

Abstract
Background
Melon (Cucumis melo L.), an important cucurbit crop, faces production limitations due to powdery mildew (PM). Developing resistant varieties offers a sustainable, genetics-based alternative to chemical treatments. Therefore, identifying PM resistance quantitative trait loci (QTL) and creating trait-associated markers are essential for efficient melon PM resistance improvement through marker-assisted backcrossing (MABC).
Results
Three F2 populations, A6, B2, and C4, were generated for QTL mapping of PM resistance. Major QTL were identified on chromosome 2 in A6, chromosome 5 in B2, and chromosomes 5 and 12 in C4. A series of TaqMan® assays targeting regions on chromosomes 2, 5, and 12 were developed and validated for foreground and recombinant selection, complemented by the double digest restriction-site associated DNA genotyping system to evaluate the recurrent parent genome recovery. Three MABC programs using resistant donor parents from A6 and C4 crossed with elite susceptible recurrent parents with green and orange fruit flesh were implemented. After two to three cycles of MABC, individual QTL was successfully introgressed into elite genetic backgrounds, giving six PM resistance lines in each green- and orange-fleshed background. PM inoculation on the twelve near-isogenic lines confirmed their resistance to PM.
Conclusions
We have identified major PM resistance QTL for melon on chromosomes 2, 5, and 12 and have introgressed individual QTL to elite genetic backgrounds using MABC in three and a half years. This study demonstrates the power of combining high-throughput genotyping with breeding efforts and showcases the efficiency of molecular breeding.
Supplementary Information
The online version contains supplementary material available at 10.1186/s40529-024-00435-x.
Keywords: Cucumis melo, Melon, Powdery mildew, Podosphaera xanthii, Quantitative trait loci (QTL), Marker assisted backcrossing (MABC), TaqMan, Double digest restriction-site associated DNA (ddRAD) sequencing
Introduction
Melon (Cucumis melo L., 2n = 2x = 24) is an economically valuable crop in the Cucurbitaceae family, cultivated widely across the world from temperate to tropical regions due to its sweet taste, flavor, and fleshy fruit. The global production of melons was around 28.56 million tonnes, making it the third most-produced cucurbit crop after watermelon and cucumber (FAOSTAT 2022). Melon is a cross-pollinated species and is an important model plant for studying sex determination. Initially, melon flowers are bisexual but can undergo abortion of either the carpel or stamen during sex determination, leading to the development of andromonoecious (producing male and bisexual flowers) or gynomonoecious (producing female and bisexual flowers) plants (Aamir et al. 2021). Typically, commercially grown melons are monoecious or andromonoecious, with male flowers on the main stem and female or hermaphrodite flowers on the proximal nodes of lateral branches. Consequently, melon breeding programs must initially assess self-pollinated breeding lines before proceeding to evaluate the hybrids.
Powdery mildew (PM) significantly impacts melon production, causing economic losses across all growing areas. This fungal disease can affect seedlings, stems, leaves, and fruit, which typically display a powdery appearance due to the abundance of conidia. It ultimately results in fruit quality degradation and yield losses due to stunting and premature plant death, limiting melon cultivation in greenhouses and fields (Egel et al. 2022). PM is caused by Podosphaera xanthii (Castagne) U. Braun & Shishkoff and Golovinomyces cichoracearum (DC.) V.P. Heluta. P. xanthii occurs more frequently in regions with high humidity and temperatures, such as subtropical and tropical areas. More than 20 races of P. xanthii have been identified based on the reactions to the differential set (McCreight 2006). Races 1, 2, and 5 are dominant races in Southern Europe; races 1 and 5 are prevalent in Japan; races 1, 2, and 3 in America; and races 1 and 2F are common in China (Zhang et al. 2013; Haonan et al. 2020). Races 1 and 5 were detected in Taiwan, with the former being predominant from 2001 to 2005, and the latter identified in 2005. From 2008 to 2010, both races were commonly identified in Tainan, the primary region for melon production in Taiwan (Huang and Wang 2007; Wang 2016). The distribution of predominant physiological races is influenced by spatiotemporal situations and the specific melon cultivars grown in these regions (López-Martín et al. 2022). PM management in melon involves cultural practices, biological controls, fungicides, and host resistance (Egel et al. 2022). Although fungicides are commonly used for melon PM control, they are not a sustainable solution since they can cause plant resistance breakdown and harm the environment. Therefore, resistant varieties remain a more sustainable and efficient option for PM control (Branham et al. 2021; Egel et al. 2022).
Identifying PM resistance quantitative trait loci (QTL) and the germplasm that carries them are essential for resistance breeding. Once the resistant materials and loci are identified, resistance loci can be effectively introduced into elite backgrounds using marker-assisted selection (Collard and Mackill 2008). Several studies have identified the genetic resources and QTL for PM resistance in melon. PM resistance QTL in melon have been identified on chromosomes 2, 4, 5, 9, 10, and 12 (Perchepied et al. 2005; Teixeira et al. 2008; Fukino et al. 2008; Yuste-Lisbona et al. 2010, 2011a, b; Wang et al. 2011, 2016; Beraldo-Hoischen et al. 2012; Zhang et al. 2013, 2023; Fazza et al. 2013; Ning et al. 2014; Kim et al. 2016; Li et al. 2017; Haonan et al. 2020; Cao et al. 2021; Branham et al. 2021; Cui et al. 2022; López-Martín et al. 2022). Regarding PM resistance materials, PI 414723, K7-1, and TARI-08874 were reported to carry a single QTL on chromosome 2 (Zhang et al. 2013; Fazza et al. 2013; Wang et al. 2016). The melon variety Ano2 carried a QTL on chromosome 5 (Wang et al. 2011), and the accession AF125Pm−1 carried resistance QTL on chromosome 9 (Teixeira et al. 2008), while the accessions wm-6, PI 124112, and MR-1 carried a single resistance QTL on chromosome 12 (Li et al. 2017; Cao et al. 2021; Zhang et al. 2023). Some accessions carried more than one resistance QTL. For instance, Edisto47 carried resistance QTL on chromosomes 2 and 5 (Ning et al. 2014); AR 5, PMR 5, and PMR 6 carried QTL on chromosomes 2 and 12 (Fukino et al. 2008; Kim et al. 2016; Haonan et al. 2020); and PI 124112 and TGR-1551 carried QTL on chromosomes 5 and 12 (Perchepied et al. 2005; Yuste‐Lisbona et al. 2010; Yuste-Lisbona et al. 2011a, b; Beraldo-Hoischen et al. 2012; López-Martín et al. 2022). Based on the abovementioned studies, the polymorphism of the PM markers are not able to correspond to the resistance consistently while applying marker-assisted selection, suggesting the investigation of high-density markers are required.
Next-generation sequencing (NGS) technologies and the release of the melon reference genome (Garcia-Mas et al. 2012) have revolutionized melon genetic analysis, offering faster methods to identify resistance QTL (Li et al. 2017; Branham et al. 2018). Genotyping-by-sequencing (GBS) techniques are a group of NGS-based high-throughput genotyping methods that use restriction enzymes to reduce genome complexity and dual-index barcoded systems to produce multiplex NGS libraries (Scheben et al. 2017). These methods include restriction site-associated DNA (RAD) sequencing (Baird et al. 2008), GBS (Elshire et al. 2011), 2-enzyme GBS (Poland et al. 2012), and double digest restriction site-associated DNA (ddRAD) sequencing (Peterson et al. 2012). NGS-based methods enable low-cost per-data-point marker discovery and high-throughput genotyping of single nucleotide polymorphisms (SNPs). Such availability enhances the construction of high-density linkage maps for QTL mapping and supports marker-assisted selection.
Marker-assisted backcrossing (MABC) utilizes the association between target phenotype and marker genotype for indirect selection (Frisch 2004). MABC has enhanced desirable characteristics in various crops, including submergence tolerance and salinity tolerance in rice (Neeraja et al. 2007; Marè et al. 2023), nutrient enrichment in maize (Singh et al. 2021; Chandrasekharan et al. 2022), and heat tolerance and fusarium head blight resistance in wheat (Zhang et al. 2021; Bellundagi et al. 2022). MABC aims to introgress favorable alleles from the donor parent into the recurrent parent’s genetic background. It involves three stages: foreground, recombinant, and background selection. Foreground and recombinant selections target the QTL-carrier chromosome with the objective of introducing an allele of interest with minimal linkage drag, while background selection aims to recover the recurrent parent genome (RPG) outside the target locus as much as possible (Collard and Mackill 2008). The effectiveness of MABC relies on adequate population size, the presence of polymorphic markers close to the target locus (Frisch et al. 1999a), and a broad distribution of markers within the genetic background (Frisch et al. 1999b). In melon, there have been cases of using markers to target disease resistance traits in backcross programs (Sousaraei et al. 2018; Palomares-Rius et al. 2018), but the implementation of MABC using markers for foreground, recombinant, and background selection has not yet been reported in the literature.
In this study, we aim to identify PM resistance QTL in the melon germplasms using high-density linkage maps, providing accurate markers for PM resistance. The objective of this study was (1) to develop high-density markers using an NGS-based high-throughput marker system, (2) to identify PM resistance QTL in melon, and (3) to introgress resistant alleles into elite melon cultivars using MABC. It took three and a half years from marker development to QTL introgression, which demonstrated the effectiveness of combining an NGS-based marker system and traditional breeding for the rapid improvement of disease resistance in melon.
Materials and methods
Plant materials
Three F2 populations for QTL mapping
In this study, we used three F2 populations, A6, B2, and C4, to map PM QTL (Table 1). The A6 population, composed of 165 F2 individuals, was derived from a cross between TARI-18-437 and TARI-18-494. The B2 population, composed of 179 F2 individuals, was derived from a cross between TARI-18-410 and TARI-18-491, while the C4 population, with a population size of 179 F2 individuals, was derived from a cross between TARI-18-491 and TARI-18-449. B2 and C4 shared TARI-18-491 as a common parent, but it served as the male parent for B2 and the female parent for C4.
Table 1.
The parental lines of the F2 population and the MABC breeding programs
| Population | Female parent | Male parent |
|---|---|---|
| F 2 populations for PM QTL mapping | ||
| A6 | TARI-18-437 | TARI-18-494 |
| B2 | TARI-18-410 | TARI-18-491 |
| C4 | TARI-18-491 | TARI-18-449 |
| Marker-assisted backcrossing | ||
| qPM2 |
TARI-18-432 TARI-18-431 |
TARI-18-437 |
| qPM5 and qPM12 |
TARI-18-432 TARI-18-431 |
TARI-18-449 |
TARI-18-432 and TARI-18-431 are the recurrent parents with green (G) and orange (O) flesh colors. The TARI-18-437 and TARI-18-449 are the PM resistance donor parents
The parental inbred lines, TARI-18-437 and TARI-18-410, were characterized by netted-rind, green-fleshed fruit, and resistance to P. xanthii race 1. TARI-18-437 was derived from a hybrid variety, and TARI-18-410 was generated from another accession after 16 and 14 generations of selfing, respectively. TARI-18-449 was a PM resistance inbred line developed through five generations of selfing of a hybrid variety with PI 124111 and PI 124112 in its pedigree. The PM resistance of the parental lines for the three F2 populations is shown in Table 2.
Table 2.
The average DI of PM for the parental lines
| Line | PM DI | |
|---|---|---|
| 2017 | 2018 | |
| TARI-18-437 | 0.0 | 0.0 |
| TARI-18-494 | 6.8 | 7.3 |
| TARI-18-410 | 1.3 | 0.0 |
| TARI-18-491 | 7.5 | 6.8 |
| TARI-18-449 | 0.0 | 0.0 |
| TARI-18-432 | 6.5 | 7.5 |
| TARI-18-431 | 2.5 | 5.8 |
The recurrent parents for MABC
Marketing preferences for new melon varieties primarily depend on fruit quality traits, particularly flesh color. Thus, we selected TARI-18-431, with orange flesh and a globular shape, and TARI-18-432, with green flesh and a rounded shape, as the elite recurrent parents (Table 1). Both parents possess desirable horticultural traits for fruit rind netting and taste quality but without PM resistance (Table 2). Therefore, the MABC breeding programs in this study aimed to improve the PM resistance of these two elite inbred lines by using them as recurrent parents.
PM evaluation
The PM reaction was assessed using leaf disc inoculation, as described by Wang et al. (2016). The experimental unit consisted of two leaf discs of 15-mm diameter per genotype, sampled from the second leaf when the melon plant was at the third-leaf stage. The leaf discs were placed on M-solution (10,000 ppm mannitol, 30 ppm benzimidazole, and 50 ppm tetracycline) in 60-mm Petri dishes. In each Petri dish, IRANH, a susceptible variety, and PMR 45, a resistant variety, were used as experimental controls. For each genotype, the disease reaction was evaluated using two replications. A conidial suspension was uniformly sprayed over the leaf discs to a density of 50–100 spores per cm2. Leaf discs were incubated at 24 °C/18 °C (day/night) with a 12-hour photoperiod for 12 days. The disease index (DI) was scored for each disc on a scale of 0 to 9, where 0 = no lesions; 1 = lesions covering 10% of the leaf area; 3 = lesions covering 50% of the leaf area; 5 = lesions covering 80% of the leaf area; 7 = lesions covering 100% of the leaf area, with thin spores on the leaf; and 9 = lesions covering 100% of the leaf area, with a thick brown disc of sporangia on the leaf discs. Plants with a mean DI < 3.0 were considered resistant, while those with a mean DI ≥ 3.0 were susceptible (Huang et al. 2002). For each batch of experiments, a non-inoculated set was set aside as a negative control. Disease rating was conducted in the fungal disease laboratory of the Plant Pathology division, Taiwan Agricultural Research Institute (TARI), Taichung, Taiwan, in spring 2017 and autumn 2018. Based on the leaf disc PM reaction evaluation on the melon differential set (McCreight 2006), the physiological race inoculated in this study is P. xanthii race 1.
Genotyping using ddRAD sequencing and its bioinformatic analysis workflow
ddRAD library preparation
Prior to library construction, we used in silico analysis on the melon reference genome DHL92 v3.5.1 (Argyris et al. 2015) and organelle genomes (Rodríguez-Moreno et al. 2011) to estimate adapter amounts and predict digested fragment sizes. Based on this analysis, the appropriate restriction enzyme sets for this study were PstI, TaqαI, and SphI. PstI and TaqαI, serving as rare and common cutters, have average recognition site distances of 11,914 bp and 482 bp, respectively, which determined the adapter input amounts. SphI was used to exclude target fragments containing the recognition site associated with rDNA sequences based on results from prior experiments.
Genomic DNA was extracted from freeze-dried young leaves using a modified 1.25% SDS method (Jobes et al. 1995). Extracted DNA was purified using the QIAquick 96 PCR Purification Kit (Qiagen, Hilden, Germany). DNA concentration was quantified using Quant-iT™ PicoGreen™ dsDNA Assay Kits (Life Technologies, Oregon, USA) and adjusted to 10–15 ng/µL for library preparation. The ddRAD library was prepared according to Peterson et al. (2012) with minor modifications as follows. The restriction enzymes, T4 DNA ligase, CutSmart Buffer, and rATP used for library construction were from New England Biolabs, Ipswich, MA, USA. Initially, 300 ng of genomic DNA from each sample was double-digested using PstI-HF and TaqαI at 37 °C and 65 °C, respectively, for 30 min. Ligation was then proceeded overnight at 16 °C, incorporating a 5-fold excess of P1 (PstI-HF) and Y (TaqαI) adapters, T4 DNA ligase, and rATP in 1X CutSmart Buffer. After inactivating the ligase, the library was treated with SphI at 37 °C for 30 min to exclude sequences and then purified using 0.8X AMPure XP beads (Beckman Coulter, Brea, CA, USA) to eliminate short fragments and adapter dimers. PCR amplification was conducted using dual-indexed primers, including Nextera XT DNA Indexes v2 and Phusion polymerase (Thermo Fisher Scientific Baltics UAB, Vilnius, Lithuania) in a 20-cycle two-step PCR protocol, which included only the denature and extension steps. The final PCR products were purified using 0.8X AMPure XP beads, quantified with Quant-iT™ PicoGreen™ dsDNA Assay Kits, and pooled in equal amounts. The ddRAD library target fragments were size-selected, ranging from 300 to 600 bp, using Blue Pippin (Sage Science, Beverly, MA, USA) in a 2% electrophoresis gel cassette. The size-selected ddRAD libraries were then sent to the Core Facility of the Cancer Progression Research Center at National Yang Ming Chiao Tung University for PE250 sequencing with extra dark cycles and a spike-in of 10% PhiX, utilizing the HiSeq2500 Rapid mode (Illumina, San Diego, CA, USA).
Bioinformatic analysis workflow for SNP calling
Paired-end FASTQ files were processed with AdapterRemoval v2.1.7 (Schubert et al. 2016) and aligned to the DHL92 v3.5.1 reference genome using Bowtie2 v2.2.9 (Langmead and Salzberg 2012). Alignments were sorted, compressed, and indexed into BAM files with SAMtools v1.3 (Li et al. 2009), and optical duplicate reads were removed using PICARD tools (http://broadinstitute.github.io/picard). Following GATK v3.8 (McKenna et al. 2010) Best Practices recommendations, base quality score recalibration refined sequence quality scores through a recalibration model. This model, typically relying on a known variants database such as dbSNP to distinguish true genetic variants from sequencing errors, utilized VCF files from HaplotypeCaller of parental BAM files as a dbSNP substitute, improving variant calling accuracy. F2 population BAM files were recalibrated, filtered with FilterSamReads, and then converted to gVCFs using HaplotypeCaller. Finally, aggregating all gVCFs through GenotypeGVCFs produced the population’s VCF file (DePristo et al. 2011; Van der Auwera et al. 2013).
The raw VCF files were subjected to filtering with specified criteria: (1) variants with more than two alleles were removed; (2) variants with a QUAL score less than 200, a Quality by Depth (QD) less than 10, and a minor allele frequency under 0.01 were excluded; and (3) genotypes with a Genotype Quality (GQ) from the VCF file’s FORMAT fields below 20 were marked as missing, increasing the missing rate threshold to 0.25.
Linkage map construction and QTL mapping
For linkage map construction and QTL mapping, we used the R/qtl package (Broman et al. 2003) implemented in R software (version 4.3.1, R Core Team 2023). Within each F2 population, the segregation distortion of each SNP was tested using the χ2 goodness-of-fit with a threshold adjusted by Bonferroni correction (α = 0.05/number of SNPs). SNPs were ordered based on physical positions, marker phasing was verified, and recombination fractions were estimated. Upon visual inspection of the recombination fraction patterns along the chromosomes, SNPs that were physically close to each other but showed no linkage were removed. Additionally, the SNP order was manually refined based on the recombination fraction patterns or physical positions, as necessary.
To prevent linkage map expansion frequently encountered in high-density maps, the Genotype Corrector (Miao et al. 2018) was applied to identify genotype errors and impute missing genotypes. Genotype Corrector used a sliding-window algorithm. The window size was set to 13 after empirical tests with the data from this study. The linkage map was then re-estimated using the Kosambi mapping function based on the corrected and imputed genotype data.
Both single QTL analysis and multiple interval mapping (Manichaikul et al. 2009) based on Haley and Knott regression (Haley and Knott 1992) were used for QTL mapping. For single QTL analysis, the empirical threshold was determined for each population based on 1,000 permutations (Churchill and Doerge 1994) at α = 0.05. For multiple interval mapping, 1,000 permutations for pairwise QTL mapping were first performed for each population. Thresholds for individual QTL and pairwise epistasis were derived from the permutation results at α = 0.01 with heavy interaction penalties only.
Conversion between physical and genetic distances
To integrate the relationship between genetic distance and physical distance from the three F2 populations, the locally estimated scatterplot smoothing (LOESS) regression was employed. The regression model for each chromosome used 10% local neighborhood data points and incorporated a quadratic term (degree of 2) in the fitting predictors (Siberchicot et al. 2017). The LOESS regression model on the carrier chromosome was used to select candidate SNPs for foreground and recombinant selection.
TaqMan® assay development for MABC
Prior to designing TaqMan® assays, MABC parental lines were genotyped using ddRAD sequencing. Polymorphic SNPs on carrier chromosomes with high quality (QUAL > 1000 and QD > 10) were selected. Each selected SNP was at least 50 bp away from any other variants and positioned at least 30 bp from the edge of the target fragments. Sequences of target fragments carrying the selected SNPs were then designed using the Custom TaqMan® Assay Design Tool (Thermo Fisher Scientific 2024). The TaqMan® assays for foreground selection were chosen based on the confidence intervals extending to the closest flanking markers. For recombinant selection, designed SNP markers were selected considering an average seed yield of approximately 200–500 per cross and the necessity of having at least one individual per cross for each MABC generation to manage risks effectively. The assays were chosen to cover at least 20 cM on both sides of the target region. TaqMan® assays were validated using parental DNA samples. Recombinant and foreground selection were made using the TaqMan® SNP Genotyping Assay Mix (Life Technologies, Marsiling, Singapore) and TaqMan™ Universal PCR Master Mix II, no UNG (Thermo Fisher Scientific Baltics UAB, Vilnius, Lithuania) according to the manufacturer’s instructions.
MABC for PM resistance introgression
The MABC aimed to enhance PM resistance in two elite recurrent parents. The process involved crossing donor parents with recurrent parents and selecting offspring according to target regions designated as qPM2, qPM5, and qPM12 (Figs. 1, 2 and 3).
Fig. 1.
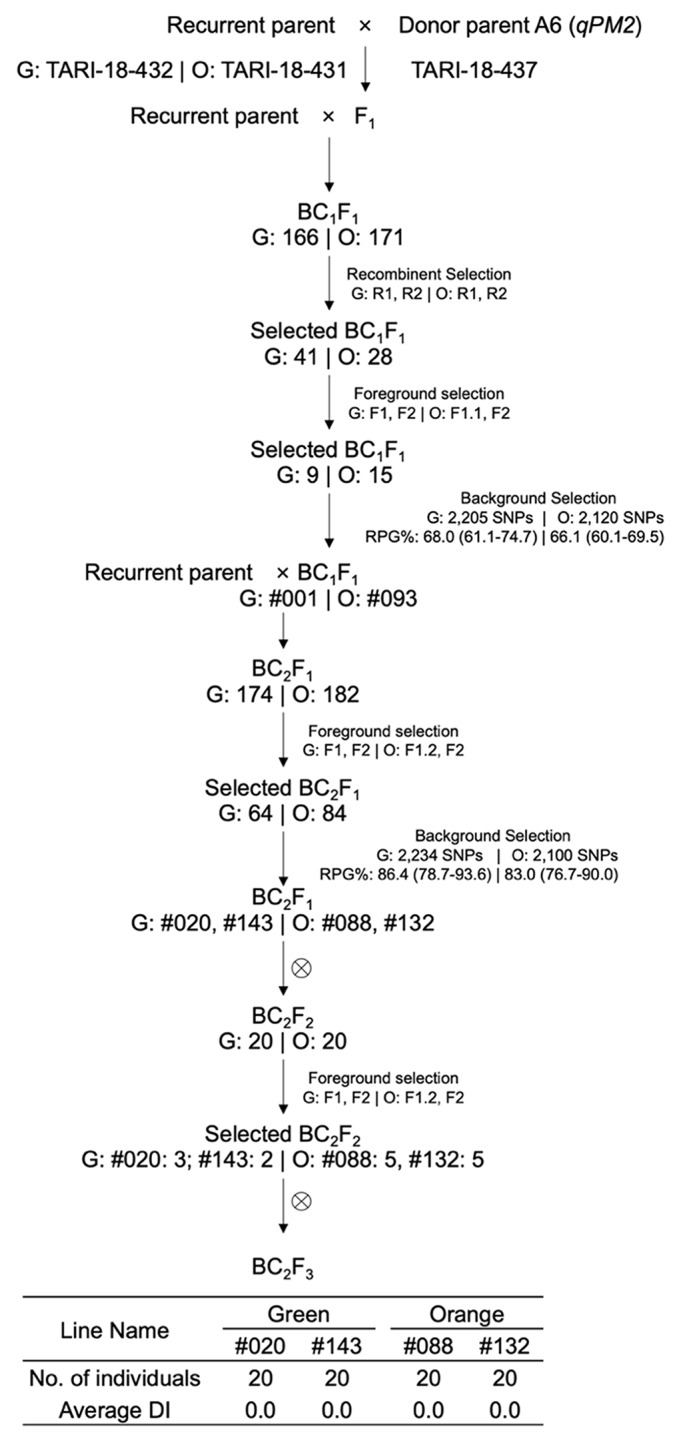
MABC scheme for qPM2. Crosses were made between the donor TARI-18-437 and recurrent parents TARI-18-432 (green-fleshed, G) and TARI-18-431 (orange-fleshed, O). G: Number | O: Number indicates the number of individuals for the green- and orange-fleshed background at each step. R1, R2, F1, F1.1, F1.2, and F2 are markers used for recombinant and foreground selection (Table 5). #Numbers indicate the selected line names derived from the selected individuals. Background selection text boxes contain the number of SNPs for RPG evaluation and average RPG recovery (%) with their ranges in brackets under green- and orange-fleshed backgrounds
Fig. 2.
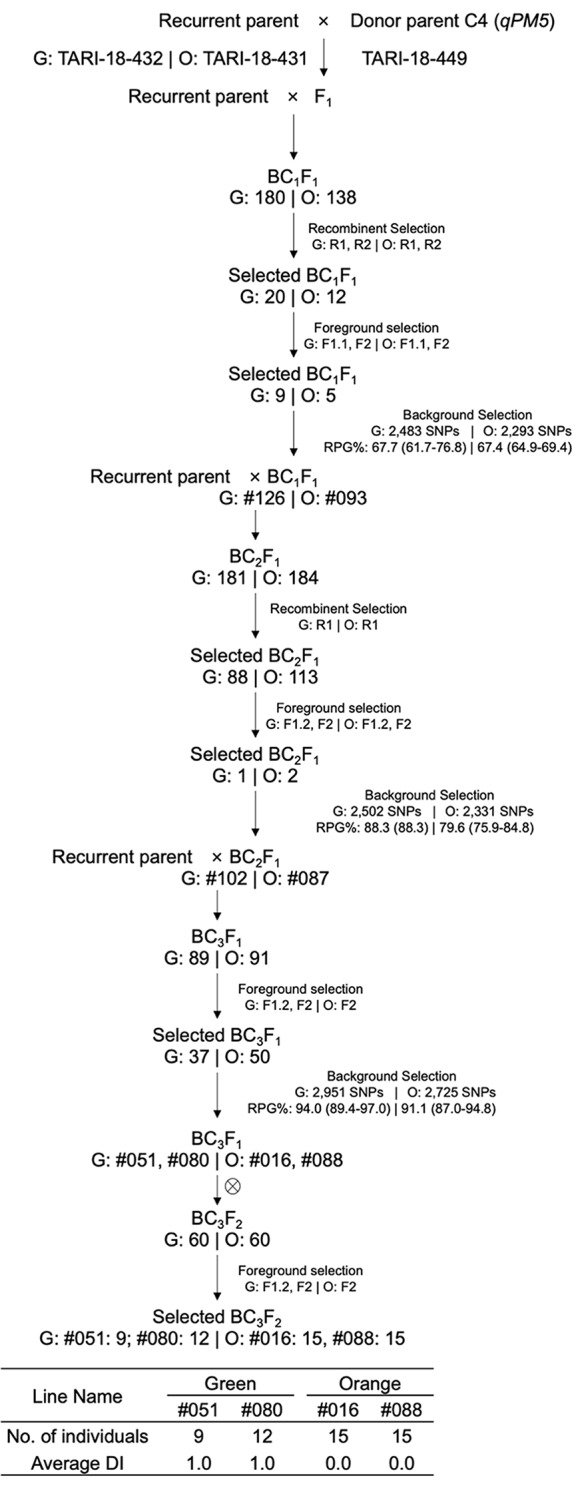
MABC scheme for qPM5. Crosses were made between the donor TARI-18-449 and recurrent parents TARI-18-432 (green-fleshed, G) and TARI-18-431 (orange-fleshed, O). G: Number | O: Number indicates the number of individuals for the green- and orange-fleshed background at each step. R1, R2, F1, F1.1, F1.2, and F2 are markers used for recombinant and foreground selection (Table 5). #Numbers indicate the selected line names derived from the selected individuals. Background selection text boxes contain the number of SNPs for RPG evaluation and average RPG recovery (%) with their ranges in brackets under green- and orange-fleshed backgrounds
Fig. 3.
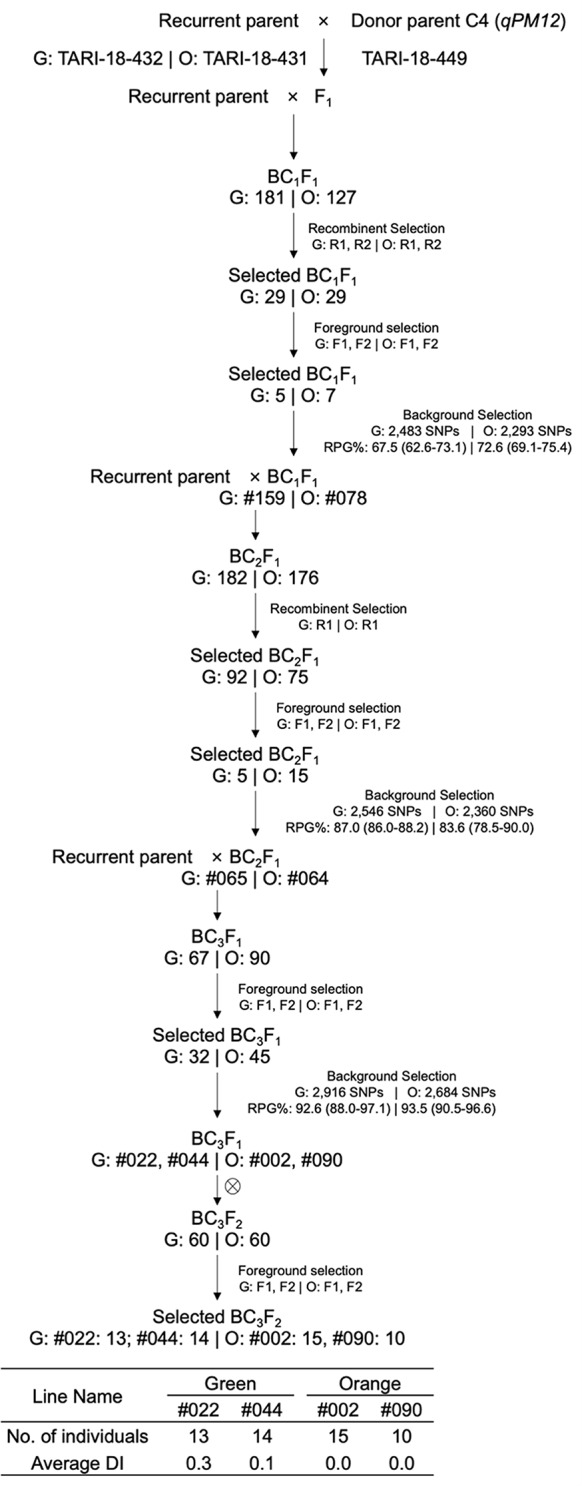
MABC scheme for qPM12. Crosses were made between the donor TARI-18-449 and recurrent parents TARI-18-432 (green-fleshed, G) and TARI-18-431 (orange-fleshed, O). G: Number | O: Number indicates the number of individuals for the green- and orange-fleshed background at each step. R1, R2, F1, F1.1, F1.2, and F2 are markers used for recombinant and foreground selection (Table 5). #Numbers indicate the selected line names derived from the selected individuals. Background selection text boxes contain the number of SNPs for RPG evaluation and average RPG recovery (%) with their ranges in brackets under green- and orange-fleshed backgrounds
At the BC1F1 generation, we aimed to first reduce the linkage drag around target loci via recombinant selection. Only individuals carrying homozygous for the recurrent parent at one recombinant marker and heterozygous genotypes at the other marker were passed to foreground selection. Those carrying resistance donor alleles showing heterozygous genotypes in two bracketed foreground markers were passed to background selection. Background selection was performed using ddRAD sequencing to well cover the whole genome, and the individuals showing high RPG recovery were then crossed with the recurrent parent to produce BC2F1 generation. The RPG recovery of the selected individuals was estimated using ddRAD genotype data. Heterozygous genotypes were assigned a value of 0.5, and homozygous genotypes of the recurrent parent were assigned a value of 1. These values were then weighted by the genetic distance between flanking markers and divided by the total genetic distance covered by the ddRAD markers on the non-carrier chromosomes.
At the BC2F1 generation, recombinant selection was first applied to screen for homozygous individuals at the second recombinant marker. Individuals showing homozygous genotypes at both recombinant markers were kept for foreground selection. Heterozygous individuals at foreground markers were selected for background selection using ddRAD sequencing. Individuals with the highest RPG recovery were backcrossed to the recurrent parents to generate BC3F1 individuals.
At the BC3F1 generation, only foreground selection was conducted. Subsequently, individuals with the highest RPG, as determined through ddRAD sequencing, were selected and self-pollinated to produce BC3F2 progeny. In the final BC3F2 generation, foreground selection was conducted to identify candidate individuals carrying homozygous PM resistance alleles. These individuals were then self-pollinated to establish the near-isogenic lines as the final outcome of the MABC in this study. The MABC schemes, as generally outlined above, were subject to minor adjustments depending on specific circumstances.
Results
The DI of PM in three F2 populations
The DI of PM in melon for the A6, B2, and C4 populations, their parental inbred lines, and F1 individuals are shown in Fig. 4. Disease reactions of most of the F2 individuals were between those of the two parents for the three populations. The F1 individuals showed a resistant phenotype, indicating that PM resistance was a dominant trait in the three populations. The phenotypic distribution of each F2 population is largely skewed toward resistance, suggesting that PM resistance is controlled by a few major QTL in the populations of interest.
Fig. 4.

The PM DI distribution of the three F2 populations. The average DI of the two parents and F1 are indicated below the histogram using red square, green triangle, and blue diamond, respectively
PM resistance QTL
For A6, B2, and C4, 127.14, 135.97, and 138.65 million paired-end reads were generated, respectively. On average, 0.76 million paired-end reads per sample were available for the three populations. The number of variants was 454,561 for A6, 444,854 for B2, and 465,971 for C4. After quality filtering, segregation distortion tests, and correction for genotyping errors, the final SNP data sets consisted of 3,466 SNPs for A6, 5,449 SNPs for B2, and 6,408 SNPs for C4. Across the full SNP data, 833 SNPs were shared by the three F2 populations, while B2 and C4 shared up to 73% or 62% common SNPs, given that the two populations had a common parent (Fig. 5). The total map lengths were 1,659.8 cM for A6, 1,586.0 cM for B2, and 1,609.2 cM for C4. SNPs were evenly distributed along the genome, except for some gaps on chromosomes 2 (B2, C4), 3 (A6), 5 (B2), 8 (A6), 9 (A6), and 10 (B2) (Fig. 6; Table 3).
Fig. 5.
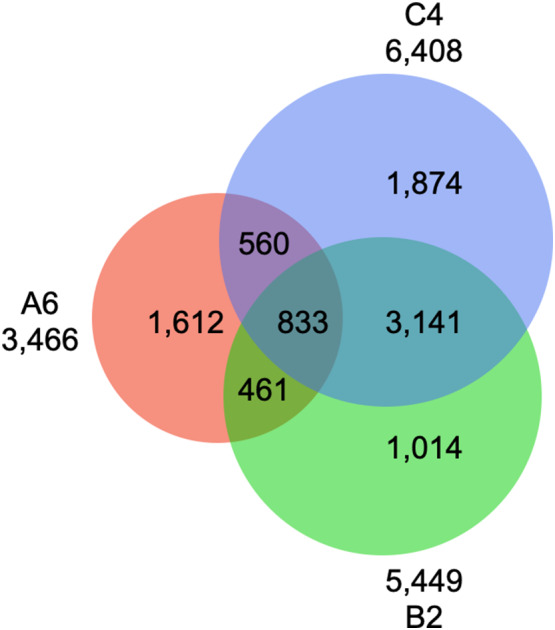
Number of SNPs across the three F2 populations
Fig. 6.

The linkage maps of the three F2 populations. The three F2 populations, A6, B2, and C4, are shown in red, green, and blue, respectively. The common markers between populations are linked using gray lines, and the black bars on the linkage groups are the QTL identified in this study. qPM5.B2 and qPM5.C4 represent the qPM5 detected in B2 and C4 population, respectively
Table 3.
Summary of linkage maps used in this study
| No. of markers | Length (cM) | Max interval (cM) | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Chr. | A6 | B2 | C4 | A6 | B2 | C4 | A6 | B2 | C4 | ||
| 1 | 294 | 761 | 873 | 157.4 | 155.4 | 142.1 | 5.8 | 4.2 | 5.1 | ||
| 2 | 250 | 199 | 296 | 126.0 | 113.8 | 128.8 | 8.2 | 13.3 | 11.2 | ||
| 3 | 345 | 518 | 601 | 138.3 | 126.0 | 130.4 | 11.8 | 4.2 | 4.8 | ||
| 4 | 401 | 644 | 721 | 179.1 | 162.5 | 181.4 | 7.9 | 5.1 | 6.5 | ||
| 5 | 281 | 334 | 430 | 131.0 | 123.3 | 123.0 | 9.7 | 24.3 | 7.2 | ||
| 6 | 309 | 488 | 588 | 154.5 | 149.2 | 163.3 | 6.8 | 9.0 | 4.2 | ||
| 7 | 324 | 428 | 511 | 126.7 | 116.3 | 112.9 | 9.9 | 3.4 | 4.5 | ||
| 8 | 283 | 414 | 543 | 166.4 | 150.5 | 164.2 | 15.5 | 8.4 | 9.3 | ||
| 9 | 245 | 399 | 425 | 127.7 | 129.3 | 121.6 | 10.9 | 7.1 | 5.4 | ||
| 10 | 169 | 277 | 327 | 93.4 | 106.4 | 85.8 | 7.8 | 10.9 | 5.4 | ||
| 11 | 267 | 485 | 595 | 137.3 | 127.4 | 141.5 | 9.5 | 8.1 | 8.9 | ||
| 12 | 298 | 502 | 498 | 122.0 | 125.7 | 114.3 | 7.2 | 5.1 | 5.7 | ||
| Overall | 3,466 | 5,449 | 6,408 | 1,659.8 | 1,586.0 | 1,609.2 | 15.5 | 24.3 | 11.2 | ||
The QTL identified in this study are summarized in Table 4 and Fig. 6. One major QTL, qPM2, was identified in A6. It was located on chromosome 2 and explained up to 93% of the total phenotypic variance. In B2, one major QTL, qPM5, was identified on chromosome 5, explaining 80% of the phenotypic variance. Two QTL, qPM5 and qPM12, were identified in C4, explaining up to 70% of the total phenotypic variance. All identified QTL exhibited dominant behavior. The confidence intervals for qPM5, extending to the closest flanking markers in the physical distance for the B2 and C4 populations on chromosome 5, were 25,267,104–25,725,099 bp and 25,203,821–25,678,875 bp, respectively. These overlapping intervals suggest that qPM5 in the B2 and C4 populations can be considered the same QTL.
Table 4.
QTL for PM resistance identified in the tree F2 populations
| Population | QTL | Chr | Pos (cM) | a | d | LOD | R 2 | Confidence interval (cM) | Flanking markers (bp) |
|---|---|---|---|---|---|---|---|---|---|
| A6 | qPM2 | 2 | 8.9 | -2.79 | -2.79 | 94.5 | 93.5 | 8.34–10.43 [2.1] | 778,333–1,142,653 |
| B2 | qPM5 | 5 | 92.7 | -2.17 | -1.28 | 62.8 | 80.1 | 91.63–94.67 [3.0] | 25,276,104–25,725,099 |
| C4 | qPM5 | 5 | 89.0 | -1.58 | -1.10 | 27.7 | 29.3 | 86.45–90.58 [4.1] | 25,203,821–25,678,875 |
| qPM12 | 12 | 77.4 | -1.52 | -1.12 | 28.9 | 31.0 | 75.15–78.44 [3.3] | 22,493,040–22,879,440 | |
| Total model | 49.2 | 71.6 |
Chr and Pos indicate the chromosome and peak position of QTL
Positions and length (between square brackets) of the confidence interval are indicated
Flanking markers (bp) is the confidence interval of QTL extending to the closest markers
a and d are the additive and the dominant effects of the QTL, respectively
R2 is the phenotypic variance explained by the QTL
TaqMan® assays for MABC
To design markers for foreground selection, the confidence interval of each QTL was extended to the closest flanking markers (Table 5 and Supplementary Table 1). The qPM2 region, located at 8.34–10.43 cM on chromosome 2 in the A6 population, had a corresponding physical distance from 778,333 to 1,142,653 bp. The qPM5 region, located at 91.63–94.67 cM on chromosome 5 in the B2 population and at 86.45–90.58 cM on the same chromosome in the C4 population, had overlapping physical distances of 25,267,104–25,725,099 bp for B2 and 25,203,821–25,678,875 bp for C4, with a common fragment at 25,203,821–25,725,099 bp. The qPM12 region, identified at 75.15–78.44 cM on chromosome 12 in the C4 population, had a corresponding physical distance from 22,493,040 to 22,879,440 bp.
Table 5.
The designed TaqMan® assays used in this study
| Target | Flesh color | Code | Assay Name | Pd.cM | BCn | Context Sequence |
|---|---|---|---|---|---|---|
| qPM2 | G | R1 | 2_382892_C-A | 7.47 | 1 | TAATAAATGTTTTAACTTTTCTAGT[C/A]TTTTTGGAAGAGGAATTGAAAGAAA |
| G | F1 | 2_621190_A-G | 9.73 | 1 + 2 | CGTCCCTAGTTACTTCTGTTTGTTT[A/G]CCTCATTATACCACTGAAGCAACAA | |
| G | F2 | 2_1603309_A-T | 19.32 | 1 + 2 | TCGGTTCTGATAAGCAGGATGCTTC[A/T]GGAAATAAGCTTCTTCAAGATATTG | |
| G | R2 | 2_2351587_A-C | 28.67 | 1 | GGCTTGTGGAGAAGATCAACAATTC[A/C]ATTGGGCAAGACCCAAATTCTAAAT | |
| O | R1 | 2_103153_C-A | 1.40 | 1 | TCTTCTCACCCTCCATCTCATTAAC[C/A]AACTGTCAGCACCCTAAGATTCCCT | |
| O | F1.2 | 2_627533_C-T | 9.78 | 2 | GAATANTATCGTTGCATTTGATGAA[C/T]TTAAGGCTCTCCTAAGTTTAGAGCT | |
| O | F1.1 | 2_778333_T-A | 10.72 | 1 | ACGAAAATGTCACATAGCCTACAAC[T/A]CCTCCATTGACGGCATCATTCTCCC | |
| O | F2 | 2_816421_C-G | 11.06 | 1 + 2 | CCATTCTCAAATCCATCCAAAACAC[C/G]AATCCAACAAAGAACAAATAAAACC | |
| O | R2 | 2_1410652_T-C | 17.03 | 1 | CTGGTATGCTCAGTTATGCAAAGAG[T/C]GTTGCAGCTGATAACTGGTTGGCAT | |
| qPM5 | G/O | R1 | 5_24520366_C-T | 86.51 | 1 | CAACTTTCCATTTTAACTATTGGAA[C/T]GTGAGTTACTATTATTCCTTTCTTT |
| G/O | F1.2 | 5_24941008_G-C | 89.02 | 2 | GCTTGTTCAATCTTGTGAGTTGGAT[G/C]AGATGATTTAGTTGAAGCGTGANTG | |
| G/O | F1.1 | 5_25264851_C-T | 89.31 | 1 | TTCTCCTAGTCACTTGTTTCTTACA[C/T]GAGATCTTTTGTTTCACTAGGAGGA | |
| G/O | F2 | 5_25862218_T-C | 96.52 | 1 + 2 | AACATGAAACACATCTCAATGATGA[T/C]GACGACGATGAATACCAGGCTGATT | |
| G/O | R2 | 5_26105586_T-A | 99.27 | 1 | AGGGACATCTTCATGGTTTGGTGGC[T/A]ACTTTAGCAAACTAAATTTTAGACA | |
| qPM12 | G | R1 | 12_21800139_A-G | 71.69 | 1 + 2 | CAACAAAAAATTTGTTTGATGGCCG[A/G]TAGGATTGTAACCTAATAAAAAAAC |
| O | R1 | 12_21116734_T-G | 63.61 | 1 + 2 | GGACTATTATTCTGAACCTACCCAG[T/G]TCACAAGAGGCCCTTGTTCTTCTGA | |
| G/O | F1 | 12_22418198_A-G | 78.87 | 1 + 2 | TGAAGTAAAGTGAAAAGAAATAATC[A/G]TGTATGGCTCGCTGTTGACGTTTGT | |
| G/O | F2 | 12_23222171_G-A | 84.38 | 1 + 2 | TGATCGCCAAAGACGAGAAACTTGC[G/A]ACTTCATACCGTGATGAGAAATGGA | |
| G/O | R2 | 12_23491653_A-G | 87.74 | 1 | CCGTGGTTTGTCTATCAAGTTTTTA[A/G]AATCAACCTATGTGACAATCGCATT |
The flesh colors G and O indicate the assays used for the green- or orange-fleshed recurrent parents, and G/O indicates the assay used for both recurrent parents in MABC
Codes R and F indicate the assays used for recombinant or foreground selection. The R1, R2, F1, and F2 indicate the physical position order of the TaqMan assays. The F1.1 and F1.2 indicate the F1 TaqMan assays but the different versions for their slight adjustment of the physical positions
Pd.cM indicates the predicted genetic position (cM) of the TaqMan assays through the LOESS regression models within each chromosome
BCn filled with 1, 2, and 1 + 2 values indicate that the TaqMan assays were used during BC1, BC2, or both BC1 and BC2 generations
Accounting for polymorphic SNPs between donor and recurrent parents, and the requirements for the design tool, the designed and validated TaqMan assays are shown in Table 5. The genetic distances between bracketed foreground selection markers ranged from 0.3 to 9.6 cM for qPM2 in the orange-fleshed parent and green-fleshed parent, respectively. The genetic distances between markers for recombinant selection and foreground selection ranged from 2.3 to 15.3 cM. The positions of markers for foreground and recombinant selection are shown in Fig. 7. Between BC1F1 and BC2F1, markers for foreground selection for qPM2 in the orange-fleshed background and those for qPM5 in orange- and green-fleshed backgrounds were changed from F1.1 to F1.2 (Table 5; Fig. 7).
Fig. 7.

Position of the TaqMan® assays used for foreground and recombinant selection. The consensus linkage groups built from A6, B2, and C4 are shown in the middle. qPM5.B2 and qPM5.C4 represent the qPM5 detected in the B2 and C4 populations, respectively. Marker positions for green- or orange-fleshed parents are indicated at the left and right sides, respectively. F1, F1.1, F1.2, F2, R1, R2 are marker names. Please refer to Table 5 and Supplementary Table 1 for detailed marker information
Generate PM resistance near-isogenic lines through MABC
MABC for qPM2
For the recombinant selection at the BC1F1 generation, 41 out of 166 (24.7%) green-fleshed individuals and 28 out of 171 (16.4%) orange-fleshed individuals showed one marker as homozygous for the recurrent parent allele and the other as heterozygous (Table 5; Figs. 1 and 7). Among the selected individuals, 9 green-fleshed and 15 orange-fleshed ones carried heterozygous genotypes for foreground markers F1, F1.1, and F2. The average RPG recovery for green- and orange-fleshed individuals was 68.0% and 66.1%, respectively. Individuals with the highest RPG recovery for green-fleshed background (#001) and orange-fleshed background (#093) were selected and crossed with the respective recurrent parent to form the BC2F1 generation. At the BC2F1 generation, a two-step selection was applied because the QTL was close to one end of chromosome 2 (Fig. 6). Among the 174 individuals in the green-fleshed BC2F1 family, 64 carried the resistance allele at the heterozygous state, while 84 out of 182 individuals in the orange-fleshed family carried heterozygous genotypes. For background selection via ddRAD sequencing, the average RPG recovery reached 86.4% and 83.0% for green- and orange-fleshed backgrounds, respectively. Individuals showing the highest RPG recovery rates, i.e., 93.6% for green-fleshed background and 89.1% for orange-fleshed background, were self-pollinated to produce the BC2F2 generation. From this step, two families were maintained: #020 and #143 for green-fleshed background and #088 and #132 for orange-fleshed background. At the BC2F2 generation, only foreground markers were used to screen 20 individuals in each family. Individuals carrying homozygous resistance genotypes were selected and self-pollinated to generate BC2F3 individuals. For each near-isogenic line, 20 BC2F3 individuals were used to evaluate the PM reaction. All the near-isogenic lines showed a DI of zero, indicating the successful introgression of PM resistance to the elite backgrounds.
MABC for qPM5
For the recombinant selection of qPM5 at the BC1F1 generation, 20 out of 180 (11.1%) green-fleshed individuals and 12 out of 138 (8.7%) orange-fleshed individuals showed one marker as homozygous for the recurrent parents allele and the other as heterozygous (Table 5; Figs. 2 and 7). Among the selected individuals, nine green-fleshed and five orange-fleshed ones carried heterozygous genotypes for foreground markers F1.1, and F2. The average RPG recovery for green- and orange-fleshed individuals was 67.7% and 67.4%, respectively. Individuals with the highest RPG recovery for green-fleshed background (#126) and orange-fleshed background (#093) were selected and crossed with the respective recurrent parent to form the BC2F1 generation. At the second recombinant selection performed at the BC2F1 generation, 88 out of 181 individuals from the green-fleshed family and 113 out of 184 individuals from the orange-fleshed family were homozygous as the recurrent parent for the marker showing heterozygous genotype at the first recombinant selection. Among these individuals, one green-fleshed and two orange-fleshed ones were heterozygous genotypes at the foreground markers F1.2 and F2. The average RPG recovery rate was 88.3% in green-fleshed individuals and 79.6% in orange-fleshed individuals. Individuals with the highest RPG recovery, i.e., 88.3% for the green-fleshed background (#102) and 84.8% for the orange-fleshed background (#087) were selected and crossed with the respective recurrent parent to form the BC3F1 generation. At the BC3F1 generation, a two-step selection was applied since the recombinant selection was done at BC2F1. Among the 89 green-fleshed BC3F1 progeny, 37 carried the resistance allele at the heterozygous state, while 50 out of 91 orange-fleshed individuals carried heterozygous genotypes. The background selection revealed an average RPG recovery of 94.0% for the green-fleshed family and 91.1% for the orange-fleshed family. The individuals with the highest RPG recovery, i.e., 95.8% and 94.8% for green- and orange-fleshed backgrounds, respectively, were self-pollinated to generate the BC3F2 generation. Two BC3F2 families were maintained for both green- and orange-fleshed backgrounds: #051 and #080 for green-fleshed families, and #016 and #088 for orange-fleshed families. At the BC3F2 generation, only foreground markers were used to select the individuals carrying homozygous resistance alleles from 60 individuals in each family. For the green- and orange-fleshed background, selected individuals were evaluated for the DI of PM. The average DI for green- and orange-fleshed near-isogenic lines were 1.0 and 0.0, indicating that the foreground marker regions effectively improved PM resistance to the elite backgrounds.
MABC for qPM12
For the recombinant selection of qPM12 at the BC1F1 generation, 29 out of 181 (16.0%) green-fleshed individuals and 29 out of 127 (22.8%) orange-fleshed individuals showed one marker as homozygous for the recurrent parents allele and the other as heterozygous (Table 5; Figs. 3 and 7). Among these individuals, five green-fleshed and seven orange-fleshed ones carried heterozygous genotypes for foreground markers F1 and F2. The average RPG recovery for green- and orange-fleshed individuals was 67.5% and 72.6%, respectively. Individuals with the highest RPG recovery for green-fleshed background (#159) and orange-fleshed background (#078) were selected and crossed with the respective recurrent parent to form the BC2F1 generation. At the second recombinant selection performed at the BC2F1 generation, 92 out of 182 green-fleshed individuals from the green-fleshed family and 75 out of 176 individuals from the orange-fleshed family were homozygous for the recurrent parent allele. Among these individuals, five green-fleshed and 15 orange-fleshed ones carried heterozygous genotypes for the foreground markers F1 and F2. The average RPG recovery rate was 87.0% in green-fleshed individuals and 83.6% in orange-fleshed individuals. Individuals with the highest RPG recovery, 88.2% for the green-fleshed background (#065) and 90.0% for the orange-fleshed background (#064), were selected and crossed with respective parents to form the BC3F1 generation. Among the 67 green-fleshed BC3F1 progeny, 32 carried the resistance allele at the heterozygous state, while 45 out of 90 orange-fleshed individuals carried heterozygous genotypes. The background selection revealed an average RPG recovery of 92.6% for the green-fleshed family and 93.5% for the orange-fleshed family. The individuals with the highest RPG recovery, nearing 97.1% and 96.1% for green- and orange-fleshed backgrounds, respectively, were self-pollinated to generate the BC3F2 families. Two BC3F2 families were maintained for both green- and orange-fleshed backgrounds: #022 and #044 for green-fleshed families, and #002 and #090 for orange-fleshed families. At the BC3F2 generation, only foreground markers were used to select the individuals carrying homozygous resistance alleles from 60 individuals in each family. For the green- and orange-fleshed background, selected individuals were evaluated for the DI of PM. The average DI for green-fleshed near-isogenic lines were 0.3 and 0.1, and each line showed a DI of zero in orange-fleshed backgrounds indicating that the resistance alleles of qPM12 was effectively introgressed into the elite backgrounds.
Discussion
In this study, we have identified three major melon PM resistance QTL located on chromosomes 2, 5, and 12 in three F2 populations and introduced these resistant QTL to the PM-susceptible elite lines. According to the results of PM resistance QTL in Table 4, both the A6 and B2 populations carried single PM resistance QTL, qPM2 and qPM5, respectively, and the C4 population carried two, qPM5 and qPM12. The phenotypic variance explained (R2) and the additive effect (a) of qPM2 were larger than qPM5. While qPM5 was accompanied by qPM12 in the C4 population, the R2 and the LOD scores of qPM12 were larger than qPM5. Although the additive effect of qPM12 was slightly lower than qPM5, the average DIs of PM in the near-isogenic lines carried qPM5 were less than those carried qPM12 under the green-fleshed genetic background, suggesting qPM2 and qPM12 contribute more PM resistance than qPM5.
To compare the resistance QTL across different studies, we blasted the reference genome DHL92 v4 (Castanera et al. 2020) using QTL flanking marker sequences and identified candidate genes within the interval between foreground markers using the DHL92 v4 annotation (Ruggieri et al. 2018). Under the interval between foreground markers, qPM2, qPM5, and qPM12 covered 128, 98, and 116 genes, respectively. Among this large number of genes, we will focus our discussion on candidate genes showing transcriptional or functional evidence from previous studies.
Several studies have identified PM resistance QTL overlapping qPM2 identified in the present study, including Pm-pxA.II and Pm-pxB.II (Fukino et al. 2008), Pm-Edisto47-1 (Ning et al. 2014), qPM2 (Wang et al. 2016) Pm-2F (Zhang et al. 2013), Pm2.1 (Haonan et al. 2020), and Pm-II (Kim et al. 2016). Among these QTL, Pm-2F and Pm2.1 were identified through PM race 2F and Pm-II through race N5, while the other PM QTL on chromosome 2 were identified through the inoculation of race 1. Therefore, there may be three different QTL co-located at this region. MELO3C015353 and MELO3C015354 may be the candidate genes because they were annotated as disease-resistance proteins with leucine-rich repeat (LRR) domains (Haonan et al. 2020). qPM5 identified in this study was co-localized with several PM QTL identified previously, including Pm-AN (Wang et al. 2011), qPx1-5 (Branham et al. 2021), Pm-R1-2, and PM-R5 (Yuste-Lisbona et al. 2011a, b). López-Martín et al. (2022) further narrowed down the region and identified the candidate genes, MELO3C004297 and MELO3C004311, which encoded a branched-chain amino-acid aminotransferase-like protein and a tomato mosaic virus resistance protein N-like gene, respectively. qPM12 identified in this study co-localized with QTL identified using different races: Pm-pxA.XII and Pm-pxB.XII (Fukino et al. 2008), BPm12.1 (Li et al. 2017), CmPMRl (Cui et al. 2022), and qPx1-12 (Branham et al. 2021), conferring resistance to race 1; Pm-R1-2-5 (Beraldo-Hoischen et al. 2012) to races 1, 2, and 5; Pm-XII to race N5 (Kim et al. 2016); pm12.1 (Haonan et al. 2020) and Cmpmr2F to race 2F (Zhang et al. 2023); and qCmPMR-12 (Cao et al. 2021) to non-specified races. López-Martín et al. (2022) further fine-mapped the QTL and identified one of the candidate genes, MELO3C002504, characterized as a cysteine-rich receptor-like protein kinase. Zhang et al. (2023) identified MELO3C002403 as a candidate gene for Cmpmr2F. The transcription level of MELO3C002403 in the resistant parent PI 124112 increased during PM inoculation. It encoded an allantoate amidohydrolase protein, primarily residing in the cytoplasm and cell membrane. qPx1-12 harbored MELO3C002392 and MELO3C002393, both were LRR receptor-like kinases (Branham et al. 2021). MELO3C002434, MELO3C002438, MELO3C002439, MELO3C002440, and MELO3C002441 were Ankyrin repeat family proteins located within at least one of the following QTL: BPm12.1, pm12.1, qCmPMR-12, and CmPMRl. The parental lines for mapping BPm12.1 and qCmPMR-12 carried non-synonymous mutations at MELO3C002434 and the expression level of MELO3C002434 was significantly higher in the resistant parent after PM inoculation (Li et al. 2017; Cao et al. 2021). Therefore, it is a potential causal gene for qPM12. The genes MELO3C002441 to MELO3C002449 were located within CmPMRl. Among these genes, six carried non-synonymous SNPs between resistant and susceptible genotypes. In addition, MELO3C002441, MELO3C002444, and MELO3C002448 were significantly upregulated after PM inoculation of resistant melon genotypes. Conversely, MELO3C002446, MELO3C002447, and MELO3C002449 were downregulated during infection. MELO3C002449 was associated with glycolytic enzyme activity but the functions of MELO3C002446 and MELO3C002437 remain unknown. Integrating results from different PM studies, it seems that the PM resistance QTL are predominantly clustered on chromosomes 2, 5, and 12. QTL detected through the use of different races co-localized at these three regions. Therefore, these regions may harbor multiple resistance loci and are promising targets for melon PM resistance MABC.
In this MABC study, we first focused on recombinant selection, followed by foreground and background selection. Performing recombinant selection before foreground selection at the BC1F1 generation optimized the use of low-throughput genotyping platforms. While this strategy was efficient, it could not entirely avoid the loss of target foreground genotypes due to potential double crossovers. Therefore, foreground selection was performed at each generation. The ddRAD sequencing system facilitated high-throughput genotyping for background selection in later generations, significantly increasing the intensity of selection with enhanced recovery of the RPG. Our MABC scheme at the BC1F1 generation reduces the genotyping effort by 17–28% due to the selection intensity for recombinant selection ranging from 8.7 to 24.7% in the qPM5 orange-fleshed and qPM2 green-fleshed MABC processes. The whole strategy was also time-effective: creating the three F2 populations, including the PM resistance QTL mapping, took one and a half years, followed by two years of MABC. This led to the production of near-isogenic PM resistance lines from creating the population for QTL mapping in 3.5 years.
The near-isogenic lines developed in this study are valuable resources for melon resistance breeding and genetic studies. On one hand, they can be used as parental lines to confer resistance in other elite backgrounds or allow the pyramiding of major QTL. On the other hand, they could be used for fine mapping of the three PM resistance QTL. Future research could explore the durability of resistance related to multiple minor QTL and identify additional genetic resources for resistance breeding in melon.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Acknowledgements
We thank Dr. Tze-Tze Liu in the Genomics Center for Clinical and Biotechnological Applications of National Core Facility for Biopharmaceuticals, Taiwan for the NGS sequencing service. We thank Technology Commons in the College of Life Science and Center for Systems Biology, National Taiwan University for the assistance in the quality control of sequencing libraries. We thank Dr. Fu-Jin Wei for his assistance in server maintenance for bioinformatic analysis. We thank Dr. Ya-Ping Lin from the World Vegetable Center Headquarters, Taiwan for her assistance and suggestion for revising the manuscript.
Author contributions
C-S W conceived the study, data analysis and prepared the original draft, editing and revised the final manuscript; S-Y L provided the experimental materials and collected the samples; J-H H evaluated the powdery mildew reactions; H-Y C conducted the sequencing library experiment; D-K L collected the samples and analyzed the data; K-K H conceived, supervised the project and led the funding acquisition; Y-H W provided guidance regarding the experiments; Y-F H drafted and revised the manuscript.
Funding
This work was funded by the Council of Agriculture, Executive Yuan (Grant No. 106AS-19.1.3-ST-a1, 107AS-15.1.3-ST-a1, 108AS-14.2.1-ST-a1, 109AS-12.2.1-ST-a1).
Data availability
The data used in this study are available from the corresponding author on reasonable request.
Declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Chun-San Wang, Email: d99621105@ntu.edu.tw.
Yung-Fen Huang, Email: huangy@ntu.edu.tw.
References
- Aamir M, Karmakar P, Singh VK, Kashyap SP, Pandey S, Singh BK, Singh PM, Singh J (2021) A novel insight into transcriptional and epigenetic regulation underlying sex expression and flower development in melon (Cucumis melo L). Physiol Plant 173:1729–1764. 10.1111/ppl.13357 [DOI] [PubMed] [Google Scholar]
- Argyris JM, Ruiz-Herrera A, Madriz-Masis P, Sanseverino W, Morata J, Pujol M, Ramos-Onsins SE, Garcia-Mas J (2015) Use of targeted SNP selection for an improved anchoring of the melon (Cucumis melo L.) scaffold genome assembly. BMC Genomics 16:4. 10.1186/s12864-014-1196-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baird NA, Etter PD, Atwood TS, Currey MC, Shiver AL, Lewis ZA, Selker EU, Cresko WA, Johnson EA (2008) Rapid SNP discovery and genetic mapping using sequenced RAD markers. PLoS ONE 3:e3376. 10.1371/journal.pone.0003376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellundagi A, Ramya KT, Krishna H, Jain N, Shashikumara P, Singh PK, Singh GP, Prabhu KV (2022) Marker-assisted backcross breeding for heat tolerance in bread wheat (Triticum aestivum L). Front Genet 13:1056783. 10.3389/fgene.2022.1056783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beraldo-Hoischen P, Gomez-Guillamon ML, Lopez-Sese AI, Sari N, Solmaz I, Aras V (2012) QTL associated with one recessive gene for powdery mildew resistance in the melon genotype TGR-1551. Cucurbitaceae 2012: Proceedings of the Xth Eucarpia meeting on genetics and breeding of cucurbitaceae
- Branham SE, Levi A, Katawczik M, Fei Z, Wechter WP (2018) Construction of a genome-anchored, high-density genetic map for melon (Cucumis melo L.) and identification of Fusarium oxysporum f. sp. melonis race 1 resistance QTL. Theor Appl Genet 131:829–837. 10.1007/s00122-017-3039-5 [DOI] [PubMed] [Google Scholar]
- Branham SE, Kousik C, Mandal MK, Wechter WP (2021) Quantitative trait loci mapping of resistance to powdery mildew race 1 in a recombinant inbred line population of melon. Plant Dis 105:3809–3815. 10.1094/PDIS-12-20-2643-RE [DOI] [PubMed] [Google Scholar]
- Broman KW, Wu H, Sen Ś, Churchill GA (2003) R/qtl: QTL mapping in experimental crosses. Bioinformatics 19:889–890. 10.1093/bioinformatics/btg112 [DOI] [PubMed] [Google Scholar]
- Cao Y, Diao Q, Chen Y, Jin H, Zhang Y, Zhang H (2021) Development of KASP markers and identification of a QTL underlying powdery mildew resistance in melon (Cucumis melo L.) by bulked segregant analysis and RNA-Seq. Front Plant Sci 11:593207. 10.3389/fpls.2020.593207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castanera R, Ruggieri V, Pujol M, Garcia-Mas J, Casacuberta JM (2020) An improved melon reference genome with single-molecule sequencing uncovers a recent burst of transposable elements with potential impact on genes. Front Plant Sci 10. 10.3389/fpls.2019.01815 [DOI] [PMC free article] [PubMed]
- Chandrasekharan N, Ramanathan N, Pukalenthy B, Chandran S, Manickam D, Adhimoolam K, Nalliappan GK, Manickam S, Rajasekaran R, Sampathrajan V, Muthusamy V, Hossain F, Gupta HS, Natesan S (2022) Development of β-carotene, lysine, and tryptophan-rich maize (Zea mays) inbreds through marker-assisted gene pyramiding. Sci Rep 12:8551. 10.1038/s41598-022-11585-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Churchill GA, Doerge RW (1994) Empirical threshold values for quantitative trait mapping. Genetics 138:963–971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collard BCY, Mackill DJ (2008) Marker-assisted selection: an approach for precision plant breeding in the twenty-first century. Philosophical Trans Royal Soc B: Biol Sci 363:557–572. 10.1098/rstb.2007.2170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Core Team (2023) R: A language and environment for statistical computing. R foundation for statistical computing, Vienna, Austria. https://www.r-project.org/
- Cui H, Fan C, Ding Z, Wang X, Tang L, Bi Y, Luan F, Gao P (2022) CmPMRl and CmPMrs are responsible for resistance to powdery mildew caused by PodospXanthiianthii race 1 in Melon. Theor Appl Genet. 10.1007/s00122-021-04025-4 [DOI] [PubMed] [Google Scholar]
- DePristo MA, Banks E, Poplin R, Garimella KV, Maguire JR, Hartl C, Philippakis AA, del Angel G, Rivas MA, Hanna M, McKenna A, Fennell TJ, Kernytsky AM, Sivachenko AY, Cibulskis K, Gabriel SB, Altshuler D, Daly MJ (2011) A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet 43:491–498. 10.1038/ng.806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egel DS, Adkins ST, Wintermantel WM, Keinath AP, D’Arcangelo KN, Parada-Rojas CH, Rennberger G, Toporek SM, Hausbeck MK, Quesada-Ocampo LM (2022) Diseases of cucumbers, melons, pumpkins, squash, and watermelons. In: Elmer WH, McGrath M, McGovern RJ (eds) Handbook of vegetable and herb diseases. Springer International Publishing, Cham, pp 1–105 [Google Scholar]
- Elshire RJ, Glaubitz JC, Sun Q, Poland JA, Kawamoto K, Buckler ES, Mitchell SE (2011) A robust, simple genotyping-by-sequencing (GBS) approach for high diversity species. PLoS ONE 6:e19379. 10.1371/journal.pone.0019379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- FAOSTAT (2022) Crops and livestock products. https://www.fao.org/faostat/en/#data/QCL. Accessed 16 Apr 2024
- Fazza AC, Dallagnol LJ, Fazza AC, Monteiro CC, de Lima BM, Wassano DT, Camargo LEA (2013) Mapping of resistance genes to races 1, 3 and 5 of Podosphaera Xanthii in melon PI 414723. Crop Breed Appl Biotechnol 13:349–355. 10.1590/S1984-70332013000400005 [Google Scholar]
- Frisch M (2004) Breeding strategies: optimum design of marker-assisted backcross programs. Molecular marker systems in plant breeding and crop improvement. Springer, pp 319–334
- Frisch M, Bohn M, Melchinger AE (1999a) Minimum sample size and optimal positioning of flanking markers in marker-assisted backcrossing for transfer of a target gene. Crop Sci 39:967–975 [Google Scholar]
- Frisch M, Bohn M, Melchinger AE (1999b) Comparison of selection strategies for marker-assisted backcrossing of a gene. Crop Sci 39:1295–1301 [Google Scholar]
- Fukino N, Ohara T, Monforte AJ, Sugiyama M, Sakata Y, Kunihisa M, Matsumoto S (2008) Identification of QTLs for resistance to powdery mildew and SSR markers diagnostic for powdery mildew resistance genes in melon (Cucumis melo L). Theor Appl Genet 118:165–175. 10.1007/s00122-008-0885-1 [DOI] [PubMed] [Google Scholar]
- Garcia-Mas J, Benjak A, Sanseverino W, Bourgeois M, Mir G, González VM, Hénaff E, Câmara F, Cozzuto L, Lowy E, Alioto T, Capella-Gutiérrez S, Blanca J, Cañizares J, Ziarsolo P, Gonzalez-Ibeas D, Rodríguez-Moreno L, Droege M, Du L, Alvarez-Tejado M, Lorente-Galdos B, Melé M, Yang L, Weng Y, Navarro A, Marques-Bonet T, Aranda MA, Nuez F, Picó B, Gabaldón T, Roma G, Guigó R, Casacuberta JM, Arús P, Puigdomènech P (2012) The genome of melon (Cucumis melo L). Proc Natl Acad Sci U S A 109:11872–11877. 10.1073/pnas.1205415109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haley CS, Knott SA (1992) A simple regression method for mapping quantitative trait loci in line crosses using flanking markers. Heredity 69:315–324 [DOI] [PubMed] [Google Scholar]
- Haonan C, Zhuo D, Chao F, Zicheng Z, Hao Z, Peng G, Feishi L (2020) Genetic mapping and nucleotide diversity of two powdery mildew resistance loci in melon (Cucumis melo). Phytopathology® 110:1970–1979. 10.1094/PHYTO-03-20-0078-R [DOI] [PubMed] [Google Scholar]
- Huang JH, Wang YH (2007) The races of Podosphaera xanthii causing melon powdery mildew in Taiwan. J Taiwan Agric Res 56:307–315 [Google Scholar]
- Huang JH, Wang YH, Lo CT (2002) Development of leaf-disk method for screening melon varieties resistant to Sphaerotheca Fuliginea race 1. J Agric Res China 51:49–56. 10.29951/JARC.200212.0005 [Google Scholar]
- Jobes DV, Hurley DL, Thien LB (1995) Plant DNA isolation: a method to efficiently remove polyphenolics, polysaccharides, and RNA. Taxon 44:379–386. 10.2307/1223408 [Google Scholar]
- Kim HT, Park JI, Robin AHK, Ishikawa T, Kuzuya M, Horii M, Yashiro K, Nou I-S (2016) Identification of a new race and development of DNA markers associated with powdery mildew in melon. Plant Breed Biotechnol 4:225–233. 10.9787/PBB.2016.4.2.225 [Google Scholar]
- Langmead B, Salzberg SL (2012) Fast gapped-read alignment with Bowtie 2. Nat Methods 9:357–359. 10.1038/nmeth.1923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, 1000 Genome Project Data Processing Subgroup (2009) The sequence Alignment/Map format and SAMtools. Bioinformatics 25:2078–2079. 10.1093/bioinformatics/btp352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li B, Zhao Y, Zhu Q, Zhang Z, Fan C, Amanullah S, Gao P, Luan F (2017) Mapping of powdery mildew resistance genes in melon (Cucumis melo L.) by bulked segregant analysis. Sci Hort 220:160–167. 10.1016/j.scienta.2017.04.001 [Google Scholar]
- López-Martín M, Pérez-de-Castro A, Picó B, Gómez-Guillamón ML (2022) Advanced genetic studies on powdery mildew resistance in TGR-1551. IJMS 23:12553. 10.3390/ijms232012553 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manichaikul A, Moon JY, Sen Ś, Yandell BS, Broman KW (2009) A model selection approach for the identification of quantitative trait loci in experimental crosses, allowing epistasis. Genetics 181:1077–1086. 10.1534/genetics.108.094565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marè C, Zampieri E, Cavallaro V, Frouin J, Grenier C, Courtois B, Brottier L, Tacconi G, Finocchiaro F, Serrat X, Nogués S, Bundó M, San Segundo B, Negrini N, Pesenti M, Sacchi GA, Gavina G, Bovina R, Monaco S, Tondelli A, Cattivelli L, Valè G (2023) Marker-assisted introgression of the salinity tolerance locus saltol in temperate Japonica Rice. Rice 16:2. 10.1186/s12284-023-00619-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCreight JD (2006) Melon-powdery mildew interactions reveal variation in melon cultigens and podosphaera xanthii races 1 and 2. J Amer Soc Hort Sci 131:59–65 [Google Scholar]
- McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, DePristo MA (2010) The genome analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res 20:1297–1303. 10.1101/gr.107524.110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miao C, Fang J, Li D, Liang P, Zhang X, Yang J, Schnable JC, Tang H (2018) Genotype-Corrector: improved genotype calls for genetic mapping in F2 and RIL populations. Sci Rep 8. 10.1038/s41598-018-28294-0 [DOI] [PMC free article] [PubMed]
- Neeraja CN, Maghirang-Rodriguez R, Pamplona A, Heuer S, Collard BCY, Septiningsih EM, Vergara G, Sanchez D, Xu K, Ismail AM, Mackill DJ (2007) A marker-assisted backcross approach for developing submergence-tolerant rice cultivars. Theor Appl Genet 115:767–776. 10.1007/s00122-007-0607-0 [DOI] [PubMed] [Google Scholar]
- Ning X, Wang X, Gao X, Zhang Z, Zhang L, Yan W, Li G (2014) Inheritances and location of powdery mildew resistance gene in melon Edisto47. Euphytica 195:345–353. 10.1007/s10681-013-1000-5 [Google Scholar]
- Palomares-Rius FJ, Garcés-Claver A, Picó MB, Esteras C, Yuste-Lisbona FJ, Gómez-Guillamón ML (2018) Carmen’, a yellow canary melon breeding line resistant to Podosphaera Xanthii, Aphis gossypii, and cucurbit yellow stunting disorder virus. Horts 53:1072–1075. 10.21273/HORTSCI13013-18 [Google Scholar]
- Perchepied L, Bardin M, Dogimont C, Pitrat M (2005) Relationship between loci conferring downy mildew and powdery mildew resistance in melon assessed by quantitative trait loci mapping. Phytopathology 95:556–565. 10.1094/PHYTO-95-0556 [DOI] [PubMed] [Google Scholar]
- Peterson BK, Weber JN, Kay EH, Fisher HS, Hoekstra HE (2012) Double Digest RADseq: an inexpensive method for de novo SNP discovery and genotyping in model and non-model species. PLoS ONE 7:e37135. 10.1371/journal.pone.0037135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poland JA, Brown PJ, Sorrells ME, Jannink J-L (2012) Development of high-density genetic maps for barley and wheat using a novel two-enzyme genotyping-by-sequencing approach. PLoS ONE 7:e32253. 10.1371/journal.pone.0032253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodríguez-Moreno L, González VM, Benjak A, Martí MC, Puigdomènech P, Aranda MA, Garcia-Mas J (2011) Determination of the melon chloroplast and mitochondrial genome sequences reveals that the largest reported mitochondrial genome in plants contains a significant amount of DNA having a nuclear origin. BMC Genomics 12:424. 10.1186/1471-2164-12-424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruggieri V, Alexiou KG, Morata J, Argyris J, Pujol M, Yano R, Nonaka S, Ezura H, Latrasse D, Boualem A, Benhamed M, Bendahmane A, Cigliano RA, Sanseverino W, Puigdomènech P, Casacuberta JM, Garcia-Mas J (2018) An improved assembly and annotation of the melon (Cucumis melo L.) reference genome. Sci Rep 8:8088. 10.1038/s41598-018-26416-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheben A, Batley J, Edwards D (2017) Genotyping-by-sequencing approaches to characterize crop genomes: choosing the right tool for the right application. Plant Biotechnol J 15:149–161. 10.1111/pbi.12645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schubert M, Lindgreen S, Orlando L (2016) AdapterRemoval v2: rapid adapter trimming, identification, and read merging. BMC Res Notes 9. 10.1186/s13104-016-1900-2 [DOI] [PMC free article] [PubMed]
- Siberchicot A, Bessy A, Guéguen L, Marais GA (2017) MareyMap online: a user-friendly web application and database service for estimating recombination rates using physical and genetic maps. Genome Biol Evol 9:2506–2509. 10.1093/gbe/evx178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh J, Sharma S, Kaur A, Vikal Y, Cheema AK, Bains BK, Kaur N, Gill GK, Malhotra PK, Kumar A, Sharma P, Muthusamy V, Kaur A, Chawla JS, Hossain F (2021) Marker-assisted pyramiding of lycopene-ε-cyclase, β-carotene hydroxylase1 and opaque2 genes for development of biofortified maize hybrids. Sci Rep 11:12642. 10.1038/s41598-021-92010-8 [DOI] [PMC free article] [PubMed]
- Sousaraei N, Ramshini H, Lotfi M, Sharzei A (2018) Marker assisted backcrossing for introgression of fusarium wilt resistance gene into melon. Euphytica 214:7. 10.1007/s10681-017-2080-4 [Google Scholar]
- Teixeira APM, Barreto FA, da Camargo S (2008) LEA An AFLP marker linked to the Pm-1 gene that confers resistance to Podosphaera xanthii race 1 in Cucumis melo. Genetics and Molecular Biology 31:547–550. 10.1590/S1415-47572008000300023
- Thermo Fisher Scientific (2024) Custom TaqMan® assay design tool. https://www.thermofisher.com/order/custom-genomic-products/tools/genotyping/. Accessed 29 May 2024
- Van der Auwera GA, Carneiro MO, Hartl C, Poplin R, del Angel G, Levy-Moonshine A, Jordan T, Shakir K, Roazen D, Thibault J, Banks E, Garimella KV, Altshuler D, Gabriel S, DePristo MA (2013) From fastq data to high-confidence variant calls: The genome analysis toolkit best practices pipeline. In: Current Protocols in Bioinformatics. John Wiley & Sons, Inc., p 11.10.1–11.10.33 [DOI] [PMC free article] [PubMed]
- Wang YH (2016) Mapping quantitative trait loci for fruit traits and powdery mildew resistance in melon (Cucumis melo L.). Doctoral Dissertation, National Taiwan University [DOI] [PMC free article] [PubMed]
- Wang X, Li G, Gao X, Xiong L, Wang W, Han R (2011) Powdery mildew resistance gene Pm-AN located in a segregation distortion region of melon LGV. Euphytica 180:421–428. 10.1007/s10681-011-0406-1 [Google Scholar]
- Wang YH, Wu DH, Huang JH, Tsao SJ, Hwu KK, Lo HF (2016) Mapping quantitative trait loci for fruit traits and powdery mildew resistance in melon (Cucumis melo). Bot Stud 57:19. 10.1186/s40529-016-0130-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuste-Lisbona FJ, López‐Sesé AI, Gómez‐Guillamón ML (2010) Inheritance of resistance to races 1, 2 and 5 of powdery mildew in the melon TGR-1551. Plant Breeding 129:72–75. 10.1111/j.1439-0523.2009.01655.x [Google Scholar]
- Yuste-Lisbona FJ, Capel C, Gómez-Guillamón ML, Capel J, López-Sesé AI, Lozano R (2011a) Codominant PCR-based markers and candidate genes for powdery mildew resistance in melon (Cucumis melo L). Theor Appl Genet 122:747–758. 10.1007/s00122-010-1483-6 [DOI] [PubMed] [Google Scholar]
- Yuste-Lisbona FJ, Capel C, Sarria E, Torreblanca R, Gómez-Guillamón ML, Capel J, Lozano R, López-Sesé AI (2011b) Genetic linkage map of melon (Cucumis melo L.) and localization of a major QTL for powdery mildew resistance. Mol Breed 27:181–192. 10.1007/s11032-010-9421-5 [Google Scholar]
- Zhang C, Ren Y, Guo S, Zhang H, Gong G, Du Y, Xu Y (2013) Application of comparative genomics in developing markers tightly linked to the Pm-2F gene for powdery mildew resistance in melon (Cucumis melo L). Euphytica 190:157–168. 10.1007/s10681-012-0828-4 [Google Scholar]
- Zhang Y, Yang Z, Ma H, Huang L, Ding F, Du Y, Jia H, Li G, Kong Z, Ran C, Gu Z, Ma Z (2021) Pyramiding of fusarium head blight resistance quantitative trait loci, Fhb1, Fhb4, and Fhb5, in modern Chinese wheat cultivars. Front Plant Sci 12(694023). 10.3389/fpls.2021.694023 [DOI] [PMC free article] [PubMed]
- Zhang T, Cui H, Luan F, Liu H, Ding Z, Amanullah S, Zhang M, Ma T, Gao P (2023) A recessive gene Cmpmr2F confers powdery mildew resistance in melon (Cucumis melo L). Theor Appl Genet 136:4. 10.1007/s00122-023-04269-2 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data used in this study are available from the corresponding author on reasonable request.