

The recently published guidelines on screening, monitoring and treatment update previous guidelines published 25 years ago (1,2). This update is timely as much has happened in the interim. Cystic fibrosis (CF) modulator therapy has dramatically improved prognosis for patients with CF, although it is not yet clear whether liver disease is improved. Our understanding of the pathophysiology of CF liver disease has also changed with the recognition of the importance of non-cirrhotic portal hypertension (3,4). Non-invasive tests for fibrosis and elastography have changed the practice of hepatology and reduced the need for liver biopsy. The guideline committee was made up of experts from North America and Europe and included adult and paediatric hepatologists and pulmonologists together with allied health practitioners and representatives of the CF community. A systematic literature search was performed and a vote of 80% was required to adopt a recommendation. The guidelines contain 7 recommendations for screening, 13 for disease monitoring and 14 for treatment.
An important change in these new guidelines is a new classification of CF liver disease (CFLD) as shown Table 1. Patients with evidence of liver disease are classified as having either advanced CFLD (aCFLD) or CF hepatobiliary involvement. This is a clear and simple classification and should prove very useful in clinical practice. Essentially any patient with evidence of cirrhosis and/or portal hypertension is classified as aCFLD. Any other features of liver or biliary disease are CF hepatobiliary involvement (CFHBI). A very similar classification was suggested in recently published joint ESPGHAN-NASPGHAN guidelines for the classification of liver abnormalities in people with CF by some of the same authors (5). In this classification, however, the term CFHBI was used to encompass the entire spectrum of liver abnormalities with aCFLD forming a subset of CFHBI. This may cause some confusion and perhaps having aCFLD and CFHBI as separate categories makes more sense.
Table 1. Suggested new classification of cystic fibrosis liver disease.
| aCFLD—one or more of the following: |
| Nodular liver |
| Advanced fibrosis (F4) |
| Multi-lobular cirrhosis ± portal hypertension |
| Non-cirrhotic portal hypertension |
| CFHBI—no features of aCFLD; but one or more of the following: |
| Hepatomegaly |
| Liver fibrosis < F4 |
| Increased liver stiffness by elastography < F4 |
| Hepatic steatosis |
| Focal biliary cirrhosis |
| Cholestasis |
| Persistently elevated liver tests >3 months |
| Abnormal liver imaging |
| Cholelithiasis |
| Sclerosing cholangitis |
| Hepatolithiasis |
aCFLD, advanced cystic fibrosis liver disease; CFHBI, cystic fibrosis hepatobiliary involvement.
The new guidelines recognise the importance of non-cirrhotic portal hypertension in CF. Recent studies suggest that this may be a major factor in the pathophysiology and may explain why the natural history of CFLD differs from the more typical cirrhotic liver diseases (3,4). Most liver diseases become more common with age and tend to progress over time. In contrast, CF liver disease generally manifests in childhood and is very unlikely to develop in later life. In a recently published, prospective, cohort study, no participants greater than 10 years of age without clinical or radiological evidence of liver disease at baseline progressed to CF liver disease (6). The main manifestations of CF liver disease are related to portal hypertension, e.g., splenomegaly, hypersplenism with thrombocytopenia, oesophageal varices and variceal bleeding. In contrast to other liver diseases, liver failure with jaundice, ascites and/or hepatic encephalopathy is relatively rare. The different natural history, compared to other chronic liver diseases, may be due to the pathophysiology. Over the past decade it has become clear that many, if not most, patients with CF liver disease have evidence of non-cirrhotic portal hypertension, characterised by obliterative portal venopathy, nodular regenerative hyperplasia and/or incomplete septal cirrhosis (7). This entity is now classified as porto-sinusoidal vascular disease (8). A possible outline of the pathophysiology is shown in Figure 1. It suggests that portal hypertension develops early due to obliterative portal venopathy and the liver becomes nodular as a result of nodular regenerative hyperplasia. Progressive portal fibrosis may ultimately cause advanced fibrosis or cirrhosis. This would explain the early development of portal hypertension and the relatively late appearance of other manifestations of liver failure. Currently there are no known treatments for porto-sinusoidal vascular disease. However, if this is the major driver of CF liver disease, therapy targeted at the vasculature rather than the biliary system may be more effective.
Figure 1.
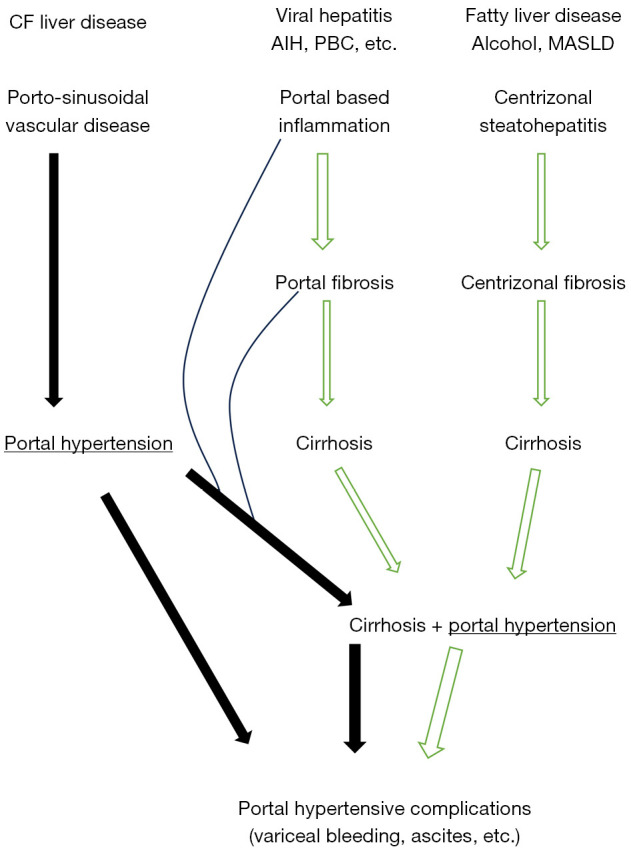
Pathophysiology of cystic fibrosis liver disease compared to other cirrhotic liver diseases. Portal hypertension occurs early due to obliterative portal venopathy and nodular regenerative hyperplasia. Cirrhosis develops late as a result of portal-based inflammation and fibrosis. CF, cystic fibrosis; AIH, autoimmune hepatitis; PBC, primary biliary cirrhosis; MASLD, metabolic associated steatotic liver disease.
The seven screening recommendations are clear. Annual physical abdominal examination and, liver blood tests and platelet counts are recommended starting at age of diagnosis. Abdominal ultrasound is recommended every 2 years from the age of 3 years till late adolescence. The age of 3 years was chosen on the basis of findings from the PUSH study (9). There is no recommendation for annual liver ultrasound in adulthood. This is sensible as de-novo liver disease is very rare in CF patients after adolescence (6). For those with evidence of liver disease, baseline elastography is recommended. It is sensible to use elastography for monitoring rather than diagnosis as elastography may not accurately assess non-cirrhotic portal hypertension and there is a degree of inherent variability particularly in children (10,11). No recommendation is made about screening in adulthood for those without known liver disease. With increasing longevity, it is important to be aware that other liver diseases may occur, e.g., metabolic associated steatotic liver disease (MASLD), alcoholic liver disease, viral hepatitis, etc. The major difficulty with screening for liver disease in CF is that there is no treatment known to alter the natural history of the condition although identification of portal hypertension may allow prevention of variceal haemorrhage.
There are 11 recommendations for monitoring. The use of non-invasive fibrosis markers of fibrosis and elastography is endorsed in addition to annual physical examination and bi-yearly liver ultrasound to monitor progression of disease. The committee recognises that there is a paucity of evidence to guide variceal surveillance or treatment. For this reason, no recommendations are made for variceal screening in children. Current American Association for the Study of Liver Disease (AASLD) and/or Baveno guidelines are recommended for adults although these do not specifically address CF related portal hypertension. This is clearly an area which would benefit from clinical studies. Annual ultrasound screening for hepatoma is recommended for children with cirrhosis or suspected cirrhosis. For adults, AASLD or European Association for the Study of the Liver (EASL) guidelines are recommended. In practice, this means every 6-month liver ultrasound. It is important to recognise that CF patients diagnosed with cirrhosis on imaging may in fact have non-cirrhotic portal hypertension. As such the non-invasive criteria for the diagnosis of hepatocellular carcinoma [Liver Imaging and Reporting & Data System (Li-RADs)] may not be applicable (12). Targeted liver biopsy may be required for confirmation.
Treatment recommendations
The treatment recommendations are not very prescriptive. This is understandable as there are no treatments proven to alter the natural history of CF liver disease. The guidelines recommend against routine treatment with ursodeoxycholic acid in all patients with CF to prevent the development of advanced liver disease. The use of CF transmembrane conductance regulator (CFTR) modulators in CFLD is a hot topic. The committee recommends treatment, with close monitoring, for CFHBI or patients who have received a liver transplant, advise against treatment in patients with decompensated liver disease and make no recommendation for patients with advanced compensated liver disease. No doubt these recommendations will be re-visited as new information becomes available. Hopefully, with the move to starting modulator treatment at an earlier age, the evolution of liver disease may be slowed or halted. The recommendations for management of portal hypertension and variceal bleeding are to follow established international guidelines, e.g., AASLD, Baveno and EASL as there is insufficient evidence to guide specific recommendations for CF. Due to a dearth of evidence, the committee could not recommend for or against beta blocker use for primary or secondary prophylaxis for varices. Given the requirement for general anaesthesia for endoscopy in children the committee could not recommend for or against screening gastroscopy for varices or banding for primary prophylaxis. Portal systemic shunts and liver transplant are endorsed as treatments for appropriate candidates. Transjugular intrahepatic portosystemic shunts (TIPS) are not mentioned, which is surprising as surgical portal systemic shunts are rarely performed now. TIPS is an excellent treatment for uncontrolled or refractory variceal bleeding. If and when elective TIPS should be used is not clear. The decision when to consider liver transplant can be difficult. As clinicians we try to avoid being too early or too late and some guidance would be appreciated, but perhaps this is not possible. The guidelines seem to suggest an international normalized ratio (INR) >1.5 as a threshold, although this seems a bit low. As the authors recognise, the guideline evaluation process highlighted many areas where clinical studies are needed to provide evidence-based advice. In particular, given the benefits of CFTR modulator therapy, it may be appropriate to look again at the safety of non-selective beta blockers.
In summary, these guidelines are a welcome update. The new classification of CF liver disease will hopefully be very useful. The use of elastography and fibrosis markers should facilitate screening and monitoring, with the caveats about non-cirrhotic portal hypertension. The guidelines identify a number of areas where there is a lack of evidence to guide recommendations and hopefully this will stimulate further research.
Supplementary
The article’s supplementary files as
Acknowledgments
Funding: None.
Ethical Statement: The authors are accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Footnotes
Provenance and Peer Review: This article was commissioned by the editorial office, HepatoBiliary Surgery and Nutrition. The article did not undergo external peer review.
Conflicts of Interest: Both authors have completed the ICMJE uniform disclosure form (available at https://hbsn.amegroups.com/article/view/10.21037/hbsn-24-442/coif). E.F. reports a grant to continue a longitudinal database of those with CF in Ireland to determine prevalence of liver disease and risk factors [payment made to institution (UCD) to employ research staff and contribute to the database]. The other author has no conflicts of interest to declare.
References
- 1.Sellers ZM, Assis DN, Paranjape SM, et al. Cystic fibrosis screening, evaluation, and management of hepatobiliary disease consensus recommendations. Hepatology 2024;79:1220-38. 10.1097/HEP.0000000000000646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sokol RJ, Durie PR. Recommendations for management of liver and biliary tract disease in cystic fibrosis. Cystic Fibrosis Foundation Hepatobiliary Disease Consensus Group. J Pediatr Gastroenterol Nutr 1999;28 Suppl 1:S1-13. 10.1097/00005176-199900001-00001 [DOI] [PubMed] [Google Scholar]
- 3.Wu H, Vu M, Dhingra S, et al. Obliterative Portal Venopathy Without Cirrhosis Is Prevalent in Pediatric Cystic Fibrosis Liver Disease With Portal Hypertension. Clin Gastroenterol Hepatol 2019;17:2134-6. 10.1016/j.cgh.2018.10.046 [DOI] [PubMed] [Google Scholar]
- 4.Witters P, Libbrecht L, Roskams T, et al. Liver disease in cystic fibrosis presents as non-cirrhotic portal hypertension. J Cyst Fibros 2017;16:e11-3. 10.1016/j.jcf.2017.03.006 [DOI] [PubMed] [Google Scholar]
- 5.Bodewes FAJA, Freeman AJ, Weymann A, et al. Towards a Standardized Classification of the Hepatobiliary Manifestations in Cystic Fibrosis (CFHBI): A Joint ESPGHAN/NASPGHAN Position Paper. J Pediatr Gastroenterol Nutr 2024;78:153-65. 10.1097/MPG.0000000000003944 [DOI] [PubMed] [Google Scholar]
- 6.Rowland M, Drummond J, Connolly L, et al. The natural history of cystic fibrosis liver disease a prospective cohort study. J Cyst Fibros 2023;22:1054-61. 10.1016/j.jcf.2023.07.002 [DOI] [PubMed] [Google Scholar]
- 7.Hermie L, Biervliet SV, Hoorens A, et al. Pre-emptive transjugular intrahepatic portosystemic shunt in pediatric cystic fibrosis-related liver disease and portal hypertension: prospective long-term results. Diagn Interv Radiol 2024;30:55-64. 10.4274/dir.2022.221818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.De Gottardi A, Rautou PE, Schouten J, et al. Porto-sinusoidal vascular disease: proposal and description of a novel entity. Lancet Gastroenterol Hepatol 2019;4:399-411. 10.1016/S2468-1253(19)30047-0 [DOI] [PubMed] [Google Scholar]
- 9.Leung DH, Ye W, Schwarzenberg SJ, et al. Long-term follow-up and liver outcomes in children with cystic fibrosis and nodular liver on ultrasound in a multi-center study. J Cyst Fibros 2023;22:248-55. 10.1016/j.jcf.2022.07.017 [DOI] [PubMed] [Google Scholar]
- 10.Klotter V, Gunchick C, Siemers E, et al. Assessment of pathologic increase in liver stiffness enables earlier diagnosis of CFLD: Results from a prospective longitudinal cohort study. PLoS One 2017;12:e0178784. 10.1371/journal.pone.0178784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rowland M, McGee A, Broderick A, et al. Repeatability of transient elastography in children. Pediatr Res 2020;88:587-92. 10.1038/s41390-020-0916-4 [DOI] [PubMed] [Google Scholar]
- 12.O'Brien C, Ramlaul N, Haughey A, et al. Hepatocellular carcinoma in cystic fibrosis liver disease: a cautionary tale. QJM 2019;112:693-4. 10.1093/qjmed/hcz150 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The article’s supplementary files as