

Abstract
Diffuse large B‐cell lymphoma (DLBCL) is the most prevalent subtype of non‐Hodgkin lymphoma (NHL) in domestic dogs, with many similarities to its human counterpart. The progression of the disease is rapid, and treatment must be initiated early to achieve cancer remission and extend life. This study examined the relationship between progression‐free survival (PFS) and microRNA (miRNA) expression in dogs with DLBCL. miRNAs are small non‐coding RNA molecules that typically regulate gene expression post‐transcriptionally. They are involved in several pathophysiological processes, including the growth and progression of cancer. Based on the analysis of small RNA sequencing (sRNA‐seq) data, we validated a group of miRNAs in lymph nodes from 44 DLBCL‐affected dogs with known outcomes. We used quantitative PCR to quantify their expression and report a specific subset of miRNAs is associated with decreased PFS in dogs with DLBCL. The miR‐192‐5p and miR‐16‐5p expression were significantly downregulated in dogs with increased PFS. These results indicate that miRNA profiling may potentially identify dogs with DLBCL that will experience poor outcomes following treatment. Identifying specific miRNAs that correlate with the progression of canine DLBCL could aid the development of individualized treatment regimens for dogs.
Keywords: cancer biomarkers, canine DLBCL, diffuse large B‐cell lymphoma, miRNAs, multicentric, sRNA sequencing
Our study explores how microRNAs (miRNAs) influence canine diffuse large B‐cell lymphoma (DLBCL) outcomes. We found specific miRNAs linked to shorter survival times, suggesting that miRNA profiling could help predict DLBCL prognosis. This research may lead to personalized dog treatments and be relevant for human lymphoma studies.
Abbreviations
- ANOVA
analysis of variance
- BOR
best overall response
- cDNA
complementary DNA
- CHOP
cyclophosphamide, doxorubicin, vincristine, and prednisone (chemotherapy regimen)
- DEMs
differentially expressed miRNAs
- DLBCL
diffuse large B‐cell lymphoma
- FDR
false discovery rate
- FFPE
formalin‐fixed paraffin‐embedded
- IPC
inter‐plate calibrator
- NHL
non‐Hodgkin lymphoma
- PFS
progression‐free survival
- RT‐qPCR
reverse transcription quantitative polymerase chain reaction
- sRNA‐seq
small RNA sequencing
- UV
ultraviolet
The most frequent form of lymphoma in dogs and humans is the diffuse large B‐cell lymphoma (DLBCL). It represents a wide range of diseases regarding morphology and subsequently, its outcomes are difficult to predict. Poor inter‐examiner consistency and limited success in outcome prediction are commonplace challenges with traditional morphologic subclassification [1]. Thus, developing and validating new prognostic tools is essential. In human studies, the International Prognostic Index (IPI) is the most crucial prognostic indicator in diffuse large B‐cell lymphoma cases in humans. All clinical trials use the IPI because of its reliability as a prognostic tool in DLBCL. Age > 65 years, high serum lactate dehydrogenase (LDH) activity, ECOG performance status ≥ 2, Ann Arbor stage III or IV for anatomical staging, and the number of involved extranodal sites are all components of this index before the use of rituximab to predict DLBCL survival [2, 3]. Although IPI is a reliable prognostic tool, it cannot foretell outcomes in patients with aggressive disease, for whom the three‐year event‐free survival rate is only about 50%. The fundamental biological disparity needs to be addressed [2].
The usefulness of canine DLBCL as a natural simulation for human DLBCL has been demonstrated by several studies [4, 5, 6], making it the most studied tumor in veterinary medicine to date [4]. Prognostic variables for dogs with canine DLBCL are poorly depicted [7]. Poorer prognoses were seen in dogs with DLBCL when certain factors were present, such as thrombocytopenia at diagnosis, higher baseline serum globulin concentration, higher baseline neutrophil count, and older age at diagnosis [8]. Tumor mitotic counts correlate with disease progression in dogs with DLBCL. Dogs with < 20 mitoses per 400 field had a median survival duration of 188 days, while those with more than 20 mitoses per 400 field had a median survival time of 31 days [9]. The percentage of neoplastic cells infiltrating the bone marrow (BM) may also predict survival and time to progression [4, 10]. A correlation was also found between canine systemic inflammatory response and decreased survival time. The inflammatory biomarker C‐reactive protein (CRP) was also linked to decreased progression‐free survival (PFS) [7].
The absence of definitive prognostic markers in canine DLBCL cases has prompted the need to investigate new biomarkers that might aid in detecting the prognosis. We aimed to investigate the prognostic significance of miRNA expression profiles in dogs with DLBCL by comparing miRNA signatures between cases with extremely short and long progression‐free survival. MicroRNAs (miRNAs) are small non‐coding RNA molecules that regulate gene expression. They are involved in several pathophysiological processes, including the growth and progression of cancer [11, 12, 13, 14]. Here, we are investigating the potential role of miRNAs as biomarkers in detecting canine DLBCL. This retrospective study with 44 archived lymph node samples of DLBCL‐affected dogs investigated the relationship between miRNA expression levels and PFS and/or best overall response (BOR) to chemotherapy. A group of differentially expressed miRNAs were identified to be linked with short PFS of dogs after chemotherapy treatment. In the future, these results may lead to models to predict treatment outcomes and help tailor treatment plans based on individualized risk assessment.
Materials and methods
Animal samples
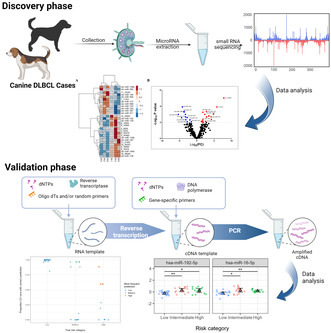
Discovery phase
Six fresh‐frozen (FF) lymph node biopsy specimens collected from dogs with DLBCL were selected for preliminary analysis (Table S1) due to their superior RNA quality over formalin‐fixed paraffin‐embedded (FFPE) samples, which is crucial for accurate small RNA sequencing. All lymph node biopsy samples were collected prior to the dogs receiving any treatment for their DLBCL. Dogs with exceptionally short (n = 3) and long (n = 3) PFS were specifically selected for this initial study to identify distinct miRNA signatures associated with these divergent outcomes. Progression‐free survival was defined as the time in days from initiation of chemotherapy until cancer progression or death from any cause occurred. After total RNA extraction from the FF lymph nodes, small RNA (sRNA) sequencing proceeded. Several studies have demonstrated comparable results between FF and FFPE samples in various molecular analyses, including gene expression profiling [15, 16, 17] and mutation detection [18], FFPE samples were used to validate the sRNA sequencing results due to their availability in a bigger well‐documented cohort.
Validation phase
Forty‐four FFPE archival lymph node biopsy specimens from dogs with DLBCL were selected to represent two phenotypic groups based on their survival times with known PFS; long PFS (L‐PFS, n = 18) and short PFS (S‐PFS, n = 26) and three phenotypic groups based on the clinical BOR classifications. The best overall response was classified according to established criteria [19]. Complete remission (CR) was defined as complete regression of all tumor lesions, with resolution of all cancer‐related clinical signs. Partial remission (PR) was defined as a reduction in the sum of the longest diameters of up to five tumor lesions by at least 50% but < 100%. Dogs that experienced partial remission (PR group n = 12) had universally short PFS times. Some who achieved complete remission still showed relatively short survival times (CR‐short PFS group n = 14), while others showed much longer PFS (Table S2). Combining the PFS and BOR for sample identification, samples with complete remission with long PFS were called low risk. Samples with complete remission with short survival were called intermediate risk, while samples that received partial remission with short survival were renamed to the high‐risk group (Fig. 1). This criterion was used to validate the sRNA sequencing using qPCR. Both FF and FFPE samples were obtained from privately owned dogs diagnosed with primary nodal DLBCL and presented to the Purdue University Veterinary Teaching Hospital (PUVTH), West Lafayette, IN, between 2006 and 2016.
Fig. 1.
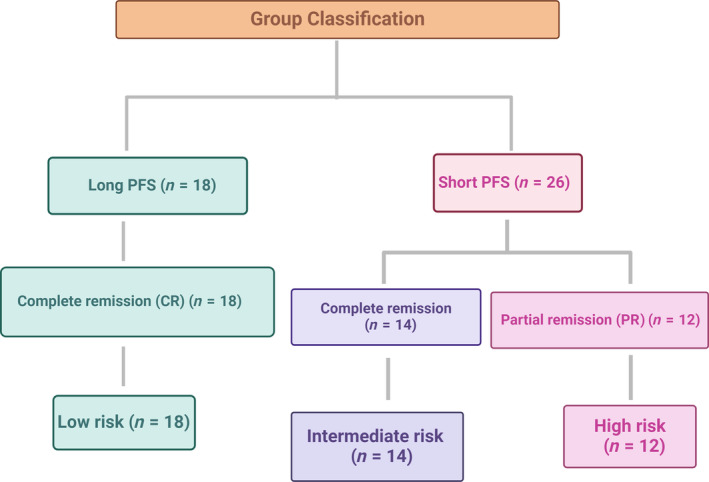
Sample classification for the validation phase (n = 44).
All samples were obtained by surgical biopsy from peripheral lymph nodes (incisional wedge biopsy or surgical extirpation of an entire lymph node) with the dogs under general anesthesia. The diagnosis of DLBCL was determined by combining histopathology and immunohistochemistry findings confirming the expression of CD79a and/or Pax‐5 and the absence of CD3 expression in malignant cells. The demographic information, medical history, and animal test results are provided (Tables S1 and S2). A board‐certified veterinary pathologist evaluated and classified the samples according to the 2008 WHO criteria. In addition to surgical lymph node biopsy, all dogs underwent clinical staging tests, including complete blood count, serum biochemistry profile, thoracic and abdominal radiography, abdominal ultrasonography, and bone marrow aspirate cytology. All dogs were assigned a clinical stage according to WHO criteria [20]. Following staging, all dogs were treated with an identical chemotherapy protocol [21] incorporating cyclophosphamide, doxorubicin, vincristine, and prednisone (i.e., CHOP). All samples for this study were collected with the owners' written informed consent, and sample collection procedures were approved by the Purdue Animal Care and Use Committee (protocols #1708001607 and #1111000308).
Total RNA extraction
The sRNA sequence samples were extracted from fresh‐frozen specimens using Monarch® Total RNA Miniprep Kit (NEB, Ipswich, MA, USA). For the archived FFPE samples, total RNA was extracted from one 20‐mm scroll from the paraffin blocks using the miRNeasy FFPE kit (QIAGEN Inc, Germantown, MD, USA) according to the manufacturer's specifications. The quantity and quality of total RNA were assessed by UV® spectrophotometry (NanoDrop; ThermoFisher Scientific, Waltham, MA, USA). The quantification of miRNA was assisted by a spectrofluorometer (Qubit 4 Fluorometer; ThermoFisher Scientific) using Invitrogen™ Qubit™ microRNA Assay Kits (ThermoFisher Scientific) as demonstrated (Table S3A,B).
Small RNA library preparation
The small RNA library preparation was guided using the sequencing libraries generated using NEBNext® Multiplex Small RNA Library Prep Set (NEB) for Illumina®. The RNA was subjected to size selection ranging from 15 to 40 nucleotides in length for small RNA. Ligation of adapters at both ends of the small RNA molecules was performed, followed by reverse transcription to produce complementary DNA (cDNA) using M‐MuLV Reverse Transcriptase (RNase H−). Subsequently, the polymerase chain reaction (PCR) amplification was employed using LongAmp Taq 2X Master Mix, SR Primer for Illumina, and index (X) primer that were designed to target the ligated adapters specifically.
Sequencing and data processing
The prepared small RNA libraries were subjected to high‐throughput sequencing for Illumina® (Novogene Co., Ltd., Beijing, China). The sequencing was performed according to the manufacturer's protocols to generate raw sequencing reads. The proportion of reads exhibiting < 1% error rate (Q20) and 0.1% error rate (Q30), as well as their GC‐content, was determined after the removal of reads containing poly‐N, 5′ adapter contaminants without 3′ adapter or the insert tag. Additionally, reads containing poly A, T, G, C, or those with low quality were excluded from the analysis, as indicated.
Read mapping and annotation
MiRNAs were counted using mirdeep2, version 2.0.1.2 [22] with the Canis lupus familiaris CanFam 3.1 genome [23] as the reference sequence. For the assignment of miR names, reference lists of dog and mouse mature and precursors were downloaded from miRbase [24] 4.35 Mreads were examined, and 445 known (276) and novel (169) miRNAs with read count > 330 (0.00001%, summed across all samples) identified. These reads account for 39.3 Mreads (90.6%) of the original counts. miR with identical mature sequences (which cannot be distinguished in the sequencing) were merged, resulting in 373 observed miRs (224 known, 149 novel). Normalized counts were determined using the median of ratios method implemented in deseq2 [25]. Predicted miRNA (novel miRNA) that match neither the known dog nor mouse miR lists are assigned names by miRDeep2 that consist of the chromosome number followed by the beginning position of the miRNA in the reference genome sequence, for example, miRNA 12_21493 is found on chromosome 12 at base 21493 (Table S4).
Differential expression analysis
Differential expression analysis of small RNAs was performed using deseq2 for dog patients with different PFS, S‐PFS, n = 3 and L‐PFS, n = 3. Small RNAs with a significant fold change of > 3 and adjusted P‐value < 0.01 were used as the threshold for significantly differential expression.
Quantitative RT‐PCR
The 44 FFPE archival lymph node biopsy specimens from dogs with DLBCL were used to extract total RNA. Following the manufacturer's protocol, the first‐strand cDNA synthesis was performed using the miRCURY LNA RT Kit (QIAGEN). The cDNA was diluted at 1 : 80 according to the manufacturer's instructions. Quantitative RT‐PCR was performed on a QuantStudio3 PCR system (ThermoFisher Scientific). The miRCURY LNA [26] miRNA Custom PCR Panel (Cat #: YCA38192; QIAGEN) was used to quantify the pre‐selected miRNAs based on the differential expressed miRNAs between short and long PFS groups in the discovery phase using FF samples showed potential prognostic value canine lymphoma and previous literature on miRNAs role in cancer prognosis [27, 28, 29, 30, 31, 32, 33, 34] (Table 1). Two samples were measured per plate in duplicate for 22 miRNAs, UniSp6 (cDNA synthesis control), UniSp3 (RT‐qPCR positive control) and inter‐plate calibrator (IPC), and non‐template control (negative control). Relative miRNA expression levels were calculated by the method [35].
Table 1.
The selected miRNAs for miRNA custom PCR panel.
| miRNA ID | miRCURY assay cat. no. | Sequence |
|---|---|---|
| hsa‐miR‐361‐5p | YP00206054 | UUAUCAGAAUCUCCAGGGGUAC |
| cfa‐miR‐101 | YP00205955 | UACAGUACUGUGAUAACUGA |
| hsa‐miR‐29c‐3p | YP00204729 | UAGCACCAUUUGAAAUCGGUUA |
| hsa‐miR‐127‐3p | YP00204048 | UCGGAUCCGUCUGAGCUUGGCU |
| hsa‐miR‐128‐3p | YP00205995 | UCACAGUGAACCGGUCUCUUU |
| hsa‐miR‐129‐5p | YP00204534 | CUUUUUGCGGUCUGGGCUUGC |
| hsa‐miR‐130b‐3p | YP00204317 | CAGUGCAAUGAUGAAAGGGCAU |
| hsa‐miR‐152‐3p | YP00204294 | UCAGUGCAUGACAGAACUUGG |
| cfa‐miR‐1840 | YP02102662 | UCACGUGACGGGCCUCGGCG |
| hsa‐miR‐187‐3p | YP00204018 | UCGUGUCUUGUGUUGCAGCCGG |
| hsa‐miR‐192‐5p | YP00204099 | CUGACCUAUGAAUUGACAGCC |
| cfa‐miR‐196b | YP02113947 | UAGGUAGUUUCCUGUUGUUGGGA |
| cfa‐miR‐23a | YP00205956 | AUCACAUUGCCAGGGAUUU |
| cfa‐miR‐889 | YP02121125 | UUAAUAUCGGACAACCAUUGUU |
| cfa‐miR‐411 | YP02114619 | AUAGUAGACCGUAUAGCGUACG |
| bta‐miR‐99a‐5p | YP00205945 | AACCCGUAGAUCCGAUCUUGU |
| hsa‐miR‐381‐3p | YP00205887 | UAUACAAGGGCAAGCUCUCUGU |
| hsa‐miR‐590‐3p | YP00205448 | UAAUUUUAUGUAUAAGCUAGU |
| cfa‐miR‐125a | YP02113289 | UCCCUGAGACCCUUUAACCUGU |
| bta‐miR‐195 | YP00205969 | UAGCAGCACAGAAAUAUUGGCA |
| hsa‐miR‐9‐5p | YP00204513 | UCUUUGGUUAUCUAGCUGUAUGA |
| hsa‐miR‐16‐5p | YP00205702 | UAGCAGCACGUAAAUAUUGGCG |
Quantitative RT‐PCR data analysis
Data analyses were performed in r, version 4.2.2 [36]. Relationships between survival times and demographics (Table 2) and clinical variables (age, sex, and cancer stage and grade at diagnosis) were analyzed by multiple regression considering 25 potential models that included all possible additive and interaction effects. Intact and neutered subjects were grouped by sex due to the small number of unneutered subjects in the sample. Differences in mean miRNA expression among the three phenotypic groups were analyzed by Welch's one‐way ANOVA for unequal variances with Bonferroni‐Holm‐corrected pairwise comparisons. In addition to the pairwise comparisons, we examined the linear contrast between the low‐risk group (with long survival) and the pooled intermediate‐ and high‐risk groups (short survival). Effect sizes for all contrasts were quantified using Cohen's d, standardized to the average variance of the groups being compared.
Table 2.
Groups number and survival period in days.
| Group | n | Progression‐free | |||
|---|---|---|---|---|---|
| Mean | SD | Min. | Max. | ||
| Short | 26 | 64.3 | 23.0 | 19 | 103 |
| BOR = PR | 12 | 48.5 | 16.3 | 19 | 75 |
| BOR = CR | 14 | 76.2 | 20.3 | 35 | 103 |
| Long | 16 | 460.8 | 107.2 | 343 | 684 |
| Very long | 2 | 1043.0 | 35.4 | 1018 | 1068 |
Multiple regression and Cox proportional hazards regression were used to investigate relationships between survival time, miRNA expression levels, and demographic and clinical variables. Due to the large number of potential explanatory variables, we used lasso feature selection in the glmnet package [37, 38] to identify the collection of predictor variables most useful for survival prediction. We used 10‐fold cross‐validation (CV) and selected the penalty weight, lambda, that yielded the simplest model with a predictive error within 1 standard error of the minimum (i.e., “lambda.1se”), as recommended by Friedman et al. [37].
Lasso selection was also used to construct a multinomial logistic regression (MLR) model to predict patient phenotypes. Performance was assessed by calculating the overall classification error rate in a 10‐fold CV. In addition, because cross‐validation results depend on the random assignment of subjects to folds, we repeated the cross‐validation analysis 200 times to assess the stability of its predictions and variable selections. miRNA expression levels were log2 transformed before their incorporation into the models.
We developed a multinomial logistic regression model to classify individual subjects as low, medium, or high‐risk based on miRNA expression levels. A subset of predictive miRNAs was identified by LASSO using 200 replicated 10‐fold cross‐validation (CV) runs. Within each run, the subjects were randomly divided into 10 groups. Then, the model was fitted to the pooled data from 9 groups, and the resulting fit was used to classify the subjects in the 10th group. This was repeated 10 times to predict each group, and then the results were evaluated at lambda.1se to obtain a final model. For details on LASSO model fitting procedures, including the lambda.1se criterion [37]. Cross‐validation results are only partly determined by model performance per se. They also depend on the training and test data sets used in the validation process. By replicating the procedure described above, we could evaluate the model's sensitivity to such sampling effects and its predictive performance for individual subjects. Predictions from a stable model (i.e., one that is not overly dependent on the specific dataset to which it is fit) should be consistent across CV runs, regardless of the predictions' correctness. Ideally, the model should classify each subject the same way every time, irrespective of the data on which it was trained. Conversely, a sensitive model may produce very different predictions if trained on a different dataset.
Results
RNA extraction and small RNA sequencing
The mean concentration of the total RNA extracted was 1390.47 ± 918.83 ng·μL−1 as measured by UV spectrophotometry. The 260/280 nm absorbance ratio was 2.1 ± 0.01 (Table S3). Total: 39.3 million raw reads were obtained from the small RNA libraries generated from fresh frozen lymph nodes. After quality control and filtering, (90.6%) of clean reads were retained for further analysis. A comprehensive overview of the cleaning procedure provides an analysis of the mapping statistics about the reads (Table S4).
Differential expression analysis of the small RNA sequencing data
Differential expression analysis revealed 30 differentially expressed small RNAs between DLBCL dog patients with different PFS, S‐PFS, n = 3 and L‐PFS, n = 3, and 18 miRNAs are upregulated. In contrast, 12 miRNAs are downregulated (fold change > 3 and adjusted P‐value < 0.01). The results are depicted in a heat map (Fig. 2A) and a volcano blot (Fig. 2B). The heatmap shows the log of the normalized expression of the selected miRNAs clustered according to correlation across samples using Ward's clustering method. Before clustering, the log‐expression values for each miRNA are converted to standard normal deviation (Z scores). The normalization method used is Z‐score normalization. Transforming the log‐expression values for each miRNA to have a mean of zero and a standard deviation of one ensures that the miRNA expression values are standardized and comparable across samples, allowing for accurate clustering and analysis of differential expression miRNA. The samples with short PFS are DLBCL1,3,5, and those with DLBCL with long PFS survival are DLBCL 2,4,6. SRNA‐seq showed over‐expressions of miR‐187‐3p, miR‐192‐5p, miR‐128, and miR‐1840 miRNAs (18_28548, 21_31463, 24_34253, 9_16069, and 9_16070) in the short PFS group (DL 1,3,5), while miR‐23a, miR‐494, miR‐381, miR‐411, miR‐152, and miR‐196b are overexpressed in the long PFS group (DL 2,4,6).
Fig. 2.
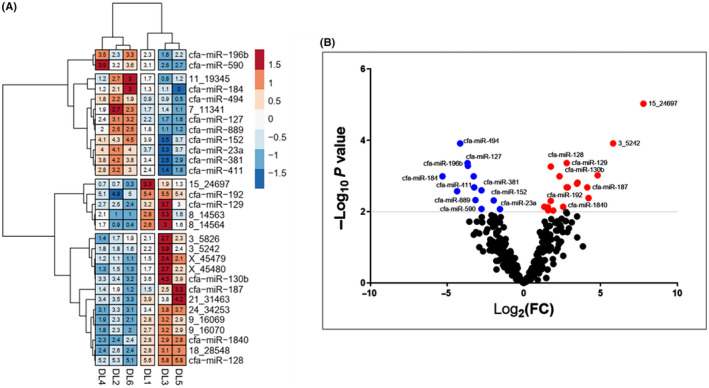
(A) Small RNA sequencing (sRNA‐Seq) heatmap of the differentially expressed miRNAs (DEMs) associated with progression‐free survival (PFS) in dogs with diffuse large B‐cell lymphoma (DLBCL). Red represents high expression; blue represents low expression. The color intensity from red to blue represents the log‐expression values for each miRNA when converted to standard normal deviation (Z scores). The samples with short PFS are DLBCL1,3,5 (n = 3) and those with DLBCL with long PFS survival are DLBCL 2,4,6 (n = 3). The miRNAs with greater than three‐fold expression differences (i.e., log FC > 1.58) and adjusted P‐values < 0.01 were selected. (B) The small RNA‐seq volcano plot of the differentially expressed miRNAs (DEMs). Red represents high expression; blue represents low expression. The miRNAs with greater than three‐fold expression differences (log2FC > 1.58) and (−log10 adj P‐values < 0.01 = 2).
Quantitative PCR analysis
The UniSp6 cDNA synthesis control was amplified in all samples at 18.4 ± 1.1 cycle quantification (C q) (Table S5). UniSp3, a positive qPCR control and inter‐plate calibrator was amplified in all samples, and C qs were calibrated to 21.64 C q across the board to reduce variability from one plate to another. Welch's 2‐sample t‐test was used to compare the relative expression of each miRNA in each sample to the relative expression level of miR‐361‐5p (Table 3) based on our previous research [39]. Normalizing miRNA data is complicated and uses diversifiable methods such as exogenous spike control [40], geometrical mean of all miRNAs expressed in one sample [41], and endogenous single or multiple miRNAs [42]. Pradervand et al. [43] underscored the importance of efficient miRNA data normalization for miRNA expression quantitation error reduction and controlling experimental technical variations.
Table 3.
Differences in mean miRNA expression among three phenotypic groups based on progression‐free survival (PFS) and clinical best overall response (BOR) classifications. Dogs were in the high‐risk (partial remission group n = 12), complete remission with low survival (intermediate‐risk group n = 14), and complete remission with high survival (low‐risk group n = 18) in 44 canine lymphoma cases.
| miRNA | df | F a | Raw P | FDR P | Low risk vs other b | Low vs high‐risk | Low vs intermediate | Intermediate vs low risk | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| d c | P d | d | P | d | P | d | P | |||||
| hsa‐miR‐192‐5p | 2, 22.9 | 7.809 | 0.0026 | 0.0299 | −1.184 | 0.001 | −1.03 | 0.032 | −1.301 | 0.005 | 0.15 | 0.708 |
| hsa‐miR‐16‐5p | 2, 25.4 | 7.521 | 0.0027 | 0.0299 | −1.137 | 0.002 | −1.25 | 0.005 | −1.107 | 0.012 | 0.325 | 0.407 |
| hsa‐miR‐29c‐3p | 2, 19.1 | 5.274 | 0.015 | 0.0886 | 0.4 | 0.336 | −0.22 | 0.595 | 1.179 | 0.021 | −0.953 | 0.08 |
| cfa‐miR‐196b | 2, 23.5 | 4.945 | 0.0161 | 0.0886 | −0.531 | 0.268 | −0.14 | 0.724 | −1.05 | 0.021 | 0.682 | 0.268 |
| cfa‐miR‐101 | 2, 22.1 | 4.401 | 0.0246 | 0.0902 | −0.774 | 0.036 | −0.56 | 0.358 | −1.047 | 0.03 | 0.16 | 0.695 |
| bta‐miR‐99a‐5p | 2, 23.5 | 4.224 | 0.0271 | 0.0902 | −0.631 | 0.079 | −1.13 | 0.038 | −0.177 | 0.624 | −0.988 | 0.07 |
| hsa‐miR‐590‐3p | 2, 23.7 | 4.138 | 0.0287 | 0.0902 | −0.651 | 0.12 | −0.36 | 0.644 | −1.009 | 0.028 | 0.412 | 0.644 |
| hsa‐miR‐152‐3p | 2, 24.6 | 3.624 | 0.0417 | 0.1045 | −0.804 | 0.046 | −0.96 | 0.058 | −0.661 | 0.154 | −0.292 | 0.466 |
| bta‐miR‐195 | 2, 26 | 3.568 | 0.0427 | 0.1045 | −0.839 | 0.041 | −0.87 | 0.079 | −0.799 | 0.079 | −0.05 | 0.9 |
| cfa‐miR‐125a | 2, 24.5 | 3.428 | 0.0487 | 0.1072 | −0.808 | 0.046 | −0.9 | 0.085 | −0.712 | 0.106 | −0.335 | 0.412 |
| cfa‐miR‐889 | 2, 25.2 | 2.993 | 0.0682 | 0.1364 | −0.755 | 0.077 | −0.68 | 0.168 | −0.816 | 0.084 | 0.059 | 0.884 |
| hsa‐miR‐187‐3p | 2, 26.1 | 2.839 | 0.0766 | 0.1405 | −0.657 | 0.132 | −0.88 | 0.092 | −0.459 | 0.384 | −0.533 | 0.384 |
| hsa‐miR‐130b‐3p | 2, 25.6 | 2.672 | 0.0883 | 0.1495 | −0.398 | 0.449 | −0.7 | 0.189 | −0.135 | 0.696 | −0.84 | 0.189 |
| cfa‐miR‐23a | 2, 23 | 2.383 | 0.1147 | 0.1802 | −0.318 | 0.572 | −0.75 | 0.223 | 0.193 | 0.592 | −0.888 | 0.169 |
| hsa‐miR‐127‐3p | 2, 24.3 | 2.129 | 0.1406 | 0.2061 | −0.639 | 0.166 | −0.64 | 0.263 | −0.631 | 0.263 | −0.087 | 0.828 |
| hsa‐miR‐129‐5p | 2, 26.8 | 1.838 | 0.1787 | 0.2457 | −0.6 | 0.259 | −0.5 | 0.33 | −0.671 | 0.259 | 0.305 | 0.439 |
| hsa‐miR‐9‐5p | 2, 26.8 | 1.56 | 0.2286 | 0.2903 | −0.191 | 1 | −0.52 | 0.555 | 0.05 | 1 | −0.595 | 0.555 |
| hsa‐miR‐361‐5p | 2, 22 | 1.536 | 0.2375 | 0.2903 | −0.245 | 0.815 | 0.057 | 0.888 | −0.615 | 0.407 | 0.473 | 0.758 |
| hsa‐miR‐128‐3p | 2, 23.6 | 1.389 | 0.2691 | 0.3116 | 0.17 | 1 | −0.18 | 1 | 0.507 | 0.592 | −0.595 | 0.592 |
| hsa‐miR‐381‐3p | 2, 24.7 | 1.27 | 0.2984 | 0.3282 | −0.408 | 0.585 | −0.61 | 0.489 | −0.211 | 0.585 | −0.491 | 0.585 |
| cfa‐miR‐411 | 2, 24.5 | 1.086 | 0.3534 | 0.3702 | −0.451 | 0.589 | −0.41 | 0.589 | −0.481 | 0.589 | −0.01 | 0.98 |
| cfa‐miR‐1840 | 2, 24.8 | 0.953 | 0.3991 | 0.3991 | −0.413 | 0.737 | −0.34 | 0.751 | −0.474 | 0.737 | 0.078 | 0.847 |
Welch's one‐way ANOVA with unequal variances.
“Other” pools intermediate risk and high risk.
Cohen's d using the average variance of the contrasted groups.
Bonferroni‐Holm adjusted P‐value for contrast in the group means.
Values in bold are statistically significant.
Survival patterns
The phenotype groups showed distinct survival patterns (Fig. 2 and Table 2). Dogs in the high and intermediate‐risk groups had rapidly progressing diseases with short PFS (≤ 103 days). Within these two groups, dogs in an intermediate risk have longer average survival rates than those in partial remission (Welch's t‐test, PFS: t = 3.855, df = 23.9, P = 0.0008). However, the low‐risk group had longer times for PFS (≥ 343 days). Two dogs in the low‐risk group belonged to a potential fourth phenotype, with PFS > 2 years. However, because this group had only two members, it was retained as part of the low‐risk group for analyses. Thus, the dogs at low risk not only had better PFS but also survived for longer periods after their cancer relapsed.
Demographic and clinical relationships with survival
We found few relationships between survival and clinical or demographic variables. Of the 25 models that we considered, only 2 had significant global F‐tests (Age: F = 4.305, df = 1 and 42, P = 0.0442; Age + Grade: F = 3.237, df = 3 and 40, P = 0.0321). The best‐performing model included additive effects for age and grade at diagnosis. Unsurprisingly, older dogs and those with intermediate‐ or high‐grade cancers did not fare as well as younger animals or those with low‐grade cancers. However, the model only accounted for a small portion of the variation in PFS (adj. R 2 = 0.135), and the decision to include grade in addition to age yielded only a marginal improvement over the Age‐only model.
Differences in miRNA expression among phenotypes
After FDR correction, only two markers, miR‐192‐5p and miR‐16‐5p, showed significant differences (P < 0.05) in mean expression in one‐way ANOVA tests. On average, both miRNAs were expressed at lower levels in the low‐risk phenotype than in the intermediate‐risk or high‐risk group. However, the values for Cohen's d (Table 3) group means were generally separated by ≤ 1.3 SD. Thus, although the group averages appear to differ, individual subjects show considerable overlap among groups for all miRNAs. It is worth noting that several additional markers had adjusted ANOVA P‐values < 0.1 and had Bonferroni‐Holm corrected P < 0.05 for at least one contrast. These included miR‐29c‐3p, miR‐196b, miR‐101, miR‐99a‐5p, miR590‐3p, miR‐195, miR‐152‐3p, and miR‐125a. Given the small size of the current sample, these markers may warrant additional future investigation. They should also be considered in potential multivariate classifiers. The miRNAs differential expression data analysis results are summarized in Table 3 and illustrated in Fig. 3.
Fig. 3.
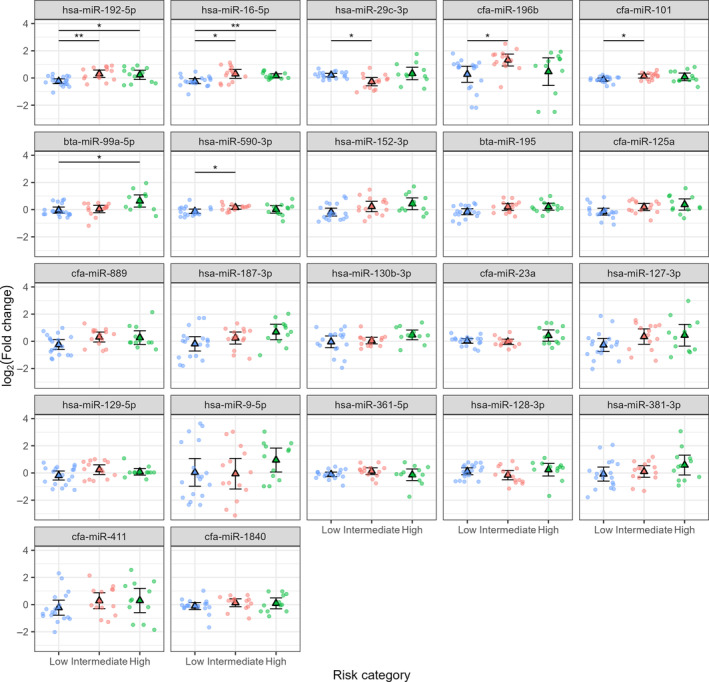
Expression of 22 miRNAs in three phenotypes of canine lymphoma using Welch's one‐way ANOVA for unequal variances with Bonferroni‐Holm‐corrected pairwise comparisons. Dogs in the Low‐risk group (n = 18) were clinically classified as being in complete remission and had relatively long progression‐free. The intermediate‐risk group (n = 14) also appeared to be in complete remission but had shorter survival rates, more similar to the high‐risk (partial remission) group (n = 12). Triangles represent group means ± 95% confidence intervals (CI). The high‐risk, partial remission group; intermediate risk, complete remission with short survival group; Low risk, complete remission with long survival group. *Significant at P < 0.05; **Significant at P < 0.01.
Prediction of survival time
We attempted to predict survival times using multiple regression and Cox models using lasso feature selection. Using linear regression, the lasso selected only one predictor, miR‐192‐5p, which yielded an R 2 = 0.253 for the relaxed model. Consistent with Fig. 3, survival times declined with increasing expression of miR‐192‐5p (t = −3.769, df = 42, P = 0.0005). However, analysis of the model residuals showed that it consistently predicted survival times within a narrow range from 124 to 165 days. In contrast to multiple regression, the Cox model selected three predictors: miR‐192‐5p, miR‐16‐5p, and miR‐152‐3p. All three miRNAs were positively related to the hazard ratio (consistent with Fig. 3).
Phenotype classification
Given the high levels of overlap among group‐wise miRNA expressions (Fig. 3), none of the miRNAs studied here can serve as a useful prognostic measure to distinguish low‐risk from intermediate‐risk or high‐risk cases on its own. Therefore, we used multinomial logistic regression with lasso feature selection to explore potential multivariate prognostic indices.
Multinomial logistic regression (MLR) generalizes the methodology of binary logistic regression to categorical responses with > 2 potential classes of outcomes. Unlike more familiar forms of the generalized linear model (GLM), which predict outcomes based on a single linear predictor, MLR generates a linear predictor for each possible outcome. In a classification setting, new observations are typically assigned to the class whose predictor has the largest value. In our analysis, miRNAs miR‐192‐5p and miR‐16‐5p consistently loaded onto the predictor for low risk (Figs S1) for cross‐validation results. Based on these variables, the model correctly classified all 18 members of the low‐risk group and correctly classified half of the members of the intermediate‐risk and high‐risk groups. In most cases, misclassified individuals were placed at low risk (Table 4). Individual classifications in the low‐risk and intermediate‐risk groups were generally consistent across CV runs (i.e., individuals were almost always classified similarly; Fig. S1). In contrast, classifications for six high‐risk group members varied among CV runs.
Table 4.
Confusion matrix for prediction of canine lymphoma phenotype using multinomial logistic regression and lasso feature selection, averaging over 200 cross‐validation sets.
| Truth | Prediction | ||
|---|---|---|---|
| Low risk | Intermediate risk | High risk | |
| Low risk | 18 | 0 | 0 |
| Intermediate risk | 7 | 7 | 0 |
| High risk | 5 | 1 | 6 |
Discussion
Despite receiving standardized CHOP chemotherapy treatment, dogs with DLBCL demonstrate significant heterogeneity in terms of survival outcomes [8, 9]. While the multidrug regime of chemotherapy CHOP has demonstrated promising effectiveness in certain dogs with DLBCL, a significant proportion of cases fail to attain complete remission or relapse following a short period of remission, contributing to poor outcomes and a significant mortality rate due to this disease. Clinical studies suggest that a subgroup of dogs with DLBCL derives only a slight benefit from CHOP chemotherapy. To date, this subpopulation is not easily identified with current prognostic variables. In human studies, the International Prognostic Index (IPI) scoring system utilizing different clinical factors, including age, disease stage, number of extranodal sites, and performance status, is used to sort patients into different risk groups. IPI is still in use after revision, although it fails to represent the cellular morphology heterogeneities precisely and tends to incline to overemphasize the high‐risk patient's percentage [44]. Unlike human studies, limited research studies have been conducted on prognostic factors in dogs with diffuse large B‐cell lymphoma.
MicroRNAs (miRNAs) are diminutive non‐coding ribonucleic acids (RNAs) that control protein synthesis at the post‐transcriptional stage. This short RNA class exhibits high conservation and binding affinity to the 3′ untranslated region of target mRNA through complementary base pairing [45], resulting in the target mRNA degradation or inhibition of protein translation [46]. Many studies have implied the important regulatory role of miRNAs interfacing with pseudogenes and various types of RNAs, including long non‐coding RNA, circular RNAs, and messenger RNAs within human cells [47]. Each microRNA (miRNA) can govern up to hundreds of genes, orchestrating various biological functions, including cell growth, differentiation, proliferation, and apoptosis in addition to pathological conditions such as tumorigenesis, drug resistance, malignant transformation, anchorage‐independent growth, new blood vessel formation, cell growth, differentiation, proliferation, and apoptosis [48]. These effects are accomplished by altering the abundance of downstream target proteins. Therefore, because of its central role in governing cancer phenotypes, miRNA exhibits significant potential as a prognostic indicator and an innovative target for cancer therapeutics [49, 50].
This study investigated miRNAs' signatures as a supplementary prognostic tool. We could differentiate between L‐PFS and S‐PFS groups of the DLBCL cases based on the survival rates using the expression level of two miRNAs: miR‐192‐5p and miR‐16‐5p. The two markers, miR‐192‐5p, and miR‐16‐5p, showed significant expression differences between low‐risk, intermediate, or high‐risk phenotypes. They also separated the short survival groups based on survival rate and BOR as they were expressed at lower levels in intermediate‐risk or high‐risk than low‐risk phenotype. Growing evidence supports the differences in miRNA expression profile observed between malignant cells and their normal counterparts; likewise, these differences correlate with prognostic factors in cancer.
The exact implications of miR‐192 in tumor development are still ambiguous in human studies. For instance, frequent studies investigated the role of miR‐192 in cancer progression and suggested its role as a tumor suppressor miRNA in human bladder cancer [51], pediatric acute lymphoblastic leukemias [52], and human hepatocellular carcinoma, [53] among others. A previous study conducted in our lab using small RNA sequencing (sRNA‐seq) and RT‐qPCR data analysis showed the upregulation of miR‐192‐5p in DLBCL cases compared to healthy groups of dogs [39]. This finding is supported by the significant overexpression of miR‐192‐5p in the low‐grade B‐cell lymphoma, Waldenstrom Macroglobulinemia [54, 55].
MiR‐16‐1 is one of the controversial miRNAs. It is a member of the miR‐15/107 superfamily, which initiates apoptosis [56], is the precursor form of the miRNA miR‐16, which comprises miR‐16‐5p and miR‐16‐3p (depending on the two arms of the precursor hairpin structure, the suffix is either “‐5p” or “‐3p”). Subsequently, both have different functions and target genes regulating different cellular processes owing to discrete sequences, even though they originate from the same precursor. In leukemia, miR‐15a/miR‐16‐1 was the first tested for tumor‐induction [57]. An experiment conducted using cell lines derived from patients with B‐cell chronic lymphocytic leukemia (B‐CLL) revealed that miR‐15a and miR‐16‐1 are deregulated and directly target the p53 protein in a positive feedback loop, wherein these miRNAs are also regulated by direct binding of p53 [57]. Chen and Zhang et al. suggested its association with the pathogenesis of lymphoma subtypes, such as mantle cell lymphoma [58, 59]. To the authors' knowledge, no study compares miR‐16‐5p in DLBCL groups with different survival and outcomes. Our study found miR‐16‐5p upregulated in the intermediate risk and high risk compared to low risk. The miR‐17‐5p and miR‐16‐5p were found to have the strongest correlations with specific mRNA targets like B‐cell lymphoma 2 (BCL2) and mothers against decapentaplegic 3 (SMAD3) genes. It is also involved in suppression of cytokine signaling 1 (SOCS1) implicated in cancer metastasis, cell cycle, adherence junctions, and interactions between the extracellular matrix and receptors leading to brain metastases in patients with advanced breast cancer [60]. Furthermore, miR‐16‐5p was detected to be overexpressed in tumor samples from breast cancer compared to healthy subjects after four validation phases following a screening phase [61]. Likewise, miR‐16‐5p showed a drop in its expression in the plasma of patients' post‐surgical removal of colorectal tumor, suggesting the source of the miRNA and the possibility of using it as an early detecting non‐invasive biomarker [62]. These findings align with miR‐16‐5p expression in tumor tissue of colorectal cancer where it is over‐expressed compared to its adjacent tissue [62]. Additionally, Lung cancer [63], esophageal squamous cell carcinoma (ESCC) [64], Kaposi's sarcoma [65], upper tract urothelial carcinoma (UTUC) [66], high‐grade serous ovarian carcinoma tissues [67], and ovarian cancer [68] are all showed overexpression in the cancerous samples compared to the healthy peers. Although there is inter‐individual variation in the expression levels of miRNAs, there is evidence that average miRNA expression differs among the phenotype groups. On the other hand, miRNA‐16 used to be known as endogenous control [69, 70] and key player in diverse malignancies development [71, 72], including neuroblastoma [73], osteosarcoma [74], hepatocellular carcinoma [75], breast cancer [76].
The MLR classifier could only correctly classify half of the intermediate‐risk and high‐risk individuals in the dataset. However, given that PR individuals are readily identified by current clinical methods, and that those methods cannot identify intermediate‐risk individuals, our results still represent a significant improvement. Assuming that the current findings can be validated, miRNA assays might complement existing clinical methods and improve the prognostic assessment of dogs with DLBCL. However, they cannot replace existing methods based on the current results. Thus, these proposed prognostic biomarkers require further investigation in prospective studies and confirmation in a larger dataset.
Despite the relatively small univariate effect sizes we have reported for miRNAs in this study, co‐expression patterns may still provide useful prognostic information. We are currently investigating the possibility that high‐risk and intermediate‐risk phenotypes are characterized not by specific changes in miRNA expression but rather by the presence of any dysregulation that carries them outside a certain range of co‐expression patterns. In other words, high‐risk and intermediate‐risk patients may be identifiable as outliers relative to the multivariate distribution of miRNA expression among low‐risk patients. Similarly, low‐risk cases may represent outliers relative to healthy individuals, but we cannot investigate this possibility with the current data set.
Having a developed prognostic index that integrates multiple biomarkers parallel to the widely utilized IPI in human medicine would represent a valuable progression in this research area. A dependable prognostic index will be significant in assisting pet owners in making well‐informed decisions regarding treatment options for their canine companions. It will also play a crucial role in interpreting and comparing clinical trial outcomes for better innovative therapeutic approaches. Effective prognostic indicators can act as a solid base for patients' stratified treatment; in that way, excessive treatment and unnecessary expenses will be avoided. The identified miRNAs in this study, together with other differentially expressed miRNAs in canine lymphoma, will be included in future studies to elucidate their roles miRNAs in DLBCL pathogenesis.
Although our study provided significant findings, the relatively small sample size of 44 dogs was a limitation for generalizing the findings. A larger cohort with a diverse population, including age, sex, breed, and response to treatment, coupled with progression‐free survival, would enhance the robustness and applicability of the findings. Although many researchers have demonstrated comparable results between FF and FFPE samples in various molecular analyses, others have depicted the potential variability between FF and FFPE samples. In this study, the lack of a larger sample size representing the intermediate‐risk group limited obtaining more data representing this group. Furthermore, investigating the newly discovered miRNA and its role in DLBCL pathogenesis is required in future studies.
Conclusions
Canine DLBCL is the most predominant form of non‐Hodgkin lymphoma (NHL) observed in dogs and is analogous to the same cancer in humans. Identifying those dogs with DLBCL that will respond most and least favorably to current therapies remains challenging. MicroRNAs can potentially enhance the prognostic assessment of dogs with this disease. This study aimed to correlate miRNA expression and progression‐free survival (PFS) in dogs with diffuse large B‐cell lymphoma (DLBCL). A discrete cluster of miRNAs exhibits a significant negative correlation with progression‐free survival (PFS) in dogs diagnosed with diffuse large B‐cell lymphoma (DLBCL). Dogs exhibiting a reduced progression‐free survival (PFS) demonstrated a statistically significant increase in the levels of miR‐192‐5p and miR‐16‐5p. The expression levels of miR‐192‐5p and miR‐16‐5p were significantly decreased in the complete remission group with a high survival rate. MicroRNA profiling can potentially predict the prognosis of dogs diagnosed with DLBCL. The identification of miRNAs that exert an impact on the progression of canine DLBCL may facilitate the development of personalized therapeutic interventions.
Conflict of interest
The authors declare no conflict of interest.
Peer review
The peer review history for this article is available at https://www.webofscience.com/api/gateway/wos/peer‐review/10.1002/2211‐5463.13887.
Author contributions
NOE, MOC, and APS contributed to conceptualization; NOE and MG contributed to data curation; NOE, NIL, and MG contributed to formal analysis; NOE, MOC, MG, and APS contributed to investigation; NOE, MG, and APS contributed to methodology; APS contributed to project administration; APS and NOE contributed to resources; NOE, MG, and EES contributed to software; MOC and APS contributed to supervision; NOE and EES contributed to validation; NOE, EES, NIL, and APS contributed to writing original draft; NOE, EES, MOC, MG, NIL, and APS contributed to writing, review and editing. All authors have read and agreed to the published version of the manuscript.
Supporting information
Fig. S1. Stability of individual‐level risk classifications for 44 dogs diagnosed with lymphoma based on miRNA expression.
Table S1. The clinical information of diffuse large B‐cell lymphoma cases used for small RNA sequencing.
Table S2. The clinical information of diffuse large B‐cell lymphoma cases used for qPCR.
Table S3. The RNA extraction information of samples used for small RNA sequencing.
Table S4. Summary of miRNA data cleaning and filtering steps.
Table S5. The calculated ΔΔCT for each miRNA in each sample.
Acknowledgements
This work was supported by the Purdue Research Foundation (PRF) of Purdue University and the Doberman Pinscher Health Foundation.
Data accessibility
The datasets used and analyzed in the current study are available from the corresponding authors upon reasonable request.
References
- 1. Hunt KE and Reichard KK (2008) Diffuse large B‐cell lymphoma. Arch Pathol Lab Med 132, 118–124. [DOI] [PubMed] [Google Scholar]
- 2. Chan A and Dogan A (2019) Prognostic and predictive biomarkers in diffuse large B‐cell lymphoma. Surg Pathol Clin 12, 699–707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Vaidya R and Witzig TE (2014) Prognostic factors for diffuse large B‐cell lymphoma in the R(X)CHOP era. Ann Oncol 25, 2124–2133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Aresu L (2016) Canine lymphoma, more than a morphological diagnosis: what we have learned about diffuse large B‐cell lymphoma. Front Vet Sci 3, 77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Richards KL, Motsinger‐Reif AA, Chen HW, Fedoriw Y, Fan C, Nielsen DM, Small GW, Thomas R, Smith C, Dave SS et al. (2013) Gene profiling of canine B‐cell lymphoma reveals germinal center and postgerminal center subtypes with different survival times, modeling human DLBCL. Cancer Res 73, 5029–5039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Seelig DM, Avery AC, Ehrhart EJ and Linden MA (2016) The comparative diagnostic features of canine and human lymphoma. Vet Sci 3, 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Childress MO, Christian JA, Ramos‐Vara JA, Rosen NK and Ruple A (2022) Greater baseline serum C‐reactive protein concentrations are associated with reduced survival in dogs receiving cyclophosphamide, doxorubicin, vincristine, and prednisone chemotherapy for primary nodal diffuse large B‐cell lymphoma. Vet J 289, 105911. [DOI] [PubMed] [Google Scholar]
- 8. Childress MO, Ramos‐Vara JA and Ruple A (2018) Retrospective analysis of factors affecting clinical outcome following CHOP‐based chemotherapy in dogs with primary nodal diffuse large B‐cell lymphoma. Vet Comp Oncol 16, E159–E168. [DOI] [PubMed] [Google Scholar]
- 9. Frantz AM, Sarver AL, Ito D, Phang TL, Karimpour‐Fard A, Scott MC, Valli VE, Lindblad‐Toh K, Burgess KE, Husbands BD et al. (2013) Molecular profiling reveals prognostically significant subtypes of canine lymphoma. Vet Pathol 50, 693–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Marconato L, Martini V, Aresu L, Sampaolo M, Valentini F, Rinaldi V and Comazzi S (2013) Assessment of bone marrow infiltration diagnosed by flow cytometry in canine large B cell lymphoma: prognostic significance and proposal of a cut‐off value. Vet J 197, 776–781. [DOI] [PubMed] [Google Scholar]
- 11. Kosaka N, Yoshioka Y, Hagiwara K, Tominaga N, Katsuda T and Ochiya T (2013) Trash or treasure: extracellular microRNAs and cell‐to‐cell communication. Front Genet 4, 173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Otmani K and Lewalle P (2021) Tumor suppressor miRNA in cancer cells and the tumor microenvironment: mechanism of deregulation and clinical implications. Front Oncol 11, 708765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Subramanian S and Steer CJ (2019) Special issue: microRNA regulation in health and disease. Genes (Basel) 10, 457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Trang P, Wiggins JF, Daige CL, Cho C, Omotola M, Brown D, Weidhaas JB, Bader AG and Slack FJ (2011) Systemic delivery of tumor suppressor microRNA mimics using a neutral lipid emulsion inhibits lung tumors in mice. Mol Ther 19, 1116–1122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hedegaard J, Thorsen K, Lund MK, Hein AM, Hamilton‐Dutoit SJ, Vang S, Nordentoft I, Birkenkamp‐Demtröder K, Kruhøffer M, Hager H et al. (2014) Next‐generation sequencing of RNA and DNA isolated from paired fresh‐frozen and formalin‐fixed paraffin‐embedded samples of human cancer and normal tissue. PLoS One 9, e98187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Iddawela M, Rueda OM, Klarqvist M, Graf S, Earl HM and Caldas C (2016) Reliable gene expression profiling of formalin‐fixed paraffin‐embedded breast cancer tissue (FFPE) using cDNA‐mediated annealing, extension, selection, and ligation whole‐genome (DASL WG) assay. BMC Med Genomics 9, 54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Zhang P, Lehmann BD, Shyr Y and Guo Y (2017) The utilization of formalin fixed‐paraffin‐embedded specimens in high throughput genomic studies. Int J Genomics 2017, 1926304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Gao XH, Li J, Gong HF, Yu GY, Liu P, Hao LQ, Liu LJ, Bai CG and Zhang W (2020) Comparison of fresh frozen tissue with formalin‐fixed paraffin‐embedded tissue for mutation analysis using a multi‐gene panel in patients with colorectal cancer. Front Oncol 10, 310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Vail DM, Michels GM, Khanna C, Selting KA and London CA (2010) Response evaluation criteria for peripheral nodal lymphoma in dogs (v1.0) – a Veterinary Cooperative Oncology Group (VCOG) consensus document. Vet Comp Oncol 8, 28–37. [DOI] [PubMed] [Google Scholar]
- 20. Owen LN, World Health Organization , Veterinary Public Health Unit and Oncology WHOCCfC (1980) TNM Classification of Tumours in Domestic Animals (Owen LN, ed.). World Health Organization, Geneva. [Google Scholar]
- 21. Garrett LD, Thamm DH, Chun R, Dudley R and Vail DM (2002) Evaluation of a 6‐month chemotherapy protocol with no maintenance therapy for dogs with lymphoma. J Vet Intern Med 16, 704–709. [DOI] [PubMed] [Google Scholar]
- 22. Friedländer MR, Mackowiak SD, Li N, Chen W and Rajewsky N (2012) miRDeep2 accurately identifies known and hundreds of novel microRNA genes in seven animal clades. Nucleic Acids Res 40, 37–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Halo JV, Pendleton AL, Shen F, Doucet AJ, Derrien T, Hitte C, Kirby LE, Myers B, Sliwerska E, Emery S et al. (2021) Long‐read assembly of a Great Dane genome highlights the contribution of GC‐rich sequence and mobile elements to canine genomes. Proc Natl Acad Sci USA 118, e2016274118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kozomara A, Birgaoanu M and Griffiths‐Jones S (2019) miRBase: from microRNA sequences to function. Nucleic Acids Res 47, D155–D162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Love MI, Huber W and Anders S (2014) Moderated estimation of fold change and dispersion for RNA‐seq data with DESeq2. Genome Biol 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Jensen SG, Lamy P, Rasmussen MH, Ostenfeld MS, Dyrskjøt L, Orntoft TF and Andersen CL (2011) Evaluation of two commercial global miRNA expression profiling platforms for detection of less abundant miRNAs. BMC Genomics 12, 435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Balatti V, Tomasello L, Rassenti LZ, Veneziano D, Nigita G, Wang HY, Thorson JA, Kipps TJ, Pekarsky Y and Croce CM (2018) miR‐125a and miR‐34a expression predicts Richter syndrome in chronic lymphocytic leukemia patients. Blood 132, 2179–2182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Chen L, Kan Y, Wang X, Ge P, Ding T, Zhai Q, Wang Y, Yu Y, Zhao Z, Yang H et al. (2020) Overexpression of microRNA‐130a predicts adverse prognosis of primary gastrointestinal diffuse large B‐cell lymphoma. Oncol Lett 20, 93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Hedström G, Thunberg U, Berglund M, Simonsson M, Amini RM and Enblad G (2013) Low expression of microRNA‐129‐5p predicts poor clinical outcome in diffuse large B cell lymphoma (DLBCL). Int J Hematol 97, 465–471. [DOI] [PubMed] [Google Scholar]
- 30. Li L, Feng T, Zhang W, Gao S, Wang R, Lv W, Zhu T, Yu H and Qian B (2020) MicroRNA biomarker hsa‐miR‐195‐5p for detecting the risk of lung cancer. Int J Genomics 2020, 7415909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Nordmo C, Glehr G, Altenbuchinger M, Spang R, Ziepert M, Horn H, Staiger AM, Ott G, Schmitz N, Held G et al. (2021) Identification of a miRNA based model to detect prognostic subgroups in patients with aggressive B‐cell lymphoma. Leuk Lymphoma 62, 1107–1115. [DOI] [PubMed] [Google Scholar]
- 32. Paydas S, Acikalin A, Ergin M, Celik H, Yavuz B and Tanriverdi K (2016) Micro‐RNA (miRNA) profile in Hodgkin lymphoma: association between clinical and pathological variables. Med Oncol 33, 34. [DOI] [PubMed] [Google Scholar]
- 33. Yazdanparast S, Huang Z, Keramat S, Izadirad M, Li YD, Bo L, Gharehbaghian A and Chen ZS (2022) The roles of exosomal microRNAs in diffuse large B‐cell lymphoma: diagnosis, prognosis, clinical application, and biomolecular mechanisms. Front Oncol 12, 904637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Yuan WX, Gui YX, Na WN, Chao J and Yang X (2016) Circulating microRNA‐125b and microRNA‐130a expression profiles predict chemoresistance to R‐CHOP in diffuse large B‐cell lymphoma patients. Oncol Lett 11, 423–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Livak KJ and Schmittgen TD (2001) Analysis of relative gene expression data using real‐time quantitative PCR and the 2(‐Delta Delta C(T)) method. Methods 25, 402–408. [DOI] [PubMed] [Google Scholar]
- 36. Alex Douglas DR, Mancini F, Couto A and Lusseau D (2023) 1.13 Citing R | An Introduction to R.
- 37. Friedman J, Hastie T and Tibshirani R (2010) Regularization paths for generalized linear models via coordinate descent. J Stat Softw 33, 1–22. [PMC free article] [PubMed] [Google Scholar]
- 38. Simon N, Friedman J, Hastie T and Tibshirani R (2011) Regularization paths for Cox's proportional hazards model via coordinate descent. J Stat Softw 39, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Elshafie NO, Gribskov M, Lichti NI, Sayedahmed EE, Childress MO and Dos Santos AP (2023) miRNome expression analysis in canine diffuse large B‐cell lymphoma. Front Oncol 13, 1238613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Vigneron N, Meryet‐Figuière M, Guttin A, Issartel JP, Lambert B, Briand M, Louis MH, Vernon M, Lebailly P, Lecluse Y et al. (2016) Towards a new standardized method for circulating miRNAs profiling in clinical studies: interest of the exogenous normalization to improve miRNA signature accuracy. Mol Oncol 10, 981–992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Mestdagh P, Van Vlierberghe P, De Weer A, Muth D, Westermann F, Speleman F and Vandesompele J (2009) A novel and universal method for microRNA RT‐qPCR data normalization. Genome Biol 10, R64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Vandesompele J, De Preter K, Pattyn F, Poppe B, Van Roy N, De Paepe A and Speleman F (2002) Accurate normalization of real‐time quantitative RT‐PCR data by geometric averaging of multiple internal control genes. Genome Biol 3, Research0034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Pradervand S, Weber J, Thomas J, Bueno M, Wirapati P, Lefort K, Dotto GP and Harshman K (2009) Impact of normalization on miRNA microarray expression profiling. RNA 15, 493–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Marcheselli R, Bari A, Tadmor T, Marcheselli L, Cox MC, Papotti R, Ferrari A, Baldini L, Gobbi P, Levy I et al. (2020) Improving the international prognostic index score using peripheral blood counts: results of a large multicenter study involving 520 patients with diffuse large B cell lymphoma. Hematol Oncol 38, 439–445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Lai EC (2002) Micro RNAs are complementary to 3' UTR sequence motifs that mediate negative post‐transcriptional regulation. Nat Genet 30, 363–364. [DOI] [PubMed] [Google Scholar]
- 46. Li Y, Ahmad A, Kong D, Bao B and Sarkar FH (2013) Targeting MicroRNAs for personalized cancer therapy. Med Princ Pract 22, 415–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Karreth FA, Reschke M, Ruocco A, Ng C, Chapuy B, Léopold V, Sjoberg M, Keane TM, Verma A, Ala U et al. (2015) The BRAF pseudogene functions as a competitive endogenous RNA and induces lymphoma in vivo. Cell 161, 319–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Moutinho C and Esteller M (2017) MicroRNAs and epigenetics. Adv Cancer Res 135, 189–220. [DOI] [PubMed] [Google Scholar]
- 49. Ni H, Wang X, Liu H, Tian F and Song G (2015) Low expression of miRNA‐224 predicts poor clinical outcome in diffuse large B‐cell lymphoma treated with R‐CHOP. Biomarkers 20, 253–257. [DOI] [PubMed] [Google Scholar]
- 50. Song G, Gu L, Li J, Tang Z, Liu H, Chen B, Sun X, He B, Pan Y, Wang S et al. (2014) Serum microRNA expression profiling predict response to R‐CHOP treatment in diffuse large B cell lymphoma patients. Ann Hematol 93, 1735–1743. [DOI] [PubMed] [Google Scholar]
- 51. Jin Y, Lu J, Wen J, Shen Y and Wen X (2015) Regulation of growth of human bladder cancer by miR‐192. Tumour Biol 36, 3791–3797. [DOI] [PubMed] [Google Scholar]
- 52. Schotte D, Akbari Moqadam F, Lange‐Turenhout EA, Chen C, van Ijcken WF, Pieters R and den Boer ML (2011) Discovery of new microRNAs by small RNAome deep sequencing in childhood acute lymphoblastic leukemia. Leukemia 25, 1389–1399. [DOI] [PubMed] [Google Scholar]
- 53. Lian J, Jing Y, Dong Q, Huan L, Chen D, Bao C, Wang Q, Zhao F, Li J, Yao M et al. (2016) miR‐192, a prognostic indicator, targets the SLC39A6/SNAIL pathway to reduce tumor metastasis in human hepatocellular carcinoma. Oncotarget 7, 2672–2683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Bouyssou JM, Liu CJ, Bustoros M, Sklavenitis‐Pistofidis R, Aljawai Y, Manier S, Yosef A, Sacco A, Kokubun K, Tsukamoto S et al. (2018) Profiling of circulating exosomal miRNAs in patients with Waldenström Macroglobulinemia. PLoS One 13, e0204589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Trino S, Lamorte D, Caivano A, De Luca L, Sgambato A and Laurenzana I (2021) Clinical relevance of extracellular vesicles in hematological neoplasms: from liquid biopsy to cell biopsy. Leukemia 35, 661–678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Parsi S, Smith PY, Goupil C, Dorval V and Hébert SS (2015) Preclinical evaluation of miR‐15/107 family members as multifactorial drug targets for Alzheimer's disease. Mol Ther Nucleic Acids 4, e256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Calin GA, Dumitru CD, Shimizu M, Bichi R, Zupo S, Noch E, Aldler H, Rattan S, Keating M, Rai K et al. (2002) Frequent deletions and down‐regulation of micro‐ RNA genes miR15 and miR16 at 13q14 in chronic lymphocytic leukemia. Proc Natl Acad Sci USA 99, 15524–15529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Chen RW, Bemis LT, Amato CM, Myint H, Tran H, Birks DK, Eckhardt SG and Robinson WA (2008) Truncation in CCND1 mRNA alters miR‐16‐1 regulation in mantle cell lymphoma. Blood 112, 822–829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Zhang X, Chen X, Lin J, Lwin T, Wright G, Moscinski LC, Dalton WS, Seto E, Wright K, Sotomayor E et al. (2012) Myc represses miR‐15a/miR‐16‐1 expression through recruitment of HDAC3 in mantle cell and other non‐Hodgkin B‐cell lymphomas. Oncogene 31, 3002–3008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Li Z, Peng Z, Gu S, Zheng J, Feng D, Qin Q and He J (2017) Global analysis of miRNA‐mRNA interaction network in breast cancer with brain metastasis. Anticancer Res 37, 4455–4468. [DOI] [PubMed] [Google Scholar]
- 61. Zou X, Xia T, Li M, Wang T, Liu P, Zhou X, Huang Z and Zhu W (2021) MicroRNA profiling in serum: potential signatures for breast cancer diagnosis. Cancer Biomark 30, 41–53. [DOI] [PubMed] [Google Scholar]
- 62. Ostenfeld MS, Jensen SG, Jeppesen DK, Christensen LL, Thorsen SB, Stenvang J, Hvam ML, Thomsen A, Mouritzen P, Rasmussen MH et al. (2016) miRNA profiling of circulating EpCAM(+) extracellular vesicles: promising biomarkers of colorectal cancer. J Extracell Vesicles 5, 31488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Reis PP, Drigo SA, Carvalho RF, Lopez Lapa RM, Felix TF, Patel D, Cheng D, Pintilie M, Liu G and Tsao MS (2020) Circulating miR‐16‐5p, miR‐92a‐3p, and miR‐451a in plasma from lung cancer patients: potential application in early detection and a regulatory role in tumorigenesis pathways. Cancers (Basel) 12, 2071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Shen Y, Ding Y, Ma Q, Zhao L, Guo X, Shao Y, Niu C, He Y, Zhang F, Zheng D et al. (2019) Identification of novel circulating miRNA biomarkers for the diagnosis of esophageal squamous cell carcinoma and squamous dysplasia. Cancer Epidemiol Biomarkers Prev 28, 1212–1220. [DOI] [PubMed] [Google Scholar]
- 65. Wu XJ, Pu XM, Zhao ZF, Zhao YN, Kang XJ, Wu WD, Zou YM, Wu CY, Qu YY, Zhang DZ et al. (2015) The expression profiles of microRNAs in Kaposi's sarcoma. Tumour Biol 36, 437–446. [DOI] [PubMed] [Google Scholar]
- 66. Tao J, Yang X, Li P, Wei J, Deng X, Cheng Y, Qin C, Ju X, Meng X, Li J et al. (2015) Identification of circulating microRNA signatures for upper tract urothelial carcinoma detection. Mol Med Rep 12, 6752–6760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Bignotti E, Calza S, Tassi RA, Zanotti L, Bandiera E, Sartori E, Odicino FE, Ravaggi A, Todeschini P and Romani C (2016) Identification of stably expressed reference small non‐coding RNAs for microRNA quantification in high‐grade serous ovarian carcinoma tissues. J Cell Mol Med 20, 2341–2348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Saral MA, Tuncer SB, Odemis DA, Erdogan OS, Erciyas SK, Saip P, Ozel S and Yazici H (2022) New biomarkers in peripheral blood of patients with ovarian cancer: high expression levels of miR‐16‐5p, miR‐17‐5p, and miR‐638. Arch Gynecol Obstet 305, 193–201. [DOI] [PubMed] [Google Scholar]
- 69. Rinnerthaler G, Hackl H, Gampenrieder SP, Hamacher F, Hufnagl C, Hauser‐Kronberger C, Zehentmayr F, Fastner G, Sedlmayer F, Mlineritsch B et al. (2016) miR‐16‐5p is a stably‐expressed housekeeping MicroRNA in breast cancer tissues from primary tumors and from metastatic sites. Int J Mol Sci 17, 156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Yang CL, Tsai FM, Chen CW, Hsiao KH, Chen JH and Kao WY (2022) Comparing miR‐16 and miR‐1228 as an optimal endogenous control for quantification of circulating microRNAs in colorectal cancer patients. Tzu Chi Med J 34, 318–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Ghafouri‐Fard S, Khoshbakht T, Hussen BM, Abdullah ST, Taheri M and Samadian M (2022) A review on the role of mir‐16‐5p in the carcinogenesis. Cancer Cell Int 22, 342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Zhang Y, Lu C and Cui H (2021) Long non‐coding RNA SNHG22 facilitates hepatocellular carcinoma tumorigenesis and angiogenesis via DNA methylation of microRNA miR‐16‐5p. Bioengineered 12, 7446–7458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Zhang X, Zhang J, Liu Q, Zhao Y, Zhang W and Yang H (2020) Circ‐CUX1 accelerates the progression of neuroblastoma via miR‐16‐5p/DMRT2 axis. Neurochem Res 45, 2840–2855. [DOI] [PubMed] [Google Scholar]
- 74. Gu Z, Li Z, Xu R, Zhu X, Hu R, Xue Y and Xu W (2020) miR‐16‐5p suppresses progression and invasion of osteosarcoma via targeting at Smad3. Front Pharmacol 11, 1324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Cheng B, Ding F, Huang CY, Xiao H, Fei FY and Li J (2019) Role of miR‐16‐5p in the proliferation and metastasis of hepatocellular carcinoma. Eur Rev Med Pharmacol Sci 23, 137–145. [DOI] [PubMed] [Google Scholar]
- 76. Wang Z, Hu S, Li X, Liu Z, Han D, Wang Y, Wei L, Zhang G and Wang X (2021) MiR‐16‐5p suppresses breast cancer proliferation by targeting ANLN. BMC Cancer 21, 1188. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Stability of individual‐level risk classifications for 44 dogs diagnosed with lymphoma based on miRNA expression.
Table S1. The clinical information of diffuse large B‐cell lymphoma cases used for small RNA sequencing.
Table S2. The clinical information of diffuse large B‐cell lymphoma cases used for qPCR.
Table S3. The RNA extraction information of samples used for small RNA sequencing.
Table S4. Summary of miRNA data cleaning and filtering steps.
Table S5. The calculated ΔΔCT for each miRNA in each sample.
Data Availability Statement
The datasets used and analyzed in the current study are available from the corresponding authors upon reasonable request.