

Abstract
Interest in the observation and characterization of organic isomers in astronomical environments has grown rapidly with an increase in the sensitivity of detection techniques. Accurate modeling and interpretation of these environments require experimental isomer-specific reactivity and spectroscopic measurements. Given the abundance of formaldehyde (H2CO) in various astrophysical objects, the properties and reactivities of its cation isomers H2CO•+ and HCOH•+ are of significant interest. However, for the hydroxymethylene radical cation HCOH•+ (and its isotopologue DCOH•+), detailed reactivity studies have been limited by the lack of suitable experimental methods to generate this isomer with high purity. Here, potential approaches to the isomer-selective generation of HCOH•+ and DCOH•+ are characterized through differential reactivity measurements. While the dissociative photoionization of cyclopropanol (c-CH2CH2CHOH) is determined to be unsuitable, the dissociation of methanol-d3 (CD3OH) allows for the formation of DCOH•+ with a fractional abundance of >99% at photon energies below 14.8 eV. These results will allow future spectroscopic and reactivity measurements of HCOH•+/DCOH•+ to be conducted, laying the groundwork for future detection and incorporation into models of the interstellar medium.
Following the determination of the relative abundances of the HCO+/HOC+ and HCN•+/HNC•+ isomeric ion pairs in the interstellar medium (ISM),1−4 there has been a growing acknowledgment of the importance of isomerism and experimental isomer-specific reactivity measurements in astrochemistry.5,6 Isomerization barriers that are sufficiently small to allow for isomers to exist in equilibrium under terrestrial conditions can be prohibitive at the lower temperatures present in many astrochemical environments, such as dark clouds. Conversely, for some species, the higher energy isomer is found to be the most abundant, a situation that is unlikely under terrestrial conditions. As such, the differing chemical and spectroscopic properties of isomers necessitate isomer-specific laboratory measurements in order to accurately model and interpret the chemistry of astronomical environments.
Isomer-selective reactivity has been shown to be of particular importance to the modeling of planetary and satellite atmospheres, with a notable example being the contribution of different [C3H3]+ isomers to the chemistry of Titan, the largest moon of Saturn.7 Furthermore, isomer-selective generation is also important for fundamental gas-phase kinetics measurements,8−11 as the differing reactivities of isomers can provide important insights into the interrelationship between structure and reactivity.12,13 This is especially significant for small radical cations where subtle changes in structure can lead to significantly different reactivity.14−19
Neutral formaldehyde (H2CO) has long been known to be present in the ISM,20 where it is routinely detected in large and varied range of astrophysical objects, from diffuse clouds21 to protoplanetary disks22 and protoplanetary nebulae23 (see Appendix A in ref (24) for a list of objects where formaldehyde and its isotopologues have been detected). With an ionization energy (IE) of 10.9 eV,25,26 it is anticipated that H2CO will be readily ionized by UV photons, cosmic rays, and high-energy electrons. As the anticipated product ions, H2CO•+ and HCOH•+, are yet to be explicitly detected in the ISM, likely due to the highly transient nature of radical cations even in such low density environments, the need for precise laboratory-based studies of these species is clear. While the formaldehyde radical cation (H2CO•+) is the most stable [CH2O]•+ isomer, the trans and cis HCOH•+ isomers are only 0.3 and 0.44 eV higher in energy, respectively.27 A third isomer, COH2•+, will probably play a minor role in cold environments since it is more than 2 eV higher in energy than the H2CO•+ and HCOH•+ isomers,28 and so it is not considered further here.
Importantly, the barrier to isomerization between H2CO•+ and trans HCOH•+ of 1.97 eV is higher in energy than that for the fragmentation into HCO+ plus H, meaning that interconversion over this barrier is highly unlikely. Furthermore, a recent computational study29 determined the tunneling lifetime of the HCOH•+ isomer to be on the order of thousands of years at representative ISM temperatures. Given this, formation mechanisms such as the protonation of HCO,30 radical hydrogen abstraction from H2COH+,31 or dissociative charge transfer reactions with methanol32,33 have the potential to form a mix of the two [CH2O]•+ isomers under the low temperature conditions typical of the ISM.
While the production of H2CO•+ (e.g., by ionization of H2CO) has been amply investigated,34,35 the generation of HCOH+• (and its isotopologue DCOH+•) is less straightforward and has been the subject of significant research interest due to its implications for both astrochemistry and fundamental reaction dynamics. Furthermore, it is notable that the characterization of the isomer-selective generation of neutral HCOH has been reported only very recently.36 While the dissociative ionizations of cyclopropanol (c-CH2CH2CH(OH)),37,38 methanol (CH3OH),38−40 and trideuterated methanol (CD3OH)17,38 have been suggested as suitable methods for generating the HCOH+• isomer, or its partially deuterated isotopologue DCOH•+, no quantitative experimental characterization of the isomeric purity has yet been performed. Here, we present experimental results on the characterization of the isomeric purity of HCOH+• and DCOH+• formed from both cyclopropanol and CD3OH for use in future ion-molecule reactivity studies.
Measurements have been performed using a guided ion beam tandem mass spectrometer, allowing for ions formed in the source chamber to be mass-selected prior to introduction into the reaction cell. By monitoring the pressure in the cell and the intensity of different parent and product mass channels, absolute cross sections (CSs) and branching ratios (BRs) can be obtained for the reactions of different parent ions. Measurements are recorded as a function of both the energy of the photons used in the dissociative photoionization process and the collision energy available to the reactants. Tunable VUV sources, in this case the DESIRS beamline41 of the SOLEIL synchrotron radiation facility, allow for a high level of control over the photon energy. This, in turn, allows for the tuning of the internal energy of the ions formed and, by extension, the fragmentation dynamics. Full experimental details, including a discussion of the uncertainties associated with the measurements reported here, are given in the Supporting Information.
For a source generating a mixture of two different species A and B (either isomers or entirely different chemical species), the observed reaction CS of the mixture, σT, can be described by the following equation
| 1 |
where nA and nB are the fractions of A and B, respectively, while σA and σB are the absolute reaction CSs of pure samples of A and B, respectively. If σA and σB are known, then nA and nB can be determined for a given σT. Here, we have employed this approach to determine the isomeric purity, i.e., the relative yields of the HCOH•+ and H2CO•+ isomers from different generation methods. However, due to the lack of isomer-selective reactivity data for HCOH•+ and DCOH•+, this quantification must be performed through the subtraction of the H2CO•+ fractional abundance, for which isomer-selective generation is well-established.
The ionization of cyclopropanol has previously been shown to produce an intense m/z 30 fragment at 1–2.5 eV above the ionization threshold of 9.10 eV.37,42 Though the m/z 30 fragment was initially assigned exclusively to the formation of HCOH•+ in combination with C2H4,37 a more recent study42 provided an alternative assigment—attributing it instead to the formation of C2H6•+ in combination with CO, though we note that the HCOH•+ isomer is not discussed in this study. The reattribution to C2H6•+ plus CO was based on the threshold energy (1.47 eV above the 9.10 eV ionization energy of cyclopropanol) being in agreement with the calculated dissociation limit of 10.57 eV, while fragmentation into H2CO•+ plus C2H4 has a higher dissociation limit of 11.35 eV. A further study38 noted that, while the m/z 30 fragment is a doublet of C2H6•+ and [CH2O]•+, the [CH2O]•+ component is consistent with the HCOH•+ isomer.
In order to benchmark our results against this previous study,42 the appearance energies (AEs) of the relevant fragments have been measured from their respective photoionization efficiency (PIE) curves, with the results for both the parent ion (m/z 58) and the m/z 29 and 30 fragments shown in Figure 1. For the m/z 29 and 30 fragments this has been performed via linear threshold extrapolation of the PIE curve,43−46 while, due to the lack of a well-defined onset, the AE for the m/z 58 parent ion has been determined as the first point above noise. We note that the AE values given here, and elsewhere in this work, are used only for comparison with AEs determined previously in the literature and not to derive relevant thermochemical values. For a complete discussion on the fitting of AE thresholds, please refer to Roithová et al.,43 Ruscic,47 and the references therein. For the m/z 58 ion, the measured AE of 9.15 ± 0.12 eV is in good agreement with the previously measured IE of 9.10 eV.42 Similarly, the AE of the m/z 30 fragment of 10.58 ± 0.04 eV is in excellent agreement with the previous measurement of 10.57 eV.42 Finally, the AE for the m/z 29 channel, which has previously been assigned to a mix of C2H5+ and HCO+, is measured as 10.94 ± 0.05 eV, in reasonable agreement with the previously reported AEs of 10.75 and 10.88 eV for the two channels, respectively.42 Notably, the m/z 29 channel shows a sharp rise in intensity above ∼12 eV, in agreement with the observation of the previous study,42 where it was assigned in part to the opening of the channel leading to the formation of C2H5+ in combination with CO and H•, which has an AE of 11.49 eV.42
Figure 1.

Photoionization efficiency (PIE) curves for the dissociative photoionization of cyclopropanol giving [C3H6O]+ (m/z 58, black squares), [CHO]+/C2H5+ (m/z 29, orange circles), and [CH2O]•+/C2H6•+ (m/z 30, blue triangles). Insets a), b), and c) show the threshold regions for the corresponding m/z values, along with the assigned appearence energies (AEs).
In order to reduce any internal energy effects while maintaining a sufficient ion yield at m/z 30, the reactivity experiments discussed below are performed at a photon energy of 11.2 eV. C2D4 is chosen as the neutral reactant to disentangle the presence of C2H6•+ from that of [CH2O]•+. The reaction of C2H6•+ with C2H4 has been studied previously,48 and the sole m/z 28 product (C2H4•+), corresponding to the charge transfer, is observed with a rate of (1.15 ± 0.12) × 10–9 cm3·molecule–1·s–1. C2D4 is chosen in preference to its undeuterated equivalent (C2H4) to simplify the analysis by partially deuterating product species that would otherwise be equivalent as, while the charge transfer reaction is also energetically accessible for the H2CO•+ ion, a previous study of the reaction of this ion with C2D4 observed not only the charge transfer product (m/z 32, C2D4•+), but also a distinctive combination of proton (m/z 33, C2D4H•+) and hydrogen atom transfer (m/z 29, HCO+) channels.16 As the BRs, and their relative collision energy dependencies, have been reported in detail previously,16 consideration of the reaction of the m/z 30 fragment with C2D4 therefore allows for discrimination between the relative contributions of the C2H6•+, H2CO•+, and HCOH•+ ions. Details of the data treatment are provided in the Supporting Information, with the resulting absolute CSs as a function of the collision energy given in Figure 2.
Figure 2.
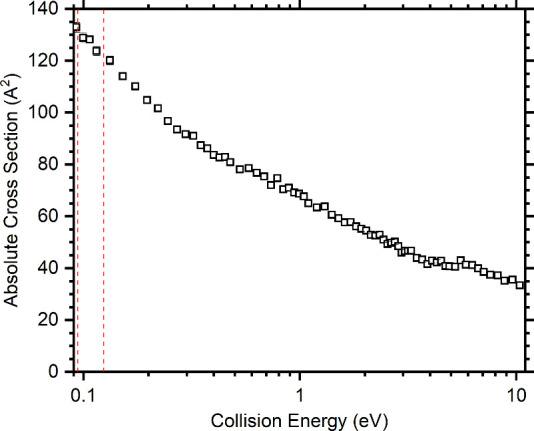
Summed absolute cross sections (CSs) as a function of the collision energy for the m/z 32 charge transfer product C2D4•+ and the corresponding secondary products at m/z 46, 62, 74, and 78, following the reaction of the m/z 30 fragment from the dissociative photoionization of cyclopropanol with C2D4 at Ephot = 11.2 eV. The red dashed lines represent the collision energy uncertainty range for the cross section used for the rate calculations, as detailed in the text.
Given CSs at a fixed collision energy, energy-dependent total rate constants can be obtained from the following expression
| 2 |
where σtot is the absolute reaction CS and ⟨v⟩ is the average relative velocity obtained from the collision energy.49,50 In order to compare the results of our measurements with those of the C2H6•+ ion recorded using an ion cyclotron resonance (ICR) mass spectrometer,48ktot(Eave) is calculated from the experimental data at a collision energy of 0.11 ± 0.02 eV, with the obtained ktot(Eave) value of (1.48 ± 0.55) × 10–9 cm3·molecule–1·s–1 being in good agreement with the literature value of (1.15 ± 0.12) × 10–9 cm3·molecule–1·s–1 for the reaction of the C2H6•+ ion.
While we do observe minor products at m/z 33, 60, and 61 that are indicative of the presence of [CH2O]•+, we conclude that the majority of the m/z 30 fragment ion signal does indeed correspond to the formation of C2H6•+. This conclusion is reached through comparison of the observed m/z 32 (and subsequent secondary reaction) CSs with those of previous studies on the reaction of C2H6•+. In this way, we are able to determine that the lower limit, within error, for the fraction of C2H6•+ present is 0.57, with an upper limit for the fraction of HCOH•+ of 0.10. The dissociative ionization of cyclopropyl alcohol is therefore disregarded as a suitable method for the selective generation of the HCOH•+ ion.
The dissociative photoionization of trideuterated methanol (CD3OH) was first suggested by Berkowitz40 as being preferable to the dissociative ionization of fully hydrogenated methanol due to the ability to separate the D2CO•+ (m/z 32) and DCOH•+ (m/z 31) fragments by their mass-to-charge ratios.17,38 However, as there is the potential for H/D exchange prior to ejection, as evidenced by the experimental observation of the D2COD+ fragment (m/z 34) with an AE of 11.85 eV that is below that of 12.60 eV for the m/z 31 fragment,51 the potential for isobaric contamination from DHCO•+ has to be considered.
As with cyclopropanol, PIE curves for the m/z 30 ([CDO]+/[13CHO]+) and 31 ([CDHO]•+/[13CDO]+) fragments have been measured and are presented in Figure 3. The AE of the m/z 31 channel has been determined, again using linear threshold extrapolation, to be 12.52 ± 0.04 eV, in reasonable agreement with the value of 12.6 eV recorded previously.51 Due to a small contamination from H2CO in the source chamber from previous measurements, the m/z 30 fragment channel has a baseline of ∼100 cps, with the appearance energy of the DCO+ channel determined from the first point above noise as 12.90 ± 0.09 eV.
Figure 3.

Photoionization efficiency (PIE) curves for the dissociative photoionization of CD3OH giving DCO+/COD+ (m/z 30, black data) and DCOH•+/DHCO•+ (m/z 31, blue data). Insets a) and b) show the threshold regions for the corresponding m/z 30 and 31 fragments, along with the assigned appearence energies (AEs).
For the characterization of any DHCO•+ impurity in the m/z 31 channel from deuterated methanol, isobutane (CH(CH3)3) has been selected as the reactant neutral because its IE of 10.68 ± 0.11 eV26 is between the IEs of DHCO (the IEs of H2CO and D2CO being 10.889 ± 0.003 eV25,26 and 10.908 ± 0.003 eV,25 respectively) and HCOH, which has an IE of 8.91 ± 0.02 eV.28 This means that the charge transfer process from DHCO•+ will be exothermic, while the charge transfer from DCOH•+ will be endothermic (and therefore closed at low collision energies in the absence of internal excitation). For this process, eq 1 can be rewritten as follows
| 3 |
where nDHCO•+ and nDCOH•+ are the isomeric fractions of DHCO•+ and DCOH•+, respectively, with σDHCO•+ and σDCOH•+ being the reaction CSs for the charge transfer reaction with CH(CH3)3 for DHCO•+ and DCOH•+, respectively. Given the endothermicity of the charge transfer process for the DCOH•+ isomer with isobutane, σDCOH•+ can be assumed to be zero in this case, thereby allowing the fraction of DHCO•+ present to be determined as follows:
| 4 |
In order to determine σDHCO•+, and under the assumption that there are no kinetic isotope effects so that σDHCO•+ is equal to σH2CO•+, we have measured CSs for the reaction of H2CO•+ with isobutane as a function of both photon and collision energy. As mentioned above, this determination of nDCOH•+ through subtraction of nDHCO•+ is necessitated by the current lack of isomer-selective DCOH•+ reactivity data.
For the purpose of a reactivity study with CH(CH3)3, H2CO•+ ions have been generated via direct ionization of neutral formaldehyde (H2CO), in turn formed via pyrolysis of paraformaldehyde at 60 °C. The obtained PIE curve around the threshold region for the m/z 30 (H2CO•+) channel is shown in Figure S3 of the Supporting Information, from which an AE for the m/z 30 mass channel of 10.88 ± 0.04 eV has been obtained via linear threshold extrapolation of the PIE curve.43−46 This is in excellent agreement with the literature IE of H2CO of 10.889 ± 0.003 eV.25,26
Absolute reaction CSs as a function of the collision energy have been measured at a photon energy of 11.0 eV in order to limit the impact of internal energy effects while ensuring sufficient reactant ion flux to allow for accurate reactivity measurements. The primary reaction product is that at m/z 58 (CH(CH3)3•+), corresponding to charge transfer, with absolute CSs for this channel, σH2CO•+, as a function of the collision energy shown in Figure 4. The step function visible in CS for both channels at a collision energy of ∼0.9 eV is due to the “L3 effect”, a key indicator of a charge transfer process, with further details given in the Supporting Information. This effect has not been corrected for here as the isomeric fraction is determined from the ratio of the two cross sections, which should remain constant. Although a range of other minor channels are also observed, these are not relevant to the characterization of the isomeric purity and so are not discussed further here.
Figure 4.
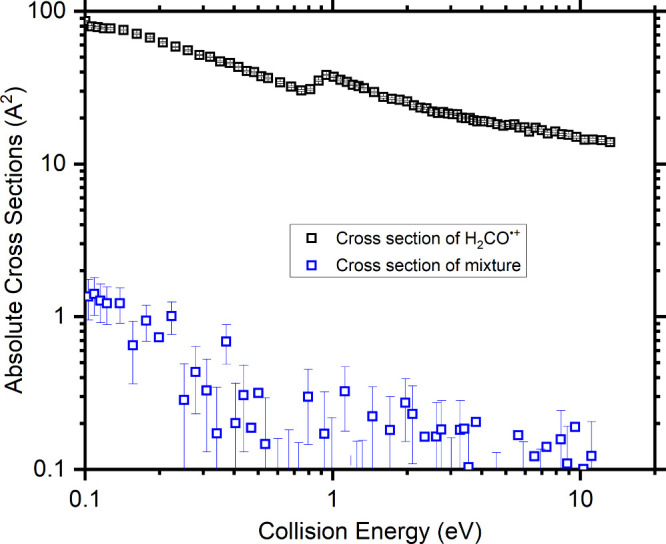
Absolute cross sections (CSs) as a function of the collision energy for the m/z 58 charge transfer product (CH(CH3)3•+) of the reaction with CH(CH3)3. Data for the reaction of H2CO•+ generated via direct photoionization of H2CO•+ (black squares) have been recorded at a photon energy of 11.0 eV, while those for the mixture of DCOH•+ and DHCO•+ ions generated from the dissociative ionization of CD3OH (blue squares) have been recorded at a photon energy of 13.07 eV.
In order to ensure that σDHCO•+ is constant over the 11–12.5 eV photon energy, the CSs for the reaction product at m/z 58 have also been recorded as a function of the photon energy, with the results shown in Figure S4 of the Supporting Information. From this, we note that the measured absolute CSs are approximately independent of the photon energy with only a very slight decrease at higher photon energies, indicating that the absolute CS for this process can be treated as constant with regard to the internal energy of the reactant ions.
Having recorded σH2CO•+, and therefore inferred σDHCO•+, σT values have been obtained as a function of the collision energy at a photon energy of 13.07 eV, above the 12.52 eV AE of the m/z 31 fragment channel, with the results shown in Figure 4. As with the reaction of H2CO•+, we have also measured the CS for this channel as a function of the photon energy, with results shown in Figure S5 of the Supporting Information. No photon energy dependence for σT is observed, allowing the isomeric purity values obtained from the σT values recorded at a photon energy of 13.07 eV to be used throughout the 13–15 eV photon energy range.
The values of nDHCO•+ obtained from the measured σDHCO•+ and σT values as a function of the collision energy are shown in Figure 5, while numerical values of nDHCO•+, σDHCO•+, and σT at selected collision energies of 0.16, 2.0, and 11.0 eV are given in Table 1. From this, we are able to determine an upper limit for the DHCO•+ fraction at this photon energy of 2.3%, with the average nDHCO•+ value over this collision energy range being 0.7 ± 0.5%.
Figure 5.
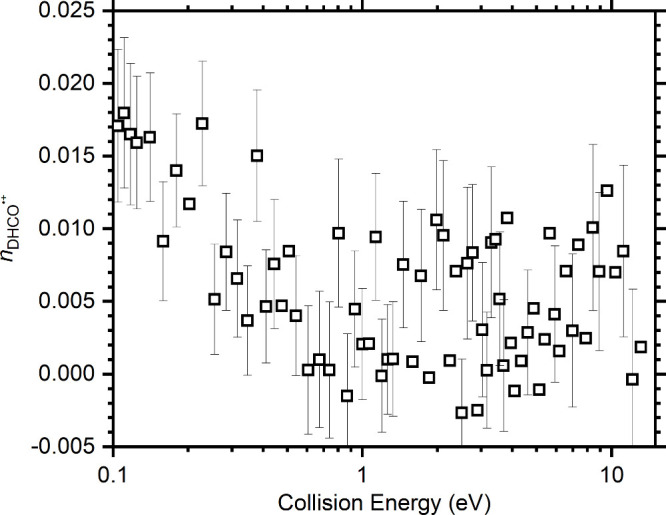
Isomeric fraction of DHCO•+, nDHCO•+, as a function of the collision energy. This is obtained from σT, recorded at a photon energy of 13.07 eV, and σDHCO•+, recorded at 11.00 eV, as detailed in the text.
Table 1. Measured σDHCO•+ and σT Recorded at Different Collision Energies, along with nDHCO•+ Values Determined Using equation 4.
| Collision Energy (eV) | σDHCO•+ (Å2) | σT (Å2) | nDHCO•+ |
|---|---|---|---|
| 0.16 ± 0.08 | 70.90 ± 0.69 | 0.65 ± 0.28 | 0.0092 ± 0.0041 |
| 2.0 ± 0.08 | 25.62 ± 0.34 | 0.27 ± 0.12 | 0.0105 ± 0.0048 |
| 11.0 ± 0.08 | 14.44 ± 0.25 | 0.12 ± 0.09 | 0.0083 ± 0.0064 |
Importantly, the mass spectra of the recorded product ions for the reaction of both H2CO•+ and the m/z 31 fragment of D3COH (see Figure S2 in the Supporting Information) indicate that the reactivity of the m/z 31 fragment is markedly different from that of the H2CO•+ ion. While detailed consideration of the various reaction processes is beyond the scope of this work, we focus here on the most intense channels from both parent ions. From H2CO•+ these are observed at m/z 58 (C4H10•+ from charge transfer) and at m/z 42 (C3H6•+ from dissociative charge transfer leading to the loss of CH4, exothermic by 0.30 eV26,28). By contrast, from the m/z 31 fragment from CD3OH (i.e., DCOH•+) the most intense products are at m/z 43 (C3H7+ from dissociative proton transfer leading to the loss of CH4, exothermic by 0.63 eV26,28), 57 (C4H9+ from dissociative proton transfer leading to the loss of H2, exothermic by about 0.84 eV26,28), and 32 (DHCOH+ formed via hydrogen atom transfer, exothermic by 0.77 eV26,28). As the reaction products for this ion are distinct from those of H2CO•+, we conclude that they arise from the reaction of a structurally distinct ionic species which, due to the lack of reasonable alternatives, must be DCOH•+.
As no photon energy dependence is observed for the recorded absolute reaction cross sections, we infer that the formation of DHCO•+ is energetically accessible from the threshold of the m/z 31 fragment channel and that the isomeric purity is therefore constant over the photon energy range considered here. We tentatively rationalize this observation as the result of efficient H/D scrambling via the interconversion between CD3OH•+ and the isotopic isomers D2COHD•+, D2HCOD•+ and DHCOD2•+, with the fragmentation step being the [1,1] ejection of D2 from DHCOD2•+. Further in-depth computational investigations are required, complementing the experimental measurements reported here, to allow for a detailed discussion of the different competing pathways. However, we note here that, due to the pathways via H/D exchange, we would expect the isomeric purity to be markedly lower in the case of nondeuterated methanol (CH3OH) due to the overlap between H2CO•+ and HCOH•+ fragmentation channels, thereby significantly removing the need for multiple rearrangements prior to fragmentation to give an isobaric contaminant.
In conclusion, we present experimental characterization of the HCOH•+ and DCOH•+ ions generated via the dissociative ionization of cyclopropanol and trideuterated methanol, respectively. In contrast to earlier suggestions,37 ,38 we find that cyclopropanol is unsuitable for this purpose. However, the generation of DCOH•+ from CD3OH is suitable, with an isomeric purity of 99.3 ± 0.5%. The key finding of this study is that DCOH•+ ions generated via the dissociative photoionization of trideuterated methanol (CD3OH) could be used in future studies to provide important isomer-specific reactivity data for various reaction systems. Such studies are not only essential for the development of accurate models of various astrochemical environments but should also allow for an understanding of the relationship between structure and reactivity that is highly valuable for fundamental reaction dynamics.
Acknowledgments
We thank SOLEIL for providing synchrotron radiation facilities as well as Laurent Nahon and the rest of the facility staff for their assistance in using the DESIRS beamline under Proposal No. 20230262. Furthermore, BRH is grateful to the European Commission (ERC grant no. 948373), and both BRH and VR thank the Leverhulme Trust (grant no. RPG-2022-264) for funding, while LA acknowledges the Department of Physics at the University of Liverpool for final year project funding. WDG and DS acknowledge support from the Swedish research council (grant no. 2019-04332). DA notes that this article is also based upon work from COST Action CA21126 - Carbon molecular nanostructures in space (NanoSpace), supported by COST (European Cooperation in Science and Technology) and acknowledges financial support from MUR PRIN 2020 project no. 2020AFB3FX “Astrochemistry beyond the second period elements” and from the National Recovery and Resilience Plan (NRRP), Mission 4, Component 2, Investment 1.1, Call for tender No. 1409 published on 14.9.2022 by the Italian Ministry of University and Research (MUR), funded by the European Union – NextGenerationEU – Project Title P20223H8CK “Degradation of space-technology polymers by thermospheric oxygen atoms and ions: an exploration of the reaction mechanisms at an atomistic level” - CUP E53D23015560001. MP acknowledges support from the Czech Ministry of Education, Youth and Sports (grant no. LTC20062). The authors would also like to thank Lucy Morris for her artistry in creating the associated cover art painting.
Data Availability Statement
Supporting data can be obtained from DataCat, the University of Liverpool Research Data Catalogue, at 10.17638/datacat.liverpool.ac.uk/2830.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jpclett.4c02374.
Further details on the experimental methods used in this work as well as the data treatment methods used to correct the data presented in this work; data plots are also provided for which summaries are given in the text (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Woods R. C.; Gudeman C. S.; Dickman R. L.; Goldsmith P. F.; Huguenin G. R.; Irvine W. M.; Hjalmarson A.; Nyman L.-A.; Olofsson H. The HCO+/HOC+ Abundance Ratio in Molecular Clouds. ApJ 1983, 270, 583–588. 10.1086/161150. [DOI] [Google Scholar]
- Dohnal P.; Jusko P.; Jiménez-Redondo M.; Caselli P. Measurements of rate coefficients of CN+, HCN+, and HNC+ collisions with H2 at cryogenic temperatures. J. Chem. Phys. 2023, 158, 244303. 10.1063/5.0153699. [DOI] [PubMed] [Google Scholar]
- Petrie S.; Freeman C. G.; McEwan M. J.; Ferguson E. E. The ion chemistry of HNC+/HCN+ isomers: astrochemical implications. Mon. Not. R. Astron. Soc. 1991, 248, 272–275. 10.1093/mnras/248.2.272. [DOI] [Google Scholar]
- Hansel A.; Glantschnig M.; Scheiring C.; Lindinger W.; Ferguson E. E. Energy dependence of the isomerization of HCN+ to HNC+ via ion molecule reactions. J. Chem. Phys. 1998, 109, 1743–1747. 10.1063/1.476748. [DOI] [Google Scholar]
- Shingledecker C. N.; Molpeceres G.; Rivilla V. M.; Majumdar L.; Kästner J. Isomers in Interstellar Environments. I. The Case of Z- and E-cyanomethanimine. ApJ 2020, 897, 158. 10.3847/1538-4357/ab94b5. [DOI] [Google Scholar]
- Garcia de la Concepción J.; Jiménez-Serra I.; Corchado J. C.; Molpeceres G.; Martinez-Henares A.; Rivilla V. M.; Colzi L.; Martín-Pintado J. A sequential acid-base mechanism in the interstellar medium: The emergence of cis-formic acid in dark molecular clouds. Astronomy & Astrophysics 2023, 675, A109. 10.1051/0004-6361/202243966. [DOI] [Google Scholar]
- Mathews L. D.; Adams N. G. Experimental study of the gas phase chemistry of C3H3+ with several cyclic molecules. Int. J. Mass Spectrom. 2011, 299, 139–144. 10.1016/j.ijms.2010.10.008. [DOI] [Google Scholar]
- Ploenes L.; Straňák P.; Mishra A.; Liu X.; Pérez-Ríos J.; Willitsch S. Collisional alignment and molecular rotation control the chemi-ionization of individual conformers of hydroquinone with metastable Neon. Nat. Chem. 2024, 10.1038/s41557-024-01590-1. [DOI] [PubMed] [Google Scholar]; Online ahead of print.
- Abma G. L.; Parkes M. A.; Horke D. A. Preparation of Tautomer-Pure Molecular Beams by Electrostatic Deflection. J. Phys. Chem. Lett. 2024, 15, 4587–4592. 10.1021/acs.jpclett.4c00768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu L.; Toscano J.; Willitsch S. Trapping and Sympathetic Cooling of Conformationally Selected Molecular Ions. Phys. Rev. Lett. 2024, 132, 083001. 10.1103/PhysRevLett.132.083001. [DOI] [PubMed] [Google Scholar]
- Mishra A.; Kim J.; Kim S. K.; Willitsch S. Isomeric and rotational effects in the chemi-ionisation of 1,2-dibromoethene with metastable neon atoms. Faraday Discuss. 2024, 251, 92–103. 10.1039/D3FD00172E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker K.; Bollis N. E.; Ryzhov V. Ion–molecule reactions of mass-selected ions. Mass Spectrom. Rev. 2024, 43, 47–89. 10.1002/mas.21819. [DOI] [PubMed] [Google Scholar]
- Liu J. K.-Y.; Niyonsaba E.; Alzarieni K. Z.; Boulos V. M.; Yerabolu R.; Kenttämaa H. I. Determination of the compound class and functional groups in protonated analytes via diagnostic gas-phase ion–molecule reactions. Mass Spectrom. Rev. 2023, 42, 1508–1534. 10.1002/mas.21727. [DOI] [PubMed] [Google Scholar]
- Richardson V.; Alcaraz C.; Geppert W. D.; Polášek M.; Romanzin C.; Sundelin D.; Thissen R.; Tosi P.; Žabka J.; Ascenzi D. The reactivity of methanimine radical cation (H2CNH•+) and its isomer aminomethylene (HCNH2•+) with methane. Chem. Phys. Lett. 2021, 775, 138611. 10.1016/j.cplett.2021.138611. [DOI] [PubMed] [Google Scholar]
- Liu J.; Van Devener B.; Anderson S. L. Reaction of formaldehyde cation with methane: Effects of collision energy and H2CO+ and methane vibrations. J. Chem. Phys. 2003, 119, 200–214. 10.1063/1.1577312. [DOI] [Google Scholar]
- Liu J.; Van Devener B.; Anderson S. L. Vibrational mode and collision energy effects on reaction of H2CO+ with C2D4. J. Chem. Phys. 2004, 121, 11746–11759. 10.1063/1.1822921. [DOI] [PubMed] [Google Scholar]
- Chamot-Rooke J.; Mourgues P.; van der Rest G.; Audier H. E. Ambient reactivity and characterization of small ionized carbenes. Int. J. Mass Spectrom. 2003, 226, 249–269. 10.1016/S1387-3806(03)00002-2. [DOI] [Google Scholar]
- Sundelin D.; Ascenzi D.; Richardson V.; Alcaraz C.; Polášek M.; Romanzin C.; Thissen R.; Tosi P.; Žabka J.; Geppert W. D. The reactivity of methanimine radical cation (H2CNH•+) and its isomer aminomethylene (HCNH2•+) with C2H4. Chem. Phys. Lett. 2021, 777, 138677. 10.1016/j.cplett.2021.138677. [DOI] [PubMed] [Google Scholar]
- Richardson V.; Ascenzi D.; Sundelin D.; Alcaraz C.; Romanzin C.; Thissen R.; Guillemin J.-C.; Polášek M.; Tosi P.; Žabka J.; Geppert W. D. Experimental and Computational Studies on the Reactivity of Methanimine Radical Cation (H2CNH•+) and its Isomer Aminomethylene (HCNH2•+) With C2H2. Front. Astron. Space Sci. 2021, 8, 752376. 10.3389/fspas.2021.752376. [DOI] [Google Scholar]
- Snyder L. E.; Buhl D.; Zuckerman B.; Palmer P. Microwave Detection of Interstellar Formaldehyde. Phys. Rev. Lett. 1969, 22, 679–681. 10.1103/PhysRevLett.22.679. [DOI] [Google Scholar]
- Gerin M.; Liszt H.; Pety J.; Faure A. H2CO and CS in diffuse clouds: Excitation and abundance. Astronomy & Astrophysics 2024, 686, A49. 10.1051/0004-6361/202449152. [DOI] [Google Scholar]
- Pegues J.; et al. An ALMA Survey of H2CO in Protoplanetary Disks. ApJ. 2020, 890, 142. 10.3847/1538-4357/ab64d9. [DOI] [Google Scholar]
- Tenenbaum E. D.; Milam S. N.; Woolf N. J.; Ziurys L. M. Molecular Survival In Evolved Planetary Nebulae: Detection of H2CO, c-C3H2, and C2H In the Helix. ApJ 2009, 704, L108–L112. 10.1088/0004-637X/704/2/L108. [DOI] [Google Scholar]
- Ramal-Olmedo J. C.; Menor-Salván C. A.; Fortenberry R. C. Mechanisms for gas-phase molecular formation of neutral formaldehyde (H2CO) in cold astrophysical regions. Astronomy & Astrophysics 2021, 656, A148. 10.1051/0004-6361/202141616. [DOI] [Google Scholar]
- Niu B.; Shirley D. A.; Bai Y. High resolution photoelectron spectroscopy and femtosecond intramolecular dynamics of H2CO+ and D2CO+. J. Chem. Phys. 1993, 98, 4377–4390. 10.1063/1.464999. [DOI] [Google Scholar]
- Linstrom P. J.; Mallard W. G.. NIST Chemistry WebBook - Standard Reference Database n. 69. [Online], accessed May 2024; http://webbook.nist.gov.
- Zanchet A.; García G. A.; Nahon L.; Bañares L.; Marggi Poullain S. Signature of a conical intersection in the dissociative photoionization of formaldehyde. Phys. Chem. Chem. Phys. 2020, 22, 12886–12893. 10.1039/D0CP01267J. [DOI] [PubMed] [Google Scholar]
- Ruscic B.; Bross D.. Active Thermochemical Tables (ATcT) values based on ver. 1.124 of the Thermochemical Network. [Online], 2024. [Google Scholar]
- Wagner J. P.; Barlett M. A.; Allen W. D.; Duncan M. A. Tunneling Isomerizations on the Potential Energy Surfaces of Formaldehyde and Methanol Radical Cations. ACS Earth Space Chem. 2017, 1, 361–367. 10.1021/acsearthspacechem.7b00068. [DOI] [Google Scholar]
- Snyder L. E.; Hollis J. M.; Ulich B. L. Radio detection of the interstellar formyl radical. ApJ 1976, 208, L91–L94. 10.1086/182239. [DOI] [Google Scholar]
- Ohishi M.; Ishikawa S. I.; Amano T.; Oka H.; Irvine W. M.; Dickens J. E.; Ziurys L. M.; Apponi A. J. Detection of A New Interstellar Molecular Ion, H2COH+ (Protonated Formaldehyde). ApJ 1996, 471, L61. 10.1086/310325. [DOI] [PubMed] [Google Scholar]
- Ball J. A.; Gottlieb C. A.; Lilley A. E.; Radford H. E. Detection of Methyl Alcohol in Sagittarius. ApJ 1970, 162, L203. 10.1086/180654. [DOI] [Google Scholar]
- Richardson V.; Valença Ferreira de Aragão E.; He X.; Pirani F.; Mancini L.; Faginas-Lago N.; Rosi M.; Martini L. M.; Ascenzi D. Fragmentation of interstellar methanol by collisions with He•+: an experimental and computational study. Phys. Chem. Chem. Phys. 2022, 24, 22437–22452. 10.1039/D2CP02458F. [DOI] [PubMed] [Google Scholar]
- Schulenburg A. M.; Meisinger M.; Radi P. P.; Merkt F. The formaldehyde cation: Rovibrational energy level structure and Coriolis interaction near the adiabatic ionization threshold. J. Mol. Spectrosc. 2008, 250, 44–50. 10.1016/j.jms.2008.04.005. [DOI] [Google Scholar]
- Liu J.; Kim H.-T.; Anderson S. L. Multiphoton ionization and photoelectron spectroscopy of formaldehyde via its 3p Rydberg states. J. Chem. Phys. 2001, 114, 9797–9806. 10.1063/1.1370943. [DOI] [Google Scholar]
- Hockey E. K.; McLane N.; Martí C.; Duckett L.; Osborn D. L.; Dodson L. G. Direct Observation of Gas-Phase Hydroxymethylene: Photoionization and Kinetics Resulting from Methanol Photodissociation. J. Am. Chem. Soc. 2024, 146, 14416–14421. 10.1021/jacs.4c03090. [DOI] [PubMed] [Google Scholar]
- Wesdemiotis C.; McLafferty F. W. Mass Spectral Evidence for the Hydroxymethylene Radical Cation. Tetrahedron Lett. 1981, 22, 3479–3480. 10.1016/S0040-4039(01)81936-5. [DOI] [Google Scholar]
- Burgers P. C.; Mommers A. A.; Holmes J. L. Ionized Oxycarbenes: [COH]+, [HCOH]+•, [C(OH)2]+•, [HCO2]+, and [COOH]+, Their Generation, Identification, Heat of Formation, and Dissociation Characteristics. J. Am. Chem. Soc. 1983, 105, 5976–5979. 10.1021/ja00357a003. [DOI] [Google Scholar]
- Mauney D. T.; Mosley J. D.; Madison L. R.; McCoy A. B.; Duncan M. A. Infrared spectroscopy and theory of the formaldehyde cation and its hydroxymethylene isomer. J. Chem. Phys. 2016, 145, 174303. 10.1063/1.4966214. [DOI] [PubMed] [Google Scholar]
- Berkowitz J. Photoionization of CH3OH, CD3OH and CH3OD: Dissociative ionization mechanisms and ionic structures. J. Chem. Phys. 1978, 69, 3044–3054. 10.1063/1.436995. [DOI] [Google Scholar]
- Nahon L.; de Oliveira N.; Garcia G. A.; Gil J.-F.; Pilette B.; Marcouillé O.; Lagarde B.; Polack F. DESIRS: a state-of-the-art VUV beamline featuring high resolution and variable polarization for spectroscopy and dichroism at SOLEIL. J. Synchrotron Radiat. 2012, 19, 508–520. 10.1107/S0909049512010588. [DOI] [PubMed] [Google Scholar]
- Bombach R.; Dannacher J.; Honegger E.; Stadelmann J.-P.; Neier R. Unimolecular Dissociations of Excited C3H6O+: A Photoelection-Photoion Coincidence Study of Cyclopropanol and Allyl Alcohol. Chem. Phys. 1983, 82, 459–470. 10.1016/0301-0104(83)85250-1. [DOI] [Google Scholar]
- Roithová J.; Schröder D.; Loos J.; Schwarz H.; Jankowiak H.-C.; Berger R.; Thissen R.; Dutuit O. Revision of the second ionization energy of toluene. J. Chem. Phys. 2005, 122, 094306. 10.1063/1.1856916. [DOI] [PubMed] [Google Scholar]
- Chupka W. A. Effect of Thermal Energy on Ionization Efficiency Curves of Fragment Ions. J. Chem. Phys. 1971, 54, 1936–1947. 10.1063/1.1675122. [DOI] [Google Scholar]
- Traeger J. C.; McLoughlin R. G. Absolute heats of formation for gas-phase cations. J. Am. Chem. Soc. 1981, 103, 3647–3652. 10.1021/ja00403a006. [DOI] [Google Scholar]
- Castrovilli M. C.; Bolognesi P.; Cartoni A.; Catone D.; O’Keefe P.; Casavola A. R.; Turchini S.; Zema N.; Avaldi L. Photofragmentation of Halogenated Pyrimidine Molecules in the VUV Range. J. Am. Soc. Mass Spectrom. 2014, 25, 351–367. 10.1007/s13361-013-0783-x. [DOI] [PubMed] [Google Scholar]
- Ruscic B. Photoionization Mass Spectroscopic Studies of Free Radicals in Gas Phase: Why and How. Res. Adv. Phys. Chem. 2000, 1, 39–75. [Google Scholar]
- Kim J. K.; Anicich V. G.; Huntress Jr. W. T. Product distributions and rate constants for the reactions of CH3+, CH4+, C2H2+, C2H3+, C2H4+, C2H5+, and C2H6+ ions with CH4, C2H2, C2H4, and C2H6. J. Phys. Chem. 1977, 81, 1798–1805. 10.1021/j100534a002. [DOI] [Google Scholar]
- Ervin K. M.; Armentrout P. B. Translational energy dependence of Ar+ + XY → ArX+ + Y (XY = H2, D2, HD) from thermal to 30 eV c.m. J. Chem. Phys. 1985, 83, 166–189. 10.1063/1.449799. [DOI] [Google Scholar]
- Nicolas C.; Alcaraz C.; Thissen R.; Žabka J.; Dutuit O. Effects of ion excitation on charge transfer reactions of the Mars, Venus, and Earth ionospheres. Plan. Space Sci. 2002, 50, 877–887. 10.1016/S0032-0633(02)00063-6. [DOI] [Google Scholar]
- Borkar S.; Sztáray B.; Bodi A. Dissociative photoionization mechanism of methanol isotopologues (CH3OH, CD3OH, CH3OD and CD3OD) by iPEPICO: energetics, statistical and non-statistical kinetics and isotope effects. Phys. Chem. Chem. Phys. 2011, 13, 13009–13020. 10.1039/c1cp21015g. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Supporting data can be obtained from DataCat, the University of Liverpool Research Data Catalogue, at 10.17638/datacat.liverpool.ac.uk/2830.