

Abstract
Acetylation of the histone H4 tail (H4Kac) has been established as a significant regulator of chromatin architecture and accessibility; however, the molecular mechanisms that underlie these observations remain elusive. Here, we characterize the ensemble features of the histone H4 tail and determine how they change following acetylation on specific sets of lysine residues. Our comprehensive account is enabled by a robust combination of experimental and computational biophysical methods that converge on molecular details including conformer size, intramolecular contacts, and secondary structure propensity. We find that acetylation significantly alters the chemical environment of basic patch residues (16–20) and leads to tail compaction that is partially mediated by transient intramolecular contacts established between the basic patch and N-terminal amino acids. Beyond acetylation, we identify that the protonation state of H18, which is affected by the acetylation state, is a critical regulator of ensemble characteristics, highlighting the potential for interplay between the sequence context and post-translational modifications to define the ensemble features of intrinsically disordered regions. This study elucidates molecular details that could link H4Kac with the regulation of chromatin architecture, illuminating a small piece of the complex network of molecular mechanisms underlying the histone code hypothesis.
1. Introduction
Intrinsically disordered regions (IDRs) are enriched within chromatin associated proteins and constitute essential topological domains of each core histone protein, highlighting that IDRs are essential for actualizing DNA-templated cellular processes.1,2 The histone IDRs, referred to as “tails”, form interactions that mediate hierarchical chromatin structure and define genome accessibility.3 The histone tails also function as regulatory hubs that can be reversibly decorated with post-translational modifications (PTMs) to recruit cellular machinery and alter chromatin structure with spatial and temporal specificity.4 The histone code hypothesis articulates that specific histone PTMs have interpretable effects on chromatin architecture and the efficiency of DNA-templated cellular processes, but the mechanistic link between a histone PTM and its regulatory outcomes can often be obscured in part due to challenges associated with structural and biophysical characterization of their disordered tails.5−7
Protein structure confers function, even within IDRs.8 Yet, IDRs sample conformational ensembles defined by free energy landscapes with shallow transition barriers between conformers, such that their structure/function relationship can be challenging to unravel.9,10 The conformational ensemble of an IDR depends on primary structure and environment, so even small perturbations to these properties can significantly alter functional behaviors including localization, compartmentalization, and interaction with molecular binding partners.8,11,12 Changes to the primary structure of an IDR can be genetically encoded through mutations or incorporated after expression through PTMs. The histone tail sequences are highly conserved across domains of life as point mutations are lethal or not well tolerated.13−16 In contrast, PTMs within histone tails are remarkably abundant.4,17 Intriguingly, the most aggressive oncohistone mutations appear to be at or near residues normally targeted for PTM.18 Structural and biophysical characterization of how PTMs alter the conformational ensemble of histone tails can provide mechanistic links between specific modification events and regulatory outcomes, elucidating the molecular details of the histone code hypothesis.4,19−21
Lysine acetylation of the histone H4 tail (H4Kac) is associated with active euchromatin and has profound impacts on chromatin architecture through unresolved mechanisms.22,23 For example, acetylation of H4K16 uniquely abolishes 30 nm fiber formation in vitro.(24) The proposed model underlying this observation is that chromatin fiber formation is mediated by electrostatic interactions between the basic patch of the H4 tail (residues 16-20) and the acidic patch formed by H2A and H2B of the next neighbor nucleosome such that neutralization of H4K16 through acetylation weakens these interactions and abrogates fiber formation.25−27 Yet, Cryo-EM studies of reconstituted chromatin fibers do not align with this model because they resolve R23 as the acidic patch anchor rather than K16 or another basic patch residue.28 Additionally, imaging of chromatin in situ suggests that 30 nm fibers may primarily be an artifact of reconstituted arrays rather than a common or functionally relevant structure in cell nuclei, further convoluting the mechanistic links between H4Kac and active euchromatin regions.3,29,30
Numerous molecular dynamics simulations have been employed to predict changes between the conformational ensembles of the unmodified and acetylated H4 tail; some have even endeavored to directly link those changes to functional outcomes by expanding the simulations to include various chromatin-like environments.31−34 These studies predict acetylation dependent changes in secondary structure propensity, conformational heterogeneity, and compaction, but disagree on specific details. Additionally, these models often utilize molecular mimics of lysine acetylation rather than chemically accurate acetyllysine moieties, and their predictions have not been interrogated by subsequent experiments.
In this study, we leveraged 13C direct-detect NMR alongside orthogonal experimental and computational biophysics approaches to characterize acetylation dependent changes to the conformational ensemble and properties of the histone H4 tail. We find that acetylation of K16 and K20 specifically changes the chemical environment of basic patch residues, and that acetylation leads to tail compaction that is partly mediated by increased sampling of intramolecular contact between the basic patch and N-terminal amino acids. Additionally, we identify a Kac dependent shift in the pKa of H18, whose protonation state informs key characteristics of the acetylated ensemble including compaction and secondary structure propensity. The results of this study derive from a conversation between experimental and computational methods, providing a cross-validated model for how acetylation influences the conformational ensemble and characteristics of the H4 tail. Ultimately, these findings provide molecular details that could link H4Kac with regulation of chromatin architecture, illuminating a small piece of the complex network of molecular mechanisms underlying the histone code hypothesis.
2. Methods
2.1. Plasmids and Construct Generation
Human histone H4(1-25) was inserted into pET His6 TEV LIC cloning vector 1 M (Addgene #29656) with ligation independent cloning. All mutagenesis was accomplished with Q5 site directed mutagenesis (NEB E0554S), including deletion of the N terminal cloning artifact (SNA) and insertion of a C-terminal exogenous W26 which was added to enable visualization by 2,2,2-Trichloroethanol (TCE) stain (BeanTown Chemical 216535) during purification.
P300HAT was originally obtained from pETduet+p300HAT (Addgene #157793) deposited by Michael Rosen, which coexpressed human p300(1284-1664) with yeast Sirt2. Additional p300HAT constructs were produced by cloning various truncations into pET His6 TEV LIC vector 1 M with ligation independent cloning.
2.2. Recombinant Protein Expression
2.2.1. Natural Abundance Histone H4 Tail
H4(1–25)W was expressed as a fusion to 6xHis tagged MBP. Transformed BL21 DE3 Escherichia coli (E. coli) were grown in Luria Broth (LB) supplemented with 50 mg/mL Kanamycin at 37 °C to OD600 of 0.8–1.0 and expression was induced with 1 mM IPTG for 3 h at 37 °C. Cells were harvested by spin centrifugation and washed in 20 mM Tris pH 7.5, 20 mM sodium chloride, 2 mM EDTA before storage of the cell pellet at −80 °C.
2.2.2. Isotope-Labeled Histone H4 Tail
For 13C/15N incorporation, expression was performed in M9 minimal media formulated with 2 g 13C d-glucose (Cambridge Isotope Laboratories CLM-481-PK) and 1 g 15N ammonium chloride (Cambridge Isotope Laboratories DNLM-8739-PK). Transformed BL21 DE3 E. coli were grown in 5 mL LB for ∼8 h, then 500 μL of starter culture was used to inoculate 50 mL M9 which was grown overnight at 37 °C. All 50 mL of the overnight was used to inoculate 1L M9 supplemented with 50 mg/mL Kanamycin. The culture was grown at 37 °C to OD600 of 0.8–1.0 and expression was induced with 1 mM IPTG for 3 h at 37 °C. Cells were harvested by spin centrifugation and washed in 20 mM Tris pH 7.5, 20 mM sodium chloride, 2 mM EDTA before storage of the cell pellet at −80 °C.
2.2.3. P300HAT
To express 9xHis tagged p300HAT from pETduet+p300HAT, transformed BL21 Rosetta E. coli were grown in Terrific Broth (TB supplemented with 0.4% w/v glycerol, 1 mM MgCl2, 100 mg/mL ampicillin, and 25 mg/mL chloramphenicol) at 37 °C to OD600 of 0.8–1.0. The growth was transferred to 18 °C for 1 h before expression was induced with 0.5 mM IPTG for 16–18 h at 18 °C. Cells were harvested by spin centrifugation and washed in 20 mM Tris pH 7.5, 20 mM sodium chloride, 2 mM EDTA before storage of the cell pellet at −80 °C. P300HAT constructs in pET His6 TEV LIC 1 M were expressed in the same manner.
2.3. Recombinant Protein Purification
2.3.1. Histone H4 Tail
The H4(1-25)W cell pellet was thawed at room temperature for 20 min then resuspended in 50 mM Tris pH 7.5, 500 mM sodium chloride, 20 mM imidazole, 2 mM β-mercaptoethanol, and 1× protease inhibitor cocktail set V (Millipore Sigma 539137-10VL). Cells were lysed by sonication and the lysate was clarified by spin centrifugation for 30 m at 14,000g and 4 °C. The soluble fraction was loaded onto NiNTA resin (G-Biosciences 786-407) equilibrated with 50 mM Tris pH 7.5, 500 mM sodium chloride, 20 mM imidazole, 2 mM β-mercaptoethanol. Bound protein was washed with 5 CV 50 mM Tris pH 7.5, 200 mM sodium chloride, 20 mM imidazole, 2 mM β-mercaptoethanol and eluted with 5 CV 50 mM Tris pH 7.5, 200 mM sodium chloride, 200 mM imidazole, 2 mM β-mercaptoethanol. The fusion tag was cleaved overnight at 4 °C with TEV (Tobacco Etch Virus) protease (a gift from Song Tan) and dialyzed in 1 kDa molecular weight cutoff (MWCO) membrane tubing (Spectrum Laboratories 132638) against 50 mM Tris pH 7.5, 100 mM sodium chloride, 1 mM dithiothreitol. Dialysate was loaded onto SP Sepharose Fast Flow resin (Cytiva 17072910) equilibrated with dialysis buffer. Bound protein was sequentially eluted with a step gradient of 4 CV fractions containing 50 mM Tris pH 7.5 and 400, 500, 700, and 1000 mM sodium chloride. The 700 mM salt fraction was dialyzed overnight at 4 °C in 1 MWCO membrane tubing against 50 mM K2HPO4 pH 7.2, 150 mM potassium chloride. The peptide was concentrated with 1 MWCO spin concentrators (Cytiva MAP001C36 and MCP001C41) at 4000g, 16 °C. The peptide does not aggregate if it is not concentrated >2 mM, typically the yield from 1 L of expression could be safely concentrated to 500 μL. Peptide can be stored at −80 °C and is stable at room temperature for >1 week.
2.3.2. Histone H4 Tail V21C
Purification followed the protocol above, however the V21C mutant elutes in the 1000 mM salt fraction of ion exchange rather than the typical 700 mM salt fraction. Additionally, reducing reagent was maintained in all buffers such that the ion exchange fractions contained 50 mM Tris pH 7.5, 2 mM β-mercaptoethanol, and 400, 500, 700, and 1000 mM sodium chloride while the final dialysis buffer contained 50 mM K2HPO4 pH 7.2, 150 mM potassium chloride, 1 mM DTT.
2.3.3. P300HAT
All p300HAT constructs were purified the same way. All purification steps were performed on ice or at 4 °C. A cell pellet was thawed at 4 °C for 30 m then resuspended in 50 mM HEPES pH 7.0, 500 mM sodium chloride, 30 mM imidazole, 10% w/v glycerol, 5 mM β-mercaptoethanol, 1 mM PMSF, 1× protease inhibitor cocktail set V. Cells were lysed by sonication and the lysate was clarified by spin centrifugation for 45 m at 20,000g and 4 °C. The soluble fraction was loaded onto NiNTA resin equilibrated with 50 mM HEPES pH 7.0, 500 mM sodium chloride, 30 mM imidazole, 10% w/v glycerol, 5 mM β-mercaptoethanol. Bound protein was washed with 5 CV 50 mM HEPES pH 7.0, 1 M sodium chloride, 30 mM imidazole, 10% w/v glycerol, 5 mM β-mercaptoethanol followed by 5 CV 50 mM HEPES pH 7.0, 150 mM sodium chloride, 30 mM imidazole, 10% w/v glycerol, 5 mM β-mercaptoethanol then eluted with 5 CV 50 mM HEPES pH 7.0, 150 mM sodium chloride, 300 mM imidazole, 10% w/v glycerol, 5 mM β-mercaptoethanol. The 9xHis or 6xHis-MBP tag was cleaved overnight at 4 °C with TEV protease and dialyzed in 3.5 MWCO membrane tubing (Fischer Scientific 08-670-5B) against 50 mM HEPES pH 7.0, 150 mM sodium chloride, 30 mM imidazole, 10% w/v glycerol, 5 mM β-mercaptoethanol. Dialysate was loaded onto NiNTA resin equilibrated with dialysis buffer, and flowthrough was chased with 2 CV dialysis buffer. Bound protein was eluted with dialysis buffer supplemented with 300 mM imidazole. The flowthrough was dialyzed overnight at 4 °C in 3 MWCO membrane tubing against 50 mM HEPES pH 7.0, 100 mM sodium chloride, 10% w/v glycerol, 1 mM DTT. The dialysate was concentrated with a 3 MWCO spin concentrator (Millipore Sigma UFC9003) so that the final concentration of p300HAT was 200 μM, then stored at −80 °C.
2.4. Reagent and Sample Preparation
Acetyl-CoA synthesis was conducted following our standard protocol,35 by combining CoA lithium salt (1 M equiv, CoALA Biosciences AC02) and 1,1′,2,2′-13C acetic anhydride (1.8 M equiv, Cambridge Isotope Laboratories CLM-1161-1) in 200 μL 0.5 M sodium bicarbonate in an Eppendorf tube, which was incubated in an ice bath for 45 min. The product was characterized by 1D 1H NMR and stored at −80 °C without further purification. A 200 μL reaction yielded approximately 13.4 mg of 13C acetyl CoA. Natural abundance acetyl-CoA was prepared in the same way, but using 12C acetic anhydride (1.8 M equiv, Sigma-Aldrich 242845).
2.4.1. Acetylation Reactions
Following dialysis into 50 mM K2HPO4 pH 7.2, 150 mM potassium chloride, the H4 tail peptide was concentrated to 2 mM with 1 MWCO spin concentrators at 4000g, 16 °C. Standard acetylation reactions were accomplished overnight at room temperature in 50 mM Tris, 50 mM Bis·Tris, 100 mM sodium acetate pH 7.5, 1 mM DTT, with 500 μM H4 tail, 10 μM p300HAT, and 5 mM acetyl-CoA (2 M equiv for each available lysine residue). Following acetylation, p300HAT was removed from the sample by heating at 90 °C for 5 m, centrifugation at 20,000g for 5 m, and transfer of the supernatant to a clean tube.
2.4.2. MALDI-MS
50 μL samples of >50 μM peptide were desalted with C18 spin columns (G-Biosciences 786-930) according to manufacturer instructions. The eluent in 70% acetonitrile was dried for 3 h in a tabletop vacuum dryer then resuspended in 5 μL chromatography grade water (Fischer L-13780). Samples were mixed 1:1 with 20 mg/mL Super-DHB matrix (Sigma-Aldritch 50862-1G-F) in 50% acetonitrile, 1% trifluoroacetic acid, 1% phosphoric acid.
2.4.3. SAXS
Recombinant peptides with 0Ac, 3Ac (on K5, K8, and K12), and 5Ac (on K5, K8, K12, K16, and K20) were synthesized by Genscript with >90% purity and guaranteed acetate substitution for trifluoracetic acid. Peptide identity and purity was confirmed by MALDI-MS (Figure S3A). Peptides were resuspended in 50 mM K2HPO4 pH 7.2, 150 mM potassium chloride at 5 mg/mL. Exact concentrations were determined by Fourier Transform Infrared Spectroscopy and samples were diluted to 2 mg/mL.
2.4.4. Chemical Shift Assignment
Following purification by ion exchange chromatography (for the unacetylated sample) or the acetylation reaction (for the acetylated sample), the 13C/15N H4 tail peptide was dialyzed overnight at 4 °C in 1 MWCO membrane tubing against 50 mM K2HPO4 pH 7.2, 150 mM potassium chloride. Samples were concentrated with 1 MWCO spin concentrators at 4000g, 16 °C to 500 μL and spiked with 0.1% sodium azide (EMD SX0299-3). Initial chemical shift assignments were collected with unacetylated (SNA)H4(1-25)(W) and a G7A mutant that enabled deconvolution between identical GKGG sequence regions (Figure S1).
2.4.5. Chemical Shift Perturbation
Following purification by ion exchange chromatography the unacetylated 13C/15N H4 tail peptide was dialyzed overnight at 4 °C in 1 MWCO membrane tubing against 50 mM K2HPO4 pH 7.2, 150 mM potassium chloride. The sample was concentrated with 1 MWCO spin concentrators at 4000g, 16 °C to 500 μL. For buffer matching purposes, unAc spectra were collected in 50 mM Tris, 50 mM Bis·Tris, 100 mM sodium acetate pH 7.5, 1 mM DTT, with 1 mM H4 tail and 10 mM acetyl-CoA. Partial (3×) acetylation of this sample was accomplished overnight at room temperature in 50 mM Tris, 50 mM Bis·Tris, 100 mM sodium acetate pH 7.5, 1 mM DTT, with 1 mM H4 tail, 20 μM p300HAT, and 10 mM acetyl-CoA (2 M equiv for each available lysine residue). We observed that the efficiency of p300HAT varied with substrate concentration such that the enzyme was unable to fully acetylate more concentrated samples, however the specifics were not reproducible. After removing the p300HAT as described above and buffer exchanging back into 50 mM K2HPO4 pH 7.2, 150 mM potassium chloride, complete (5×) acetylation was accomplished overnight at room temperature in 50 mM Tris, 50 mM Bis·Tris, 100 mM sodium acetate pH 7.5, 1 mM DTT, with 500 μM H4 tail, 10 μM p300HAT, and 5 mM acetyl-CoA, then the sample was concentrated back to 1 mM for data collection.
2.4.6. MTSSL Labeling
Following purification by ion exchange chromatography (for the unacetylated sample) or the acetylation reaction (for the acetylated sample), the 13C/15N H4 V21C tail peptide was dialyzed overnight at 4 °C in 1 MWCO membrane tubing against 50 mM K2HPO4 pH 7.2, 150 mM potassium chloride, 1 mM DTT. The sample was concentrated with 1 MWCO spin concentrators at 4000g, 16 °C to 600 μL such that the peptide concentration was approximately 1.7 mM. Ten M equivalents of DTT were added and the sample incubated at room temperature for 30 m. For the unacetylated sample, desalting was accomplished using a PD-10 desalting column (Cytiva 17085101) according to manufacturer instructions. For the acetylated sample, peptide compaction prevented efficient separation from small molecules with a PD-10 column. Therefore, desalting was accomplished using a polyacrylamide desalting column (ThermoScientific 43426) and collection of the first 3 mL of elution. Peptides were eluted into a black out 5 mL Eppendorf tube. Immediately following elution, 10 mg MTSSL (VWR 89151-196) resuspended in 500 mL acetonitrile acetonitrile was added to the eluent and the sample was incubated at 4 °C for 1 h. The sample was transferred to a 1 MWCO spin concentrator and diluted to 25 mL with 50 mM K2HPO4 pH 7.2, 150 mM potassium chloride. Buffer exchange was accomplished by concentrating the sample to <5 mL and refilling to 25 mL with fresh buffer twice before bringing the final sample volume to 600 μL.
2.4.7. pKa Titrations
Following purification by ion exchange chromatography (for the unacetylated sample) or the acetylation reaction (for the acetylated sample), the 13C/15N H4 tail peptide was dialyzed overnight at 4 °C in 1 MWCO membrane tubing against 50 mM K2HPO4 pH 8.25, 150 mM potassium chloride. The sample was concentrated with 1 MWCO spin concentrators at 4000g, 16 °C to 700 μL.
2.5. Molecular Dynamics (MD) Simulations
The initial H4 peptide configurations were selected from three distinct coil structures generated via the Flexible Meccano algorithm36 using the ProtSA Web server.37 Force field parameters for lysine acetylation were sourced from the Force field_PTM developed by Khoury et al.38 System topologies were modeled employing the Amber03ws force field,39 available at https://bitbucket.org/jeetain/all-atom_ff_refinements. The protonation state of H18 was modeled interactively in Gromacs during structure preparation with the pdb 2gmx tool. An octahedral box with a side length of 6 nm was chosen to ensure at least 1.5 nm of water molecules surrounding the H4 peptide. Initially, the system energy was minimized in vacuum, followed by another energy minimization, after solvation with TIP4P/2005 water molecules,40 using the steepest descent algorithm in Gromacs-2021.6.41 To replicate physiological salt concentrations (150 mM), Na+ and Cl– ions were added, along with additional counterions, to achieve charge neutrality. Improved salt parameters from Lou and Roux42 were utilized for all simulations. The system underwent initial equilibration in a canonical ensemble (NVT) using a Nose-Hoover thermostat43 with a coupling constant of 1.0 ps at 300 K. This was followed by further equilibration in an isothermal–isobaric ensemble (NPT) using a Berendsen barostat44 with an isotropic coupling constant of 5.0 ps to maintain a pressure of 1 bar. The Gromacs files were converted to Amber inputs using the “gromber” function in Parmed,45,46 and hydrogen mass repartitioning47 to 1.5 amu was performed during conversion to facilitate a 4 fs time step for production runs.
Subsequent simulations were performed using the Amber22 MD simulation package. A tabulated list of all simulations performed in this study and their associated time-dependent secondary structure plots are included in Figure S7. Initially, a minimization run was conducted using both the steepest descent and conjugate gradient algorithms, with all non-hydrogen atoms restrained by a 5 kcal/(mol·Å2) force constant. This was followed by two NVT simulations with a time step of 4 fs: a 5 ns equilibration at 300 K with a reduced force constant of 1 kcal/(mol·Å2) and an additional 5 ns equilibration at 300 K with all restraints removed. Further equilibration was performed for 10 ns under an isothermal–isobaric ensemble (NPT) using a 4 fs time step, a Monte Carlo barostat with an isotropic coupling constant of 1.0 ps, and a pressure of 1.0 bar. Temperature control was maintained using Langevin dynamics with a friction coefficient of 1.0 ps–1, while the SHAKE algorithm48 was applied to hydrogen atoms. A nonbonded cutoff of 0.9 nm was applied for short-range interactions, and long-range electrostatic interactions were managed using the Particle Mesh Ewald (PME) method.49,50 Finally, a 4 μs NPT production run for each configuration was carried out, utilizing the same parameters as those used for equilibration.
2.6. Data Collection and Analysis
2.6.1. MALDI-MS
Peptide samples were evaluated on a Bruker Ultraflextreme MALDI TOF-TOF instrument equipped with a 355 nm frequency-tripled NdYAG smartbeam-II laser. The mass spectra were acquired using a factory-configured instrument method for reflector positive-ion detection over the 700–3500 m/z range. Laser power attenuation and pulsed ion extraction time were optimized to achieve the best signal-to-noise. The instrument was calibrated with a bovine serum albumin tryptic peptide mixture (Protea, p/n PS-204-1). Mass spectra were opened in FlexAnalysis, smoothed (SavitzkyGolay, 0.2 m/z, 1 cycle), baseline-subtracted (TopHat), and the mass lists were generated using a Snap peak detection algorithm with signal-to-noise threshold set at 6 and using the Averagine SNAP average composition. Data normalization and plotting was accomplished in R-Studio (1.4.11).
2.6.2. SEC-MALS
Samples were evaluated at 2 mg/mL and 25 °C on an Agilent 1260 Infinity II HPLC system equipped with an autosampler and Superdex 200 Increase 10/300 GL column (Cytiva 28990944). Wyatt Technology DAWN MALS, Wyatt Optilab refractive index detector, and Agilent UV detector were used for analyzing the molar mass of peaks that eluted from the column. The SEC–MALS system was equilibrated for 5 h with 50 mM K2HPO4 pH 7.2, 150 mM potassium chloride. A 40 μL sample volume was injected at a flow rate of 1.0 mL/min and a chromatogram run time of 24 min. Normalization and alignment of the MALS and refractive index detectors were carried out on standard BSA. Data were analyzed using Wyatt ASTRA software. The chromatogram showed a single monodisperse monomer peak in the light scattering and the refractive index (data not shown).
2.6.3. SAXS
Samples were evaluated at 2 mg/mL and 25 °C at a wavelength of 1.54 Å on in-house home source X-rays generated by a Rigaku MM007 rotating anode housed with the BioSAXS2000nano Kratky camera system. The system includes OptiSAXS confocal max-flux optics designed specifically for SAXS and a HyPix-3000 Hybrid Photon Counting detector. The sample capillary-to-detector distance was 495.5 μm and calibrated using silver behenate powder (The Gem Dugout, State College, PA). The useful momentum transfer scattering vector q-space range (4π sin θ/λ with 2θ being the scattering angle) was generally from qmin = 0.008 Å–1 to qmax = 0.6 Å–1 (q = 4π sin (θ)/λ, where 2θ is the scattering angle). The energy of the X-ray beam was 1.2 keV, with the Kratky block attenuation of 22% and beam diameter of ∼100 μm. Protein samples were loaded using the Rigaku autosampler on to a quartz capillary flow cell mounted on a sample stage and aligned in the X-ray beam. The sample cell and full X-ray flight path, including beam stop, were kept in vacuo <1 × 10–3 Torr to eliminate air scatter. The Rigaku SAXSLAB software was programmed for automated data collection of each protein with elaborate cleaning cycles between samples. Data reduction including image integration and normalization, and background buffer data subtraction were carried out using the SAXSLAB software. Six 10 min images and five replicates from protein and buffer samples were collected and averaged after ensuring that no X-ray radiation damage had occurred. Guinier and p(r) analysis was accomplished in ATSAS Primus (3.0) after averaging the five buffer subtracted replicates for each sample. Best practices for SAXS analysis of IDPs were observed in accordance with guidelines published by Kragelund and Skriver.51 Error estimates provided for Gunier Rg values reported in Figure 3A are from the Primus Gunier Wizard and account for error in the best fit interval as well as error associated with other possible intervals within the acceptable Gunier range. No error estimates are provided by the Primus Distance Distribution Wizard.
Figure 3.
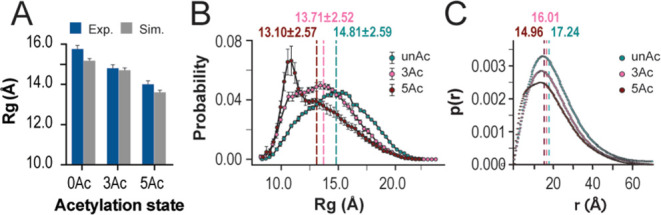
H4 tail acetylation results in a more compact ensemble. (A) Histogram displaying average Rg values for experimental Guinier analysis (blue) and MD computational Crysol (gray)scattering profiles for unAc, 3Ac, and 5Ac. Associated error estimates were provided by Primus Gunier Wizard and Crysol. (B) Rg probability distributions from MD with mean Rg values and their standard deviations indicated. Errors of the Rg distributions are estimated using block averages with five blocks. (C) P(r) distributions from experimental SAXS data with P(r) derived Rg values indicted.
2.6.4. Chemical Shift Assignments
All experiments were conducted on a Bruker AVIII-500 MHz spectrometer equipped with a triple resonance TCI single-axis gradient cryoprobe. The sample temperature was maintained at 25 °C. The 13C/15N (SNA)H4(1-25)(W) was supplemented with 5% D2O and transferred to a 5 mm NMR tube. Entirely “protonless” chemical shift assignments were generated following published protocols.52 Briefly, [15N, 13C]-CON and [15N, 13C]-HACACON experiments were collected using the standard pulse program from the Bruker Topspin library with a matrix size of 1024(C′) × 512(N), spectral width of 20 × 30 ppm, 16 scans, and a recycle delay of 1 s. [15N, 1H]-HSQC experiments were collected using the standard pulse program from the Bruker Topspin library with a matrix size of 2048(H′) × 256(N), spectral width of 16 × 44 ppm, 18 scans, and a recycle delay of 1 s. [15N, 13C]-HACANCACON experiments were collected using the standard pulse program from the Bruker Topspin library with a matrix size of 1024(C′) × 64(N) × 256(C), spectral width of 20 ppm × 30 ppm × 30 ppm, 8 scans, and a recycle delay of 1 s. [15N, 13C]-CCCON experiments were collected using the standard pulse program from the Bruker Topspin library with a matrix size of 1024(C′) × 64(N) × 256(C), spectral width of 20 × 30 × 80 ppm, 8 scans, and a recycle delay of 1 s. In all cases, virtual decoupling in the 13C direct dimension was achieved through utilization of in-phase antiphase (IPAP) spectra processed to yield virtual decoupling using the standard “splitcomb” AU program distributed with the Bruker Topspin library. Phase correction was accomplished in Topspin (4.2.0) and spectra were exported as UCSF files using the ‘easy_bruk2ucsf’ EXE program distributed with the NMRFAM-Sparky library. Peak picking and chemical shift assignments were performed in NMRFAM-SPARKY (1.470) powered by Sparky (3.190) after referencing carbon dimensions to DSS (methyl 1H = −2.60 ppm) in 50 mM K2HPO4, 150 mM KCl. As this original sequence was modified (substitution, deletion, labeling, acetylation), 2D and 3D experiments were repeated as necessary to confidently transfer chemical shift assignments. Assignments have been deposited in the BioMagResBank53 (unacetylated BMRB ID 52585, 5Ac BMRB ID 52586).
2.6.5. Chemical Shift Perturbations
For each acetylation state, the 13C/15N H4 sample was supplemented with 5% D2O and transferred to a 5 mm NMR tube. [15N, 13C]-CON and [15N, 13C]-CCCON spectra were collected for unAc, 3Ac, and 5Ac species with the same parameters and initial processing as described above. After import into NMRFAM-SPARKY, the identity of the acetylated lysines in each sample was confirmed by comparing chemical shift information in the [15N, 13C]-CCCON spectra (Figure S2). Initial processing was accomplished in Topspin, peak picking was performed in NMRFAM-SPARKY54 and chemical shift information was exported for calculation of chemical shift perturbation for each residue according to the equation δ = √0.4 × (ω1A – ω1B)2 + 0.7 × (ω2A – ω2B)2 where ω1 is the indirect dimension (15N), ω2 is the direct dimension (13C), and A and B are the two acetylation states being compared.55
2.6.6. Paramagnetic Relaxation Enhancement
The MTSSL labeled 13C/15N H4 V21C sample was supplemented with 5% D2O and transferred to a 5 mm NMR tube. Paramagnetic [15N, 13C]-CON experiments were collected using the standard pulse program from the Bruker Topspin library with a matrix size of 1024(C′) × 256(N), spectral width of 20 × 30 ppm, 64 scans, and a recycle delay of 1 s. The nitroxide radical was quenched by addition of 5 M equiv of sodium ascorbate. The diamagnetic [15N, 13C]-CON was collected with the same parameters as the paramagnetic [15N, 13C]-CON, as were the paramagnetic and diamagnetic 5Ac experiments. Initial processing was accomplished in Topspin, peak picking and integration using Gaussian fit was performed in NMRFAM-SPARKY after setting NC_PROC to −14 for all experiments. The intensity ratio was determined by taking diamagnetic/paramagnetic intensity for each residue. Due to the partially degenerate positions of R3, K5, K8, and K12 in the CON, those four residues were evaluated as a group under a single peak (Figure S4B).
2.6.7. pKa Titrations
The 700 μL 13C/15N H4 sample was supplemented
with 50 μL D2O such that the final volume was 750
μL and peptide concentration was approximately 1.3 mM. The sample
was transferred to a 5 mm NMR tube. [15N, 13C]-CON experiments were collected using the standard pulse program
from the Bruker Topspin library with a matrix size of 1024(C′)
× 256(N), spectral width of 20 ppm × 30 ppm, 8 scans, and
a recycle delay of 1 s. For each pH adjustment, the sample was removed
from the NMR tube with an elongated glass pipet and transferred to
an Eppendorf tube, the pH was adjusted by 0.25 units using 1 M HCl
and monitored with a micro pH probe (ThermoFischer Scientific 9110DJWP),
then transferred back to the NMR tube for collection of the next [15N, 13C]-CON. Initial processing was accomplished
in Topspin, peak picking was performed in NMRFAM-SPARKY and chemical
shift information was exported for CSP determination according to
the equation δ = √0.4 × (ω1A –
ω1B)2 + 0.7 × (ω2A – ω2B)2 where ω1 is the indirect dimension (15N), ω2 is
the direct dimension (13C), and A and B are the two pH
states being compared. For each residue where start and end positions
did not overlap (G2, R3, G4, H18, R19, K20, V21, N25, and W26), CSP
as a function of experimental pH was fit in R-Studio to determine
pKa with the nlme: Linear and Nonlinear
Mixed Effects Models package according to the function 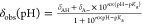 where δAH and δA– are the calculated 15N, 13C CSP for the protonated and deprotonated species, the Hill
coefficient, n, represents the slope of the titration
curve in the transition region, and the pKa value represents the pH at which the ionizable group is protonated
halfway. The N25 and W26 titrations were not fittable because their
δAH was not measured in the pH range of these experiments.
where δAH and δA– are the calculated 15N, 13C CSP for the protonated and deprotonated species, the Hill
coefficient, n, represents the slope of the titration
curve in the transition region, and the pKa value represents the pH at which the ionizable group is protonated
halfway. The N25 and W26 titrations were not fittable because their
δAH was not measured in the pH range of these experiments.
2.6.8. NMR Secondary Structure Prediction
Chemical shift information for Cα (side chain), Cβ (side chain), CO (backbone), and N (backbone) for each residue were compiled into SHIFTY file format and submitted to the Δ2D web tool.56 This tool provides no error estimate, however it marks residues with “less reliable” predictions; none of our residues were marked as less reliable. As for chemical shift assignments, chemical shift information submitted to Δ2D was referenced in the carbon dimensions to DSS (methyl 1H = −2.60 ppm) in 50 mM K2HPO4 pH 7.2, 150 mM potassium chloride.
2.6.9. MD Trajectory Analysis
The Gromacs utility gmx gyrate was employed to calculate the radius of gyration (Rg) over the entire trajectory. SAXS scattering curves were computed for conformations in each ensemble using the CRYSOL program.57 Ensemble-averaged scattering curves were generated for the H4 tail simulations with unacetylated, three-acetylated, and five-acetylated lysine residues. The software ATLAS Primus was utilized to calculate the Rg from the scattering curves using Guinier analysis. Secondary structure calculations were conducted with the gmx do_dssp program, which leverages the DSSP library.58 For contact analysis, two residues (i and j) were considered in contact if their sequence separation exceeded three (|i – j| > 3) and any pair of heavy atoms from these residues were within 0.6 nm of each other. This distance cutoff, extensively tested in our previous studies,59−61 adequately encompasses various contact modes such as van der Waals interactions, hydrogen bonds, and salt bridges. Solvent-accessible surface areas were calculated using the gmx sasa utility, which implements the algorithm developed by Eisenhaber et al.62
3. Results and Discussion
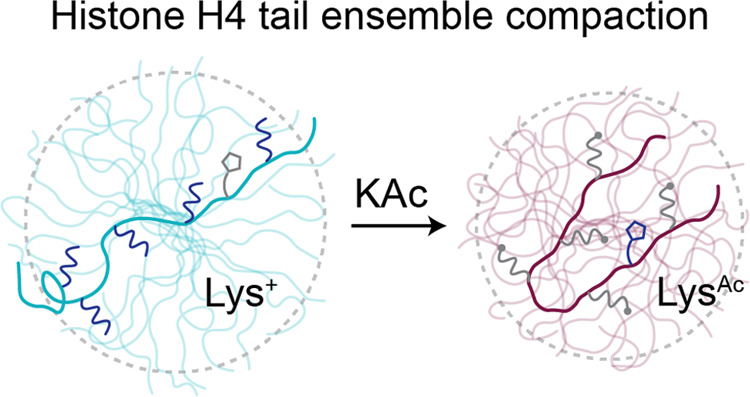
We aimed to generate an H4 tail construct that could be used to study the effects of H4Kac on ensemble characteristics. To accomplish this, we recombinantly expressed and purified human H4(1–25) from E. coli and added an exogenous C-terminal tryptophan to facilitate detection during purification such that the final working construct was H4(1-25)W (Figure 1A). We then used the human p300HAT module to enzymatically acetylate the peptide at K5, K8, K12, K16, and K20- the 5 available lysine residues. We were also able to stop the reaction at an intermediately acetylated state with marks mainly on K5, 8, and 12. With this system in hand, we used NMR, SAXS, and MD simulations to investigate the conformational ensemble and properties of the unacetylated (unAc), intermediately acetylated (3Ac), and uniformly acetylated (5Ac) H4 tail constructs, probing for acetylation dependent changes to chemical environment, compactness, secondary structure propensity, intramolecular contacts, and side chain pKa.
Figure 1.
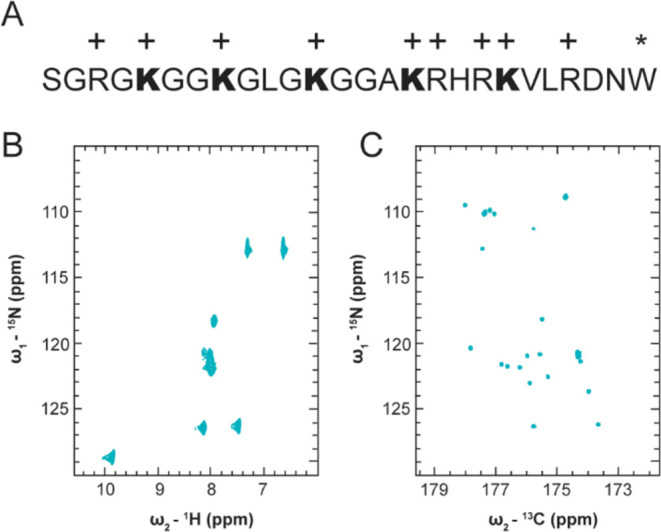
H4 tail chemical shift information resolved using 13C direct-detect NMR spectroscopy. (A) Schematic of H4(1-25)W experimental construct. All experiments were performed with this construct unless a mutant is specifically indicated. Lysine residues are indicated in bold, positively charged residues with +, and the exogenous tryptophan with *. (B) 15N, 1H-HSQC spectrum of H4(1-25)W (C) 15N, 13C–CON spectrum of H4(1-25)W. All NMR experiments were performed in 50 mM K2HPO4, pH 7.2, 150 mM KCl unless otherwise indicated.
3.1. 15N, 13C–CON Enables Straightforward Chemical Shift Assignment of the Histone H4 Tail
NMR spectroscopy remains the only experimental technique that provides high resolution structural information for IDRs, making it an ideal platform for characterization of acetylation dependent changes in the conformational ensemble of the H4 tail.7,63 The near atomic resolution of NMR relies on acquisition of a sufficient inventory of chemical shift information to facilitate unique mapping of observable resonances back to the nuclear correlation(s) from which they arise. Available 1H-detect spectra of the H4 tail are challenging to interpret as they suffer from poor spectral resolution in the direct dimension, which is typical for IDRs due to high internal flexibility and can prevent unique resonance mapping or assignment.63,64 Of the four histone tails, the histone H4 tail has the most crowded and spectrally degenerate 15N, 1H-HSQC spectrum. While nucleosome context marginally improves spectral degeneracy, it also limits the number of observable resonances in the H4 tail to just 15.65 Application of 13C direct-detect NMR typically overcomes poor spectral dispersion for IDRs, so we wanted to establish whether 13C-detect NMR could be leveraged to improve spectral resolution and the utility of chemical shift information for the H4 tail.52,66
In agreement with prior literature reports, resonances in the 15N, 1H-HSQC of our construct are quite degenerate, especially around 8 ppm 1H (Figure 1B). Additionally, some resonances are exchange broadened into the noise due to high solvent exposure, such as those around 110 ppm 15N. These spectral characteristics obscure confident assignment and complicate downstream interpretation. The spectral resolution for the H4 tail improves significantly using 15N, 13C–CON compared to 15N, 1H-HSQC NMR spectroscopy, enabling unique assignment of observable resonances in the CON to specific peptide bonds in H4(1-25)W (Figure 1C). In addition to improved resolution, the 13C-detected spectrum facilitates detection of the residues around 110 ppm 15N which were exchange broadened in the 1H-detected spectrum. The H4 tail’s easily interpretable 15N, 13C–CON spectrum, and the underlying resolution of 13C chemical shift information, provides the necessary foundation for characterization of acetylation dependent changes in the conformational ensemble of the H4 tail with atomic resolution.
3.2. Lysine Acetylation Changes the Chemical Environment of Glycine and Basic Patch Residues
The conformational ensemble of IDRs depends on primary structure and is significantly influenced by charge patterning.67,68 We expected that neutralization of positive lysine side chains through acetylation would perturb the chemical environment of the H4 tail and effect changes in the conformational ensemble.8 Changes in chemical environment for specific amino acids can be reported through chemical shift perturbations (CSPs) of resonances in NMR spectra.69 Following enzymatic acetylation of the H4 tail peptide with p300HAT, we evaluated per residue CSP to determine which amino acids experienced the most significant changes in chemical environment.
We found that H4Kac results in backbone CSPs for many H4 tail residues. We generated two uniformly 15N, 13C enriched H4Kac constructs with 3Ac and 5Ac modifications. The 3Ac species is primarily modified on K5, 8, and 12 while the 5Ac is uniformly modified on K5, 8, 12, 16, and 20 (Figure 2A). The number of acetyl marks was determined by MALDI-MS (Figure 2B) and the identity of the modified lysine residues was determined by lysine side chain CSPs in the 15N, 13C, 13C–CCCON (Figure S2A). Surprisingly, the backbone CSPs of acetylated lysines are negligible, while more significant CSPs occur for glycine residues preceded by modified lysines. However, the most significant CSPs occur in the basic patch, H4(16-20), only after acetylation of K16 and K20. These significant CSPs cannot be attributed to lysine side chain neutralization alone as the frequency of acetylation in the basic patch is no higher than for any other 5 amino acid k-mer, so this local phenomenon invites closer examination of the basic patch’s unique chemical features which are explored in later sections. It is worth noting that these details would not be interpretable if we had used 1H-detect rather than 13C direct-detect NMR, as emphasized by overlay of the unAc and 5Ac 15N, 1H-HSQC spectra (Figure S2B).
Figure 2.
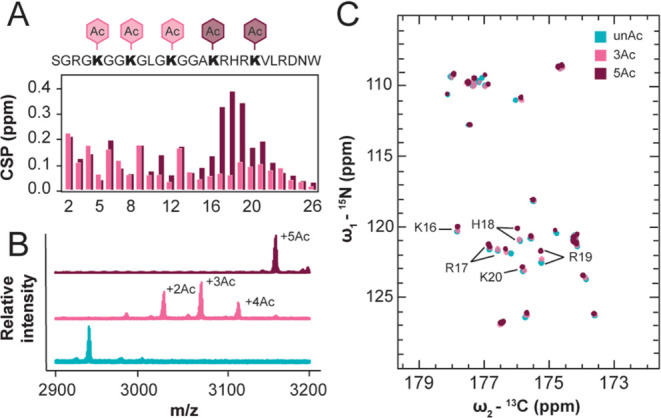
Chemical shift information changes in a biphasic manner for the 3Ac and 5Ac constructs compared to unAc. (A) Schematic of H4(1-25)W with different acetylation states, histogram of backbone CSPs for 3Ac and 5Ac with respect to unAc. (B) MALDI-MS of unAc, 3Ac, and 5Ac. (C) Overlay of unAc, 3Ac, and 5Ac 15N, 13C–CON spectra with basic patch residues indicated. NMR experiments were performed in 50 mM K2HPO4, pH 7.2, 150 mM KCl.
In addition to CSPs, a new cluster of resonances centered at 126.9 ppm 15N, 176.5 ppm 13C appear in the 3Ac and 5Ac 15N, 13C–CON spectra which can be attributed to the ε-nitrogen conjugated acetyl moieties since the acetyl-CoA donor is 13C labeled on both carbons of the acetyl group (Figure 2C). We attempted to resolve these resonances in the 5Ac 15N, 13C–CON spectrum by narrowing sweep width parameters and observed marginal improvement in resonance dispersion, but the resolution was not sufficient to enable deconvolution and unique assignment of each Kac resonance (Figure S2D). However, we were able to establish that no acetyl modifications were installed at the N-terminus of the WT H4-tail, a potential off target site for p300HAT, such that all observed CSPs can be confidently attributed to modification of the ε-nitrogen in lysine side chains. To accomplish this, we produced an H4(1-25)W(S1G) mutant that was acetylated at the N-terminus with ∼50% efficiency and observed emergence of a resonance centered at 26.1 ppm 13C, 184.1 ppm 13C’ in the 13C, 13C-CaCO spectrum attributable to the N-terminally conjugated acetyl moiety which was distinct from the resonances attributable to ε-nitrogen conjugated acetyl moieties centered at 24.6 ppm 13C, 176.5 ppm 13C’. The N-terminal acetyl resonance does not appear in the acetylated wildtype 13C, 13C-CaCO spectrum, confirming that there is no N-terminal acetylation of the wildtype H4 tail (Figure S2C). Together, these results demonstrate that H4Kac alters the chemical environment of many H4 tail residues, especially those in the basic patch, and establishes the foundation for further characterization of acetylation dependent changes to the conformational ensemble.
3.3. Acetylated H4 Tail Favors More Compact Conformers
Changes in chemical environment are strong predictors of changes in conformational ensemble which can present as measurable differences in average molecule size.70 Neutralization of lysine residues should reduce electrostatic repulsion, favoring more compact conformers in the acetylated H4 tail. With this in mind, we aimed to determine whether H4Kac leads to a change in molecule size by comparing the probability distribution and average radius of gyration (Rg) obtained through SAXS experiments and MD simulations for differentially acetylated H4 tail constructs. Note that while all experiments were performed with H4 tail constructs that included the exogenous tryptophan, all simulation constructs did not include the tryptophan.
The 3Ac and 5Ac tail ensembles favor more compact conformers compared to unAc. Guinier analysis of experimental SAXS profiles finds that unAc, 3Ac, and 5Ac have average Rg of 15.75 ± 0.17, 14.86 ± 0.18, and 14.02 ± 0.15 Å respectively. Crysol analysis to generate theoretical SAXS profiles reports comparable values with average Rg of 15.17 ± 0.08, 14.70 ± 0.10, and 13.60 ± 0.07 Å respectively (Figures 3A and S3B). This trend is also shown in the Rg probability distributions derived from the MD simulations (Figure 3B). Interestingly, the shape of the 5Ac probability distribution is uniquely skewed to the left. This suggests that the 5Ac ensemble may uniquely favor compact conformers that are mostly absent in the unAc and 3Ac ensembles. To a lesser extent, this is also reflected in the shape of the SAXS derived P(r) distributions, which show left-hand skewedness in the 5Ac distribution that is not as pronounced in 3Ac or unAc (Figure 3C). Existing predictions from MD simulations regarding changes in the Rg of the H4 tail with different acetylation states have shown disagreement in both direction and magnitude of the size change.31−34 Here, the direction and magnitude of acetylation dependent change in molecule size reported by our MD simulations are experimentally validated, which highlights the significance of improvements that were made to force field parameters in the years following prior simulations and establishes grounds for confidence in the finer molecular details that can be provided by these modern simulations.
3.4. Acetylation Increases Distal Intramolecular Contacts
We next wanted to consider the molecular mechanism(s) that lead to compaction of the H4 tail following acetylation. The decrease in molecule size predicted by MD and measured by SAXS could be attributed to globally distributed compaction, differential sampling of tertiary contacts, or altered propensity toward formation of secondary structure element.
With respect to tertiary contacts, acetylation increases intramolecular contacts between H4 tail residues separated by sequence distance. The MD simulations report an increase in distant intramolecular contacts for residues H4(5-22) in 3Ac compared to unAc, and these interactions are strengthened as well as expanded over the full tail sequence for 5Ac (Figure 4A). These data predict that H4KAc causes the tail to increase sampling of wrapped conformations that bring the N and C terminus in proximity. Informed by these predictions, we generated and acetylated uniformly 15N, 13C enriched H4(1-25)W V21C for conjugation with paramagnetic MTSSL and collection of 15N, 13C–CON paramagnetic relaxation enhancement experiments to determine which residues are proximal to the basic patch in unAc and 5Ac. We validated that the unAc and 5Ac 15N, 13C–CON assignments were transferrable to the V21C mutant and demonstrated complete MTSSL labeling efficiency of both samples by MALDI-MS (Figure S4A). The intensity of each resonance in the paramagnetic and diamagnetic spectra for unAc and 5Ac were then evaluated and compared (Figure S4B). When signal is lower in the paramagnetic sample than in the corresponding diamagnetic sample, this is attributed to line broadening due to magnetization transfer between the detected nuclei and the paramagnetic spin label, the magnitude of which increases as a function of proximity.
Figure 4.
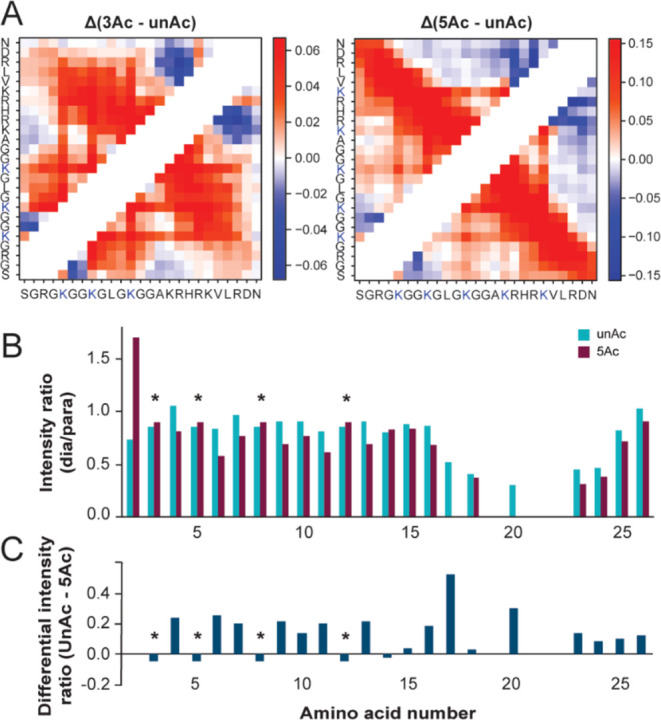
H4 tail acetylation increases intramolecular contacts. (A) Intramolecular contact map differences for 3Ac and 5Ac MD simulations compared to unAc. Color gradient values refer to the change in the fraction of simulation time each residue pair spent in contact, with positive values indicating increased contact. (B) PRE histograms of signal intensity ratios between paramagnetic and diamagnetic samples for unAc and 5Ac NMR samples. Asterisks mark resonances for which the reported intensity ratios are indistinguishable due to degenerate resonance positions. (C) Difference between the intensity ratios reported in panel B for unAc compared to 5Ac, positive values indicate increased proximity to the probe in the 5Ac NMR sample.
In the unAc experiments, there is a concerted decrease in the intensity ratio for residues 17, 18, 20, 23, and 24 as well as signal disappearance of 19, 21, and 22 in the paramagnetic spectrum (Figure 4B). This can be attributed to these amino acids’ closeness in sequence to the probe location at position 21. In the 5Ac experiments, residues 17, 19, 20, 21, and 22 disappear from the paramagnetic spectrum and the intensity ratio for 16, 18, 23, and 24 is decreased (Figure 4B). This indicates that the basic patch is more compact in the 5Ac sample. Additionally, many resonances N-terminal to the basic patch show slightly increased relaxation enhancement in the 5Ac sample (Figure 4C).
Note that the differential intensity ratio for G2 was excluded from Figure 4C, as its irregular line shape in the 5Ac spectra likely led to inaccurate integration. The resonances for R3, K5, K8 and K12 are not well enough resolved in these spectra to enable accurate quantitation, so the displayed intensity ratio and differential intensity ratio for these residues only represent their bulk properties (Figure S4C). Together, these computational and experimental data support the idea that acetylation increases tertiary contacts between sequentially distant amino acids.
3.5. Acetylation Minimally Alters Sampled Secondary Structure Elements
Acetylation-dependent nucleation of secondary structure could contribute to peptide compaction in addition to the tertiary contacts described above. Backbone and side chain chemical shift information can be used to predict secondary structure content in IDRs by referencing databases populated with chemical shift information from known disordered and structured protein regions. The chemical shift information arising from a nuclear correlation for a specific amino acid will differ in a predictable way depending on whether the amino acid is positioned in an unstructured region or a region with propensity for secondary structure. Considering the available chemical shift information, we were able to apply Δ2D to predict secondary structure in unAc and 5Ac (Figure S5A).56 Note that Δ2D does not provide quantitative error estimates associated with the predicted secondary structure propensity, but predictions for all residues were marked as reliable. This method did not predict differential sampling of secondary structure elements between unAc and 5Ac ensembles, and both ensembles were primarily random coil (Figure 5A). This means that if secondary structure elements are formed, their population is negligible such that the contribution to average chemical shift values is not distinguishable.
Figure 5.

Unacetylated and acetylated H4 tail ensembles are primarily disordered. (A) Average secondary structure fractions predicted by Δ2D based on unAc and 5Ac chemical shift information. (B) Mean fraction of total residues involved in the formation of various secondary structure elements calculated over three independent MD simulations for unAc, 3Ac, and 5Ac. Error bars denote standard error of the mean. (C) Secondary structure variation as a function of time in representative unAc (top) and 5Ac (bottom) simulations.
Accordingly, our MD simulations predict that the unacetylated and acetylated ensembles are primarily composed of random coil conformers, but the acetylated ensembles show marginal increases in propensity toward formation of α helix and β sheet secondary structure elements (Figures 5B and S5B). This result is consistent with the chemical shift-based predictions, which return primarily random coil assignment and show no meaningful difference between unAc and 5Ac fractions (Figures 5A and S5A). Examination of Dictionary of Secondary Structure of Proteins (DSSP) plots reveals that parallel β sheets are a persistent feature of the 5Ac ensemble as they are briefly sampled many times throughout the simulation (Figure 5C). This structural element is consistent with the PRE, where N-terminal residues are closer to the basic patch on average in the 5Ac sample. In contrast, the α helix is sampled infrequently and for a longer duration- suggesting that this conformer may be artificially trapped at a local minimum (Figure 5C). This isolated α helix is composed of amino acids 16-19, which have frequently been predicted to form more persistent helices (on the order of 20–80% content) in prior H4 tail simulations (Figure S5B).31,32 Comparatively, our simulations report low secondary structure populations (with more emphasis on β sheet than α helix character) that strongly agree with our experimental observations. Together with the PRE, these data support that 5Ac uniquely favors wrapped and β-strand-like conformations where the N-terminus transiently contacts the basic patch.
3.6. Protonation State of H18 Depends on the Acetylation State and Influences Ensemble Characteristics
The H4 tail basic patch contains four positively charged amino acid side chains surrounding H18 (KRHRK), two of which are neutralized following uniform H4Kac. We reasoned that the high positive charge density in unAc may cause the proton on the d-nitrogen of H18 to be more acidic compared to 5Ac so that H18 is more protonated in 5Ac. This could contribute to the significant CSPs in the 5Ac basic patch, as crosstalk between acetylation and histidine protonation would amplify the difference in chemical environment.
To determine whether the pKa of H18 differs between unAc and 5Ac, we performed NMR based pKa titrations across a pH range of 4.5–8.5. We collected a 15N, 13C–CON spectrum for each ∼0.25 pH increment in this range (Figure 6A). These data enabled us to fit CSP as a function of pH and estimate side chain pKa (Figure S6A). Three groups of CSPs were observed that are attributable to titration of the N-terminal amine (reported by CSP of residues 2-4), titration of the C-terminal carboxylic acid (reported by CSP of residues 25 and 26), and titration of the H18 side chain (reported by CSP of residues 18-21). The apparent pKa of H18 in the unAc context is 6.45, and 6.60 in 5Ac context (Figure 6B). Considering residues 19, 20, and 21 as additional reporters of the same pKa, the average shift is +0.1 upon acetylation, so H18 is slightly less acidic in the 5Ac sample (Figure S6A).
Figure 6.

H4 tail acetylation changes the pKa of H18, which in turn further informs ensemble characteristics. (A) 15N, 13C–CON spectra of unAc (left) and 5Ac (right) pH titrations. NMR experiments were performed in 50 mM K2HPO4, 150 mM KCl, with variable pH throughout the titration. (B) CSP vs pH fits for H18 in the unAc and 5Ac context. (C) Proton adjusted Rg probability distributions from MD simulations with average Rg values indicated. Errors of the Rg distributions are estimated using block averages with five blocks. (D) Mean fractions of total residues involved in the formation of various secondary structure elements calculated over three independent MD simulations for unAc, 5Ac, and protonated H18 of 5Ac. Error bars denote standard error of the mean. (E) Per residue difference in SASA from MD simulations, comparing the H18 protonated and deprotonated states. Errors represent the standard error of the mean calculated over three independent MD simulations.
Considering this, we revisited the MD simulations which had previously defined H18 as deprotonated in all cases (as would primarily be expected at biological pH). Intriguingly, protonation of H18 changes many previously established characteristics of the 5Ac ensemble; the average Rg increases from 13.10 to 14.31, the Rg probability distribution shifts to a more uniform shape, the low population secondary structure features are diminished, and the intramolecular contacts between the N-terminus and the basic patch are mitigated (Figures 6C,D and S6A, S6B). Thus, the ensemble features that result from 5Ac are partially mitigated by the corresponding increase in H18 pKa. This may also provide a conformational buffering mechanism, where the collapsed conformations favored by the 5Ac ensemble are buffered by the shifted pKa. This exposes an additional degree of complexity in the 5Ac ensemble behavior, as acetylated tails with a protonated H18 side chain can access more open conformations than those with the deprotonated side chain. Furthermore, analysis of solvent-accessible surface area (SASA) showed that H18 and other basic patch residues maintain more accessibility when H18 is protonated (Figures 6E and S6D). This is also true for several N-terminal residues, which further demonstrates that changes to basic patch chemistry translate to the N-terminus, potentially through the tertiary contacts established between the N-terminus and basic patch when the basic patch is acetylated.
4. Conclusions
Elucidating structure–function paradigms within IDRs has historically been challenging due to methodological limitations; the same is true for studying the biophysical implications of PTMs. Here, we demonstrate rigorous implementation of biophysical and computational methods to investigate how specific ensemble features of an IDR change when it is acetylated. We applied SAXS to investigate bulk ensemble properties and 13C direct-detect NMR to deconvolute spectral information for the disordered and repetitive histone H4-tail, enabling residue specific mapping of changes in the chemical environment, intramolecular contacts, and secondary structure propensity that arise when the tail is acetylated. Orthogonal to these powerful experimental techniques, we performed MD simulations with force field parameters optimized for disordered and acetylated systems which corroborated our experimental findings and elaborated on key molecular details. These details enable us to present a biophysical model for how acetylation state defines important ensemble features of the H4 tail.
Progressive acetylation of the H4 tail leads to increased compaction, with mild CSPs presenting in residues 2-13 when K5, K8, and K12 are acetylated compared to more pronounced CSPs in residues 16-21 when K16 and K20 are additionally acetylated. MD provided insight that intramolecular contacts are favored between N-terminal residues and the basic patch when the tail is acetylated at all five lysine residues compared to just three, so acetylation of K16 and K20 specifically promote folding of the tail back on itself, reminiscent of a β-strand conformation. PRE experiments support this model, though chemical shift and DSSP based structure predictions agree that the population of strict β sheet elements is diminutive even in the 5Ac ensemble such that the compact conformers remain loose and labile. Beyond acetylation at K16 and K20, we found that presentation of the 5Ac-specific ensemble features depends on the protonation state of H18, such that key features are ablated if H18 is protonated. Interestingly, the pKa of H18 shifts higher when the H4-tail is acetylated, so the presence of H18 in the center of the basic patch may present a conformational buffer against entrapment in collapsed conformations, which is supported by the increase in solvent-accessible surface area when H18 is protonated. This provides an elegant example of the chemical complexity that can define ensemble behavior in post-translationally modified IDRs where the interplay between a modification and sequence context can have unexpected outcomes.
While these experiments were performed on the H4 tail in isolation from the nucleosome context, these findings have interesting implications for chromatin structure if the principles translate. In particular, the compact conformations favored by the 5Ac ensemble could underly established observations that H4KAc abrogates assembly of chromatin arrays into higher order structures- the acetylated tail may not readily establish assembly mediating interactions with adjacent nucleosomes due to electrostatics, distance constraints, or competitive intramolecular interactions. Thus, the biophysical characteristics described here may provide the molecular details that link H4Kac with regulation of chromatin architecture and enrichment in open chromatin regions.
Acknowledgments
This work was supported by the National Institutes of Health through awards R21-GM137129 to S.A.S., R35GM153388 to J.M. Additionally, J.M. was supported by Welch Foundation grant A-2113-20220331. Support for S.M.D. was provided by the National Institutes of Health training grant T32GM102057. The small-angle X-ray scattering research was supported by instrumentation purchased through National Institutes of Health grant S10-OD028589 to Dr. Neela Yennawar. Mass spectrometric analyses were performed at the Penn State Proteomics and Mass Spectrometry Core Facility, University Park, PA (RRID:SCR_024462). The authors thank Dr. Tatiana Laremore for providing access to the equipment and for technical advice. SAXS and SEC-MALS experiments were performed at the Penn State X-ray Crystallography Core Facility. The authors thank Dr. Neela Yennawar and Julia Fecko for providing access to the equipment, technical advice, and assistance with data collection and analysis. Atomistic MD simulations were conducted with the computing resources provided by Texas A&M High Performance Research Computing (HPRC).
Glossary
Abbreviations
- IDR
intrinsically disordered region
- PTM
post-translational modification
- H4Kac
histone H4 tail acetylation
- NMR
nuclear magnetic resonance
- Kac
lysine acetylation
- TEV
tobacco etch virus
- LIC
ligation independent cloning
- E. coli
Escherichia coli
- LB
Luria broth
- TB
terrific broth
- MWCO
molecular weight cutoff
- SAXS
small-angle X-ray scattering
- SEC
size exclusion chromatography
- MALS
multiangle light scattering
- MD
molecular dynamics
- CSP
chemical shift perturbation
- Rg
radius of gyration
- MALDI-MS
matrix-assisted laser desorption ionization mass spectrometry
- PRE
paramagnetic resonance enhancement
- SASA
solvent-accessible surface area
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jpcb.4c05701.
Additional experimental results including NMR and MALDI-MS, titration summary tables, per residue secondary structure fractions, and raw SASA and contact maps (DOC). DSS referenced chemical shift information for unAc and 5Ac H4(1-25)W in 50 mM K2HPO4, 150 mM potassium chloride (NMR-STAR) (PDF)
Author Contributions
S.M.D. provided the first version of the manuscript. The manuscript was revised through contributions of all authors. S.M.D. performed NMR, MS, and SAXS experiments and corresponding analyses. E.J.K. performed NMR experiments and corresponding analyses. T.M.P. performed simulations and corresponding analyses. S.A.S. and J.M. provided mentorship and funding support for this work. All authors have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Xie H. B.; Vucetic S.; Iakoucheva L. M.; Oldfield C. J.; Dunker A. K.; Obradovic Z.; Uversky V. N. Functional Anthology of Intrinsic Disorder. 3. Ligands, Post-Translational Modifications, and Diseases Associated with Intrinsically Disordered Proteins. J. Proteome Res. 2007, 6, 1917–1932. 10.1021/pr060394e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cermakova K.; Hodges H. C. Interaction Modules That Impart Specificity to Disordered Protein. Trends Biochem. Sci. 2023, 48, 477–490. 10.1016/j.tibs.2023.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maeshima K.; Iida S.; Tamura S. Physical Nature of Chromatin in the Nucleus. Cold Spring Harbor Perspect. Biol. 2021, 13, a040675 10.1101/cshperspect.a040675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sehrawat P.; Shobhawat R.; Kumar A. Catching Nucleosome by Its Decorated Tails Determines Its Functional States. Front. Genet. 2022, 13, 903923 10.3389/fgene.2022.903923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strahl B. D.; Allis C. D. The Language of Covalent Histone Modifications. Nature 2000, 403, 41–45. 10.1038/47412. [DOI] [PubMed] [Google Scholar]
- Rothbart S. B.; Strahl B. D. Interpreting the Language of Histone and DNA Modifications. Biochim. Biophys. Acta 2014, 1839, 627–643. 10.1016/j.bbagrm.2014.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Musselman C. A.; Kutateladze T. G. Characterization of Functional Disordered Regions within Chromatin-Associated Proteins. iScience 2021, 24, 102070 10.1016/j.isci.2021.102070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holehouse A. S.; Kragelund B. B. The Molecular Basis for Cellular Function of Intrinsically Disordered Protein Regions. Nat. Rev. Mol. Cell Biol. 2023, 25, 187–211. 10.1038/s41580-023-00673-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camacho-Zarco A. R.; Schnapka V.; Guseva S.; Abyzov A.; Adamski W.; Milles S.; Jensen M. R.; Zidek L.; Salvi N.; Blackledge M. Nmr Provides Unique Insight into the Functional Dynamics and Interactions of Intrinsically Disordered Proteins. Chem. Rev. 2022, 122, 9331–9356. 10.1021/acs.chemrev.1c01023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chong S. H.; Ham S. Folding Free Energy Landscape of Ordered and Intrinsically Disordered Proteins. Sci. Rep. 2019, 9, 14927 10.1038/s41598-019-50825-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das R. K.; Ruff K. M.; Pappu R. V. Relating Sequence Encoded Information to Form and Function of Intrinsically Disordered Proteins. Curr. Opin. Struct. Biol. 2015, 32, 102–112. 10.1016/j.sbi.2015.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moses D.; Ginell G. M.; Holehouse A. S.; Sukenik S. Intrinsically Disordered Regions Are Poised to Act as Sensors of Cellular Chemistry. Trends Biochem. Sci. 2023, 48, 1019–1034. 10.1016/j.tibs.2023.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai J.; Hyland E. M.; Yuan D. S.; Huang H.; Bader J. S.; Boeke J. D. Probing Nucleosome Function: A Highly Versatile Library of Synthetic Histone H3 and H4Mutants. Cell 2008, 134, 1066–1078. 10.1016/j.cell.2008.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang H.; Maertens A. M.; Hyland E. M.; Dai J.; Norris A.; Boeke J. D.; Bader J. S. Histonehits: A Database for Histone Mutations and Their Phenotypes. Genome Res. 2009, 19, 674–681. 10.1101/gr.083402.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Espinoza Pereira K. N.; Shan J.; Licht J. D.; Bennett R. L. Histone Mutations in Cancer. Biochem. Soc. Trans. 2023, 51, 1749–1763. 10.1042/BST20210567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nacev B. A.; Feng L.; Bagert J. D.; Lemiesz A. E.; Gao J.; Soshnev A. A.; Kundra R.; Schultz N.; Muir T. W.; Allis C. D. The Expanding Landscape of ’Oncohistone’ Mutations in Human Cancers. Nature 2019, 567, 473–478. 10.1038/s41586-019-1038-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowman G. D.; Poirier M. G. Post-Translational Modifications of Histones That Influence Nucleosome Dynamics. Chem. Rev. 2015, 115, 2274–2295. 10.1021/cr500350x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amatori S.; Tavolaro S.; Gambardella S.; Fanelli M. The Dark Side of Histones: Genomic Organization and Role of Oncohistones in Cancer. Clin. Epigenet. 2021, 13, 71 10.1186/s13148-021-01057-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrison E. A.; Bowerman S.; Sylvers K. L.; Wereszczynski J.; Musselman C. A. The Conformation of the Histone H3 Tail Inhibits Association of the Bptf Phd Finger with the Nucleosome. eLife 2018, 7, e31481 10.7554/eLife.31481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jennings C. E.; Zoss C. J.; Morrison E. A. Arginine Anchor Points Govern H3 Tail Dynamics. Front. Mol. Biosci. 2023, 10, 1150400 10.3389/fmolb.2023.1150400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furukawa A.; Wakamori M.; Arimura Y.; Ohtomo H.; Tsunaka Y.; Kurumizaka H.; Umehara T.; Nishimura Y. Acetylated Histone H4 Tail Enhances Histone H3 Tail Acetylation by Altering Their Mutual Dynamics in the Nucleosome. Proc. Natl. Acad. Sci. U.S.A. 2020, 117, 19661–19663. 10.1073/pnas.2010506117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kundu T. K.; Palhan V. B.; Wang Z.; An W.; Cole P. A.; Roeder R. G. Activator-Dependent Transcription from Chromatin in Vitro Involving Targeted Histone Acetylation by P300. Mol. Cell 2000, 6, 551–561. 10.1016/S1097-2765(00)00054-X. [DOI] [PubMed] [Google Scholar]
- Shia W. J.; Pattenden S. G.; Workman J. L. Histone H4 Lysine 16 Acetylation Breaks the Genome’s Silence. Genome Biol. 2006, 7, 217. 10.1186/gb-2006-7-5-217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shogren-Knaak M.; Ishii H.; Sun J. M.; Pazin M. J.; Davie J. R.; Peterson C. L. Histone H4-K16 Acetylation Controls Chromatin Structure and Protein Interactions. Science 2006, 311, 844–847. 10.1126/science.1124000. [DOI] [PubMed] [Google Scholar]
- Dorigo B.; Schalch T.; Bystricky K.; Richmond T. J. Chromatin Fiber Folding: Requirement for the Histone H4 N-Terminal Tail. J. Mol. Biol. 2003, 327, 85–96. 10.1016/S0022-2836(03)00025-1. [DOI] [PubMed] [Google Scholar]
- Sinha D.; Shogren-Knaak M. A. Role of Direct Interactions between the Histone H4 Tail and the H2a Core in Long Range Nucleosome Contacts. J. Biol. Chem. 2010, 285, 16572–16581. 10.1074/jbc.M109.091298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalashnikova A. A.; Porter-Goff M. E.; Muthurajan U. M.; Luger K.; Hansen J. C. The Role of the Nucleosome Acidic Patch in Modulating Higher Order Chromatin Structure. J. R. Soc., Interface 2013, 10, 20121022 10.1098/rsif.2012.1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song F.; Chen P.; Sun D.; Wang M.; Dong L.; Liang D.; Xu R. M.; Zhu P.; Li G. Cryo-Em Study of the Chromatin Fiber Reveals a Double Helix Twisted by Tetranucleosomal Units. Science 2014, 344, 376–380. 10.1126/science.1251413. [DOI] [PubMed] [Google Scholar]
- Derenzini M.; Olins A. L.; Olins D. E. Chromatin Structure in Situ: The Contribution of DNA Ultrastructural Cytochemistry. Eur. J. Histochem. 2014, 58, 2307. 10.4081/ejh.2014.2307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ou H. D.; Phan S.; Deerinck T. J.; Thor A.; Ellisman M. H.; O’Shea C. C. Chromemt: Visualizing 3d Chromatin Structure and Compaction in Interphase and Mitotic Cells. Science 2017, 357 (6349), aag0025 10.1126/science.aag0025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang D.; Arya G. Structure and Binding of the H4 Histone Tail and the Effects of Lysine 16 Acetylation. Phys. Chem. Chem. Phys. 2011, 13, 2911–2921. 10.1039/C0CP01487G. [DOI] [PubMed] [Google Scholar]
- Winogradoff D.; Echeverria I.; Potoyan D. A.; Papoian G. A. The Acetylation Landscape of the H4 Histone Tail: Disentangling the Interplay between the Specific and Cumulative Effects. J. Am. Chem. Soc. 2015, 137, 6245–6253. 10.1021/jacs.5b00235. [DOI] [PubMed] [Google Scholar]
- Zhang R.; Erler J.; Langowski J. Histone Acetylation Regulates Chromatin Accessibility: Role of H4k16 in Inter-Nucleosome Interaction. Biophys. J. 2017, 112, 450–459. 10.1016/j.bpj.2016.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shabane P. S.; Onufriev A. V. Significant Compaction of H4 Histone Tail Upon Charge Neutralization by Acetylation and Its Mimics, Possible Effects on Chromatin Structure. J. Mol. Biol. 2021, 433, 166683 10.1016/j.jmb.2020.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraser O. A.; Namitz K. E. W.; Showalter S. A. Advances in Direct Detection of Lysine Methylation and Acetylation by Nuclear Magnetic Resonance Using (13)C-Enriched Cofactors. Methods 2023, 218, 72–83. 10.1016/j.ymeth.2023.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozenne V.; Bauer F.; Salmon L.; Huang J. R.; Jensen M. R.; Segard S.; Bernado P.; Charavay C.; Blackledge M. Flexible-Meccano: A Tool for the Generation of Explicit Ensemble Descriptions of Intrinsically Disordered Proteins and Their Associated Experimental Observables. Bioinformatics 2012, 28, 1463–1470. 10.1093/bioinformatics/bts172. [DOI] [PubMed] [Google Scholar]
- Estrada J.; Bernado P.; Blackledge M.; Sancho J. Protsa: A Web Application for Calculating Sequence Specific Protein Solvent Accessibilities in the Unfolded Ensemble. BMC Bioinf. 2009, 10, 104 10.1186/1471-2105-10-104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khoury G. A.; Thompson J. P.; Smadbeck J.; Kieslich C. A.; Floudas C. A. Forcefield_Ptm: Ab Initio Charge and Amber Forcefield Parameters for Frequently Occurring Post-Translational Modifications. J. Chem. Theory Comput. 2013, 9, 5653–5674. 10.1021/ct400556v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Best R. B.; Zheng W.; Mittal J. Balanced Protein-Water Interactions Improve Properties of Disordered Proteins and Non-Specific Protein Association. J. Chem. Theory Comput. 2014, 10, 5113–5124. 10.1021/ct500569b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abascal J. L. F.; Vega C. A General Purpose Model for the Condensed Phases of Water: Tip4p/2005. J. Chem. Phys. 2005, 123, 234505 10.1063/1.2121687. [DOI] [PubMed] [Google Scholar]
- Abraham M. J.; Murtola T.; Schulz R.; Pall S.; Smith J. C.; Hess B.; Lindahl E. Gromacs: High Performance Molecular Simulations through Mulit-Level Parallelism from Laptops to Supercomputers. SoftwareX 2015, 1–2, 19–25. 10.1016/j.softx.2015.06.001. [DOI] [Google Scholar]
- Luo Y.; Roux B. Simulation of Osmotic Pressure in Concentrated Aqueous Salt Solutions. J. Phys. Chem. Lett. 2010, 1, 183–189. 10.1021/jz900079w. [DOI] [Google Scholar]
- Evans D. J.; Holian B. L. The Nose-Hoover Thermostat. J. Chem. Phys. 1985, 83, 4069–4074. 10.1063/1.449071. [DOI] [Google Scholar]
- Berendsen H. J. C.; Postma J. P. M.; Vangunsteren W. F.; Dinola A.; Haak J. R. Molecular-Dynamics with Coupling to an External Bath. J. Chem. Phys. 1984, 81, 3684–3690. 10.1063/1.448118. [DOI] [Google Scholar]
- Shirts M. R.; Klein C.; Swails J. M.; Yin J.; Gilson M. K.; Mobley D. L.; Case D. A.; Zhong E. D. Lessons Learned from Comparing Molecular Dynamics Engines on the Sampl5 Dataset. J. Comput.-Aided Mol. Des. 2017, 31, 147–161. 10.1007/s10822-016-9977-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shirts M. R.; Klein C.; Swails J. M.; Yin J.; Gilson M. K.; Mobley D. L.; Case D. A.; Zhong E. D. Erratum To: Lessons Learned from Comparing Molecular Dynamics Engines on the Sampl5 Dataset. J. Comput.-Aided Mol. Des. 2017, 31, 777. 10.1007/s10822-017-0043-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hopkins C. W.; Le Grand S.; Walker R. C.; Roitberg A. E. Long-Time-Step Molecular Dynamics through Hydrogen Mass Repartitioning. J. Chem. Theory Comput 2015, 11, 1864–1874. 10.1021/ct5010406. [DOI] [PubMed] [Google Scholar]
- Ryckaert J. P.; Ciccotti G.; Berendsen H. J. C. Numerical-Integration of Cartesian Equations of Motion of a System with Constraints - Molecular-Dynamics of N-Alkanes. J. Comput. Phys. 1977, 23, 327–341. 10.1016/0021-9991(77)90098-5. [DOI] [Google Scholar]
- Darden T.; York D.; Pedersen L. Particle Mesh Ewald - an N.Log(N) Method for Ewald Sums in Large Systems. J. Chem. Phys. 1993, 98, 10089–10092. 10.1063/1.464397. [DOI] [Google Scholar]
- Essmann U.; Perera L.; Berkowitz M. L.; Darden T.; Lee H.; Pedersen L. G. A Smooth Particle Mesh Ewald Method. J. Chem. Phys. 1995, 103, 8577–8593. 10.1063/1.470117. [DOI] [Google Scholar]
- Skriver K.; Kragelund B. B.. Intrinsically Disordered Proteins Methods and Protocols. 2020; Vol. 2141. [DOI] [PubMed]
- Cook E. C.; Usher G. A.; Showalter S. A. The Use of (13)C Direct-Detect Nmr to Characterize Flexible and Disordered Proteins. Methods Enzymol. 2018, 611, 81–100. 10.1016/bs.mie.2018.08.025. [DOI] [PubMed] [Google Scholar]
- Markley J. L.; Ulrich E. L.; Berman H. M.; Henrick K.; Nakamura H.; Akutsu H. Biomagresbank (Bmrb) as a Partner in the Worldwide Protein Data Bank (Wwpdb): New Policies Affecting Biomolecular Nmr Depositions. J. Biomol. NMR 2008, 40, 153–155. 10.1007/s10858-008-9221-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee W.; Tonelli M.; Markley J. L. Nmrfam-Sparky: Enhanced Software for Biomolecular Nmr Spectroscopy. Bioinformatics 2015, 31, 1325–1327. 10.1093/bioinformatics/btu830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harms M. J.; Castaneda C. A.; Schlessman J. L.; Sue G. R.; Isom D. G.; Cannon B. R.; Garcia-Moreno E. B. The Pk(a) Values of Acidic and Basic Residues Buried at the Same Internal Location in a Protein Are Governed by Different Factors. J. Mol. Biol. 2009, 389, 34–47. 10.1016/j.jmb.2009.03.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camilloni C.; De Simone A.; Vranken W. F.; Vendruscolo M. Determination of Secondary Structure Populations in Disordered States of Proteins Using Nuclear Magnetic Resonance Chemical Shifts. Biochemistry 2012, 51, 2224–2231. 10.1021/bi3001825. [DOI] [PubMed] [Google Scholar]
- Franke D.; Petoukhov M. V.; Konarev P. V.; Panjkovich A.; Tuukkanen A.; Mertens H. D. T.; Kikhney A. G.; Hajizadeh N. R.; Franklin J. M.; Jeffries C. M.; Svergun D. I. Atsas 2.8: A Comprehensive Data Analysis Suite for Small-Angle Scattering from Macromolecular Solutions. J. Appl. Crystallogr. 2017, 50, 1212–1225. 10.1107/S1600576717007786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabsch W.; Sander C. Dictionary of Protein Secondary Structure: Pattern Recognition of Hydrogen-Bonded and Geometrical Features. Biopolymers 1983, 22, 2577–2637. 10.1002/bip.360221211. [DOI] [PubMed] [Google Scholar]
- Zheng W.; Dignon G. L.; Jovic N.; Xu X.; Regy R. M.; Fawzi N. L.; Kim Y. C.; Best R. B.; Mittal J. Molecular Details of Protein Condensates Probed by Microsecond Long Atomistic Simulations. J. Phys. Chem. B 2020, 124, 11671–11679. 10.1021/acs.jpcb.0c10489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Her C.; Phan T. M.; Jovic N.; Kapoor U.; Ackermann B. E.; Rizuan A.; Kim Y. C.; Mittal J.; Debelouchina G. T. Molecular Interactions Underlying the Phase Separation of Hp1alpha: Role of Phosphorylation, Ligand and Nucleic Acid Binding. Nucleic Acids Res. 2022, 50, 12702–12722. 10.1093/nar/gkac1194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phan T. M.; Kim Y. C.; Debelouchina G. T.; Mittal J. Interplay between Charge Distribution and DNA in Shaping Hp1 Paralog Phase Separation and Localization. eLife 2024, 12, RP90820 10.7554/eLife.90820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisenhaber F.; Lijnzaad P.; Argos P.; Sander C.; Scharf M. The Double Cubic Lattice Method - Efficient Approaches to Numerical-Integration of Surface-Area and Volume and to Dot Surface Contouring of Molecular Assemblies. J. Comput. Chem. 1995, 16, 273–284. 10.1002/jcc.540160303. [DOI] [Google Scholar]
- Gibbs E. B.; Cook E. C.; Showalter S. A. Application of Nmr to Studies of Intrinsically Disordered Proteins. Arch. Biochem. Biophys. 2017, 628, 57–70. 10.1016/j.abb.2017.05.008. [DOI] [PubMed] [Google Scholar]
- Marunde M. R.; Fuchs H. A.; Burg J. M.; Popova I. K.; Vaidya A.; Hall N. W.; Weinzapfel E. N.; Meiners M. J.; Watson R.; Gillespie Z. B.; et al. Nucleosome Conformation Dictates the Histone Code. eLife 2024, 13, e78866 10.7554/eLife.78866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou B. R.; Feng H.; Ghirlando R.; Kato H.; Gruschus J.; Bai Y. Histone H4 K16q Mutation, an Acetylation Mimic, Causes Structural Disorder of Its N-Terminal Basic Patch in the Nucleosome. J. Mol. Biol. 2012, 421, 30–37. 10.1016/j.jmb.2012.04.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahu D.; Bastidas M.; Showalter S. A. Generating Nmr Chemical Shift Assignments of Intrinsically Disordered Proteins Using Carbon-Detected Nmr Methods. Anal. Biochem. 2014, 449, 17–25. 10.1016/j.ab.2013.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mittal A.; Holehouse A. S.; Cohan M. C.; Pappu R. V. Sequence-to-Conformation Relationships of Disordered Regions Tethered to Folded Domains of Proteins. J. Mol. Biol. 2018, 430, 2403–2421. 10.1016/j.jmb.2018.05.012. [DOI] [PubMed] [Google Scholar]
- Taneja I.; Holehouse A. S. Folded Domain Charge Properties Influence the Conformational Behavior of Disordered Tails. Curr. Res. Struct. Biol. 2021, 3, 216–228. 10.1016/j.crstbi.2021.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kosol S.; Contreras-Martos S.; Cedeno C.; Tompa P. Structural Characterization of Intrinsically Disordered Proteins by Nmr Spectroscopy. Molecules 2013, 18, 10802–10828. 10.3390/molecules180910802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sibille N.; Bernado P. Structural Characterization of Intrinsically Disordered Proteins by the Combined Use of Nmr and Saxs. Biochem. Soc. Trans. 2012, 40, 955–962. 10.1042/BST20120149. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.