

Hippo–YAP1 signaling maintains stemness in gastric cancer by upregulating FOXP4, identifying FOXP4 as a stemness biomarker and therapeutic target that could help improve patient outcomes.
Abstract
The Hippo–YAP1 pathway is an evolutionally conserved signaling cascade that controls organ size and tissue regeneration. Dysregulation of Hippo–YAP1 signaling promotes initiation and progression of several types of cancer, including gastric cancer. As the Hippo–YAP1 pathway regulates expression of thousands of genes, it is important to establish which target genes contribute to the oncogenic program driven by YAP1 to identify strategies to circumvent it. In this study, we identified a vital role of forkhead box protein 4 (FOXP4) in YAP1-driven gastric carcinogenesis by maintaining stemness and promoting peritoneal metastasis. Loss of FOXP4 impaired gastric cancer spheroid formation and reduced stemness marker expression, whereas FOXP4 upregulation potentiated cancer cell stemness. RNA sequencing analysis revealed SOX12 as a downstream target of FOXP4, and functional studies established that SOX12 supports stemness in YAP1-induced carcinogenesis. A small-molecule screen identified 42-(2-tetrazolyl) rapamycin as a FOXP4 inhibitor, and targeting FOXP4 suppressed gastric cancer tumor growth and enhanced the efficacy of 5-fluorouracil chemotherapy in vivo. Collectively, these findings revealed that FOXP4 upregulation by YAP1 in gastric cancer regulates stemness and tumorigenesis by upregulating SOX12. Targeting the YAP1–FOXP4–SOX12 axis represents a potential therapeutic strategy for gastric cancer.
Significance: Hippo–YAP1 signaling maintains stemness in gastric cancer by upregulating FOXP4, identifying FOXP4 as a stemness biomarker and therapeutic target that could help improve patient outcomes.
Introduction
Cancer stemness is one of the cancer hallmarks (1). It refers to the ability of cancer cells to exhibit stem cell–like properties such as self-renewal, the ability to differentiate into diverse cell types, and dormancy (2, 3). Cancer stem cells (CSC), also known as tumor-initiating cells, represent a cell fractionation within the tumor and are thought to be responsible for tumorigenicity, metastasis, recurrence, and therapy resistance in various types of cancers. CSCs promote survival and proliferation and maintain stemness properties by driving tumor growth and heterogeneity, evading immune surveillance, and responding to environmental cues (4–7). According to the expression of surface markers such as CD44, CD24, CD29, CD90, and CD133, CSCs can be isolated from multiple cancers and cell lines (8, 9).
As most CSCs harbor genetic and epigenetic alterations that lead to dysregulation of signaling pathways, like TP53 mutation, β-catenin overactivation, or DNA methylation (4, 10). Several studies also indicated that cancer stemness could be regulated by aberrant activation or repressing of normal stem cell pathways, such as Wnt, NF-κB, Notch, hedgehog, JAK–STAT, PI3K/AKT/mTOR, and Hippo–YAP1 signaling pathways (11, 12).
The Hippo pathway, also known as the Hippo–YAP1 signaling pathway, was initially discovered in Drosophila melanogaster by mutation screening. It is an evolutionally highly conserved signaling cascade that serves as a master controller of organ size, development, and cell-contact inhibition, and it also maintains the balance between cell proliferation, apoptosis, and tissue homeostasis (13–16). Furthermore, dysregulation of the Hippo–YAP1 pathway has been demonstrated to empower several significant cancer characteristics, like control of cell proliferation, proliferation, resistant cancer cell death, and endowing cancer stem cell traits. In addition, it is implicated in the development of various human cancer types, including gastric, breast, colorectal, liver, and esophageal cancers (17–20). Previously, we have revealed the driving role of the Hippo–YAP1 signaling pathway in gastric carcinogenesis (19). In the core of this pathway, as a transcriptional coactivator, YAP1 mainly binds to the TEAD transcription factor family to promote cell proliferation (21, 22) and exerts an oncogenic role in gastric cancer. We also summarized the functional roles of dysregulated Hippo–YAP1 signaling in gastric cancer (20). Mechanistically, the Hippo–YAP1 pathway regulates thousands of target genes in promoting tumorigenesis. However, the direct YAP1 target genes in gastric cancer with stemness properties have not been identified.
This study unveiled forkhead box protein 4 (FOXP4) as a novel stemness marker that promotes tumorigenesis by modulating YAP1 signaling in gastric cancer. Furthermore, we confirmed that FOXP4 maintains stemness properties primarily through SOX12. Lastly, we have developed potential small molecules that specifically target FOXP4.
Materials and Methods
Cell lines and human primary samples
Human gastric cancer cell lines (AGS, BGC823, KATO III, MGC803, MKN1, MKN7, MKN28, MKN45, NCI-N87, SGC7901, SNU1, and SNU16) were obtained from either ATCC or RIKEN Cell Bank. These cells were routinely maintained in RPMI 1640 media (Gibco) supplemented with 10% FBS (Gibco) and 1% penicillin/streptomycin (Gibco) in a humidified incubator at 37°C in 5% CO2. All cell lines underwent a maximum of 15 passages after revival from frozen stocks and were tested periodically to confirm free of Mycoplasma contamination. The Hong Kong and Beijing independent gastric cancer cohorts on tissue microarrays were used, containing 278 patients collected between 1998 and 2006 at Prince of Wales Hospital (23) and 166 cases collected at Peking University Cancer Hospital. The clinical information of each case, together with survival status and survival time, was collected. Frozen gastric cancer tumors and paired nontumor tissues were collected after surgical resection at Prince of Wales Hospital. Samples were stored at −80°C in a refrigerator or liquid nitrogen. Briefly, to extract proteins from tumor tissue samples, the frozen tissue was thawed and cut into small pieces. These tissue pieces were then subjected to lysis using a cell lysis buffer containing protease inhibitors to break down the cells and release the proteins. After centrifugation to remove cellular debris, the supernatant containing the extracted proteins was collected. The protein concentration was determined, and subsequent Western blot analysis was performed on the extracted proteins. Human primary sample usage was approved by the Joint Chinese University of Hong Kong-New Territories East Cluster Clinical Research Ethics Committee (CREC Ref. No.: 2022-060).
Cell proliferation and viability assays
Briefly, gastric cancer cell lines were reverse-transfected by Lipofectamine RNAiMAX (Invitrogen) or FuGENE HD (Promega) and seeded into 96-well or six-well plates. Cell viability was determined by cell counting kit 8 (MCE Chemicals). The data shown are presented in at least three independent experiments.
Cell migration and invasion assays
Cell migration and invasion assays were generated after treatment with siScramble or siRNA, using 24-well Transwell inserts (Corning) and Matrigel Invasion chambers (Corning), respectively. We adopted mitomycin C (Merck) in knockdown and overexpression experiments involving migration/invasion assay to inhibit the proliferation of cancer cells. A measure of 1 μg/mL mitomycin C was added and placed in an incubator at 37°C in 5% CO2 for 2 to 4 hours after cell transfection, and then the medium was removed and washed with PBS three times. Briefly, gastric cancer cells suspended in a serum-free medium were added to the inner chamber, and the medium containing 10% FBS was added to the bottom. After incubating for 16 to 20 hours, migrated or invaded cells to the lower membrane surface were stained with 0.4% (w/v) crystal violet (Sigma-Aldrich) and then viewed under the 20× objective lens to take images of at least three random fields. The quantification was carried out as follows: three randomly selected, equally sized views from the original figures of each group were used for manual cell counting. The average cell count of siScramble from each assay was then normalized to 1, and the cell count of the corresponding groups was adjusted accordingly. Subsequently, the average and SD were calculated and presented using bar charts. Statistical analysis was performed using GraphPad Prism with a Student t test to demonstrate significance. The data shown are presented in at least three independent experiments.
Colony formation and gastric spheroid assays
Cells were plated into six-well plates at a lower density of 1,000 cells per well and cultured for 10 days. Then the cells were stained with 0.4% (w/v) crystal violet and quantified. Gastric sphere assays were generated after treatment with siScramble or siRNA, 1,000 cells/well cultured in serum-free Advanced DMEM/F-12 (Life Technologies), containing B27 (1:50, Life Technologies), 50 ng/mL EGF (Life Technologies), 100 ng/mL FGF10 (Life Technologies), and 1% penicillin/streptomycin (Gibco) in a 24-well plate. Cells were monitored for 14 days to calculate spheroid formation. The sequence information on siRNAs, short hairpin RNAs (shRNA; Vector backbone: pLKO.1), and chromatin immunoprecipitation (ChIP)-qPCR primers used in this study were recorded and are shown in Supplementary Tables S1–S3. The expression plasmid for FOXP4 and SOX12 was obtained from VectorBuilder. The expression plasmid for YAP1 was obtained from p2xFlaghYAP1 (#17791, Addgene) and pEGFP-C3-hYAP1 (#17843, Addgene). The expression plasmid for YAP (5SA) was obtained from pCMV-Flag-YAP2-5SA (Plasmid #27371). The data shown are presented in at least three independent experiments.
Organoid culture
The organoid harvest and culture procedures were conducted according to the previously instituted protocol (23). Briefly, gastric cancer samples were collected from patients undergoing gastrectomy at Prince of Wales Hospital, Hong Kong. Tumor tissues were collected in DMEM and then cut into 0.5-mm (or smaller) pieces. Then the pieces were incubated with collagenase–accutase digestion solution for 60 minutes at 37°C while shaking vigorously every 10 minutes. After centrifugation, the supernatant was collected, and the remaining cell suspension was filtered over a 40-μm cell strainer. Then, it was neutralized by culture medium. Cell pellets were resuspended with 200 µL Matrigel matrix and seeded in the well to establish a 3D culture model. The culture medium was changed twice a week. The data shown are presented in at least three independent experiments.
qRT-PCR
Total RNA extraction was extracted using RNAiso Plus (Takara). PrimeScript RT Master Mix (Takara) was used for cDNA synthesis. qPCR was performed using TB Green Premix Ex Taq (Takara) according to the manufacturer’s instructions. The results were normalized and calculated by using the 2−ΔΔCt method. The sequences of primers for each gene were as follows: β-actin (forward 5′-AGA GCT ACG AGC TGC CTG AC-3′; reverse 5′-AGC ACT GTG TTG GCG TAC AG-3′); YAP1 (forward 5′-TAG CCC TGC GTA GCC AGT TA-3′; reverse 5′-TCA TGC TTA GTC CAC TGT CTG T-3′); FOXP4 (forward 5′-ACC AGG ATG TTC GCC TAT TTC C-3′; reverse 5′-CGG CAC CCT TGA CGT TCT C-3′); and SOX12 (forward 5′-AAG AGG CCG ATG AAC GCA TT-3′; reverse 5′-TAG TCC GGG TAA TCC GCC AT-3′). The data shown are presented in at least three independent experiments.
Western blot analysis
The primary YAP1 antibody (ab52771) was purchased from Abcam, and FOXP4 (HPA001196) antibody was obtained from Sigma-Aldrich. Simultaneously, antibodies against SOX12 (PA5-103280) and SOX2 (14-9811-82) were achieved from Life Technology. Other primary antibodies were obtained from Cell Signaling Technology, including p21 (#2946), p27 (#2552), p-Rb (Ser807/811; #9308), cyclin D1 (#2978), c-Myc (#9402), KLF4 (ABclonal), Nanog (#4903), and active β-catenin (#8814s). Anti–mouse IgG-HRP (Dako P0260, 1:3,000) and anti–rabbit IgG-HRP (Dako P0448, 1:2,500) were used as secondary antibodies. The data shown are presented in at least two independent experiments.
Apoptotic assay
After cells were treated with siRNA or inhibitors, apoptosis was analyzed with FITC Annexin V and propidium iodide staining using a FITC Annexin V Apoptosis Detection Kit (BD Pharmingen). Apoptotic cells were determined by flow cytometry using BD LSRFortessa (BD Biosciences), and the results were analyzed using FlowJo V10 software (FlowJo LLC).
FISH
A FOXP4 probe (Empire Genomics) was employed to detect the copy-number changes. Pretreatment kit 1 (KA2375, Abnova) was performed to pretreat formalin-fixed, paraffin-embedded tissue sections. Copy-number gain (3–5 spots per cell) and gene amplification (copy number > 5 per cell) were defined depending on the probe signal.
IHC staining
After dewaxing and re-dehydration, tissue sections were put into Tris-EDTA (pH 8.0) and boiled in a pressure cooker for 3 minutes for antigen retrieval. Then 3% H2O2 solution covered the sections to eliminate the effect of endogenous peroxidase and incubated for 15 minutes. At room temperature, the sections were blocked with 5% BSA for 1 hour, incubated with primary antibodies for 3 hours, then incubated with secondary antibodies for 1 hour, and stained with DAB (Dako; ref. 24). Primary antibodies include FOXP4 (HPA001196), YAP1 (ab52771, Abcam), SOX12 (ab54371, Abcam).
Luciferase reporter assay
The wild-type or mutation of the putative transcription factor–binding motifs was subcloned into the pGL3-Basic luciferase reporter vector. Dual-Luciferase Reporter Assay System (Promega) was performed to detect luciferase activity according to the manufacturer’s instructions. The data shown are presented in at least three independent experiments.
ChIP
The ChIP assay was performed according to the SimpleChIP Enzymatic Chromatin IP Kit (Cell Signaling Technology, #9003) standard protocol. NCI-N87 cells were transfected with YAP1 or FOXP4 for 48 hours. Cells were fixed with 1% formaldehyde for 10 minutes at room temperature and then a quenching step by glycine for 5 minutes. Cell pellets were lysed in SDS lysis buffer containing proteasome inhibitor cocktail, and the chromatin was sonicated to shear DNA to 100 to 1000 bp. Then, ChIP was carried out using an anti-YAP1 antibody (Cell Signaling Technology, #14074) or anti-FOXP4 antibody (HPA001196) and appropriate IgG. All immunoprecipitated chromatin DNA, as well as input DNA, was quantified by qPCR.
Assessment of drug synergy
Drug combination synergy was evaluated with the highest single-agent model utilizing the SynergyFinder web tool (25).
Xenograft formation assays
Animal experiments were carried out according to a protocol approved by the Animal Experimentation Ethics Committee of CUHK (Ref. No. 22-034-NSF). Five-week-old Balb/c nude mice were injected subcutaneously in the dorsal flank with a 100 μL suspension of 2 × 106 NCI-N87 cells transduced with lentiviral carrying shControl, shFOXP4-1, and shFOXP4-2. For assessing the synergistic effect of FOXP4 depletion and 5-fluorouracil (5-FU), we inoculated mice with NCI-N87 cells intraperitoneally, then randomly divided into four groups (five mice/group), and treated with vehicle (0.1% DMSO), shFOXP4, 5-FU (5 mg/kg/day), and shFOXP4 + 5-FU for 4 weeks, respectively. The combinational administration of 42-(2-tetrazolyl) rapamycin and 5-FU was also performed to investigate gastric cancer peritoneal dissemination. Gastric cancer peritoneal metastases were monitored using the IVIS 200 in vivo imaging system (Xenogen). Before imaging, the mice were injected with D‐fluorescein (150 mg/kg; Promega) and anesthetized with 2.5% isoflurane (Zoetis). The luminescence signal was analyzed using Living Image software (Xenogen) as photon emission/seconds/cm2.
Public dataset–based bioinformatic analysis
Two public datasets were adopted in this work, namely, The Cancer Genome Atlas (TCGA)-stomach adenocarcinoma (STAD) dataset (https://xenabrowser.net/) and the Asian Cancer Research Group (ACRG) dataset (26). The genomic alteration analysis was conducted using the online tool cBioPortal (https://www.cbioportal.org/) according to data from TCGA-STAD (https://gdc.cancer.gov/about-data/publications/pancanatlas). For the functional enrichment analysis with regard to the FOXP4 expression level, we first evaluated the whole genomic expression level alteration (as in fold change) by comparing the 10% samples with the lowest FOXP4 expression (n = 37) and the 10% samples with the highest FOXP4 expression (n = 37). The differently expressed genes (DEG) in the FOXP4-high group against the FOXP4-low group were identified using the R package “DESeq2.” The Gene Ontology (GO) enrichment analysis and gene set enrichment analysis (GSEA) were subsequently performed based on the gene expression alterations using the R package “clusterProfiler.” The same grouping method was adopted according to the expression level of SOX12, revealing the expression profile of the stem cell function gene set [visualized using the R package “pheatmap” (v1.0.12)]. Spearman correlation of the expression level of FOXP4 and SOX12 was analyzed according to the expression data from TCGA and ACRG cohorts [calculated using the R package “stats” (v4.1.3)]. The binding motifs of YAP1/TEAD4 and FOXP4 on the corresponding promoter region of FOXP4 and SOX12 were predicted by the Eukaryotic Promoter Database (https://epd.epfl.ch//index.php) and JASPAR 2022 database (https://jaspar.genereg.net).
Single-cell RNA sequencing analysis
The single-cell RNA sequencing (scRNA-seq) analysis was carried out based on a public dataset (https://dna-discovery.stanford.edu/research/datasets/) using the R package “Seurat” (v4.0.2). After data normalization and cluster labeling, gene expression in individual cells was visualized using “FeaturePlot.” The gene set variation analysis was conducted using the R package “GSVA” (v1.46.0) to evaluate the activation level of biological processes and pathways in cells of the “cancer cell” cluster of the Seurat object. Furthermore, the results were visualized using the R package “pheatmap” (v1.0.12) and “FeaturePlot.”
RNA sequencing analysis
Cultured cells were harvested, and total RNA was extracted using the RNeasy kit. RNA quality was assessed using TapeStation (Agilent). The library was prepared using the Illumina TruSeq RNA Kit (Illumina). RNA sequencing (RNA-seq) was conducted after YAP1 or FOXP4 knockdown using the NovaSeq 6000 platform (Illumina) with 100-bp single-end reads. Reads were quality-checked using FastQC (v0.12.0), and Cutadapt (v4.2) was used for sequence trimming. The raw sequencing reads were aligned to the Homo sapiens genome assembly GRCh37 (hg19) from the NCBI database using HISAT2 (v2.1.0). Gene expressions were quantified using featureCount (v1.6.4) in “count” and then transferred as transcript per million using the R package “convertCounts.” The DEGs were identified using the R package “DESeq2.” The functional enrichment analysis was performed based on the determined DEGs using the R package “clusterProfiler.” The processed bulk RNA-seq data generated in this work are presented in Supplementary Table S4.
Cellular thermal shift assay
The cellular thermal shift assay was performed to validate the binding of FOXP4 and candidate-targeted inhibitor 42-(2-tetrazolyl) rapamycin. The cell lysis was prepared as follows: first, harvested and washed with PBS, then resuspended in PBS supplemented with protease inhibitor cocktail (Roche), and freeze-thawed three times using liquid nitrogen for complete cell lysis. The cell lysate–containing supernatants were centrifuged at 20,000 g for 20 minutes at 4°C to eliminate cell debris. The lysates were then diluted in PBS and divided into the treated group [mixed with 42-(2-tetrazolyl) rapamycin (10 µmol/L)] and the control group (mixed with an empty vehicle). After a 30-minute incubation at room temperature, the lysates were divided into 50 μL aliquots, and each aliquot was heated to the designated temperatures for 3 minutes using a thermal cycler (Applied Biosystems), followed by cooling for 3 minutes at room temperature. The heated lysates were centrifuged at 20,000 g for 20 minutes at 4°C to collect supernatants containing the soluble protein fraction for subsequent Western blot analysis.
Molecular docking
The 3D structure files of FOXP4 were retrieved from AlphaFold (AF-Q8IVH2-F1). The potential ligand binding sites of FOXP4 protein were conducted using online tools PrankWeb (https://prankweb.cz/) and DeepSite (https://www.playmolecule.com/deepsite/) based on the 3D structure of FOXP4. The compound library was composed of 4,511 small molecules with literature evidence proving their anticancer effectiveness. The 3D structure files of the compounds in “sdf” format were downloaded from PubChem (https://pubchem.ncbi.nlm.nih.gov/). Molecular docking was performed using AutoDock 4.2.6 to predict the binding affinity between the predicted sites among the small molecules and FOXP4. The Lamarckian genetic algorithm was applied for the docking procedure, and the detailed parameters were set as follows: energy grid box, 50 × 50 × 50 Å; energy grid spacing, 0.375 Å; the number of individuals in a population, 150; the maximum number of energy evaluations, 2.5 × 106; the maximum number of generations, 2.7 × 104; and the rate of gene mutation, 0.02. The docking results were evaluated using the calculated binding energy. The candidates were screened out by four independent docking predictions with a strict screening criterion (binding energy < −12.16 kcal/mol, referring to a nanomolar Ki of the ligand–receptor combination). The interacting patterns of FOXP4 with the top four predicted candidates were visualized using PyMOL 2.3.
Statistical analysis
All data were analyzed using PRISM8 software (GraphPad Software, Inc.). Statistical analysis was performed using an unpaired t test, Mann–Whitney U test, one-way ANOVA, or two-way ANOVA. Results were considered as statistically significant when P < 0.05.
Ethical approval and consent to participate
The use of human samples was approved by the Joint Chinese University of Hong Kong-New Territories East Cluster Clinical Research Ethics Committee, Hong Kong.
Data availability
The RNA-seq data generated in this study are publicly available in Gene Expression Omnibus at GSE267854. TCGA-STAD RNA-seq data analyzed in this study were obtained from https://www.cancer.gov/tcga. The scRNA-seq data analyzed in this study from former research (DOI: 10.1158/1078-0432.CCR-19-3231) are available at https://dna-discovery.stanford.edu/research/datasets/. The ACRG dataset analyzed in this study was obtained from Gene Expression Omnibus at GSE66229. All other raw data generated in this study are available upon request from the corresponding author.
Results
FOXP4 is overexpressed in gastric cancer and correlated with poor survival
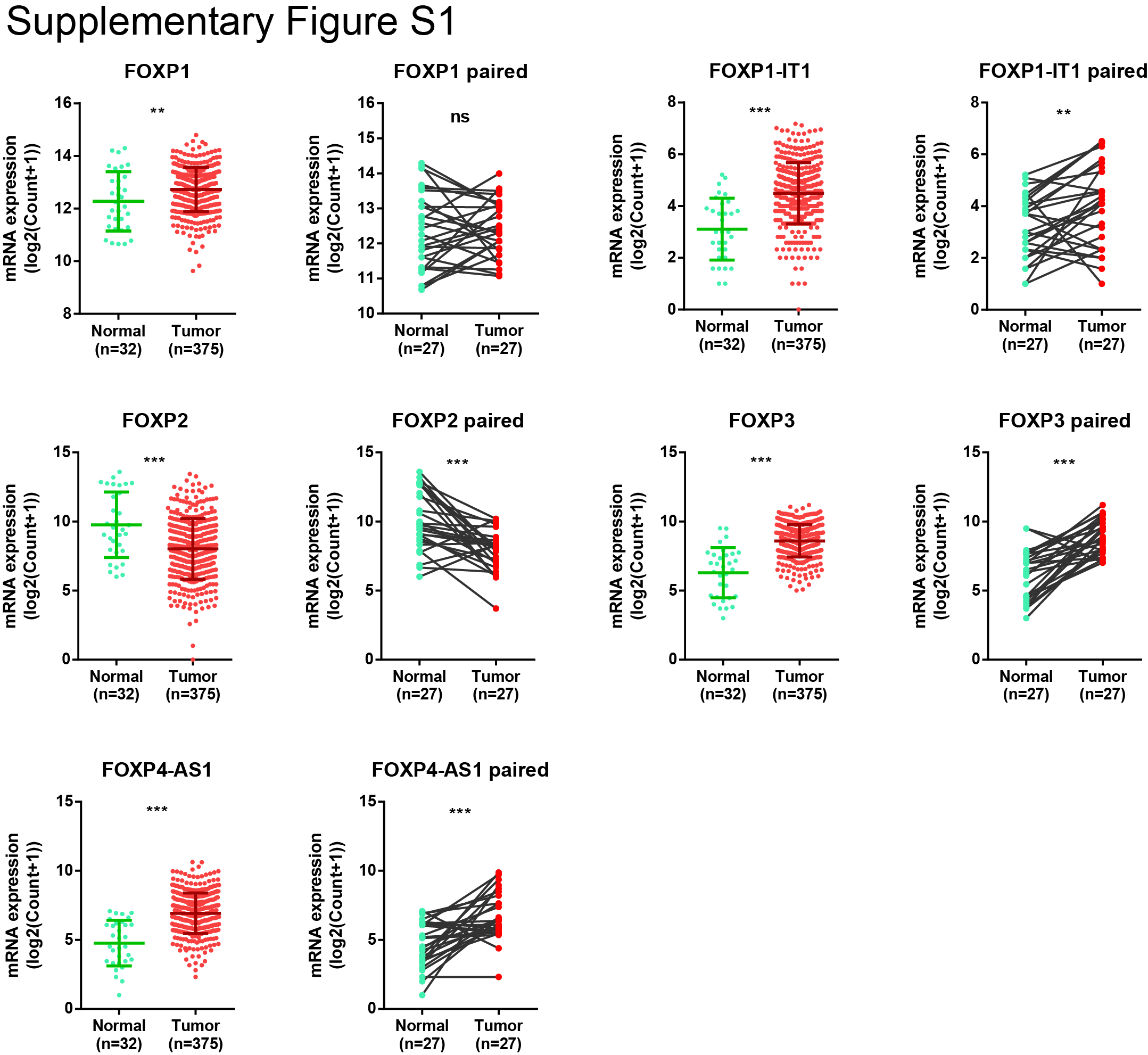
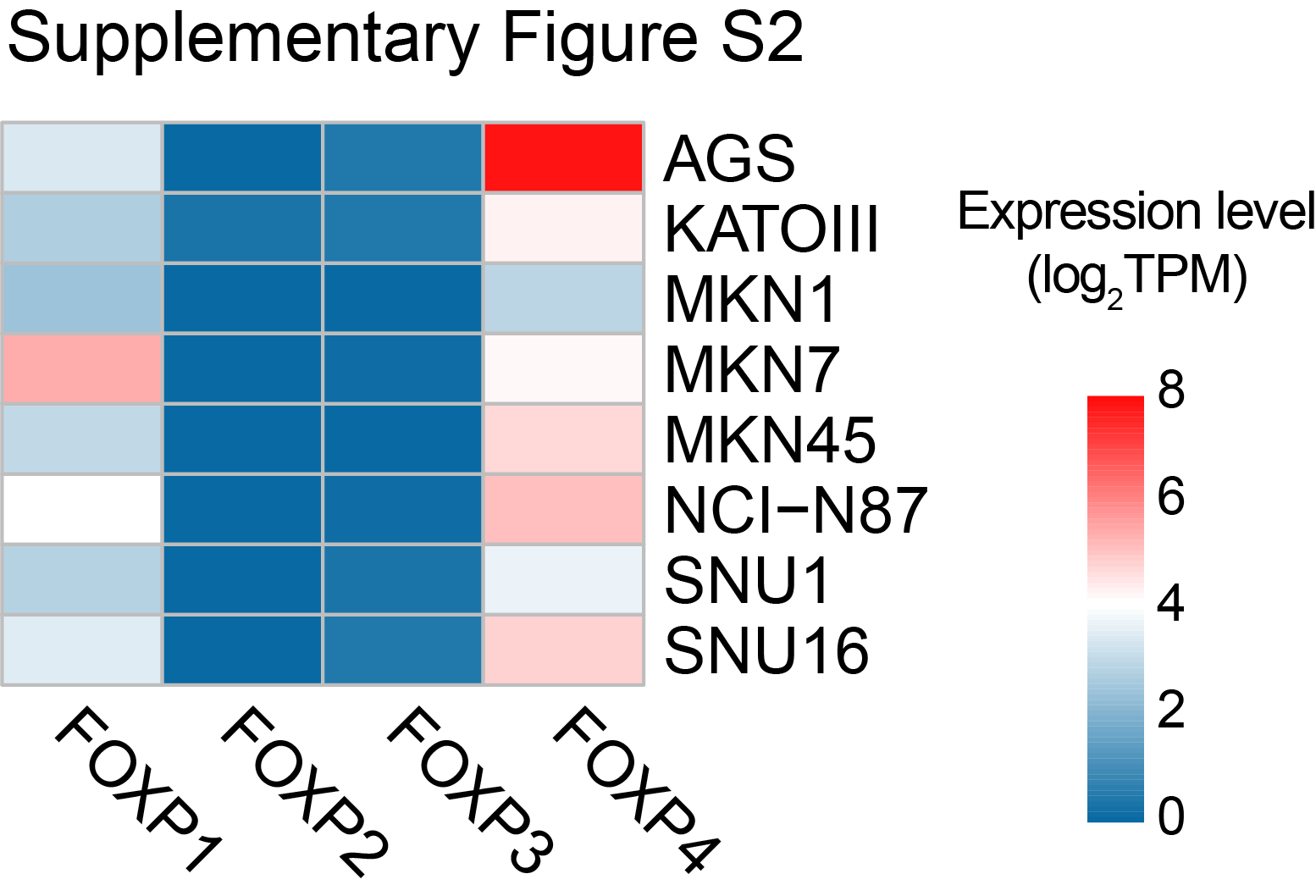
The genetic and epigenetic alterations of FOXP members in primary gastric cancer cases were demonstrated by TCGA cBioPortal analysis. Genetic or mRNA alterations in FOXP1 to FOXP4 account for 6.9%, 5.2%, 4.9%, and 16.2% of gastric cancer cases, respectively (Fig. 1A). FISH analysis revealed 4.0% and 6.8% of primary cases (n = 278) with FOXP4 copy-number gain (3–5 copies) and amplification (more than five copies), respectively (Fig. 1B). In TCGA cohort, FOXP4 mRNA levels were significantly upregulated in gastric cancer samples (n = 415) compared with normal tissues (n = 35; P < 0.001; Fig. 1C). The upregulation of FOXP4 was also detected in tumor tissues compared with paired normal tissues (n = 32; P < 0.001; Fig. 1D). Similarly, the upregulation of FOXP1, FOXP1-IT1, FOXP3, and FOXP-AS1 was also observed in tumor tissues, whereas FOXP2 demonstrated a decreased expression (Supplementary Fig. S1). By IHC staining, we found that FOXP4 was predominantly localized in the nuclei of the cancer cells, whereas no FOXP4 was detected in the adjacent normal tissues (Fig. 1E). Western blot analysis of paired tissues also verified that the FOXP4 protein level was upregulated in tumor tissues (Fig. 1F). In most of the gastric cancer cell lines, FOXP4 demonstrated abundant expression, such as MKN28 and NCI-N87, which were selected for loss-of-function assays (Fig. 1G). The expression level of FOXP4 was revealed to be higher than those of other FOXP family proteins in most of the gastric cancer cell lines (Supplementary Fig. S2). High FOXP4 expression cases (FOXP4+ in >25% cancer cells) were found in both intestinal and diffuse types, and these cases were associated with poor prognosis in both Hong Kong (n = 278) and Beijing cohorts (n = 166; Fig. 1H and I).
Figure 1.

FOXP4 is overexpressed in gastric cancer and is correlated with poor survival. A, Clinical cases with genetic or mRNA alterations in FOXPs in TCGA cohort. The genetic and mRNA alterations in FOXP1 to FOXP4 account for 4.9%, 5.2%, 6.9%, and 16.2% of gastric cancer cases, respectively. B, Copy number gain or amplification of FOXP4 in primary gastric cancer samples (n = 2) was detected by FISH analysis. C and D,FOXP4 mRNA levels in nonpaired and paired samples from TCGA-STAD dataset. E, IHC staining of the expression and cellular localization of FOXP4 in cancer cells and adjacent normal tissues (n = 3). F and G, Western blot analysis (n = 2) of FOXP4 protein expression in paired gastric cancer tissues and cell lines. N, adjacent nontumor; T, tumor. H and I, High FOXP4 expression was associated with poor prognosis in both Hong Kong and Beijing cohorts.
FOXP4 depletion exerts antitumor effects on gastric cancer
Considering the abundance of FOXP4 in gastric cancer, we first employed loss-of-function tests to investigate its functional role. Two independent siRNAs were transfected into MKN28 and NCI-N87 cells for basic functional tests. Knocking down FOXP4 suppressed gastric cancer cell proliferation and colony formation (Fig. 2A and B). Meanwhile, the cell migration and invasion were significantly inhibited by FOXP4 depletion (Fig. 2C). FOXP4 knockdown led to the downregulation of cell-cycle regulators, CDK6, CCND1, and p-Rb, whereas the p27 was activated (Fig. 2D), implying G1 cell-cycle arrest. Meanwhile, silencing FOXP4 induced upregulation of cleaved-PARP, caspase-7, and caspase-8 (Fig. 2E). The FOXP4 knockdown–induced apoptosis was further confirmed by flow cytometry analysis (Fig. 2F). Moreover, the knockdown of FOXP4 by shRNAs could suppress the growth of patient-derived organoid models of gastric cancer (Fig. 2G). In a subcutaneous tumor growth model, FOXP4 knockdown in NCI-N87 cells formed smaller xenografts compared with the control group (Fig. 2H). To further evaluate whether FOXP4 expression was related to first-line anticancer drug resistance, gastric cancer cells with either siFOXP4 or siScramble control were treated with escalating concentrations of 5-FU. IC50 was significantly decreased in the siFOXP4 transfectants compared with the siScramble control cells (Fig. 2I). The combination of FOXP4 knockdown and 5-FU treatment significantly inhibited peritoneal metastasis when compared with vehicle or single treatment with prolonged survival of mice (Fig. 2J). All these results indicated that FOXP4 might function as an oncogene in gastric cancer and its depletion exerted antitumor effects.
Figure 2.

FOXP4 depletion exerts antitumor effects in gastric cancer. A and B, siFOXP4 inhibited cancer cell proliferation and colony formation (n = 3). C, siFOXP4 suppressed gastric cancer cell invasiveness (n = 3). D and E, Western blot analysis of cell cycle–associated and apoptosis-associated proteins after FOXP4 knockdown. F, FOXP4 knockdown–induced apoptosis was confirmed by flow cytometry (n = 3). G, Representative patient-derived organoid images with shFOXP4-mediated knockdown. Scale bar, 50 μm. H, Subcutaneous injection of FOXP4-depleted gastric cancer cells formed smaller xenografts than the control group mice. I, According to IC50 displayed, siFOXP4 increased the 5-FU sensitivity. J, FOXP4 deletion and 5-FU combination suppressed peritoneal metastasis and prolonged survival time of mice. *, P < 0.05; **, P < 0.01; ***, P < 0.001.
FOXP4 is a direct downstream target of YAP1
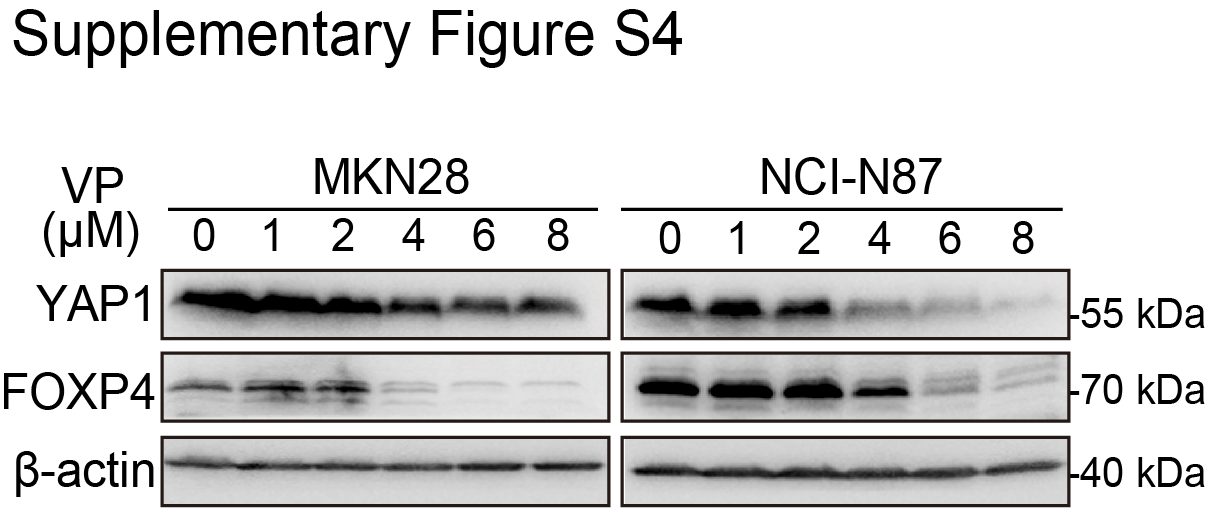
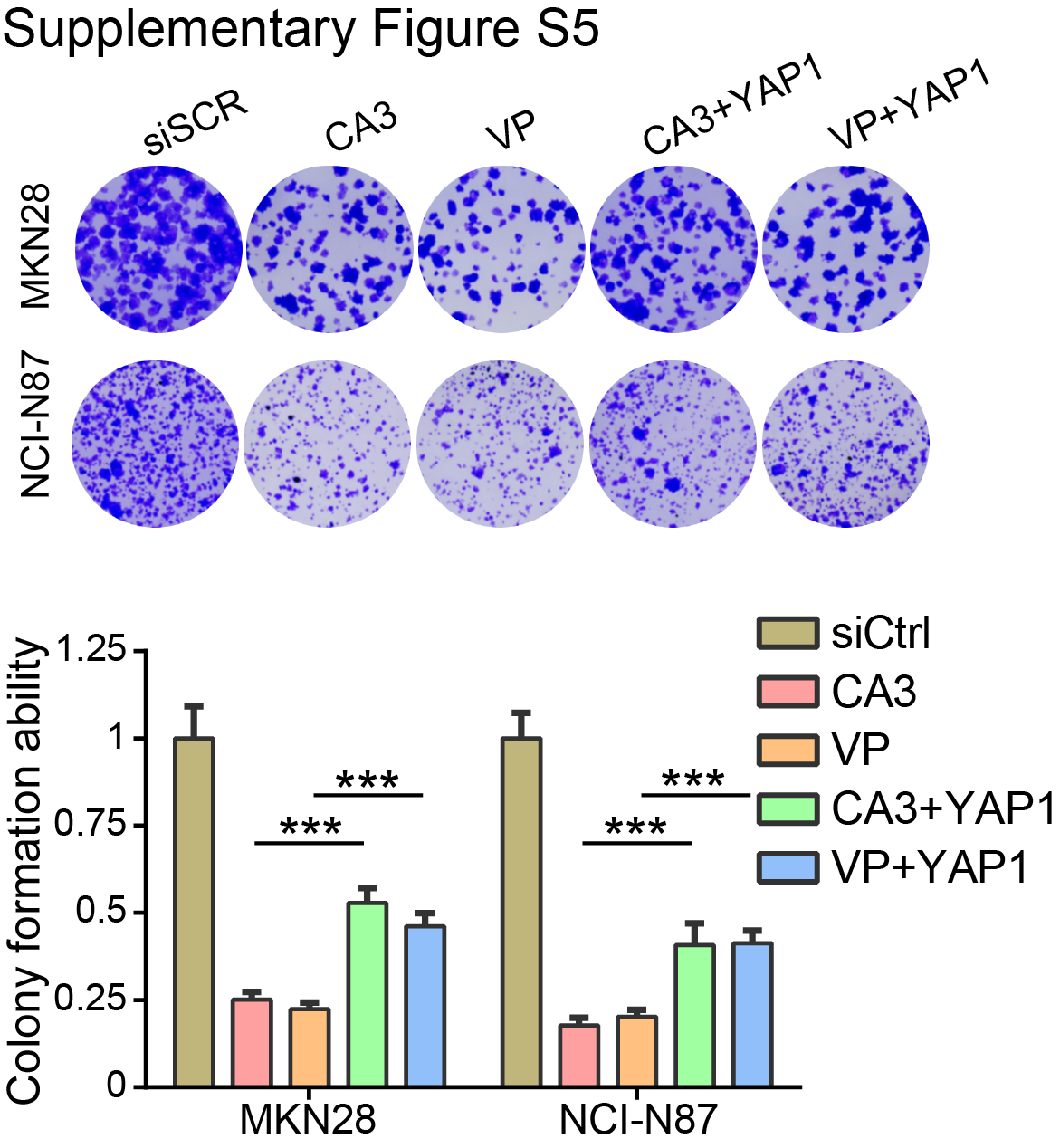
YAP1 plays a driving role in gastric cancer (19). On the basis of our long record on the mechanism of YAP1 in gastric carcinogenesis, we tentatively explored whether YAP1 is related to the overexpression of FOXP4 in gastric cancer. We performed an RNA-seq assay after knocking down YAP1 in NCI-N87 cells. Among the FOXP family members, FOXP4 is the most downregulated one (Fig. 3A). The Eukaryotic Promoter Database showed a potential YAP1/TEAD4-binding motif on the FOXP4 promoter (−647 bp; P < 0.001; Fig. 3B), whereas no binding motif was found on the promoters of FOXP2, FOXP3, and FOXP4-AS1 (Supplementary Fig. S3). By ChIP-qPCR assay, we confirmed the direct binding of YAP1/TEAD4 on the FOXP4 promoter (Fig. 3C). Luciferase reporter assays further validated that YAP1 binds with the FOXP4 promoter to activate its expression. In contrast, there was no difference in YAP1-knockdown cells for the binding site–mutated reporter compared with the control cells (Fig. 3D). siRNA-mediated YAP1 depletion led to a significant decrease of FOXP4 protein in both gastric cancer cell lines (Fig. 3E). Moreover, overexpression of wild-type YAP1 or constitutively active YAP (YAP5SA) increased FOXP4 expression (Fig. 3F). Consistently, we employed YAP1/TEAD inhibitors to evaluate the regulation of YAP1 on FOXP4 expression, namely, CA3, VT107, and verteporfin (20). The results demonstrated that all three inhibitors could attenuate FOXP4 expression dose-dependently (Fig. 3G and H; Supplementary Fig. S4). Meanwhile, YAP1 overexpression partially rescued the decreased colony formation ability of MKN28 and NCI-N87 after CA3/verteporfin treatment (Supplementary Fig. S5). We next assessed the expression correlation of YAP1 and FOXP4. In the N-methyl-N’-nitro-N-nitrosoguanidine–induced gastric cancer model (27–29), Yap1/Taz double-knockout (Yap1−/−Taz−/−) mice demonstrated lower FOXP4 expression (Fig. 3I). Furthermore, the positive association between YAP1 and FOXP4 protein expression was confirmed in primary samples by IHC staining (Fig. 3J and K). In a YAP1-knockout organoid constructed by shRNAs, the FOXP4 expression was decreased, accompanied by shrunk organoid size (Fig. 3L and M). The data in this part support FOXP4 as a direct downstream target of YAP1 in gastric cancer.
Figure 3.

FOXP4 is a direct downstream of YAP1. A, RNA-seq revealed that among the FOXP family, FOXP4 exhibits significant downregulation after the knockdown of YAP1. B, The Eukaryotic Promoter Database showed a putative YAP1/TEAD4-binding site on the FOXP4 promoter (−647 bp; P < 0.001). C, ChIP-qPCR assay (n = 3) confirmed the direct binding of YAP1/TEAD4 complex to the FOXP4 promoter. TSS, transcription start site. D, Luciferase reporter assays (n = 3) verified that YAP1 can bind with the FOXP4 promoter (wild-type binding motif). E, Western blot analysis showed that siRNA-mediated YAP1 depletion led to a significant decrease in FOXP4 protein in both gastric cancer cell lines. F, Western blot analysis revealed that the overexpression of wild-type YAP1 or constitutively active YAP (YAP5SA) increased FOXP4 expression. G and H, Administration of CA3 or VT107 inhibited the expression of YAP1 and FOXP4 dose-dependently. I, Left, workflow for generating an MNNG (N-methyl-N’-nitro-N-nitrosoguanidine)-induced gastric cancer model. d.w., drinking water. Middle and right, IHC staining showed that Yap1/Taz double-knockout mice (Yap1−/−Taz−/−) exhibited low FOXP4 expression in the MNNG-induced gastric cancer model. Scale bar, 50 μm. J and K, IHC staining confirmed a significant correlation between YAP1 and FOXP4 in both intestinal and diffuse gastric cancer types. Scale bar, 50 μm. L and M, Patient-derived organoid models further depicted that FOXP4 expression is regulated by YAP1. Scale bar, 50 μm. ***, P < 0.001.
FOXP4 overexpression promotes tumor growth and partially rescues the suppressive effects of YAP1 knockdown
To further confirm the oncogenic role of FOXP4 in gastric cancer, a low FOXP4 expression cell line, MKN7, was chosen for gain-of-function assays. FOXP4 overexpression enhances MKN7 cell proliferation and colony formation (Fig. 4A and B) and also promotes migration and invasion abilities (Fig. 4C). In the tumor formation assays, MKN7 cells overexpressing FOXP4 exhibited a higher propensity for tumor formation and demonstrated accelerated tumor growth compared with the control group (Fig. 4D and E). Western blot analysis revealed that the stemness markers, Nanog, SOX2, and KLF4, were upregulated in the FOXP4 overexpression cells (Fig. 4F) and downregulated in FOXP4-deleted cells (Supplementary Fig. S6), suggesting that FOXP4 might be associated with cancer stemness. Furthermore, we established an intraperitoneal metastasis NOD/SCIDγ (NSG) mouse model to assess the efficacy of targeting FOXP4 on gastric cancer metastasis. As predicted, overexpression of FOXP4 significantly increased the peritoneal metastasis of gastric cancer (Fig. 4G). To determine that FOXP4 is a functional downstream effector of YAP1 that mediates YAP1 signal transduction, rescue experiments were performed by re-introduction of FOXP4 into gastric cancer cells with YAP1 depletion (Fig. 4H). Functional tests revealed that siYAP1 transfectants with FOXP4 overexpression grew faster than those with YAP1 depletion (Fig. 4I and J). Meanwhile, the tumorsphere formation assays confirmed that the siYAP1-induced impaired self-renewal abilities were recovered by FOXP4 overexpression (Fig. 4K). Furthermore, the inhibited cell migration and invasion were partially rescued by FOXP4 overexpression (Fig. 4L). Taken together, FOXP4 is required for YAP1 to promote gastric cancer progression.
Figure 4.

FOXP4 overexpression promotes tumor growth and partially rescues the suppressive effects of YAP1 knockdown. A and B, FOXP4 overexpression enhanced the proliferation and colony formation ability of MKN7 cells. C, FOXP4-overexpressed MKN7 cells showed enhanced migration and invasion abilities. D and E, FOXP4 overexpression significantly increased the tumor formation ability of MKN7 cells. F, Western blot analysis revealed that the expression level of stemness markers was upregulated in the FOXP4 overexpression cells. G, Stronger peritoneal metastasis signals were detected in mice injected with FOXP4-overexpressed MKN7 cells compared with the empty vector group. H, Rescue experiments were performed by re-introducing FOXP4 into gastric cancer cells with YAP1 depletion. Re-overexpressing FOXP4 partially abolished the suppressive effects of YAP1 knockdown. I and J, The suppressed proliferation and colony formation ability from YAP1 knockdown were rescued by FOXP4 overexpression. K, The decreased spheroid-forming ability induced by siYAP1 was rescued by FOXP4 overexpression. L, FOXP4 overexpression partially rescued the suppressed cell migration and invasion induced by YAP1 deletion. *, P < 0.05; **, P < 0.01; ***, P < 0.001.
Maintaining cancer stemness is the principal function of FOXP4
Maintaining stemness is one of the prominent hallmarks of cancer cells. By GSEA in TCGA dataset, we found that FOXP4 was positively associated with stem cell proliferation and upregulation (Fig. 5A and B). FOXP4 knockdown in MKN28 and NCI-N87 cells significantly impairs the tumorsphere formation (Fig. 5C), indicating that FOXP4 plays a crucial role in maintaining cancer stemness. RNA-seq analysis revealed that a total of 724 genes that co-downregulated in both YAP1-knockout and FOXP4-knockout samples (Supplementary Fig. S7), and multiple stemness markers were downregulated after knocking down YAP1 and FOXP4, including SOX12, listed in the top ranks (Fig. 5D). scRNA-seq analysis also demonstrated that YAP1 and FOXP4 were co-overexpressed in the same gastric cancer cell population (Fig. 5E). Interestingly, this cell population was also enriched by stem cell maintenance and proliferation, implying that YAP1 and FOXP4 might preserve stemness characteristics (Supplementary Fig. S8). GO analysis by TCGA cohort revealed that high FOXP4 expression was associated with the Wnt signaling pathway and cellular response to TGFβ stimulus (Supplementary Fig. S9A). GO enrichment analysis demonstrated that gland development and regulation of cell morphogenesis were correlated with FOXP4 expression (Supplementary Fig. S9B). Among the downregulated stemness markers, SOX12 has a binding motif for FOXP4 in its promoter (−91 bp, P < 0.001; Fig. 5F). The direct binding affinity of FOXP4 with the SOX12 promoter was further confirmed by ChIP-qPCR assay (Fig. 5G). Knocking down FOXP4 decreased the SOX12 mRNA and protein levels in both gastric cancer lines (Fig. 5H and I). In both TCGA and ACRG cohorts, FOXP4 and SOX12 demonstrated a positive correlation (Fig. 5J and K), suggesting that FOXP4 tightly regulates SOX12 expression. Meanwhile, there is a significant but not close correlation between SOX12 and YAP1 in TCGA and ACRG cohorts but no significant correlation between SOX12 and classical YAP1/TEAD1 downstream targets CTGF/CYR61 (Supplementary Fig. S10). To add up, the expression of SOX12 could be activated by both YAP1 and FOXP4 overexpression, whereas SOX2 levels were solely induced by FOXP4 overexpression but not by YAP1 overexpression (Supplementary Fig. S11). These results suggest a nondirect regulation of YAP1 on SOX12, implying the necessity of FOXP4 as a signal transducer for the Hippo signaling pathway to participate in cancer cell stemness acquisition and maintenance. We further conducted rescue assays to confirm the direct regulation of FOXP4 on SOX12, and the results showed that the overexpression of FOXP4 promoted the proliferation and metastasis ability of gastric cancer cell lines, whereas the knockdown of SOX12 attenuated the malignancy of cancer cells. Furthermore, SOX12 depletion significantly blocked the promoting functions of FOXP4 overexpression, as there was no difference between the siSOX2 groups and oeFOXP4+siSOX2 groups in colony formation and invasion activity of gastric cancer cells (Supplementary Fig. S12). The data in this part highlight the role of FOXP4 in maintaining stemness by regulating SOX12 expression.
Figure 5.

Maintaining cancer stemness is the major role of FOXP4. A and B, GSEA demonstrated a positive correlation between FOXP4 and stem cell proliferation and upregulation. C, FOXP4 depletion compromises tumorsphere formation. D, RNA-seq revealed that various stemness markers were downregulated after knocking down YAP1 and FOXP4. E, scRNA-seq analysis demonstrated that YAP1/FOXP4 was co-upregulated with biological processes correlated with stem cell maintenance and proliferation. F and G, FOXP4 directly regulates SOX12 expression in gastric cancer, which was confirmed by the ChIP-qPCR assay. H and I, qRT-PCR and Western blot analysis of SOX12 expression in siFOXP4 transfectants. J and K, Both TCGA and ACRG cohorts demonstrated a positive association between FOXP4 and SOX12. TPM, transcript per million. ***, P < 0.001.
SOX12 is a novel stemness marker in gastric cancer progression
To evaluate the potential role of SOX12 in gastric cancer, we first examined the expression of SOX12 in the N-methyl-N’-nitro-N-nitrosoguanidine–induced gastric cancer model and patients with gastric cancer. The IHC staining results revealed that SOX12 expression was hardly detected in Yap1−/−Taz−/− mice (Fig. 6A). In the Hong Kong cohort, the upregulated SOX12 (SOX12+ in >25% cancer cells deemed as high expression) was also associated with poor prognosis of the patients with gastric cancer (Fig. 6B and C). We then employed siRNA-mediated knockdown to investigate the SOX12 function (Fig. 6D). The cell proliferation indicated by cell counting kit 8 and colony formation assays, together with the cell migration/invasion abilities, was all significantly repressed by SOX12 depletion (Fig. 6E–G). In addition, SOX12 knockdown effectively inhibited the self-renewal potential of the gastric cancer cells (Fig. 6H) and promoted apoptosis (Fig. 6I). Western blot analysis further confirmed the cell-cycle arrest and apoptosis, indicated by the activation of p27 and cleaved-PARP but downregulation of p-Rb (Fig. 6J). Overexpressing SOX12 in MKN7 cells enhanced the expression of stemness markers such as SOX2, Nanog, and KLF4 (Fig. 6K). Multiple stemness markers were upregulated significantly in high SOX12 expression samples in TCGA cohort (Fig. 6L; Supplementary Fig. S13). The results in this part indicate that SOX12 is a novel stemness and prognostic biomarker in gastric cancer.
Figure 6.

SOX12 is a novel stemness marker in gastric cancer progression. A, IHC staining showed that Yap1−/−Taz−/− mice exhibited low SOX12 expression in the MNNG (N-methyl-N’-nitro-N-nitrosoguanidine)-induced gastric cancer model. B and C, Highly expressed SOX12 gastric cancer cases were associated with unfavorable outcomes compared with the low-expression group. D–H, SOX12 depletion by siRNA inhibited cell proliferation, colony formation, cell metastasis abilities, and tumorsphere formation. I and J, Flow cytometry and Western blot analysis confirmed that SOX12 knockdown induced cell apoptosis. K, SOX12 overexpression in MKN7 cells increased the expression levels of canonical stemness markers. L, Heatmap displays multiple stemness markers that were upregulated in SOX12 high expression cases (from TCGA dataset). *, P < 0.05; **, P < 0.01; ***, P < 0.001.
Targeting FOXP4 by high-content screened small molecules
As FOXP4 is highly expressed in part of the gastric cancer cell lines and primary samples and associated with an aggressive phenotype, we performed high-content small molecule screening from 4,511 anticancer drugs to select the potent FOXP4 inhibitors. We used four modeling tools to screen the most potent small molecules from the library (Fig. 7A). Four small molecules were proposed to inhibit FOXP4 activity potentially (Fig. 7B). The small molecule 42-(2-tetrazolyl) rapamycin displayed a markedly inhibitory effect in the FOXP4 highly expressed gastric cancer lines MKN28 and NCI-N87. However, the IC50 of 42-(2-tetrazolyl) rapamycin treatment was relatively high in gastric cancer cells with a low expression level of FOXP4 (Fig. 7C). The 2D binding pattern analysis revealed strong intermolecular interactions in the binding site of the drug–target complex (Supplementary Fig. S14A). The cellular thermal shift assay experiment demonstrated the direct binding of 42-(2-tetrazolyl) rapamycin and FOXP4 by enhanced stability (Supplementary Fig. S14B). 42-(2-Tetrazolyl) rapamycin inhibited FOXP4 expression and cell colony formation ability dose-dependently (Fig. 7D and E). Dose–response combination assays of two drugs confirmed synergy among all 10 combinations, and the red peak of 3D plots indicates the average highest single-agent synergy scores (Fig. 7F). In addition, 42-(2-tetrazolyl) rapamycin treatment enhanced the sensitivity of gastric cancer cells to 5-FU–induced apoptosis (Fig. 7G). To further investigate the antitumor effect of the combination of 5-FU and 42-(2-tetrazolyl) rapamycin in vivo, immunodeficient NSG mice bearing xenografted NCI-N87 cells by subcutaneous inoculation were established. Then the mice were treated with vehicle control, 5-FU, 42-(2-tetrazolyl) rapamycin, or a combination of 5-FU and 42-(2-tetrazolyl) rapamycin for 28 days. The combination treatment with 5-FU and 42-(2-tetrazolyl) rapamycin significantly inhibited tumor volume and weight (Fig. 7H). The intraperitoneal metastasis NSG mouse model consistently demonstrated synergistic efficacies for inhibiting peritoneal dissemination with prolonged survival (Fig. 7I).
Figure 7.

Targeting FOXP4 by high-content screened small molecule. A, High-content small molecule screening was performed from 4,511 anticancer drugs to select the potent FOXP4 inhibitors. Four modeling tools were used to screen the most potent small molecules from the library. B, Four small molecules were deduced to potentially inhibit FOXP4 activity. C, The small molecule 42-(2-tetrazolyl) rapamycin displayed an inhibitory effect in the FOXP4 highly expressed gastric cancer lines MKN28 and NCI-N87. D and E, 42-(2-Tetrazolyl) rapamycin inhibited FOXP4 expression and cell colony formation ability in a dose-dependent manner. F, Dose–response combination assays of two drugs confirmed synergy among all the combinations of 5-FU and 42-(2-tetrazolyl) rapamycin, and the red peak of 3D plots represent the average highest single-agent model synergy scores. HSA, highest single agent. G, 42-(2-Tetrazolyl) rapamycin treatment enhanced the sensitivity towards 5-FU–induced apoptosis. H and I, Coadministration of 5-FU and 42-(2-tetrazolyl) rapamycin resulted in robust antitumor activity in gastric cancer xenografts and restrained gastric cancer peritoneal metastasis. The combination administration exhibited better survival in the mice model. *, P < 0.05; **, P < 0.01; ***, P < 0.001.
Discussion
Gastric cancer is the most common malignant tumor globally, with remarkably elevated incidence and mortality. GLOBOCAN 2020 indicated more than 1 million estimated new cases of gastric cancer and 768,793 deaths annually, making it the fifth most diagnosed malignancy and fourth leading cause of mortality worldwide (30). Despite the diverse treatment options available, the prognosis for patients with gastric cancer remains poor (31). Therefore, urgent efforts are needed to develop effective biomarkers and therapeutic targets to combat gastric cancer.
The family of the FOXP transcription factors contains four members, FOXP1 to FOXP4, which play crucial roles in various physiologic and pathologic processes (32–34). Growing evidence suggested a potential correlation between the FOXP family and gastric cancer. Specifically, FOXP1 expression was significantly reduced in gastric cancer tissues compared with adjacent nontumor tissues (35). FOXP1 serves as a tumor suppressor gene. It binds to PDL1 enhancers and inhibits PDL1 transcription, suppressing tumor immune escape (36). Indeed, FOXP2-induced oncogenesis results in the upregulation of miR-190, which alters cell behavior regarding proliferation and metastasis (37). On the other hand, FOXP3 expression was upregulated in gastric cancer, and its abundance was associated with tumor invasion and lymph node metastasis (38). These findings suggest that the FOXP family could be a potential diagnostic and therapeutic target for gastric cancer. FOXP4 plays a cancer-promoting role in colorectal cancer (39), non–small cell lung cancer (40), and breast cancer, especially in breast cancer, in which FOXP4 promotes epithelial–mesenchymal transition through Snail (41), yet the expression and functional roles of FOXP4 in gastric cancer remain largely unknown.
In gastric cancer, it has been reported that YAP1 can activate MYC transcription by the binding promoter and enhancer (42), and YAP1 can maintain cell stemness in peritoneal metastasis of gastric cancer (associated with SOX9, HES1, EGR3, and ALDH3A1; ref. 43). This study demonstrated that the YAP1–FOXP4–SOX12 axis exerts an oncogenic function in gastric carcinogenesis with stemness maintenance. These findings are consistent with our previously mentioned cancer stemness–associated signaling pathways, including the Hippo–YAP1 signaling pathway. In this study, we unraveled that FOXP4 and SOX12 are novel stemness factors in gastric cancer with clinical and prognostic significance.
According to a single-cell sequencing analysis of gastric cancer, YAP1 and FOXP4 were abundantly expressed in a specific population of gastric cancer cells, showing significant stemness characteristics. Meanwhile, knocking down YAP1 and FOXP4 significantly suppressed tumorsphere formation, implying that YAP1 and FOXP4 are involved in stemness maintenance. Maintenance of stemness is one of the hallmarks of cancer cells, and SOX12 was identified as an effector for the oncogenic YAP1–FOXP4 signal transduction. SOX12 is reported as a novel stem cell marker in liver cancer (44). In colorectal cancer, SOX12 facilitates the proliferation of cancer cells through asparagine synthesis (45). In gastric cancer, SOX12 has been reported to increase the upregulation of matrix metalloproteinase 7 and insulin-like growth factor 1 to promote gastric cancer metastasis (46).
In summary, our study demonstrated that FOXP4 serves as an oncogene in promoting the progression of YAP1-driven gastric cancer. Mechanistically, FOXP4 acted as a mediator for YAP1 to elevate the SOX12 expression and maintain gastric cancer stemness. Additionally, we revealed that the combination of FOXP4 inhibition by a small molecule, named 42-(2-tetrazolyl) rapamycin, and 5-FU (first-line chemotherapy) exhibited a more powerful antitumor effect (47), suggesting that targeting FOXP4 by adding 5-FU might serve as a prospective therapeutic approach for gastric cancer. Besides, our study provides novel insights into stemness maintenance in YAP1-driven gastric cancer. The aberrant activation of the YAP1–FOXP4–SOX12 axis serves as a promising therapeutic target in gastric cancer.
The oncogenic YAP1–FOXP4–SOX12 signaling with stemness property was elucidated in gastric cancer (Fig. 8). YAP1 directly binds to the FOXP4 promoter and upregulates FOXP4 transcription. Subsequently, FOXP4 drives gastric carcinogenesis mainly by regulating SOX12 and maintaining the stemness of the cancer cells. Our study provides novel insights into stemness maintenance in YAP1-driven gastric cancer. The aberrant activation of the YAP1–FOXP4–SOX12 axis serves as a promising therapeutic target in gastric cancer.
Figure 8.

Overall schematic presentation of the YAP1–FOXP4–SOX12 axis in gastric cancer. YAP1 directly regulates FOXP4 expression via binding its promoter. FOXP4 exerts oncogenic properties by upregulating SOX12. As a novel therapeutic target, depleting FOXP4 suppresses cancer cell stemness, reverses drug resistance, and reduces peritoneal metastasis. Small molecule targeting FOXP4 quenches YAP1-driven gastric tumor progression and enhances the 5-FU efficacy. (Created with BioRender.com.)
Supplementary Material
Supplementary Figure S1
{kind=link}
Supplementary Figure S2
{kind=link}
Supplementary Figure S3
{kind=link}
Supplementary Figure S4
{kind=link}
Supplementary Figure S5
{kind=link}
Supplementary Figure S6
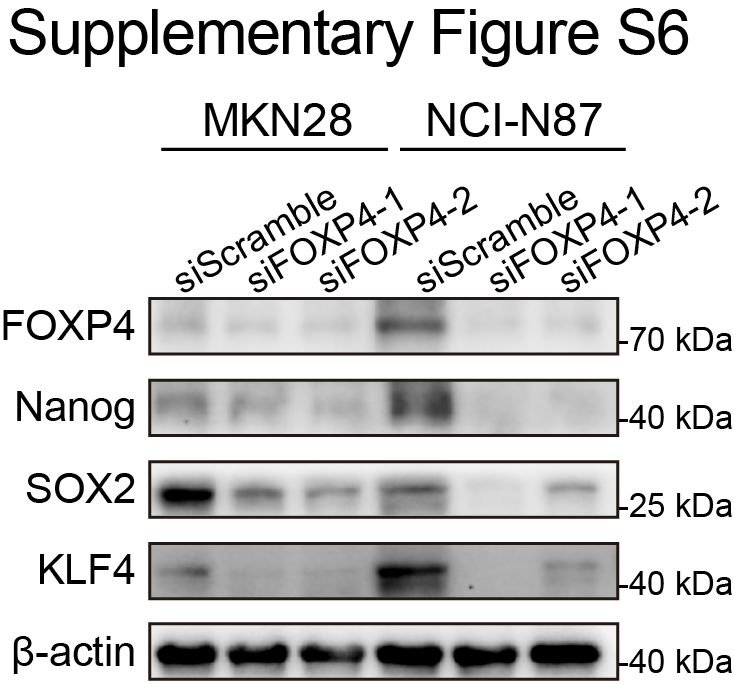{kind=link}
Supplementary Figure S7
{kind=link}
Supplementary Figure S8
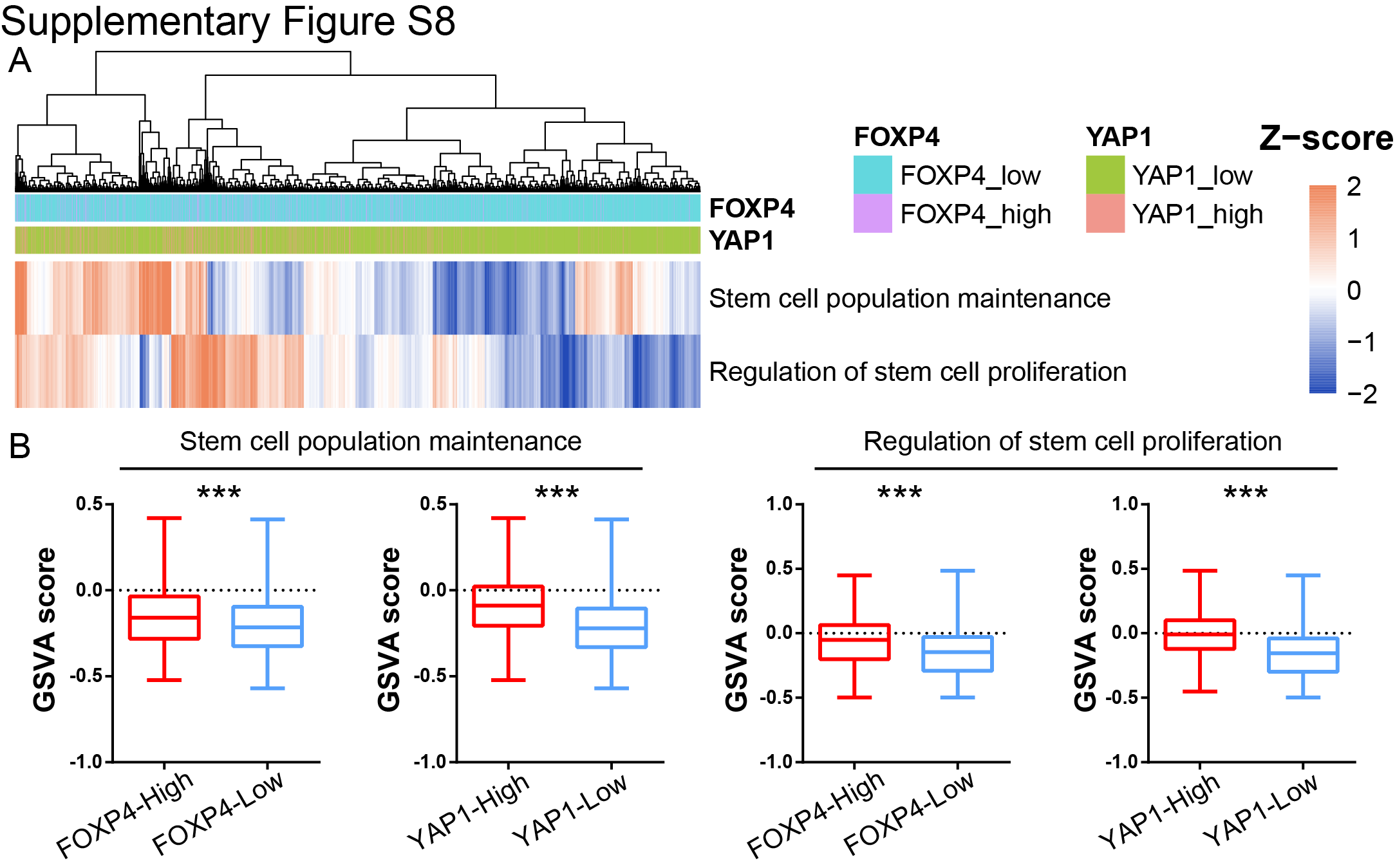{kind=link}
Supplementary Figure S9
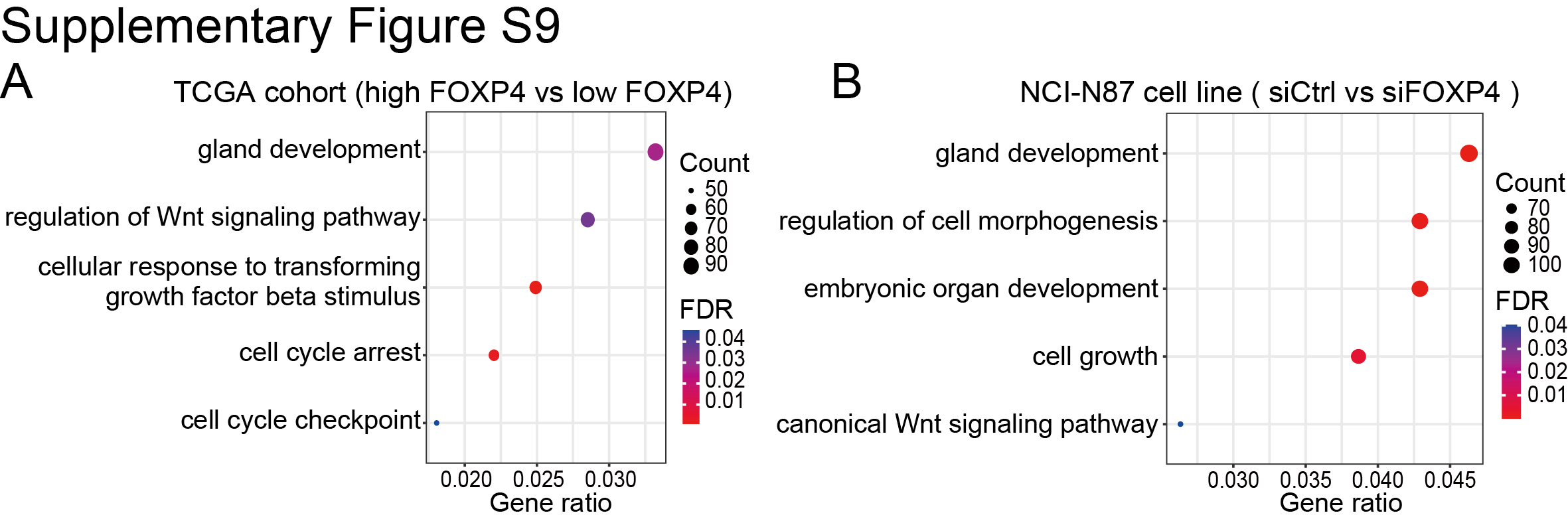{kind=link}
Supplementary Figure S10
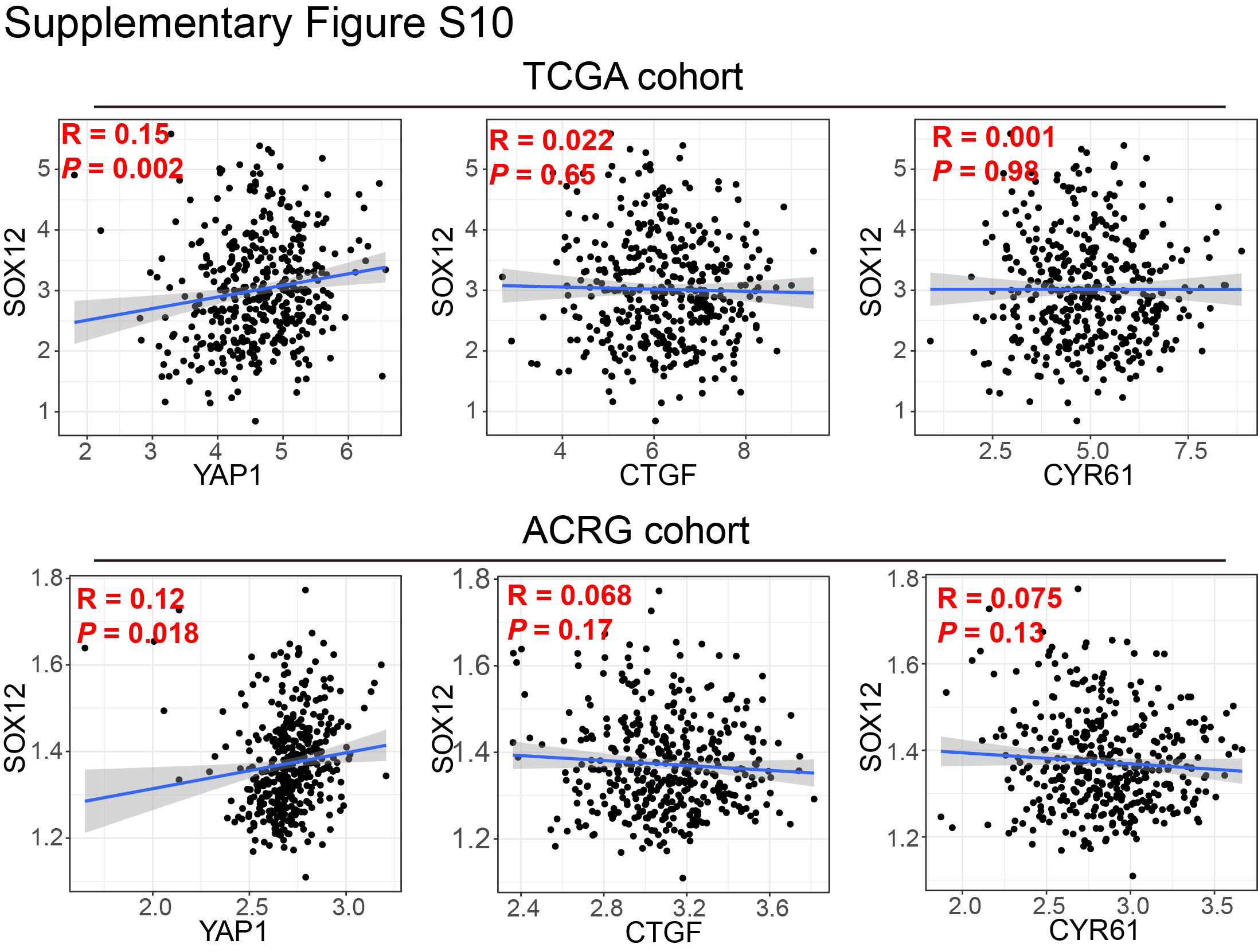{kind=link}
Supplementary Figure S11
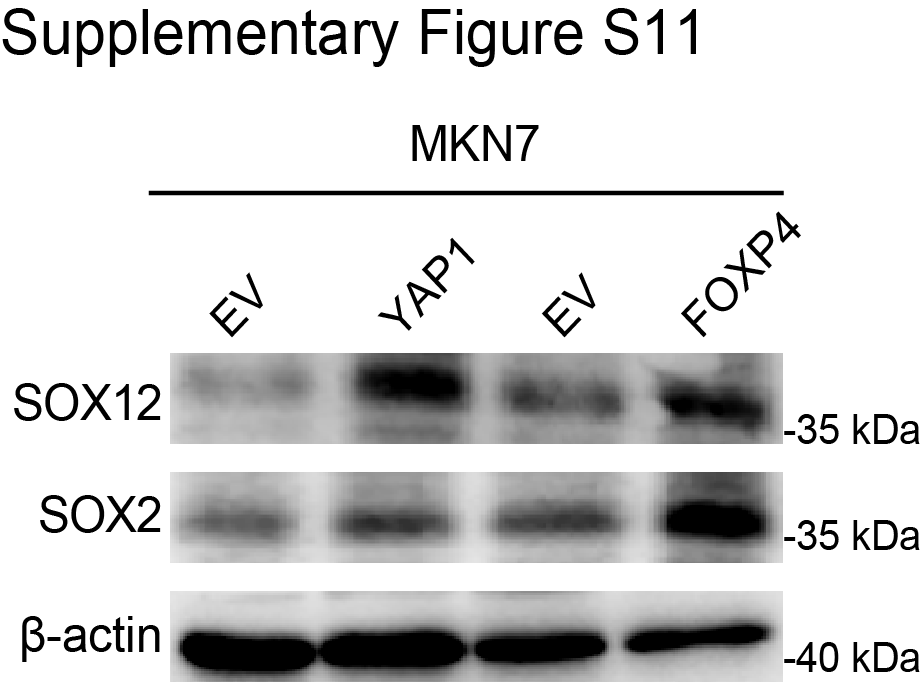{kind=link}
Supplementary Figure S12
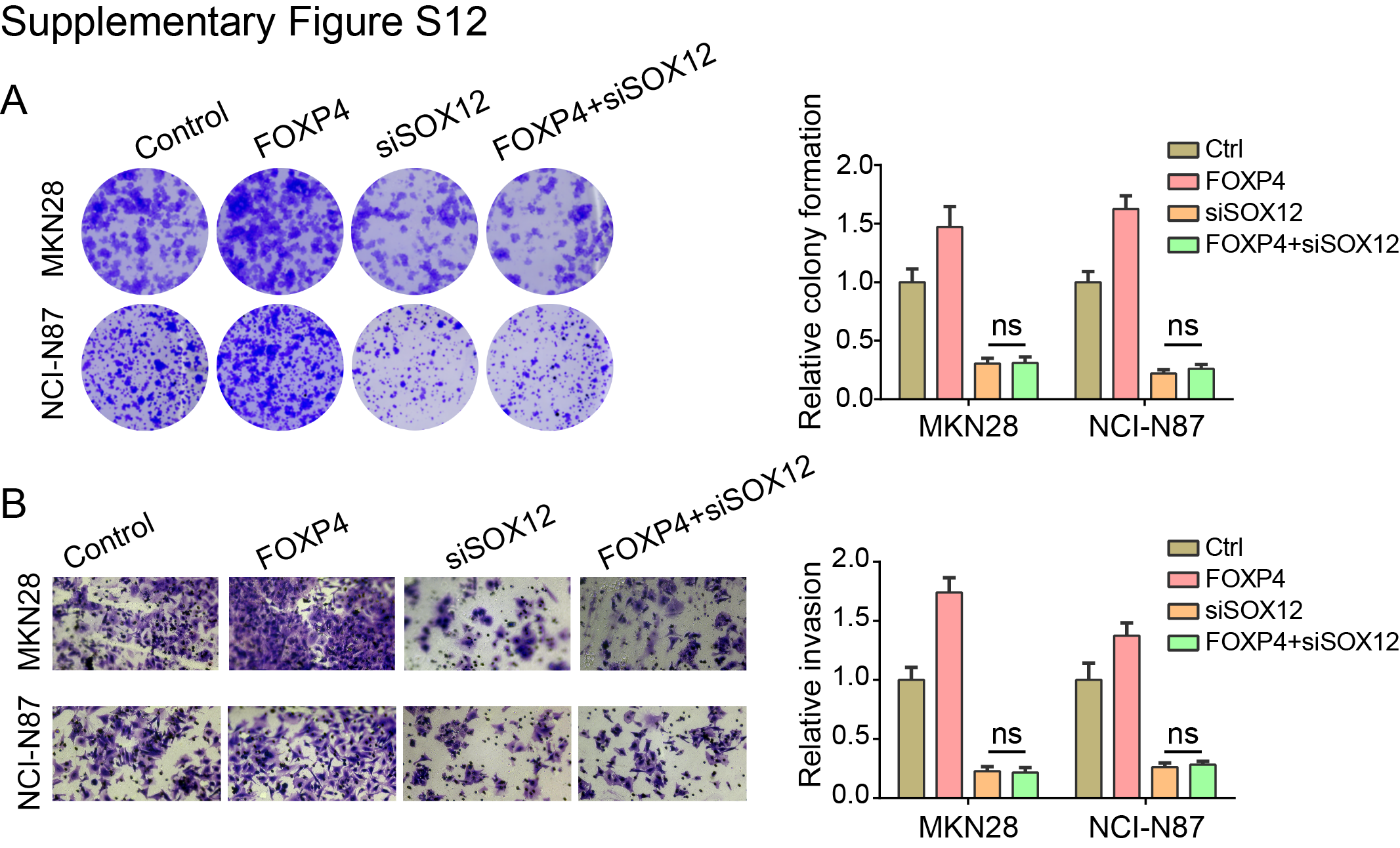{kind=link}
Supplementary Figure S13
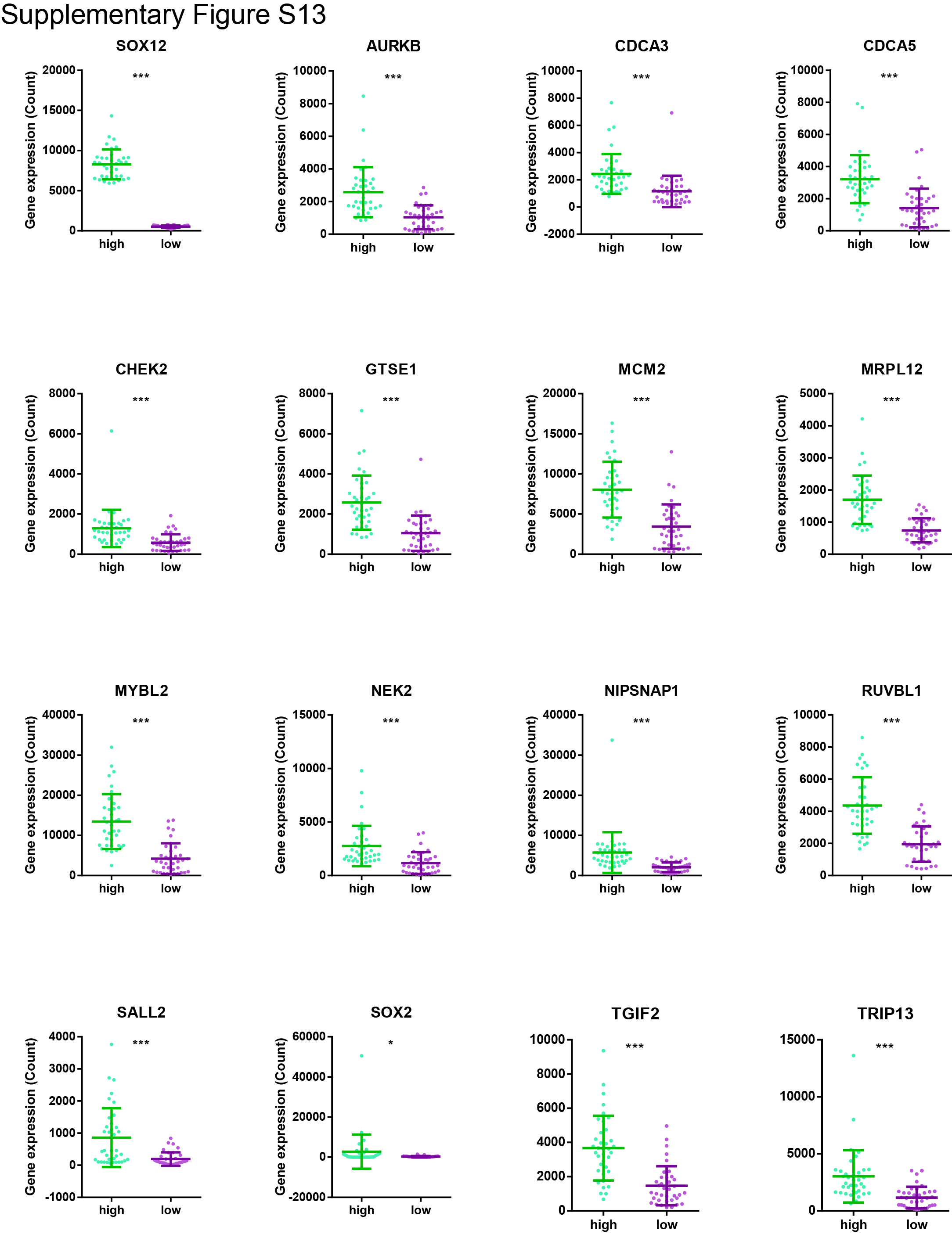{kind=link}
Supplementary Figure S14
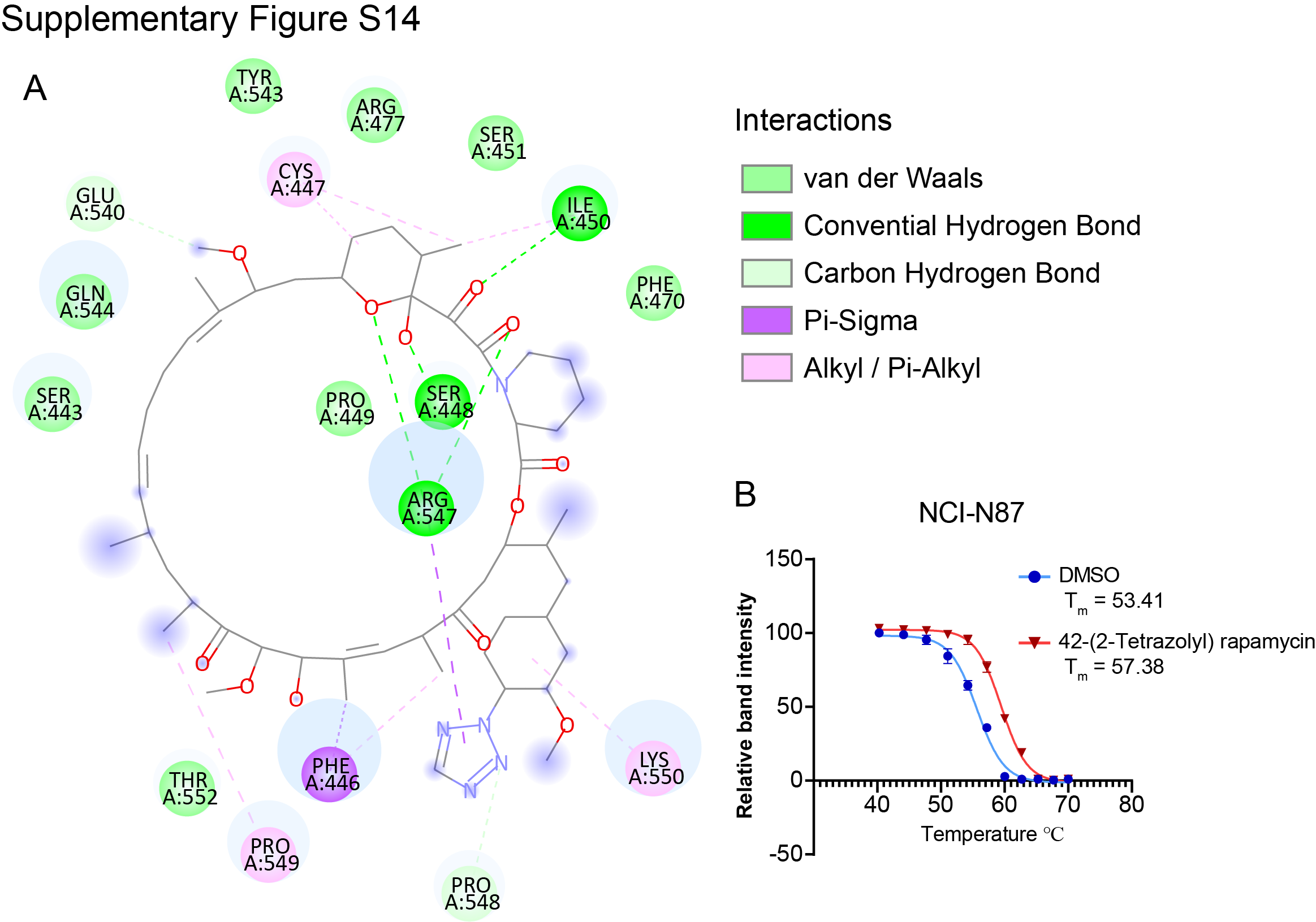{kind=link}
Supplementary Table S1-S4
Acknowledgments
We thank TCGA Research Network (http://cancergenome.nih.gov/) for the datasets and analysis. We appreciated the generous provision of the transgenic mice from Prof. Zhou Zhaocai (Fudan University). We also appreciate the technical support from the Core Utilities of Cancer Genomics and Pathobiology of the Department of Anatomical and Cellular Pathology at the Chinese University of Hong Kong. This study is supported by the National Natural Science Foundation of China (2022, No. 82272990), Health and Medical Research Fund (08190586), Chinese University of Hong Kong Direct Research Grant (2022.001 and 2020.004), and Cheng Yue Pui Charity Foundation.
Footnotes
Note: Supplementary data for this article are available at Cancer Research Online (http://cancerres.aacrjournals.org/).
Authors’ Disclosures
K.W. Lo reports grants from Viracta Therapeutics and ScinnoHub Pharmaceutical Co., Ltd., outside the submitted work. No disclosures were reported by the other authors.
Authors’ Contributions
X. Liu: Methodology, writing–original draft, writing–review and editing. B. Chen: Software, methodology. F. Xie: Software, methodology. K.Y. Wong: Methodology. A.H.K. Cheung: Methodology. J. Zhang: Methodology. Q. Wu: Methodology. C. Fang: Methodology. J. Hu: Methodology. S. Wang: Writing–review and editing. D. Xu: Writing–review and editing. J. Chen: Writing–review and editing. Y. Wang: Writing–review and editing. C.C. Wong: Writing–review and editing. H. Chen: Writing–review and editing. W.K.K. Wu: Writing–review and editing. J. Yu: Resources, writing–review and editing. M.W.Y. Chan: Writing–review and editing. C.M. Tsang: Writing–review and editing. K.W. Lo: Writing–review and editing. G.M.K. Tse: Writing–review and editing. K.-F. To: Writing–review and editing. W. Kang: Conceptualization, resources, funding acquisition, methodology, writing–original draft.
References
- 1. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell 2011;144:646–74. [DOI] [PubMed] [Google Scholar]
- 2. Reya T, Morrison SJ, Clarke MF, Weissman IL. Stem cells, cancer, and cancer stem cells. Nature 2001;414:105–11. [DOI] [PubMed] [Google Scholar]
- 3. Aponte PM, Caicedo A. Stemness in cancer: stem cells, cancer stem cells, and their microenvironment. Stem Cell Int 2017;2017:5619472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Saygin C, Matei D, Majeti R, Reizes O, Lathia JD. Targeting cancer stemness in the clinic: from hype to hope. Cell Stem Cell 2019;24:25–40. [DOI] [PubMed] [Google Scholar]
- 5. Chen P, Hsu W-H, Han J, Xia Y, DePinho RA. Cancer stemness meets immunity: from mechanism to therapy. Cell Rep 2021;34:108597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Clevers H. The cancer stem cell: premises, promises and challenges. Nat Med 2011;17:313–9. [DOI] [PubMed] [Google Scholar]
- 7. Miranda A, Hamilton PT, Zhang AW, Pattnaik S, Becht E, Mezheyeuski A, et al. Cancer stemness, intratumoral heterogeneity, and immune response across cancers. Proc Natl Acad Sci U S A 2019;116:9020–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Medema JP. Cancer stem cells: the challenges ahead. Nat Cell Biol 2013;15:338–44. [DOI] [PubMed] [Google Scholar]
- 9. Nassar D, Blanpain C. Cancer stem cells: basic concepts and therapeutic implications. Annu Rev Pathol 2016;11:47–76. [DOI] [PubMed] [Google Scholar]
- 10. Tsui Y-M, Chan L-K, Ng IO-L. Cancer stemness in hepatocellular carcinoma: mechanisms and translational potential. Br J Cancer 2020;122:1428–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Takebe N, Miele L, Harris PJ, Jeong W, Bando H, Kahn M, et al. Targeting Notch, Hedgehog, and Wnt pathways in cancer stem cells: clinical update. Nat Rev Clin Oncol 2015;12:445–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Yang L, Shi P, Zhao G, Xu J, Peng W, Zhang J, et al. Targeting cancer stem cell pathways for cancer therapy. Signal Transduct Target Ther 2020;5:8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Harvey KF, Pfleger CM, Hariharan IK. The Drosophila Mst ortholog, Hippo, restricts growth and cell proliferation and promotes apoptosis. Cell 2003;114:457–67. [DOI] [PubMed] [Google Scholar]
- 14. Staley BK, Irvine KD. Hippo signaling in Drosophila: recent advances and insights. Dev Dyn 2012;241:3–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Yu FX, Zhao B, Guan KL. Hippo pathway in organ size control, tissue homeostasis, and cancer. Cell 2015;163:811–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Miranda MM, Lowry WE. Hip to the game: YAP/TAZ is required for nonmelanoma skin cancers. EMBO J 2018;37:e99921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Zanconato F, Cordenonsi M, Piccolo S. YAP/TAZ at the roots of cancer. Cancer Cell 2016;29:783–803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kapoor A, Yao W, Ying H, Hua S, Liewen A, Wang Q, et al. Yap1 activation enables bypass of oncogenic Kras addiction in pancreatic cancer. Cell 2014;158:185–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kang W, Tong JH, Chan AW, Lee TL, Lung RW, Leung PP, et al. Yes-associated protein 1 exhibits oncogenic property in gastric cancer and its nuclear accumulation associates with poor prognosis. Clin Cancer Res 2011;17:2130–9. [DOI] [PubMed] [Google Scholar]
- 20. Liu X, Wang Y, Chen B, Chan WN, Mui CW, Cheung AHK, et al. Targeting the Hippo pathway in gastric cancer and other malignancies in the digestive system: from bench to bedside. Biomedicines 2022;10:2512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Zhao B, Ye X, Yu J, Li L, Li W, Li S, et al. TEAD mediates YAP-dependent gene induction and growth control. Genes Dev 2008;22:1962–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Wu S, Liu Y, Zheng Y, Dong J, Pan D. The TEAD/TEF family protein scalloped mediates transcriptional output of the Hippo growth-regulatory pathway. Dev Cell 2008;14:388–98. [DOI] [PubMed] [Google Scholar]
- 23. Huch M, Gehart H, van Boxtel R, Hamer K, Blokzijl F, Verstegen MM, et al. Long-term culture of genome-stable bipotent stem cells from adult human liver. Cell 2015;160:299–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kang W, Huang T, Zhou Y, Zhang J, Lung RWM, Tong JHM, et al. miR-375 is involved in Hippo pathway by targeting YAP1/TEAD4-CTGF axis in gastric carcinogenesis. Cell Death Dis 2018;9:92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ianevski A, He L, Aittokallio T, Tang J. SynergyFinder: a web application for analyzing drug combination dose–response matrix data. Bioinformatics 2017;33:2413–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Cristescu R, Lee J, Nebozhyn M, Kim K-M, Ting JC, Wong SS, et al. Molecular analysis of gastric cancer identifies subtypes associated with distinct clinical outcomes. Nat Med 2015;21:449–56. [DOI] [PubMed] [Google Scholar]
- 27. Jiao S, Wang H, Shi Z, Dong A, Zhang W, Song X, et al. A peptide mimicking VGLL4 function acts as a YAP antagonist therapy against gastric cancer. Cancer Cell 2014;25:166–80. [DOI] [PubMed] [Google Scholar]
- 28. Jiao S, Guan J, Chen M, Wang W, Li C, Wang Y, et al. Targeting IRF3 as a YAP agonist therapy against gastric cancer. J Exp Med 2018;215:699–718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Tang Y, Fang G, Guo F, Zhang H, Chen X, An L, et al. Selective inhibition of STRN3-containing PP2A phosphatase restores Hippo tumor-suppressor activity in gastric cancer. Cancer Cell 2020;38:115–28.e9. [DOI] [PubMed] [Google Scholar]
- 30. Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, et al. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin 2021;71:209–49. [DOI] [PubMed] [Google Scholar]
- 31. Smyth EC, Nilsson M, Grabsch HI, van Grieken NCT, Lordick F. Gastric cancer. Lancet 2020;396:635–48. [DOI] [PubMed] [Google Scholar]
- 32. Kim J-H, Hwang J, Jung JH, Lee H-J, Lee DY, Kim S-H. Molecular networks of FOXP family: dual biologic functions, interplay with other molecules and clinical implications in cancer progression. Mol Cancer 2019;18:180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Myatt SS, Lam EWF. The emerging roles of forkhead box (Fox) proteins in cancer. Nat Rev Cancer 2007;7:847–59. [DOI] [PubMed] [Google Scholar]
- 34. Bach DH, Long NP, Luu TT, Anh NH, Kwon SW, Lee SK. The dominant role of forkhead box proteins in cancer. Int J Mol Sci 2018;19:3279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Koon HB, Ippolito GC, Banham AH, Tucker PW. FOXP1: a potential therapeutic target in cancer. Expert Opin Ther Targets 2007;11:955–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Wang J, Ge J, Wang Y, Xiong F, Guo J, Jiang X, et al. EBV miRNAs BART11 and BART17-3p promote immune escape through the enhancer-mediated transcription of PD-L1. Nat Commun 2022;13:866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Jia WZ, Yu T, An Q, Yang H, Zhang Z, Liu X, et al. MicroRNA-190 regulates FOXP2 genes in human gastric cancer. Onco Targets Ther 2016;9:3643–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Ma GF, Miao Q, Liu YM, Gao H, Lian JJ, Wang YN, et al. High FoxP3 expression in tumour cells predicts better survival in gastric cancer and its role in tumour microenvironment. Br J Cancer 2014;110:1552–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Wu H, Tao Y, Zhang W, Wang G, Zhang Q. circ-0000212 promotes cell proliferation of colorectal cancer by sponging miR-491 and modulating FOXP4 expression. Mol Med Rep 2021;23:300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Yang T, Li H, Thakur A, Chen T, Xue J, Li D, et al. FOXP4 modulates tumor growth and independently associates with miR-138 in non-small cell lung cancer cells. Tumor Biol 2015;36:8185–91. [DOI] [PubMed] [Google Scholar]
- 41. Ma T, Zhang J. Upregulation of FOXP4 in breast cancer promotes migration and invasion through facilitating EMT. Cancer Manag Res 2019;11:2783–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Choi W, Kim J, Park J, Lee DH, Hwang D, Kim JH, et al. YAP/TAZ initiates gastric tumorigenesis via upregulation of MYC. Cancer Res 2018;78:3306–20. [DOI] [PubMed] [Google Scholar]
- 43. Ajani JA, Xu Y, Huo L, Wang R, Li Y, Wang Y, et al. YAP1 mediates gastric adenocarcinoma peritoneal metastases that are attenuated by YAP1 inhibition. Gut 2021;70:55–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Zou S, Wang C, Liu J, Wang Q, Zhang D, Zhu S, et al. Sox12 is a cancer stem-like cell marker in hepatocellular carcinoma. Mol Cell 2017;40:847–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Du F, Chen J, Liu H, Cai Y, Cao T, Han W, et al. SOX12 promotes colorectal cancer cell proliferation and metastasis by regulating asparagine synthesis. Cell Death Dis 2019;10:239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Du F, Feng W, Chen S, Wu S, Cao T, Yuan T, et al. Sex determining region Y-box 12 (SOX12) promotes gastric cancer metastasis by upregulating MMP7 and IGF1. Cancer Lett 2019;452:103–18. [DOI] [PubMed] [Google Scholar]
- 47. Xu ZY, Tang JN, Xie HX, Du YA, Huang L, Yu PF, et al. 5-Fluorouracil chemotherapy of gastric cancer generates residual cells with properties of cancer stem cells. Int J Biol Sci 2015;11:284–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3
Supplementary Figure S4
Supplementary Figure S5
Supplementary Figure S6
Supplementary Figure S7
Supplementary Figure S8
Supplementary Figure S9
Supplementary Figure S10
Supplementary Figure S11
Supplementary Figure S12
Supplementary Figure S13
Supplementary Figure S14
Supplementary Table S1-S4
Data Availability Statement
The RNA-seq data generated in this study are publicly available in Gene Expression Omnibus at GSE267854. TCGA-STAD RNA-seq data analyzed in this study were obtained from https://www.cancer.gov/tcga. The scRNA-seq data analyzed in this study from former research (DOI: 10.1158/1078-0432.CCR-19-3231) are available at https://dna-discovery.stanford.edu/research/datasets/. The ACRG dataset analyzed in this study was obtained from Gene Expression Omnibus at GSE66229. All other raw data generated in this study are available upon request from the corresponding author.