated in dishes, supplemented with 5% FBS or pools of healthy donor or GSD sera, and cultured for 2 days. The experiment was repeated three times using independent osteoblast cultures, RNA was extracted using the acid phenol technique. For RT-PCR, 1 μg total RNA was reverse-transcribed using the RevertAid H Minus First Strand cDNA Synthesis Kit. Real time PCR reaction was performed loading 0.1 μg of cDNA using the Luna® Universal qPCR Master Mix. Gene expression data were represented as fold change over the control and normalised by Gapdh, unless otherwise stated. PCR conditions and primer pairs used are listed in Supplementary Table 1. For quantitative analysis, all amplification signals were normalised against the signal of the housekeeping gene, GAPDH.
Supplementary Information
Below is the link to the electronic supplementary material.
Acknowledgements
This study was supported by the grant AIFA-2016-02364539 from the Agenzia Italiana del Farmaco (AIFA). We are indebted to Prof. Maja Di Rocco, Rare Metabolic Diseases Unit, Department of Paediatrics, IRCCS Institute Giannina Gaslini, Genova, Italy, for her contribution in patient recruitment.
Funding
Agenzia Italiana del Farmaco, Ministero della Salute, AIFA-2016-02364539, Maria Luisa Bianchi
Declarations
Conflict of interest
Professor Sabrina Corbetta has received research grants from Ascendis Pharma, Theramex, Abiogen Pharma, Gedeon Richter and Savio Pharma Italia, from Italian Ministry of Health, and Fondazione Cariplo to her Institution. All other authors declare no conflict of interest.
Informed consent
The clinical protocol and the informed consent were approved by the Ethics Committee of all participating Centres (n° 31/2018 for the University of L’Aquila and n° 2017_12_19_05 for Istituto Auxologico Italiano).
Human and animal rights
The study involved standard clinical follow-up procedures for patients already taken care of at the participating Centres, with no experimental drugs used. Sera and cells from healthy individuals were collected for cellular studies with their informed consent. The study was conducted in accordance with the Declaration of Helsinki, adhering to the procedures and principles of Good Clinical Practice. No animals were involved in the study.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Silvia Vai and Alberto Falchetti contributed equally to this work.
References
- 1.Hannah WB, Derks TGJ, Drumm ML, Grünert SC, Kishnani PS, Vissing J (2023) Glycogen storage diseases. Nat Rev Dis Primers 9:46 [DOI] [PubMed] [Google Scholar]
- 2.Gümüş E, Özen H (2023) Glycogen storage diseases: an update. World J Gastroenterol 29(25):3932–3963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Soon GST, Torbenson M (2023) The liver and glycogen: in sickness and in health. Int J Mol Sci 24(7):6133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kishnani PS, Austin SL, Abdenur JE, Arn P, Bali DS, Boney A, Chung WK, Dagli AI, Dale D, Koeberl D, Somers MJ, Wechsler SB, Weinstein DA, Wolfsdorf JI, Watson MS, American College of Medical Genetics and Genomics (2014) Diagnosis and management of glycogen storage disease type I: a practice guideline of the American College of Medical Genetics and Genomics. Genet Med. 16(11):e1 [DOI] [PubMed] [Google Scholar]
- 5.Odievre M (1991) Clinical presentation of metabolic liver disease. J Inherit Metab Dis 14(4):526–530 [DOI] [PubMed] [Google Scholar]
- 6.Melis D, Cozzolino M, Minopoli G, Balivo F, Parini R, Rigoldi M, Paci S, Dionisi-Vici C, Burlina A, Andria G, Parenti G (2015) Progression of renal damage in glycogen storage disease type I is associated to hyperlipidemia: a multicenter prospective Italian study. J Pediatr 166(4):1079–1082 [DOI] [PubMed] [Google Scholar]
- 7.Bianchi L (1993) Glycogen storage disease I and hepatocellular tumours. Eur J Pediatr 152:S63-70 [DOI] [PubMed] [Google Scholar]
- 8.Bali DS, El-Gharbawy A, Austin S, Pendyal S, Kishnani PS. Glycogen Storage Disease Type I. 2006 Apr 19 [updated 2021 Oct 14]. In: Adam MP, Feldman J, Mirzaa GM, Pagon RA, Wallace SE, Bean LJH, Gripp KW, Amemiya A, editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993–2024. [PubMed]
- 9.Lee PJ, Patel JS, Fewtrell M, Leonard JV (1995) Bishop NJ bone mineralisation in type I glycogen storage disease. Eur J Pediatr 154(6):483–487 [DOI] [PubMed] [Google Scholar]
- 10.Minarich LA, Kirpich A, Fiske LM, Weinstein DA (2013) Bone mineral density in glycogen storage disease type Ia and Ib. Genet Med 14(8):737–741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rake JP, Visser G, Huismans D, Huitema S, van der Veer E, Piers DA, Smit GP (2003) Bone mineral density in children, adolescents and adults with glycogen storage disease type Ia: a cross-sectional and longitudinal study. J Inherit Metab Dis 26(4):371–384 [DOI] [PubMed] [Google Scholar]
- 12.Verbeek RJ, Sentner CP, Smit GP, Maurits NM, Derks TG, van der Hoeven JH, Sival DA (2016) Muscle ultrasound in patients with glycogen storage disease types I and III. Ultrasound Med Biol 42(1):133–142 [DOI] [PubMed] [Google Scholar]
- 13.Schwahn B, Rauch F, Wendel U, Schönau E (2002) Low bone mass in glycogen storage disease type I is associated with reduced muscle force and poor metabolic control. J Pediatr 141(3):350–356 [DOI] [PubMed] [Google Scholar]
- 14.Jacoby JT, Bento Dos Santos B, Nalin T, Colonetti K, Farret Refosco L, de Souza CFM, Spritzer PM, Poloni S, Hack-Mendes R, Schwartz IVD (2021) Bone mineral density in patients with hepatic glycogen storage diseases. Nutrients. 13(9):2987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sharma GB, Robertson DD, Laney DA, Gambello MJ, Terk M (2016) Machine learning based analytics of micro-MRI trabecular bone microarchitecture and texture in type I Gaucher disease. J Biomech 49(9):1961–1968 [DOI] [PubMed] [Google Scholar]
- 16.Froissart R, Piraud M, Boudjemline AM, Vianey-Saban C, Petit F, Hubert-Buron A, Eberschweiler PT, Gajdos V, Labrune P (2011) Glucose-6-phosphatase deficiency. Orphanet J Rare Dis 20(6):27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Stone WL, Basit H, Adil A. Glycogen storage disease. 2023. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2024 [PubMed]
- 18.Chou JY, Jun HS, Mansfield BC (2010) Glycogen storage disease type I and G6Pase-beta deficiency: etiology and therapy. Nat Rev Endocrinol 6(12):676–688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Derks TGJ, Rodriguez-Buritica DF, Ahmad A, de Boer F, Couce ML, Grünert SC, Labrune P, López Maldonado N, Moura Fischinger, de Souza C, Riba-Wolman R, Rossi A, Saavedra H, Gupta RN, Valayannopoulos V, Mitchell J (2021) Glycogen storage disease type Ia: current management options, burden and unmet needs. Nutrients. 13(11):3828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rafeeq MM, Murad HAS (2017) Cystic fibrosis: current therapeutic targets and future approaches. J Transl Med 15(1):84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sun C, Shen L, Zhang Z, Xie X (2020) Therapeutic strategies for Duchenne muscular dystrophy: an update. Genes (Basel). 11(8):837 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Brudvik C, Hove LM (2003) Childhood fractures in Bergen, Norway: identifying high-risk groups and activities. J Pediatr Orthop. 23(5):629–634 [DOI] [PubMed] [Google Scholar]
- 23.Cooper C, Dennison EM, Leufkens HG, Bishop N, van Staa TP (2004) Epidemiology of childhood fractures in Britain: a study using the general practice research database. J Bone Miner Res 19(12):1976–1981 [DOI] [PubMed] [Google Scholar]
- 24.Rennie L, Court-Brown CM, Mok JY, Beattie TF (2007) The epidemiology of fractures in children. Injury 38(8):913–922. 10.1016/j.injury.2007.01.036 [DOI] [PubMed] [Google Scholar]
- 25.Hedström EM, Svensson O, Bergström U, Michno P (2010) Epidemiology of fractures in children and adolescents. Acta Orthop 81(1):148–153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.van den Berg LE, Zandbergen AA, van Capelle CI, de Vries JM, Hop WC, van den Hout JM, Reuser AJ, Zillikens MC, van der Ploeg AT (2010) Low bone mass in Pompe disease: muscular strength as a predictor of bone mineral density. Bone 47(3):643–649. 10.1016/j.bone.2010.06.021 [DOI] [PubMed] [Google Scholar]
- 27.Marmol-Perez A, Gil-Cosano JJ, Ubago-Guisado E, Llorente-Cantarero FJ, Pascual-Gázquez JF, Ness KK, Martinez-Vizcaino V, Ruiz JR, Gracia-Marco L (2024) Muscle strength deficits are associated with low bone mineral density in young pediatric cancer survivors: the iBoneFIT project. J Sport Health Sci. 10.1016/j.jshs.2024.01.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Robling AG, Bonewald LF (2020) The osteocyte: new insights. Annu Rev Physiol 10(82):485–506. 10.1146/annurev-physiol-021119-034332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Moretti A, Iolascon G (2023) Sclerostin: clinical insights in muscle-bone crosstalk. J Int Med Res. 10.1177/03000605231193293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Magarò MS, Bertacchini J, Florio F, Zavatti M, Potì F, Cavani F, Amore E, De Santis I, Bevilacqua A, Reggiani Bonetti L, Torricelli P, Maurel DB, Biressi S, Palumbo C (2021) Identification of sclerostin as a putative new myokine involved in the muscle-to-bone crosstalk. Biomedicines 9(1):71. 10.3390/biomedicines9010071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lewiecki EM, Gordon CM, Baim S, Leonard MB, Bishop NJ, Bianchi ML, Kalkwarf HJ, Langman CB, Plotkin H, Rauch F, Zemel BS, Binkley N, Bilezikian JP, Kendler DL, Hans DB, Silverman S (2008) International society for clinical densitometry 2007 adult and pediatric official positions. Bone 43:1115–1121. 10.1016/j.bone.2008.08.106 [DOI] [PubMed] [Google Scholar]
- 32.Gama EMF, Mendonça LMC, Paranhos-Neto FP, Vieira Neto L, Madeira M, Farias MLF (2024) TBS correlates with bone density and microstructure at trabecular and cortical bone evaluated by HR-pQCT. J Bone Miner Metab. 10.1007/s00774-024-01508-4 [DOI] [PubMed] [Google Scholar]
- 33.Roux JP, Duboeuf F, Sornay-Rendu E, Rinaudo L, Ulivieri FM, Wegrzyn J, Chapurlat R (2024) The relationship between bone strain index, bone mass, microarchitecture and mechanical behavior in human vertebrae: an ex vivo study. Osteoporos Int. 10.1007/s00198-024-07066-9 [DOI] [PubMed] [Google Scholar]
- 34.Giovanni A, Luisa BM, Carla C, Ernesto C, Francesco C, Marco DP, Angelo F, Davide G, Francesca G, Stefano G, Anna LF (2024) Bone health status evaluation in men by means of REMS technology. Aging Clin Exp Res. 36(1):74. 10.1007/s40520-024-02728-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pitukcheewanont P, Austin J, Chen P, Punyasavatsut N (2013) Bone health in children and adolescents: risk factors for low bone density. Pediatr Endocrinol Rev. 10(3):318–35 [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.