

Abstract
HER2-positive and triple-negative breast cancers (TNBC) are difficult to treat and associated with poor prognosis. Despite showing initial response, HER2-positive breast cancers often acquire resistance to HER2-targeted therapies, and TNBC lack effective therapies. To overcome these clinical challenges, we evaluated the therapeutic utility of co-targeting TrkA and JAK2/STAT3 pathways in these breast cancer subtypes. Here, we report the novel combination of FDA-approved TrkA inhibitors (Entrectinib or Larotrectinib) and JAK2 inhibitors (Pacritinib or Ruxolitinib) synergistically inhibited in vitro growth of HER2-positive breast cancer cells and TNBC cells. The Entrectinib-Pacritinib combination inhibited the breast cancer stem cell subpopulation, reduced expression of stemness genes, SOX2 and MYC, and induced apoptosis. The Entrectinib-Pacritinib combination suppressed orthotopic growth of HER2-positive Trastuzumab-refractory breast cancer xenografts and basal patient-derived xenograft (PDXs), reduced tumoral SOX2 and MYC, and induced apoptosis in both mouse models. The Entrectinib-Pacritinib combination inhibited overall metastatic burden, and brain and bone metastases of intracardially inoculated TNBC cells without toxicity. Together, our results demonstrate for the first time that co-inhibition of TrkA and JAK2 synergistically suppresses breast cancer growth and metastasis, thereby providing preclinical evidence that supports future clinical evaluations.
Keywords: Breast cancer, TrkA, JAK2, Breast cancer metastasis, Combined targeted therapy
1. Introduction
Breast cancer is the second-leading cause of cancer-related deaths in American women [1]. While the National Cancer Institute reports a five-year survival of 85 % following initial diagnosis, the survival rate drops to 27 % upon development of distant metastases [2]. Breast cancers are classified largely based upon the expression of receptors for estrogen (ER), progesterone (PR), or human epidermal growth factor 2 (HER2). Of the five major clinical breast cancer subtypes, HER2-positive breast cancers and triple-negative breast cancers (TNBC; ER−/PR−/HER2−) are more aggressive and are associated with poorer clinical outcomes due to increased rates of relapse and metastases [3]. The current standard of care implements surgical resection, as well as radiation, neoadjuvant or adjuvant therapies, or hormone therapies [4]. However, breast cancers persist as a therapeutic challenge due to activation of redundant signaling pathways that sustain tumor cell growth, proliferation, invasion, and metastasis. HER2-positive breast cancers and TNBC frequently metastasize to the brain [5,6]; however, there is a lack of targeted therapies capable of penetrating the blood-brain-barrier (BBB) and blood-tumor-barrier (BTB). Therefore, there is an urgent and unmet need to enhance efficacy of anti-tumor therapies in metastatic breast cancers.
Tropomyosin receptor tyrosine kinase A (TrkA) is encoded by the NTRK1 gene and is prone to forming oncogenic NTRK1 fusions in many solid cancer types [7,8]. However, overexpression of wild-type TrkA is also sufficient to enhance growth, migration, and metastasis of breast cancer cells [9]. TrkA is activated upon binding to its cognate ligand, β-nerve growth factor, leading to TrkA homodimerization. Activated TrkA phosphorylates its target substrates to activate downstream pathways, such as PI3K and RAS/MAPK, ultimately promoting cell proliferation and survival [7]. Aberrant TrkA signaling is implicated in malignant progression and metastasis of breast cancers and other solid tumor types [7,9]. Thus, TrkA is an important therapeutic target in several cancers. Two orally active TrkA small molecule inhibitors (TrkAis), Larotrectinib and Entrectinib, are FDA-approved for JVTRiC-altered solid tumors. Larotrectinib (Vitravki®) is a pan-TRK inhibitor with enhanced selectivity for TrkA and was the first tissue-agnostic molecularly targeted anti-cancer therapy to receive FDA approval [10]. Larotrectinib is currently in phase II clinical trials in patients with NTRK-altered tumors, including breast cancers (NCT02576431, NCT02465060, NCT03834961). Entrectinib (Rozlytrek®) is also a pan-TRK inhibitor with increased activity against TrkA [11] and received tumor-agnostic FDA approval in 2019 for NTRK gene fusion-positive solid tumors [12]. Entrectinib is currently in phase II clinical trials as a single agent in solid tumors bearing NTRK, ROS1, or ALK1 fusions (NCT02568267) or in combination with endocrine therapies in lobular breast carcinomas (NCT04551495). Importantly, both Entrectinib and Larotrectinib are BBB-penetrant compounds, and may be of clinical benefit to patients with brain tumors or brain metastases [13]. Accordingly, Entrectinib and Larotrectinib are also being evaluated in patients with primary central nervous system malignancies (NCT02650401, NCT04655404).
Janus kinase 2 (JAK2) is a non-receptor tyrosine kinase frequently amplified or hyperactivated in HER2-enriched breast cancers and TNBC [14,15]. JAK2 serves as a central signaling hub that propagates extracellular signals from receptor tyrosine kinases and interleukin receptors to transcription factor, signal transducer and activator of transcription 3 (STAT3) [16]. JAK2 phosphorylates STAT3-Y705, promoting STAT3 transcriptional activity and enhances expression of genes associated with cell proliferation, oncogenesis, angiogenesis, and metastasis [17]. Aberrant JAK2-STAT3 activity is found in many cancers including breast cancers, myeloproliferative neoplasms, and acute myeloid leukemias [17]. Several orally active JAK2 small molecule inhibitors (JAK2is), such as Ruxolitinib and Pacritinib, are FDA-approved for varying indications. Ruxolitinib (Jakafi ®) is a JAK1/JAK2i FDA-approved for myelofibrosis [18] and polycythemia vera [19]. While Ruxolitinib can inhibit JAK1 and JAK2, it demonstrates enhanced selectivity for JAK2 [20]. Unfortunately, clinical trials for Ruxolitinib in breast cancer patients (NCT02120417, NCT01562873) demonstrated minimal clinical benefit and suggested that Ruxolitinib may not be effective as a single agent therapy in breast cancer patients. Another JAK2i, Pacritinib (Vonjo®) [21], demonstrated anti-cancer efficacy in patients with leukemia and glioblastoma when administered with other chemotherapies [22,23]. Pacritinib is approved by the FDA for primary and secondary myelofibrosis [24] and is currently being evaluated for safety and efficacy in breast cancer patients bearing 1q21.3 amplifications (NCT04520269). Notably, Ruxolitinib and Pacritinib are both capable of penetrating the BBB/BTB, suggesting their therapeutic utility in JAK1/2-altered brain malignancies [23,25].
We recently reported a novel signaling crosstalk between TrkA and JAK2-STAT3 pathways in which these pathways are co-activated in HER2-enriched breast cancers and TNBC [26]. We showed that TrkA interacts with and phosphorylates STAT3 at Y705, to activate STAT3’s transcriptional activity [26]. We further reported that the TrkA and JAK2-STAT3 crosstalk enhanced transcription of STAT3 target genes associated with cancer cell stemness, namely SOX2 and c-MYC which, in turn, promoted breast cancer stem cell (CSC) phenotypes [26]. Since both TrkA and JAK2 phosphorylate STAT3 and activate its transcriptional activity, and co-activation of TrkA and JAK2 promotes CSCs, co-targeting TrkA and JAK2 using FDA-approved small molecule inhibitors could be a novel viable treatment strategy for HER2-positive breast cancers and TNBC. Combined targeting of TrkA and JAK2 has never been investigated in any cancer or cell types. This novel combination was evaluated for the first time in this study using HER2-positive breast cancers and TNBC.
2. Materials and methods
2.1. Cell lines and cell culture
Brain-seeking SKBR3 (SKBRM) cells were provided by Dr. Kounosuke Watabe [27]. Trastuzumab-resistant BT474 (BT474-TtzmR) were gifted by Dr. Dihua Yu [28]. Brain, bone, and lung-seeking MDA-MB-231 cells (MDA-MB-231-BrM, -BoM, and -LM, respectively) were provided by Dr. Joan Massagué [29–31]. All other cell lines were purchased from American Type Culture Collection (ATCC, Manassas, VA), maintained in appropriate growth media, and validated using the standard method. Cell lines were confirmed as mycoplasma negative using a mycoplasma detection kit (InvivoGen; San Diego, CA).
2.2. Mammary fat pad (orthotopic) implantation and treatment study
All animal studies were completed with approval from the Institutional Animal Care and Use Committee. All colonies of female nude mice (Athymic Nude-Foxn1nu, Charles River; Wilmington, MA, USA) were housed in a pathogen-free facility under a 12:12-h light/dark cycle and fed irradiated rodent chow ad libitum. Anesthetized mice were inoculated with 5x106 actively growing BT474-TtzmR cells expressing luciferase resuspended in Matrigel (Corning) or 1 mm3 viable fragment of PDX-3887 tumor (Baylor College of Medicine PDX resource; Houston, TX, USA). Digital caliper measurements were taken twice weekly to measure tumor volume, which was estimated using the following equation:
where width and length are the shorter and longer diameters of the tumor, respectively. Formation of any metastases were monitored by bi-weekly bioluminescent imaging (BLI) in which mice were intraperitoneally injected with 100 mg/kg d-luciferin (PerkinElmer; Waltham, MA, USA) and imaged using the IVIS Lumina LT series III imager (PerkinElmer). Once average tumor volume reached 75–100 mm3, animals were randomized into four treatment groups: vehicle (100 % PEG), Entrectinib (25 mg/kg), Pacritinib (50 mg/kg) or the Entrectinib-Pacritinib combination such that tumor volumes were approximately equal across groups. Entrectinib and Pacritinib were purchased from Adooq BioScience (Irvine, CA). Treatments were orally administered twice per day (b.i.d.), 5 days/week until study termination. Animal weights were monitored bi-weekly.
2.3. Intracardiac inoculation and treatment of TNBC metastasis
Mice were inoculated with 2x105 exponentially growing MDA-MB-231 cells expressing luciferase in 100 μL ice-cold PBS into the left ventricle. Successful inoculations were confirmed by visualization of brain bioluminescent signal within 1 h of inoculation; otherwise, mice were immediately sacrificed. Tumor progression was monitored with bi-weekly BLI in which mice were intraperitoneally injected with 100 mg/kg d-luciferin (PerkinElmer) and imaged using the IVIS Lumina LT Series III imager (PerkinElmer). Mice were randomized into vehicle (100 % PEG), Entrectinib (25 mg/kg), Pacritinib (50 mg/kg), or Entrectinib-Pacritinib combination. Treatments were administered orally b.i.d., 5 days/week until study termination. Animal weights were recorded twice-weekly. Tumor burden was analyzed by quantifying the BLI signal in each region-of-interest (ROI) measured in total flux (p/s) with the Living Image software version 4.7.2 (PerkinElmer).
3. Results
3.1. TrkA and JAK2 inhibitors synergize to reduce cell viability of HER2-positive breast cancer cells and TNBC in vitro
We previously reported that TrkA and JAK2-STAT3 signaling axes are co-activated in HER2-enriched and TNBC patient samples [26]. Here, we further examined the extent of TrkA/JAK2-STAT3 co-activation across breast cancer subtypes using Western blot analyses and found that TrkA and JAK2 are expressed in breast cancer cell lines regardless of molecular subtypes (Fig. 1A). However, expression of activated STAT3 (p-STAT3 Y705), a common phosphorylation substrate for TrkA and JAK2, is enhanced in HER2-positive and TNBC cells compared to most luminal breast cancer lines. Interestingly, levels of TrkA, JAK2 and p-STAT3 were substantially elevated in BT474-TtzmR cells, HER2-positive BT474 cell that have acquired resistance to FDA-approved HER2 therapeutic antibody Trastuzumab, compared to the parental BT474 cells. Notably, activated STAT3 is abundantly found in BT474-TtzmR, SKBRM (brain-tropic variant of HER2-positive SKBR3), and in TNBC cells. Given these observations and our previous findings [26], we sought to investigate if co-inhibition of TrkA and JAK2 may result in enhanced tumor cell death of HER2-positive and TNBC cells. TrkAis Entrectinib and Larotrectinib, and JAK2is Ruxolitinib and Pacritinib, were tested as single agents in HER2-positive BT474 and BT474-TtzmR, SKBRM, and TNBC MDA-MB-231, to determine the half-maximal inhibitory concentration (IC50) of each drug. Our results indicated that Entrectinib and Pacritinib were the more potent TrkAi and JAK2i, respectively, as indicated by their lower IC50 values (Fig. 1B). Interestingly, BT474-TtzmR cells, which expressed relatively higher levels of TrkA and JAK2 than BT474, were more sensitive to TrkAi or JAK2i treatments compared to BT474 cells. We subsequently evaluated, for the first time, TrkAis and JAK2i combinations at various ratios and determined their combination indices (CI) using the Chou-Talalay method (CompuSyn) (see Supplementary Materials). A CI of less than 1 indicates synergy, whereas a CI value greater than one indicates antagonism between inhibitors. We observed that multiple different combinations yielded synergistic effects on all five cell lines evaluated (Fig. 1C). Based on these results, we reasoned that Entrectinib and Pacritinib would be the best candidate TrkAi and JAK2i, respectively, to use in combination therapy when administered at 1:2 ratio. Representative cell viability in BT474-TtzmR (Fig. 1D), MDA-MB-231 (Fig. 1E), and SKBRM (Fig. 1F) are shown. These data suggest, for the first time, that dual targeting of TrkA and JAK2 is synergistic and more effective than monotherapies against HER2-positive breast cancer cells and TNBC cells.
Fig. 1. TrkA and JAK2 inhibitors synergize to reduce cell viability of HER2-positive breast cancer cells and TNBC in vitro.

(A) Western blot panel of 2 normal mammary epithelial cells and 14 breast cancer cell lines to examine relative expression of TrkA and JAK2. Densitometry values (normalized to β-actin) are displayed below each blot. LumA, luminal A. LumB, luminal B. Representative IC50 values of JAK2is (Pacritinib or Ruxolitinib) and TrkAis (Larotrectinib or Entrectinib) derived from treatment of parental BT474 and its trastuzumab-resistant variant (BT474-TtzmR), TNBC cell line MDA-MB-231, and SKBRM (brain-metastatic variant of HER2-enriched SKBR3). Lower IC50 values for each inhibitor are highlighted in red (B). Representative combination index (CI) values for combinations of TrkA and JAK2 inhibitors. CI < 1 indicates synergy, CI = 1 indicates additive and CI > 1 indicates antagonism. Synergistic CI values are highlighted in red (C). Representative cell viability plots of BT474-TtzmR (D), MDA-MB-231 (E), and SKBRM (F) after treatment with Entrectinib-Pacritinib combination at fixed 1:2 ratio. Experiments were repeated at least three times to derive averages. Data are presented as mean ± SEM. One-way ANOVA with Tukey’s multiple comparison post hoc test was used to compare p values.
3.2. Breast CSCs are inhibited by the combination of TrkA and JAK2 inhibitors
Breast CSCs are associated with breast tumor development, metastasis, and tumor recurrence [32]. We previously showed that TrkA overexpression enhances mammosphere formation of breast cancer cells, which is exacerbated upon co-overexpression of constitutively active STAT3 (STAT3-CA) [26]. Therefore, we posited that co-inhibition of TrkA and JAK2 would effectively suppress the breast CSC population. To this end, we evaluated changes in mammosphere formation of HER2-positive SKBRM and BT474-TtzmR, which expressed high levels of TrkA, JAK2, and p-STAT3 (Y705) (Fig. 1A), following treatment with Entrectinib-Pacritinib combination therapy. We found that SKBRM cells (Fig. 2A) formed a significantly lower number of mammospheres upon combination therapy treatment when compared to vehicle or single agents, suggesting that the combination therapy is more effective in suppressing the breast CSC subpopulation in vitro. Similarly, Trastuzumab-resistant BT474-TtzmR cells treated with combination therapy formed significantly lower number of mammospheres when compared to vehicle (Fig. 2B). While single agent treatments also reduced mammosphere formation of BT474-TtzmR, this did not reach statistical significance compared to the vehicle control.
Fig. 2. Breast CSCs are inhibited by the combination of TrkA and JAK2 inhibitors.

Mammosphere formation assay of HER2-positive SKBRM (A) or BT474-TtzmR (B) after treatment with vehicle, Entrectinib, Pacritinib, or combination. Representative mammosphere images for each cell line are shown. Scale bar represents 200 μm. CD44high/CD24low flow cytometry of SKBRM (C) and BT474-TtzmR (D) after treatment with vehicle, Entrectinib, Pacritinib, or combination. Representative flow cytometry plots are shown. Western blot analysis of SKBRM (E) and BT474-TtzmR (F) after treatment with vehicle, Entrectinib, Pacritinib, or combination. Densitometry values (normalized to β-actin) are displayed below each blot. ALDH activity in SKBRM (G) or BT474-TtzmR (H) after treatment with vehicle, Entrectinib, Pacritinib, or combination. Data of three to four experimental repeats are presented as mean ± SEM. One-way ANOVA with Tukey’s multiple comparison post hoc test was used to compare p values.
Breast CSCs express cell-surface markers such as CD44 and CD24 [33]. CD44high/CD24low cancer cells are considered the CSC subpopulation [34]. We previously showed that co-overexpression of TrkA and STAT3-CA significantly increased the CD44high/CD24low CSC subpopulation [26]. Therefore, we next evaluated whether dual targeting TrkA and JAK2 would suppress the CD44high/CD24low CSCs using flow cytometry. Results showed that the combination therapy significantly reduced the CD44high/CD24low CSCs population in SKBRM (Fig. 2C) and BT474-TtzmR (Fig. 2D) cells compared to the vehicle group. In contrast, single agent treatments did not significantly alter the CD44high/CD24low population in either cell line. These results suggest that the Entrectinib-Pacritinib combination therapy effectively suppresses the breast CSCs in vitro.
Since co-overexpression of TrkA and STAT3-CA enhances expression of stemness genes SOX2 and MYC [26], we evaluated SOX2 and MYC expression levels upon co-inhibition of TrkA and JAK2 using Western blot analyses on SKBRM and BT474-TtzmR cells. Results show that expression of SOX2 and MYC in SKBRM (Fig. 2E) and BT474-TtzmR cells (Fig. 2F) was potently reduced in cells treated with combination therapy compared to those treated with vehicle. Though Pacritinib single therapy suppressed SOX2 and MYC expression compared to vehicle, we found that expression of these proteins was further reduced by the combination therapy. CD44 expression was also reduced in both cell lines upon combination treatment, which supports our earlier findings (Fig. 2C and D). While SKBRM cells treated with combination therapy did not show robust reduction in p-STAT3 (Y705) compared to single agent treatment, BT474-TtzmR cells showed a marked reduction in p-STAT3 (Y705) by the combination therapy, but not with Entrectinib or Pacritinib alone. Lastly, we evaluated changes in aldehyde dehydrogenase (ALDH) activity upon co-inhibition of TrkA and JAK2. ALDH activity is enhanced in stem-like cancer cells and is considered a universal marker for identification of CSCs [35]. We observed a modest but significant reduction in ALDH activity of SKBRM cells treated with combination therapy compared to vehicle or single agent treatments (Fig. 2G). ALDH activity of BT474-TtzmR cells was significantly suppressed by the combination therapy, but not by either monotherapy treatment (Fig. 2H). Collectively, results in Fig. 2 demonstrate that the Entrectinib-Pacritinib combination therapy more effectively suppresses breast CSCs than single agent treatments.
3.3. Co-inhibition of TrkA and JAK2 enhances apoptosis of breast cancer cells
We examined whether the Entrectinib-Pacritinib combination therapy enhances breast cancer cell apoptosis. SKBRM and Trastuzumab-resistant BT474-TtzmR cells were treated with the combination therapy and analyzed for the extent of apoptosis using Annexin V staining followed by flow cytometry. Annexin V stains phosphatidylserine residues, which are exposed to the extracellular surface during early apoptosis [36]. Our results demonstrated that the combination therapy significantly enhanced Annexin V-positivity in SKBRM cells (19.82 %) compared to vehicle (10.91 %) or single agents (9.2 % or 12.93 % for Entrectinib or Pacritinib, respectively) (Fig. 3A). Similarly, BT474-TtzmR cells treated with the combination therapy had significantly more Annexin V-positivity (5.53 %) compared to vehicle (3.74 %), Entrectinib (3.71 %), or Pacritinib (3.4 %) (Fig. 3B). Monotherapy treatments did not significantly alter apoptosis in either cell line evaluated, suggesting that the combination therapy most significantly induces apoptosis of breast cancer cells in vitro.
Fig. 3. Co-inhibition of TrkA and JAK2 enhances apoptosis of breast cancer cells.
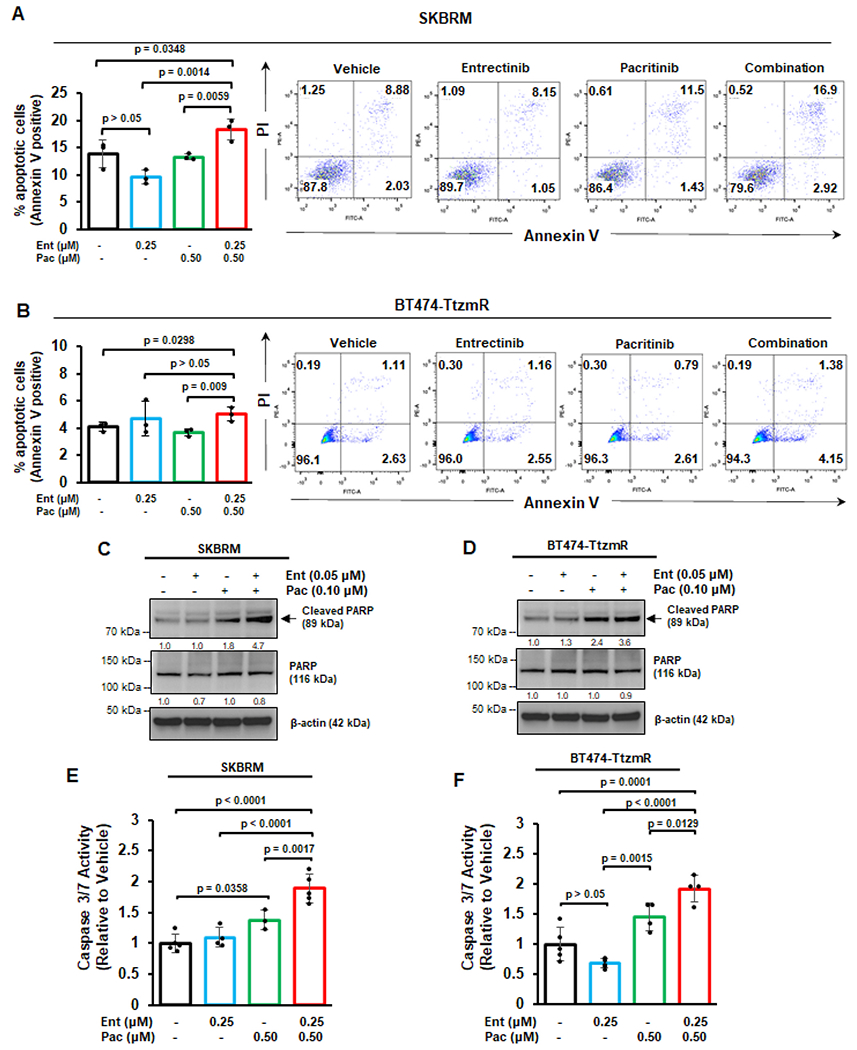
Annexin V flow cytometry of SKBRM (A) or BT474-TtzmR (B) cells after treatment with vehicle, Entrectinib, Pacritinib, or combination. Cells were counterstained with propidium iodide (PI). Representative flow cytometry plots are displayed for each treatment condition. Western blot analysis of SKBRM (C) or BT474-TtzmR (D) for cleaved and total PARP after single agent or combination therapy treatment. Densitometry values (normalized to β-actin) are displayed below each blot. Caspase 3/7 activity of SKBRM (E) and BT474-TtzmR (F) after single agent or combination therapy treatment. Data of at least three experimental repeats are presented as mean ± SEM. One-way ANOVA with Tukey’s multiple comparison post hoc test was used to compare p values.
To further assess the ability of the treatment combination to induce apoptosis, we evaluated cleavage of Poly ADP-ribose polymerase (PARP). PARP is involved in DNA damage and programmed cell death, and is cleaved by activated caspases, such as caspases 3/7, to promote cellular disassembly [37]. PARP cleavage produces two distinct fragments: an 89-kDa catalytic fragment, which is frequently used as an apoptosis marker, and a 24-kDa DNA binding fragment [38]. Our Western blot analyses indicated that, while single agent treatment induced modest increases in PARP cleavage, the combination therapy robustly enhanced PARP cleavage in SKBRM cells (4.7-fold higher than vehicle, as indicated by densitometry) (Fig. 3C) and BT474-TtzmR cells (3.6-fold higher than vehicle) (Fig. 3D).
Due to the observed increase in PARP cleavage following combination therapy, we also measured caspase 3/7 activity. We found that the combination therapy induced the highest increase in caspase 3/7 activity compared to vehicle or monotherapy groups in SKBRM (Fig. 3E) and BT474-TtzmR (Fig. 3F) cells. Together, these findings demonstrate that the Entrectinib-Pacritinib combination effectively induces apoptosis of breast cancer cells in vitro.
3.4. Orthotopic growth of HER2-positive trastuzumab-resistant breast cancer xenografts is suppressed by the Entrectinib-Pacritinib combination therapy
Based on our findings that Entrectinib and Pacritinib synergize to reduce breast cancer cell viability and promote apoptosis, we reasoned that the combination therapy could effectively reduce breast tumor growth in vivo. Luciferase-expressing Trastuzumab-resistant BT474-TtzmR cells were inoculated into the 4th inguinal mammary fat pad (MFP) of female nude mice. Tumors were allowed to reach ~100 mm3 at Day 9 and then randomized to receive oral administration of vehicle, Entrectinib (25 mg/kg), Pacritinib (50 mg/kg), or combination five times a week. Tumor volume was determined by caliper measurements twice a week (Fig. 4A). Results showed that BT474-TtzmR MFP tumor growth was significantly suppressed by the combination therapy compared to vehicle and monotherapy treatments; single agent treatments did not alter MFP tumor growth (Fig. 4B). Mirroring our in vivo data, the average ex vivo tumor mass was significantly reduced in the combination group compared to vehicle or single agent groups while the monotherapy did not significantly affect tumor mass compared to vehicle (Fig. 4C). Analysis of the tumor growth inhibition (TGI) further demonstrated that the combination therapy was more effective in inhibiting growth of the MFP tumors (TGI: 43.1 %) when compared to entrectinib or pacritinib alone (TGI: 2.2 % and 1.6 %, respectively) (Supplementary Fig. 1).
Fig. 4. Orthotopic growth of HER2-positive trastuzumab-resistant breast cancer xenografts is suppressed by the Entrectinib-Pacritinib combination therapy.

(A) Scheme for the mammary fat pad (MFP) tumor treatment model. Trastuzumab-refractory BT474 (BT474-TtzmR) cells stably expressing luciferase were inoculated into the right inguinal MFP of female nude mice and assessed bi-weekly for tumor volume via caliper measurement. Once MFP tumors reached an average tumor volume of ~100 mm3, mice were randomized and began receiving vehicle, Entrectinib (25 mg/kg), Pacritinib (50 mg/kg), or combination orally b.i.d. 5 times per week (N = 10–11/group). (B) MFP tumor growth curve for each treatment condition. V: vehicle; E: Entrectinib; P: Pacritinib; C: combination. Tx: treatment. (C) Average ex vivo MFP tumor mass at study endpoint. (D-J) Percent nuclear positivity or H-score quantitation of MFP tumors after IHC staining. For all analyses, at least 3 fields were quantified per tumor section (N = 3–5 tumors/group). (K) Representative IHC images of MFP tumors for each treatment group. All images were captured at 40x magnification. Scale bar, 50 μm. (L) Average animal weights for each treatment group throughout the course of the study. (M) Liver toxicity following systemic therapy administration was measured using ALT activity assay. Data of at least three experimental repeats are presented as mean ± SEM. One-way ANOVA with Tukey’s multiple comparison post hoc test was used to compare p values.
To determine the effects of the combination therapy on the BT474-TtzmR MFP tumors, we performed immunohistochemical (IHC) analyses to evaluate proliferation, apoptosis, and activation of TrkA/JAK2-STAT3 pathways. The combination therapy resulted in the most significant reduction in tumoral Ki-67 compared to vehicle or single agents, in which percent (%) Ki-67 nuclear positivity indicates the degree of cell proliferation (Fig. 4D). Entrectinib monotherapy did not significantly reduce cell proliferation compared to vehicle although Pacritinib significantly reduced cell proliferation compared to vehicle or Entrectinib. Furthermore, tumors treated with combination therapy had the most reduction in p-STAT3 (Y705) compared to other treatment groups while we observed the expected reduction in p-STAT3 (Y705) expression in tumors treated with Entrectinib or Pacritinib monotherapy (Fig. 4E). Next, we analyzed the tumors for the expression levels of two CSC markers SOX2 and MYC using IHC. Results showed that the combination therapy, but not monotherapies, significantly reduced nuclear positivity of SOX2 (Fig. 4F) and MYC (Fig. 4G) compared to vehicle or monotherapies. Moreover, combination therapy but not monotherapies, significantly induced tumoral apoptosis, as indicated by % cleaved PARP nuclear positivity, compared to vehicle or single agent treatments (Fig. 4H). As expected, expression of p-TrkA (Y490) (Fig. 4I) and p-JAK2 (Y1007) (Fig. 4J) were significantly reduced upon treatment with Entrectinib or Pacritinib, respectively, thereby confirming the inhibitory activity of the inhibitors used. Representative IHC images are shown in Fig. 4K. The treatments were well-tolerated as the mouse body weights remained consistent throughout the study (Fig. 4L). Furthermore, none of the treatments administered caused a significant increase in alanine transaminase (ALT) activity (Fig. 4M), indicating there is no hepatotoxicity. Additionally, ALT activity in all treatment groups was below the published ALT activities found in nude mice treated with thioacetamide, a compound known to induce liver injury [39]. Together, these observations show, for the first time, that co-inhibition of TrkA and JAK2 using the combination of Entrectinib and Pacritinib significantly suppresses orthotopic growth of HER2-positive trastuzumab-refractory breast cancer xenografts and enhances tumoral apoptosis with no toxicity.
3.5. The Entrectinib-Pacritinib combination suppresses the orthotopic growth of TNBC PDX MFP tumors
We next evaluated the therapeutic efficacy of the Entrectinib-Pacritinib combination therapy using a patient-derived xenograft (PDX) MFP tumor treatment study. PDX tumors recapitulate primary tumor biology and provide a more translational research model for evaluating treatment efficacy. We implanted ~1 mm3 fragment of BCM-3887 PDX into the right inguinal MFP of 6–7 week old female nude mice, and randomized the mice into four treatment groups: vehicle, Entrectinib (25 mg/kg), Pacritinib (50 mg/kg) or combination (5X/week) when the tumors reached about 100 mm3 at Day 21 (Fig. 5A). BCM-3887 PDX was selected for in vivo treatment study due to its high TrkA-JAK2 co-activation score, basal subtype, and the propensity to form brain metastases (Fig. 5B). Importantly, BCM-3887 expresses abundant levels of TrkA and JAK2 (Fig. 5C). Tumor growth curves showed that the mice receiving combination therapy had the slowest tumor growth compared to vehicle treatment (Fig. 5D). While Entrectinib significantly suppressed tumor growth, Pacritinib did not significantly reduce tumor growth. Tumor cell proliferation was significantly reduced in tumors that received combination therapy treatment, but not in tumors that received single agent therapies (Fig. 5E). The combination therapy significantly suppressed expression of tumoral p-STAT3 (Y705) compared to vehicle or single agents (Fig. 5F). Concordantly, nuclear positivity of SOX2 (Fig. 5G) and MYC (Fig. 5H) was most significantly reduced in tumors treated with combination therapy. The combination therapy significantly induced tumoral apoptosis compared to other treatment groups (Fig. 5I). As expected, tumoral p-TrkA (Y490) (Fig. 5J) and p-JAK2 (Y1007) (Fig. 5K) levels were significantly reduced in tumors that received Entrectinib or Pacritinib, respectively. Representative IHC images are shown in Fig. 5L. Evaluation of animal weights (Fig. 5M) and ALT activities in mouse sera collected at study endpoint (Fig. 5N) indicated no acute toxicity. Collectively, these findings suggest that co-inhibition of TrkA and JAK2 using the Entrectinib-Pacritinib combination more effectively suppresses TNBC PDX tumor growth in vivo compared to monotherapies.
Fig. 5. The Entrectinib-Pacritinib combination therapy suppresses the orthotopic growth of TNBC PDX MFP tumors.
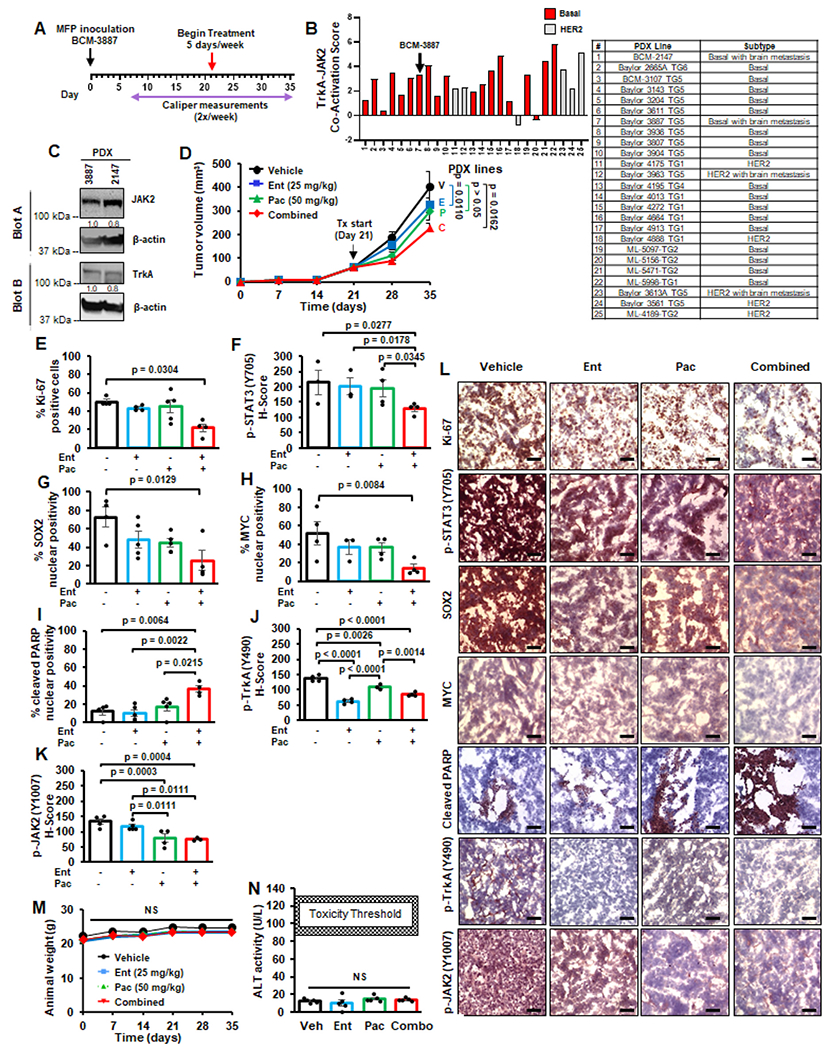
(A) Schema of PDX MFP tumor treatment model. 1-mm3 fragment of BCM-3887 PDX tissue was implanted into the right inguinal mammary fat pad of female nude mice. Once tumors reached an average volume of ~100 mm3, animals were randomized based on tumor volume and began receiving vehicle, Entrectinib only (25 mg/kg), Pacritinib (50 mg/kg), or combination via oral gavage b.i.d., 5 times/week (N = 11/group). BCM-3887 was chosen due to its basal subtype, relatively higher level of JAK2-TrkA pathway co-activation score, and propensity to develop brain metastases (B). (C) Western blot analysis of basal PDX tissues BCM-3887 BCM-2147. Densitrometry values (normalized to β-actin) are displayed below each blot. (D) MFP tumor growth curve for each treatment condition. V: vehicle; E: Entrectinib; P: Pacritinib; C: combination, Tx: treatment. (E-K) Percent nuclear positivity or H-score quantitation of MFP tumors after IHC staining. For all analyses, at least 3 fields were quantified per tumor section (N = 3–5 tumors/group). (L) Representative IHC images of MFP tumors for each treatment group. All images were captured at 40x magnification. Scale bar, 50 μm. (M) Average animal weights throughout the course of the study. (N) Liver toxicity was measured using ALT activity assay. Data are represented as mean ± SEM. One-way ANOVA with Tukey’s multiple comparison post hoc test was used to compare p values.
3.6. Combined treatment with entrectinib and pacritinib reduces TNBC metastatic burden in vivo
TNBC frequently metastasize to the brain, lung, bone, and liver [3]. To evaluate the efficacy of the Entrectinib-Pacritinib combination against multi-organ metastasis of TNBC, luciferase-expressing MDA-MB-231 TNBC cells were intracardially inoculated into female nude mice. When brain metastases were detected on day 11, mice were randomized into four treatment groups: vehicle, Entrectinib (25 mg/kg), Pacritinib (50 mg/kg), or combination, and treated as described earlier (Fig. 6A). Bioluminescent imaging (BLI) indicated that the combination therapy, but not single agent treatments, significantly reduced metastatic burden (Fig. 6B and C). Ex vivo organ analyses indicated that combination therapy, but not single agent treatments, significantly reduced brain metastases (Fig. 6D and E). Bone metastasis burden was also significantly reduced in mice that received monotherapies or combination treatment compared to vehicle (Fig. 6D and F). Liver metastases (Fig. 6D and G) were not affected by monotherapies or combination therapies. All treatments were well-tolerated as average animal weights did not significantly change throughout the study (Fig. 6H). Moreover, ALT activity was not significantly enhanced in any of the treatment groups compared to vehicle (Fig. 6I). Together, these data indicate, for the first time, that the Entrectinib-Pacritinib combination therapy reduces TNBC brain and bone metastases, and overall metastatic burden in vivo with no toxicity.
Fig. 6. Combined treatment with Entrectinib and Pacritinib reduces TNBC metastatic burden in vivo.

(A) Scheme for the intracardiac inoculation treatment model. Luciferase-expressing MDA-MB-231 cells were inoculated into the left ventricle of athymic nude mice and assessed for metastasis burden using bi-weekly bioluminescent imaging (BLI). Mice were randomized and began receiving oral administration (b.i.d.) of vehicle, Entrectinib (25 mg/kg), Pacritinib (50 mg/kg), or combination, 5 days/week (N = 12/group). (B) Average metastatic burden for each treatment group throughout the study as measured by BLI. One-way ANOVA with Tukey’s multiple comparison post-hoc test was used to compute p-values for differences in metastatic burden at study endpoint (Day 25). (C) Representative bioluminescent images of mice 25 days post-inoculation (study endpoint). (D) Representative ex vivo organ images are shown. Ex vivo brain (E), bone (F), and liver (G) bioluminescence was quantified. (H) Average mouse weights throughout the course of the study. (I) ALT assay was performed on serum collected at study endpoint to assess liver toxicity. Data are represented as mean ± SEM. One-way ANOVA with Tukey’s multiple comparison post-hoc test was used to compute p-values.
4. Discussion
In this study we report, for the first time, that TrkAis and JAK2is synergize to suppress the growth and metastasis of HER2-positive breast cancers and TNBC. We have made the following novel observations [1]: Co-inhibition of TrkA and JAK2 synergistically reduces cell viability of HER2-positive and TNBC cells in vitro [2]. Combination of TrkAis and JAK2is targets breast CSCs [3]. The Entrectinib-Pacritinib combination therapy significantly enhances apoptosis of breast cancer in vitro and in vivo [4]. Orthotopic tumor growth of trastuzumab-refractory HER2--positive BT474-TtzmR cells and basal PDX tumors is suppressed by the Entrectinib-Pacritinib combination. [5] Overall metastatic burden and brain and bone metastases of TNBC is reduced by the Entrectinib-Pacritinib combination. Through these significant and novel observations, our study identifies a novel therapeutic strategy for HER2-positive breast cancers and TNBC.
TrkA and JAK2 have been established as important therapeutic targets for several cancer types including breast cancer [7,9,15]. To date, the combination of TrkAis and JAK2is has never been investigated in any tumor or cell type although they have been used as single agent treatments or in combination with other chemotherapies [13,23,40]. Though Ruxolitinib and Pacritinib also have activity against other kinases such as JAK1 and TYK, they were selected for our studies due to their FDA-approved status and enhanced selectivity for JAK2 [20]. While Ruxolitinib, alone or in combination with chemotherapy, was evaluated for safety and efficacy in TNBC, these clinical trials reported no significant patient benefit (NCT01562873, NCT02041429, NCT02120417). Moreover, clinical evaluation of Ruxolitinib, in combination with trastuzumab, did not yield improved progression-free survival of HER2-positive breast cancer patients (NCT02066532) [41]. However, Ruxolitinib treatment frequently elicits myelosuppressive effects and led to a search for JAKis with reduced risk for adverse events. Pacritinib, another JAK2i, is FDA-approved for myelofibrosis and thrombocytopenia [24] and lacks myelosuppressive activity previously observed with Ruxolitinib. Earlier clinical trials investigating Pacritinib were terminated due to holds placed by the FDA in response to reported side effects and drug shortages, respectively (NCT02277093, NCT02342353). Pacritinib is currently being evaluated in clinical trials for graft-versus-host disease (NCT05531786), prostate cancer (NCT04635059), breast cancers (NCT04520269), and in myelofibrosis patients previously treated with Ruxolitinib (NCT03165734).
TrkAis, Entrectinib and Larotrectinib, received tissue-agnostic FDA approval for NTRK fusion-positive cancers [12,42]. Notably, Larotrectinib was the first molecularly targeted therapy to receive tissue-agnostic FDA approval [42]. Breast cancer patients who received Larotrectinib had an overall response rate (ORR) of 83 % (NCT02576431) [43]. Similarly, Entrectinib elicited an ORR of up to 71 % in patients with NTRK fusion-positive breast cancers (NCT02568267, NCT02097810) [44]. Importantly, TrkAis show potent intracranial activity in patients with brain metastases [45,46]. These clinical findings strongly suggest that TRKis may benefit patients with locally advanced or metastatic NTRK fusion-positive breast cancers. Entrectinib continues to be evaluated in breast cancer patients bearing oncogenic NTRK, ROS1, or ALK gene fusions (NCT02568267, NCT02650401). Larotrectinib is being investigated in solid tumors (NCT02576431) or in refractory solid tumors (NCT02465060). Interestingly, NTRK gene fusions are rare in breast cancers (<1 %) [47], and supports an oncogenic role for wild-type TrkA in breast cancer.
Our previous findings demonstrated a functional TrkA/JAK2-STAT3 crosstalk in which TrkA directly interacts with and phosphorylates STAT3 (Y705), thereby promoting oncogenic gene transcription and breast CSCs in tandem with JAK2 [26]. These findings provided the rationale for co-inhibition of the TrkA and JAK2 in HER2-enriched breast cancers and TNBC with TrkA/JAK2-STAT3 pathway co-activation. In agreement with previous reports that Entrectinib or Pacritinib can inhibit cell proliferation [11,21] and promote apoptosis [40], we observed that the Entrectinib-Pacritinib combination therapy most significantly reduced cell proliferation and enhanced apoptosis when compared to vehicle or single agent therapies. Moreover, the combination therapy significantly reduced orthotopic tumor growth and suppressed brain and bone metastases of TNBC cells in vivo.
Given that TrkA and JAK2 pathways can promote breast CSCs, we posited that the reduction in primary tumor growth and suppression of multi-organ metastases in vivo is due to the inhibition of CSCs by the combination therapy. Previous reports indicate that inhibition of the IL-6/JAK2/STAT3 pathway reduces the CD44high/CD24low subpopulation and reduces tumor growth in vivo [48]. While the effect of Entrectinib on breast CSCs is currently unknown, our current study demonstrates that dual targeting of TrkA and JAK2 significantly reduced the CD44high/CD24low subpopulation, reduced expression of SOX2 and MYC, and effectively suppressed the CSC subpopulation. Moreover, our data shows that the Entrectinib-Pacritinib combination therapy significantly reduced ALDH activity of breast cancer cells (Fig. 2G and H). Our Western blot findings were (Fig. 2E and F) were mirrored by IHC of MFP xenograft tumors in which mice treated with the combination therapy had significantly reduced expression of stemness markers SOX2 and MYC (Figs. 4K and 5K). These findings strongly indicate the applicability of the Entrectinib-Pacritinib combination therapy against the CSC population to inhibit growth of HER2-positive breast cancers and TNBC. To further support our findings, a future study using limiting dilution transplantation assay would demonstrate the effect of the combination therapy on the breast CSC function, and their ability to elicit tumor induction, growth, and metastasis. Similarly, a limiting dilution transplantation assay of breast cancer cells with co-overexpression of TrkA and JAK2 may be used to determine the effect of the pathway crosstalk on the CSC function in breast tumorigenesis and metastasis.
In our MFP treatment models, we found that dual inhibition of TrkA and JAK2 significantly inhibited MFP tumor growth (Fig. 4B and 5D), though the impact of the combination therapy on overall survival remains unclear. Though our MFP treatment models showed significant therapeutic efficacy when compared to monotherapies, the study was designed such that the primary endpoint is the MFP tumor volume and our samples were collected on the day of study endpoint, which would not be an appropriate model for survival analyses. A future treatment study to evaluate animal survival, wherein the primary endpoint is animal death, would serve as a more suitable model for this analysis. Alternatively, another potential study would be to evaluate whether animals inoculated with breast cancer cells co-overexpressing TrkA and JAK2 would demonstrate significantly shorter overall survival when compared to those inoculated with cells that express either TrkA or JAK2 alone.
In our metastasis treatment model, we reported that the Entrectinib-Pacritinib combination most significantly reduced brain metastases of the MDA-MB-231 cells when compared to vehicle or single agent therapies (Fig. 6). The reduction of brain metastases by either Entrectinib or Pacritinib monotherapies supports previous reports that these inhibitors are capable of penetrating the BBB [23,49]. We also observed that Entrectinib and Pacritinib, alone or in combination, significantly reduced bone metastases of MDA-MB-231 cells when compared to vehicle (Fig. 6D and F). This finding is in agreement with a previous study in which Larotrectinib reduced bone metastases in a case of pediatric congenital mesoblastic nephroma, a renal tumor that can metastasize to the bone [50]. However, while trending, we did not achieve significant differences in bone metastasis between our single agent and combination treatment groups. Though we did not observe significant differences between monotherapy and combination treatments in our metastasis model, our data demonstrated that mice that received combination therapy trended towards having the lowest overall metastatic burden (Fig. 6B and C) and lowest organ-specific metastases (Fig. 6D–G). Importantly, our combination therapy did not elicit acute hepatotoxicity with prolonged administration (Fig. 6H and I). These findings suggests that the combination therapy could be administered to patients for enhanced anti-cancer efficacy and minimal toxicity when compared to monotherapies. Moreover, our findings suggest that the Entrectinib-Pacritinib combination therapy will still benefit patients with metastatic breast cancers by reducing overall metastatic burden and has the potential to prolong patient survival. A future metastasis treatment mouse study to evaluate the effect of the Entrectinib-Pacritinib combination therapy on overall survival would further support these findings.
In conclusion, we provide novel evidence that dual targeting of TrkA and JAK2 signaling pathways using TrkAi Entrectinib and JAK2i Pacritinib is more effective than monotherapies in suppressing orthotopic tumor growth of TNBC and trastuzumab-refractory HER2-positive breast cancers, and reducing the metastatic potential of TNBC cells in vivo. The Entrectinib-Pacritinib combination therapy elicited potent anti-tumor efficacy with no hepatotoxicity. The data provide the preclinical foundation for future clinical development of novel combinations of two FDA-approved targeted therapies for TNBC and trastuzumab-refractory HER2-positive breast cancers.
Supplementary Material
Acknowledgements
We would like to acknowledge the Massagué laboratory for gifting the MDA-231-BoM, MDA-231-BrM, and MDA-231-LM breast cancer cell lines, Dr. Dihua Yu for gifting the BT474-TtzmR cell line, and Dr. Kounosuke Watabe for providing the SKBRM cell line. We acknowledge Dongqin Zhu for performing Annexin V and CD44/CD24 flow cytometry analyses. We acknowledge funding support for this project from NIH grants R01CA228137 (HWL) and 1T32CA247819-03 (KW, ATR), and DoD grants W81XWH-19-1-0072 (HWL, KW), W81XWH-20-1-0044 (HWL, HW), and W81XWH-19-1-0753 (HWL), as well as, MetaVivor Translational Research Grant (HWL), MetaVivor Early Career Investigator Award (ATR), and AACR Victoria’s Secret Global Fund for Women’s Cancers Career Development Award (ATR).
Footnotes
CRediT authorship contribution statement
Angelina T. Regua: Writing – original draft, Writing – review & editing, Methodology, Investigation, Formal analysis, Data curation, Conceptualization. Shivani Bindal: Writing – review & editing. Mariana K. Najjar: Writing – review & editing, Methodology, Formal analysis, Data curation. Chuling Zhuang: Writing – review & editing. Munazza Khan: Writing – review & editing. Austin B.J. Arrigo: Writing – review & editing, Methodology, Investigation, Formal analysis, Data curation. Anneliese O. Gonzalez: Writing – review & editing, Formal analysis. Xinhai R. Zhang: Writing – review & editing, Formal analysis. Jay-Jiguang Zhu: Writing – review & editing. Kounosuke Watabe: Writing – review & editing, Resources, Methodology. Hui-Wen Lo: Writing – review & editing, Visualization, Validation, Supervision, Resources, Project administration, Methodology, Investigation, Funding acquisition, Conceptualization.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Appendix A. Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.canlet.2024.217023.
References
- [1].Siegel RL, Miller KD, Fuchs HE, Jemal A, Cancer statistics, CA A Cancer J. Clin 72 (2022) 7–33, 2022. [DOI] [PubMed] [Google Scholar]
- [2].Howlader N, Noone AM, Krapcho M, Miller D, Brest A, Yu M, et al. SEER Cancer Statistics Review, 1975-2016, National Cancer Institute. Bethesda, MD, https://seer.cancer.gov/csr/1975_2016/, based on November 2018 SEER data submission, posted to the SEER web site, April 2019. [Google Scholar]
- [3].Perou CM, Sorlie T, Eisen MB, van de Rijn M, Jeffrey SS, Rees CA, et al. , Molecular portraits of human breast tumours, Nature 406 (2000) 747–752. [DOI] [PubMed] [Google Scholar]
- [4].Waks AG, Winer EP, Breast cancer treatment: a review, JAMA 321 (2019) 288–300. [DOI] [PubMed] [Google Scholar]
- [5].Kim YJ, Kim JS, Kim IA, Molecular subtype predicts incidence and prognosis of brain metastasis from breast cancer in SEER database, J. Cancer Res. Clin. Oncol 144 (2018) 1803–1816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Soni A, Ren Z, Hameed O, Chanda D, Morgan CJ, Siegal GP, et al. , Breast cancer subtypes predispose the site of distant metastases, Am. J. Clin. Pathol 143 (2015) 471–478. [DOI] [PubMed] [Google Scholar]
- [7].Regua AT, Doheny D, Arrigo A, Lo HW, Trk receptor tyrosine kinases in metastasis and cancer therapy, Discov. Med 28 (2019) 195–203. [PubMed] [Google Scholar]
- [8].Lange AM, Lo HW, Inhibiting TRK proteins in clinical cancer therapy, Cancers 10 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Kyker-Snowman K, Hughes RM, Yankaskas CL, Cravero K, Karthikeyan S, Button B, et al. , TrkA overexpression in non-tumorigenic human breast cell lines confers oncogenic and metastatic properties, Breast Cancer Res. Treat 179 (2020) 631–642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Scott LJ, Larotrectinib: first Global approval, Drugs 79 (2019) 201–206. [DOI] [PubMed] [Google Scholar]
- [11].Ardini E, Menichincheri M, Banfi P, Bosotti R, De Ponti C, Pulci R, et al. , Entrectinib, a pan-TRK, ROS1, and ALK inhibitor with activity in multiple molecularly Defined cancer Indications, Mol. Cancer Therapeut 15 (2016) 628–639. [DOI] [PubMed] [Google Scholar]
- [12].Entrectinib OK’d for cancers with NTRK fusions, NSCLC, Cancer Discov. 9 (2019) OF2. [DOI] [PubMed] [Google Scholar]
- [13].Hong DS, DuBois SG, Kummar S, Farago AF, Albert CM, Rohrberg KS, et al. , Larotrectinib in patients with TRK fusion-positive solid tumours: a pooled analysis of three phase 1/2 clinical trials, Lancet Oncol. 21 (2020) 531–540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Balko JM, Giltnane JM, Wang K, Schwarz LJ, Young CD, Cook RS, et al. , Molecular profiling of the residual disease of triple-negative breast cancers after neoadjuvant chemotherapy identifies actionable therapeutic targets, Cancer Discov. 4 (2014) 232–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Balko JM, Schwarz LJ, Luo N, Estrada MV, Giltnane JM, Davila-Gonzalez D, et al. , Triple-negative breast cancers with amplification of JAK2 at the 9p24 locus demonstrate JAK2-specific dependence, Sci. Transl. Med 8 (2016) 334ra53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Loh CY, Arya A, Naema AF, Wong WF, Sethi G, Looi CY, Signal transducer and activator of transcription (STATs) proteins in cancer and Inflammation: Functions and therapeutic implication, Front. Oncol 9 (2019) 48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Huang B, Lang X, Li X, The role of IL-6/JAK2/STAT3 signaling pathway in cancers, Front. Oncol 12 (2022) 1023177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Mascarenhas J, Hoffman R, Ruxolitinib: the first FDA approved therapy for the treatment of myelofibrosis, Clin. Cancer Res 18 (2012) 3008–3014. [DOI] [PubMed] [Google Scholar]
- [19].Raedler LA, Jakafi (ruxolitinib): first FDA-approved Medication for the treatment of patients with polycythemia vera, Am Health Drug Benefits 8 (2015) 75–79. [PMC free article] [PubMed] [Google Scholar]
- [20].Quintas-Cardama A, Vaddi K, Liu P, Manshouri T, Li J, Scherle PA, et al. , Preclinical characterization of the selective JAK1/2 inhibitor INCB018424: therapeutic implications for the treatment of myeloproliferative neoplasms, Blood 115 (2010) 3109–3117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Hart S, Goh KC, Novotny-Diermayr V, Tan YC, Madan B, Amalini C, et al. , Pacritinib (SB1518), a JAK2/FLT3 inhibitor for the treatment of acute myeloid leukemia, Blood Cancer J. 1 (2011) e44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Jeon JY, Zhao Q, Buelow DR, Phelps M, Walker AR, Mims AS, et al. , Preclinical activity and a pilot phase I study of pacritinib, an oral JAK2/FLT3 inhibitor, and chemotherapy in FLT3-ITD-positive AML, Invest. N. Drugs 38 (2020) 340–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Jensen KV, Cseh O, Aman A, Weiss S, Luchman HA, The JAK2/STAT3 inhibitor pacritinib effectively inhibits patient-derived GBM brain tumor initiating cells in vitro and when used in combination with temozolomide increases survival in an orthotopic xenograft model, PLoS One 12 (2017) e0189670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Lamb YN, Pacritinib: first approval, Drugs 82 (2022) 831–838. [DOI] [PubMed] [Google Scholar]
- [25].Haile WB, Gavegnano C, Tao S, Jiang Y, Schinazi RF, Tyor WR, The Janus kinase inhibitor ruxolitinib reduces HIV replication in human macrophages and ameliorates HIV encephalitis in a murine model, Neurobiol. Dis 92 (2016) 137–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Regua AT, Aguayo NR, Jalboush SA, Doheny DL, Manore SG, Zhu D, et al. , TrkA interacts with and phosphorylates STAT3 to enhance gene transcription and promote breast cancer stem cells in triple-negative and HER2-enriched breast cancers, Cancers 13 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Xing F, Liu Y, Sharma S, Wu K, Chan MD, Lo HW, et al. , Activation of the c-Met pathway Mobilizes an Inflammatory Network in the brain Microenvironment to promote brain metastasis of breast cancer, Cancer Res. 76 (2016) 4970–4980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Zhang S, Huang WC, Li P, Guo H, Poh SB, Brady SW, et al. , Combating trastuzumab resistance by targeting SRC, a common node downstream of multiple resistance pathways, Nat. Med 17 (2011) 461–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Bos PD, Zhang XH, Nadal C, Shu W, Gomis RR, Nguyen DX, et al. , Genes that mediate breast cancer metastasis to the brain, Nature 459 (2009) 1005–1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Minn AJ, Gupta GP, Siegel PM, Bos PD, Shu W, Giri DD, et al. , Genes that mediate breast cancer metastasis to lung, Nature 436 (2005) 518–524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Kang Y, Siegel PM, Shu W, Drobnjak M, Kakonen SM, Cordon-Cardo C, et al. , A multigenic program mediating breast cancer metastasis to bone, Cancer Cell 3 (2003) 537–549. [DOI] [PubMed] [Google Scholar]
- [32].Butti R, Gunasekaran VP, Kumar TVS, Banerjee P, Kundu GC, Breast cancer stem cells: biology and therapeutic implications, Int. J. Biochem. Cell Biol 107 (2019) 38–52. [DOI] [PubMed] [Google Scholar]
- [33].Senbanjo LT, Chellaiah MA, CD44: a Multifunctional cell surface Adhesion receptor is a Regulator of progression and metastasis of cancer cells, Front. Cell Dev. Biol 5 (2017) 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Sheridan C, Kishimoto H, Fuchs RK, Mehrotra S, Bhat-Nakshatri P, Turner CH, et al. , CD44+/CD24− breast cancer cells exhibit enhanced invasive properties: an early step necessary for metastasis, Breast Cancer Res. 8 (2006) R59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Toledo-Guzman ME, Hernandez MI, Gomez-Gallegos AA, Ortiz-Sanchez E, ALDH as a stem cell marker in solid tumors, Curr. Stem Cell Res. Ther 14 (2019) 375–388. [DOI] [PubMed] [Google Scholar]
- [36].Koopman G, Reutelingsperger CP, Kuijten GA, Keehnen RM, Pals ST, van Oers MH, Annexin V for flow cytometric detection of phosphatidylserine expression on B cells undergoing apoptosis, Blood 84 (1994) 1415–1420. [PubMed] [Google Scholar]
- [37].Kaufmann SH, Desnoyers S, Ottaviano Y, Davidson NE, Poirier GG, Specific proteolytic cleavage of poly(ADP-ribose) polymerase: an early marker of chemotherapy-induced apoptosis, Cancer Res. 53 (1993) 3976–3985. [PubMed] [Google Scholar]
- [38].Lazebnik YA, Kaufmann SH, Desnoyers S, Poirier GG, Earnshaw WC, Cleavage of poly(ADP-ribose) polymerase by a proteinase with properties like ICE, Nature 371 (1994) 346–347. [DOI] [PubMed] [Google Scholar]
- [39].Ackerman Z, Pappo O, Link G, Glazer M, Grozovski M, Liver toxicity of thioacetamide is increased by hepatocellular iron overload, Biol. Trace Elem. Res 163 (2015) 169–176. [DOI] [PubMed] [Google Scholar]
- [40].Sohn SH, Sul HJ, Kim BJ, Kim HS, Zang DY, Entrectinib induces apoptosis and inhibits the epithelial-Mesenchymal Transition in Gastric cancer with NTRK overexpression, Int. J. Mol. Sci 23 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Kearney M, Franks L, Lee S, Tiersten A, Makower DF, Cigler T, et al. , Phase I/II trial of ruxolitinib in combination with trastuzumab in metastatic HER2 positive breast cancer, Breast Cancer Res. Treat 189 (2021) 177–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Larotrectinib OK’d for cancers with TRK fusions, Cancer Discov. 9 (2019) 8–9. [DOI] [PubMed] [Google Scholar]
- [43].Rosen EY, Italiano A, Juric D, Reeves JA, Dima L, Brega N, et al. , Abstract PS11-06: efficacy and safety of larotrectinib in patients with TRK fusion breast cancer, Cancer Res. 81 (2021). PS11–06. [Google Scholar]
- [44].Lu J, Blakely CM, Le Tourneau C, Waqar SN, Bauer TM, de Braud F, et al. , Abstract PS11-28: efficacy and safety of entrectinib in NTRK fusion-positive (NTRK-fp) breast cancer, Cancer Res. 81 (PS11–28–PS11–28) (2021). [Google Scholar]
- [45].Rosen EY, Schram AM, Young RJ, Schreyer MW, Hechtman JF, Shu CA, et al. , Larotrectinib demonstrates CNS efficacy in TRK fusion-positive solid tumors, JCO Precis Oncol 3 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Doebele RC, Drilon A, Paz-Ares L, Siena S, Shaw AT, Farago AF, et al. , Entrectinib in patients with advanced or metastatic NTRK fusion-positive solid tumours: integrated analysis of three phase 1-2 trials, Lancet Oncol. 21 (2020) 271–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Westphalen CB, Krebs MG, Le Tourneau C, Sokol ES, Maund SL, Wilson TR, et al. , Genomic context of NTRK1/2/3 fusion-positive tumours from a large real-world population, npj Precis. Oncol 5 (2021) 69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Marotta LL, Almendro V, Marusyk A, Shipitsin M, Schemme J, Walker SR, et al. , The JAK2/STAT3 signaling pathway is required for growth of CD44(+)CD24(−) stem cell-like breast cancer cells in human tumors, J. Clin. Invest 121 (2011) 2723–2735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Menichincheri M, Ardini E, Magnaghi P, Avanzi N, Banfi P, Bossi R, et al. , Discovery of entrectinib: a New 3-Aminoindazole as a potent Anaplastic Lymphoma kinase (ALK), c-ros Oncogene 1 kinase (ROS1), and pan-Tropomyosin receptor kinases (Pan-TRKs) inhibitor, J. Med. Chem 59 (2016) 3392–3408. [DOI] [PubMed] [Google Scholar]
- [50].Halalsheh H, McCarville MB, Neel M, Reynolds M, Cox MC, Pappo AS, Dramatic bone remodeling following larotrectinib administration for bone metastasis in a patient with TRK fusion congenital mesoblastic nephroma, Pediatr. Blood Cancer 65 (2018) e27271. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.