

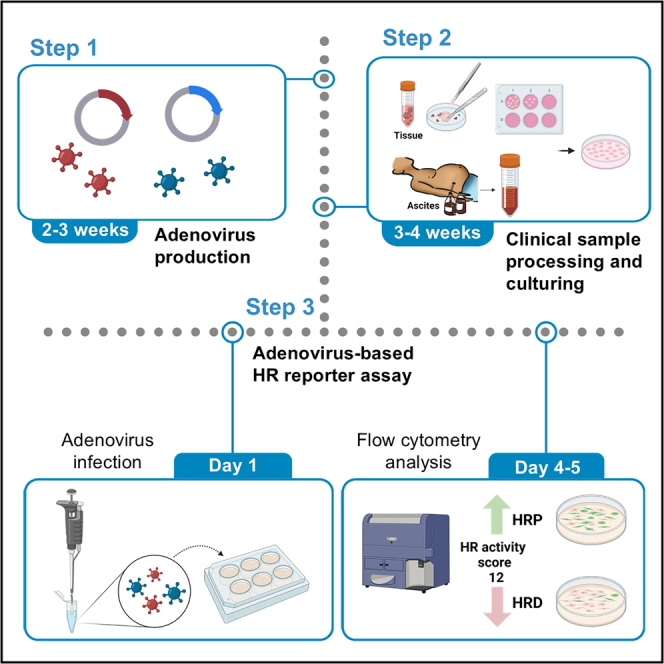
Summary
Cancer cells with homologous recombination deficiency (HRD) exhibit a distinctive vulnerability to poly(ADP-ribose) polymerase inhibitors (PARPis). To address the limitations of existing methodologies incapable of providing real-time insights into homologous recombination (HR) status, we present an adenovirus-based functional assay designed to quantify cellular HR activity. Here, we delineate the step-by-step procedure for producing the adenovirus harboring an HR reporter, processing primary cells, and assessing HR activity in primary ovarian cancer cells.
For complete details on the use and execution of this protocol, please refer to Lee et al.1
Subject areas: cell-based assays, cancer, clinical protocol
Graphical abstract
Highlights
-
•
A high-quantity adenovirus reporter is generated from plasmid
-
•
Clinical samples from ovarian cancer patients and primary cell culture are processed
-
•
Analysis of homologous recombination via an adenovirus reporter and flow cytometry
Publisher’s note: Undertaking any experimental protocol requires adherence to local institutional guidelines for laboratory safety and ethics.
Cancer cells with homologous recombination deficiency (HRD) exhibit a distinctive vulnerability to poly(ADP-ribose) polymerase inhibitors (PARPis). To address the limitations of existing methodologies incapable of providing real-time insights into homologous recombination (HR) status, we present an adenovirus-based functional assay designed to quantify cellular HR activity. Here, we delineate the step-by-step procedure for producing the adenovirus harboring an HR reporter, processing primary cells, and assessing HR activity in primary ovarian cancer cells.
Before you begin
To expedite timely and user-friendly assessments of the homologous recombination (HR) status of cancer cells, applicable to both clinical and research contexts, we have developed a novel functional test. This test involves integrating a conventional DR-GFP cassette into an adenovirus, expanding the utility of DR-GFP, which was previously limited to a few cell lines.2 Our methodology is broadly adaptable to primary cells and diverse cell lines, significantly expanding the scope of HR status determination. Furthermore, our assay not only complements existing techniques, such as analyses of RAD51 foci and DNA sequencing, but also overcomes their limitations by faithfully capturing real-time HR activity.1,3
By assessing HR activity in ovarian, colon, and breast cancer cell lines, coupled with implementation of genomic scar-based, HR activity-based, and RAD51 foci-based HRD tests, as well as Kaplan-Meier analysis, in primary ovarian cells, we have successfully validated our functional test. We have established a threshold of 12 for homologous recombination deficiency (HRD) through our investigations. This achievement underscores the robustness and reliability of our approach, positioning it as a valuable tool for precisely determining HR status in both clinical and research settings.
Leveraging the high infection efficiency of adenovirus across multiple cell types without genomic integration, we tailored two adenoviral vectors to precisely measure HR rates. One vector encompasses the I-SceI endonuclease to instigate double-strand breaks, while the second vector harbors a DR-GFP cassette. Following the Adeno-X Adenoviral System 3 user manual, we have independently produced and purified these adenoviruses.
Cultivation of primary cells presents significant challenges in terms of both isolation and maintenance, with an emphasis on retaining high viability. Our protocol adeptly processed 46 tissue and ascitic fluid samples, incorporating refined tissue digestion techniques derived from existing tumor dissociation kits. Below, we detail the methodology for ascitic fluid sample handling and establish guidelines for cell density and optimal culture media for primary cell growth. Recognizing the necessity for some cancer patients to undergo routine ascitic fluid drainage, our protocol is strategically designed to enable longitudinal monitoring of HR status. Additionally, we outline cryopreservation strategies for clinical specimens when immediate sample processing is impractical.
Prior to deploying our HR functional assay, it is critical to ascertain cellular health, standard proliferation rates, and the correct adenovirus concentration for infection to ensure the validity of the results. We meticulously delineate the procedures for cell seeding, viral transduction, and flow cytometry, equipping users to gauge HR activity in their cells of interest. Furthermore, utilizing our cutting-edge assay enables investigations of the potential effects of uncharacterized genes or therapeutic agents on HR activity.
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Bacterial and virus strains | ||
| I-SceI-mCherry | This paper | N/A |
| DRGFP-ECFP | This paper | N/A |
| Critical commercial assays | ||
| Adeno-X adenoviral system 3 | Clontech | Cat# 632266 |
| Adeno-X Maxi purification kit | Clontech | Cat# 631533 |
| CalPhos mammalian transfection kit | Clontech | Cat# 631312 |
| Tumor dissociation kit | Miltenyi Biotec | Cat# 130-095-929 |
| Ammonium chloride solution | STEMCELL Technologies | Cat# 07850 |
| Tumor cell isolation kit | Miltenyi Biotec | Cat# 130-108-339 |
| Recombinant DNA | ||
| pAdenoX-pCMV-I-SceI-mCherry | This paper | N/A |
| pAdenoX-pCMV-DR-GFP-pPGK-ECFP | This paper | N/A |
Materials and equipment
DMEM culture medium for Adeno-X 293 cells (500 mL)
| Reagent | Final concentration (v/v) | Amount |
|---|---|---|
| Dulbecco’s modified Eagle’s medium (DMEM) | 89% | 445 mL |
| 10,000 units/mL Penicillin-10,000 μg/mL Streptomycin | 1% | 5 mL |
| Fetal bovine serum | 10% | 50 mL |
| Total | – | 500 mL |
Storage conditions: Store at 4°C for up to 1 month.
Ovarian tumor tissue medium (before tissue processing) (50 mL)
| Reagent | Final concentration | Amount |
|---|---|---|
| DMEM/F-12 | 89% | 45 mL |
| 10,000 units/mL Penicillin-10,000 μg/mL Streptomycin | 1% | 0.5 mL |
| 250 μg/mL Amphotericin B | 2.5 μg/mL | 0.5 mL |
| 50 mg/mL Gentamicin sulfate | 50 μg/mL | 0.05 mL |
| Fetal bovine serum | 10% | 5 mL |
| Total | – | 50 mL |
Storage conditions: Store at 4°C for up to 2 months.
Ovarian Carcinoma Modified Ince (OCMI) culture medium (450 mL)
| Reagent | Final concentration | Amount |
|---|---|---|
| DMEM/F12 | 45% | 204.21 mL |
| Medium 199 | 45% | 204.21 mL |
| Fetal bovine serum | 5% | 22.5 mL |
| 200 mM Glutamine | 2 mM | 4.5 mL |
| 2 mg/mL Insulin | 20 μg/mL | 4.5 mL |
| 50 mg/mL Transferrin | 10 μg/mL | 0.09 mL |
| 4 ng/mL Triiodothyronine | 0.2 pg/mL | 0.0225 mL |
| 25 mg/mL o-Phosphoryl ethanolamine | 5 μg/mL | 0.09 mL |
| 8 μg/mL Selenous acid | 8 ng/mL | 0.45 mL |
| 500 μg/mL all-Trans retinoic acid | 25 ng/mL | 0.0225 mL |
| 50 μg/mL Hydrocortisone | 500 ng/mL | 4.5 mL |
| 500 μg/mL Cholera toxin | 25 ng/mL | 0.0225 mL |
| 200 μg/mL EGF | 10 ng/mL | 0.0225 mL |
| 50 mg/mL Linoleic acid | 5 μg/mL | 0.045 mL |
| 1 mM 17-β-Estradiol | 100 nM | 0.045 mL |
| 10,000 units/mL Penicillin-10,000 μg/mL Streptomycin | 1% | 4.5 mL |
| 250 μg/mL Amphotericin B | 2.5 μg/mL | 4.5 mL |
| 50 mg/mL Gentamicin sulfate | 50 μg/mL | 0.45 mL |
| Total | – | 450 mL |
Storage conditions: Store at 4°C for up to 1 month. Based on Ince et al.4
Step-by-step method details
Adenovirus production
Timing: 2–3 weeks
To streamline the production of adenoviruses to measure HR rates, our protocol encompasses the generation, amplification, and purification of two adenoviral vectors: pAdenoX-pCMV-I-SceI-mCherry and pAdenoX-pCMV-DR-GFP-pPGK-ECFP. To mitigate cytotoxicity in target cells, we ensured that we used adenoviruses of high purity. These adenoviral vectors are available from the corresponding author upon request.
-
1.Plasmid Preparation (Figure 1A).
-
a.To expose the adenoviral genome from the plasmid backbone, the plasmid DNA containing the adenovirus-based HR reporters must be linearized using PacI enzyme digestion. The PacI enzyme digestion mixture contains 5 μg plasmid DNA, 4 μL 10X rCutSmart Buffer, 20 units Pacl restriction enzyme, and sterile deionized H2O made up to a total digestion reaction of 40 μL.Note: Each 40 μL digestion reaction is sufficient to transfect one 60-mm dish.
-
b.Incubate the 40 μL digestion reaction at 37°C for 2 h, and check enzyme digestion by 1% agarose gel electrophoresis. The pAdenoX vector will run to ∼3 kb against a size standard, and the adenoviral genome will be > 10 kb.
-
c.Remove the enzymes, as per the Adeno-X Adenoviral System 3 user manual, and resuspend the purified DNA in 10 μL 1X Tris-EDTA (TE) buffer (pH 8.0). Store the purified DNA at −20°C or use immediately.
 Pause point: The PacI-digested DNA can be stored at −20°C.
Pause point: The PacI-digested DNA can be stored at −20°C.
-
a.
-
2.Cell Transfection (Figure 1B).
-
a.Prepare Adeno-X 293 cells to 50%–70% confluency (1.1 × 106 cells) in 60-mm dishes using Dulbecco’s modified Eagle medium (DMEM) (a detailed recipe is presented in materials and equipment). Culture the cells at 37°C in a 5% CO2 environment.
-
b.After 24 h, transfect the cells with the linearized plasmid DNA using the CalPhos Mammalian Transfection Kit.
-
c.After 16 h, replace the transfection mixture with fresh culture medium after a 1X phosphate-buffered saline (PBS) wash.
-
a.
-
3.Primary Virus Stock Preparation (Figure 1B).
-
a.Monitor the cells until more than 50% either detach from the dish or remain attached but become rounded, indicating late cytopathic effects (CPE) (Figure 2), which are typically observed within 5–7 days. Directly collect both the detached and adherent cells in culture medium by means of gentle pipetting.Note: Avoid using trypsin. Given that adenovirus possesses a non-enveloped, protein-based capsid, it is recommended to avoid using trypsin for cell detachment to prevent potential damage to the virus.
-
b.Centrifuge (1,500 × g for 5 min) the harvested cells, resuspend them in 500 μL 1X PBS and then lyse them via three freeze-thaw cycles in liquid nitrogen and a 37°C water bath. Vortex the cell lysate after each thawing step.
-
c.Centrifuge (1,500 × g for 5 min) again, and collect the supernatant containing the primary virus stock in a new sterile centrifuge tube.Note: Freeze the cell lysate in liquid nitrogen for approximately two to three minutes, then thaw it at 37°C for approximately one to two minutes. Keep the suspension cold. Ensure the suspension does not reach 37°C during the freeze-thaw cycles.Pause point: The primary virus stock can be stored at −20°C.
-
a.
-
4.Secondary Virus Stock Preparation (Figure 1C).
-
a.Prepare Adeno-X 293 cells to 50%–70% confluency (1.1 × 106 cells) in 60-mm dishes, and culture in culture medium.
-
b.After 24 h, infect the cells with 250 μL of the primary virus stock.
-
c.After 16 h, replace the medium with fresh culture medium.
-
d.Monitor the cells until the appearance of late CPE. Typically, CPE is observed within 3 or 8 days post-infection for I-SceI-mCherry and DRGFP-ECFP, respectively.
-
e.Repeat harvesting as described in Steps 3b and 3c.
-
a.
Note: The titers for our secondary virus stocks are as follows: I-Sce1-mCherry at 9.3 × 108 copies/mL and DRGFP-ECFP at 2.5 × 109 copies/mL, both measured from a 6-cm dish lysate in 500 μL. This volume and titer are typically sufficient for approximately ten HR assays. However, cytotoxicity due to cell debris from the host Adeno-X 293 cells limits the volume of secondary virus stocks that can be used. To mitigate cytotoxicity, we employ the Adeno-X Maxi Purification Kit, which efficiently removes host cell proteins and toxins. The kit's user guide recommends processing lysates from five 15-cm dishes per purification column. Consequently, we scale up production to a third round, followed by purification, to ensure we obtain a sufficient quantity of purified virus for effective infection without damaging the target cells.
-
5.Final Virus Stock Preparation and Purification (Figures 1D and 1E).
-
a.Prepare Adeno-X 293 cells to 50%–70% confluency (107 cells) in 150-mm dishes for 5 dishes, and culture in culture medium.Note: For the virus purification step, the capacity of one Adeno-X Maxi Purification assembly is able to purify the lysate collected from five 15-cm plates.
-
b.After 24 h, infect the cells with 256 μL of the secondary virus stock from each plate for 90 min at 37°C. Replace the culture supernatant with 25 mL of fresh culture medium. Harvest the cells once late CPE is evident, approximately 3–5 days later.
-
c.Directly collect cells in culture medium by means of gentle pipetting.Note: Avoid using trypsin.
-
d.Centrifuge (500 × g for 10 min) the harvested cells, resuspend them in 5 mL fresh culture medium, and then lyse them via three freeze-thaw cycles in liquid nitrogen and a 37°C water bath. Vortex the cell lysate after each thawing step.
 CRITICAL: Do not resuspend the cells in PBS as doing so will inhibit the Benzonase nuclease used in the subsequent virus purification step.Note: It is not necessary to centrifuge between each freeze-thaw cycle.
CRITICAL: Do not resuspend the cells in PBS as doing so will inhibit the Benzonase nuclease used in the subsequent virus purification step.Note: It is not necessary to centrifuge between each freeze-thaw cycle. -
e.Centrifuge (1,500 × g for 5 min) and collect the supernatant in a new sterile centrifuge tube.
-
f.Add 5 μL 25 U/μL Benzonase nuclease (provided in the Adeno-X Maxi Purification Kit) for 30 min at 37°C.
-
g.Dilute the lysate with an equal volume (approximately 5 mL) of 1X Dilution buffer and filter it through a 0.45 μm filter (provided in the Adeno-X Maxi Purification Kit).
-
h.Purify the virus utilizing the Adeno-X Maxi Purification Kit, as per the instructions provided by the manufacturer.
-
i.Aliquot the purified virus (50–100 μL in individual Eppendorf tubes) and store at −80°C for long-term preservation, but avoid further repeated freeze-thaw cycles.
-
a.
Figure 1.

Schematic of the procedure for adenovirus production
(A) Plasmid preparation: linearize the plasmid DNA containing the adenovirus-based HR reporters using PacI enzyme digestion for transfection.
(B) Transfection and primary virus production: transfect the Adeno-X 293 cells with PacI-digested DNA in 6-cm dishes and, after the appearance of cytopathic effects (CPE) (5–7 days), collect and lyse the cell lysate as the primary virus (1st virus).
(C) Secondary virus production: infect the Adeno-X 293 cells with 250 μL 1st virus in 6-cm dishes, and after observing CPE (3 or 8 days), collect and lyse the cell lysate as the secondary virus (2nd virus).
(D) Final virus production: infect the Adeno-X 293 cells with 256 μL 2nd virus in five 15-cm dishes for 90 min, and after observing CPE (3–5 days), collect and lyse the cell lysate as the final virus (3rd virus).
(E) Virus purification: purify the final (3rd) virus with an Adeno-X Maxi Purification Kit, and then aliquot and store at −80°C.
Figure 2.

Observation of cytopathic effects (CPE) following adenovirus infection
Adeno-X 293 cells were infected with DRGFP-ECFP virus and monitored using a fluorescence microscope. This figure displays representative bright-field and fluorescence images of early CPE (A) and late CPE (B).
Processing of clinical samples
Note: Below, we outline the protocol for culturing primary ovarian cancer cells derived from tumor tissue and ascites. If users intend to test their specific cells of interest, please omit Steps 6–12 and proceed directly to conducting the HR activity described in Step 13.
Optimal tissue processing and digestion are critical to maximizing the yield of viable target primary cells from clinical samples. Our procedure, detailed below, includes steps for both immediate processing and preservation by freezing, thereby facilitating clinical applications.
-
6.Tissue Sample Processing (Figure 3) (Troubleshooting 1).
-
a.Collect ovarian tumor tissues in sterile 15-mL conical tubes containing 5 mL ice-cold DMEM/F-12 medium (details of the ingredients are presented in materials and equipment).
-
b.Rinse the tissues with 1X PBS to remove blood. Remove fat from the biopsy and cut the tumor into ∼2 mm3 pieces using sterile fine-tip forceps and a scalpel blade.Note: Efficient digestion necessitates small tissue pieces. The milky-white ovaries can have a texture ranging from hard to soft. Avoid cutting into blood vessels.
-
c.Prepare the tissue digestion mixture according to the instructions for the Tumor Dissociation Kit. The mixture contains 4.7 mL RPMI 1640 medium (Gibco), 200 μL enzyme H, 100 μL enzyme R, 25 μL enzyme A, and 10 μM Y-27632 (an inhibitor of Rho-associated coiled-coil-containing protein kinases, STEMCELL) for a 0.2–1 g tumor sample. If the amount of tissue exceeds 1 g, adjust the quantity of the mixture proportionately.
-
d.Divide the tissue pieces equally among the wells of 6-well plates, and then add 1.5 mL of digestion mix to each well. Incubate at 37°C, gently swirling the plates every 30 min for a total of 90 min.CRITICAL: Typically, distributing 10–20 tissue pieces, each of 2 mm³, per well across a 6-well plate achieves optimal coverage. It is critical to ensure that each tissue piece is fully immersed in the digestion mix. Avoid overloading each well with too many tissue pieces as doing so can result in incomplete digestion, especially for dense tissue sections. Additionally, if the tissue pieces contain fat, an excessive number in the well may lead to a high fat content in the culture medium, which in turn can reduce the efficiency of tissue digestion.Note: Monitor progress of digestion under a microscope every 30 min. Properly digested tissues will appear less opaque, with cells becoming individualized. Aggregated tissues can be gently teased apart with pipette tips.
-
e.Pipette the digested tissue suspension and pass it through a 70-μm cell strainer, before rinsing the cell strainer with 20 mL RPMI 1640 medium.
-
f.Centrifuge the strained cell mixture at 300 × g for 5 min at 25°C, and discard the supernatant. Wash the pellet in 10 mL 1X PBS.
-
g.Centrifuge the strained cell mixture at 300 × g for 5 min at 25°C, discard the supernatant, resuspend the cell pellet in Ovarian Carcinoma Modified Ince (OCMI) culture medium4 (details of ingredients are presented in materials and equipment), and seed onto new 6-well plates so each well attains ∼70%–80% cell confluency in 1.5 mL OCMI medium.Note: Replace the culture medium daily until the cells exhibit stable growth suitable for subculturing, typically at ∼2 weeks.
-
a.
-
7.Ascites Fluid Processing (Figure 4).
-
a.Centrifuge collected ascites in sterile 50-mL conical tubes at 300 × g for 5 min at 25°C, discard the supernatant, and quantify the remaining fluid.
-
b.Add a 4X volume of red blood cell lysis buffer (ammonium chloride solution) to the ascites, incubate for 10 min at 4°C, centrifuge at 300 × g for 5 min at 25°C and discard the lysed supernatant.
-
c.Strain the cell suspension through a 70-μm strainer, wash with 10 mL 1X PBS, centrifuge at 300 × g for 5 min at 25°C, and discard the wash.
-
d.Resuspend the final pellet in OCMI medium and seed onto new 6-well plates, so that each well attains ∼70%–80% cell confluency with 1.5 mL OCMI medium.
-
a.
Note: Replace the culture medium daily until the cells exhibit stable growth suitable for subculturing, typically at ∼2 weeks.
-
8.Freezing Tissue and Ascite Samples.
-
a.For tissue preservation, mince and resuspend tissue pieces in OCMI medium with 10% dimethyl sulfoxide (DMSO) and store overnight in cryovials within an isopropanol chamber at −80°C prior to liquid nitrogen storage. Thawed samples can be processed as fresh samples.
-
b.For ascites, cells post-processing should be suspended in OCMI medium with 10% DMSO for cryopreservation.
-
a.
Figure 3.

Schematic of a step-by-step procedure for tissue sample processing
(A) Rinse the tumor tissue with PBS to eliminate residual blood and trim it into smaller sections, meticulously avoiding fatty tissue and blood vessels.
(B) Subject 10–20 tissue fragments to enzymatic digestion or, alternatively, (C) proceed with tissue cryopreservation.
(D) Agitate the culture dish gently and perform periodic microscopic examinations at 30-min intervals.
(E) After 1.5 h of enzymatic treatment, observe the tissue transition from opaque to translucent, indicating cell dispersion.
(F) Filter the enzymatic digestion mixture through a cell strainer.
(G) Centrifuge the strained cells and perform a single PBS wash.
(H) Finally, resuspend the cell pellet in OCMI medium to establish cell culture.
Figure 4.

Schematic of a step-by-step procedure for ascites fluid processing
(A) Transfer ascites fluid into sterile conical tubes and centrifuge to separate cells. Carefully remove the supernatant without disturbing the cell pellet.
(B) Record the volume of the cell pellet.
(C) Introduce a quadruple volume of red blood cell (RBC) lysis buffer to the pellet and incubate at 4°C for 10 min.
(D) Centrifuge the mixture and dispose of the supernatant, leaving the ascitic cells.
(E) Resuspend the cells in PBS, filter through a cell strainer, and perform a single PBS wash.
(F) Centrifuge again, then resuspend the cell pellet in OCMI medium for subsequent culture, or (G) proceed to cryopreservation.
These carefully calibrated procedures ensure sample integrity for downstream analyses, providing reliable and reproducible conditions for the cultivation and preservation of primary cells derived from clinical specimens.
Primary culture for cells derived from tumor tissue and ascites
Our primary cell subculture protocol encompasses three distinct phases to propagate cells dissociated from tumor tissues and ascites for subsequent HR activity assays or for cryopreservation. The first phase involves amplifying the cells before tumor cell isolation. The second phase pertains to seeding cells immediately after isolation. The third phase addresses post-isolation subculturing. In addition to tumor cells, the initial cell mix contains a heterogeneous assortment of lymphocytes, fibroblasts, and endothelial cells. Following specimen processing and successful cell expansion, tumor cells are isolated using a Tumor Cell Isolation Kit to separate them from non-tumor cells1 It is critical to monitor cell density during the subculturing process, as both sparse and over-confluent conditions can adversely affect cell growth rates. Subsequent amplification of the isolated tumor cells prepares them for detailed HR activity analysis or for preservation by freezing.
-
9.Amplifying the Cells Prior to Isolation.Note: If, after processing the specimen, there are more than six wells with 70% cell confluency, skip cell amplification and directly conduct cell isolation using a Tumor Cell Isolation Kit to separate tumor and non-tumor cells. If there are fewer than six wells, amplify the cells by following Steps 9a to 9c until one or more 10-cm dishes attain 70% cell confluency for isolation.
-
a.Upon achieving 70% confluency, wash the cells with 1X PBS, trypsinized with 0.05% trypsin/EDTA for 3 min at 37°C, neutralize the cells with 5 mL OCMI medium, and then count the cells.CRITICAL: Due to the heterogeneity of primary cell types, it is crucial to confirm under microscopy that all cells have detached to prevent unintended cell loss.
-
b.Cell counts should be conducted to determine the appropriate seeding density. As per our guidelines:
-
c.Centrifuge at 300 × g for 5 min at 25°C, resuspend the cell pellet with OCMI medium, and seed onto the plate based on our guidelines (Step 9b, Table 1).
-
a.
-
10.Seeding Isolated Post-Primary Cells.
-
a.Immediately following cell isolation using a Tumor Cell Isolation Kit to separate tumor and non-tumor cells, perform a cell count to ascertain seeding volumes consistent with the cell densities mentioned in Step 9b above.
-
a.
Note: Conduct daily medium changes until cells show stable growth and achieve sufficient density for subsequent subculturing.
-
11.Subculturing After Isolation.
-
a.When cells achieve 70% confluency, wash them with 1X PBS, then trypsinize them with 0.05% trypsin/EDTA for 3 min at 37°C, and finally neutralize them with 5 mL OCMI medium.
-
b.Count cells to determine the seeding density.Note: When subculturing after isolation, compared to the above two phases, i.e., Amplifying the Cells Prior to Isolation and Seeding Isolated Post-Primary Cells, the growth and survival of cells is much more stable. Typically, when cell numbers exceed 7 × 10⁴, using a 10-cm dish for seeding is sufficient for successful cell expansion.
-
c.Centrifuge at 300 × g for 5 min at 25°C, resuspend the cell pellet with OCMI medium, and seed onto the culture dish. Maintain the cells at 37°C with 5% CO2 and normal oxygen levels.Note: Medium should be refreshed every 2–3 days to maintain optimal cell conditions.
-
a.
-
12.Cryopreservation of Primary Cells.
-
a.Prepare cells for freezing at 70% confluency using the trypsinization and centrifugation method described above.
-
b.Resuspend the cell pellet in OCMI medium supplemented with 10% DMSO, aiming for a minimum of 5 × 105 cells per cryovial for freezing.
-
a.
Table 1.
The seeding density of primary cells
| Surface area (cm2) | Seeding density (per dish or well) | |
|---|---|---|
| Culture dishes | ||
| 35 mm | 8.8 | 7.8 × 104 |
| 60 mm | 21.5 | 1.9 × 105 |
| 100 mm | 56.7 | 5 × 105 |
| 150 mm | 145 | 1.3 × 106 |
| Culture plates | ||
| 6-well | 9.6 | 8.5 × 104 |
| 12-well | 3.5 | 3.1 × 104 |
| 24-well | 1.9 | 1.7 × 104 |
| 48-well | 1.1 | 9.7 × 103 |
| 96-well | 0.32 | 2.8 × 103 |
These steps are designed to promote the healthy propagation of primary cells for downstream applications while preserving the integrity and viability of the cells throughout the process.
Adenovirus-based HR reporter assay
Our adenovirus-based HR reporter assay is designed to evaluate HR activity using two recombinant adenoviruses. One adenovirus expresses I-SceI endonuclease and mCherry, a red fluorescent protein, while the other contains a DR-GFP cassette alongside an enhanced cyan fluorescent protein (ECFP). The fluorescence emitted by mCherry and ECFP is instrumental in confirming co-infection via flow cytometry. Successful HR-mediated repair of I-SceI-induced DNA breaks results in EGFP expression; the percentage of EGFP-positive cells is correlated with HR activity. This assay can be adapted for diverse cancer cell types, albeit requiring meticulous attention to cell density, optimal multiplicity of infection (MOI), and flow cytometry gating for reliable outcomes.
-
13.Cell Infection Process (Figure 5A).
-
a.Seed cells in 6-well plates to achieve 60% confluency.
-
b.After 24 h, infect the cells with adenoviruses at an optimal MOI ranging from 10 to 100 infectious units/cell. Set up controls including non-infected cells, cells infected with each adenovirus individually, and co-infected cells.CRITICAL: Prior to the HR assay, determine the adenovirus titer (infectious units/mL) by assessing the number of fluorescent-positive infected cells under a microscope. Calculate infectious units/mL as follows: ((infected cells/field)(field/well))(volume of diluted virus added (mL)(dilution factor)). After calculating the infectious units/mL, test a range of MOIs from 10 to 100 infectious units/cell to determine the optimal virus amount that achieves efficient cell infection with minimal toxicity.CRITICAL: After establishing the optimal MOI for the I-SceI-mCherry and DRGFP-ECFP viruses individually, we advise setting additional infection conditions at half of optimal MOI for initial flow cytometry experiments. This adjustment is recommended due to the increased toxicity observed with co-infection for some cell lines. Consequently, we propose the establishment of seven experimental groups for quantifying HR activity in new cell lines: non-infected cells, cells infected individually with I-SceI-mCherry and DRGFP-ECFP at both the optimal MOI and half of it, and two groups of co-infected cells at optimal MOI and half, respectively (Figure 5A). If the co-infection at optimal MOI does not induce notable cell toxicity, there is no need to include infection at half of the optimal MOI afterward.
-
c.After 16 h, replace with fresh OCMI medium and incubate for an additional 2–3 days.Note: We consistently assess the HR scores of breast, colon, cervical cancers, and osteosarcoma cell lines two days after replacing with fresh culture medium. However, we noticed that the adenovirus infection in ovarian cancer cells showed much fewer fluorescence-positive cells two days after medium change compared with the above-mentioned cell lines, with less than 10% positive cells. Thus, we determine the HR activity of both ovarian cancer cell lines and primary ovarian cancer cells after three days, which show an increase in co-infection rates higher than at least 20%. Thus, we recommend first quantifying HR activity two days after medium change. If the co-infection rate is lower than 10%, extend it to three days after replacing medium. Nevertheless, under these conditions, we have observed a robust correlation between HR scores and sensitivity to PARP inhibitors.1 Notably, a standard HR activity threshold can identify HRD cells among different cell lines.1
-
d.Harvest cells using 0.05% trypsin, wash the cell pellet with 1X PBS, centrifuge at 300 × g for 5 min at 25°C, and remove the supernatant. Wash the cells twice.
-
e.Resuspend the cells in 500 μL 1X PBS to prepare them for flow cytometry analysis.
-
a.
-
14.Flow Cytometry Analysis for HR Activity (Figures 5B–5E) (Troubleshooting 2).
-
a.Utilize excitation lasers and corresponding filters for ECFP (405 nm, 525/50 filter), mCherry (561 nm, 610/20 filter), and EGFP (488 nm, 530/30 filter) detection.
-
b.For the non-infected negative control group, analyze 10,000 cells, adjusting the 405-, 488-, and 561-nm laser intensities to position cell autofluorescence in a negative fluorescence region.
-
c.Define the fluorescence thresholds for mCherry-, ECFP-, and EGFP-positive cells by analyzing 10,000 cells from each virus infection group. Set the fluorescence threshold for ECFP- and EGFP-negative cells using cells infected with I-SceI-mCherry virus alone. Establish the fluorescence threshold for the mCherry-negative cells by analyzing the cells infected with DRGFP-ECFP alone.CRITICAL: ECFP signal partially overlaps with the EGFP fluorescence spectrum. Thus, to alleviate the false positive readout of HR activity (percentage of EGFP+ cells) due to the ECFP fluorescence spillover, we have designed a weak PGK promoter and a strong CMV promoter to express ECFP and EGFP, respectively, in the virus vector DRGFP-ECFP. Importantly, although ECFP is expressed weakly in cells, the percentage of ECFP-positive cells should still exceed the number of EGFP-positive cells; the former indicates the infection rates of DRGFP-ECFP, the latter defines the HR activity, which is restricted to the S/G2 phases of positive-infected cells. To verify that the fluorescence spillover from ECFP in the EGFP channel is low and does not interfere with HR activity assessment, the percentage of EGFP-positive cells should be one-quarter to a half that of ECFP-positive cells in infection with DRGFP-ECFP alone, based on our previous study1 in which we identified HRD across cancer types.
-
d.In the co-infected cell group, set two gates for ECFP+ mCherry+ double-positive and EGFP+ mCherry+ double-positive cells. Collect 1,000 to 10,000 ECFP+ mCherry+ double-positive cells and determine the proportion of EGFP-positive cells within this gate to determine HR activity.
-
e.Interpretation: The percentage of EGFP+ cells in the ECFP+ mCherry+ double-positive group is defined as the HR activity score. HR activity scores above 12 indicate homologous recombination proficiency (HRP), whereas those below 12 denote homologous recombination deficiency (HRD).Note: In instances where the number of available cells is limited, a minimum of 200 co-infected ECFP+ mCherry+ double-positive cells are required for analysis to achieve consistent and reliable results.This detailed protocol facilitates accurate HR activity assessment, ensuring that experimental conditions are optimized for reliable data acquisition.
-
a.
Figure 5.

Flow cytometric evaluation of homologous recombination (HR) activity
This schematic outlines the procedure for assessing HR activity in HeLa cells using flow cytometry.
(A) Diagram of the HR assay workflow.
(B) Control group without infection. Laser settings are adjusted to mitigate autofluorescence, ensuring the accuracy of cell population gating: blue box for ECFP-positive, purple for mCherry-positive, and green for EGFP-positive cells. The red box (P1) identifies cells positive for both mCherry and ECFP, indicating successful co-infection; yellow within P1 demarcates EGFP-positive cells, reflecting HR activity.
(C) Group with single adenoviral infection showcasing mCherry expression (purple box), indicating successful viral entry expressing I-SceI-mCherry.
(D) Group with a different single adenoviral infection showing ECFP expression (blue box), indicative of cells carrying the DR-GFP reporter; the proportion of ECFP-positive cells is expected to exceed the baseline EGFP signal (green box) by 2–4 fold.
(E) Co-infection group: dual-positive ECFP/mCherry cells (red box, P1) confirm co-infection. (E−1) Analysis of P1 reveals HR activity of 32%, as indicated by the proportion of EGFP-positive cells (yellow box).
Expected outcomes
The anticipated outcomes of implementing our HR functional test are twofold in terms of both clinical and research applications. By amassing a broader collection of HR-associated cancer samples, expanding the possibilities for clinical studies, and enabling systematic analyses of patient-derived ascites, our assay is poised to enhance the precision of HR status evaluations. This, in turn, will inform and refine treatment strategies involving PARP inhibitors and other therapeutic modalities.
From a research perspective, our assay offers a versatile platform for scientists to assess HR activity within their specific cellular models of interest. This functionality is invaluable for probing the effects of novel genetic factors or experimental therapeutics on DNA repair mechanisms. Critically, the scope of measuring HR activity now extends to previously restricted cell types, broadening the potential for discoveries across diverse cell populations.
Limitations
In addition to enlarging the cohort of clinical samples and conducting methodical comparisons between our functional assay and genomic-scar- or RAD51 foci-based tests, it is critical to monitor patients' longitudinal responses. Observing patient outcomes over an extended period will be integral to assessing the long-term efficacy of therapies tailored to HRD status. Doing so will enable us to correlate HR activity levels with clinical outcomes, further refining the predictive power of our assay and improving the personalization of treatment plans for enhanced patient care.
Troubleshooting
Problem 1
Excessive blood cells in a tissue sample. (Step 6, Tissue Sample Processing).
Potential solution
-
•
Carefully dissect and select primarily white tissue areas for processing, avoiding regions dense in blood vessels.
-
•
If a significant number of blood cells are observed following tissue dissociation, introduce red blood cell lysis buffer to eliminate these cells effectively.
-
•
If blood cells continue to be a concern after cell attachment to culture plates, regular washing with PBS combined with frequent changes of the culture medium can help reduce their presence, thereby facilitating growth of the target primary cells.
Problem 2
Strategies for reliable HR rate quantification by flow cytometry (Adenovirus-based HR reporter assay).
Potential solution
-
•
Recognize that HR predominantly occurs during the S/G2 phases of the cell cycle, and HR activity can be influenced by cell health and confluency. Ensure that cell confluency at the time of harvest does not surpass 80%, which could result in G1 phase cell cycle arrest and subsequently reflect false-negative HR status.
-
•
Another factor contributing to non-reproducible results could be the inappropriate MOI value. An overly high MOI can be cytotoxic, leading to cell death and, consequently, unreliable HR activity scores. It is beneficial to evaluate a range of MOIs to determine the optimal infectious units per cell for your assay. Fluorescence signal in response to increasing MOI can be monitored and an optimal MOI chosen based on evaluating the balance between infection efficiency and cell viability.
-
•
Lastly, repeated freeze-thaw cycles can compromise adenoviral stock quality and reduce infection efficiency, so limit virus stocks to no more than three freeze-thaw cycles.
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Peter Chi (peterhchi@ntu.edu.tw).
Technical contact
Technical questions on executing this protocol should be directed to and will be answered by the technical contacts, Shih-Han Huang (bettyh1727@gmail.com) or Chih-Ying Lee (chih-ying.lee@dkfz-heidelberg.de).
Materials availability
Access to plasmids and materials associated with this protocol are restricted by the need for a Material Transfer Agreement (MTA). For requests, please reach out to the lead contact as mentioned above.
Data and code availability
No datasets or code was generated by this study.
Acknowledgments
We extend our gratitude to the Flow Cytometric Analyzing and Sorting Core of the First Core Laboratory at the National Taiwan University College of Medicine for their invaluable service. Figures were created with BioRender. This research received support from Academia Sinica, National Taiwan University, and the National Science and Technology Council.
Author contributions
S.-H.H. composed the initial draft of the manuscript. C.-Y.L. and P.C. provided edits to the text. All authors collaboratively discussed the protocol and made contributions to the manuscript.
Declaration of interests
The authors declare no competing interests.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.xpro.2024.103403.
Contributor Information
Shih-Han Huang, Email: bettyh1727@gmail.com.
Chih-Ying Lee, Email: chih-ying.lee@dkfz-heidelberg.de.
Peter Chi, Email: peterhchi@ntu.edu.tw.
Supplemental information
References
- 1.Lee C.Y., Cheng W.F., Lin P.H., Chen Y.L., Huang S.H., Lei K.H., Chang K.Y., Ko M.Y., Chi P. An activity-based functional test for identifying homologous recombination deficiencies across cancer types in real time. Cell Rep. Med. 2023;4 doi: 10.1016/j.xcrm.2023.101247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pierce A.J., Johnson R.D., Thompson L.H., Jasin M. XRCC3 promotes homology-directed repair of DNA damage in mammalian cells. Genes Dev. 1999;13:2633–2638. doi: 10.1101/gad.13.20.2633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lin P.H., Chen M., Tsai L.W., Lo C., Yen T.C., Huang T.Y., Chen C.K., Fan S.C., Kuo S.H., Huang C.S. Using next-generation sequencing to redefine BRCAness in triple-negative breast cancer. Cancer Sci. 2020;111:1375–1384. doi: 10.1111/cas.14313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ince T.A., Sousa A.D., Jones M.A., Harrell J.C., Agoston E.S., Krohn M., Selfors L.M., Liu W., Chen K., Yong M., et al. Characterization of twenty-five ovarian tumour cell lines that phenocopy primary tumours. Nat. Commun. 2015;6:7419. doi: 10.1038/ncomms8419. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
No datasets or code was generated by this study.