

Abstract
Cerebrovascular injury (CVI) is a common pathology caused by infections, injury, stroke, neurodegeneration and autoimmune disease. Rapid resolution of a CVI requires a coordinated innate immune response. In the present study, we sought mechanistic insights into how central nervous system-infltrating monocytes program resident microglia to mediate angiogenesis and cerebrovascular repair after an intracerebral hemorrhage. In the penumbrae of human stroke brain lesions, we identifed a subpopulation of microglia that express vascular endothelial growth factor A. These cells, termed ‘repair-associated microglia’ (RAMs), were also observed in a rodent model of CVI and coexpressed interleukin (IL)-6Ra. Cerebrovascular repair did not occur in IL-6 knockouts or in mice lacking microglial IL-6Ra expression and single-cell transcriptomic analyses revealed faulty RAM programming in the absence of IL-6 signaling. Infltrating CCR2+ monocytes were the primary source of IL-6 after a CVI and were required to endow microglia with proliferative and proangiogenic properties. Faulty RAM programming in the absence of IL-6 or infammatory monocytes resulted in poor cerebrovascular repair, neuronal destruction and sustained neurological defcits that were all restored via exogenous IL-6 administration. These data provide a molecular and cellular basis for how monocytes instruct microglia to repair damaged brain vasculature and promote functional recovery after injury.
Cerebrovascular injuries (CVIs) can be caused by infections, strokes, intracerebral hemorrhages, traumatic brain injuries (TBIs), neurodegenerative diseases and autoimmune diseases, resulting in potentially serious damage to surrounding central nervous sytem (CNS) tissue1–4. Vascular hemorrhage is one of the most serious types of CVI, which is caused by massive blood–brain barrier (BBB) breakdown and deposition of blood products in the parenchyma4. This can lead to brain swelling, hypoxia, inflammation and neural cell death. The kinetics of vascular remodeling and BBB closure have a major influence on the recovery trajectory, for example, after a stroke, spontaneous recovery usually occurs within the first few weeks or months5. However, stroke survivors often demonstrate varying recovery trajectories even after experiencing comparable initial injuries6. Although some patients suffer from lasting neurological deficits due to incomplete recovery, others regain functionality and show notable improvement over time. One key to functional recovery after a stroke is preserving and restoring brain tissue in the penumbra region surrounding the heavily injured necrotic lesion core7. The factors that influence spontaneous recovery trajectories are largely unknown. Microglia have been shown to favorably influence outcomes after a CVI8,9, whereas pathogens, and the type I interferon (IFN) signaling that they induce, impede recovery10. Observations in humans and rodent models demonstrate that the brain has a remarkable capacity to remodel after a CVI; however, the process is sometimes suboptimal. It is therefore important to identify and understand all the mechanisms responsible for rebuilding damaged blood vessels and promoting functional recovery after a CVI.
The BBB represents an important interface between the CNS and periphery that helps remove unwanted metabolites from the CNS while also regulating the entry of oxygen, nutrients and immune cells11. The BBB is maintained by a neurovascular unit consisting of endothelial cells, mural cells, neurons, astrocytes and microglia11, but sometimes becomes the target of insults from degenerative factors12,13 and cerebrovascular diseases including stroke14. Microglia are among the first responders to a CVI and BBB damage15–18. Using purinergic receptors, microglia extend processes toward astrocyte-derived purines within minutes of injury and help to seal damaged vessels18,19. This is usually followed by recruitment of myelomonocytic cells from the blood18,20,21. Massive myelomonocytic cell extravasation in the early stages of a CVI can worsen vascular pathology and promote potentially fatal brain swelling; however, these cells are eventually required for successful cerebrovascular repair18,20. After a CVI, monocyte-derived macrophages (MDMs) are known to facilitate neural tissue repair, but the mechanisms by which they accomplish this are not entirely clear18,22. Some studies have suggested that the phagocytic uptake of debris by MDMs is an important part of the repair process22.
Although studies have shown a role for microglia in brain homeostasis, acute injury responses, antimicrobial immunity, neurodegeneration and autoimmunity23–25, our understanding of how these cells participate in neural tissue repair after a CVI is lacking. This is attributed partly to heterogeneity in the functional programming of microglial subtypes that develop in response to neural tissue damage and during different CNS disease states26. In addition, it is often difficult to separate the roles of microglia from MDMs that invade the CNS. In response to a CVI, we recently identified a microglial subpopulation, referred to as repair-associated microglia (RAMs), that emerged in the damaged brain during the phase of angiogenesis and cerebrovascular remodeling18. These cells express vascular endothelial growth factor A (VEGFA), a potent proangiogenic factor27, and are juxtaposed to areas of injured cerebrovasculature. The functional relevance of RAMs to cerebrovascular repair is unknown. It is also unclear how these cells are instructed to become proangiogenic and whether they receive signals from peripheral monocytes, which are also required for successful cerebrovascular repair18.
In the present study, we sought insights into the dialog between peripheral monocytes and microglia that is required to repair and rebuild damaged blood vessels after a CVI. We examined the distribution of proangiogenic VEGFA+ RAMs in the penumbrae of human stroke brain lesions, as well as their proximity to damaged cortical blood vessels in a rodent CVI model. We then identified a cytokine (IL-6) released by monocytes/macrophages that endowed RAMs with their proangiogenic and proliferative properties. This cytokine induced signaling directly in microglia to promote RAM generation, thereby facilitating successful cerebrovascular repair and functional recovery. We also examined the transcriptomic landscape in purified cortical myeloid cells at the single-cell level to define RAM/MDM programming and determine mechanistically how CVI-induced IL-6 signaling triggers microglia to become reparative. These studies provide molecular insights into how monocytes coordinate with microglia to rebuild damaged cerebrovasculature and offer potential therapeutic opportunities to facilitate RAM generation after a CVI.
Results
CVI induces generation of repair-associated microglia
To monitor the microglial response to a CVI, we injected microbubbles intravenously (i.v.) into Tmem119-CreERT2 Stopfl/+ TdTomato mice (hereafter referred to as Tmem119-TdTo) and exposed them to low-intensity pulse ultrasound through a thinned-skull window (35- to 40-μm thickness) as described previously18 (Fig. 1a). This induces an immediate CVI beneath the thinned-skull window, causing varying degrees of vascular damage, including BBB breakdown and hemorrhage18. Before injury, adult Tmem119-TdTo mice were fed tamoxifen chow for 1 month and then regular chow for an additional 3 weeks to allow short-lived TdTomato+ cells like monocytes to turn over28 (Fig. 1a). This approach was shown previously to drive reporter protein expression in microglia, but not other CNS macrophages such as those residing in the meninges, choroid plexus or perivascular spaces28. We confirmed that ~95% of microglia expressed TdTomato in these mice (Extended Data Fig. 1a,b). This allowed us to monitor the acute microglial response to a CVI as well as how they participated in vascular remodeling and repair. Immediately after CVI, we observed by two-photon microscopy (TPM) that microglia quickly extended processes toward and wrapped injured vessels as reported previously18,19 (Fig. 1b, white arrowheads and Supplementary Video 1). Microglia also interacted with tomato lectin-labeled material derived from the vasculature (Fig. 1b, cyan arrowheads and Supplementary Video 1). These data confirm that microglia can wrap processes around damaged blood vessels and interact with debris after an acute CVI18. Less is known, however, about how these cells are programmed to participate in cerebrovascular repair, which is completed by day 10 in this model of CVI18. We therefore assessed the immune cell composition by flow cytometry in the injured brains of Tmem119-TdTo mice at a time point (day 6) when the vascular repair process is underway. CD11b+ myeloid cells expressing high levels of TdTomato were observed primarily in the brain, but not the spleen or meninges, of mice at day 6 post-CVI (Extended Data Fig. 1c,e) and had the phenotypic characteristics of microglia, consistent with the original study of Tmem119-TdTo reporter mice (Extended Data Fig. 1f)28. Notably, TdTomatohi microglia increased by ~150% in injured relative to uninjured mice at day 6 (Extended Data Fig. 1e), suggesting a profound microglial response to a CVI. We also observed a sizeable increase (~140%) in the number of TdTomatoneg macrophages (assumed to be peripherally derived) at this same time point but no changes in the number of neutrophils, B cells or T cells (Extended Data Fig. 1f–h). These data indicate that microglia can be distinguished from peripherally derived myeloid cells in Tmem119-TdTo mice and that microglia/macrophages are the two major immune subsets found in the brains of CVI mice at day 6.
Fig. 1 |. CVI induces RAM generation.
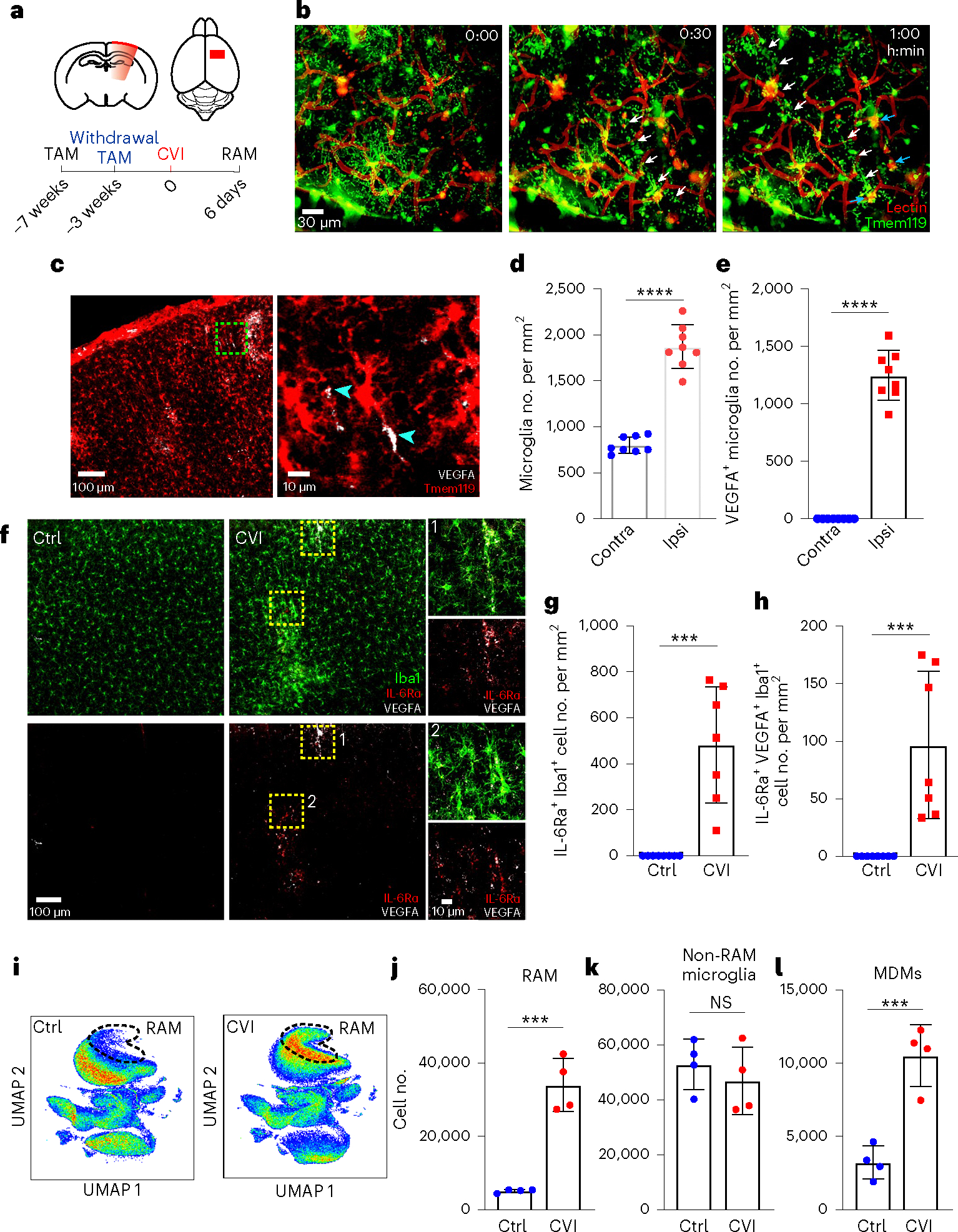
a, Schematic representation of CVI induction (red) in the mouse brain from a coronal (top left) and axial (top right) view. The timeline shows when tamoxifen (TAM) chow was administered and withdrawn in relation to CVI and RAM analysis. b, Time-lapse images from a Tmem119-TdTo mouse (n = 4) imaged by TPM through a thinned-skull window showing the acute microglial response to a CVI. Fluorescent tomato lectin (red, pseudocolored) was injected i.v. to visualize vessels and extravasated material from the blood. Microglia (green, pseudocolored) rapidly extended processes toward a damaged vessel (white arrowheads) and interacted with extravasated, lectin-derived material within minutes post-CVI (cyan arrowheads). The data represent two independent experiments (see Supplementary Video 1). c, Immunofluorescent staining for VEGFA (white) in the neocortex of a Tmem119-TdTo mouse on day 6 post-CVI. The magnified image in the right panel is denoted by a dotted green box in the left panel. Microglia (red) express VEGFA in their cell bodies and distal processes (cyan arrowheads). d,e, Bar graphs show the number of microglia (d) and VEGFA-expressing microglia (e) in the injured ipsilateral (Ipsi) versus uninjured contralateral (Contra) hemisphere at day 6 post-CVI in Tmem119-TdTo mice (n = 8). Combined data from two independent experiments are shown in c–e. f, Immunofluorescent staining for IL-6Ra (red), VEGFA (white) and Iba1 (green) in the neocortex of B6 mice on day 6 post-CVI relative to uninjured controls (Ctrl). CVI induced IL-6Ra expression in microglia, some of which coexpressed VEGFA (yellow boxes 1 and 2). Boxed areas are enlarged on the far right. g,h, Bar graphs depicting quantification of IL-6Ra+ (g) and IL-6Ra+VEGFA+ (h) microglia in uninjured Ctrl (n = 8) versus CVI (n = 7) mice. Combined data from two independent experiments are shown in f–h. i, UMAP plots showing high-parameter flow cytometry data of leukocytes extracted from punch-biopsied brains of injured (n = 4) versus uninjured (Ctrl, n = 4) Tmem119-TdTo mice at day 6 post-CVI. Dashed lines delineate RAMs based on the expression of markers identified using Cluster Explorer (see Extended Data Fig. 1k). j–l, Bar graphs showing the absolute number of RAMs (TdTo+VEGFA+IL-6Ra+CD24+CD44+CD206+CD11c+Ly6clo/intMHCIIloCX3CR1+CD11b+CD45lo/int) (j), non-RAMs (TdTo+VEGFA−IL-6Ra−CD24−CD44−CD206−CD11c−Ly6c−MHCIIloCX3C R1+CD11b+CD45lo) (k) and MDMs (TdTo−CD24+CD44+CD206+CD11c+Ly6c+CX3CR 1+CD11b+CD45+) (l) for the groups denoted in i. Data represent two independent experiments. All graphs show mean ± s.e.m. and dots represent individual mice. ***P < 0.0005, ****P < 0.0001; two-tailed, unpaired Student’s t-test. NS, not significant.
Further examination of TdTomato+ microglia in the brains of CVI mice at day 6 revealed cluster formation in areas of damage (Fig. 1c and Extended Data Fig. 1i). In fact, microglia number increased by ~100% in the CVI-injured cortex relative to the uninjured contralateral hemisphere (Fig. 1d). Some of these microglia also expressed VEGFA, a proangiogenic molecule27,29 and signature RAM protein18, in their processes (Fig. 1c, arrowhead,e). In fact, ~37% of all microglia expressed VEGFA at day 6 post-CVI (Extended Data Fig. 1j). We further analyzed myeloid clusters, which also contained macrophages, in injured brain tissue using Iba1 staining and revealed that VEGFA+ myeloid cells expressed IL-6Ra, a major component of the IL-6 receptor complex30 that is induced in injured peripheral tissues31 (Fig. 1f–h). These cells, not observed in uninjured brain tissue, increased considerably after a CVI (Fig. 1g,h). We performed unbiased flow cytometric studies in Tmem119-TdTo mice at day 6 post-CVI to confirm the identity of these cells and found that membrane-bound IL-6Ra was indeed expressed by TdTomato+ RAMs (VEGFA+CD24+CD44+CD206+CD11c+L y6clo/intMHCIIloCX3CR1+CD11b+CD45lo/int), which correlated positively with VEGFA expression (Fig. 1i,j and Extended Data Fig. 1k,l). In addition, RAM and MDM numbers increased significantly in CVI mice relative to uninjured controls, whereas the number of non-RAM microglia remained unchanged (Fig. 1j–l).
RAM number is linked to brain injury severity
To determine whether RAM numbers are influenced by brain injury severity, we next evaluated two different injury paradigms. CVI severity can be easily altered by changing skull thickness and ultrasound duration. We therefore compared RAM generation and pathology in a more severe CVI model (10-s sonication; ~20- to 25-μm skull thickness) to that observed in a milder model (5-s sonication; ~35- to 40-μm skull thickness). The severe model creates more substantial vascular damage and leaky vessels than the mild model. We performed flow cytometric studies using brain tissue from injured Tmem119-TdTo mice to, again, distinguish between MDMs (TdTo−) and microglia (TdTo+) (Extended Data Fig. 2a,b). These studies revealed an additional increase in both RAMs and MDMs in mice that experienced the more severe injury relative to mild CVI (Fig. 2a–c). In addition, a substantial increase in the number of MHCIIhi RAMs was observed after a severe CVI (Fig. 2d and Extended Data Fig. 2a–c). These increases corresponded with elevated passage of i.v. injected tomato lectin (Fig. 2g) and Evans blue (EB; Extended Data Fig. 2d,e) after severe versus mild CVI, whereas vascular coverage (Fig. 2e,f) remained comparable between the two groups. These data indicate that RAM and MDM numbers are influenced by CVI lesion severity.
Fig. 2 |. RAMs increase with lesion severity and are observed in human stroke brains.
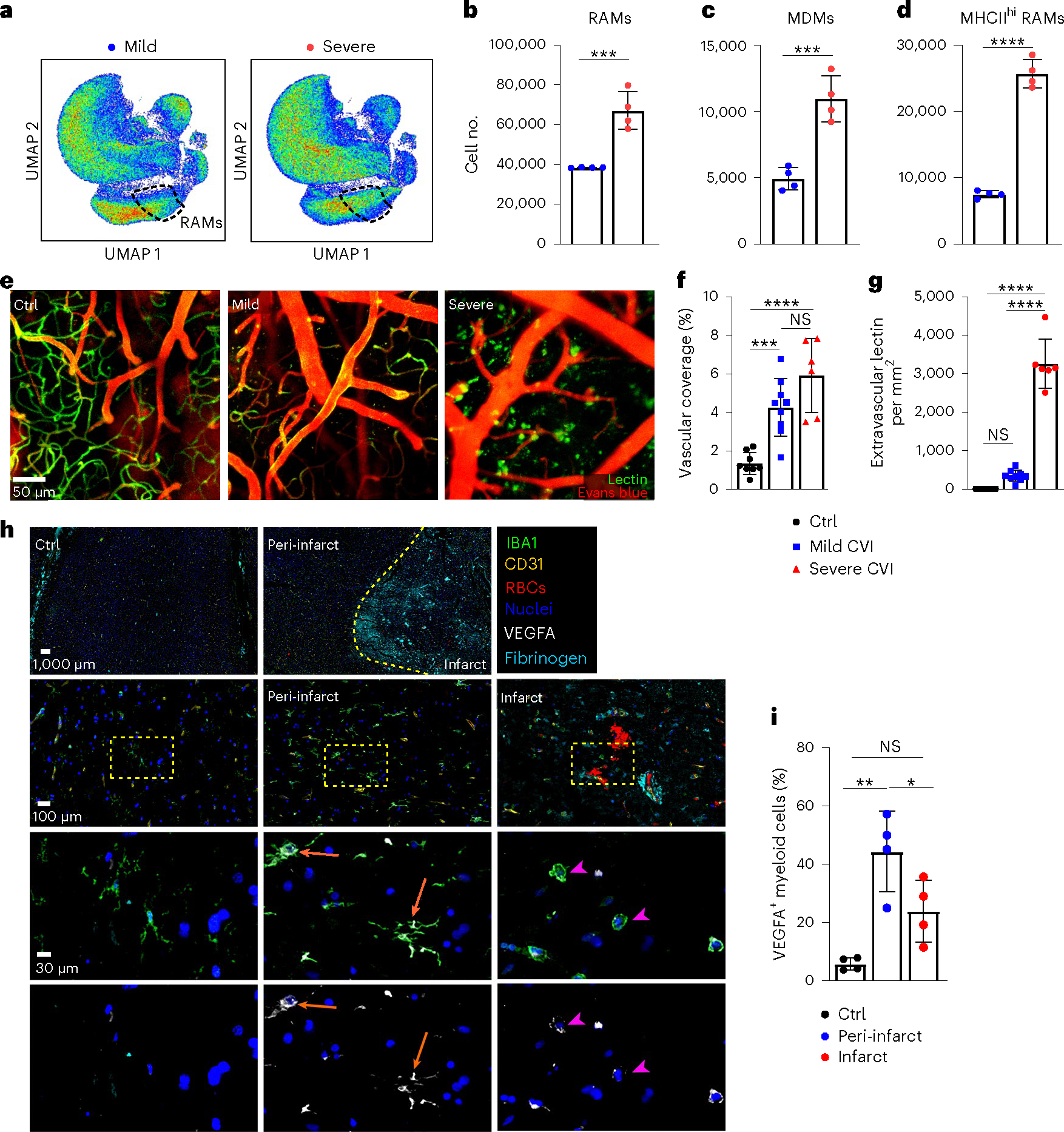
a, Representative UMAPs depicting high-parameter flow cytometry data of leukocytes isolated from the brains of day 6 mild (n = 4) versus severe (n = 4) CVI Tmem119-TdTo mice by punch biopsy. The dashed lines denote RAMs. b–d, Bar graphs show the absolute number of RAMs (TdTo+VEGFA+IL-6Ra+CD24+CD44+CD206+CD11c+Ly6clo/intMHCIIloCX3CR1+CD11b+CD45lo/int) (b), MHCIIhi RAMs (TdTo+VEGFA+IL-6Ra+CD24+CD44+CD206+CD11c+Ly6c+MHCIIhiCX3 CR1+CD11b+CD45+) (c) and MDMs (TdTo−CD24+CD44+CD206+CD11c+Ly6c+CX3CR 1+CD11b+CD45+) (d) for the groups indicated in a. Data represent two independent experiments. ***P < 0.0005, ****P < 0.0001; two-tailed, unpaired Student’s t-test. e, Representative maximal projection images captured by intravital TPM showing vasculature in the superficial neocortex of uninjured Ctrl (n = 8) versus day 10 mild (n = 9) and severe (n = 6) CVI mice. Tomato lectin (green) and EB (red) were injected i.v. before imaging. f,g, Bar graphs showing quantification of percentage vascular coverage (f) and the amount of extravasated tomato lectin (g) in the mice from e. Data are combined from two independent experiments. ***P < 0.0005, ****P < 0.0001; one-way ANOVA with Tukey’s test. In b–g, the dots represent individual animals. h, Representative multiplex immunostaining of human stroke (n = 4) brain tissue (patient ID AN-060–37) versus Ctrl (n = 4) brain tissue (patient ID 85747) without neurological disease (Supplementary Table 1). Tissue was stained with antibodies directed against VEGFA (white), IBA1 (green), RBCs, CD31 (yellow) and fibrinogen (cyan). Cell nuclei are blue. The dashed yellow line denotes a border between infarct core and peri-infarct penumbrae. The dotted yellow boxes denote the location of enlarged images below. The orange arrows point to IBA1+ cells with a ramified microglial morphology that express VEGFA in peri-infarct stroke tissue. The pink arrowheads denote spherical IBA1+ cells expressing low amounts of VEGFA in the stroke lesion core. i, Bar graph showing quantification of the percentage VEGFA+IBA1+ myeloid cells in Ctrl, peri-infarct and infarct ROIs from Ctrl (n = 4) and stroke (n = 4) brain tissues. Individual dots denote the average of percentage VEGFA+IBA1+ myeloid cells from one to three ROIs per specimen analyzed (see Extended Data Fig. 2f). *P < 0.05, **P < 0.005; one-way ANOVA with Tukey’s test. All graphs show mean ± s.e.m.
VEGFA-expressing myeloid cells are found in human stroke brain tissue
To establish the relevance of our findings and determine whether RAM-like cells emerge in the human brain after a CVI, we examined brain tissue from stroke patients versus neurologically healthy controls for the presence of VEGFA+ myeloid cells using multiplex immunostaining (Fig. 2h,i, Extended Data Fig. 2f and Supplementary Table 1). Parenchymal IBA1+ myeloid cells in control brain tissue were ramified and had small-cell bodies, resembling naive microglia. These cells expressed VEGFA at a low level, if present, and the vasculature in this tissue was determined to be anatomically intact based on the confinement of red blood cells (RBCs) and fibrinogen within CD31+ vessels (Fig. 2h(left),i). By contrast, the core infarct region of brain tissue from stroke patients showed profound evidence of vascular disruption, including the presence of RBCs and fibrinogen throughout the associated parenchyma (Fig. 2h(right)). IBA1+ myeloid cells within the core were spherical in morphology and ~24% of those were positive for VEGFA (Fig. 2h(right),i). However, in peri-infarct areas (penumbrae), VEGFA was expressed in the cell bodies and/or processes of ~47% of ramified IBA1+ cells resembling microglia (Fig. 2h(middle),i). Collectively, these data suggest that microglia acquire proangiogenic properties after a stroke in humans.
Infiltrating inflammatory monocytes promote RAM generation
Given that proangiogenic microglia develop after a CVI in mice and humans, we were next interested in whether infiltrating peripheral immune cells are required to endow microglia with this property. Monocytes are the main infiltrating immune cells found in the injured brain parenchyma after a CVI (Fig. 3a–e and Extended Data Fig. 1f–h). We observed that the frequency of Tmem119-TdTomato−Iba1+ myeloid cells increased significantly in the brain at day 6 relative to uninjured controls (Fig. 3a,b). In addition, intravital TPM showed evidence of Ly6C+ inflammatory monocytes extravasating directly from parenchymal blood vessels into the CVI-injured brain (Fig. 3c and Supplementary Video 2). These data were supported by analyses of brain sections demonstrating that the number of infiltrating CCR2+ monocytes rose between days 1 and 3 post-CVI and then decreased slightly by day 6 (Fig. 3d,e). To evaluate the role of monocyte infiltration in RAM generation, we utilized Ccr2−/− mice that have a reduced number of classic monocytes in the blood as well as impaired monocyte recruitment to sites of inflammation32,33. Flow cytometric analyses revealed that Ccr2−/− mice have a significant reduction in RAM (~60% reduction) and MDM (~80% reduction) numbers relative to wild-type (WT) controls at day 6 post-CVI (Fig. 3f–h), suggesting that proinflammatory monocytes play an important role in RAM generation.
Fig. 3 |. Infiltrating monocytes produce IL-6 and are critical for RAM generation.
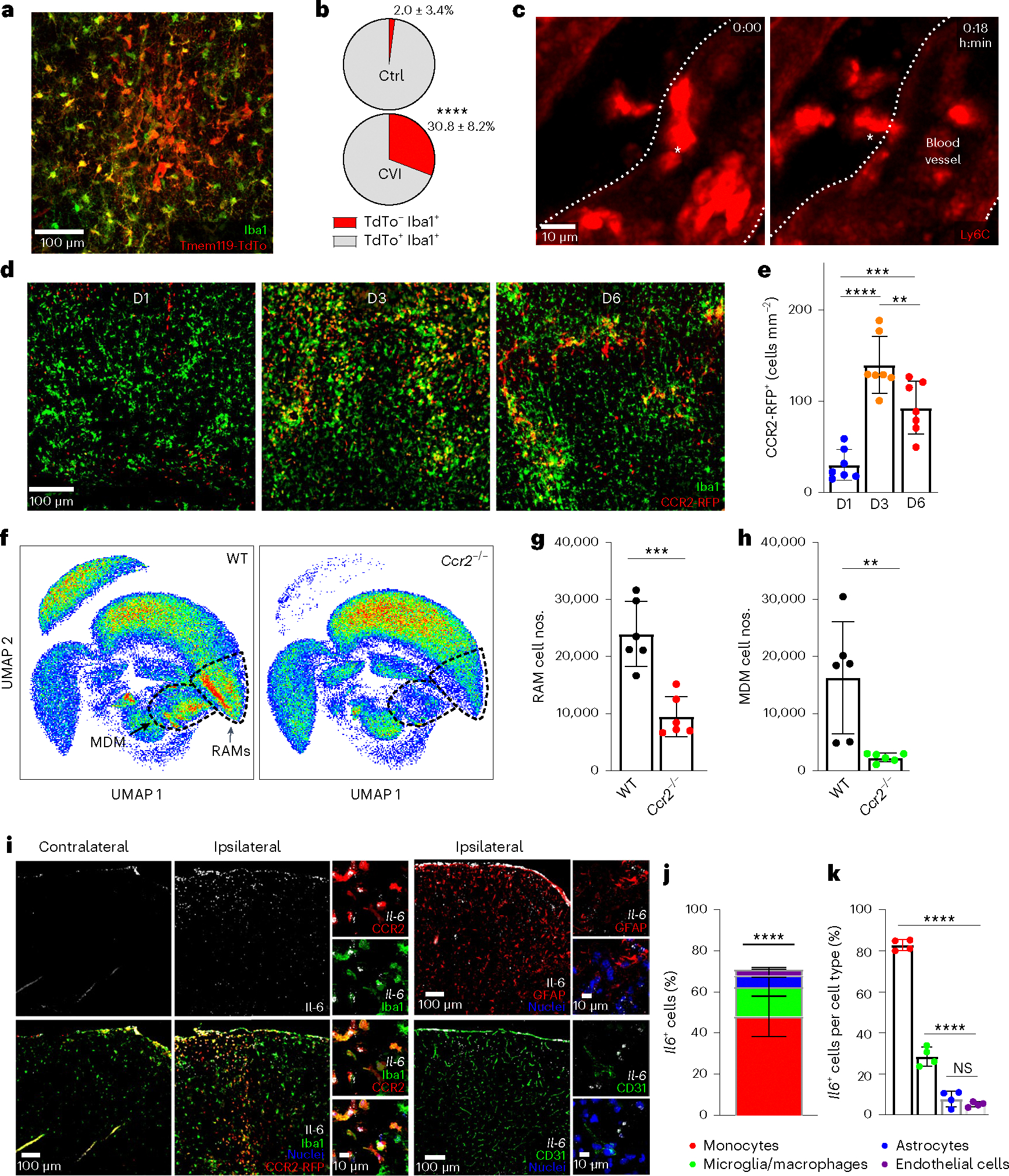
a, Immunofluorescent staining for Iba1 (green) and TdTo (red) in the neocortex of a Tmem119-TdTo mouse on day 6 post-CVI. Data represent two independent experiments. b, Pie charts depicting the proportion of TdTo−Iba1+ (monocytes/macrophages, red) and TdTo+ Iba1+ (microglia, gray) in the brains of uninjured Ctrl (n = 8) versus injured (n = 7) mice. Data represent two independent experiments. ****P < 0.0001; two-tailed, unpaired Student’s t-test. c, Time-lapse intravital TPM images showing monocyte extravasation into the parenchyma at day 2 post-CVI (n = 3 mice). The white asterisk denotes a Ly6c+ monocyte (red) extravasating from a blood vessel (demarcated by white dotted lines) into the parenchyma (Supplementary Video 2). d, Immunofluorescent staining for Iba1 (green) and RFP (red) in brains of CX3CR1gfp/+CCR2rfp/+ (n = 7 mice per group) at days 1, 3 and 6 (D1, D3, D6) post-CVI. Green fluorescent protein (GFP) and RFP were photobleached before anti-Iba1 and RFP staining. e, Bar graph depicting the absolute number of CCR2-RFP+ monocytes per mm2 of brain tissue at the denoted time points after a CVI. Combined data from two independent experiments are shown in d and e. **P < 0.005, ***P < 0.0005, ****P < 0.0001; one-way ANOVA with Tukey’s test. f, UMAPs showing high-parameter flow cytometric data of leukocytes isolated via punch biopsy from WT (n = 6) and Ccr2−/− (n = 6) mice on day 6 post-CVI. The dashed lines delineate RAMs and MDMs. g,h, Bar graphs showing the absolute number of RAMs (g) and MDMs (h) in WT versus Ccr2−/− mice derived from f. The cell cluster in the top left of the UMAPs corresponds to CD45− nonimmune cells. Data represent two independent experiments. **P < 0.005, ***P < 0.0005; two-tailed, unpaired Student’s t-test. i, Representative images of RNAscope in situ hybridization for Il-6 transcripts (white) combined with immunofluorescent staining for Iba1, RFP, GFAP and CD31 in ipsilateral (injured) versus contralateral (uninjured) neocortex of CX3CR1gfp/+CCR2rfp/+ mice (n = 4) at day 3 post-CVI. GFP and RFP were photobleached before anti-Iba1 and RFP staining. The following cell populations were identified: microglia/macrophages (Iba1+CCR2-RFP−), monocytes (CCR2-RFP+), astrocytes (GFAP+) and endothelial cells (CD31+). Cell nuclei are shown in blue. j,k, Bar graphs showing the percentage of each cell type expressing Il-6 (j) and the percentage of Il-6-expressing cells within each cell type (k) from the mice described in i. Combined data from two independent experiments are shown in i–k. The asterisks in j denote a statistically significant difference between monocytes and all other IL-6-expressing cells: ****P < 0.0001; one-way ANOVA with Tukey’s test. All graphs in this figure show mean ± s.e.m. The dots represent individual animals.
IL-6 signaling is required for RAM generation
Infiltrating innate immune cells can modulate the function of tissue resident cells via the production of cytokines. IL-6 is a proinflammatory cytokine known to be rapidly expressed after infections and injuries34. We therefore quantified cells expressing Il-6 transcripts in CVI-damaged brain tissue at day 3—a time point preceding the cerebrovascular remodeling phase. At this time point, IL-6 transcripts were prominent throughout the injured ipsilateral hemisphere but not the uninjured contralateral hemisphere of mice (Fig. 3i). Colabeling studies revealed that infiltrating CCR2-RFP+Iba1+ monocytes were the primary source of Il-6, accounting for ~48% of the Il-6-expressing cells followed by macrophages/microglia (CCR2-RFP−Iba1+, ~15%), astrocytes (GFAP+, ~6%) and endothelial cells (CD31+, ~3%) (Fig. 3j). Within specific cell types, ~83% of monocytes expressed Il-6 (Fig. 3k). Collectively, these data demonstrate that infiltrating monocytes are the major source for IL-6 after a CVI.
To gain additional insights into RAM programming and the contribution of IL-6 signaling to this programming, we analyzed purified CD11b+ myeloid cells obtained from the cortex of WT versus Il6−/− mice by single-cell RNA-sequencing (scRNA-seq) at day 6 post-CVI (Fig. 4a and Supplementary Tables 6–26). This sequencing experiment yielded a total of 9,027 single cells. Unsupervised partition analysis uncovered seven partitions (Ps) and cellular identities were assigned based on canonical cell-type gene expression, revealing populations of microglia (P1, P2, P3, P6 and P7), monocytes/macrophages (P4) and contaminating endothelial cells (P5) (Fig. 4b,c). CVI induced notable changes in the transcriptional profiles of WT myeloid cells compared with uninjured WT and injured Il6−/− cells (Extended Data Fig. 3a–d). Myeloid cell P1–P4 each contained enough cells to allow for more in-depth analyses (Extended Data Fig. 3c). A heatmap of P1 differentially expressed genes (DEGs) showed a profound upregulation of gene expression in microglia from CVI mice relative to uninjured controls (Fig. 4d). To examine the influence of IL-6 on microglial responses during CVI, Enrichr35 was used to evaluate DEGs between WT versus Il6−/− CVI P1 microglia. Gene set enrichment analyses (GSEAs) showed an elevated expression of angiogenesis genes (Spp1, Pf4 and Igf1) in WT microglia compared with Il6−/− microglia (Fig. 4e and Extended Data Fig. 3e–g). In addition, based on expression of ten classic proangiogenic factors (Vegfa, Vegfb, Il6, Fgf2, Pdgfa, Pdgfb, Hgf, Igf1, Igf2 and Tnf)36, microglia within P1 and P6 have the greatest proangiogenic potential (Fig. 4f,g). Importantly, this expression module was reduced after a CVI in Il6−/− mice relative to WT controls (Fig. 4h).
Fig. 4 |. ScRNA-seq reveals that IL-6 deficiency impairs proangiogenic microglial programming.
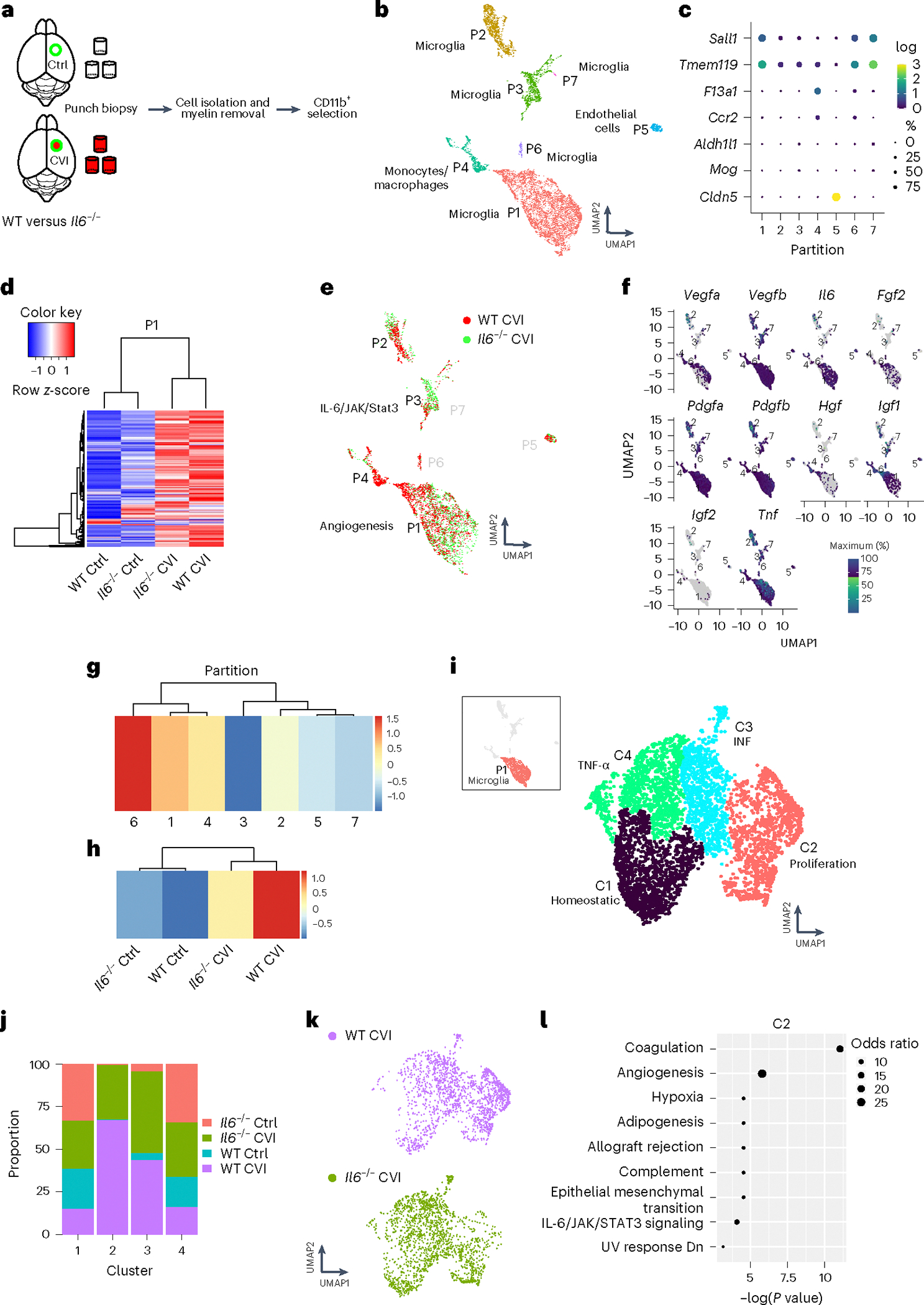
a, Schematic of how purified single-cell suspensions of CD11b+ cells were isolated for scRNA-seq via punch biopsy from the neocortex of WT versus Il6−/− mice at day 6 post-CVI. b, The UMAP depicts 7 different partitions based on 9,027 cells processed by scRNA-seq. Each dot represents a single cell and each color denotes a partition that is labeled with the corresponding cell type. c, Dot plots showing differential expression of canonical cell type markers for each partition. The dot size represents the percentage of cells within the partition that express the denoted gene and the color reflects the average expression level. d, A Pearson’s correlation-based clustered heatmap showing DEGs (q < 0.05) in P1 among uninjured (Ctrl) versus injured WT and Il6−/− mice. e, UMAP showing an all-partition comparison of WT versus Il6−/− CVI mice. The top pathway expressed in WT versus Il6−/− CVI based on DEGs is shown for P1 and P3. f, Expression of classic proangiogenic factors shown in each partition from all groups combined. g,h, Heatmaps showing the aggregated expression of the ten proangiogenic genes (denoted in f) per partition (g) and per experimental group (h). i, UMAP depicting drill-down analysis of P1, which revealed four clusters labeled with different colors. The top weighted signaling pathway for each cluster is listed. These pathways were identified using GSEA of DEGs (q < 0.05) between injured and uninjured mice. j, Bar graph showing the proportion of cells in each cluster of P1 that are attributed to the four denoted groups of mice. k, UMAPs depicting a side-by-side comparison of microglia P1 for WT versus Il6−/− CVI mice. l, Pathways revealed by GSEA of DEGs (q < 0.05) between WT and Il6−/− CVI mice in microglial P1 C2. Dot size corresponds to the odds ratio. Only statistically significant pathways (P < 0.05) are shown on the graph. Pathways are ordered based on −log(P value) multiplied by the corresponding z-score.
To construct a more detailed P1 microglial activation map, we performed drill-down cluster analysis (K = 9), which yielded four clusters (Cs) within P1 (Fig. 4i). C1 represented ~57% of the microglia in uninjured WT controls and, based on gene expression, were designated as homeostatic microglia. To identify clusters within P1 that had a proangiogenic gene expression signature, we first analyzed differential expression in each cluster relative to all the other clusters in the partition. GSEAs using DEGs from this differential testing uncovered upregulation of genes associated with mitotic spindle assembly (Trio, Numa1 and Epb41) in C2, IFN signaling (Iftm3, Rsad2 and Ifit3) in C3 and a TNF-α response (Gch1, Tnfaip2 and Tnf) in C4 (Fig. 4i and Extended Data Fig. 3h–j). Moreover, many of the proangiogenic genes in P1 (Fig. 4e and Extended Data Fig. 3e–g) were elevated in C2 compared with C1, C3 and C4 (Extended Data Fig. 3k,l), suggesting that C2 contains proangiogenic microglia. C2 microglia were found almost exclusively in the brains of injured mice and their proportion increased by a factor of 2 in WT (890 out of 1,776 total P1 cells) versus Il6−/− (424 out of 1,765 total P1 cells) mice (Fig. 4j,k). There were also proportionally fewer homeostatic C1 microglia in WT relative to Il6−/− CVI mice, suggesting reduced activation in Il6 knockouts (Fig. 4j,k). Further analysis of WT versus Il6−/− DEGs in C2 microglia after CVI revealed increased expression of coagulation (Apoc2, Apoc1 and Mmp8) and angiogenesis response genes (Spp1, Pf4 and Igf1) in WT cells (Fig. 4l), whereas few DEGs were observed in C3 and C4 CVI microglia between WT and Il6−/− mice.
Based on DEG expression, P2 and P3 microglia also showed evidence of activation in injured versus uninjured WT mice. Among the top upregulated pathways in both microglia clusters from WT CVI mice were IL-6/JAK/STAT3-mediated signaling (Stat1, Il1b and Tlr2) and IFN responses (Iftm2, Rtp4 and Ifi35) (Extended Data Fig. 4a,b). Notably, genes involved in IL-6 production and/or IL-6 signaling were upregulated in microglia P1, P2 and P3 from injured versus uninjured WT mice (Extended Data Fig. 4c). In addition, trajectory analysis revealed that P1, P2 and P3 represent three distinct transcriptional profiles (Extended Data Fig. 4d). P4 consisted primarily of monocytes and MDMs (Extended Data Fig. 5). Heatmap analysis demonstrated profound differences in P4 gene expression in CVI mice relative to the other groups (Extended Data Fig. 5c). Examination of differentially regulated pathways between Il6−/− and WT CVI mice revealed upregulated TNF-α-mediated signaling in the absence of IL-6 (Extended Data Fig. 5d–f). Together these data indicate that CVI promotes complex remodeling of the myeloid cell landscape in the brain that is mediated in part by IL-6 signaling. Three key features of this program in microglia are the induction of gene-associated IL-6 signaling, proliferation and angiogenesis.
Given the influence of IL-6 on microglia programming after CVI, we next assessed whether this cytokine plays a role in RAM generation. To do this, we performed a flow cytometric study in WT versus Il6−/− mice at day 6 post-CVI and included Ccr2−/− mice as a positive control for deficient RAM generation. Relative to WT, Il6−/− mice showed a significant reduction in RAM (Fig. 5b) and MDM (Fig. 5c) numbers comparable to that observed in Ccr2−/− mice. To test whether monocyte-derived IL-6 is required for RAM generation, we adoptively transferred monocytes (CD11b+Ly6C+CX3CR1+CD3−Siglec F−NK1.1−Ly6G−CD117−CD220−) obtained from the bone marrow of WT or Il6−/− mice into Ccr2−/− mice on days 0–4 post-CVI and quantified RAM generation on day 6 (Fig. 5a and Extended Data Fig. 5g). WT and Il6−/− monocytes infiltrated the injured brain after adoptive transfer and elevated the number of MDMs (Fig. 5e). However, only WT monocytes were able to significantly increase RAM numbers at day 6 post-CVI. IL-6-deficient monocytes failed to promote RAM generation and these recipients had RAM numbers comparable to those observed in uninjured controls or injured Ccr2−/− mice that received no monocytes (Fig. 5d,e). These data indicate that monocyte-derived IL-6 is required for RAM generation.
Fig. 5 |. Monocytes and IL-6 are required for RAM generation, vascular repair and functional recovery.
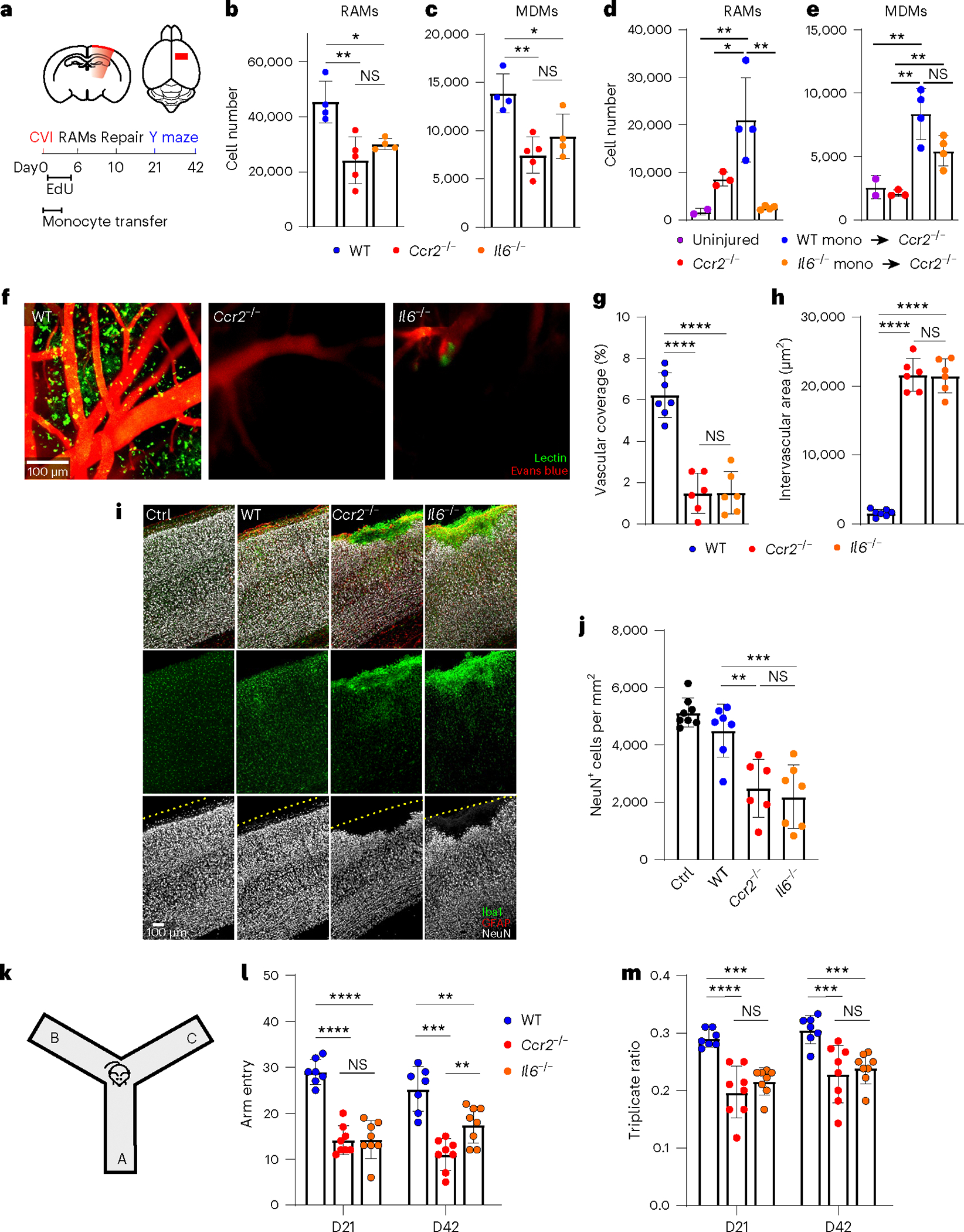
a, Schematic depicting the experimental timeline. b,c, Bar graphs depicting the absolute number of RAMs (b) and MDMs (c) obtained from cortical punch biopsies of WT (n = 4), Ccr2−/− (n = 5) and Il6−/− (n = 4) mice at day 6 post-CVI. Data represent two independent experiments. *P < 0.05, **P < 0.005; one-way ANOVA with Tukey’s test. d,e, Bar graphs showing the absolute number of RAMs (d) and MDMs (e) obtained from cortical punch biopsies of uninjured Ccr2−/− mice (n = 2) as well as Ccr2−/− mice at day 6 post-CVI that received no transfer (n = 3) or adoptive transfer of WT (n = 4) versus Il6−/− (n = 4) monocytes. Data represent two independent experiments. *P < 0.05, **P < 0.005; one-way ANOVA with Tukey’s test. f, Representative intravital TPM images captured through the thinned skull of WT (n = 7), Ccr2−/− (n = 6) and Il6−/− (n = 6) mice at day 10 post-CVI. Tomato lectin (green) and EB (red) were injected i.v. to visualize vasculature. g,h, Bar graphs showing quantification of vascular coverage (g) and intervascular area (h) in the denoted groups of mice in f. Data are combined from two independent experiments. ****P < 0.0001; one-way ANOVA with Tukey’s test. i, Immunofluorescent staining for Iba1 (green), GFAP (red) and NeuN (white) in neocortex from uninjured WT mice (Ctrl; n = 8), as well as WT (n = 7), Ccr2−/− (n = 6) and Il6−/− (n = 7) mice at day 10 post-CVI. The dashed yellow lines denote the brain surface. j, Bar graph showing the number of NeuN+ neurons per mm2 of tissue for the denoted groups in i. Data are combined from two independent experiments. **P < 0.005, ***P < 0.0005; one-way ANOVA with Tukey’s test. k, Schematic showing Y-maze behavioral test used to evaluate mouse cognitive–motor function. l,m, Bar graphs demonstrating the total number of Y-maze arm entries (l) and the triplicate ratio (m) for WT (n = 8), Ccr2−/− (n = 8) and Il6−/− (n = 7) at days 21 and 42 post-CVI. Combined data from two independent experiments are shown in l and m. **P < 0.005, ***P < 0.0005, ****P < 0.0001; two-way ANOVA with Holm–Šidák test. All graphs show mean ± s.e.m. The dots represent individual animals.
As a result of our scRNA-seq data suggesting that IL-6 signaling contributed to microglia proliferation, we pulsed WT, Il6−/− and Ccr2−/− mice with 5-ethynyl-2′-deoxyuridine (EdU) from day 1 to day 5 post-CVI to label proliferating cells (Fig. 5a). The number of EdU+Iba1+ myeloid cells was significantly reduced to a comparable degree in the brains of Il6−/− and Ccr2−/− mice at day 6 post-CVI relative to WT controls (Extended Data Fig. 6a,b). Myeloid cell proliferation was largely attributed to microglia (EdU+Tmem119-TdTomato+) rather than infiltrating monocytes (EdU+CCR2-RFP+) (Extended Data Fig. 6e–g). We also observed a marked decrease in EdU+CD31+ endothelial cells in both Il6−/− and Ccr2−/− mice when compared with WT controls (Extended Data Fig. 6c,d). These data demonstrate that IL-6 promotes RAM generation as well as microglial and endothelial cell proliferation.
IL-6 deficiency impedes cerebrovascular repair and neurological recovery after CVI
Given the functional role of IL-6 in RAM generation, we evaluated whether cerebrovascular repair and functional recovery were impeded in Il6−/− mice after CVI. In WT mice, we previously observed significant vascular remodeling in CVI-damaged brain tissue by day 10 (Fig. 2e–g) and functional improvement in a Y-maze assay by day 20 (ref. 18). However, Il6−/− and Ccr2−/− mice showed a comparable reduction in vascular coverage at day 10 post-severe CVI (Fig. 5f,g) and a corresponding increase in intervascular area (the space between blood vessels; Fig. 5f,h), demonstrating that vascular remodeling failed in these mice relative to WT controls. We also performed a similar study using our mild model of CVI, which generates less severe vascular damage (Fig. 2e–g). Quantification of extravasated tomato lectin and EB as measures of BBB breakdown at day 10 post-CVI revealed that WT mice had markedly improved BBB integrity by this time point, whereas vascular barriers in Il6−/− and Ccr2−/− mice continued to leak (Extended Data Fig. 6h–k). Moreover, the failed vascular remodeling and leaky barriers in Il6−/− and Ccr2−/− mice were associated with fibrosis consisting of substantial type 1 collagen deposits (Extended Data Fig. 7a–d) as well as a marked reduction in NeuN+ neurons (Fig. 5i,j). Functional recovery as assessed by Y maze was also impaired in Il6−/− and Ccr2−/− mice (Fig. 5k–m), although a slight improvement in arm entries (a measure of motor function) was observed in Il6−/− mice at day 42 post-CVI. Uninjured WT, Il6−/− and Ccr2−/− mice all performed comparably in the Y-maze assay (Extended Data Fig. 7e,f). These data demonstrate that IL-6 and monocyte deficiency lead to faulty cerebrovascular repair, neuronal damage, fibrosis and impaired functional recovery after a CVI.
Microglial IL-6 signaling is required for RAM generation and vascular repair
To determine whether IL-6 is required specifically in microglia for RAM generation and vascular repair, we used a conditional knockout approach by generating Tmem119-CreERT2 Il-6rafl/fl mice (hereafter referred to as ΔmIL-6Ra; Fig. 6a). Mice with Tmem119-CreERT2, Il-6rafl/fl or Il-6rafl/+ alleles were all used as controls (Ctrl IL-6Ra). No differences were observed among any of the different control mice. After 1 month on tamoxifen chow and a 3-week rest period on regular chow, we observed, by flow cytometry, that membrane-bound IL-6Ra was reduced by ~50% on brain microglia from ΔmIL-6Ra mice relative to Ctrl IL-6Ra mice at day 6 post-CVI (Fig. 6b). In addition, flow cytometric studies at day 6 post-CVI revealed that ΔmIL-6Ra mice had ~53% in RAM numbers compared with Ctrl IL-6Ra mice (Fig. 6c), whereas no statistically significant difference in the number of MDMs was observed between the two groups (Fig. 6d), suggesting that microglial IL-6 signaling is required for RAM generation. To examine the contribution of microglial IL-6 signaling to vascular repair, we quantified vascular coverage and integrity between ΔmIL-6Ra and Ctrl IL-6Ra mice at day 10 post-CVI (Fig. 6e–g). The ΔmIL-6Ra mice showed a significant reduction in vascular coverage and a marked increase in intervascular area relative to Ctrl IL-6Ra mice (Fig. 6e–g). Supporting these data, ΔmIL-6Ra mice also had reduced BBB integrity as evidenced by EB leakage (Fig. 6h,i) and a reduced number of NeuN+ neurons (Fig. 6j,k). In addition, when assessed by Y maze, ΔmIL-6Ra mice showed a significant reduction in arm entries and triplicate ratio compared with Ctrl IL-6Ra mice at day 21 post-CVI (Fig. 6l,m). Together these data demonstrate that microglial IL-6 signaling plays an important role in RAM generation, cerebrovascular repair, neuronal preservation and functional restoration after CVI.
Fig. 6 |. Microglial IL-6 signaling contributes to RAM generation and vascular repair.
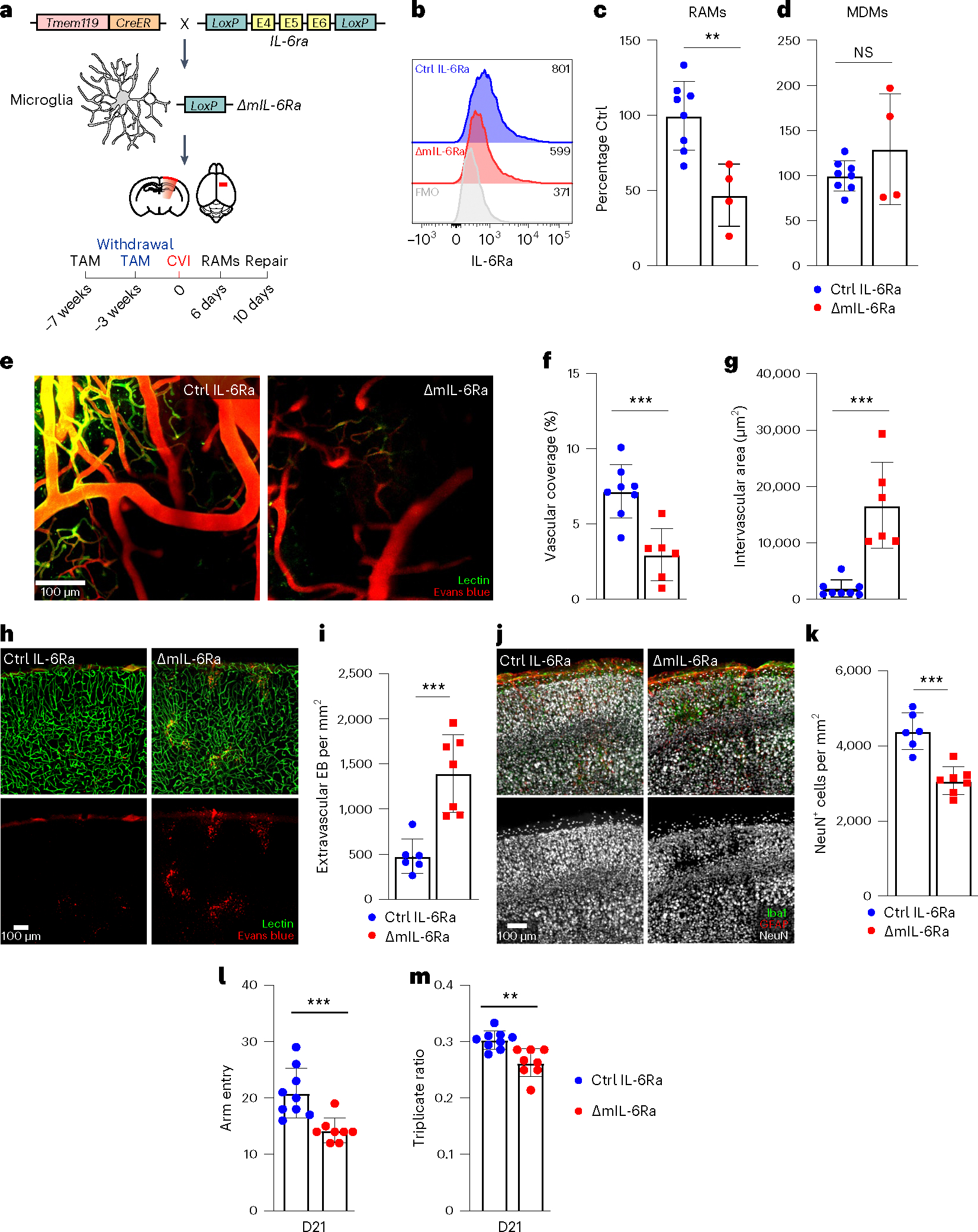
a, Schematic showing the experimental timeline and strategy used conditionally to delete IL-6Ra from microglia (ΔmIL-6Ra mice). b, Histograms showing the expression of membrane-bound IL-6Ra on CD45+CD11b+ cells obtained from the brains of Ctrl IL-6Ra (n = 3) and ΔmIL-6Ra (n = 1) mice on day 6 post-CVI relative to the FMO control (gray). Values in the histograms represent the median fluorescent intensity for each condition. Data represent two independent experiments. c,d, Bar graphs showing the percentage of RAMs (TdTo+VEGFA+IL-6Ra+CD24+CD44+CD206+CD11c+Ly6clo/int MHCIIloCX3CR1+CD11b+CD45lo/int) (c) and MDMs (TdTo−CD24+CD44+CD206+C D11c+Ly6c+CX3CR1+CD11b+CD45+) (d) determined by flow cytometric analysis of leukocytes isolated from cortical punch biopsies of ΔmIL-6Ra mice (n = 4) percentage normalized to their littermate Ctrl counterparts (n = 8) at day 6 post-CVI. Data are combined from three independent experiments. **P < 0.005; two-tailed, unpaired Student’s t-test. e, Representative TPM images showing vasculature through the thinned skull of Ctrl IL-6Ra (n = 8) versus ΔmIL-6Ra (n = 6) mice at day 10 post-CVI. Lectin (green) and EB (red) were injected i.v. to visualize vasculature. f,g, Bar graphs showing quantification of vascular coverage (f) and intervascular area (g) in the denoted groups in e. Data are combined from two independent experiments. ***P < 0.0005; two-tailed unpaired Student’s t-test. h, Representative confocal images of neocortex from Ctrl IL-6Ra (n = 6) versus ΔmIL-6Ra (n = 7) mice on day 10 post-CVI. Lectin (green) and EB (red) were injected i.v. to visualize vessels and measure BBB integrity. i, Bar graph showing the quantity of extravascular EB per mm2 of tissue in the denoted mice in h. Data are combined from two independent experiments. ***P < 0.0005; two-tailed, unpaired Student’s t-test. j, Representative confocal images of the neocortex from Ctrl IL-6Ra (n = 6) versus ΔmIL-6Ra (n = 7) mice at day 10 post-CVI stained for Iba1 (green), GFAP (red) and NeuN (white). k, Bar graph demonstrating the number of NeuN+ neurons per mm2 of tissue in the denoted mice in j. Data are combined from two independent experiments. ***P < 0.0005; two-tailed, unpaired Student’s t-test. l,m, Bar graphs depicting the total number of Y-maze arm entries (l) and the triplicate ratio (m) for Ctrl IL-6Ra (n = 9) and ΔmIL-6Ra (n = 8) mice at day 21 post-CVI. Combined data from two independent experiments are shown. **P < 0.005; two-tailed, unpaired Student’s t-test. All graphs show mean ± s.e.m. The dots represent individual animals.
Exogenous IL-6 compensates for monocyte deficiency and restores cerebrovascular repair
We established that CCR2+ monocyte infiltration and IL-6 signaling in microglia are important for RAM generation and cerebrovascular repair. However, it remained possible that CCR2+ monocytes contributed to this repair process via mechanisms not involving IL-6. We therefore addressed whether exogenous IL-6 could compensate for the reduction in inflammatory monocytes observed in Ccr2−/− mice. Recombinant IL-6 (rIL-6) was administered transcranially and i.v. daily from day 1 to day 5 post-CVI in Ccr2−/− mice, and we quantified RAM generation on day 6 (Fig. 7a). Vehicle-treated WT mice served as a positive control for RAM generation and successful cerebrovascular remodeling in this line of experimentation. At day 6 post-CVI, vehicle-treated Ccr2−/− mice showed an expected reduction in RAM numbers, whereas rIL-6 administration increased their numbers to a level almost similar to that observed in WT mice without significantly elevating the number of MDMs (Fig. 7b,c). To assess vascular repair, we administered rIL-6 daily from day 1 to day 9 post-CVI and quantified vascular coverage at day 10 (Fig. 7a). This rIL-6 treatment significantly improved the extent of parenchymal revascularization in Ccr2−/− mice relative to vehicle-treated controls, which showed very little vascular repair relative to WT mice (Fig. 7d–f). Treatment over this same time window also reduced the amount of NeuN+ neuronal loss (Fig. 7g,h) and improved neurological function, as assessed via the Y-maze test (Fig. 7i,j). Importantly, rIL-6-treated Ccr2−/− mice demonstrated a partial improvement in locomotive function (Fig. 7i) and a complete restoration of cognitive function (Fig. 7j) by day 21 post-CVI. This improvement in function was maintained at day 42, demonstrating a lasting functional recovery promoted by rIL-6 (Fig. 7i,j). Collectively, these data indicate that exogenous IL-6 can restore RAM programming, improve vascular repair and promote neurological recovery in inflammatory monocyte-deficient mice.
Fig. 7 |. Exogenous IL-6 restores RAM, vascular repair and functional recovery in Ccr2−/− mice.
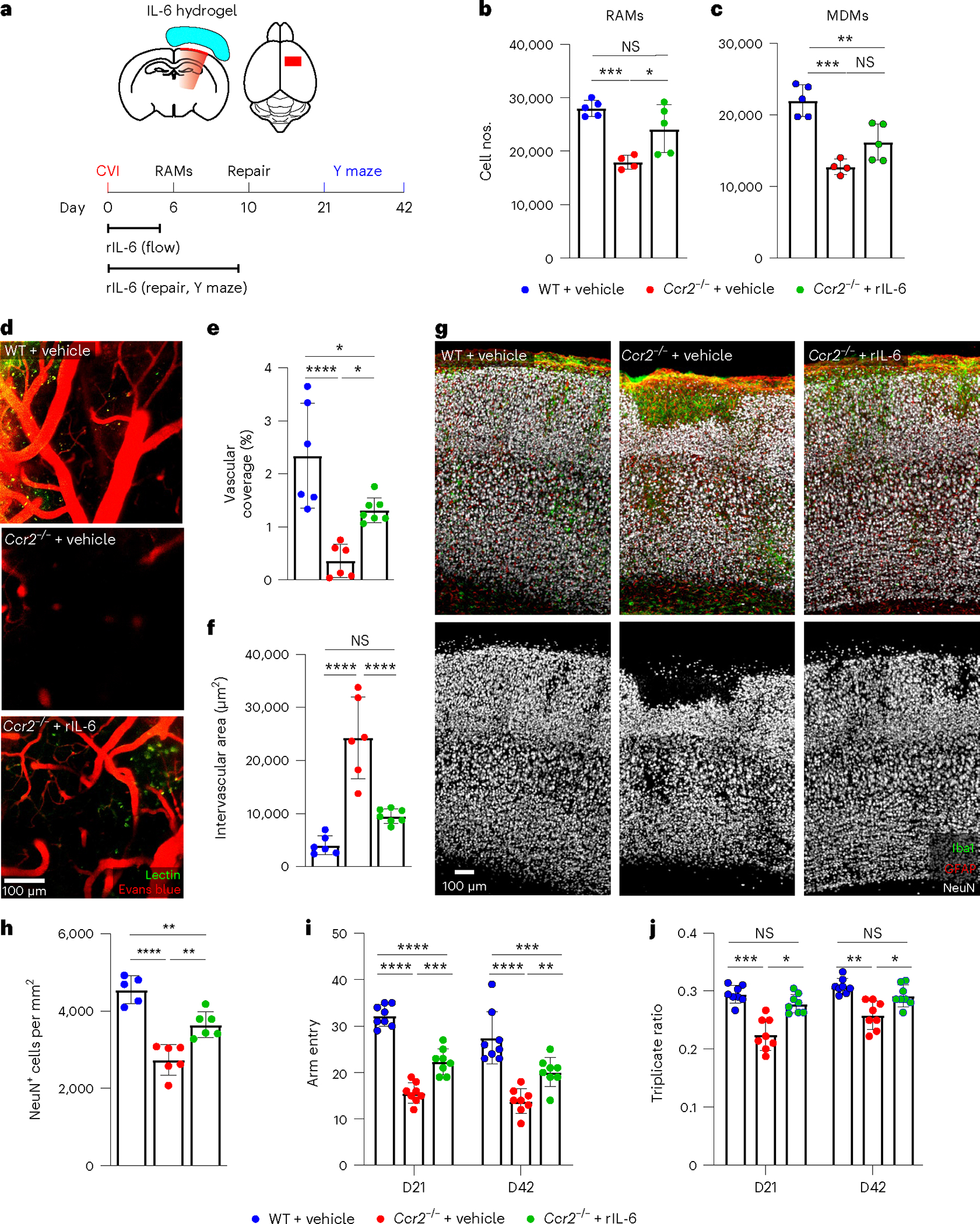
a, Schematic showing experimental timeline and transcranial application of rIL-6. b,c, Bar graph showing the absolute number of RAMs (b) and MDMs (c) determined by flow cytometric analysis of leukocytes isolated from cortical punch biopsies of WT + vehicle (n = 5), Ccr2−/− + vehicle (n = 4) and Ccr2−/− + rIL-6 (n = 5) at day 6 post-CVI. Data represent two independent experiments. *P < 0.05, **P < 0.005, ***P < 0.0005; one-way ANOVA with Tukey’s multiple comparisons test. d, Representative intravital TPM images captured through the thinned skull of WT + vehicle (n = 6), Ccr2−/− + vehicle (n = 6) and Ccr2−/− + rIL-6 (n = 7) at day 10 post-CVI. Tomato lectin (green) and EB (red) were injected i.v. to visualize vasculature. e,f, Bar graphs showing quantification of vascular coverage (e) and intervascular area (f) in the denoted groups of mice in d. Data are combined from two independent experiments. *P < 0.05, ****P < 0.0001; one-way ANOVA with Tukey’s multiple comparison test. g, Immunofluorescent staining for Iba1 (green), GFAP (red) and NeuN (white) in the neocortex from WT + vehicle (n = 5), Ccr2−/− + vehicle (n = 6) and Ccr2−/− + rIL-6 (n = 6) mice at day 10 post-CVI. h, Bar graph showing the number of NeuN+ neurons per mm2 of tissue for the denoted groups in g. Data are combined from two independent experiments. **P < 0.005, ****P < 0.0001; one-way ANOVA with Tukey’s test. i,j, Bar graphs demonstrating the total number of Y-maze arm entries (i) and the triplicate ratio (j) for WT + vehicle (n = 8), Ccr2−/− + vehicle (n = 8) and Ccr2−/− + rIL-6 (n = 8) at days 21 and 42 post-CVI. Data are combined from two independent experiments. *P < 0.05, **P < 0.005,***P < 0.0005, ****P < 0.0001; two-way ANOVA with Tukey’s multiple comparisons test. All graphs show mean ± s.e.m. The dots represent individual animals.
Discussion
In the present study, we uncovered a monocyte–microglia dialog that is crucial for cerebrovascular repair and functional brain recovery after a CVI. Our data demonstrate that proangiogenic RAMs are an important part of the intrinsic brain repair program that is established in response to cerebrovascular injuries in humans and mice. Using a murine CVI model, we established that monocytes are the major peripheral immune cells recruited into damaged brain tissue and the main source of IL-6 required for RAM generation. We also examined the transcriptomic landscape in myeloid cells at the single-cell level, demonstrating that CVI induces multiple signaling pathways in microglia and MDMs that include IL-6, TNF-α and IFN-I signaling. Notably, we observed a microglial subpopulation (RAMs) that expresses a proangiogenic gene signature and is highly proliferative. In the absence of IL-6 signaling or CCR2+ monocytes, RAM generation is impeded, resulting in failed cerebrovascular repair, a leaky BBB, neural cell death, fibrosis and diminished functional recovery. Importantly, exogenous IL-6 treatment was able to compensate for CCR2+ monocyte deficiency by restoring RAM generation and cerebrovascular remodeling. We therefore propose that IL-6-induced VEGFA+ RAMs are required for successful vascular repair and functional brain recovery after a CVI.
Our data demonstrate that proangiogenic RAMs appear as a result of cerebrovascular damage in mice and humans. In our rodent model, RAM numbers increased in response to lesion severity, demonstrating that local programming can be scaled up to deal with a larger amount of vascular damage. Moreover, we also observed Iba1+VEGFA+ myeloid cells with a ramified microglial morphology in the peri-infarct zone of human stroke brain tissue. VEGFA is a potent proangiogenic neurovascular remodeling factor27,29 and its expression in peri-infarct microglia suggests an attempt at revascularization of the brain after a stroke. Stroke recovery typically occurs within a few weeks to months post-injury and is associated with restoration of BBB integrity and cerebral blood flow, absorption of edematous exudates, clearance of debris and remodeling of the neural network6. Importantly, angiogenesis and revascularization of the stroke penumbra, which can begin as early as 3 d post-injury37, are often a measure of a favorable recovery that is associated with restored blood flow38. Our data in the present study demonstrate that interference with the timing or programming of this cerebrovascular repair process can have a major negative impact on the recovery trajectory, resulting in neural cell death, fibrosis and neurological dysfunction. We, therefore, postulate that enhancing RAM numbers and function could potentially improve the cerebrovascular repair and neurological outcomes, especially in situations where the repair trajectory is suboptimal.
Our study has also highlighted an important role for infiltrating CCR2+ monocytes in RAM genesis through their production of IL-6. Monocyte infiltration into the brain usually occurs in the subacute phase after a CVI18,39,40. CCR2+ inflammatory monocytes have been shown to worsen tissue edema and damage in the acute phase of CVI18,20 and then promote tissue repair in later phases18,41,42. We observed that infiltrating monocytes and MDMs are the main source of IL-6 after CVI and that this cytokine is required for RAM generation, cerebrovascular repair and functional recovery. RAMs express membrane IL-6Ra, which was upregulated in response to CVI, and conditional knockout of this receptor in microglia (ΔmIL-6Ra) resulted in decreased RAM generation. Moreover, therapeutic administration of exogenous rIL-6 to Ccr2−/− mice restored RAM generation, demonstrating that an important function of infiltrating monocytes after a CVI is to provide IL-6. These data also demonstrate that IL-6 alone can restore RAM programming and function in CCR2+ monocyte-deficient mice. IL-6 is a pleiotropic innate immune cytokine with a wide range of biological activities in health and disease that include B- and T-cell proliferation43, wound healing44, pathogenesis of inflammatory disorders and cancer45. Moreover, IL-6 has been shown to play an important role in promoting angiogenesis, neuroprotection and functional outcomes in murine models of stroke46–48. In human CVI patients, IL-6 levels increase markedly in the blood and cerebrospinal fluid (CSF) after injury49,50. Together with macrophage colony-stimulating factor and transforming growth factor-β1, CSF IL-6 levels were reported to correlate positively with functional outcomes in patients with TBI49. By contrast, IL-6 in the blood, considered to be an inflammatory biomarker, correlated positively with larger stroke lesion volumes and poor neurological outcomes at 1-year post-injury51. These data do not necessarily signify that IL-6 causes poor outcomes in stroke patients. Elevated IL-6 may instead reflect enhanced lesion severity and an attempt at repair, similar to how RAM numbers increase in response to lesion severity.
We propose that IL-6 signaling plays a fundamental role in RAM generation and function. Single-cell transcriptomic analysis of purified myeloid cells at day 6 post-CVI allowed us to assess the complexity of microglia and MDM programming at a time when the cerebrovascular repair process was underway. Our scRNA-seq analyses revealed multiple CVI-induced microglial subpopulations which reflected varying states of activation and signaling, including proliferation, angiogenesis, coagulation, IL-6 signaling, TNF-α signaling and IFN-I signaling, among others. Of particular interest was a CVI-induced microglial subpopulation (P1 C2 in Fig. 4i) with proangiogenic and proliferative properties that were decreased in Il6−/− relative to WT CVI mice. Our scRNA-seq and EdU labeling studies suggest that this RAM pool is established by induction of IL-6-dependent microglia proliferation and acquisition of proangiogenic functionality that is beneficial to the cerebrovascular repair process. In support of this conclusion, studies have demonstrated that selective ablation of proliferating microglia worsened stroke pathology8, and stimulation or addition of microglia promoted post-stroke regeneration via production of neurotrophic factors such as insulin-like growth factor 1, brain-derived neurotrophic factor and glial cell line-derived neurotrophic factor8,9,52. Future studies are required to determine how IL-6 induces both proliferative and reparative programming in microglia. IL-6 is known to promote proliferation of bone marrow progenitors and T cells in the presence of growth factors53–55.
In human brains with CVI, microglia and MDMs exist in varying functional activation states21,56,57 and the specific contribution of microglial subsets to vascular repair is largely unknown. Our data in CVI mice indicate that at least one microglial subpopulation (RAMs), possibly others, is committed to vascular repair and neural tissue recovery. RAM generation was impeded equivalently in Ccr2−/− and Il6−/− mice, resulting in diminished cerebrovascular remodeling, profound BBB leakage, reduced endothelial cell proliferation, fibrosis and neural cell death, which all explain the failure to recover neurological function. Although faulty cerebrovascular repair was observed in ΔmIL-6Ra mice and exogenous rIL-6 administration restored the repair program in Ccr2−/− mice, we acknowledge that rIL-6-mediated improvements may not be linked solely to the actions of RAMs on vascular repair. IL-6-mediated intracellular signaling transduction requires binding of IL-6 to its cognate receptor complex, consisting of IL-6Ra and gp130 (ref. 30). Both membrane-bound and secreted IL-6Ra have been shown to associate with gp130 and exert the biological functions of IL-6 (ref. 58). It is therefore conceivable that RAM-mediated vascular repair and neurogenesis promoted by the secreted form of IL-6Ra59,60 both contribute to functional brain recovery after a CVI. The contributions of membrane-bound versus secreted IL-6 signaling to the CVI repair process need to be addressed in future studies.
In conclusion, our findings offer important mechanistic insights into the CVI recovery process by demonstrating how monocytes promote the generation of RAMs via IL-6 signaling. The recovery trajectory after a CVI in humans is probably influenced by many different factors; however, our data highlight a straightforward dialog between monocytes and microglia that has the potential to be manipulated therapeutically. The rIL-6 may shift microglia programming toward a more proangiogenic state in situations of suboptimal cerebrovascular repair, possibly linked to insufficient monocyte infiltration or competing cytokine signals. IFN-I signaling, for example, is known to impede repair after TBI and CVI10,61, and we found evidence of this signaling pathway by scRNA-seq in specific CVI-induced microglia subsets. It will be important in future studies to determine whether repair can be enhanced by offering competing IL-6 signaling to microglia as a therapeutic treatment. Development of treatments that enhance the CVI recovery trajectory has the potential to improve outcomes in many different human CNS disorders.
Online content
Any methods, additional references, Nature Portfolio reporting summaries, source data, extended data, supplementary information, acknowledgements, peer review information; details of author contributions and competing interests; and statements of data and code availability are available at https://doi.org/10.1038/s41590-023-01521-1.
Methods
Mice
All mice used in the present study were handled in accordance with the guidelines established by the National Institutes of Health (NIH) Animal Care and Use Committee (ACUC) and the recommendations of the Association for Assessment and Accreditation of Laboratory Animal Care International (AAALAC) for the Care and Use of Laboratory Animals. The animal study protocol was approved by the National Institute of Neurological Disorders and Stroke (NINDS) ACUC. Mice were bred and maintained within individually ventilated cages in the specific pathogen-free animal facilities at the NIH. Housing conditions included a temperature of 22 °C, humidity of 50% and a 12-h light:dark cycle. C57BL/6J (WT; catalog no. 000664), B6.129(Cg)-Ccr2tm2.1Ifc/J (ref. 32) (Ccr2RFP/RFP; Ccr2−/−; catalog no. 017586), B6.129S2-Il6tm1Kopf/J (ref. 62) (Il6−/−; catalog no. 002650), B6;SJL-Il6ratm1.1Drew/J (ref. 63) (Il6rafl/fl; catalog no. 012944), C57BL/6-Tmem119em1(cre/ERT2)Gfng/J (ref. 28) (Tmem119-CreERT2; catalog no. 031820), B6.129P2(Cg)-Cx3cr1tm1Litt/J (ref. 64) (Cx3cr1GFP/GFP; catalog no. 005582), B6.Cg-Gt(ROSA)26Sortm14(CAG-tdTomato)Hze/J (ref. 65) (Stopfl/fl TdTomato; catalog no. 007914) were purchased from Jackson Laboratory. Tmem119-CreERT2Stopfl/+TdTomato mice (Tmem119-TdTo) were generated from an F1 cross between Tmem119-CreERT2 and Stopfl/flTdTomato mice. Tmem119-CreERT2Il6rafl/fl mice (ΔmIL-6Ra) and Tmem119-CreERT2-Il6ra+/+ or -Il6rafl/+ mice (IL-6Ra Ctrl) were obtained in the F2 generation after an original cross of Tmem119-CreERT2 and Il6rafl/fl mice. Cx3cr1GFP/+Ccr2RFP/+ mice were generated from an F1 cross between Cx3cr1GFP/GFP and Ccr2RFP/RFP mice. Male and female mice aged 8–10 weeks were used in the present study, except for Tmem119-TdTo, ΔmIL-6Ra and IL-6Ra Ctrl mice, which were used at age 11–12 weeks.
Human brain tissue
Postmortem, formalin-fixed, paraffin-embedded human brain tissue was obtained from the Mt. Sinai Brain Bank, University of Maryland Brain and Tissue Bank, and the Human Brain and Spinal Fluid Resource Center through the NIH Neurobiobank. Brain tissue was also obtained from the National Cancer Institute Laboratory of Pathology at the NIH. We examined eight human brain specimens from four stroke patients and four neurologically healthy subjects. There was no significant difference in age of death between the two groups (P = 0.3513, two-tailed, unpaired Student’s t-test). Additional information about the human brain tissue, including neuropathology and clinical diagnosis, is provided in Supplementary Table 1.
Human brain multiplex fluorescence immunohistochemistry and analysis
Multiplex fluorescence immunohistochemistry was performed on 5-μm-thick paraffin sections cut from postmortem normal and stroke human cortical brain tissue. Briefly, sections were first deparaffinized using a standard xylene/ethanol/rehydration protocol followed by antigen unmasking with a 10-min heat-mediated, antigen retrieval step in 10 mM Tris base/1 mM EDTA buffer, pH 9 using an 800-W microwave set at 100% power. The sections were then incubated with Human BD Fc Blocking solution (BD Biosciences) for 15 min at room temperature (RT, 20–25 °C) to saturate endogenous Fc receptors, followed by treatment with True Black Reagent (Biotium) for 5 min at RT to quench intrinsic tissue autofluorescence. Sections were subsequently immunoreacted for 1 h at RT using 1 μg ml−1 of a cocktail mixture of immunocompatible primary antibodies against IBA1, CD31, VEGFA, fibrinogen and glycophorin A for RBCs, as listed in Supplementary Table 2. This step was followed by washing off excess primary antibodies in phosphate-buffered saline (PBS) supplemented with 1 mg ml−1 of bovine serum albumin (BSA), followed by incubation of the sections using a 1-μg ml−1 cocktail mixture of the appropriately crossadsorbed secondary antibodies (purchased from Thermo Fisher Scientific, Jackson ImmunoResearch or Li-Cor Biosciences), conjugated to one of the following spectrally compatible fluorophores: Alexa Fluor-488, Alexa Fluor-546, Alexa Fluor-594, Alexa Fluor-647 or IRDye 800CW. After washing off excess secondary antibodies, sections were counterstained using 1 μg ml−1 of DAPI (Thermo Fisher Scientific) for visualization of cell nuclei. Slides were then coverslipped using Immu-Mount medium (Thermo Fisher Scientific) and imaged using a multi-channel, wide-field epifluorescence microscope (see below).
For multispectral imaging of multiplex immunohistochemistry-stained sections, images were acquired from whole-specimen sections using the Axio Imager.Z2 slide scanning fluorescence microscope (Zeiss) equipped with a ×20/0.8 numerical aperture (NA) Plan-Apochromat (Phase-2) nonimmersion objective (Zeiss), a high-resolution ORCA-Flash4.0 sCMOS digital camera (Hamamatsu), a 200-W X-Cite 200DC broadband lamp source (Excelitas Technologies) and six customized filter sets (Semrock) optimized to detect the following fluorophores: DAPI, Alexa Fluor-488, Alexa Fluor-546, Alexa Fluor-594, Alexa Fluor-647 and IRDye 800CW. Image tiles (600 × 600 μm2 viewing area) were individually captured at 0.325-μm pixel−1 spatial resolution and the tiles seamlessly stitched into whole-specimen images using the ZEN 2 image acquisition and analysis software program (Zeiss), with an appropriate color table having been applied to each image channel to either match its emission spectrum or set a distinguishing color balance. Pseudocolored stitched images were then exported to Adobe Photoshop and overlaid as individual layers to create multicolored merged composites or exported as 8-bit grayscale BigTIFF files and imported in Imaris v.9.8 (Bitplane) for image analysis.
One to three regions of interest (ROIs) (2.25 mm2 per ROI) were analyzed for Ctrl, peri-infarct and infarct areas of human brain tissue samples. To quantify VEGFA+ myeloid cells, surfaces were created for IBA1+ cells and then VEGFA+IBA1+ cells. Percentage VEGFA+ myeloid cells was calculated by dividing the average number of VEGFA+IBA1+ cells by the total number of IBA1+ cells and then multiplying by 100.
Ultrasound injury
Mice were subjected to either CVI or sham surgery (Ctrl) as previously described with a slight modification18. Briefly, 8- to 12-week-old mice were anesthetized using a cocktail of ketamine (85 mg kg−1), xylazine (13 mg kg−1) and acepromazine (2 mg kg−1) in PBS and placed on hot pads to maintain body temperature throughout the surgery and during the recovery phase. After an incision over the midline of the scalp, the skull was exposed and a metal bracket was glued on to the skull, leaving a circular area of the skull exposed, the center of which was 3 mm posterior to bregma and 2.5 mm from the sagittal suture. A cranial window (2 × 3 mm2) was carefully thinned to ~30–35 μm (mild CVI) or ~20–25 μm (severe CVI). Afterwards, microspheres (Optison, Perflutren Protein-Type A, GE Healthcare) were injected i.v. via retro-orbital injection (1 μl per g of body weight) and a drop of artificial CSF (Harvard Apparatus, catalog no. 597316) was placed over the cranial window. Low-intensity pulsed ultrasound was then applied at 1 MHz, with a peak negative pressure of ~200 kPa (2.2 W cm−2), a burst length of 1 ms, 10% duty cycle and a 5-s (mild CVI) or 10-s (severe CVI) exposure to the thinned skull using a Sonicato (catalog no. RS-9255) and a wound clipper (Roboz, catalog no. RS-9250); mice were provided with 1 ml of prewarmed saline (i.v.) and diet gel during the post-surgery recovery period. The more severe model of CVI was used throughout the present study unless otherwise stated.
Tamoxifen, EdU and rIL-6 administration
Beginning at age ~4–5 weeks, Tmem119-TdTo, ΔmIL-6Ra and IL-6Ra Ctrl mice were fed tamoxifen chow (500 mg per kg of diet; Envigo) for 1 month to induce expression of Cre recombinase. Mice were then switched to regular feed for another 3 weeks to allow turnover of circulating monocytes before CVI induction. To label cells that underwent proliferation, EdU (Invitrogen, catalog no. A10044) was injected into mice intraperitoneally at a concentration of 1 mg ml−1 in PBS twice daily with at least 8 h between each injection. EdU administration was initiated on day 1 post-CVI and continued for 5 d consecutively (5 μg per g body weight). Then 3 μg of rIL-6 (R&D systems, catalog no. 7270-IL-100/CF) or vehicle was administered both transcranially and i.v. to mice once daily, beginning on day 1 post-CVI. For transcranial administration, 1.5 μg of rIL-6 in PBS or PBS alone (control; 200 μl) was formulated into a hydrogel (1% (v:v) carboxymethylcellulose sodium; Sigma-Aldrich, catalog no. C9481) and injected beneath the scalp after stitching the opening shut from the CVI procedure. For intravenous injections, 1.5 μg of rIL-6 in PBS or PBS alone (100 μl) was injected into the retro-orbital plexus. Daily injections were given for 6 d for flow cytometric studies and 9 d for all other studies.
Monocyte adoptive transfer
Bone marrow cells were obtained under sterile conditions from the femurs and tibias of WT and Il6−/− mice (aged 8–9 weeks). Monocytes were purified by negative selection using EasySep Mouse Monocyte Isolation Kit (StemCell Technologies, catalog no. 19861). We quantified the number of isolated cells using a hemocytometer and determined cell purity and viability by flow cytometry (Extended Data Fig. 5g). After intravenous administration of 106 WT or Il6−/− monocytes (CD11b+Ly6C+CX3CR1+CD3−Siglec F−NK1.1−Ly6G−CD117−CD45R−) in 100 μl of PBS per Ccr2−/− recipient, mice immediately experienced a CVI and then received bone marrow-derived monocytes once daily on days 1, 2 and 3 post-CVI. Surface antibodies used for all flow cytometry experiments are listed in Supplementary Table 4.
Intravital TPM
Mice with a skull-thinned cranial window (~20 μm) were imaged using a Leica SP8 two-photon microscope (Leica Application Suite X) equipped with an 8,000-Hz resonant scanner, a ×25 collar-corrected water-dipping objective (1.0 NA) or a ×20 water-dipping objective (1.0 NA) and a quad HyD external detector array. To image Dylight 488, a Mai Tai HP DeepSee Laser (Spectra-Physics) was tuned to 905 nm. An Insight DS laser (Spectra-Physics) was tuned to 1,040 nm for imaging EB and TdTomato or 1,100 nm for Alexa Fluor-647. Three-dimensional (3D) time-lapse movies were captured at 1-min and 2-min intervals for 30 min or 1.5-h duration. Images were acquired from neocortical layers I and II below the pia mater using z-stacks of 80- to 100-μm depth (2.5- or 3-μm step size) at 1,024 × 1,024 pixel2 resolution (350 × 350 μm2 area). To visualize blood vessels, 30 μl of 0.5 mg ml−1 EB (Sigma-Aldrich) and 100 μl of tomato lectin Dylight 488 (Vector Labs) were injected i.v. before imaging. To capture dye extravasation, mice injected with a mixture of EB and tomato lectin were imaged within 1 h. Ly6C+ monocytes and blood vessels were visualized by injecting 100 μg of Alexa Fluor-647 anti-mouse Ly-6C antibody (clone HK1.4; BioLegend, catalog no. 128010) 2 h before imaging. Adobe Premiere Pro (Adobe Systems) was used to create the final videos.
Mouse brain immunohistochemistry
Anesthetized mice received an intracardiac perfusion with PBS followed by 4% paraformaldehyde (PFA; Electron Microscopy Sciences, catalog no. 15714). After post-fixation with 4% PFA overnight, tissue samples were placed in 30% sucrose for at least 3 d. Brain tissue was then embedded in Tissue-Tek OCT (Optical Coherence Tomography) compound (VWR, catalog no. 25608–930), frozen on dry ice and sectioned coronally (100 μm) with a Leica CM1860 cryostat. Free-floating brain sections were incubated in blocking solution (5% normal fetal serum, 0.3% Triton X-100 in PBS) for 2 h at RT (20–25 °C). Tissue sections were then incubated with primary antibodies against Iba1, Tmem119, IL-6Ra, VEGF-A, NeuN, glial fibrillary acidic protein (GFAP), CD31, mCherry (for TdTomato and red fluorescent protein (RFP) detection) and collagen type 1 overnight at 4 °C. After washing with PBS, secondary antibodies were added and incubated overnight at 4 °C. Sections were mounted on to slides with mounting reagent (Polysciences, catalog no. 18606–20). To identify microglia and monocytes, brain sections from CX3CR1GFP/+CCR2RFP/+ double-reporter mice were photobleached at RT (20–25 °C) for at least 8 h before incubation with anti-Iba1 and mCherry antibodies. To assess dye extravasation in deep cortical layers, mice were injected with EB and tomato lectin Dylight 488, as described above, and then perfused within 1 h of injection. The development of EdU was carried out using Click-iT plus EdU imaging kit (Thermo Fisher Scientific, catalog no. C10640) according to the manufacturer’s instructions. All primary and secondary antibodies used for immunohistochemistry are provided in Supplementary Table 3. Images were acquisitioned from the cortices of five to eight animals per group. Cortical images corresponding to motor and somatosensory areas were acquired using an Olympus FV1200 laser-scanning confocal microscope fitted with ×4/0.16 NA, ×20/0.75 NA and ×40/0.95 NA objectives. All confocal images were analyzed using Imaris v.9.7 and v.9.8.
Flow cytometry
Single cells for flow cytometry experiments were prepared using multi-tissue dissociation kit 1 (Miltenyi Biotec, catalog no. 130–110-201). Briefly, after perfusion of animals at day 6 post-CVI with ice-cold PBS, brain tissue from injured and uninjured control mice was obtained using a 1-mm biopsy punch (Thermo Fisher Scientific, catalog no. 12–460-410). Biopsied brain tissue was dissociated into single cells using a cocktail of enzymes according to the manufacturer’s instructions. After using the debris removal solution (Miltenyi Biotec, catalog no. 130–109-398), single cells were resuspended in 50 μl of staining buffer (PBS with 2% fetal bovine serum) with purified rat anti-mouse CD16/CD32 antibody (1:200; Fc receptor block; clone 93; BioLegend) and incubated at 4 °C for 10 min. A cocktail mix of antibodies for surface proteins (50 μl) was added for 15 min at 4 °C and then for 30 min at RT (20–25 °C). Subsequently, cells were washed twice with staining buffer and resuspended in 4% PFA for 10 min and stored in PBS. For intracellular staining of VEGF, fixed single-cell suspensions were incubated with 50 μl of Cytofix/Cytoperm (BD, catalog no. 554714) containing mouse VEGF120/164 antibody (1:400; monoclonal rat IgG2B; clone 39917; R&D Systems) that was directly conjugated to Alexa Fluor-647 using the Alexa Fluor-647 Antibody Labeling Kit (1:300; Invitrogen, catalog no. A20186). Surface antibodies used for all flow cytometry experiments are listed in Supplementary Table 4. Fluorescence minus one (FMO) controls were generated to determine the appropriate gating for positive cells. Before sample acquisition, 5,000 beads (Invitrogen, catalog no. C36950) were added into stained samples to calculate the absolute number of cells in each sample. Dead cells were stained using the Live/Dead Fixable Blue Dead Cell Stain kit (1:1,000; Thermo Fisher Scientific, catalog no. L34962) and excluded from the analysis. Flow cytometric samples were acquired using a BD FACSymphony flow cytometer (BD Biosciences) equipped with 355-nm, 405-nm, 488-nm, 532-nm and 628-nm laser lines, and FACS Diva v.8.0.1 software. Data analysis was performed using FlowJo v.10.5.3 (BD Biosciences). Doublets were excluded via FSC-A versus FSC-H gating. After gating on live CD45+ cells, we performed multiple unbiased analyses with FlowJo, including generation of uniform manifold approximation and projection (UMAP)66 plots for dimensionality reduction and data visualization, as well as PhenoGraphs67 to partition high-parameter single-cell data into subpopulations (clusters) based on phenotypic coherence. Cluster Explorer was used to visualize the PhenoGraph subpopulations on UMAP plots. To identify clusters containing RAMs, we first identified microglia based on CD11b, P2RY12 and Tmem119-TdTomato expression, and then defined RAM clusters based on VEGFA, CD24, CD44, CD11c, CD206 and major histocompatibilty complex (MHC)-II expression.
Single-cell isolation for scRNA-seq
WT and Il6−/− mice that were either uninjured (control) or at day 6 post-CVI were anesthetized and perfused using ice-cold PBS. Control and CVI-damaged brain tissue was collected using a 4-mm biopsy punch (Thermo Fisher Scientific, catalog no. 12–460-410). Single cells were dissociated from the pooled punch biopsies (three mice per group) using a multi-tissue dissociation kit 1 (Miltenyi Biotec, catalog no. 130–110-201). After myelin removal using myelin removal beads (Miltenyi Biotec, catalog no. 130–096-733), CD11b+ myeloid cells were positively selected using CD11b microbeads (Miltenyi Biotec, catalog no. 130–093-636). Cell viability and counting were assessed using Acridine Orange. Single-cell samples in PBS containing 0.05% BSA with viabilities >80% were used for sequencing.
ScRNA-seq library preparation, sequencing and analysis
Complementary DNA libraries were prepared from single-cell suspensions following the 10x Genomics 3′ v.3 protocol (www.10xgenomics.com) and sequenced on NovaSeq (www.illumina.com) with the aim of generating 150 million reads per sample for 4,000 cells per sample. Post-sequencing, the 10x Genomics ‘cellranger’ tool suite (v.6.0.1) was used to generate expression counts per known gene (refdata-gex-mm10-2020-A) per sample. These counts were then imported into R (https://cran.r-project.org) for analysis using functions supported in the Monocle3 v.1.3.1 package (https://cole-trapnell-lab.github.io). For import, the ‘load_cellranger_data’ function was used followed by the ‘combine_cds’ function to collapse the count data across samples into one analysis-ready object. To select the number of principal components to use for analysis (n = 50), the ‘plot_pc_variance_explained’ function was used. To generate cell-level metrics, the ‘reprocess_cds’ function was used. Cells observed to have a total number of features and/or total number of genes >99.9th quantile or <0.01th quantile were removed from the analysis. Cells having a total percentage read mapping to ‘mt-’ genes >15% were also removed. The ‘preprocess_cds’ function was then used again with the ‘residual_model_formula_str’ parameter included to correct for differences in total percentage reads that map to ‘mt-’ genes for cells not removed. The ‘align_cds’, ‘reduce_dimension’ and ‘cluster_cells’ functions were then used to produce initial groupings of cells by partition and cluster. Doublet clusters and doublet cells were then tested for and removed using the ‘findDoubletClusters’ and ‘computeDoubletDensity’ functions supported in the ‘scDblFinder’ package. Cells falling in clusters having <100 cells were also removed. The same functions used to generate the initial groupings of cells was then repeated, starting with the ‘preprocess_cds’ function, producing final groupings of cells by partition and cluster. Annotation of these groupings per cell type was accomplished using the ‘SingleR’ package together with the ‘MouseRNAseqData’ and ‘ImmGenData’ reference datasets. Visual inspection of the final groupings of cells was accomplished using the ‘plot_cells’ function (reduction_method = ’UMAP’). To describe the number of cells versus group versus sample, bar plots were generated using the ‘ggplot’ function together with different options (position = ’fill’, position = ’stack’). To test for and identify marker genes per group, the ‘top_markers’ function was used. This function models the expression by gene for cell membership in a group versus all others using generalized linear regression. The function reports in table form various statistics per marker, including: an R2 value that describes the model fit, a specificity value based on the Jensen–Shannon divergence method, an overall maker score that represents the fraction of cells expressing the marker scaled by the specificity value and both an uncorrected and Benjamini– Hochberg-corrected P value (that is, q value) that describes the marker significance under the likelihood ratio test. To identify differential genes within a group between samples, the ‘fit_models’ function was used. This function models the expression by gene between samples using generalized linear regression. To summarize and report the modeling results, the ‘coefficient_table’ function was used. This function reports in table form various statistics per gene, including: the coefficient estimate from the regression model and both an uncorrected and Benjamini–Hochberg-corrected P value (that is, q value) that describes whether the estimate differs significantly from zero under Wald’s test. To visualize markers and differential genes, both dot plots and violin plots were generated using the ‘plot_genes_by_group’ and ‘plot_genes_violin’ functions, respectively. To identify and visualize modules of coregulated genes per group, the ‘graph_test’, ‘find_gene_modules’, ‘aggregate_gene_expression’ and ‘pheatmap’ functions were used. For groups of cells observed to have substructure, cells for the group were subset and the same functions used to generate the final groupings of cells repeated. Trajectory fitting was also explored for these subset groups of cells using the ‘learn_graph’ and ‘order_cells’ functions, with the resulting fits visualized using the previously mentioned ‘plot_cells’ function (show_trajectory_graph = TRUE). All analysis was performed on NIH’s Biowulf high-performance computing (HPC) system (x86_64-pc-linux-gnu (64-bit) plate form and running under CentOS Linux 7 (Core)). As our scRNA-seq data were based on individual cells from pooled brain biopsies, crosscondition comparisons (for example, WT versus Il6−/− and Ctrl versus CVI) should be considered to be preliminary.
Fluorescence in situ hybridization
Brain tissue from uninjured or day 3 post-CVI CX3CR1GFP/+CCR2RFP/+ mice was embedded in OCT compound as described above. The tissue was sectioned into 16-μm-thick coronal slices using a Leica CM1860 and then placed on positively charged slides. Slides were dried at RT (20–25 °C) for at least 2 h and then stored at −80 °C. Fluorescence in situ hybridization (FISH) was performed to detect Il-6 transcripts on brain slices (ACD, catalog no. 15891) and combined with immunohistochemistry to determine the identity of the Il-6-expressing cells. Antibodies used for immunohistochemistry are listed in Supplementary Table 3. FISH coupled with immunostaining was performed using the Multiplex fluorescent reagent kit v.2 (ACD, catalog no. 323100) according to the manufacturer’s instructions for fixed sections. Briefly, sections were treated with Protease III for 30 min and hybridized for 2 h at 40 °C with Il-6 probe. After hybridization with amplifiers followed by horseradish peroxidase, sections were incubated in the blocking solution and then with primary and secondary antibodies as described under Mouse brain immunohistochemistry. The probe target signals were visualized using TSA Plus Cyanine (Akoya, catalog no. SKU#NEL745001KT). Sections were counterstained with DAPI (Sigma-Aldrich, catalog no. D9542). Neocortical images were acquired using an Olympus FV1200 laser-scanning confocal microscope and a ×20/0.75 NA objective. All confocal images were visualized and analyzed using Imaris v.9.8.
Y maze
To measure cognitive and locomotive brain functions, mice were subjected to the Y-maze task at the indicated days post-CVI. We used age-matched male WT, Ccr2−/− and Il6−/− mice for the behavior studies and conducted our studies between 6pm and 10pm under infrared light. The Y maze is made up of three arms, each positioned at a 120° angle from each other. Mice were placed in the center of the maze and permitted to freely explore each arm with spatial cues for 5 min per session. An entry was defined when all the limbs entered an arm. The number of total arm entries and triplicate entries (alteration) were recorded during the session. Alteration was achieved when an animal visited three consecutive arms clockwise or counterclockwise. The triplicate ratio was calculated as the number of triplicate entries divided by the number of total arm entries.
Two-photon image analysis
Two-photon images of superficial neocortical vessels were acquired on day 10 post-CVI and analyzed using Imaris v.9.8. Vascular coverage and intervascular area were analyzed as described previously18. Briefly, to calculate the volume of vascular coverage, a 3D volumetric surface was created in Imaris using the ‘surface’ function that corresponded to EB+ vasculature. The vascular coverage was calculated as a fraction of vascular volume divided by the volume of the 3D image. The intervascular area was defined as the area not occupied by vessels in the maximal intensity projection for each image. The intervascular area was obtained by masking the areas negative for EB and tomato lectin, using the surfaces function. To assess dye extravasation, the number of tomato lectin+ spots outside EB-labeled blood vessels was calculated in Imaris using the spot function and then divided by the area for each ROI using the maximum intensity projection. For areas with extensive EB permeation, surfaces were created for extravascular EB signals and then EB+ areas were normalized by the area of each ROI.
Immunohistochemistry and FISH image analysis
All 3D image analysis was conducted in neocortical areas below the pial surface using Imaris. Specific cell types were first identified based on marker expression using the ‘spot’ (for VEGFA, extravascular EB, CCR2-RFP, Iba1, Il-6, IL-6Ra, NeuN, EdU) or ‘surface’ (for TdTo+ microglia, Iba1, GFAP, CD31, collagen type I) function in Imaris. To quantify EdU+ microglia, surfaces were generated for TdTo+Iba1+ microglia (seed point = 10 μm). Surfaces that were negative for Iba1 were excluded from the analysis. The number of TdTo+Iba1+ microglia containing EdU+ ‘spots’ was then quantified using a MATLAB tool: ‘find spots close to surface’. The number of microglia positive for EdU was divided by the surface area (mm2) for each ROI. A similar approach was used to quantify Iba1+ microglia expressing VEGFA and/or IL-6Ra. CCR2-RFP+ monocytes were quantified by generating ‘spots’ for RFP+ cells (spot diameter = 7 μm) and dividing by the surface area (mm2) for each ROI. Quantification of Il6 transcripts in monocytes or microglia was performed by first generating ‘spots’ for CCR2-RFP or Iba1 signal (spot diameter = 10 μm). DAPI− spots were excluded from the analysis. The number of CCR2-RFP+ or Iba1+ spots containing Il6 signal was then calculated. A similar approach was used to quantify Il-6 transcripts in astrocytes or endothelial cells, except that ‘surfaces’ were generated that corresponded to GFAP or CD31 signal, respectively. Il-6+ astrocytes or endothelial cells that were negative for DAPI were excluded. Percentages of monocytes, microglia, astrocytes and endothelial cells expressing Il-6 were calculated for each ROI by dividing the number of Il-6+ cells by the total number of each cell type and multiplying by 100. Stratification of Il-6-expressing cell types was performed by dividing the number of Il-6-expressing cells within a specific cell type by the total number of Il-6-expressing cells and multiplying by 100. To quantify dye extravasation, EB signal outside tomato lectin-labeled blood vessels was calculated using the ‘spots’ function (seed point = 4 μm) and divided by the ROI area (mm2). For some experiments, severe EB leakage was quantified using the ‘surface’ function instead. Areas of collagen type I expression were quantified using the ‘surface’ function and divided by the tissue area (mm2) for each ROI. NeuN+ cells were quantified in cortical layers 2 and 3 using the ‘surface’ function (split objects = 7 μm) and divided by the ROI area (mm2).
Statistical analysis
Statistical analyses and graphing were performed using Prism v.9 (GraphPad Software) for all experiments except scRNA-seq, which is described above. All image analysis was performed by a blinded investigator. Experiments containing two groups were analyzed using unpaired, two-tailed Student’s t-test. Experiments involving more than two groups were analyzed by one- or two-way analysis of variance (ANOVA) followed by a Tukey’s multiple comparison test or Šídák’s multiple comparison test. Groups were considered statistically different when P < 0.05. All data are displayed as the mean ± s.e.m. No statistical methods were used to predetermine sample sizes, which were based on our previous studies of this murine CVI model10,18. Mouse littermates were randomly assigned to each group, but no active randomization protocol was used. In addition, samples were randomly selected for data acquisition and analysis, but no active randomization protocol was used.
Extended Data
Extended Data Fig. 1 |. Identification of microglia in Tmem119-TdTo reporter mice.
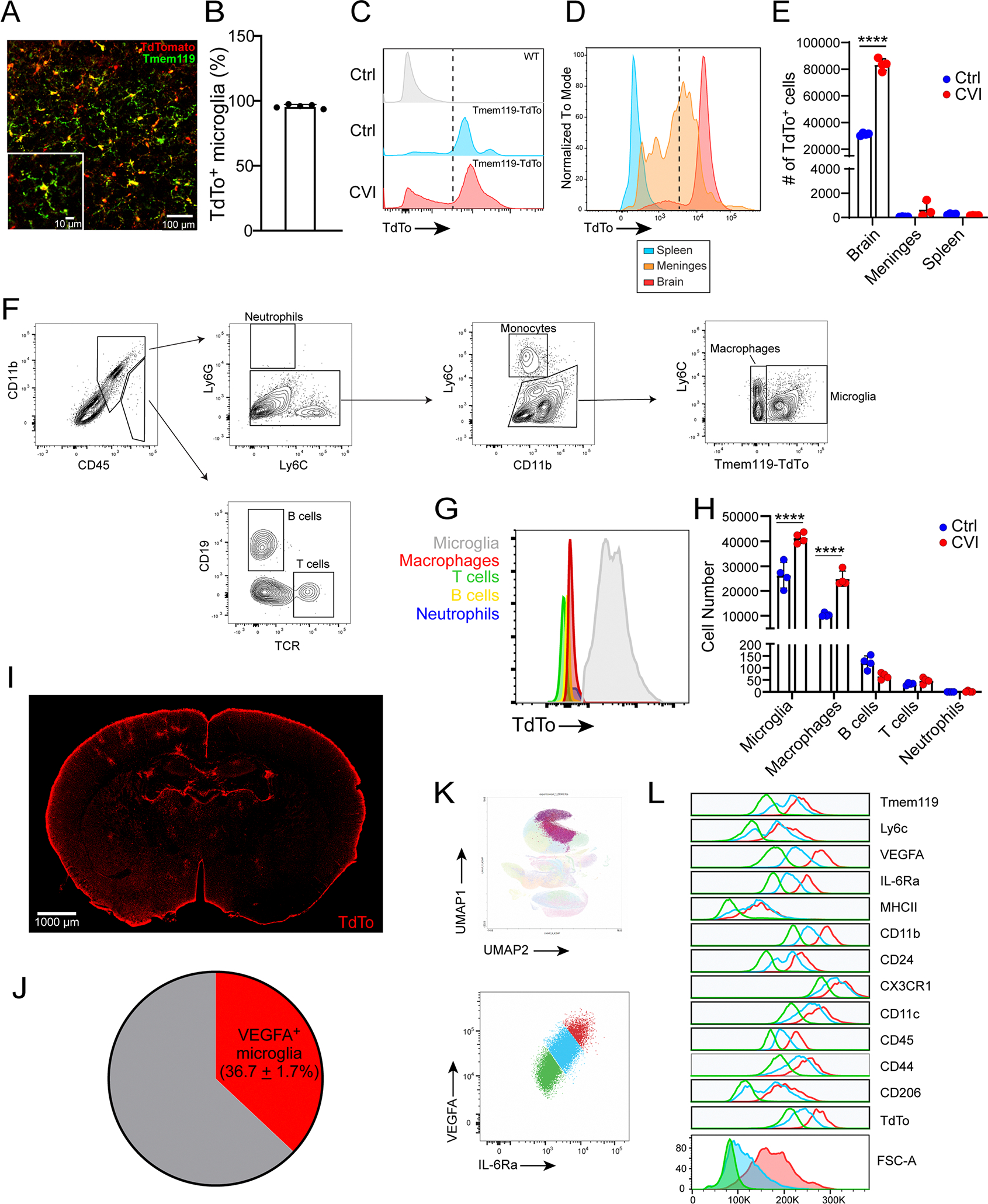
a, Immunofluorescent staining for Tmem119 (green) and TdTo (red) of neocortex from a naïve Tmem119-TdTo mouse. Inset depicts an enlarged area of the image. b, Bar graph shows quantification of TdTo+ Tmem119+ microglia after tamoxifen treatment (n = 5 mice). c, Histograms show TdTo expression on CD45+ CD11b+ cells harvested from the brains of uninjured (Ctrl) WT mice (n = 4) as well as uninjured (n = 4) and D6 post-CVI (n = 4) Tmem119-TdTo mice. Each histogram represents concatenated samples for the entire group. Data are representative of two independent experiments. d, Histograms show TdTo expression in CD45+ CD11b+ myeloid cells isolated from the brain (n=8), meninges (n=6) and spleen (n=8) of uninjured and D6 post-CVI Tmem119-TdTo mice. The histogram represents a concatenation of both uninjured and injured samples. Data are representative of two independent experiments. Dashed lines in panels c and d denote the cut-off value for cells with TdTo positivity. Note that meningeal macrophages have more background fluorescence in this channel than splenic macrophages. e, Bar graph shows the absolute number of TdTo+ cells in the denoted tissues from uninjured Ctrl (blue) and D6 post-CVI (red) Tmem119-TdTo mice in panel c and d. (****P<0.0001; two-way ANOVA with Šídák’s multiple comparisons test). f, Gating strategy for flow cytometric analysis of leukocytes isolated from the brains of Tmem119-TdTo mice following CVI. g, Histogram shows TdTo expression in the indicated cell populations, identified using the gating strategy from panel f, from the brains of Tmem119-TdTo mice (n = 5) at D6 post-CVI. h, Bar graph shows the absolute number of the denoted cell types from the brains of uninjured Ctrl (n=4) versus D6 post-CVI (n = 4) Tmem119-TdTo mice. (****P<0.0001; two-way ANOVA with Šídák’s multiple comparisons test). Data are representative of two independent experiments. i, Representative TdTo expression (red) on a coronal brain section from a Tmem119-TdTo mouse (n=3) at D6 post CVI. Data are representative of two independent experiments. j, Pie chart depicts the percentage of VEGFA+ microglia (mean+SEM, red) among total CD11b+ CX3CR1+ TdTo+ microglia obtained from the punch biopsies of injured brains (n=4) at D6 post CVI described in Fig. 1i. k, A dimensionality reduction plot (top) generated using Cluster Explorer depicts three RAM clusters (shades of purple) identified in the brains of Tmem119-TdTo mice (n=4) at D6 post-CVI. Data are representative of two independent experiments. A representative dot plot (bottom) shows expression of VEGFA and IL-6Ra by the three RAM clusters identified in D6 Tmem119-TdTo mice. Arbitrary gating was used to analyze marker expression in panel l. Specifically, RAM were stratified based on low, intermediate, and high VEGFA and IL-6Ra co-expression: VEGFAlo IL-6Ralo (green), VEGFAint IL-6Raint (cyan), VEGFAhi IL-6Rahi (red). l, Histograms show expression of the indicated markers and FSC-A by the three RAM subpopulations identified in panel k. All graphs show mean ± SEM. Dots represent individual mice.
Extended Data Fig. 2 |. Mild versus severe CVI in mice and immunohistochemical staining of human brain tissue.
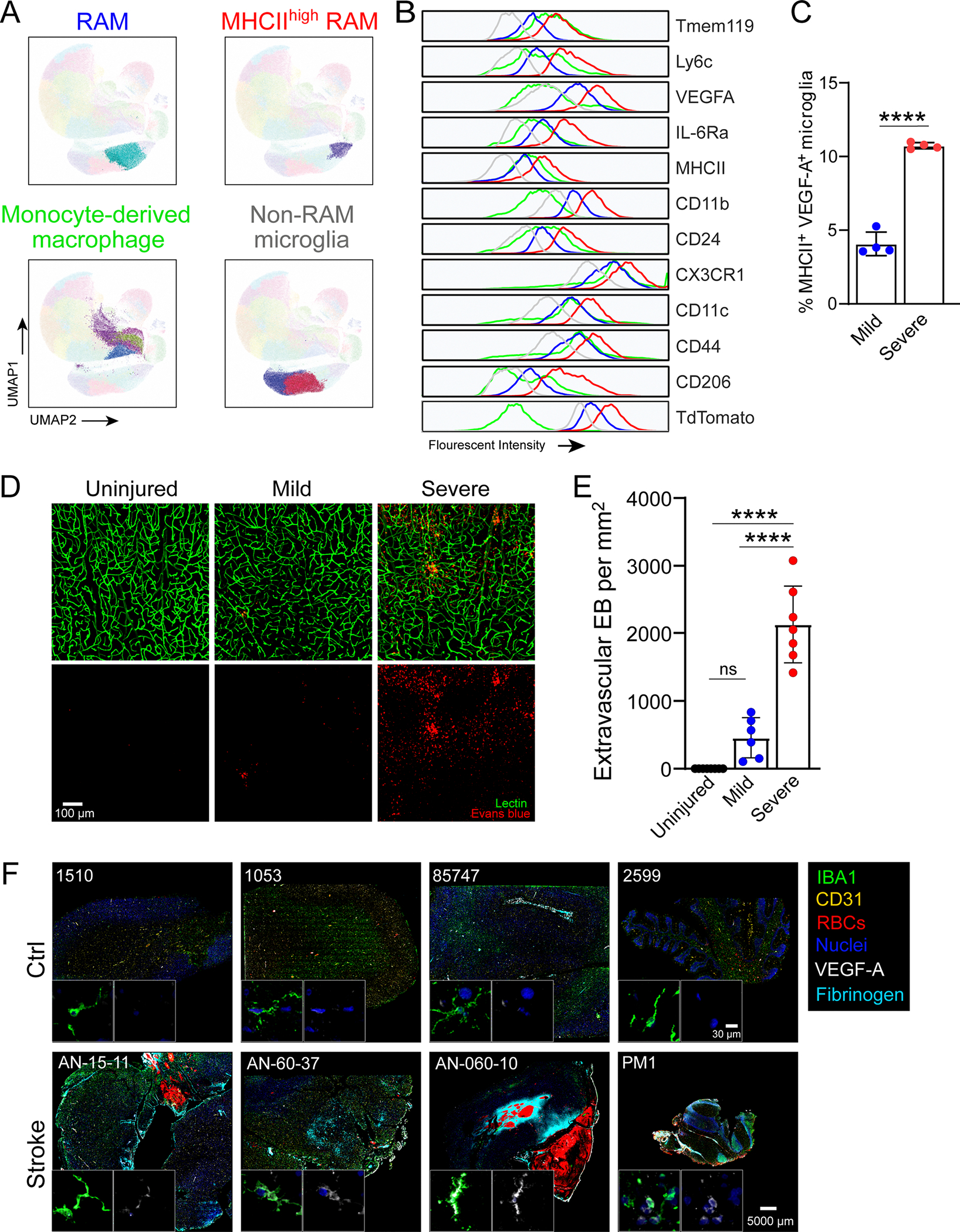
a, Dimensionality reduction plots generated using Cluster Explorer show RAM (blue), MHC IIhi RAM (red), non-RAM microglia (gray), and MDM (green) in Tmem119-TdTo mice on D6 post CVI. Plots show concatenated samples obtained from mild (n=4) and severe (n=4) WT CVI mice described in Fig. 2. The gating strategy for the different myeloid subsets is described in Fig. 2b–d. Data are representative of two independent experiments. b, Histograms show expression of the indicated markers by the different myeloid cell populations (a). c, Bar graph shows the percentage of MHCII+ VEGFA+ microglia in the brains of D6 Tmem119-TdTo mice described in Fig. 2a–d following mild versus severe CVI (****P<0.0001; two-tailed unpaired t test). d, Immunofluorescent staining of neocortical vasculature in uninjured Ctrl (n=8) as well as mild (n=6) and severe (n=7) CVI mice at day 10. Lectin (green) and Evans blue (red) were injected i.v. to visualize vessels and BBB disruption. e, Quantification showing numbers of extravasated dye among the groups of animals (Ctrl=8, mild CVI=6, and severe CVI=7). Data are combined from two independent experiments. (****P<0.0001; one-way ANOVA with Tukey’s test). All graphs show mean ± SEM. Dots represent individual mice. f, Multiplex immunostaining of human stroke (n=4) versus Ctrl (n=4) brain tissue (see Supplementary Table 1) showing expression of IBA1, CD31, glycophorin A (red blood cells, RBCs), VEGF-A, fibrinogen, and DAPI (cell nuclei). All images are comparably scaled. The magnified images in the bottom of each image depict VEGF-A (white) expression in IBA1+ myeloid cells (green).
Extended Data Fig. 3 |. Single cell RNA-seq analysis of WT vs Il6−/− CVI mice.
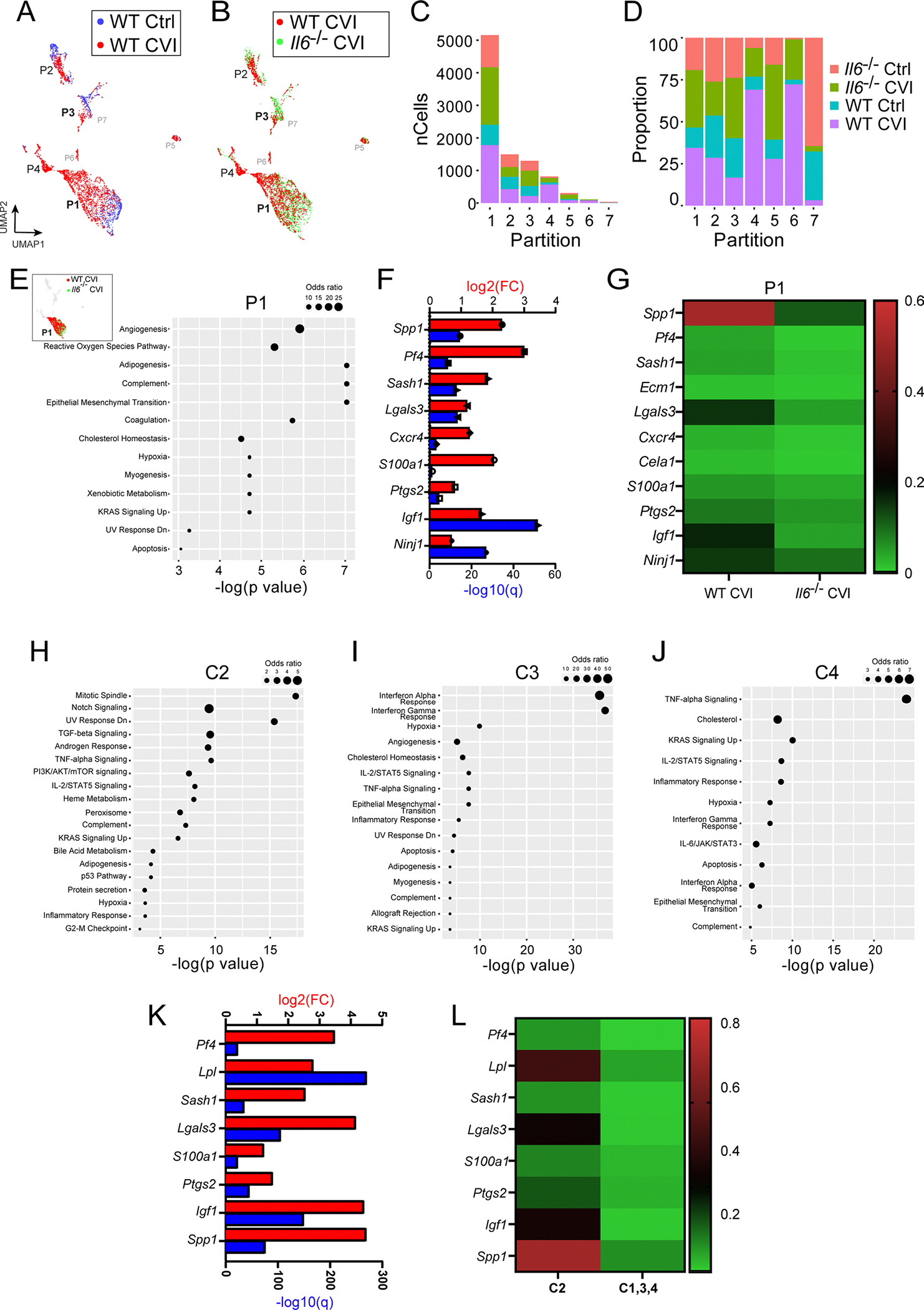
a-b, UMAPs show overlays of all single cell partitions for uninjured Ctrl versus WT D6 CVI (a) and WT CVI versus Il6−/− CVI mice (b). c-d, Bar graphs depict the number (c) and percentage (d) of cells corresponding to each mouse group for the 7 different partitions. e, UMAP inset shows an overlay of WT CVI versus Il6−/− CVI mice for all partitions. Top pathways are also shown based on gene set enrichment analysis of DEGs (Q<0.05) between injured WT versus Il6−/− mice in P1. Only statistically significant pathway terms are plotted on the graph (P<0.05; Fisher exact test), and dot size represents the odds ratio. f, Bar graph shows statistical significance (−log q value; blue) and the expression level (log2 fold change; red) for genes involved in angiogenesis, the top-rated signaling pathway from gene set enrichment analysis in (e). g, Heatmap shows the mean expression of genes associated with angiogenesis for injured WT and Il6−/− mice. The color legend (right) depicts the gene expression level. h-j, Top pathways are shown based on gene set enrichment analysis of DEGs (Q<0.05) between uninjured and injured WT mice in P1 C2 (h), 3 (i), and 4 (j). Only statistically significant pathway terms are plotted on the graphs (P<0.05; Fisher exact test), and dot size represents the odds ratio. k, Bar graph shows statistical significance (−log q value; blue) and the expression level (log2 fold change; red) for genes involved in angiogenesis for C2 vs C1, 3, and 4 within P1. l, Heatmap shows the mean expression of genes associated with angiogenesis in panel k.
Extended Data Fig. 4 |. Analysis of signaling pathways in microglia partitions identified by scRNA-seq.
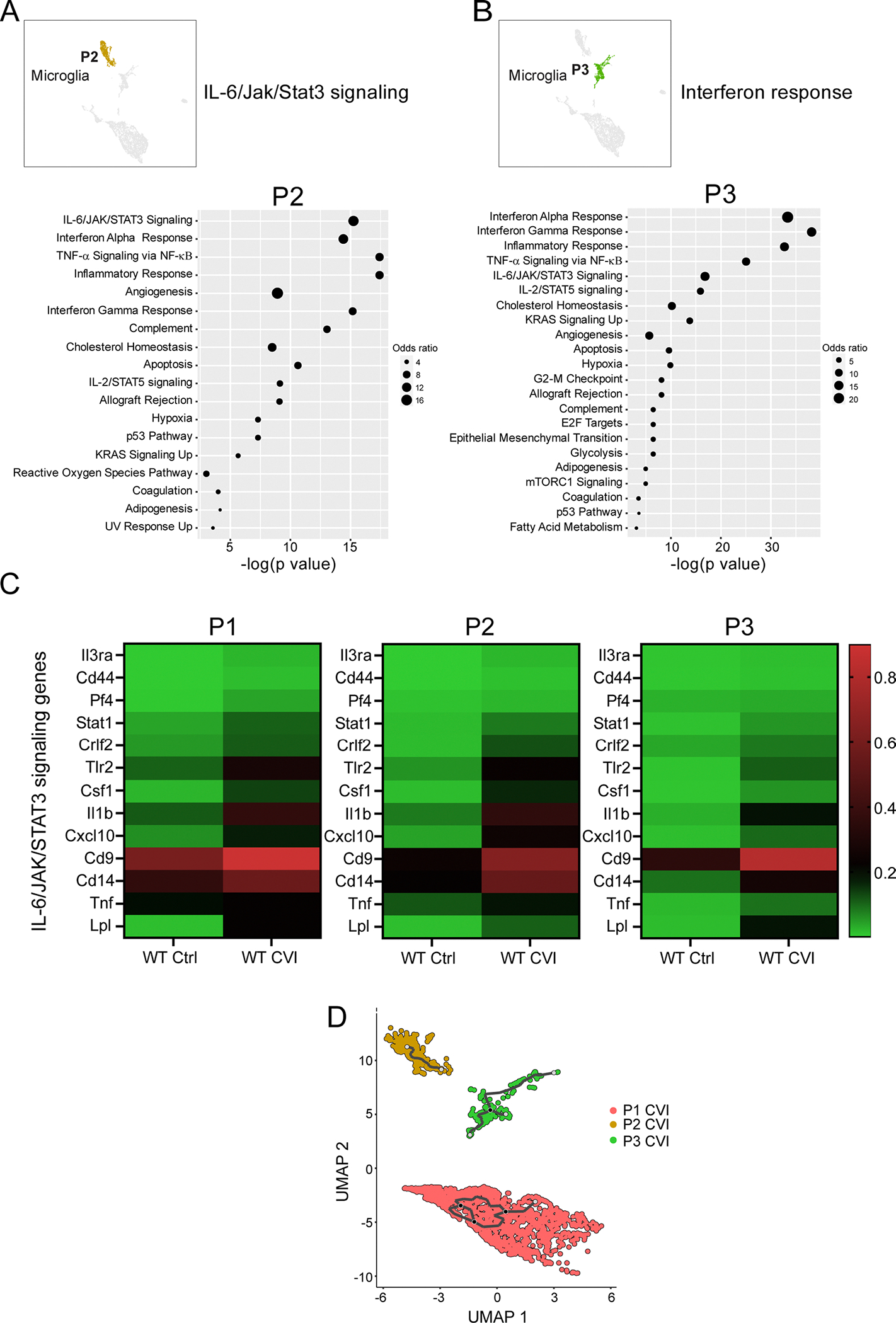
a-b, UMAP insets show all partitions and the location of P2 (a) and 3 (b). Dot plots depict the top pathways in the respective partitions based on gene set enrichment analysis of DEGs (Q<0.05) between WT Ctrl and CVI mice. Dot size corresponds to the odds ratio. Only statistically significant pathways (P<0.05) are shown on the graph. Pathways are ordered based on the negative log of the P value multiplied by the corresponding Z score. The top ranked pathways are listed next to the UMAP insets. c, Heatmaps show mean expression levels of genes associated with IL-6/JAK/Stat3 signaling between WT and Il6−/− CVI mice for P1, 2, and 3. The color legend (right) depicts the gene expression level. d, UMAP plot shows differentiation trajectories for P1, 2, and 3 microglia. Individual dots represent single cells and the distance between two cells infers transcriptional similarity. Black traces represent trajectory branches. Each light grey circle denotes a different outcome of the trajectory. Black circles indicate branch nodes, from which cells can move to one of the denoted outcomes.
Extended Data Fig. 5 |. scRNA-seq analysis of macrophages and flow cytometric purity of sorted monocytes.
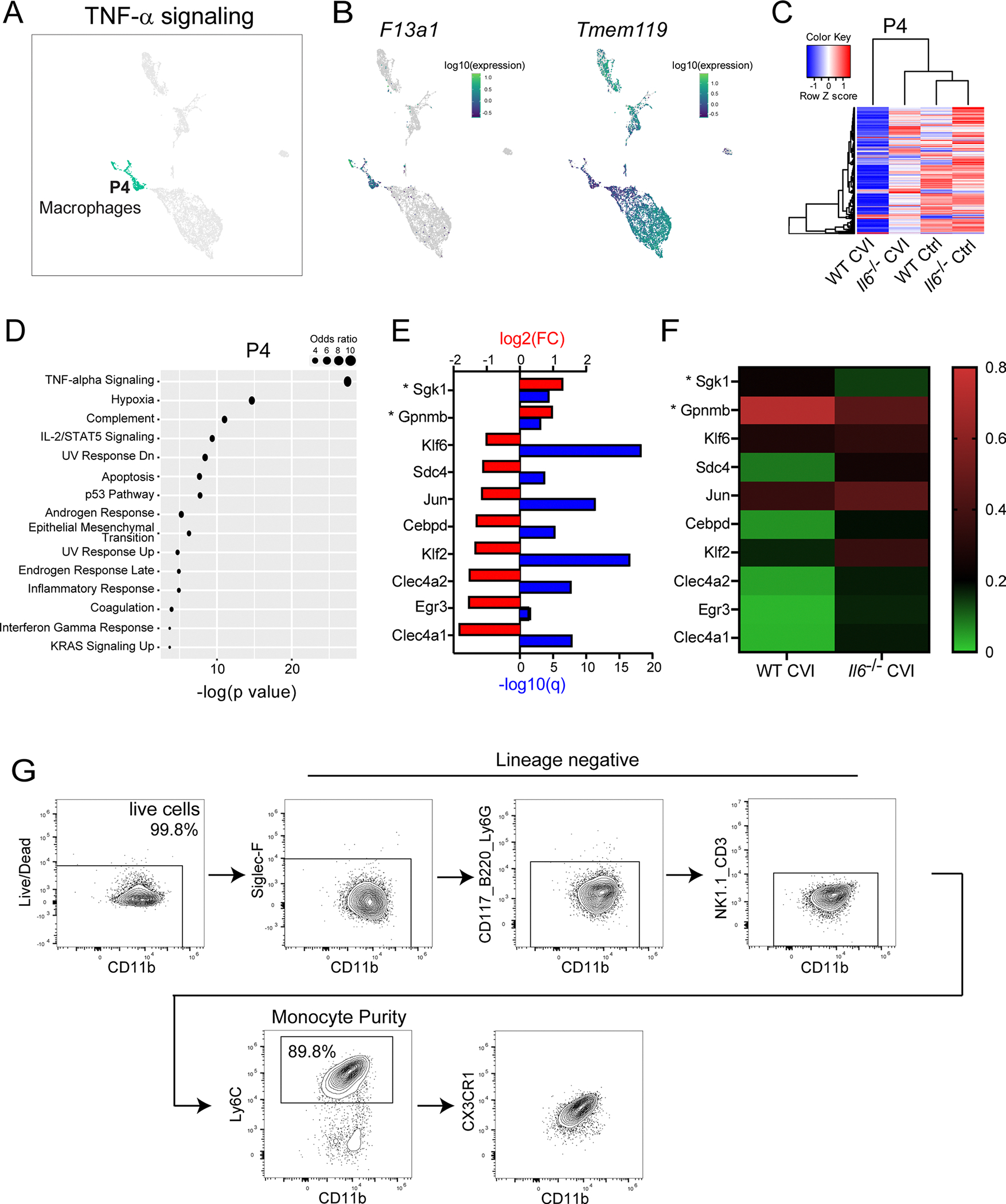
a, UMAP shows all partitions and the location of P4, which corresponds to macrophages. The top ranked pathway in P4 is noted above the UMAP. b, UMAPs depict expression of F13a1 and Tmem119 in all partitions. c, Pearson correlation-based clustered heatmap using DEGs (Q<0.05) between WT Ctrl and CVI mice in P4. The color legend reflects the Z score. d, Dot plot depicts the top pathways in P4 based on gene set enrichment analysis of DEGs (q<0.05) between WT and Il6−/− CVI mice. Dot size corresponds to the odds ratio. Only statistically significant pathways (P<0.05) are shown on the graph. Pathways are ordered based on the negative log of the P value multiplied by the corresponding Z score. e, Bar graph shows statistical significance (−log q value; blue) and the expression level (log2 fold change; red) for genes involved in TNFα signaling, the top-rated signaling pathway from gene set enrichment analysis in (d). Asterisks denote suppressor genes. f, Heatmap shows mean expression levels of genes in (e) for WT and Il6−/− CVI mice. The color legend (right) depicts the gene expression level. g, Gating strategy used for flow cytometric analysis of purified bone marrow-derived monocytes (CD11b+ Ly6C+ CX3CR1+ CD3− Siglec F− NK1.1− Ly6G− CD117− CD220−) from WT or Il6−/− mice that were adoptively transferred into Ccr2−/− recipients as described in Fig. 5d,e.
Extended Data Fig. 6 |. Quantification of cell proliferation and vascular leakage after CVI.
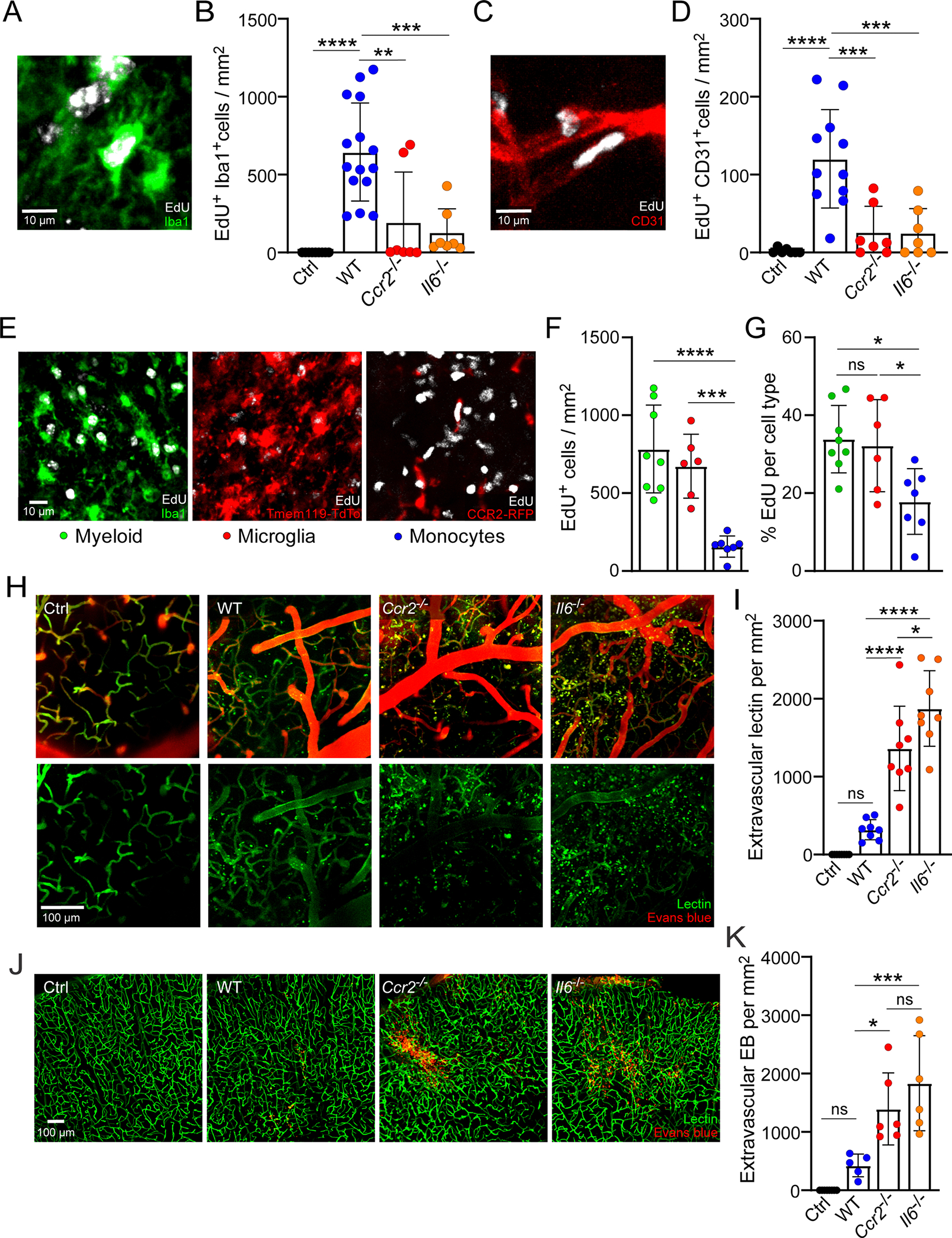
a-d, Representative confocal images from the neocortex stained for Iba1 (green) and EdU (white) (a) or CD31 (red) and EdU (white) (c). Bar graphs show the number of EdU+ Iba1+ cells (b) and EdU+ CD31+ cells (d) in uninjured WT Ctrl (n=9) versus D6 post-CVI WT (n=15), Ccr2−/− (n=7), and Il6−/− (n=7) mice at D6 post-CVI. Data are combined from two independent experiments. (**P<0.01, ***P<0.001, ****P<0.0001; one-way ANOVA with Tukey’s test). e-g, Representative confocal images from the neocortex stained for Iba1 (green), Tmem119-TdTo (red), CCR2-RFP (red), and EdU (white). Bar graphs show the number (f) and percentage (g) of EdU+ cells that are myeloid (green), microglia (red), and monocytes (blue) based on analysis of WT (n=8), Tmem119-TdTo (n=6), and CX3CR1gfp/+ CCR2rfp/+ (n=7) mice at D6 post-CVI. Data are combined from two independent experiments. (*P<0.05, ***P<0.001, ****P<0.0001; one-way ANOVA with Tukey’s test). h, Representative intravital TPM images captured through the thinned skull of uninjured WT Ctrl (n=8) versus CVI WT (n=8), Ccr2−/− (n=8), and Il6−/− (n=8) mice at D10 post-mild CVI. Tomato lectin (green) and Evans blue (red) were injected i.v. to visualize vasculature and dye extravasation. i, Bar graph depicts quantification of extravasated tomato lectin based on the dataset in (h). Data are combined from two independent experiments. (*P<0.05, ****P<0.0001; one-way ANOVA with Tukey’s test). j, Representative confocal images from the neocortex of uninjured WT Ctrl (n=7) versus D6 post-CVI WT (n=5), Ccr2−/− (n=6), and Il6−/− (n=6) mice at D10 post-CVI show i.v. injected tomato lectin (green) and Evans blue (red). k, Bar graph shows quantification of extravasated Evans blue (EB) based on the dataset shown in (j). Data are combined from two independent experiments. (*P<0.05, ***P<0.001; one-way ANOVA with Tukey’s test). All graphs show mean ± SEM. Dots represent individual mice.
Extended Data Fig. 7 |. Evaluation of Y-maze performance in uninjured mice and quantification of type 1 collagen deposition and Evans blue leakage after CVI.
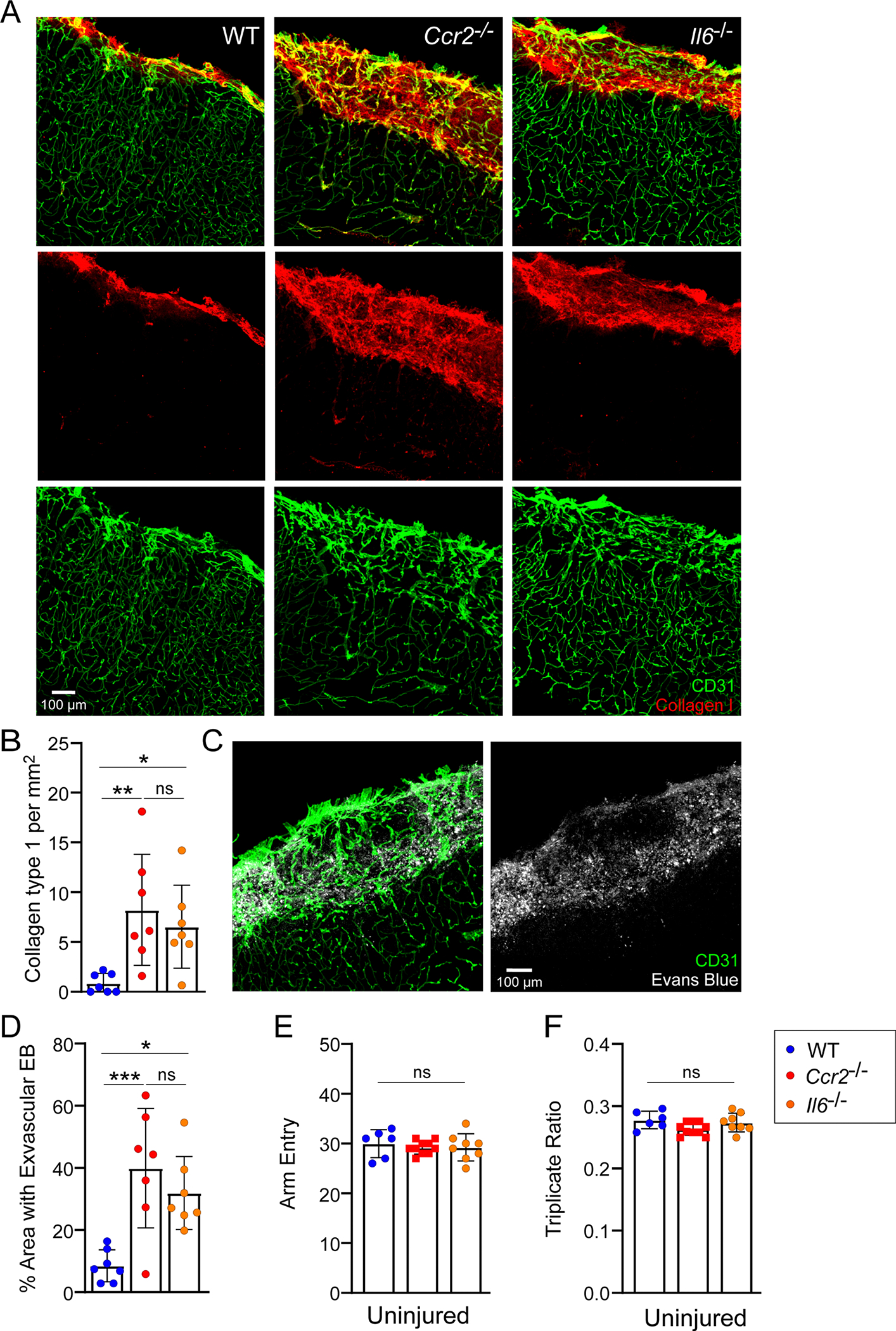
a, Representative confocal images from the neocortex of WT (n=7), Ccr2−/− (n=7), and Il6−/− (n=7) mice at D10 post-CVI show staining for CD31 (green) and collagen type 1 (red). b, Bar graph shows quantification of collagen type 1 based on the dataset shown in (a). Data are combined from two independent experiments. (*P<0.05, **P<0.01; one-way ANOVA with Tukey’s test). c, Representative confocal images from the neocortex of WT (n=7), Ccr2−/− (n=7), and Il6−/− (n=7) mice at D10 post-CVI show staining for CD31 (green) and i.v. injected Evans blue (white). d, Bar graph shows quantification of extravasated Evans blue (EB) based on the dataset shown in (c). Data are combined from two independent experiments. (*P<0.05, ***P<0.001; one-way ANOVA with Tukey’s test). All graphs show mean ± SEM. Dots represent individual mice. e-f, Bar graphs demonstrate the total number of Y maze arm entries (e) and the triplicate ratio (f) for uninjured WT (n=6), Ccr2−/− (n=10), and Il6−/− (n=8) mice. Data are from a single experiment. All graphs show mean ± SEM (one-way ANOVA with Tukey’s test). Dots represent individual mice.
Supplementary Material
Acknowledgements
This research was supported by the intramural program at the NINDS, NIH. We thank A. Elkahloun and B. Sisay in the National Human Genome Research Institute Microarray core for their assistance with the scRNA-seq experiment. The present study utilized the computational resources of the NIH Biowulf HPC cluster (http://hpc.nih.gov).
Footnotes
Competing interests
The authors declare no competing interests.
Ethics
The present study was carried out in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the NIH. The protocol was approved by the NINDS ACUC (protocol no. 1295–20).
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Additional information
Extended data is available for this paper at https://doi.org/10.1038/s41590-023-01521-1.
Peer review information Nature Immunology thanks the anonymous reviewer(s) for their contribution to the peer review of this work. Primary Handling Editor: L. A. Dempsey, in collaboration with the Nature Immunology team.
Reprints and permissions information is available at www.nature.com/reprints.
Supplementary information The online version contains supplementary material available at https://doi.org/10.1038/s41590-023-01521-1.
Data availability
The data that support the findings of the present study are available from the corresponding author upon request. There are no restrictions on data availability. ScRNA-seq data are available in the National Center for Biotechnology Information Gene Expression Omnibus under accession no. GSE215312. The mouse genome database used in our scRNA-seq analysis was mm10 (https://www.ncbi.nlm.nih.gov/assembly/GCF_000001635.20). Source data are provided with this paper.
References
- 1.Parikh NS, Merkler AE & Iadecola C Inflammation, autoimmunity, infection, and stroke: epidemiology and lessons from therapeutic intervention. Stroke 51, 711–718 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kenney K et al. Cerebral vascular injury in traumatic brain injury. Exp. Neurol. 275, 353–366 (2016). [DOI] [PubMed] [Google Scholar]
- 3.Schneider JA & Bennett DA Where vascular meets neurodegenerative disease. Stroke 41, S144–S146 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Badjatia N & Rosand J Intracerebral hemorrhage. Neurologist 11, 311–324 (2005). [DOI] [PubMed] [Google Scholar]
- 5.Twitchell TE The restoration of motor function following hemiplegia in man. Brain 74, 443–480 (1951). [DOI] [PubMed] [Google Scholar]
- 6.Rossini PM, Calautti C, Pauri F & Baron JC Post-stroke plastic reorganisation in the adult brain. Lancet Neurol. 2, 493–502 (2003). [DOI] [PubMed] [Google Scholar]
- 7.Furlan M, Marchal G, Viader F, Derlon JM & Baron JC Spontaneous neurological recovery after stroke and the fate of the ischemic penumbra. Ann. Neurol. 40, 216–226 (1996). [DOI] [PubMed] [Google Scholar]
- 8.Lalancette-Hebert M, Gowing G, Simard A, Weng YC & Kriz J Selective ablation of proliferating microglial cells exacerbates ischemic injury in the brain. J. Neurosci. 27, 2596–2605 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Narantuya D et al. Human microglia transplanted in rat focal ischemia brain induce neuroprotection and behavioral improvement. PLoS ONE 5, e11746 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mastorakos P, Russo MV, Zhou T, Johnson K & McGavern DB Antimicrobial immunity impedes CNS vascular repair following brain injury. Nat. Immunol. 22, 1280–1293 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schaeffer S & Iadecola C Revisiting the neurovascular unit. Nat. Neurosci. 24, 1198–1209 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Park L et al. Brain perivascular macrophages initiate the neurovascular dysfunction of Alzheimer abeta peptides. Circ. Res. 121, 258–269 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nortley R et al. Amyloid beta oligomers constrict human capillaries in Alzheimer’s disease via signaling to pericytes. Science 365, eaav9518 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mastorakos P & McGavern D The anatomy and immunology of vasculature in the central nervous system. Sci. Immunol. 4, eaav0492 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Davalos D et al. ATP mediates rapid microglial response to local brain injury in vivo. Nat. Neurosci. 8, 752–758 (2005). [DOI] [PubMed] [Google Scholar]
- 16.Nimmerjahn A, Kirchhoff F & Helmchen F Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science 308, 1314–1318 (2005). [DOI] [PubMed] [Google Scholar]
- 17.Roth TL et al. Transcranial amelioration of inflammation and cell death after brain injury. Nature 505, 223–228 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mastorakos P et al. Temporally distinct myeloid cell responses mediate damage and repair after cerebrovascular injury. Nat. Neurosci. 24, 245–258 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lou N et al. Purinergic receptor P2RY12-dependent microglial closure of the injured blood-brain barrier. Proc. Natl Acad. Sci. USA 113, 1074–1079 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dimitrijevic OB, Stamatovic SM, Keep RF & Andjelkovic AV Absence of the chemokine receptor CCR2 protects against cerebral ischemia/reperfusion injury in mice. Stroke 38, 1345–1353 (2007). [DOI] [PubMed] [Google Scholar]
- 21.Perego C, Fumagalli S & De Simoni MG Temporal pattern of expression and colocalization of microglia/macrophage phenotype markers following brain ischemic injury in mice. J. Neuroinflam. 8, 174 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wattananit S et al. Monocyte-derived macrophages contribute to spontaneous long-term functional recovery after stroke in mice. J. Neurosci. 36, 4182–4195 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Prinz M, Jung S & Priller J Microglia biology: one century of evolving concepts. Cell 179, 292–311 (2019). [DOI] [PubMed] [Google Scholar]
- 24.Keren-Shaul H et al. A unique microglia type associated with restricting development of Alzheimer’s disease. Cell 169, 1276–1290 e1217 (2017). [DOI] [PubMed] [Google Scholar]
- 25.Nayak D, Roth TL & McGavern DB Microglia development and function. Annu. Rev. Immunol. 32, 367–402 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Stratoulias V, Venero JL, Tremblay ME & Joseph B Microglial subtypes: diversity within the microglial community. EMBO J. 38, e101997 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Reitmeir R et al. Vascular endothelial growth factor induces contralesional corticobulbar plasticity and functional neurological recovery in the ischemic brain. Acta Neuropathol. 123, 273–284 (2012). [DOI] [PubMed] [Google Scholar]
- 28.Kaiser T & Feng G Tmem119-EGFP and Tmem119-CreERT2 transgenic mice for labeling and manipulating microglia. eNeuro 6, ENEURO.0448–18.2019 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ferrara N & Henzel WJ Pituitary follicular cells secrete a novel heparin-binding growth factor specific for vascular endothelial cells. Biochem. Biophys. Res. Commun. 161, 851–858 (1989). [DOI] [PubMed] [Google Scholar]
- 30.Van Snick J Interleukin-6: an overview. Annu. Rev. Immunol. 8, 253–278 (1990). [DOI] [PubMed] [Google Scholar]
- 31.Peters M et al. Combined interleukin 6 and soluble interleukin 6 receptor accelerates murine liver regeneration. Gastroenterology 119, 1663–1671 (2000). [DOI] [PubMed] [Google Scholar]
- 32.Saederup N et al. Selective chemokine receptor usage by central nervous system myeloid cells in CCR2-red fluorescent protein knock-in mice. PLoS ONE 5, e13693 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Serbina NV & Pamer EG Monocyte emigration from bone marrow during bacterial infection requires signals mediated by chemokine receptor CCR2. Nat. Immunol. 7, 311–317 (2006). [DOI] [PubMed] [Google Scholar]
- 34.Tanaka T, Narazaki M & Kishimoto T Interleukin (IL-6) Immunotherapy. Cold Spring Harb. Perspect. Biol. 10, a028456. (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chen EY et al. Enrichr: interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinf. 14, 128 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Marech I et al. Classical and non-classical proangiogenic factors as a target of antiangiogenic therapy in tumor microenvironment. Cancer Lett. 380, 216–226 (2016). [DOI] [PubMed] [Google Scholar]
- 37.Hayashi T, Noshita N, Sugawara T & Chan PH Temporal profile of angiogenesis and expression of related genes in the brain after ischemia. J. Cereb. Blood Flow Metab. 23, 166–180 (2003). [DOI] [PubMed] [Google Scholar]
- 38.Krupinski J, Kaluza J, Kumar P, Kumar S & Wang JM Role of angiogenesis in patients with cerebral ischemic stroke. Stroke 25, 1794–1798 (1994). [DOI] [PubMed] [Google Scholar]
- 39.Kato H, Kogure K, Liu XH, Araki T & Itoyama Y Progressive expression of immunomolecules on activated microglia and invading leukocytes following focal cerebral ischemia in the rat. Brain Res. 734, 203–212 (1996). [PubMed] [Google Scholar]
- 40.Schroeter M, Jander S, Huitinga I, Witte OW & Stoll G Phagocytic response in photochemically induced infarction of rat cerebral cortex. The role of resident microglia. Stroke 28, 382–386 (1997). [DOI] [PubMed] [Google Scholar]
- 41.Gliem M et al. Macrophages prevent hemorrhagic infarct transformation in murine stroke models. Ann. Neurol. 71, 743–752 (2012). [DOI] [PubMed] [Google Scholar]
- 42.Gliem M et al. Macrophage-derived osteopontin induces reactive astrocyte polarization and promotes re-establishment of the blood brain barrier after ischemic stroke. Glia 63, 2198–2207 (2015). [DOI] [PubMed] [Google Scholar]
- 43.Hirano T, Akira S, Taga T & Kishimoto T Biological and clinical aspects of interleukin 6. Immunol. Today 11, 443–449 (1990). [DOI] [PubMed] [Google Scholar]
- 44.Lin ZQ, Kondo T, Ishida Y, Takayasu T & Mukaida N Essential involvement of IL-6 in the skin wound-healing process as evidenced by delayed wound healing in IL-6-deficient mice. J. Leukoc. Biol. 73, 713–721 (2003). [DOI] [PubMed] [Google Scholar]
- 45.Atreya R & Neurath MF Involvement of IL-6 in the pathogenesis of inflammatory bowel disease and colon cancer. Clin. Rev. Allergy Immunol. 28, 187–196 (2005). [DOI] [PubMed] [Google Scholar]
- 46.Gertz K et al. Essential role of interleukin-6 in post-stroke angiogenesis. Brain 135, 1964–1980 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Clark WM et al. Lack of interleukin-6 expression is not protective against focal central nervous system ischemia. Stroke 31, 1715–1720 (2000). [DOI] [PubMed] [Google Scholar]
- 48.Gronhoj MH, Clausen BH, Fenger CD, Lambertsen KL & Finsen B Beneficial potential of intravenously administered IL-6 in improving outcome after murine experimental stroke. Brain Behav. Immun. 65, 296–311 (2017). [DOI] [PubMed] [Google Scholar]
- 49.Li Z et al. M-CSF, IL-6, and TGF−beta promote generation of a new subset of tissue repair macrophage for traumatic brain injury recovery. Sci. Adv. 7, eabb6260 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ormstad H, Aass HC, Lund-Sorensen N, Amthor KF & Sandvik L Serum levels of cytokines and C-reactive protein in acute ischemic stroke patients, and their relationship to stroke lateralization, type, and infarct volume. J. Neurol. 258, 677–685 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Waje-Andreassen U et al. IL-6: an early marker for outcome in acute ischemic stroke. Acta Neurol. Scand. 111, 360–365 (2005). [DOI] [PubMed] [Google Scholar]
- 52.Kitamura Y et al. Recovery of focal brain ischemia-induced behavioral dysfunction by intracerebroventricular injection of microglia. J. Pharm. Sci. 97, 289–293 (2005). [DOI] [PubMed] [Google Scholar]
- 53.Koike K et al. Synergism of BSF-2/interleukin 6 and interleukin 3 on development of multipotential hemopoietic progenitors in serum-free culture. J. Exp. Med. 168, 879–890 (1988). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bot FJ, van Eijk L, Broeders L, Aarden LA & Lowenberg B Interleukin-6 synergizes with M-CSF in the formation of macrophage colonies from purified human marrow progenitor cells. Blood 73, 435–437 (1989). [PubMed] [Google Scholar]
- 55.Uyttenhove C, Coulie PG & Van Snick J T cell growth and differentiation induced by interleukin-HP1/IL-6, the murine hybridoma/plasmacytoma growth factor. J. Exp. Med. 167, 1417–1427 (1988). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hu X et al. Microglia/macrophage polarization dynamics reveal novel mechanism of injury expansion after focal cerebral ischemia. Stroke 43, 3063–3070 (2012). [DOI] [PubMed] [Google Scholar]
- 57.Askenase MH et al. Longitudinal transcriptomics define the stages of myeloid activation in the living human brain after intracerebral hemorrhage. Sci. Immunol. 6, eabd6279 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Garbers C & Rose-John S Dissecting interleukin-6 classic- and trans-signaling in inflammation and cancer. Methods Mol. Biol. 1725, 127–140 (2018). [DOI] [PubMed] [Google Scholar]
- 59.Willis EF et al. Repopulating microglia promote brain repair in an IL-6-dependent manner. Cell 180, 833–846 e816 (2020). [DOI] [PubMed] [Google Scholar]
- 60.Leibinger M et al. Transneuronal delivery of hyper-interleukin-6 enables functional recovery after severe spinal cord injury in mice. Nat. Commun. 12, 391 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Barrett JP et al. Interferon-beta plays a detrimental role in experimental traumatic brain injury by enhancing neuroinflammation that drives chronic neurodegeneration. J. Neurosci. 40, 2357–2370 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
References
- 62.Kopf M et al. Impaired immune and acute-phase responses in interleukin-6-deficient mice. Nature 368, 339–342 (1994). [DOI] [PubMed] [Google Scholar]
- 63.McFarland-Mancini MM et al. Differences in wound healing in mice with deficiency of IL-6 versus IL-6 receptor. J. Immunol. 184, 7219–7228 (2010). [DOI] [PubMed] [Google Scholar]
- 64.Jung S et al. Analysis of fractalkine receptor CX(3)CR1 function by targeted deletion and green fluorescent protein reporter gene insertion. Mol. Cell. Biol. 20, 4106–4114 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Madisen L et al. A robust and high-throughput Cre reporting and characterization system for the whole mouse brain. Nat. Neurosci. 13, 133–140 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Becht E et al. Dimensionality reduction for visualizing single-cell data using UMAP. Nat. Biotechnol. 37, 38–44 (2019). [DOI] [PubMed] [Google Scholar]
- 67.Levine JH et al. Data-driven phenotypic dissection of AML reveals progenitor-like cells that correlate with prognosis. Cell 162, 184–197 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that support the findings of the present study are available from the corresponding author upon request. There are no restrictions on data availability. ScRNA-seq data are available in the National Center for Biotechnology Information Gene Expression Omnibus under accession no. GSE215312. The mouse genome database used in our scRNA-seq analysis was mm10 (https://www.ncbi.nlm.nih.gov/assembly/GCF_000001635.20). Source data are provided with this paper.