

Abstract
In recent years, major advances have been made in the understanding of the cellular and molecular mechanisms driving pulmonary vascular remodelling in various forms of pulmonary hypertension, including pulmonary arterial hypertension, pulmonary hypertension associated with left heart disease, pulmonary hypertension associated with chronic lung disease and hypoxia, and chronic thromboembolic pulmonary hypertension. However, the survival rates for these different forms of pulmonary hypertension remain unsatisfactory, underscoring the crucial need to more effectively translate innovative scientific knowledge into healthcare interventions. In these proceedings of the 7th World Symposium on Pulmonary Hypertension, we delve into recent developments in the field of pathology and pathophysiology, prioritising them while questioning their relevance to different subsets of pulmonary hypertension. In addition, we explore how the latest omics and other technological advances can help us better and more rapidly understand the myriad basic mechanisms contributing to the initiation and progression of pulmonary vascular remodelling. Finally, we discuss strategies aimed at improving patient care, optimising drug development, and providing essential support to advance research in this field.
Shareable abstract
This task force report delves into recent developments in the field of pathology and pathophysiology in pulmonary hypertension, prioritising them while questioning their relevance to different subsets of PH https://bit.ly/4ewUuzw
Pathology and pathobiology: a brief overview
Pulmonary vascular remodelling is a hallmark shared by all forms of pulmonary hypertension (PH), characterised by a spectrum of structural and functional changes occurring primarily in the distal part of the pulmonary circulation. These changes can predominantly affect the pulmonary arterioles in the case of pulmonary arterial hypertension (PAH) or the small-to-medium-sized veins and capillaries in conditions like pulmonary veno-occlusive disease, pulmonary capillary haemangiomatosis, or PH associated with left heart disease. When these changes affect a substantial portion of the pulmonary circulation, they lead to increased pulmonary vascular resistance (PVR), subsequently influencing pulmonary artery pressure, and affecting the structure and function of the right ventricle [1].
At the time of diagnosis, PAH patients present established, potentially irreversible pulmonary vascular lesions. However, at present, no causal connection or association has been identified to link a specific type of lesion to the marked increase in PVR observed in PAH [2–4]. Some mapping studies have localised certain obliterative lesions to branching points or supernumerary arteries; however, the distribution of vascular lesions along the fractal pulmonary artery remains too unclear to rely on these elements. Another challenge is related to the histological heterogeneity of PAH and across different PH groups. This heterogeneity stems not only from inherent clinical factors specific to each patient, such as age at onset, clinical presentation, rate of progression, treatment utilisation and combination for different patients, treatment response and comorbidities, but potentially also from the diversity of potentially possible aetiologies suspected to lead to PAH or PH. This heterogeneity, combined with the rarity of the disease and limited access to lung tissue, clearly hampers therapy development, but it increasingly directs us towards personalised and targeted therapy, which requires the identification of reliable and reproducible biomarkers, and perhaps the use of emerging technologies, as discussed in this article. Despite these limitations, it is clear that a key characteristic of pulmonary vascular remodelling is the accumulation of vascular and immune cells inside and around the vascular walls, orchestrated by a dynamic network of pathogenic molecular mechanisms [2, 5–8]. This process is driven by changes in the secretion of vasoconstrictors and vasodilators by pulmonary endothelial cells (ECs), as well as imbalances in factors regulating pulmonary artery (PA) smooth muscle cells (SMCs) and fibroblast growth, thrombotic and inflammatory mediators, and signalling pathways such as the transforming growth factor (TGF)-β family affecting bone morphogenetic proteins (BMP)–small mothers against decapentaplegic (Smad)1/5/8 and activin–Smad2/3 signalling pathways. High shear stress, chronic hypoxia, genetic predispositions and dysregulation of the TGF-β family all serve as crucial contributing factors to this process. Currently, several novel therapies are undergoing evaluation at various stages of the drug development pipeline, offering promising prospects for improving outcomes in PAH/PH patients [9].
The progress in understanding and treating different forms of PH, including those associated with chronic lung diseases, is hindered by a relative scarcity of available comparative studies between characteristic histological lesions across these different PH groups. In chronic high-altitude PH, assumed to be primarily mediated by hypoxia, autopsies of long-term high-altitude dwellers have revealed hypertrophy of the media of proximal pulmonary arteries and increased muscularisation of distal arterioles, resulting in an increased ratio of distal to proximal arteries [10]. Similarly, patients with PH associated with COPD (COPD-PH) exhibit hypertrophy of the tunica media layer and the development of a smooth muscle layer with a new internal elastic lamina in smaller arterioles [11, 12]. The pathology of COPD-PH is also characterised by an accumulation of vascular cells in the intima and media. As observed in animals exposed to cigarette smoke or bleomycin, COPD and idiopathic pulmonary fibrosis (IPF) patients undergoing lung transplantation with severe PH exhibit increased microvessel remodelling compared to those with moderate PH or no PH, which differs from the characteristic PAH remodelling patterns [1]. The reason why the presence of plexiform lesions is generally not observed in pathological studies of PH in various lung diseases, including IPF-PH, COPD-PH, and others, remains a true mystery and questions our current perceptions of their functional role and origin. Indeed, their frequent presence in lungs of idiopathic and heritable PAH patients could indicate the predominance of distinct pathomechanisms: Galambos et al. [13] have reported evidence for bronchopulmonary anastomoses, and thus shunting, within plexiform lesions, connecting the systemic lung vasculature to the pulmonary vasculature. Similar observations were published by Ghigna et al. [2], who observed large plexiform-like lesions, mostly in PAH patients carrying BMPR2 mutations, connecting vasa vasorum and bronchial arteries to pulmonary veins in a bypassing fashion. Recent publications have advanced our understanding of vascular lesions by employing synchrotron-based micro-computed tomography (CT) for three-dimensional (3D) analysis, revealing the potential presence of four types of plexiform lesions [14]. Illustrated in figure 1, these lesions demonstrate the versality of synchrotron-based micro-CT. Additionally, this imaging technique can be complemented with subsequent sectioning and methods such as histology, immunohistochemistry or in situ hybridisation, offering a comprehensive approach to exploring disease mechanisms. It is suggested that certain types of plexiform lesions may be exclusive to PAH, while others could be present in different PH groups [14]. Likewise, there is growing body of evidence indicating vascular rarefaction in PAH, contributing to elevated PVR, which is also observed to varying degrees in other types of PH [14–16]. However, many of these studies lack lung stereology and high-resolution 3D pulmonary vascular imaging, which is essential for validating their findings and ensuing their accuracy. This limitation could potentially be mitigated in the future with the ongoing advancement of computer-assisted morphometric analyses, as discussed later in the article. To discern the subtle distinctions in pulmonary vascular remodelling across various form of PH, it is essential to conduct further rigorous studies that can lead to definitive conclusions.
FIGURE 1.
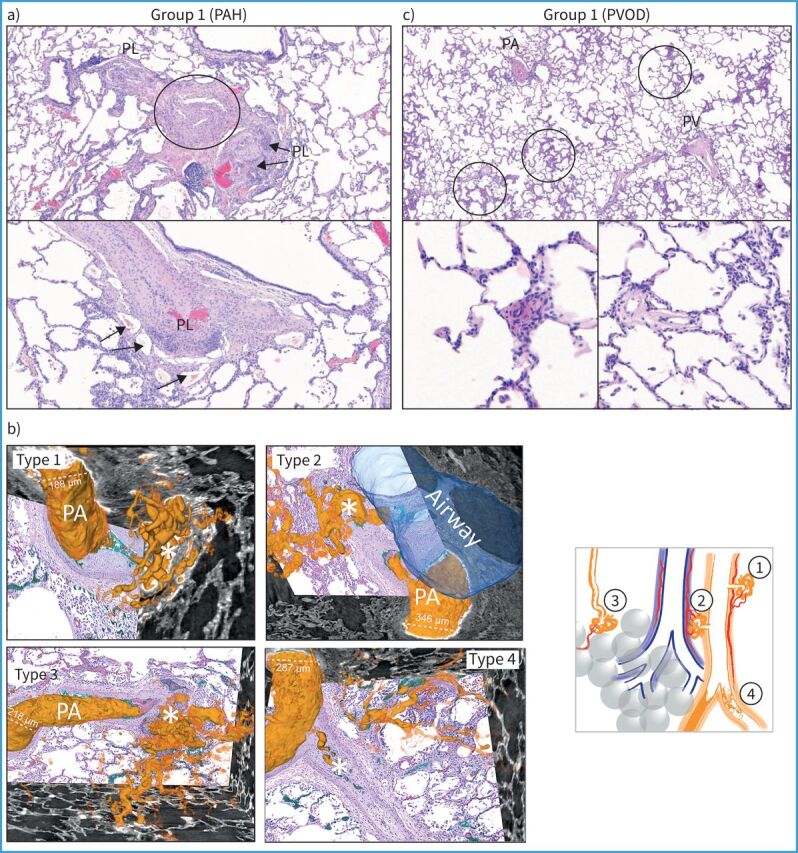
Spectrum of pulmonary vascular diseases in pulmonary hypertension (PH): histology of lung samples from the five groups of the clinical classification. a) In pulmonary arterial hypertension (PAH), plexiform lesions (PL) are a hallmark of the disease; the upper photo is centred on a pulmonary artery (PA, circled) which shows dichotomous branching and development of PL on both branches, to the left and to the right. Note that the plexiform core of the right-sided lesion appears to involve the outer layer (adventitia) of the PA (arrows). The lower image highlights this adventitial involvement and depicts the PL's vicinity to vasa vasorum (arrows). b) Schematic illustration of four types of plexiform lesions (*) identified with synchrotron-based imaging [14]. Type 1 are present in supernumerary arteries and typically connect to the vasa vasorum and type 2 connect pulmonary arteries and peribronchial vessels. Connections to the bronchial circulation from those two types of lesions may relieve suprasystemic pressure and thereby protect the right ventricle, but shunting will cause desaturation. Type 3 lesions are present at abrupt ends of distal pulmonary arteries with runoff through dilated pulmonary venules, and type 4 are obstructed pulmonary arteries with recanalisation. c) Lungs from patients with pulmonary veno-occlusive disease (PVOD) show remodelling of pulmonary veins (PV), PAs, microvessels <70 µm in diameter and capillaries. Note the patchy distribution of pulmonary capillary haemangiomatosis (upper image, circled), which corresponds to characteristic centrilobular ground-glass opacities on high-resolution computed tomography of the chest. The lower left image depicts a muscularised arteriole (<70 µm diameter); the lower right image a venule with intimal fibrosis of the same patient. (Continues on following page.)
FIGURE 1.

Continued. d) Lungs from a patient with PH associated with heart failure with preserved ejection fraction (group 2) showing lung oedema, hemosiderin-laden macrophages (HM) and important muscularisation of PV and venules that may reach the degree of arterialisation on PVs. e) In PH associated with idiopathic pulmonary fibrosis (IPF), lung parenchyma shows extensive collagen-rich fibrosis, destruction of the elastic fibres and distortion of the alveoloseptal architecture (upper image; collagen is orange, elastic fibres are black;Elastica–van Gieson–saffron staining). Smooth muscle actin staining highlights the increase in interstitial smooth muscle cells and myofibroblasts, as well as heavily remodelled PA in this area (lower left image); note that interstitial and PA remodelling are often associated in IPF. Staining with collagen 1A1 highlights typical fibroblastic foci in this same area, which are frequently located in the interstitial space of small airways and considered to be the activity hotspots of IPF. Note their vicinity to remodelled PA. f) Lungs from chronic thromboembolic pulmonary hypertension (CTEPH) patients typically show PA with thrombotic lesions that are partially organised/recanalised (upper image). PV may show remodelling, too, which is probably due to functional bronchopulmonary anastomoses and increase in venous drainage of systemic blood from bronchial arteries and vasa vasorum, in analogy to PAH (see earlier). Of note, pre- and post-capillary microvessels (arterioles and venules, lower image) show secondary remodelling that is not due to embolism or thrombosis, but to increase of shear stress and pressure. g) An example of PH group 5 (sarcoidosis-associated PH (SAPH)). Several mechanisms are responsible for the increase of pulmonary vascular resistance in this condition, including hypoxaemia, interstitial fibrosis, vascular wall remodelling resembling PAH and PVOD, but also compression of pulmonary vessels by chronic granulomatous inflammation, as shown. Note at least three granulomas with epithelioid histiocytes and Langhans giant cells (★) surrounding the PA, which displays medial thickening and near-occlusive intimal fibrosis. The lower image depicts an endothelial staining focusing on a PV visibly compressed by granulomas. All photographed cases were collected from different institutions and with courtesy of Anton Vonk-Nordegraaf (University of Amsterdam, Amsterdam, the Netherlands), Marc Humbert (University Paris-Saclay, Le Kremlin-Bicetre, France) and Werner Seeger (University of Giessen, Giessen, Germany), and their pathology departments. LHD: left heart disease; CLD: chronic lung disease.
Exploring innovative pathways for advancing the understanding of PH pathophysiology
Unlocking pulmonary vascular lesions in PH with single-cell and spatial transcriptomics
The advancements in transcriptomic analyses, at both the single-cell (scRNAseq) and single-nucleus (snRNASeq) levels, have led to substantial progress in recent years [17, 18]. These developments have fundamentally reshaped our traditional histological atlases of the lung and pulmonary vasculature. As exemplified by the pioneering work in creating the first comprehensive molecular cell atlas of the human lung from scRNAseq [19], these atlases provide sets of molecular markers. They not only help in categorising different cell types and uncovering the molecular identities of the constituent lung cells, but also offer insights into their functions and alterations in the context of pulmonary vascular pathologies [4–6]. Subsequent studies delved further into this realm [20–28], unveiling numerous distinct subpopulations of cells previously categorised as endothelial, smooth muscle, or fibroblasts, highlighting cellular diversity even within the same cell category [26, 29]. This level of analytical precision provides an unprecedented understanding of the composition and modifications of various cellular microenvironments. For example, using a multi-omics integrative approach, an initial analysis of 96 explant lungs with PH alongside 52 control lungs was conducted, revealing a novel gene subnetwork associated with clinicopathological severity, genetic risk, specific vascular cell types and potentially new therapeutic targets in PAH. [20]. Another study focused on the structural cells of vessels associated with PH using multicolour immunofluorescent staining combined with confocal imaging of precision-cut human lung tissue sections [21]. This approach revealed an intermediate phenotype between PA-SMCs and adventitial fibroblasts, along with an imbalance in their communication networks with immune cells. In this context, an important step forward has been made through the application of multiplexed ion beam imaging by time-of-flight, leading to the creation of the first architectural map of PAH lungs. This work has pinpointed neutrophils as pivotal in exacerbating the severity of vascular pathology in patients with BMPR2 mutations, shedding light on alterations in endothelial cells. Additionally, it has underscored the involvement of TIM-3+ T-cells, IDO-1+TIM-3+SAMHD1+ dendritic cells and monocyte-derived dendritic cells as novel contributors to immune dysregulation, thus propelling the progression of PAH [30]. Numerous publicly available scRNAseq datasets, sometimes accompanied by multi-omic approaches at the proteomic level, are of significant value in comprehending the complex interactions within various PH groups. A compelling study examined the influence of hypoxia and shear stress on cultured pulmonary ECs, with a specific focus on COPD [31]. These in vitro analyses revealed 55 proteins that appear to play a role in modulating the responses of pulmonary microvascular endothelium to hypoxia under shear stress. To validate the relevance of these findings for COPD, publicly available scRNAseq data from healthy lungs and lungs of COPD patients were analysed, confirming the dysregulation of five mRNAs out of the 55 proteins in COPD [31]. This correlation between in vitro results and publicly accessible scRNAseq datasets could significantly emphasise the pathological relevance in human diseases. The recently unveiled integrated Human Lung Cell Atlas has accentuated the impact of individual variations on molecular traits within lung tissues. This promising development holds the potential to pinpoint valuable biomarkers for enhancing the prediction of patient responses to therapy, thereby underscoring the potential of personalised medical interventions in improving treatment outcomes [32].
While single-cell analysis continues to be pivotal in mapping lung cellular composition and profiling cell-specific expression in disease conditions, understanding tissue organisation within its spatial context is equally indispensable. Spatial transcriptomics exhibits substantial promise for high-throughput, spatially resolved profiling of proteins and RNAs, even in formalin-fixed, paraffin-embedded samples. This technique enables the identification and mapping of gene expression patterns within individual cells in their native tissue milieu [33, 34]. Studying these aspects enhances our understanding of tissue organisation, especially in conditions like PH, which exhibit significant diversity in pulmonary vascular remodelling. In idiopathic PAH (IPAH), four distinct lesions (plexiform, obliterative, intimal medial hypertrophy and adventitial remodelling) are consistently observed [2, 8]. Digital spatial profiling analysis of IPAH lesions reveals significant molecular heterogeneity, particularly in plexiform and adventitial lesions, which exhibit the highest number of differentially expressed genes compared to intima-media hypertrophy and obliterative lesions. Plexiform lesions and IPAH adventitia are enriched for genes associated with or mutated in IPAH, notably those related to BMP/TGF-β signalling, extracellular matrix and endothelial–mesenchymal transition [35]. Cellular deconvolution of the transcriptomically profiled lesions reveals variations in the number and signature of vascular and inflammatory cells, contributing to the diversity of lesional gene expression.
In conclusion, it is crucial to emphasise the importance of a precise understanding of the clinical phenotypes of patients studied through these technologies, as well as protein-scale validation. Ongoing studies in the field of spatial transcriptomics are poised to provide invaluable working foundations using carefully characterised patient data (figure 2). These significant advances in single-cell analysis are reshaping our comprehension of pathophysiology and paving the way for promising prospects in this emerging post-omics era. However, when discussing the prospects of these technologies, it is essential to consider their limitations, such as the descriptive nature of omics and the influence of tissue digestion protocols on the derived cell types.
FIGURE 2.

Paving the way for precision medicine in pulmonary hypertension. Schematic illustration underscoring the imperative of bridging omics data, be it genomic, epigenomic, transcriptomic, proteomic, metabolomic or lipidomic, with comprehensive clinical and demographic information. This approach, whether conducted in bulk, at single-cell, single-nuclei or spatial resolution, enhances data readability and interpretation, steering medicine closer to precise and individualised practices.
Exploring genetic and environmental risk factors with iPSCs and in vitro models in PH research
Many genes, particularly those coding for TGF-β superfamily members, are implicated in mutations that elevate the risk of PAH development [36, 37]. However, understanding the intricacies of these genes, their association with PAH, and why certain mutations do not consistently lead to PAH remains a significant challenge [38]. In this context, delving deeper into potential modifiers associated with common genetic variants, which could provide protection or compensate for known mutations [39, 40], is crucial. Alongside genetic modifiers, environmental factors play a significant role in influencing pulmonary vascular remodelling. These factors encompass haemodynamic forces, shear stress, drugs including those implicated in cancer [41–43], toxins, hormones, inflammation, oxidative stress and the ageing of the pulmonary circulation. Understanding these areas is essential for unravelling the complexity of PAH and the underlying processes contributing to its development.
Various experimental strategies, including in vitro microfluidic systems, cell co-cultures and animal models, are routinely employed to unravel the intricate mechanisms underlying the pulmonary vascular remodelling associated to PAH. The integration of novel technologies holds significant potential to advance such research, with induced pluripotent stem cells (iPSCs) emerging as a particularly promising tool for uncovering the genetic and environmental factors that influence the penetrance of a mutation in causing disease. These iPSCs, derived from somatic cells of patients, whether harbouring mutations or not, offer versatility by differentiating into diverse cell types, including ECs, SMCs, pericytes, fibroblasts, cardiomyocytes and myeloid cells. Manipulating iPSCs using techniques such as CRISPR/Cas9 or viral infection enhances their adaptability. Cutting-edge in vitro models, both 2D and 3D, have been established using iPSCs derived from both PAH patients and healthy controls, specifically transforming them into iPSC-cardiomyocytes (CMs). Notably, these models have unveiled alterations in beating frequency, contraction and relaxation times in iPSC-CMs from patients with mutations in the BMPR2 gene [44]. Moreover, iPSC-ECs from seven PAH patients underwent screening with >4500 United States Food and Drug Administration-approved drugs and bioactive compounds, leading to the identification of the AG1296 agent. This agent, by enhancing BMP receptors and downstream signalling while suppressing PAH PA-SMC proliferation, demonstrates the potential for targeted and personalised PAH therapy [45]. This study emphasises the significance of disease modelling through iPSCs in the context of targeted and personalised PAH therapy, emphasising the role of high-throughput libraries.
In recent years, 3D cell-culture models have emerged as a groundbreaking development, surpassing traditional 2D monolayers. These models not only enhance cell interactions but also closely mimic in vivo architecture, effectively bridging the gap between 2D cultures and animal models. Their potential spans a wide range of applications, including drug discovery and the investigation of signalling pathways, among others. For instance, while 2D and 3D cell cultures are suitable for exploring cellular responses to drugs, toxins and biomaterials, it is essential to note that only 3D cultures exhibit a hierarchical structure and cellular heterogeneity that can faithfully replicate in vivo cell morphology and function. This includes vital aspects like proliferation, differentiation, gene expression, cell–cell interactions and diffusion barriers. One innovative 3D approach is the application of organoids. Organoids, miniature organs grown from cells, offer versatility and have played a key role in various research areas, including the study of respiratory virus infections [46]. They can represent both healthy and diseased states, with potential applications in understanding lung development and personalised medical approaches. However, challenges such as technology maturity and cost remain, requiring further progress to enhance reproducibility and enable high-throughput analysis. While organoids show promise for advancing our understanding of lung physiology, their application in PAH research is currently limited. This is primarily due to the fact that most studies focus on replicating hollow organs like the gut and alveoli, and many organoids lack vascularisation. Approaches to vascularise lung organoids, such as by incorporating vascular ECs into developing lung organoids or by co-culturing lung organoids with blood vessel organoids, will enable us to further develop this model to recapitulate PH in the context of chronic lung disease and hypoxia. For PAH, bio-engineered models such as artery-on-a-chip and lung microvascular networks on a chip are considered more relevant [47–49]. Therefore, future research may benefit from focusing on vessel-on-a-chip models to address the complexities of lung vascular research.
Another unique method to study intact lung tissue in detail in vitro is lung tissue organ culture [45] and more recently precision-cut lung slices (PCLS) [50, 51]. PCLS captures the multifaceted nature of lung tissue, giving a closer look at how various cells interact in both healthy and diseased states. It is like obtaining a snapshot of the lung's condition at the exact moment of slicing, allowing researchers to delve deep into disease mechanisms at different stages. Moreover, these slices preserve the intricate cellular interplays and structural nuances of the lung, facilitating meticulous study of various lung components and their responses. However, caution is needed, as processing may induce unwanted responses, which can be mitigated with careful handling and preparation. PCLS hold vast potential, being adaptable to mimic various diseases and serve as a framework for complex 3D cell studies [51, 52]. Illustrating its effectiveness, Chelladurai et al. [53] demonstrated the utility of PCLS in evaluating drug efficacy in human PAH, while Jandl et al. [54] showcased its relevance in PH associated with lung fibrosis. Jandl et al. [54] aimed to validate the therapeutic potential of collagen resolution in the fibrotic human lung through ex vivo experiments on end-stage fibrotic lung tissue. PCLS cultured with KRN7000-treated (natural killer T-cell activation) peripheral blood mononuclear cells (PBMCs) for 3 days revealed a visibly lighter and looser lung structure under stereomicroscopy. Masson's trichrome staining indicated reduced collagen in the parenchyma and remodelled pulmonary arteries, with a detailed vessel examination revealing decreased collagen-I deposition between SMCs via modulation of tissue inhibitors of metalloproteinases in PBMCs treated with KRN7000. Recently, Grobs et al. [55] successfully induced specific aspects of vascular remodelling in PCLS from non-PAH patients using a growth factor cocktail, showcasing the potential of utilising PCLS from non-PAH patients to study specific features of PAH. Moreover, PCLS can be cryopreserved for convenient, on-demand usage in regulatory contexts, and these slices can undergo treatment with small-molecule inhibitors or be subjected to genetic manipulation using short interfering RNAs/gapmers, further expanding their utility and potential in diverse research applications [56]. Thus, in the context of PH research, these innovative ex vivo approaches represent a significant advance from traditional methods and hold the promise of opening new horizons.
Unveiling molecular pathways, growth factors and epigenetic modifications
Enhancing the comprehension of cellular compartments is not only crucial for unravelling the intricacies of life, but also forms the bedrock for gaining profound insights into disease pathophysiology. The cell membrane, signalling pathways, the nucleus, mitochondria and epigenetic processes are the foundational pillars of cellular biology, providing a window into the intricate mechanisms through which cells communicate, replicate and regulate themselves. In recent years, significant progress has been made in the field of PAH/PH at each of these levels.
There has been a growing recognition that dysregulation in the expression and interaction of various receptors is centrally implicated in the observed accumulation of cells within arterial walls. The upregulation and overactivation of diverse receptor tyrosine kinases (RTKs), including epidermal growth factor receptors, vascular endothelial growth factor receptors, platelet-derived growth factor receptors, and fibroblast growth factor receptors, have been documented in various vascular wall cells in both experimental models and patients with PAH [1]. The inhibition of some of these RTKs has demonstrated benefits against the progression of the disease by reducing mitogen-activated protein kinase, Janus kinase/signal transducer and activator of transcription and phosphoinositide 3-kinase/Akt. However, this field currently lacks tools tailored to PAH that normalise without completely blocking pathways essential for the proper functioning of pulmonary vascular cells, especially those that do not affect the hypertrophic adaptive response of the right ventricle [1]. Addressing this gap requires significant efforts to identify or create the most effective tyrosine kinase inhibitors, antibodies, or other agents capable of normalising the excessively robust signals of growth factors in PAH. Considering the shared nature of these pathways across multiple cells and organs, a more targeted administration to the lungs, such as through inhalation, may offer a promising approach to overcome the challenges faced by our field. Equally crucial is gaining a deeper understanding of the roles played by phosphatases and various G protein-coupled receptors [57–60]. Exploring potential pathways on that front may provide valuable insights in the future.
Beyond RTKs, disturbances in various serine-threonine kinases, particularly those associated with the BMP/TGF-β family (figure 3), play a significant role in the pulmonary vascular remodelling associated to PAH and warrant thorough investigation. In the BMP/TGF-β family, these malfunctions encompass defects affecting the expression of several type I, II and III receptors [61]. Among the widely accepted alterations, there is a loss of expression of BMPR-II, and an overabundance of ALK2, ALK4, ALK5, ACTRIIA and ACTRIIB, which is linked to a notable increase in phospho-Smad2/3 [62]. While the precise mechanisms by which pathogenic BMPR2 variants predispose to PAH remain elusive, recent progress has been made. Effectors of BMPR-II signalling, such as FOXF1, a transcription factor involved in DNA repair and angiogenesis [63, 64], have been identified. However, there is still much work to be done in this aspect.
FIGURE 3.

Signalling by the bone morphogenetic proteins (BMP) and transforming growth factor (TGF)-β signalling. GDF: growth differentiation factor; AMH: anti-Müllerian hormone; BMPRII: BMP receptor type II; AMHRII: AMH receptor type II; ACTRII: activin receptor type II; TGFβRII: TGF-β receptor type II; ALK: activin receptor-like kinase; Smad: small mothers against decapentaplegic; MAPK: mitogen-activated protein kinase; JNK: C-Jun N-terminal kinase; ERK: extracellular signal-regulated kinase. Reproduced and modified with permission [61].
In addition to BMPR-II dysfunction, there appears to be compromised abundance of several ligands in the BMP/TGF-β pathway, including those of the activin pathway members. Indeed, PAH is associated with increased serum and pulmonary levels of activin A, activin B, follistatin and follistatin-like 3 (FSTL)3, along with a reduced abundance of the inhibin-α subunit of inhibin A and B proteins [62]. Interestingly, elevated serum levels of activin A and FSTL3 have been shown to predict death or lung transplantation in patients with idiopathic, heritable or anorexigen-associated PAH [62]. This evidence, combined with anomalies in the expression of certain key receptors of the BMP/TGF-β family, supports the idea that, in PAH, the activin pathway is overactive in pulmonary vascular walls and contributes to their progressive remodelling. Consistent with this notion, the deletion of the INHBA gene, which encodes one of the constituent chains of activins, protects mice against the development of pulmonary vascular remodelling induced by chronic hypoxia. Conversely, its overexpression induces mild PH at baseline and exacerbates hypoxia-induced PH [65]. Similarly, the inhibition of the activin pathway using a soluble form of the ACTRIIA receptor (ACTRIIA-Fc), capable of interacting with key ligands such as activins, certain growth differentiation factors (GDFs) such as GDF11, and BMPs such as BMP-10 and BMP-9, also protects rats and mice from various inducers of PH such as monocrotaline or Sugen/hypoxia [66, 67]. Despite these strategic pre-clinical demonstrations, the precise mechanisms driven by the activin and these key pathways remain to be specified. Currently, it appears that the beneficial action of the ACTRIIA-Fc in vivo appears to involves a reduction in the phosphorylation of Smad2/3, rather than modifications in the activities of Smad1/5/8 [67], which would result in the expression of a diverse panel of genes restoring the quiescence of ECs and their capacity to form tubes in vitro, as well as influencing the contractile status of PA-SMCs and their accumulation, and reducing the activation and recruitment of various inflammatory and immune cells [65–67]. Furthermore, the blockade of BMP-9 and BMP-10 by ACTRIIA-Fc could also contribute to its beneficial effect, as the loss or inhibition of BMP-9, either independently or particularly in conjunction with BMP-10, is associated with pulmonary capillary vasodilation [29, 68, 69] and acts directly on vascular smooth muscle cells to induce and maintain their contractile state [70]. These adjustments likely contribute to slowing down the progression of the disease, but validations at the human scale are still necessary.
This strategy, aiming to sequester excess ACTRIIA ligands, is a central focus of the soluble ACTRIIA IgG-Fc fusion protein sotatercept. The drug has recently completed successful phase 2 and 3 trials [71–73]. Nonetheless, numerous questions remain regarding the treatment of other forms of PH, understanding the mechanisms of action, and assessing how this molecule restores the complex network of signalling pathways within pulmonary arterial walls. The precision of addressing these questions could potentially enhance these strategies and contribute to the development of even more effective therapeutic tools. In addition, these investigations could help to determine whether its action truly leads to the reversal of the characteristic pulmonary vascular lesions associated with the disease.
Recent research evaluated the potential reversibility of pulmonary vascular remodelling in a monocrotaline-induced rat model with aortocaval shunts [74]. Researchers transplanted the left lungs of these rats, which exhibited less advanced pulmonary vascular remodelling, into healthy rats, creating an environment devoid of haemodynamic stress. They observed reversibility of the remodelling 3 weeks later, particularly when the transplanted lungs were harvested at early disease stages, in contrast to those taken from rats with advanced remodelling [74]. To delve deeper into this investigation, the team conducted an RNAseq analysis to compare the lungs that exhibited reversibility with those that did not. They identified differentially expressed genes, including those associated with inflammation, DNA damage accumulation, and senescence. Interestingly, they tested the effect of a senolytic and found that it improved reversibility [74]. However, it is important to note that senolytics could adversely affect endothelial integrity and function, depending on factors like dosage, timing of administration and the nature of remodelling [75]. These findings suggest that the accumulation of dysfunctional cells within vascular walls, alongside inflammation and haemodynamic stress, may play a critical role in determining the potential for reversibility. In line with these data, studies have demonstrated that reducing blood flow through pulmonary artery ligation can prevent and reverse pulmonary vascular remodelling induced by the Sugen/hypoxia model. This reduction in blood flow leads to a decrease in cell proliferation and inflammation [76]. Overall, these findings suggest that interventions focused on reducing haemodynamic or oxidative stress, in combination with anti-inflammatory strategies, have the potential to enhance reversibility in the context of PAH.
Studies highlight the crucial role of ion channels in PAH progression. However, a comprehensive understanding of their involvement remains elusive due to complex regulatory mechanisms. Notably, KCNK3/TASK-1 signalling in PAH has garnered attention for its role in determining PA-SMC membrane potential and modulating pulmonary arterial tone [77–79]. Analysis of chloride channels in IPAH revealed significant regulation of TMEM16A and cystic fibrosis transmembrane conductance regulator (CFTR), with subsequent research implicating CFTR loss in PAH pathogenesis, suggesting a potential therapeutic target [80, 81]. Dysregulated cellular metabolism, particularly in acetyl-CoA generation, also plays a crucial role in PAH. Upregulation of metabolic enzymes like ATP citrate lyase (ACLY) and aldehyde dehydrogenase 1A3 (ALDH1A3) leads to an increased pool of acetyl-CoA. This surplus acetyl-CoA drives histone acetylation, regulating gene expression and promoting cell proliferation and vascular remodelling in PAH [55, 82]. Targeting ACLY and ALDH1A3 may offer therapeutic potential, but precise strategies are needed considering the differing demands of PA-SMCs and PA-ECs for acetyl-CoA. Enhanced acetyl-CoA production by PFKFB3 downstream of Notch signalling in PA-ECs, as well as ALDH1A3, may be significant for their potential regeneration [55, 83]. Recent research suggests that the stiffening of blood vessels in PAH may be linked to excessive metabolism of glutamine and serine. This process appears to lead to collagen overproduction, causing the vessels to stiffen. Medications that inhibit the absorption of these amino acids, combined with dietary restrictions, have shown promising results in reducing collagen production and could potentially pave the way for new therapies [84].
Our understanding of epigenetic modifications in the cells contributing to the pathology of the hypertensive vascular wall has significantly improved, revealing alterations in the expression of key enzymes involved in genome-wide chromatin changes and extensive networks of transcription factor interactions [85]. Recent findings indicate that physiological levels of shear stress play a crucial role in preserving endothelial identity by regulating critical transcription factors and chromatin remodellers [86]. Notably, increased expression of histone acetyltransferases (P300, CREB-binding protein (CBP), and p300/CBP-associated factor (PCAF)), general control nondepressible 5 (GCN5), acetylation readers (bromodomain-containing protein (BRD)-4), histone deacetylases (HDACs) and histone acetylation modifications (histone 3 lysine 9 acetylation (H3K9ac), H3K27ac and H4K5/8/12/16ac) have been observed in PAH vascular cells and remodelled pulmonary vessels in PAH lungs [53, 55, 87–89]. Additionally, there is an overexpression of the histone methyltransferases enhancer of zeste homolog (EZH2) and G9a in the PA-SMCs of PAH [90]. Conversely, the loss of Suv4–20h1, a protein lysine methyltransferase that initiates di- and trimethylation of H4K20 in cardiopulmonary progenitor cells, has been associated with a COPD-like/PH phenotype in mice [91]. Emerging evidence also underscores a strong correlation between DNA methylation and vascular remodelling in PAH. Global DNA methylation increases and upregulation of DNA methyltransferase 3B (DNMT3B) have been observed in both PH patients and rodent models [92]. Another critical enzyme, Tet methylcytosine dioxygenase-2 (TET2), has been identified with mutations or reduced levels in patients with IPAH. Tet2 knockout mice spontaneously develop PAH, adverse pulmonary vascular remodelling, and inflammation [93].
Consistent with these observations, the reversal of epigenetic modifications, whether through apabetalone (RVX-208), a bromodomain (BRD2-4) inhibitor [87], or with two different P300/CBP inhibitors, CCS1477 and SGC-CBP30 [53], improves pulmonary haemodynamics, reduces the muscularisation of distal pulmonary vessels and mitigates gene expression patterns associated with PAH. Based on these observations, a multicentre pre-clinical validation of BET inhibition for the treatment of PAH [87] has transitioned to an open-label pilot study, assessing the feasibility of a future early-stage clinical trial evaluating BRD inhibition in PAH. Apabetalone was well tolerated, with patients completing all study-related procedures, including optional cardiac magnetic resonance imaging, without dose reduction or interruption. An exploratory analysis also suggested potential decreases in PVR and improved cardiac function [94]. It should also be noted that a recent study by Chen et al. [95] delving into mechanisms contributing to persistent inflammation in PAH found that PBMCs from patients with PH showed increased expression of class II HDACs (HDACs 5, 6 and 9) compared with controls, and the proportion of FOXP3+ T regulatory cells (Tregs) was reduced consistent with previous observation. They showed that in vitro exposure of isolated T-cells to suberoylanilide hydroxamic acid (SAHA), a class I/II HDAC inhibitor, led to increased expression of FOXP3 in PH cells and a higher conversion from conventional T-cells to Tregs than in controls. Studies of PBMCs from monocrotaline-treated rats showed similar results, with increased expression of HDACs 1, 6 and 9; lower FOXP3 expression; and lower amounts of Tregs. These were reversed by treatment with SAHA, but not valproic acid. These results were validated in the Sugen-hypoxia model of PH. This study and others have led to an international multicentre study using an HDAC inhibitor (clinicaltrials.gov identifier NCT05224531). While interpreting these results with caution is essential, these studies undoubtedly support the need for future evaluation of the safety and efficacy of BRD and HDAC inhibitors for PAH treatment.
Additional pathways can contribute to abnormalities of gene and protein expression in PH. Elevated levels of the RNA modification N6-methyladenosine in IPAH patients have been demonstrated to influence multiple RNA pathways and are associated with the immune environment [96, 97]. Furthermore, the elucidation of the role of eukaryotic initiation factor 5A (eIF5A), along with its isoforms eIF5A1 and eIF5A2, in selectively translating mRNAs encoding proline-rich proteins such as collagen and bromodomain-containing protein, implicated in PAH, together with certain long noncoding RNAs (lncRNAs), including MALAT1, MANTIS, H19, and TYKRIL, play a crucial role in pulmonary vascular remodelling and the development of PAH [56, 98–100]. All these factors and their associated pathways hold promise as potential therapeutic targets in clinical applications.
Advancing biomarkers and precision medicine in PH with artificial intelligence and machine learning
In recent years, a multitude of biomarkers have emerged, providing invaluable insights to refine PAH risk stratification. Currently, B-type natriuretic peptide (BNP) and/or N-terminal pro-BNP are the only circulating biomarkers employed in clinical practice. However, other potential candidates, such as uric acid, creatinine, latent TGF-β binding protein 2 (LTBP-2), GDF-15, interleukin (IL)-6, β-nerve growth factor (NGF), tumor necrosis factor-related apoptosis-inducing ligand (TRAIL), chemokine (C-X-C motif) ligand 9 (CXCL9), activin-A, FSTL3, axis inhibition protein 1 (AXIN-1), IL-15RA, neutrophil elastase, and more have been identified [62, 101–106]. Although significant work remains to incorporate these biomarkers into existing tools, the search for novel biological markers to predict response to therapy and outcomes is crucial to expedite the development of precision medicine tools for PH and PAH. Furthermore, these biomarkers can offer deeper insights into the significance of each disease component in its various forms. In this context, the progress of new technologies, including the advancement of artificial intelligence and machine learning, has the potential to expedite these research endeavors. Indeed, these techniques are increasingly gaining traction in biomarker identification and have already revealed distinctions among patients with PAH who exhibit distinct immune phenotypes [104]. Further, new concepts in the use of AI may allow more specific interrogation of markers relevant to more patient specific predictions of responses to therapy and outcome. A recent study presented a novel computational pipeline for clinician–algorithm interaction in PAH risk assessment using a case study of prospectively analysed patients. This approach can be expanded to consider hundreds and even thousands of patient measurements, thus introducing a critical step towards implementing big data and artificial intelligence into clinical decision-making and entering the era of personalised medicine [95, 107, 108].
Molecular imaging, despite being in its early stages of development, holds the promise of a significant breakthrough in both medical imaging and molecular biology. The digitalisation of medical images, along with the ready availability of powerful computing resources and advanced algorithms, has positioned the field of medical imaging at the forefront of AI integration [109]. This integration not only has the potential to enhance patient stratification and care by identifying pathological indicators within these images but may also enable non-invasive, real-time monitoring of pulmonary vascular remodelling in PAH. Advancements in biomarkers and molecular imaging are crucial in this field and are likely to revolutionise our understanding of the disease by providing insight into its dynamic progression in the same patient (or animal) over time (figure 2).
Going beyond the identification of key genes predisposing to PAH
The number of genes associated with predisposition to PAH has expanded since the initial linkage studies on the BMPR2 gene in familial PAH in 2000 [36, 37]. While these new predisposing genes are rare, found in <5% of heritable PAH cases, and have low penetrance, they have generated significant interest in the research community. Researchers are striving to gain a better understanding of their potential involvement, connections with the disease, and interactions among themselves and with the BMPR-II pathway.
Among the genes associated with PAH, some have a clear connection to the BMPR-II pathway, including ENG, ACVRL1, CAV1, SMAD9 and GDF2. Mutations in GDF2, which codes for the key BMPR-II ligand BMP-9 produced by the stellate cells in the liver, are rare, occurring in 0.8–1.2% of PAH cases in people of European descent [36, 110, 111], and in 6.7% of PAH cases in individuals with Asian descent [112]. These mutations primarily affect the furin cleavage site at the interface of the prodomain and the growth factor, preventing the proper processing and secretion of BMP-9 [113]. Consequently, BMP-9 plasma levels are decreased or undetectable in patients with these GDF2 mutations. However, in PAH patients without mutations and without major liver issues such as portopulmonary hypertension (PoPH) and patients with hepatic pulmonary syndrome (HPS), BMP-9 levels are similar to controls [114–116]. Interestingly, BMP-9 levels were slightly higher in females than in males, observed in both healthy controls and PAH patients, suggesting a potential link between sex differences and GDF2 [110, 114]. BMP-9 primarily triggers BMPR-II/Smad1/5/8 signalling in pulmonary ECs, enhancing BMPR-II expression and reducing apoptosis and barrier dysfunction. BMP-9 treatment has been shown to enhance vascular barrier integrity and reverse PAH in animal models [117]. However, BMP-9 can also activate Smad-2/3 signalling, resulting in pro-inflammatory and remodelling effects [19]. In certain circumstances, BMP-9 may induce features of endothelial-to-mesenchymal transition, particularly in PAH-derived ECs with low BMPR-II expression [21]. Recent studies have shown that BMP-9-deficient animals do not develop spontaneous PH and are partially protected against experimental PH [29, 68, 69]. This result is consistent with observations that inhibiting BMP-9 using neutralising antibodies or soluble ALK1ECD partially improves various PH models [29, 68, 69]. The complexity in BMP-9 signalling may be attributed to factors like the monomer/dimer status of BMP-9, differences in receptor availability and dimerisation in healthy versus diseased states, and more. Future research should investigate the specific contextual role of BMP-9 using cell-specific knockout models and conduct detailed histological analysis in GDF2 mutation carriers to fully understand BMP-9 biology.
Other genes, including KDR, TBX4 and SOX17, have recently emerged as genes associated with PAH, although their direct connections to the BMPR-II signalling pathway is less clear. These genes are widely recognised for their pivotal roles in both the development and maintenance of the pulmonary circulatory system, rendering them as potential candidates contributing to the predisposition of PAH. Gain- and loss-of-function mutations in TBX4 suggest differences in PAH-associated phenotypes [118] although both may represent developmental abnormalities [53]. While ATP13A3 has been identified as a novel gene associated with PAH, our knowledge regarding its biological mechanisms and resulting phenotype is limited. Genes such as AQP1 and PDGFD are currently under consideration, although they have yet to attain a high level of confidence in their association with PAH.
A significant number of PAH cases remain genetically enigmatic, and common genomic variations may also contribute to PAH pathobiology. Common variants, present in over 5% of the general population, typically have mild to moderate effects. Complex interactions between these common variants, environmental factors, and epigenetic factors may contribute to hereditary mechanisms in PAH. A comprehensive genome-wide association study (GWAS) involving 5895 individuals, including 847 with PAH, identified single nucleotide polymorphisms (SNPs) in two loci linked to PAH [40, 119]. These loci were found to be enhancers for the SOX17 promoter and within the HLA-DPA1/DPB1 gene. Notably, certain alleles within these loci demonstrated a protective effect against the development of PAH. Enhancers are known to be cell type-specific and are influenced by various factors, including blood flow [86]. This highlights the importance of mapping variants to cell-specific enhancers for vascular or immune cells, a strategy that has proven effective in understanding conditions such as congenital heart disease [120]. Another innovative approach involves the utilisation of long-read sequencing technology, capable of detecting genetic variations, including alternative isoforms that may not be apparent in short-read whole-genome sequencing. Furthermore, this method allows for the examination of mitochondrial DNA variations and acquired somatic mutations, which could provide valuable insights into the genetic underpinnings of PAH [121]. As research in genetics and genomics continues to advance, these approaches hold promise in unravelling the complex genetic landscape of PAH, shedding light on previously mysterious aspects of the condition.
Translating research insights into therapeutic strategies
Revealing sex differences and gene–environment interactions
PAH exhibits a higher prevalence in women than in men, with female-to-male ratios ranging from 2.3:1 in IPAH to 9:1 in PAH related to connective tissue diseases [122]. Moreover, the penetrance of PAH-related mutations is greater in females compared to males. Among BMPR2 mutation carriers, the penetrance is three times higher in females, with 42% of females developing PAH, compared to 14% of males [123]. This ratio aligns with the incidence rates found in prospective follow-up studies of asymptomatic BMPR2 carriers, revealing an incidence of 0.99% per year in males and 3.5% per year in females [37]. Interestingly, despite a lower incidence in men, the prognosis for males diagnosed with PAH is less favourable [124]. This difference in prognosis is primarily attributed to variations in the adaptation of the right ventricle to afterload. When comparing right ventricular afterload, women have shown superior adaptation, as indicated by functional parameters such as contractility, coupling and structural parameters [125–127]. Several studies have attempted to replicate these gender differences in pre-clinical models of PH and unveil the underlying mechanisms. However, the female predominance is not recapitulated in most PH animal models.
What appears evident is that oestrogens play a dual role in PAH, exerting local effects that drive lung vascular remodelling contributing to PAH, while simultaneously offering systemic protection and improving right ventricular adaptation. This intricate interplay underscores the multifaceted impact of oestrogens in the context of PAH. A phase 3 randomised controlled trial (PHANTOM study) assessed the efficacy of anastrozole in reducing 17β-oestradiol levels in patients with PAH. The results showed that treatment with anastrozole did not affect the primary outcome, which was the 6-min walk distance, nor did it influence secondary outcomes [128]. This indicates that targeting this pathway alone is insufficient to alter the course of PAH. Reflecting the complexity of this approach, newly discovered genes related to PAH have been demonstrated to interact with oestrogens. The expression of the SOX17 protein was found to be reduced in the lungs of PH-afflicted rats [129]. In this study, SOX17 was lower in the lungs of female rats, an effect attributed to the repression of the SOX17 promoter by the pathological oestrogen metabolite 16αOHE. In recent years, alternative mechanisms have emerged as possible explanations for sex differences in PAH. Notably, molecular agents derived from sex chromosomes, including the X-chromosome long noncoding RNA and the sex-determining region Y gene on the Y chromosome, have been found to induce phenotypic sex differences in PAH [130–133].
In conclusion, understanding the complex mechanisms underlying the gender differences in PAH is crucial. However, the challenges lie in accurately reproducing these phenomena in animal models due to the intricate interactions between genes, hormones and the environment.
Identifying gaps to enhance patient care
The growing utilisation of sophisticated yet expensive technologies that yield extensive datasets becomes meaningful only when complemented by a thorough understanding and control of the clinical and demographic data associated with the samples. This is pivotal for minimising variations and biases, serving as the bedrock not only for ongoing research but also as a cornerstone for a multitude of future studies. Equally important is the need to consider and address interindividual variabilities more comprehensively and enhance patient stratification, moving us towards precision medicine. For this, a better understanding of the effects related to age, including its impact on the inflammatory system, pulmonary circulation ageing, hormonal changes, decreased cellular regeneration capacity and increased prevalence of associated chronic diseases, is essential. This nuanced approach not only advances our understanding of the intricate variations in implicated pathophysiological mechanisms in PAH and other PH forms, which can differ in intensity from one patient to another, but also unveils additional mechanisms, especially in the setting of comorbidities, associated diseases and predisposing mutations. In light of emerging therapeutic strategies, there is a critical need to expand our repertoire of biomarkers. This expansion could allow for the meticulous selection and customisation of strategies for each patient, culminating in a more personalised and efficacious approach to treatment. Moreover, it is crucial to better understand the mechanisms of action of novel PAH therapies, with activin signalling inhibitors being at the forefront, and to evaluate them in other forms of PH. This also requires a clarification of the development of side-effects such as bleeding and telangiectasia. Special attention will need to be given to PAH patients who already present with a higher risk of epistaxis and telangiectasia, such as those carrying ACVRL1, BMP10, GDF2 and ENG mutations.
Alternative models and animal studies are pivotal in unravelling these aspects, underscoring the imperative need to persist in our endeavours to elevate and reinforce their relevance. A key approach involves maximising their integration to amalgamate the insights gleaned from our research. While inherent limitations exist in modelling PAH, there are several strategies to enhance predictive accuracy and translational efficiency. These encompass a meticulous definition of the research problem, a comprehensive evaluation of the strengths and weaknesses inherent in each available model, and strategic planning for their optimal utilisation, including robust haemodynamic analyses. Refinements can be implemented by incorporating randomisation of animals, considering both sexes, and introducing blinding protocols for observations related to haemodynamic and structural end-points.
Translating pre-clinical discoveries into the clinical setting poses unique challenges for a rare, complex and multifactorial disease like PAH. However, recent discoveries have proven that this is indeed possible. It is for this reason that efforts must be continued, building upon these advancements and better integrating new technologies with the clinical data of each patient. Through this approach, we can refine treatment approaches, improve quality of life, and potentially reverse or stop the progression of this devastating condition.
Shareable PDF
Acknowledgement
C. Guignabert and M. Humbert are supported by a State funding managed by the National Research Agency according to the Investments for the Future program integrated in France 2030, under the reference ANR-18-RHUS-0006 (DESTINATION 2024).
Footnotes
Conflict of interest: C. Guignabert reports grants from Acceleron Pharma, MSD, Corteria Pharmaceuticals, Structure Therapeutics (ex-ShouTi) and Gossamer Bio, payment or honoraria for lectures, presentations, manuscript writing or educational events from MSD, and patents planned, issued or pending (WO/2024/023139, WO/2018/011376). J. Aman has no potential conflicts of interest to disclose. S. Bonnet reports grants from Morphic Therapeutic, Sunshine Bio and Janssen, consultancy fees from Morphic Therapeutic and Chiesi, and participation on a data safety monitoring board or advisory board with Morphic Therapeutic and Allienaire. P. Dorfmüller reports payment or honoraria for lectures, presentations, manuscript writing or educational events from AstraZeneca. A.J. Olschewski reports grants from Austrian Science Fund (FWF) (10.55776/I6299 and 10.55776/KLI1153), payment or honoraria for lectures, presentations, manuscript writing or educational events from MSD, patents pending (PCT/EP2017/055440), and stock (or stock options) with Bayer. S. Pullamsetti reports grants and consultancy fees from Gossamer Bio. M. Rabinovitch reports consultancy fees from Pfizer, Amgen, Merck and Tiakis, payment or honoraria for lectures, presentations, manuscript writing or educational events from NIH, patents planned, issued or pending (FK506: tacrolimus in pulmonary hypertension), participation on a data safety monitoring board or advisory board with Amgen and NIH, is associate editor for JACC BTS, receipt of equipment, materials, drugs, medical writing, gifts or other services from Tiakis (tiprelestat). R.T. Schermuly reports grants from Chiesi and Attgeno, and consultancy fees from Gossamer, Attgeno and Chiesi. M. Humbert reports grants from Gossamer and Merck, consultancy fees from 35 Pharma, Aerovate, AOP Orphan, Chiesi, Ferrer, Gossamer, Janssen, Keros, Liquidia, Merck, Novartis, Respira, Roivant and United Therapeutics, payment or honoraria for lectures, presentations, manuscript writing or educational events from Janssen and Merck, and participation on a data safety monitoring board or advisory board with 35 Pharma, Aerovate, Janssen, Keros, Merck, Novartis and United Therapeutics. K.R. Stenmark reports grants from NIH/NHLBI and DoD, and a leadership role with PVRI.
References
- 1.Humbert M, Guignabert C, Bonnet S, et al. . Pathology and pathobiology of pulmonary hypertension: state of the art and research perspectives. Eur Respir J 2019; 53: 1801887. doi: 10.1183/13993003.01887-2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ghigna MR, Guignabert C, Montani D, et al. . BMPR2 mutation status influences bronchial vascular changes in pulmonary arterial hypertension. Eur Respir J 2016; 48: 1668–1681. doi: 10.1183/13993003.00464-2016 [DOI] [PubMed] [Google Scholar]
- 3.Tuder RM, Archer SL, Dorfmuller P, et al. . Relevant issues in the pathology and pathobiology of pulmonary hypertension. J Am Coll Cardiol 2013; 62: D4–D12. doi: 10.1016/j.jacc.2013.10.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gu S, Goel K, Forbes LM, et al. . Tensions in taxonomies: current understanding and future directions in the pathobiologic basis and treatment of group 1 and group 3 pulmonary hypertension. Compr Physiol 2023; 13: 4295–4319. doi: 10.1002/cphy.c220010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Huertas A, Perros F, Tu L, et al. . Immune dysregulation and endothelial dysfunction in pulmonary arterial hypertension: a complex interplay. Circulation 2014; 129: 1332–1340. doi: 10.1161/CIRCULATIONAHA.113.004555 [DOI] [PubMed] [Google Scholar]
- 6.Huertas A, Tu L, Humbert M, et al. . Chronic inflammation within the vascular wall in pulmonary arterial hypertension: more than a spectator. Cardiovasc Res 2020; 116: 885–893. doi: 10.1093/cvr/cvz308 [DOI] [PubMed] [Google Scholar]
- 7.Rabinovitch M, Guignabert C, Humbert M, et al. . Inflammation and immunity in the pathogenesis of pulmonary arterial hypertension. Circ Res 2014; 115: 165–175. doi: 10.1161/CIRCRESAHA.113.301141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Stacher E, Graham BB, Hunt JM, et al. . Modern age pathology of pulmonary arterial hypertension. Am J Respir Crit Care Med 2012; 186: 261–272. doi: 10.1164/rccm.201201-0164OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Humbert M, Sitbon O, Guignabert C, et al. . Treatment of pulmonary arterial hypertension: recent progress and a look to the future. Lancet Respir Med 2023; 11: 804–819. doi: 10.1016/S2213-2600(23)00264-3 [DOI] [PubMed] [Google Scholar]
- 10.King CS, Shlobin OA. The trouble with group 3 pulmonary hypertension in interstitial lung disease: dilemmas in diagnosis and the conundrum of treatment. Chest 2020; 158: 1651–1664. doi: 10.1016/j.chest.2020.04.046 [DOI] [PubMed] [Google Scholar]
- 11.Magee F, Wright JL, Wiggs BR, et al. . Pulmonary vascular structure and function in chronic obstructive pulmonary disease. Thorax 1988; 43: 183–189. doi: 10.1136/thx.43.3.183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wright JL, Petty T, Thurlbeck WM. Analysis of the structure of the muscular pulmonary arteries in patients with pulmonary hypertension and COPD: National Institutes of Health Nocturnal Oxygen Therapy Trial. Lung 1992; 170: 109–124. doi: 10.1007/BF00175982 [DOI] [PubMed] [Google Scholar]
- 13.Galambos C, Sims-Lucas S, Abman SH, et al. . Intrapulmonary bronchopulmonary anastomoses and plexiform lesions in idiopathic pulmonary arterial hypertension. Am J Respir Crit Care Med 2016; 193: 574–576. doi: 10.1164/rccm.201507-1508LE [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Westöö C, Norvik C, Peruzzi N, et al. . Distinct types of plexiform lesions identified by synchrotron-based phase-contrast micro-CT. Am J Physiol Lung Cell Mol Physiol 2021; 321: L17–L28. doi: 10.1152/ajplung.00432.2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sakao S, Voelkel NF, Tatsumi K. The vascular bed in COPD: pulmonary hypertension and pulmonary vascular alterations. Eur Respir Rev 2014; 23: 350–355. doi: 10.1183/09059180.00007913 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nossent EJ, Smits J, Seegers C, et al. . Clinical correlates of a nonplexiform vasculopathy in patients with a diagnosis of idiopathic pulmonary arterial hypertension. Chest 2024; 166: 190–200. doi: 10.1016/j.chest.2024.02.046 [DOI] [PubMed] [Google Scholar]
- 17.Gawad C, Koh W, Quake SR. Single-cell genome sequencing: current state of the science. Nat Rev Genet 2016; 17: 175–188. doi: 10.1038/nrg.2015.16 [DOI] [PubMed] [Google Scholar]
- 18.Reyfman PA, Walter JM, Joshi N, et al. . Single-cell transcriptomic analysis of human lung provides insights into the pathobiology of pulmonary fibrosis. Am J Respir Crit Care Med 2019; 199: 1517–1536. doi: 10.1164/rccm.201712-2410OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Travaglini KJ, Nabhan AN, Penland L, et al. . A molecular cell atlas of the human lung from single-cell RNA sequencing. Nature 2020; 587: 619–625. doi: 10.1038/s41586-020-2922-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hong J, Wong B, Rhodes CJ, et al. . Integrative multiomics to dissect the lung transcriptional landscape of pulmonary arterial hypertension. bioRxiv 2023; preprint [ 10.1101/2023.01.12.523812v1]. doi: 10.1101/2023.01.12.523812v1 [DOI] [Google Scholar]
- 21.Crnkovic S, Valzano F, Fließer E, et al. . Single-cell transcriptomics reveals skewed cellular communication and phenotypic shift in pulmonary artery remodeling. JCI Insight 2022; 7: e153471. doi: 10.1172/jci.insight.153471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Viswanathan G, Kirshner HF, Nazo N, et al. . Single-cell analysis reveals distinct immune and smooth muscle cell populations that contribute to chronic thromboembolic pulmonary hypertension. Am J Respir Crit Care Med 2023; 207: 1358–1375. doi: 10.1164/rccm.202203-0441OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kumar S, Mickael C, Kumar R, et al. Single cell transcriptomic analyses reveal diverse and dynamic changes of distinct populations of lung interstitial macrophages in hypoxia-induced pulmonary hypertension. Front Immunol 2024; 15: 1372959. 10.3389/fimmu.2024.1372959 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Thomas S, Manivannan S, Garg V, et al. . Single-cell RNA sequencing reveals novel genes regulated by hypoxia in the lung vasculature. J Vasc Res 2022; 59: 163–175. doi: 10.1159/000522340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rodor J, Chen SH, Scanlon JP, et al. . Single-cell RNA sequencing profiling of mouse endothelial cells in response to pulmonary arterial hypertension. Cardiovasc Res 2022; 118: 2519–2534. doi: 10.1093/cvr/cvab296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Asosingh K, Comhair S, Mavrakis L, et al. . Single-cell transcriptomic profile of human pulmonary artery endothelial cells in health and pulmonary arterial hypertension. Sci Rep 2021; 11: 14714. doi: 10.1038/s41598-021-94163-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schupp JC, Adams TS, Cosme C Jr, et al. . Integrated single-cell atlas of endothelial cells of the human lung. Circulation 2021; 144: 286–302. doi: 10.1161/CIRCULATIONAHA.120.052318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Muhl L, Genové G, Leptidis S, et al. . Single-cell analysis uncovers fibroblast heterogeneity and criteria for fibroblast and mural cell identification and discrimination. Nat Commun 2020; 11: 3953. doi: 10.1038/s41467-020-17740-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Berrebeh N, Mbouamboua Y, Thuillet R, et al. . Bone morphogenetic protein-9 controls pulmonary vascular growth and remodeling. medRxiv 2023; preprint [ 10.1101/2023.06.02.23290910]. doi: 10.1101/2023.06.02.23290910 [DOI] [Google Scholar]
- 30.Ferrian S, Cao A, McCaffrey EF, et al. . Single -cell imaging maps inflammatory cell subsets to pulmonary arterial hypertension vasculopathy. Am J Respir Crit Care Med 2024; 209: 206–218. doi: 10.1164/rccm.202209-1761OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kostyunina DS, Rowan SC, Pakhomov NV, et al. . Shear stress markedly alters the proteomic response to hypoxia in human pulmonary endothelial cells. Am J Respir Cell Mol Biol 2023; 68: 551–565. doi: 10.1165/rcmb.2022-0340OC [DOI] [PubMed] [Google Scholar]
- 32.Sikkema L, Ramírez-Suastegui C, Strobl DC, et al. . An integrated cell atlas of the lung in health and disease. Nat Med 2023; 29: 1563–1577. doi: 10.1038/s41591-023-02327-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Merritt CR, Ong GT, Church SE, et al. . Multiplex digital spatial profiling of proteins and RNA in fixed tissue. Nat Biotechnol 2020; 38: 586–599. doi: 10.1038/s41587-020-0472-9 [DOI] [PubMed] [Google Scholar]
- 34.Neubert L, Borchert P, Stark H, et al. . Molecular profiling of vascular remodeling in chronic pulmonary disease. Am J Pathol 2020; 190: 1382–1396. doi: 10.1016/j.ajpath.2020.03.008 [DOI] [PubMed] [Google Scholar]
- 35.Tuder RM, Gandjeva A, Williams S, et al. . Digital spatial profiling identifies distinct molecular signatures of vascular lesions in pulmonary arterial hypertension. Am J Respir Crit Care Med 2024; 210: 329–342. doi: 10.1164/rccm.202307-1310OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Welch CL, Aldred MA, Balachandar S, et al. . Defining the clinical validity of genes reported to cause pulmonary arterial hypertension. Genet Med 2023; 25: 100925. doi: 10.1016/j.gim.2023.100925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Montani D, Girerd B, Jaïs X, et al. . Screening for pulmonary arterial hypertension in adults carrying a BMPR2 mutation. Eur Respir J 2021; 58: 2004229. doi: 10.1183/13993003.04229-2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Aldred MA, Morrell NW, Guignabert C. New mutations and pathogenesis of pulmonary hypertension: progress and puzzles in disease pathogenesis. Circ Res 2022; 130: 1365–1381. doi: 10.1161/CIRCRESAHA.122.320084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gu M, Shao NY, Sa S, et al. . Patient-specific iPSC-derived endothelial cells uncover pathways that protect against pulmonary hypertension in BMPR2 mutation carriers. Cell Stem Cell 2017; 20: 490–504. doi: 10.1016/j.stem.2016.08.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Walters R, Vasilaki E, Aman J, et al. . SOX17 enhancer variants disrupt transcription factor binding and enhancer inactivity drives pulmonary hypertension. Circulation 2023; 147: 1606–1621. doi: 10.1161/CIRCULATIONAHA.122.061940 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Riou M, Seferian A, Savale L, et al. . Deterioration of pulmonary hypertension and pleural effusion with bosutinib following dasatinib lung toxicity. Eur Respir J 2016; 48: 1517–1519. doi: 10.1183/13993003.01410-2016 [DOI] [PubMed] [Google Scholar]
- 42.Guignabert C, Phan C, Seferian A, et al. . Dasatinib induces lung vascular toxicity and predisposes to pulmonary hypertension. J Clin Invest 2016; 126: 3207–3218. doi: 10.1172/JCI86249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nagaraj C, Tang B, Bálint Z, et al. . Src tyrosine kinase is crucial for potassium channel function in human pulmonary arteries. Eur Respir J 2013; 41: 85–95. doi: 10.1183/09031936.00211811 [DOI] [PubMed] [Google Scholar]
- 44.Lluciá-Valldeperas A, Smal R, Bekedam FT, et al. . Development of a 3-dimensional model to study right heart dysfunction in pulmonary arterial hypertension: first observations. Cells 2021; 10: 3595. doi: 10.3390/cells10123595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gu M, Donato M, Guo M, et al. . iPSC-endothelial cell phenotypic drug screening and in silico analyses identify tyrphostin-AG1296 for pulmonary arterial hypertension. Sci Transl Med 2021; 13: eaba6480. doi: 10.1126/scitranslmed.aba6480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sachs N, Papaspyropoulos A, Zomer-van Ommen DD, et al. . Long-term expanding human airway organoids for disease modeling. EMBO J 2019; 38: e100300. doi: 10.15252/embj.2018100300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bai H, Ingber DE. What can an organ-on-a-chip teach us about human lung pathophysiology? Physiology 2022; 37: 242–252. doi 10.1152/physiol.00012.2022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Vo Q, Benam KH. Advancements in preclinical human-relevant modeling of pulmonary vasculature on-chip. Eur J Pharm Sci 2024; 195: 106709. doi: 10.1016/j.ejps.2024.106709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mandrycky CJ, Howard CC, Rayner SG, et al. . Organ-on-a-chip systems for vascular biology. J Mol Cell Cardiol 2021; 159: 1–13. doi: 10.1016/j.yjmcc.2021.06.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Liu G, Betts C, Cunoosamy DM, et al. . Use of precision cut lung slices as a translational model for the study of lung biology. Respir Res 2019; 20: 162. doi: 10.1186/s12931-019-1131-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Alsafadi HN, Staab-Weijnitz CA, Lehmann M, et al. . An ex vivo model to induce early fibrosis-like changes in human precision-cut lung slices. Am J Physiol Lung Cell Mol Physiol 2017; 312: L896–L902. doi: 10.1152/ajplung.00084.2017 [DOI] [PubMed] [Google Scholar]
- 52.Van Dijk EM, Culha S, Menzen MH, et al. . Elastase-induced parenchymal disruption and airway hyper responsiveness in mouse precision cut lung slices: toward an ex vivo COPD model. Front Physiol 2016; 7: 657. doi: 10.3389/fphys.2016.00657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Chelladurai P, Kuenne C, Bourgeois A, et al. . Epigenetic reactivation of transcriptional programs orchestrating fetal lung development in human pulmonary hypertension. Sci Transl Med 2022; 14: eabe5407. doi: 10.1126/scitranslmed.abe5407 [DOI] [PubMed] [Google Scholar]
- 54.Jandl K, Marsh LM, Mutgan AC, et al. . Impairment of the NKT-STAT1-CXCL9 axis contributes to vessel fibrosis in pulmonary hypertension caused by lung fibrosis. Am J Respir Crit Care Med 2022; 206: 981–998. doi: 10.1164/rccm.202201-0142OC [DOI] [PubMed] [Google Scholar]
- 55.Grobs Y, Romanet C, Lemay S-E, et al. . ATP citrate lyase drives vascular remodeling diseases development through metabolic-epigenetic reprograming. bioRxiv 2024; preprint [ 10.1101/2024.02.02.578545]. doi: 10.1101/2024.02.02.578545 [DOI] [Google Scholar]
- 56.Zehendner CM, Valasarajan C, Werner A, et al. . Long noncoding RNA TYKRIL plays a role in pulmonary hypertension via the p53-mediated regulation of PDGFRβ. Am J Respir Crit Care Med 2020; 202: 1445–1457. doi: 10.1164/rccm.201910-2041OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Alastalo TP, Li M, Perez Vde J, et al. . Disruption of PPARγ/β-catenin-mediated regulation of apelin impairs BMP-induced mouse and human pulmonary arterial EC survival. J Clin Invest 2011; 121: 3735–3746. doi: 10.1172/JCI43382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bordenave J, Thuillet R, Tu L, et al. . Neutralization of CXCL12 attenuates established pulmonary hypertension in rats. Cardiovasc Res 2020; 116: 686–697. doi: 10.1093/cvr/cvz153 [DOI] [PubMed] [Google Scholar]
- 59.Bordenave J, Tu L, Berrebeh N, et al. . Lineage tracing reveals the dynamic contribution of pericytes to the blood vessel remodeling in pulmonary hypertension. Arterioscler Thromb Vasc Biol 2020; 40: 766–782. doi: 10.1161/ATVBAHA.119.313715 [DOI] [PubMed] [Google Scholar]
- 60.Wang L, Moonen JR, Cao A, et al. . Dysregulated smooth muscle cell BMPR2-ARRB2 axis causes pulmonary hypertension. Circ Res 2023; 132: 545–564. doi: 10.1161/CIRCRESAHA.121.320541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Guignabert C, Humbert M. Targeting transforming growth factor-β receptors in pulmonary hypertension. Eur Respir J 2021; 57: 2002341. doi: 10.1183/13993003.02341-2020 [DOI] [PubMed] [Google Scholar]
- 62.Guignabert C, Savale L, Boucly A, et al. . Serum and pulmonary expression profiles of the activin signaling system in pulmonary arterial hypertension. Circulation 2023; 147: 1809–1822. doi: 10.1161/CIRCULATIONAHA.122.061501 [DOI] [PubMed] [Google Scholar]
- 63.Isobe S, Nair RV, Kang HY, et al. . Reduced FOXF1 links unrepaired DNA damage to pulmonary arterial hypertension. Nat Commun 2023; 14: 7578. doi: 10.1038/s41467-023-43039-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Day RW, Lai KV, Baisch HN, et al. . Descriptive images and features of pulmonary vascular disease in a child with a FOXF1 variant. Pediatr Pulmonol 2023; 58: 2383–2385. doi: 10.1002/ppul.26480 [DOI] [PubMed] [Google Scholar]
- 65.Ryanto GRT, Ikeda K, Miyagawa K, et al. . An endothelial activin A-bone morphogenetic protein receptor type 2 link is overdriven in pulmonary hypertension. Nat Commun 2021; 12: 1720. doi: 10.1038/s41467-021-21961-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Joshi SR, Liu J, Bloom T, et al. . Sotatercept analog suppresses inflammation to reverse experimental pulmonary arterial hypertension. Sci Rep 2022; 12: 7803. doi: 10.1038/s41598-022-11435-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Yung LM, Yang P, Joshi S, et al. . ACTRIIA-Fc rebalances activin/GDF versus BMP signaling in pulmonary hypertension. Sci Transl Med 2020; 12: eaaz5660. doi: 10.1126/scitranslmed.aaz5660 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Bouvard C, Tu L, Rossi M, et al. . Different cardiovascular and pulmonary phenotypes for single- and double-knock-out mice deficient in BMP9 and BMP10. Cardiovasc Res 2022; 118: 1805–1820. doi: 10.1093/cvr/cvab187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Tu L, Desroches-Castan A, Mallet C, et al. . Selective BMP-9 inhibition partially protects against experimental pulmonary hypertension. Circ Res 2019; 124: 846–855. doi: 10.1161/CIRCRESAHA.118.313356 [DOI] [PubMed] [Google Scholar]
- 70.Wang L, Rice M, Swist S, et al. . BMP9 and BMP10 act directly on vascular smooth muscle cells for generation and maintenance of the contractile state. Circulation 2021; 143: 1394–1410. doi: 10.1161/CIRCULATIONAHA.120.047375 [DOI] [PubMed] [Google Scholar]
- 71.Hoeper MM, Badesch DB, Ghofrani HA, et al. . Phase 3 trial of sotatercept for treatment of pulmonary arterial hypertension. N Engl J Med 2023; 388: 1478–1490. doi: 10.1056/NEJMoa2213558 [DOI] [PubMed] [Google Scholar]
- 72.Humbert M, McLaughlin V, Gibbs JSR, et al. . Sotatercept for the treatment of pulmonary arterial hypertension. N Engl J Med 2021; 384: 1204–1215. doi: 10.1056/NEJMoa2024277 [DOI] [PubMed] [Google Scholar]
- 73.Humbert M, McLaughlin V, Gibbs JSR, et al. . Sotatercept for the treatment of pulmonary arterial hypertension: PULSAR open-label extension. Eur Respir J 2023; 61: 2201347. doi: 10.1183/13993003.01347-2022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.van der Feen DE, Bossers GPL, Hagdorn QAJ, et al. . Cellular senescence impairs the reversibility of pulmonary arterial hypertension. Sci Transl Med 2020; 12: eaaw4974. doi: 10.1126/scitranslmed.aaw4974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Born E, Lipskaia L, Breau M, et al. . Eliminating senescent cells can promote pulmonary hypertension development and progression. Circulation 2023; 147: 650–666. doi: 10.1161/CIRCULATIONAHA.122.058794 [DOI] [PubMed] [Google Scholar]
- 76.Abe K, Shinoda M, Tanaka M, et al. . Haemodynamic unloading reverses occlusive vascular lesions in severe pulmonary hypertension. Cardiovasc Res 2016; 111: 16–25. doi: 10.1093/cvr/cvw070 [DOI] [PubMed] [Google Scholar]
- 77.Olschewski A, Li Y, Tang B, et al. . Impact of TASK-1 in human pulmonary artery smooth muscle cells. Circ Res 2006; 98: 1072–1080. doi: 10.1161/01.RES.0000219677.12988.e9 [DOI] [PubMed] [Google Scholar]
- 78.Antigny F, Hautefort A, Meloche J, et al. . Potassium channel subfamily K member 3 (KCNK3) contributes to the development of pulmonary arterial hypertension. Circulation 2016; 133: 1371–1385. doi: 10.1161/CIRCULATIONAHA.115.020951 [DOI] [PubMed] [Google Scholar]
- 79.Lambert M, Capuano V, Boet A, et al. . Characterization of Kcnk3-mutated rat, a novel model of pulmonary hypertension. Circ Res 2019; 125: 678–695. doi: 10.1161/CIRCRESAHA.119.314793 [DOI] [PubMed] [Google Scholar]
- 80.Papp R, Nagaraj C, Zabini D, et al. . Targeting TMEM16A to reverse vasoconstriction and remodelling in idiopathic pulmonary arterial hypertension. Eur Respir J 2019; 53: 1800965. doi: 10.1183/13993003.00965-2018 [DOI] [PubMed] [Google Scholar]
- 81.Le Ribeuz H, To L, Ghigna MR, et al. . Involvement of CFTR in the pathogenesis of pulmonary arterial hypertension. Eur Respir J 2021; 58: 2000653. doi: 10.1183/13993003.00653-2020 [DOI] [PubMed] [Google Scholar]
- 82.Li D, Shao NY, Moonen JR, et al. . ALDH1A3 coordinates metabolism with gene regulation in pulmonary arterial hypertension. Circulation 2021; 143: 2074–2090. doi: 10.1161/CIRCULATIONAHA.120.048845 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Miyagawa K, Shi M, Chen PI, et al. . Smooth muscle contact drives endothelial regeneration by BMPR2-Notch1-mediated metabolic and epigenetic changes. Circ Res 2019; 124: 211–224. doi: 10.1161/CIRCRESAHA.118.313374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Rachedi NS, Tang Y, Tai Y-Y, et al. Dietary intake and glutamine-serine metabolism control pathologic vascular stiffness. Cell Metab 2024; 36: 1335–1350. 10.1016/j.cmet.2024.04.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Reyes-Palomares A, Gu M, Grubert F, et al. . Remodeling of active endothelial enhancers is associated with aberrant gene-regulatory networks in pulmonary arterial hypertension. Nat Commun 2020; 11: 1673. doi: 10.1038/s41467-020-15463-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Moonen JR, Chappell J, Shi M, et al. . KLF4 recruits SWI/SNF to increase chromatin accessibility and reprogram the endothelial enhancer landscape under laminar shear stress. Nat Commun 2022; 13: 4941. doi: 10.1038/s41467-022-32566-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Van der Feen DE, Kurakula K, Tremblay E, et al. . Multicenter preclinical validation of BET inhibition for the treatment of pulmonary arterial hypertension. Am J Respir Crit Care Med 2019; 200: 910–920. doi: 10.1164/rccm.201812-2275OC [DOI] [PubMed] [Google Scholar]
- 88.Hu CJ, Zhang H, Laux A, et al. . Mechanisms contributing to persistently activated cell phenotypes in pulmonary hypertension. J Physiol 2019; 597: 1103–1119. doi: 10.1113/JP275857 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Chelladurai P, Boucherat O, Stenmark K, et al. . Targeting histone acetylation in pulmonary hypertension and right ventricular hypertrophy. Br J Pharmacol 2021; 178: 54–71. doi: 10.1111/bph.14932 [DOI] [PubMed] [Google Scholar]
- 90.Habbout K, Omura J, Awada C, et al. . Implication of EZH2 in the pro-proliferative and apoptosis-resistant phenotype of pulmonary artery smooth muscle cells in PAH: a transcriptomic and proteomic approach. Int J Mol Sci 2021; 22: 2957. doi: 10.3390/ijms22062957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Qi H, Liu H, Pullamsetti SS, et al. . Epigenetic regulation by Suv4-20h1 in cardiopulmonary progenitor cells is required to prevent pulmonary hypertension and chronic obstructive pulmonary disease. Circulation 2021; 144: 1042–1058. doi: 10.1161/CIRCULATIONAHA.120.051680 [DOI] [PubMed] [Google Scholar]
- 92.Yan Y, He YY, Jiang X, et al. . DNA methyltransferase 3B deficiency unveils a new pathological mechanism of pulmonary hypertension. Sci Adv 2020; 6: eaba2470. doi: 10.1126/sciadv.aba2470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Potus F, Pauciulo MW, Cook EK, et al. . Novel mutations and decreased expression of the epigenetic regulator TET2 in pulmonary arterial hypertension. Circulation 2020; 141: 1986–2000. doi: 10.1161/CIRCULATIONAHA.119.044320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Provencher S, Potus F, Blais-Lecours P, et al. . BET protein inhibition for pulmonary arterial hypertension: a pilot clinical trial. Am J Respir Crit Care Med 2022; 205: 1357–1360. doi: 10.1164/rccm.202109-2182LE [DOI] [PubMed] [Google Scholar]
- 95.Chen CN, Hajji N, Yeh FC, et al. . Restoration of Foxp3+ regulatory T cells by HDAC-dependent epigenetic modulation plays a pivotal role in resolving pulmonary arterial hypertension pathology. Am J Respir Crit Care Med 2023; 208: 879–895. doi: 10.1164/rccm.202301-0181OC [DOI] [PubMed] [Google Scholar]
- 96.Huang T, Zeng Y, Yang Y, et al. . Comprehensive analysis of m6A methylomes in idiopathic pulmonary arterial hypertension. Epigenetics 2023; 18: 2242225. doi: 10.1080/15592294.2023.2242225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Gao G, Chen A, Gong J, et al. . Comprehensive analyses of m6A RNA methylation patterns and related immune microenvironment in idiopathic pulmonary arterial hypertension. Front Genet 2023; 14: 1222368. doi: 10.3389/fgene.2023.1222368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Liu J, Sun Y, Zhu B, et al. . Identification of a potentially novel LncRNA-miRNA-mRNA competing endogenous RNA network in pulmonary arterial hypertension via integrated bioinformatic analysis. Life Sci 2021; 277: 119455. doi: 10.1016/j.lfs.2021.119455 [DOI] [PubMed] [Google Scholar]
- 99.Omura J, Habbout K, Shimauchi T, et al. . Identification of long noncoding RNA H19 as a new biomarker and therapeutic target in right ventricular failure in pulmonary arterial hypertension. Circulation 2020; 142: 1464–1484. doi: 10.1161/CIRCULATIONAHA.120.047626 [DOI] [PubMed] [Google Scholar]
- 100.Lemay SE, Grobs Y, Romanet C, et al. . Hypusine signaling promotes pulmonary vascular remodeling in pulmonary arterial hypertension. Am J Respir Crit Care Med 2024; 209: 1376–1391. doi: 10.1164/rccm.202305-0909OC [DOI] [PubMed] [Google Scholar]
- 101.Boucly A, Tu L, Guignabert C, et al. . Cytokines as prognostic biomarkers in pulmonary arterial hypertension. Eur Respir J 2023; 61: 2201232. doi: 10.1183/13993003.01232-2022 [DOI] [PubMed] [Google Scholar]
- 102.Garfield BE, Crosby A, Shao D, et al. . Growth/differentiation factor 15 causes TGFβ-activated kinase 1-dependent muscle atrophy in pulmonary arterial hypertension. Thorax 2019; 74: 164–176. doi: 10.1136/thoraxjnl-2017-211440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Savale L, Akagi S, Tu L, et al. . Serum and pulmonary uric acid in pulmonary arterial hypertension. Eur Respir J 2021; 58: 2000332. doi: 10.1183/13993003.00332-2020 [DOI] [PubMed] [Google Scholar]
- 104.Sweatt AJ, Hedlin HK, Balasubramanian V, et al. . Discovery of distinct immune phenotypes using machine learning in pulmonary arterial hypertension. Circ Res 2019; 124: 904–919. doi: 10.1161/CIRCRESAHA.118.313911 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Sweatt AJ, Miyagawa K, Rhodes CJ, et al. . Severe pulmonary arterial hypertension is characterized by increased neutrophil elastase and relative elafin deficiency. Chest 2021; 160: 1442–1458. doi: 10.1016/j.chest.2021.06.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Boucherat O, Yokokawa T, Krishna V, et al. . Identification of LTBP-2 as a plasma biomarker for right ventricular dysfunction in human pulmonary arterial hypertension. Nat Cardiovasc Res 2022; 1: 748–760. doi: 10.1038/s44161-022-00113-w [DOI] [PubMed] [Google Scholar]
- 107.Kheyfets VO, Sweatt AJ, Gomberg-Maitland M, et al. . Computational platform for doctor-artificial intelligence cooperation in pulmonary arterial hypertension prognostication: a pilot study. ERJ Open Res 2023; 9: 00484-2022. doi: 10.1183/23120541.00484-2022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Bordag N, Nagy BM, Zügner E, et al. . Lipidomics for diagnosis and prognosis of pulmonary hypertension. medRxiv 2023; preprint [ 10.1101/2023.05.17.23289772]. doi: 10.1101/2023.05.17.23289772 [DOI] [Google Scholar]
- 109.Alabed S, Alandejani F, Dwivedi K, et al. . Validation of artificial intelligence cardiac MRI measurements: relationship to heart catheterization and mortality prediction. Radiology 2022; 305: 68–79. doi: 10.1148/radiol.212929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Eyries M, Montani D, Nadaud S, et al. . Widening the landscape of heritable pulmonary hypertension mutations in paediatric and adult cases. Eur Respir J 2019; 53: 1801371. doi: 10.1183/13993003.01371-2018 [DOI] [PubMed] [Google Scholar]
- 111.Grynblat J, Bogaard HJ, Eyries M, et al. . Pulmonary vascular phenotype identified in patients with GDF2 (BMP9) or BMP10 variants: an international multicentre study. Eur Respir J 2024; 63: 2301634. doi: 10.1183/13993003.01634-2023 [DOI] [PubMed] [Google Scholar]
- 112.Wang XJ, Lian TY, Jiang X, et al. . Germline BMP9 mutation causes idiopathic pulmonary arterial hypertension. Eur Respir J 2019; 53: 1801609. doi: 10.1183/13993003.01609-2018 [DOI] [PubMed] [Google Scholar]
- 113.Gräf S, Haimel M, Bleda M, et al. . Identification of rare sequence variation underlying heritable pulmonary arterial hypertension. Nat Commun 2018; 9: 1416. doi: 10.1038/s41467-018-03672-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Hodgson J, Swietlik EM, Salmon RM, et al. . Characterization of GDF2 mutations and levels of BMP9 and BMP10 in pulmonary arterial hypertension. Am J Respir Crit Care Med 2020; 201: 575–585. doi: 10.1164/rccm.201906-1141OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Nikolic I, Yung LM, Yang P, et al. . Bone morphogenetic protein 9 is a mechanistic biomarker of portopulmonary hypertension. Am J Respir Crit Care Med 2019; 199: 891–902. doi: 10.1164/rccm.201807-1236OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Robert F, Certain MC, Baron A, et al. . Disrupted BMP-9 signaling impairs pulmonary vascular integrity in hepatopulmonary syndrome. Am J Respir Crit Care Med 2024; 210: 648–661 doi: 10.1164/rccm.202307-1289OC [DOI] [PubMed] [Google Scholar]
- 117.Long L, Ormiston ML, Yang X, et al. . Selective enhancement of endothelial BMPR-II with BMP9 reverses pulmonary arterial hypertension. Nat Med 2015; 21: 777–785. doi: 10.1038/nm.3877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Prapa M, Lago-Docampo M, Swietlik EM, et al. . First genotype-phenotype study in TBX4 syndrome: gain-of-function mutations causative for lung disease. Am J Respir Crit Care Med 2022; 206: 1522–1533. doi: 10.1164/rccm.202203-0485OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Rhodes CJ, Batai K, Bleda M, et al. . Genetic determinants of risk in pulmonary arterial hypertension: international genome-wide association studies and meta-analysis. Lancet Respir Med 2019; 7: 227–238. doi: 10.1016/S2213-2600(18)30409-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Ameen M, Sundaram L, Shen M, et al. . Integrative single-cell analysis of cardiogenesis identifies developmental trajectories and non-coding mutations in congenital heart disease. Cell 2022; 185: 4937–4953. doi: 10.1016/j.cell.2022.11.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Frascarelli C, Zanetti N, Nasca A, et al. . Nanopore long-read next-generation sequencing for detection of mitochondrial DNA large-scale deletions. Front Genet 2023; 14: 1089956. doi: 10.3389/fgene.2023.1089956 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Wagenvoort CA, Wagenvoort N. Primary pulmonary hypertension. Circulation 1970; 42: 1163–1184. doi: 10.1161/01.CIR.42.6.1163 [DOI] [Google Scholar]
- 123.Larkin EK, Newman JH, Austin ED, et al. . Longitudinal analysis casts doubt on the presence of genetic anticipation in heritable pulmonary arterial hypertension. Am J Respir Crit Care Med 2012; 186: 892–896. doi: 10.1164/rccm.201205-0886OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Humbert M, Sitbon O, Chaouat A, et al. . Survival in patients with idiopathic, familial, and anorexigen-associated pulmonary arterial hypertension in the modern management era. Circulation 2010; 122: 156–163. doi: 10.1161/CIRCULATIONAHA.109.911818 [DOI] [PubMed] [Google Scholar]
- 125.Jacobs W, van de Veerdonk MC, Trip P, et al. . The right ventricle explains sex differences in survival in idiopathic pulmonary arterial hypertension. Chest 2014; 145: 1230–1236. doi: 10.1378/chest.13-1291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.van Wezenbeek J, Groeneveldt JA, Lluciá-Valldeperas A, et al. . Interplay of sex hormones and long-term right ventricular adaptation in a Dutch PAH-cohort. J Heart Lung Transplant 2022; 41: 445–457. doi: 10.1016/j.healun.2021.11.004 [DOI] [PubMed] [Google Scholar]
- 127.Tello K, Richter MJ, Yogeswaran A, et al. . Sex differences in right ventricular-pulmonary arterial coupling in pulmonary arterial hypertension. Am J Respir Crit Care Med 2020; 202: 1042–1046. doi: 10.1164/rccm.202003-0807LE [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Kawut SM, Feng R, Ellenberg SS, et al. Pulmonary Hypertension and Anastrozole (PHANTOM): a randomized, double-blind, placebo-controlled trial. Am J Respir Crit Care Med 2024; in press [ 10.1164/rccm.202402-0371OC]. 10.1164/rccm.202402-0371OC [DOI] [PubMed] [Google Scholar]
- 129.Sangam S, Sun X, Schwantes-An TH, et al. . SOX17 deficiency mediates pulmonary hypertension: at the crossroads of sex, metabolism, and genetics. Am J Respir Crit Care Med 2023; 207: 1055–1069. doi: 10.1164/rccm.202203-0450OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Arnold AP. X chromosome agents of sexual differentiation. Nat Rev Endocrinol 2022; 18: 574–583. doi: 10.1038/s41574-022-00697-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Qin S, Predescu D, Carman B, et al. . Up-regulation of the long noncoding RNA X-inactive-specific transcript and the sex bias in pulmonary arterial hypertension. Am J Pathol 2021; 191: 1135–1150. doi: 10.1016/j.ajpath.2021.03.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Cunningham CM, Li M, Ruffenach G, et al. . Y-chromosome gene, Uty, protects against pulmonary hypertension by reducing proinflammatory chemokines. Am J Respir Crit Care Med 2022; 206: 186–196. doi: 10.1164/rccm.202110-2309OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Yan L, Cogan JD, Hedges LK, et al. . The Y chromosome regulates BMPR2 expression via SRY: a possible reason “why” fewer males develop pulmonary arterial hypertension. Am J Respir Crit Care Med 2018; 198: 1581–1583. doi: 10.1164/rccm.201802-0308LE [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
This one-page PDF can be shared freely online.
Shareable PDF ERJ-01095-2024.Shareable (530.6KB, pdf)