

Abstract
Metastasis to the liver is a leading cause of death in patients with colorectal cancer. To investigate the characteristics of cancer cells prone to metastasis, we utilized an isogenic model of BALB/c and colon tumor 26 (C26) cells carrying an active KRAS mutation. Liver metastatic (LM) 1 cells were isolated from mice following intrasplenic transplantation of C26 cells. Subsequent injections of LM1 cells generated LM2 cells, and after four cycles, LM4 cells were obtained. In vitro, using a perfusable capillary network system, we found comparable extravasation frequencies between C26 and LM4 cells. Both cell lines showed similar growth rates in vitro. However, C26 cells showed higher glucose consumption, whereas LM4 cells incorporated more fluorescent fatty acids (FAs). Biochemical analysis revealed that LM4 cells had higher cholesterol levels than C26 cells. A correlation was observed between fluorescent FAs and cholesterol levels detected using filipin III. LM4 cells utilized FAs as a source for cholesterol synthesis through acetyl‐CoA metabolism. In cellular analysis, cholesterol accumulated in punctate regions, and upregulation of NLRP3 and STING proteins, but not mTOR, was observed in LM4 cells. Treatment with a cholesterol synthesis inhibitor (statin) induced LM4 cell death in vitro and suppressed LM4 cell growth in the livers of nude mice. These findings indicate that colorectal cancer cells prone to liver metastasis show cholesterol‐dependent growth and that statin therapy could help treat liver metastasis in immunocompromised patients.
Keywords: cholesterol, colorectal cancer, extravasation, fatty acid, liver metastasis
In this study, liver metastasis‐prone LM4 cells were isolated from C26 mouse colorectal cancer cells, and metabolic pathways were found to be altered. Although C26 cells utilized glucose, LM4 cells incorporated FAs and contained higher cholesterol levels. De novo cholesterol synthesis is required for LM4 cell growth in vitro. In mice without T cells, statin treatment suppressed LM4 cell growth in the liver.
1. INTRODUCTION
Colorectal cancer (CRC) is the third most common cancer worldwide. 1 In 2019 and 2021, CRC showed the highest incidence and the second‐highest mortality rates in Japan. Generally, mortality is dependent on metastasis. In CRC, the liver is the most frequent site of metastasis 2 because CRC cells predominantly spread through the mesenteric circulation to the liver. 3 During initial diagnosis, approximately 25% of patients have synchronous liver metastasis, and liver metastasis develops within 5 years in an additional 25% of patients. 2 , 4 Therefore, understanding the characteristics of CRC cells that metastasize to the liver is crucial for developing inhibitors against them. 5
Various animal models have been established to understand the mechanisms of liver metastasis. 6 As an isogenic model, colon tumor 26 (CT26) is commonly used to investigate metastatic mechanisms in vivo. In a previous study, CT26, an undifferentiated colon carcinoma cell line, was derived from female BALB/c mice treated with N‐nitroso‐N‐methylurea intrarectally. 7 Subsequently, C‐C26, a cultured cell line derived from CT26 (hereafter referred to as cell line C26), was established, 8 revealing a G12D mutation in KRAS. 9 , 10 Implanting C26 cells in the subcutaneous region resulted in lung metastasis in 1 out of 11 mice. 8 Injecting these cells into the rectal smooth muscle layer led to metastasis in the lungs, liver, and lymph nodes. 11 These findings suggest that C26 cells contain subclones that preferentially metastasize to various organs. Despite numerous studies, the characteristics of CRC cells prone to liver metastasis remain unresolved. Given that CRC cells travel from the colon to the liver by way of the portal vein, this route was mimicked by transplanting cancer cells into the spleen. 12 , 13 Using this model, we herein attempted to isolate CRC cells that preferentially metastasize to the liver.
2. MATERIALS AND METHODS
Reagents, cells, Abs, and plasmids used in this study, along with their respective sources, are listed in Table S1. Methods are described in Document S1.
2.1. Cells
C26 and LM4 cells were cultured in RPMI‐1640 supplemented with 10% FBS, 100 units/mL penicillin, and 100 μg/mL streptomycin in a 5% CO2 humidified incubator at 37°C. Retroviruses were used to establish stable cell lines expressing green or cyan fluorescent proteins (GFP or CFP, respectively). These viruses were generated in BOSC23 cells transfected with expression vectors (pCX4neo‐mGFP or pCX4bsr‐m1CFP), a retroviral packaging plasmid pGP, 14 and an envelope plasmid pCMV‐VSV‐G‐Rsv‐Rev. Following infection, cells expressing GFP or CFP were selected using antibiotics G418 or blasticidin and subsequently sorted using an SH800S Cell Sorter (Sony Corporation).
2.2. Isolation of liver metastatic cells
To isolate liver metastatic (LM)1 cells, 1 × 106 C26 cells were injected into the spleen of female BALB/c mice (Figure 1). The spleen was removed 1 min after tumor injection. After specified intervals, mice were euthanized, and their livers were excised. Tumor colonies in the liver were isolated, minced with scissors, and plated onto plastic dishes. These cells were designated as LM1. Similarly, LM2–LM4 cells were isolated by injecting LM1–LM3 cells sequentially.
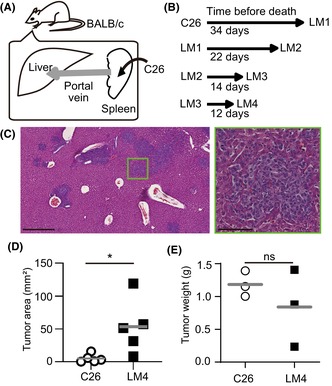
FIGURE 1.
Mouse model for liver metastasis in colorectal cancer. (A) Schematic view of intrasplenic transplantation of C26 cells. (B) Duration of transplantation of C26 and liver metastatic (LM) 4 cells until autopsy. (C) Representative H&E staining image of LM4 cells in the liver. Scale bars, 500 μm (left panel) and 100 μm (right panel). (D) Comparison of the tumor area in the liver. (E) Comparison of the tumor weight transplanted in the skin (14 days after transplantation). Bars indicate the mean of each group. *p < 0.05; t‐test. ns, not significant.
2.3. Histopathological analysis
BALB/c mice injected with 1.0 × 106 C26 or LM4 cells into the spleen were euthanized after 6 or 7 days. The excised livers were fixed in neutralized 10% formalin (Mildform, Fujifilm Wako Chemicals) and processed for H&E staining. Slides were scanned using a Nanozoomer (Hamamatsu Photonics), and the tumor area was quantified manually.
2.4. In vitro observation of the extravasation process
Perfusable capillary networks were established in vitro following a previously reported method. 15 Briefly, 150 μL of a fibrin/collagen gel solution containing 5.0 × 106 HUVECs expressing red fluorescent protein was placed in a 35 mm glass‐bottom tissue culture dish with a glass separator (Figure 2A) and allowed to solidify. Before applying the cell‐containing fibrin gel, two thin polydimethylsiloxane pieces, approximately 6–8 mm in size, were placed on the glass bottom. Subsequently, 100 μL of the fibrin gel solution containing 5.0 × 105 human lung fibroblasts (Lonza) was added to the edge of the dish and allowed to solidify. The cells were cultured for 7 days in Endothelial Cell Growth Medium 2 (EGM‐2, Lonza) supplemented with 10 μM tranexamic acid. Once the vascular network was established, both ends of the preformed network were cut using a sharp scalpel to open the channels. Live C26‐CFP cells were stained with 20 μM (CellTracker Blue CMAC, ThermoFisher) for 30 min at 37°C. Following removal of the medium from both wells, 2 mL of EGM‐2 medium containing a mixture of 5.0 × 105 LM4‐GFP cells and 5.0 × 105 CMAC‐stained C26‐CFP cells (1.0 × 106 cells in total) was added to one of the wells. Time‐lapse imaging was carried out for 24 h using a Nikon A1R microscope. Z‐stack images were acquired after 24 h of perfusion to quantify the number of extravasated cells.
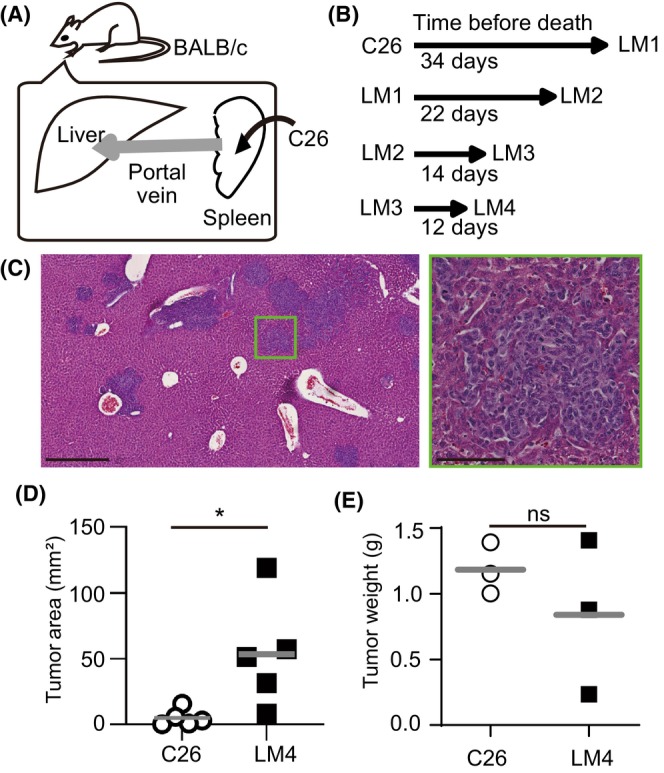
FIGURE 2.
In vitro extravasation of C26 and liver metastatic (LM) 4 cells. (A) Schematic view of in vitro capillary networks. A 35 mm glass‐bottom culture dish (blue) was divided. HUVECs were cultured in fibrin gel (yellow) on the glass bottom to form vessel structures (red lines). Dashed lines in the fibrin indicate cutting lines. Right images show the formed vessels. (B) Representative time‐lapse images of the extravasation process of C26‐CFP and LM4‐GFP cells. LM4 cells (arrows) flowed within the vascular lumen, protruded toward the extravascular region, and migrated outside the lumen. C26 cells transitioned (arrowhead) into LM4 cells. Scale bar, 10 μm. (C) Quantification of extravasated cells per dish. Data were obtained from 12 independent dishes per group (three dishes per four experiments). The lower graph is displayed on a different scale from the upper graph. p = 0.068; Wilcoxon signed‐rank test.
2.5. Live cell imaging
Cells (2.0 × 104) were plated on a CELLview quadrant glass‐bottom 35 mm dish (Greiner Bio‐One), cultured in 500 μL CO2‐independent medium supplemented with 10% FBS and penicillin/streptomycin, and overlaid with mineral oil. The cells were illuminated using pE400max (COOL LED; Andover) and imaged using an IX81 inverted microscope (Evident, Tokyo, Japan) equipped with a UPlan FLN 10× PH/0.3 objective lens (Evident, 2 × 2 binning), maintained at 37°C throughout the 3‐day imaging period. The position of each stage was captured every 20 min. Images were processed using MetaMorph software (Molecular Devices).
2.6. Crystal violet cell viability assay
Viable cells were quantified as previously described with some modifications. 16 Briefly, C26 or LM4 cells were plated in 12‐well dishes and cultured in RPMI‐1640 or CO2‐independent medium supplemented with 10% FBS and penicillin/streptomycin. In certain experiments, various inhibitors were added to the media 24 h later, and cells were cultured for an additional 48 h. After washing with PBS, cells were fixed with Mildform, stained with 0.5% crystal violet, washed with water, and then dissolved in 2% SDS. The absorbance at 550 nm was measured using a Multiscan GO spectrophotometer (Thermo Fisher Scientific) to quantify the number of viable cells.
2.7. Extracellular flux analysis
XF24 (Agilent) was used to measure the oxygen consumption rate (OCR) as previously described with some modifications. 17 Briefly, 8 × 104 cells were plated onto an XF24 cell culture plate (#100777‐004; Agilent) with complete medium. For the mitochondrial stress test, the medium was changed to unbuffered DMEM containing 25 mM glucose, 1× GlutaMAX (Thermo Fisher Scientific K.K.), and 1 mM pyruvate, and cells were incubated at 37°C for 1 h without CO2 control. Oligomycin A (1 μM) was injected to inhibit ATP synthase. Maximum OCR was induced by exposing cells to the mitochondrial uncoupler carbonyl cyanide‐p‐trifluoromethoxyphenylhydrazone (FCCP, 0.5 μM). To disrupt mitochondria‐dependent respiration, antimycin A (2 μM) and rotenone (2 μM) were added. Measurements were carried out using a 3 min protocol, followed by 2 min of mixing and 3 min of incubation.
2.8. Fluorescence fatty acid and filipin III quantification
Cells plated on 35 mm glass‐bottom dishes (Iwaki Glass) were cultured in M199 media without phenol red for 10 min and treated with fluorescent fatty acids (FAs). After another 10 min, cells were washed with PBS and fixed in 4% paraformaldehyde (PFA) for 20 min at room temperature. For cholesterol staining, after fixation with 4% PFA, cells were treated with 50 mM NH4Cl for 10 min, blocked with 10% FBS for 20 min, and then incubated with filipin III (100 μg/mL in blocking buffer). Fluorescent images were captured using an IX81 inverted epi‐fluorescent microscope (Evident) equipped with either a UPlanApo 20×/0.7 or UPlan FLN 10× PH/0.3 objective lens (Evident). For imaging of FAs and filipin III, cells were illuminated using pE400max at 490 and 365 nm, respectively, with exposure times of 10 and 100 ms, respectively. The cooled charge‐coupled device camera Cool SNAP‐HQ (Roper Scientific) was set to 4 × 4 binning. Excitation filters FF01‐485/20 (Semrock) and 365WB50 (Omega Optical), dichroic mirror 86006bs (Chroma), and emission filters FF01‐542/27 (Semrock) and FF01‐482/25 (Semrock) were used for FA and filipin III imaging, respectively. Images were processed using MetaMorph software (Molecular Devices). The area of individual cells or clusters was manually outlined. After subtracting background values, the intensities of each area were measured and exported to Excel (Microsoft Japan, Tokyo).
2.9. Protein separation and western blotting
Cells were lysed in 1× SDS sample buffer containing 2‐mercaptoethanol, sonicated, and incubated at 95°C. Proteins were separated by SDS‐PAGE on 5%–12% SuperSep precast gels (Fujifilm Wako Chemicals) and transferred to PVDF membranes (Immobilon; Millipore). After blocking with Intercept blocking buffer for 30 min, the membranes were incubated with various Abs (Table S1) diluted in Intercept blocking buffer (LI‐COR) solution and TBS, followed by incubation with secondary Abs. The membranes were then scanned using an Odyssey IR scanner (LI‐COR; Lincoln, NE, USA). Quantification was undertaken using Image Studio Lite software (LI‐COR Biosciences).
2.10. Bioluminescent analysis of liver metastasis
To establish LM4 cells expressing Akaluc, 18 LM4 cells were transfected with the expression vectors pPBbsr2_Venus‐Akaluc and pCMV‐mPBase. Several days later, cells were trypsinized and isolated using an SH800S Cell Sorter (Sony Corporation) with the corresponding filter sets. LM4 cells stably expressing Akaluc (2.5 × 105 cells/100 μL PBS in total) were injected into the spleens of anesthetized 13‐ or 11‐week‐old female BALB/c or BALB/c nu/nu mice. The mice were then anesthetized, injected s.c. with Aka Lumine (0.45 mg/mouse), and imaged using an IVIS Lumina XRMS (PerkinElmer). 19 After imaging, the mice were injected i.p. with DMSO or mevastatin (12.5 mg/kg) in PEG400. Following the final imaging session, the mice were euthanized, and their livers were processed for H&E staining.
2.11. Statistical analysis
Data were analyzed using GraphPad Prism 8 (GraphPad Prism Software).
3. RESULTS
3.1. Establishment of mouse model for CRC metastasis
To isolate cells that efficiently metastasize to the liver, C26 cells were injected into the spleen, and cells from the liver were isolated (Figure 1A). The isolated cells were named LM1, cultured for several passages in vitro, and reinjected into the spleen. LM4 cells were obtained after four cycles of injection and isolation. The autopsy date was determined based on noticeable cachexia changes in mice. As shown in Figure 1B, the time to autopsy was shortened after multiple cycles of injection and isolation.
To confirm the higher metastatic ability of LM4 cells, C26 and LM4 cells were injected into the spleens of mice. The liver tissues were analyzed by H&E staining (Figure 1C). As C26 cells lacked E‐cadherin expression and showed mesenchymal features (data not shown), LM4 cells showed similar morphology to C26. The tumor areas in livers transplanted with LM4 cells were significantly larger than those with C26 cells (Figure 1D). In contrast, when transplanted into the skin, C26 and LM4 cells showed comparable tumor weights (Figure 1E). These results suggest that LM4 cells possess enhanced metastatic potential or growth capability in the liver compared to C26 cells.
3.2. Interaction between CRC and endothelial cells
Two possible reasons were proposed to explain why more LM4 cells metastasize to the liver compared to C26: LM4 cells might show better extravasation or better proliferation in the liver than C26 cells. Recently, a microfluidic device with a perfusable vascular network was developed to observe immune cell dynamics in vitro. 15 In this system, HUVECs cultured with fibrin and fibroblasts form vessel structures (Figure 2A). In our study, we observed individual events of the extravasation process of C26‐CFP and LM4‐GFP cells in the same dish (Figure 2B, Movie S1). Contrary to our expectations, quantification indicated that LM4 cells showed fewer extravasation events compared to C26 cells (Figure 2C). This result suggests that extravasation might not be the primary cause of liver metastasis by LM4 cells.
3.3. Cell survival ability
The growth rates of C26 and LM4 cells in vitro were examined. As shown in Figure 3A, C26 and LM4 cells showed comparable growth rates. Subsequently, time‐lapse imaging of C26 and LM4 cells was carried out over 3 days without changing the medium (Movie S2). LM4 cells continued to proliferate, whereas C26 cells began to show signs of cell death at around 48 h. Quantitative analysis indicated that LM4 cells showed significantly better survival than C26 cells (Figure 3B). Furthermore, following addition of 100 mM glucose, more C26 cells survived at 48 and 72 h (Figure 3C). These results suggest that C26 cells consume more glucose compared to LM4 cells; that is, LM4 cells require less glucose for growth.
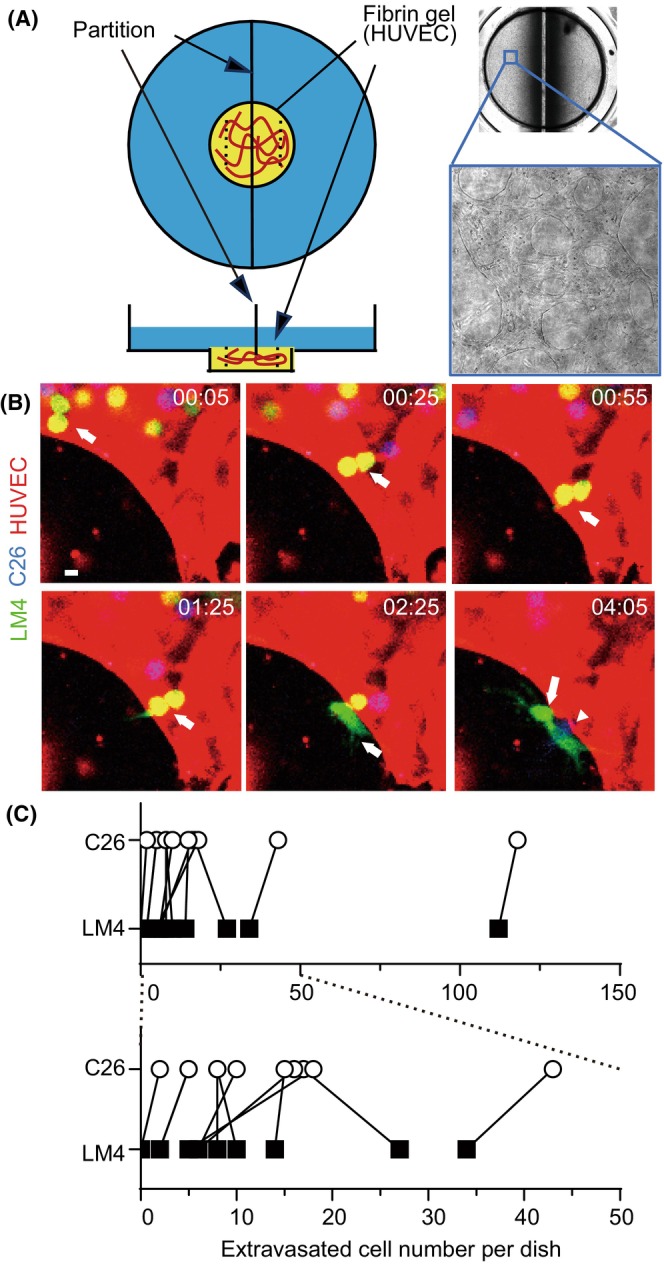
FIGURE 3.
In vitro cell growth of C26 and liver metastatic (LM) 4 cells. (A) Growth curves of C26 and LM4 cells in vitro. (B, C) Quantification of cell growth by crystal violet (CV) staining. Cells were cultured for the indicated periods, fixed, and stained with CV. Live imaging was captured for 72 h. (D) Representative images of the culture media from dishes without cells (m), C26 (C), and LM4 (L) cells. After 72 h, the culture media were collected, and pH was directly measured using a pH meter. pH values of C26 and LM4 cell cultures were subtracted from that of media alone (Δ pH). **p < 0.005; paired t‐test, 0.0016. (E) Oxygen consumption rate measured by XF24. Results represent the average of 10 independent experiments. Inverted triangles indicate the addition of reagents. *p < 0.05, **p < 0.005, ****p < 0.0001; Mann–Whitney U‐test. FCCP, Carbonyl cyanide‐p‐trifluoromethoxyphenylhydrazone.
3.4. Oxidative metabolism change in LM4 cells
During the experiments, we observed that the culture media of C26 cells, but not LM4 cells, turned yellowish after 72 h of cultivation (Figure 3D). Glucose, once incorporated into cells, is metabolized to pyruvate, which is further metabolized to lactate. Lactate, along with protons, is transported out of cells, leading to media acidification. 20 As shown in Figure 3D, the pH of the C26 media was lower than that of LM4 media. Oncogenic KRAS controls metabolic programming in cancer cells, promoting glycolysis over oxidative respiration. 21 , 22 Because C26 cells harbor a KRAS mutation and consume more glucose than LM4 cells, it is plausible that LM4 cells might have switched from glycolysis to oxidative respiration. To investigate this hypothesis, metabolic analysis was carried out. As expected, LM4 cells showed increased basal and maximal respirations, indicating a metabolic shift toward oxidative respiration (Figure 3E).
3.5. Fatty acid intake and cholesterol levels in LM4 cells
When cells do not consume glucose, they utilize FAs and amino acids for growth 23 (Figure 4A). C26 and LM4 cells were treated with fluorescence‐labeled FAs and washed, and the intensities of the FAs in the cells were measured. As expected, LM4 incorporated more FA compared to C26 cells (Figure 4B,C). Fatty acids are transported to the mitochondria where they are metabolized to acetyl‐CoA, a precursor of cholesterol. 24 When cells were labeled with filipin III, which binds to cholesterol without ester (free cholesterol), overall fluorescence was higher in LM4 cells compared to C26 (Figure 4D). Quantitative analysis of intensity in single cells or small clusters confirmed that LM4 cells had significantly higher cholesterol levels than C26 cells (Figure 4E). When LM4 cells treated with fluorescent FAs were fixed and stained with filipin III, intensities showed a parallel correlation (Figure 4F), suggesting that cholesterol production correlates with higher FA intake in cells. Biochemical analysis also indicated that total and free cholesterol were significantly higher in LM4 (Figure 4G). Low‐density lipoprotein (LDL) contains cholesterol synthesized in the liver and binds to LDL receptors on the plasma membrane, facilitating crucial cholesterol uptake necessary for CRC cell growth in the liver. 25 Although the amount of LDL receptor was higher in LM4 cells than in C26 cells (Figure 4H), the levels of total cholesterol did not decrease significantly when cells were cultured with LDL‐depleted serum (Figure 4I). This result suggests that de novo cholesterol synthesis is the primary source of cholesterol for both C26 and LM4 cells. It is known that the expression of the LDL receptor is regulated by sterol regulatory element‐binding transcription factors 1 and 2 (Srebf1/2), which also upregulate the expression of key enzymes for cholesterol synthesis, namely 3‐hydroxy‐3‐methylglutaryl‐coenzyme A synthase (Hmgcs) and 3‐hydroxy‐3‐methylglutaryl‐coenzyme A reductase (Hmgcr) (Figure S1A). 26 Free cholesterol in the cytoplasm is esterified by acetyl‐coenzyme A acetyltransferase 1 (Acat1) and transported out of the cell by scavenger receptor class B members 1 and 2 (Scarb1/2). 27 We compared the mRNA expression levels of these genes and found that Srebf1, LDL receptor, Hmgcs, and Hmgcr tended to be upregulated in LM4 cells, whereas Scarb1/2 and Acat1 were downregulated (Figure S1B). Although we did not confirm the protein expression, these results suggest that in LM4 cells, cholesterol levels are elevated due to an increase in cholesterol synthesis and downregulation of efflux pathways.
FIGURE 4.
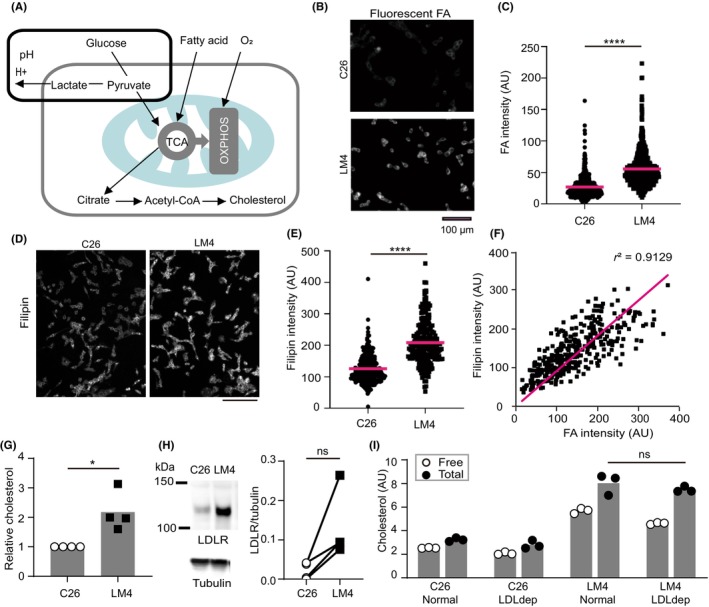
Quantification of fluorescent fatty acid (FA) intake and cholesterol in vitro. (A) Schematic depiction of glucose, FA, and O2 utilization in cells. Gray and light blue lines represent the plasma membrane and mitochondrion, respectively. (B, D) Representative images of cells treated with fluorescent FA (B, captured with a 20× objective lens) and filipin (D, captured with a 10× objective lens). Scale bar, 100 μm. (C) Fluorescent intensities of individual cells or clusters were measured. Averages were obtained from three independent experiments. n = 479 (C26) and 613 (LM4). (E) Fluorescent intensities of individual cells or clusters were measured. n = 238 (C26) and 229 (LM4). ****p < 0.0001; Mann–Whitney U‐test. (F) Fluorescent intensities of FA (x‐axis) and filipin (y‐axis) from individual cells or clusters were measured. The magenta line indicates the approximation curve. (G) Cholesterol measurement. LM4 values were normalized to C26 values. *p < 0.05; t‐test. (H) Western blot analysis of the low‐density lipoprotein (LDL) receptor (LDLR) and tubulin. Band intensity of LDLR was normalized to that of tubulin. p = 0.067 by paired t‐test (ns). (I) Cholesterol measurement with normal and LDL‐deficient (LDLdep) sera. p = 0.7 by Mann–Whitney U‐test (ns). OXPHOS, oxidative phosphorylation.
3.6. Downregulation of cholesterol and cell survival following treatment with various inhibitors
Cholesterol is a major lipid found in both the plasma membrane and organelles, particularly enriched in lysosomes. 28 In C26 and LM4 cells, cholesterol accumulated in dot‐like structures within the cytoplasm (Figure 5A). Among the various pathways activated by cholesterol accumulation in these cells, we focused on three: NLRP3, 29 STING, 30 and mTORC 31 (Figure 5B). The levels of NLRP3 protein tended to be higher in LM4 cells, and nuclear factor‐κB, a downstream target of NLRP3, was repeatedly phosphorylated in LM4 cells. STING proteins were also upregulated consistently in LM4 cells (Figure 5C). Regarding mTORC signaling, the expression of mTOR protein was comparable between C26 and LM4 cells. S6 kinase, a downstream protein of mTORC1, was not phosphorylated in LM4 cells (Figure 5C). Although the mRNA expression of mTOR increased in LM4 cells (Figure S2), protein analysis indicated that the mTORC1 pathway is not involved in the growth of LM4 cells.
FIGURE 5.
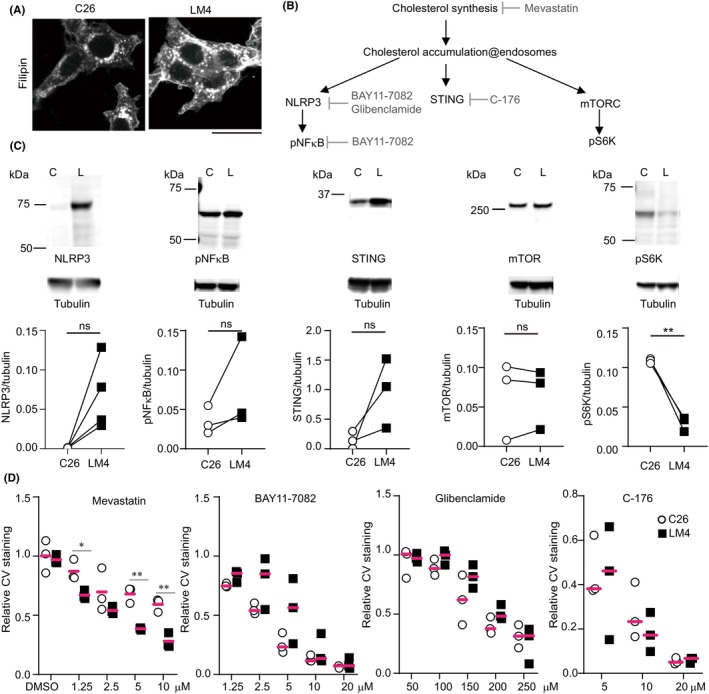
Expression of various proteins in cholesterol‐mediated signaling. (A) C26 and liver metastatic (LM) 4 cells were fixed, stained with filipin, and imaged using confocal microscopy. Scale bar, 25 μm. (B) Schematic representation of cholesterol‐mediated signaling. (C) Western blot analysis of various proteins. p = 0.068 (NLRP3), 0.227 (pNF‐κB), 0.157 (STING), mTOR (0.924), and 0.007 (pS6K) by paired t‐test. (D) LM4 cells were cultured in the presence of the inhibitors mentioned above. After 48 or 72 h, cells were fixed and stained with crystal violet. The values were normalized to those without reagent treatment. *p < 0.05, **p < 0.005; t‐test. C, C26 cells; L, LM4 cells; ns, not significant.
To examine the effects of cholesterol, NLRP3, and STING on cell growth, C26 and LM4 cells were treated with inhibitors targeting HMG CoA reductase (mevastatin), NLRP3 (glibenclamide and BAY‐7082), and STING (C‐176). Mevastatin preferentially killed LM4 cells, whereas BAY‐7082, glibenclamide, and C‐176 did not show the same effect to C26 or LM4 cells. These results suggest that LM4 cell growth is dependent on cholesterol synthesis.
To confirm the effect of cholesterol synthesis on cell growth in the liver, LM4 cells expressing Akaluc 18 were injected into the spleen, and mevastatin was administered (Figure 6A). A recent study suggested that the significance of cholesterol is cell‐type specific; when mice were treated with statins, CD8+ T cells were specifically depleted, allowing cancer cells transplanted in the mice to grow well. 32 We therefore utilized both WT and nu/nu BALB/c (nude) mice for transplantation. As expected, mevastatin enhanced LM4 cell growth in the liver of WT mice during the early phases (Figure 6B, white boxes, days 2–4). In nude mice treated with DMSO or mevastatin, LM4 cells showed less growth compared to those in WT mice (Figure 6B, middle graph). However, upon adjusting the graph scale (right graph), LM4 cells in DMSO‐treated mice showed enhanced growth (Figure 6B, black circles), whereas those in mevastatin‐treated mice showed reduced growth (Figure 6B, white circles). In the later phases (Figure 6C), LM4 cells did not grow well in WT mice (Figure 6C, left and right graphs), whereas they showed increased growth in nude mice (Figure 6C, middle graph). Notably, in two nude mice treated with mevastatin, LM4 cell numbers decreased (Figure 6C, white circles, days 9–10 and 10–11). When the values were compared with those from the previous day (Figure 6D), mevastatin slightly increased LM4 cell numbers in WT mice (Figure 6D, left graph, days 4/3, 9/8, and 11/10). In contrast, mevastatin treatment reduced LM4 cell growth in nude mice (Figure 6D, right graph, days 3/2, 4/3, 9/8, and 11/10). Because our experimental intensities were not dependent on the time after injection, these values were summarized regardless of the days. As shown in Figure 6E, mevastatin treatment tended to reduce growth in nude mice but not in WT mice. This is because LM4 cells grew better in DMSO‐treated nude mice than in DMSO‐treated WT mice.
FIGURE 6.
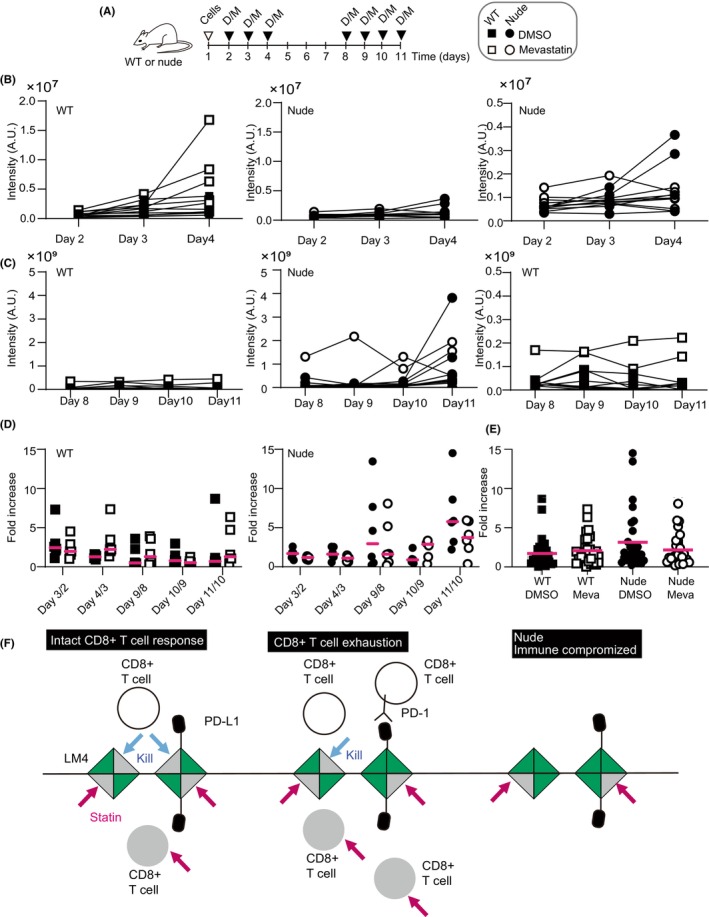
Statin treatment of mice with intrasplenic transplantation of liver metastatic (LM) 4 cells. (A) Schematic overview of the experiments. BALB/c (WT) or BALB/c nu/nu (nude) mice were transplanted with LM4 cells expressing Akaluc on day 1 (indicated by inverted triangle “cells”). Filled inverted triangles denote mice injected i.p. with DMSO (D) or mevastatin (M). Luciferase activities were measured before injection. (B, C) Intensities at the indicated days summarized in graphs. Right graphs show smaller y‐axis values. Analysis included six mice per group. (D) The intensity of each mouse on day 3 was normalized to that on day 2 (day 3/2). Similarly, relative values for day 4/3, day 9/8, day 10/9, and day 11/10 were obtained. Magenta bars represent averages. (E) Relative increase values for each group summarized in a graph. n = 30 (5 points × 6 mice). Magenta bars indicate averages. (F) Working model of statin treatment in the liver. LM4 cells are represented as squares. Green and gray triangles denote dead and live populations of LM4 cells, respectively. White and gray circles denote live and dead CD8+ T cells, respectively. Magenta arrows indicate statin treatment; blue arrows indicate killing by CD8+ T cells. PD‐1, programmed cell death‐1; PD‐L1, PD‐1 ligand.
To explain these results, we propose a working hypothesis (Figure 6F). In WT mice, CD8+ T cells eliminate LM4 cells (Figure 6F, left). Statin treatment kills both LM4 and CD8+ T cells (Figure 6F, lower left, gray triangles and circle). When exhausted CD8+ T cells expressing programmed cell death‐1 (PD‐1) increase (Figure 6F, middle), 33 LM4 cells survive and grow better, but statin treatment diminishes LM4 growth to a level similar to that observed in mice without exhausted CD8+ T cells. In nude mice (Figure 6F, right), LM4 cells proliferate without statin treatment. However, with statin treatment, LM4 cells are eliminated, similar to the effect observed in the presence of CD8+ T cells. Although it is unclear whether LM4 cells killed by CD8+ T cells are statin‐resistant or vice versa, this model explains the increased proliferation of LM4 cells in nude mice without statin treatment. It also explains that variations in LM4 growth in mice may depend on the numbers of CD8+ T cells expressing PD‐1 and LM4 cells expressing PD‐1 ligand (PD‐L1). We confirmed higher PD‐L1 mRNA expression in LM4 cells compared to C26 cells (Figure S3). It has been reported that in KRAS‐mutated lung cancer, PD‐L1 mRNA stability increases when tristetraprolin (TTP) activity is low. 34 We confirmed lower Ttp mRNA levels in LM4 compared to C26, suggesting that LM4 uses a similar mechanism to upregulate PD‐L1 expression and evade CD8+ T cell attack.
4. DISCUSSION
In this study, we isolated liver metastasis‐prone LM4 cells from mouse CRC cell line C26, and revealed metabolic pathways were altered in LM4 cells. C26 cells utilized glucose, whereas LM4 cells preferred FAs and showed elevated cholesterol levels. De novo cholesterol synthesis was essential for LM4 cell growth in vitro. In mice lacking T cells, statin treatment effectively suppressed LM4 cell growth in the liver.
Previous studies have underscored the importance of cholesterol synthesis in CRC cell growth in the absence of T cells. Statin treatment in SCID mice has been shown to decrease the growth of human CRC cells metastasized to the liver 35 or injected directly into it. 36 Additionally, statin treatment in nude mice reduced the number of CRC cells transplanted into the skin. 37 In our study, statin treatment slightly suppressed cancer cell growth in nude mice but not in WT mice (Figure 6E). According to our hypothesis (Figure 6F), statins could prove effective when patients experience a decline in CD8 cell numbers or their cytotoxic activity. Although the mechanisms have not been elucidated, statins have been shown to enhance the killing activity of oxaliplatin against C26 cells transplanted in the skin of WT mice. 38 A clinical study reported that statin treatment, in combination with XELOX (capecitabine and oxaliplatin) plus bevacizumab, improved progression‐free survival and overall survival in patients with metastatic CRC. 39 Further elucidating the relationship between drug‐resistant tumor cells and CD8 activity will help identify the optimal application of statins for liver‐metastasized CRC.
In this study, we examined KRAS‐mutated CRC cells, although previous reports have indicated that cholesterol synthesis is altered in various cancers regardless of KRAS mutations. The results from phase II and III studies on statin treatment in patients with KRAS mutations remain controversial. 40 , 41 Another clinical study suggested that statin treatment following diagnosis reduced the risk of death in patients whose primary lesions expressed SMAD4 and BMP receptors, irrespective of KRAS mutations. 42 Statins have also been found to inhibit the growth of APC‐mutated CRC cell lines. 43 In other cancers, p53 mutations upregulate genes associated with the mevalonate pathway in breast cancers, 44 whereas ovarian cancer with p53 mutation shows increased cholesterol synthesis. 45 Direct pathways linking KRAS, BMP, or p53 signaling to cholesterol synthesis have not yet been determined, suggesting that cholesterol synthesis might be a characteristic of specific subpopulations of cancer cells, such as stem cells. Supporting this notion, CRISPR screening in stem cell‐enriched spheroids of human CRCs has identified the cholesterol synthesis pathway as crucial for stem cell growth. 37
As a mechanism involving cholesterol in cells, various pathways downstream of cholesterol accumulation were examined. However, none of these pathways effectively inhibited cell growth in vitro (Figure 5). These results do not negate the importance of these signaling pathways in the interaction between LM4 cells with interstitial cells for cell growth and survival in vitro. In addition to cholesterol synthesis, the mevalonate pathway produces various end products crucial for posttranslational modifications of small GTPases. 46 Cholesterol is essential for the signaling platform in the plasma membrane 47 and contributes to cancer progression in various ways. 47 Our gene set enrichment analysis revealed upregulation of pathways involving FAs in LM4 cells, whereas those related to ribosomes and ribosomal subunits were downregulated (data not shown). This difference in the balance between protein and lipid synthesis between C26 and LM4 cells affected their growth both in vitro and in vivo. Cholesterol production primarily occurs in the liver, with the intestine being the second organ with a high synthetic rate of cholesterol. 27 It is intriguing that colon cancer cells, by producing cholesterol similarly to hepatocytes, may adapt to the liver environment. Further experiments are necessary to understand the role of cholesterol synthesis in survival. LM4 cells can provide insights into the molecular mechanisms of cancer survival in the liver.
AUTHOR CONTRIBUTIONS
Kumiko Taniguchi: Data curation; formal analysis; resources. Kei Sugihara: Data curation; formal analysis; investigation; methodology; resources; writing – original draft. Takashi Miura: Data curation; formal analysis; investigation; methodology; supervision; writing – original draft. Daisuke Hoshi: Conceptualization. Susumu Kohno: Conceptualization; data curation; formal analysis; methodology; resources; validation; writing – original draft. Chiaki Takahashi: Conceptualization; supervision; writing – original draft. Eishu Hirata: Conceptualization; visualization. Etsuko Kiyokawa: Conceptualization; data curation; formal analysis; funding acquisition; investigation; methodology; project administration; resources; validation; visualization; writing – original draft.
FUNDING INFORMATION
This work was supported by a Grant for Assist KAKEN from Kanazawa Medical University (K2022‐4), a Grant‐in‐Aid for Scientific Research on Innovative Areas from the Japan Society for the Promotion of Science (JSPS) KAKENHI (Grant No. 26112719), and a Grant‐in‐Aid for Scientific Research (C) from JSPS KAKENHI (Grant No. 17K08774).
CONFLICT OF INTEREST STATEMENT
Chiaki Takahashi is an editorial board member of Cancer Science. The other authors declare no conflict of interest.
ETHICS STATEMENT
Approval of the research protocol by an Institutional Review Board: N/A.
Informed consent: N/A.
Registry and Registration No. of the study/trial: N/A.
Animal studies: Animal experiments were carried out in accordance with the guidelines established by the Institutional Animal Care and Use Committee of Kanazawa Medical University (approval nos. 2014‐13, 2015‐13, 2016‐20, 2019‐22, 2018‐7, 2019‐11, 2020‐73, 2021‐6, and 2022‐9).
Supporting information
Figure S1. Cholesterol metabolism and gene expression of related proteins.
Figure S2. mRNA expression of Nlrp3, Sting, and mTOR in C26 and LM4 cells.
Figure S3. mRNA expression of Pd‐l1, Ttp, and Ksrp in C26 and LM4 cells.
Table S1. Reagents, cells, antibodies, and plasmids used in this study.
Movie S1. Live imaging of extravasation processes (corresponding to Figure 2B).
Movie S2. Live imaging of C26 and LM4 cells for 3 days (corresponding to Figure 3B).
ACKNOWLEDGMENTS
We thank A. Kinebuchi, T. Kanitani, Y. Nakashima, Y. Mizuochi, and A. Nakayama for their technical assistance.
Taniguchi K, Sugihara K, Miura T, et al. Cholesterol synthesis is essential for the growth of liver metastasis‐prone colorectal cancer cells. Cancer Sci. 2024;115:3817‐3828. doi: 10.1111/cas.16331
REFERENCES
- 1. Zarour LR, Anand S, Billingsley KG, et al. Colorectal cancer liver metastasis: evolving paradigms and future directions. Cell Mol Gastroenterol Hepatol. 2017;3:163‐173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Wang Y, Zhong X, He X, et al. Liver metastasis from colorectal cancer: pathogenetic development, immune landscape of the tumour microenvironment and therapeutic approaches. J Exp Clin Cancer Res. 2023;42:177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Hess KR, Varadhachary GR, Taylor SH, et al. Metastatic patterns in adenocarcinoma. Cancer. 2006;106:1624‐1633. [DOI] [PubMed] [Google Scholar]
- 4. Hayashi M, Inoue Y, Komeda K, et al. Clinicopathological analysis of recurrence patterns and prognostic factors for survival after hepatectomy for colorectal liver metastasis. BMC Surg. 2010;10:27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Milette S, Sicklick JK, Lowy AM, Brodt P. Molecular pathways: targeting the microenvironment of liver metastases. Clin Cancer Res. 2017;23:6390‐6399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Oh BY, Hong HK, Lee WY, Cho YB. Animal models of colorectal cancer with liver metastasis. Cancer Lett. 2017;387:114‐120. [DOI] [PubMed] [Google Scholar]
- 7. Corbett TH, Griswold DP, Roberts BJ, Peckham JC, Schabel FM. Tumor induction relationships in development of transplantable cancers of the colon in mice for chemotherapy assays, with a note on carcinogen structure. Cancer Res. 1975;35:2434‐2439. [PubMed] [Google Scholar]
- 8. Sato N, Michaelides MC, Wallack MK. Characterization of tumorigenicity, mortality, metastasis, and splenomegaly of two cultured murine colon lines. Cancer Res. 1981;41:4107‐4110. [PubMed] [Google Scholar]
- 9. Zhang B, Halder SK, Zhang S, Datta PK. Targeting transforming growth factor‐β signaling in liver metastasis of colon cancer. Cancer Lett. 2009;277:114‐120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Castle JC, Loewer M, Boegel S, et al. Immunomic, genomic and transcriptomic characterization of CT26 colorectal carcinoma. BMC Genomics. 2014;15:190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Sonoshita M, Aoki M, Fuwa H, et al. Suppression of colon cancer metastasis by Aes through inhibition of notch signaling. Cancer Cell. 2011;19:125‐137. [DOI] [PubMed] [Google Scholar]
- 12. Yu Y, Khan J, Khanna C, Helman L, Meltzer PS, Merlino G. Expression profiling identifies the cytoskeletal organizer ezrin and the developmental homeoprotein Six‐1 as key metastatic regulators. Nat Med. 2004;10:175‐181. [DOI] [PubMed] [Google Scholar]
- 13. He Y, Hara H, Núñez G. Mechanism and regulation of NLRP3 inflammasome activation. Trends Biochem Sci. 2016;41:1012‐1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Akagi T, Shishido T, Murata K, Hanafusa H. v‐Crk activates the phosphoinositide 3‐kinase/AKT pathway in transformation. Proc Natl Acad Sci USA. 2000;97:7290‐7295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Sugihara K, Yamaguchi Y, Usui S, et al. A new perfusion culture method with a self‐organized capillary network. PLoS One. 2020;15:e0240552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kiyokawa E, Shoji H, Daikoku T. The supression of DOCK family members by their specific inhibitors induces the cell fusion of human trophoblastic cells. Biochem Biophys Res Commun. 2020;529:1173‐1179. [DOI] [PubMed] [Google Scholar]
- 17. Kohno S, Linn P, Nagatani N, et al. Pharmacologically targetable vulnerability in prostate cancer carrying RB1‐SUCLA2 deletion. Oncogene. 2020;39:5690‐5707. [DOI] [PubMed] [Google Scholar]
- 18. Iwano S, Sugiyama M, Hama H, et al. Single‐cell bioluminescence imaging of deep tissue in freely moving animals. Science. 2018;359:935‐939. [DOI] [PubMed] [Google Scholar]
- 19. Hirata E, Ishibashi K, Kohsaka S, et al. The brain microenvironment induces DNMT1 suppression and indolence of metastatic cancer cells. iScience. 2020;23:101480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Webb BA, Chimenti M, Jacobson MP, Barber DL. Dysregulated pH: a perfect storm for cancer progression. Nat Rev Cancer. 2011;11:671‐677. [DOI] [PubMed] [Google Scholar]
- 21. Ying H, Kimmelman AC, Lyssiotis CA, et al. Oncogenic Kras maintains pancreatic tumors through regulation of anabolic glucose metabolism. Cell. 2012;149:656‐670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Rozeveld CN, Johnson KM, Zhang L, Razidlo GL. KRAS controls pancreatic cancer cell lipid metabolism and invasive potential through the lipase HSL. Cancer Res. 2020;80:4932‐4945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Broadfield LA, Pane AA, Talebi A, Swinnen JV, Fendt SM. Lipid metabolism in cancer: new perspectives and emerging mechanisms. Dev Cell. 2021;56:1363‐1393. [DOI] [PubMed] [Google Scholar]
- 24. Martin‐Perez M, Urdiroz‐Urricelqui U, Bigas C, Benitah SA. The role of lipids in cancer progression and metastasis. Cell Metab. 2022;34:1675‐1699. [DOI] [PubMed] [Google Scholar]
- 25. Xu C, Gu L, Kuerbanjiang M, et al. Adaptive activation of EFNB2/EPHB4 axis promotes post‐metastatic growth of colorectal cancer liver metastases by LDLR‐mediated cholesterol uptake. Oncogene. 2023;42:99‐112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Goldstein JL, DeBose‐Boyd RA, Brown MS. Protein sensors for membrane sterols. Cell. 2006;124:35‐46. [DOI] [PubMed] [Google Scholar]
- 27. Shen WJ, Azhar S, Kraemer FB. SR‐B1: a unique multifunctional receptor for cholesterol influx and efflux. Annu Rev Physiol. 2018;80:95‐116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Meng Y, Heybrock S, Neculai D, Saftig P. Cholesterol handling in lysosomes and beyond. Trends Cell Biol. 2020;30:452‐466. [DOI] [PubMed] [Google Scholar]
- 29. Razani B, Feng C, Coleman T, et al. Autophagy links inflammasomes to atherosclerotic progression. Cell Metab. 2012;15:534‐544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Chu TT, Tu X, Yang K, Wu J, Repa JJ, Yan N. Tonic prime‐boost of STING signalling mediates Niemann‐pick disease type C. Nature. 2021;596:570‐575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Davis OB, Shin HR, Lim C‐Y, et al. NPC1‐mTORC1 signaling couples cholesterol sensing to organelle homeostasis and is a targetable pathway in Niemann‐pick type C. Dev Cell. 2021;56:260‐276.e267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Yan C, Zheng L, Jiang S, et al. Exhaustion‐associated cholesterol deficiency dampens the cytotoxic arm of antitumor immunity. Cancer Cell. 2023;41:1276‐1293. [DOI] [PubMed] [Google Scholar]
- 33. Wherry EJ, Kurachi M. Molecular and cellular insights into T cell exhaustion. Nat Rev Immunol. 2015;15:486‐499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Coelho MA, Carné Trécesson S, Rana S, et al. Oncogenic RAS signaling promotes tumor immunoresistance by stabilizing PD‐L1 mRNA. Immunity. 2017;47:1083‐1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Juneja M, Kobelt D, Walther W, et al. Statin and rottlerin small‐molecule inhibitors restrict colon cancer progression and metastasis via MACC1. PLoS Biol. 2017;15:e2000784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Zhang KL, Zhu WW, Wang SH, et al. Organ‐specific cholesterol metabolic aberration fuels liver metastasis of colorectal cancer. Theranostics. 2021;11:6560‐6572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Gao S, Soares F, Wang S, et al. CRISPR screens identify cholesterol biosynthesis as a therapeutic target on stemness and drug resistance of colon cancer. Oncogene. 2021;40:6601‐6613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Tsubaki M, Takeda T, Matsuda T, et al. Statins enhances antitumor effect of oxaliplatin in KRAS‐mutated colorectal cancer cells and inhibits oxaliplatin‐induced neuropathy. Cancer Cell Int. 2023;23:73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Kim Y, Kim TW, Han SW, et al. A single arm, phase II study of simvastatin plus XELOX and bevacizumab as first‐line chemotherapy in metastatic colorectal cancer patients. Cancer Res Treat. 2019;51:1128‐1134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Lee J, Hong YS, Hong JY, et al. Effect of simvastatin plus cetuximab/irinotecan for KRAS mutant colorectal cancer and predictive value of the RAS signature for treatment response to cetuximab. Investig New Drugs. 2014;32:535‐541. [DOI] [PubMed] [Google Scholar]
- 41. Krens LL, Simkens LHJ, Baas JM, et al. Statin use is not associated with improved progression free survival in cetuximab treated KRAS mutant metastatic colorectal cancer patients: results from the CAIRO2 study. PLoS One. 2014;9:e112201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Voorneveld PW, Reimers MS, Bastiaannet E, et al. Statin use after diagnosis of colon cancer and patient survival. Gastroenterology. 2017;153:470‐479.e474. [DOI] [PubMed] [Google Scholar]
- 43. Shailes H, Tse WY, Freitas MO, Silver A, Martin SA. Statin treatment as a targeted therapy for APC‐mutated colorectal cancer. Front Oncol. 2022;12:12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Freed‐Pastor William A, Mizuno H, Zhao X, et al. Mutant p53 disrupts mammary tissue architecture via the mevalonate pathway. Cell. 2012;148:244‐258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Kobayashi Y, Kashima H, Wu R‐C, et al. Mevalonate pathway antagonist suppresses formation of serous tubal intraepithelial carcinoma and ovarian carcinoma in mouse models. Clin Cancer Res. 2015;21:4652‐4662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Patelli G, Tosi F, Amatu A, et al. Strategies to tackle RAS‐mutated metastatic colorectal cancer. ESMO Open. 2021;6:100156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Liu X, Lv M, Zhang W, Zhan Q. Dysregulation of cholesterol metabolism in cancer progression. Oncogene. 2023;42:3289‐3302. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Cholesterol metabolism and gene expression of related proteins.
Figure S2. mRNA expression of Nlrp3, Sting, and mTOR in C26 and LM4 cells.
Figure S3. mRNA expression of Pd‐l1, Ttp, and Ksrp in C26 and LM4 cells.
Table S1. Reagents, cells, antibodies, and plasmids used in this study.
Movie S1. Live imaging of extravasation processes (corresponding to Figure 2B).
Movie S2. Live imaging of C26 and LM4 cells for 3 days (corresponding to Figure 3B).