

Abstract
The cerebrospinal fluid (CSF) is a key matrix for discovery of biomarkers relevant for prognosis and the development of therapeutic targets in pediatric central nervous system malignancies. However, the wide range of protein concentrations and age-related differences in children makes such discoveries challenging. In addition, pediatric CSF samples are often sparse and first prioritized for clinical purposes. The present work focused on optimizing each step of the proteome analysis workflow to extract the most detailed proteome information possible from the limited CSF resources available for research purposes. The strategy included applying sequential ultracentrifugation to enrich for extracellular vesicles (EV) in addition to analysis of a small volume of raw CSF, which allowed quantification of 1351 proteins (+55% relative to raw CSF) from 400 μL CSF. When including a spectral library, a total of 2103 proteins (+240%) could be quantified. The workflow was optimized for CSF input volume, tryptic digestion method, gradient length, mass spectrometry data acquisition method and database search strategy to quantify as many proteins a possible. The fully optimized workflow included protein aggregation capture (PAC) digestion, paired with data-independent acquisition (DIA, 21 min gradient) and allowed 2989 unique proteins to be quantified from only 400 μL CSF, which is a 340% increase in proteins compared to analysis of a tryptic digest of raw CSF.
Keywords: cerebrospinal fluid, extracellular vesicle, biomarker, CSF input volume, protein aggregation capture
Introduction
The cerebrospinal fluid (CSF) provides structural and functional support essential to the central nervous system (CNS).1 Functions attributed to this compartment include the clearance of excreted waste products and providing a cytotoxic environment for invading microorganisms, which is facilitated by anaerobic conditions with limited nutrients and growth factors.1 The state of the CNS is often reflected in the CSF, and examination of CSF is integrated into the diagnostic workflow of several diseases, including infectious CNS disease,2 neurodegenerative diseases,3 subarachnoid hemorrhage (SAH),4 and CNS cancer.5 Cytological evaluation using microscopy remains the primary approach for the identification of cancer cells in CSF, in contrast to other diseases with established noncellular biomarkers, such as xanthochromia caused by hemoglobin catabolism in SAH or β-amyloid in Alzheimer’s disease.6
The lack of accurate biomarker-based risk categorization in pediatric CNS cancer patients implies that treatment with CNS-directed chemotherapy is administered to children, adolescents, and young adults, even when there is no detection of circulating or invasive tumor cells.7−9 CNS-directed chemotherapy is associated with substantial short- and long-term adverse neurotoxic effects, and this treatment may be unwarranted in low-risk cancer patients. A deeper understanding of the CSF proteome could advance our knowledge of CNS malignancies. Identification of unique proteome profiles may allow for reliable risk categorization, complement disease progression monitoring, and early detection of neurotoxicity in pediatric CNS cancer patients.7
However, the pediatric CSF proteome remains to be extensively studied and the current proteome research is limited to diverse experimental workflows and small cohort sizes, which consequently affect the quality of evidence synthesis.10 The rapid technological advancement of mass spectrometry (MS)-based proteomics continues to enable more in-depth proteome analysis through an increasing collection of different methods.11 Despite these advancements, several challenges often persist in proteomic studies of pediatric CSF: first, the rarity of childhood cancer often necessitates long-term inclusion to obtain adequately sized cohorts; second, the challenge of obtaining adequate CSF volumes due to the limited natural reserves in children; and third, the diversity in age of pediatric cancer patients may obscure distinctions between natural variations in CNS development and disease.10 Consequently, CSF samples from pediatric patients constitute a unique but scarce resource that emphasizes the need for cost-efficient analyses.
We previously published a review with a meta-analysis of individual patient data, synthesizing methods and evidence from previous LC-MS/MS-based studies on CSF from pediatric patients with CNS malignancies.10 We refer to this review for more detailed information, as it covered: (1) methods of CSF collection, preparation before storage, and storage conditions; (2) the analytical workflow applied (including CSF volume, discovery process, and proteome depth); and (3) the validation process (validation cohort, analytical method, and data availability) for each individual study. Results in brief, CSF proteome studies focusing on children with ALL have quantified between 366 and 635 proteins.12−15 In contrast, studies that have focused on solid brain tumors have quantified in the range of 729 to 1526 unique proteins.16−18
The protein concentration in CSF is substantially less than that in plasma, with CSF protein levels being 50 to 100 times lower. Typically, plasma protein concentrations range from approximately 60 to 70 mg/mL, whereas CSF protein concentrations normally range between 0.2 and 0.7 mg/mL.19 Despite this reduced concentration, CSF retains many properties similar to plasma, including a wide range of protein concentrations (referred to as “high dynamic range”). A persistent but significant challenge for biomarker discovery is encompassed by this high dynamic range of protein concentrations present in CSF.20 The presence of highly abundant proteins can mask the detection of low-abundance, yet potentially critical, biomarkers. Overcoming this challenge requires methods that can either deplete high-abundance proteins, but carries a risk of codepletion of other proteins, or enrich low-abundance ones without compromising the integrity of the sample. In plasma proteomics, strategies such as sequential ultracentrifugation for the enrichment of extracellular vesicles (EVs) have proven effective in addressing the high dynamic range in CSF.21 By selectively enriching these EVs, the relative concentration of proteins within the EVs is increased, thus facilitating the identification of potential biomarkers that would otherwise be below the detection limit of MS.
The aim of this study is to provide a comprehensive assessment of analytical workflows for deep CSF proteome analysis involving different sample preparation methods and MS data acquisition strategies through application of high-resolution MS-based proteomics. Sample preparation methodologies included ultracentrifugation-based fractionation of the CSF proteome to enrich different EV fractions as well as testing different protein extraction and digestion protocols. Here, we used a unique CSF sample set collected in our biobank to create a homogeneous pool that represented a diverse cohort that includes noncancer controls, CNS cancers of hematological and solid origin, and a wide age range extending from infancy to young adulthood. The optimized protocols were ultimately applied to eight individual ALL patient samples to evaluate their technical performance at the individual level rather than on a homogeneous pool, exclusively.
Materials and Methods
Cerebrospinal Fluid Samples
Since 2012, CSF has been collected from pediatric patients with malignancies involving or potentially involving CNS. Our biobank contains CSF samples from pediatric patients treated for acute lymphoblastic leukemia (ALL) and solid brain tumors, including all pathological subtypes, at the Department of Pediatric and Adolescent Medicine, Copenhagen University Hospital, Rigshospitalet.
In this study, cases that encompassed the ALL subgroup included patients with precursor B-ALL, exclusively. Samples were collected at diagnosis (Day 0) from 22 patients of which 11 were without detectable leukemic cells in the CSF by flow cytometry (FCM-negative) and 11 were with detectable leukemic cells by flow cytometry (FCM-positive) and Day 15 during induction therapy from 8 patients (four FCM-negative and four FCM-positive). Flow cytometry positive was defined as ≥10 leukemic blasts detected.22,23 The brain tumor subgroup included patients with a variety of different pathologies (Table 1 for detailed overview). The perioperative sampling was achieved immediately after opening of the dura and prior to excision of the tumor and as part of the surgical procedure. Care was taken not to pollute the sample with blood although not completely avoidable. All samples were received in our laboratory within 30 min from collection and underwent a standardized protocol, primarily to remove cell pellet by centrifugation at 652g for 5 min, thereby obtaining a cell-free supernatant for further analysis (which correspond to the Raw fraction, as elaborated later). All samples were stored at −80 °C.
Table 1. Overview of Samples Included in Pool Formation.
| acute lymphoblastic leukemia | primary brain tumor | nontumor control | |
|---|---|---|---|
| subgroup | 30 unique patients | 18 unique patients | 30 unique individuals |
| age | median: 5.3 years | median: 8.5 years | median: 15.3 years |
| interquartile: 3.4–8.2 years | interquartile: 4.6–15.4 years | interquartile: 3.0–26.0 years | |
| range: 0.1–17.1 years | range: 0.4–17.7 years | range: 0.0–35.2 years | |
| sampling method | 30 lumbar puncture | 8 extraventricular drainage | 30 lumbar puncture |
| 10 perioperatively | |||
| pathology | day 0 | anaplastic ependymoma | sterile CSF |
| • FCM-positive: n = 11 | • n = 4 | • erythrocyte count <300 × 106/L | |
| • FCM-negative: n = 11 | atypical teratoid rhabdoid tumor | • nucleated cells <4 × 106/L | |
| • n = 1 | |||
| craniopharyngioma | |||
| • n = 2 | |||
| day 15 (induction therapy) | diffuse intrinsic pontine glioma | ||
| • FCM-positive n = 4 | • n = 2 | ||
| gliomas | |||
| • FCM-negative: n = 4 | • n = 2 | ||
| medulloblastoma | |||
| • group 3, n = 1 | |||
| • not WNT/not SHH | |||
| n = 2 | |||
| pilocytic astrocytoma | |||
| n = 4 | |||
| total volume | total 15 mL: | total 15 mL: | total 15 mL: |
| day 0 | anaplastic ependymoma | ||
| • FCM-positive: 5.5 mL | • 3.5 mL | ||
| • FCM-negative: 5.5 mL | atypical teratoid rhabdoid tumor | ||
| • 0.5 mL | |||
| craniopharyngioma | |||
| • 1.5 mL | |||
| day 15 (induction therapy) | diffuse intrinsic pontine glioma | ||
| • FCM-positive: 2 mL | • 2 mL | ||
| • FCM-negative: 2 mL | gliomas | ||
| • 2 mL | |||
| medulloblastoma | |||
| • group 3, 1 mL | |||
| • not WNT/not SHH 1.5 mL | |||
| pilocytic astrocytoma | |||
| • 3.0 mL |
Cases, or their legal guardian, provided written consent, with approvals obtained from the Capital Regional Committee on Biomedical Research Ethics (IRB) (H-1–2012–077 and H-20000845) and the Danish Data Protection Agency (j. nr. 30.8094). The nontumor control group included children, adolescents, and young adults initially suspected of neuro-infectious diseases. Following a microbiological examination of the CSF at the Department of Clinical Microbiology, Copenhagen University Hospital, these individuals were cleared of having a CNS infection (IRB approval: H-20000845\84459). Finally, other healthy individuals voluntarily enrolled as nontumor controls (IRB approval: H-18007272).
Pooled CSF
The limited sample availability from individual patients necessitated the pooling of CSF to achieve a homogeneous sample pool suitable for the optimization experiments. First, we created CSF pools for each subgroup, i.e., a pool of nontumor controls, patients with ALL or patients with a brain tumor, respectively. These subgroup-specific pools were stored as aliquots of 0.1, 0.2, or 1 mL in polypropylene Eppendorf tubes. For each of the optimization experiments below, the required amount of CSF was thawed on slushed ice water and subsequently pooled together in a 1:1:1 ratio before use in the individual experiments.
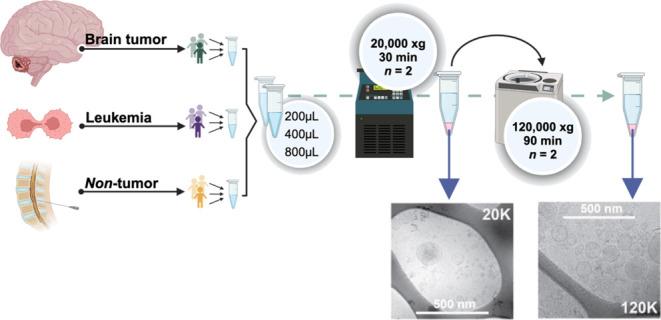
Optimizing Sample Input for EV Enrichment by Ultracentrifugation
EVs were isolated from CSF pools of increasing volume (200, 400, and 800 μL). Each sample was supplemented with phosphate-buffered saline (PBS, cat #20012–019, Gibco) to a final volume of 800 μL. The CSF samples were subsequently centrifuged at 20,000g at 4 °C for 30 min and 780 μL supernatant was removed leaving a small pellet with 20 μL supernatant. The 20K pellet was washed by resuspending in PBS (final volume of 800 μL) and then subjected to recentrifugation at the same conditions. The supernatant was discarded, thereby yielding a 20 μL pellet washed and enriched with 20K EVs. The supernatant, which was removed after the first centrifugation, was subjected to ultracentrifugation at 120,000g at 4 °C for 90 min. This step also resulted in a pellet with the remaining 20 μL of the supernatant, this time concentrated with 120K EVs. The supernatant, that was removed after the 120,000g centrifugation was included as the EV-depleted (EV-depl.) fraction. Finally, the 20 μL pellet containing 120K EVs was washed following resuspension in PBS, and recentrifuged under identical 120,000g conditions. In total, four fractions from CSF were collected and included Raw CSF, 20K EVs, 120K EVs and the EV-depl. fractions. The Raw fraction refers to the cell-free CSF fraction obtained by initial laboratory centrifugation.
For the comparative analysis of different sample input volumes and EV fractions obtained through sequential ultracentrifugation, we used the urea-based in-solution digestion as described below.
Optimizing Protein Digestion
In this experiment, we used a fixed sample volume of 400 μL and subjected it to the fractionation steps described above. The four 20 μL fractions were subsequently proteolytically digested evaluating three different protocols in parallel (urea-based in-solution digestion, protein aggregation capture (PAC)-based on-bead digestion, and direct digestion).
For urea-based in-solution digest, each sample underwent reduction and alkylation for 30 min at room temperature in a solution with final concentrations comprising 4 M urea, 5 mM tris(2-carboxyethyl)phosphine (TCEP), and 10 mM 2-chloroacetamide (CAA). Following this, endoproteinase Lys-C (Cat# 129–02541, FUJIFILM Wako Pure Chemical Corporation, Richmond, VA) was added at a 1:250 enzyme-to-protein ratio and the mixture was incubated for an hour at 22 °C on a thermoshaker. Subsequently, the samples were diluted 4-fold with Milli-Q water to 1 M urea and trypsin (Cat# V5111, Promega) was added at a 1:100 enzyme-to-protein ratio before the samples were left for an overnight digestion at 22 °C on a thermoshaker. The digestion process was terminated by acidification with 10% v/v trifluoroacetic acid (TFA) to a 1% final concentration. The sample digest was stored at −80 °C until further analysis.
For PAC-based on-bead digestion, samples were supplemented with 1:3 volume 4% SDS, 100 mM Tris pH 7.5 supplemented with TCEP and CAA resulting in a final concentration of 5 mM TCEP, 10 mM CAA, and 1% Sodium dodecyl sulfate (SDS).24 Then MagReSyn hydroxyl beads (cat# MR-HYX2L, ReSyn Biosciences, Ltd., South Africa) were supplied in a protein-to-bead ratio of 1:10 for the 20K EV fraction (due to a low protein amount) and 1:2 for the Raw, 120K EV and EV-depl. fractions. Acetonitrile was then added to a final concentration of 70% v/v before incubation for 20 min at room temperature. Then, the beads underwent a washing process: first, with 100% ACN and then with 70% ethanol. The washing process was repeated twice for each solvent to ensure complete SDS removal. Afterward, Lys-C was added in a 1:250 enzyme-to-protein mass ratio and incubated for 1 h at 37 °C on a thermoshaker in a 50 mM 4-(2-hydroxyethyl)piperazine-1-ethanesulfonic acid (HEPES) buffer at pH 7.5. This step was followed by an overnight incubation with trypsin supplemented at a 1:100 enzyme-to-protein mass ratio, also at 37 °C on a thermoshaker. Postincubation, the beads were separated on a magnetic rack, and the supernatant was transferred to a new tube and acidified using 10% v/v TFA to a final concentration of 1%.24 The final sample digest was stored at −80 °C until further analysis.
In the direct digestion method, each sample was supplemented with 1:3 volume 50 mM Hepes pH 7.5 supplemented with TCEP and CAA (5 mM TCEP, 10 mM CAA final concentration) and incubated for 30 min at room temperature. Following this, Lys-C was added to the samples at a ratio of 1:250 (enzyme to protein mass) and were incubated on a thermoshaker at 37 °C for 1 h. Trypsin was subsequently supplemented at a 1:100 enzyme-to-protein ratio, and the samples underwent overnight incubation at 37 °C on the thermoshaker. All samples were acidified with 10% v/v TFA to a final concentration of 1% TFA terminate the digestion. The sample digest was stored at −80 °C until further analysis.
LC-MS/MS Analysis
All samples were prepared in workflow triplicates. Samples were loaded on EvoTips (Cat# EV2001; EvoSep Biosystems, Odense, Denmark) for desalting and preparation for liquid chromatography tandem mass spectrometry (LC-MS/MS) analysis. A subset of experiments utilized volume-based loading, and 20, 40, and 80 μL digest were loaded on EvoTips. In the remaining experiments peptide amount -based loading was applied and here each EvoTip was loaded with 750 ng peptide. All samples, regardless of loading strategy, were subjected to A280 measurements using a NanoDrop spectrophotometer (ThermoFisher Scientific). In mass-based loading, guided by the A280 measurements, the samples were diluted with 0.1% formic acid to obtain equal volumes with the intended loading of 750 ng peptide. For the peptide elution, an EvoSep One LC system (EvoSep Biosystems) was applied with the following specifications: 8 cm × 150 μm analytical performance column packed with 1.5 μm ReproSil Saphir C18 beads (cat# EV-1109, EvoSep Biosystems).25 Solvent A (0.1% formic acid in water) was loaded into the column and peptides were eluted by gradually increasing the concentration of solvent B (99.9% ACN, 0.1% formic acid) over gradient length corresponding to 12, 21, or 45 min (30, 60, or 100 samples-per-day [SPD]). The peptides were electrospray-ionized into an Orbitrap Exploris-480 Mass Spectrometer (Thermo Fisher Scientific, Bremen, Germany) at a 2 kV spray voltage, with the funnel RF level set at 40 and a capillary temperature of 275 °C.26 The mass spectrometer operated in positive ion mode, where mass spectrometry data was obtained through data-independent acquisition (DIA). In a subexperiment, we compared the results obtained from DIA to those from data dependent acquisition (DDA). In DDA mode, full-scan precursor spectra from 350 to 1400 Da were recorded at a resolution of 60,000 at mz 200, with a normalized automatic gain control (AGC) target of 300% and a maximum injection time of 25 ms. The fragment spectra, captured at a resolution of 15,000, were based on a precursor intensity threshold of 2× 105. The top 12 most intense precursors in each cycle were fragmented by higher-energy collisional dissociation (HCD) in the Ion-Routing Multipole (IRM) using 30% normalized collision energy, a normalized AGC target of 200%, and a maximum injection time of 22 ms with a dynamic exclusion of 30 s to prevent repeated fragmentation.27 In DIA mode, the full-scan precursor spectra were recorded at a higher resolution of 120,000 at mz 200, maintaining the same AGC target and a maximum injection time of 45 ms. Fragment spectra covered 56 sequential 13 Da windows (with 1 mz overlap) across a range of 361–1033 Da, at a resolution of 30,000. Precursors were fragmented in the HCD cell using 27% normalized collision energy, a higher normalized AGC target of 1000%, and a maximum injection time of 54 ms.
Additionally, to construct a project-specific spectral library, leftover samples from each subexperiment were repeatedly injected, utilizing gas-phase fractionation combined with DIA in a series of staggered, narrow windows (thus, resembling a DDA-like mode).28 The sample was injected seven times, covering precursor mz ranges of 350–450, 449–549 Da, and so on, up to 944–1044 Da. These were recorded in profile mode at a resolution of 120,000 at mz 200, with a normalized AGC target of 300% and a maximum injection time of 45 ms. Fragment spectra were then captured, fragmenting 50 consecutive 2 Da windows (each spanning 100 Da) at a resolution of 15,000. Precursors underwent fragmentation in the HCD cell with 27% normalized collision energy, a normalized AGC target of 1000%, and a maximum injection time of 22 ms.
Nanoparticle Tracking
We determined the concentration and size distributions of EVs in the Raw, 20K EV and 120K EV fractions using nanoparticle tracking analysis (NTA). This analysis was performed using a ZetaView PMX-220 nanoparticle analyzer (Particle Metrix GmbH; Inning am Ammersee, Germany) equipped with a 520 nm laser enabling detection of Brownian motion of EVs. Instrument calibration was performed using 100 nm polystyrene beads prior to the analysis of the samples. We then evaluated pooled CSF samples from children diagnosed with ALL, from children with brain tumors, and from nontumor control subjects. The analysis was carried out at 22 °C by scanning 11 different positions twice. For the EV concentration and size distribution analysis, we included data from the positions specified as reliable by the software version (8.05.14 SP7), exclusively.
Cryo-Transmission Electron Microscopy
We used Cryo-transmission electron microscopy (cryoTEM) to visualize EVs isolated from both 20K and 120K EV fractions. We assessed the EVs separately for each of the three groups: (1) ALL patients, (2) brain tumor patients, and (3) nontumor controls. Using sequential ultracentrifugation, as described, EVs were isolated from 2.5 mL of CSF for each pool from these groups.
The initial preparation step encompassed treating lacey Formvar-coated copper grids (300 mesh Cu, Ted Pella Inc., p/n 01887-F) with a glow discharge at 10 mA for 30 s using a Leica EM ACE 200 (Leica Microsystems, Wetzlar, Germany). For sample application, 3 μL of the sample was placed on the carbon-coated side of the grid, which was set up in a Vitrobot Mark IV (FEI Instruments, Oregon). The grid, maintained at 4 °C and 100% humidity, was blotted for 2.5 s (blot force 0) to eliminate excess sample. It was then rapidly frozen in liquid ethane cooled by liquid nitrogen, a process that swiftly forms amorphous ice, thus preserving the EVs in their morphological state. Following this, the grid was moved to a Tecnai G2 electron microscope (FEI Instruments, Oregon) fitted with a Megaview II camera (Olympus Soft Imaging Systems, Münster, Germany). The microscope operated at an acceleration voltage of 200 kV, capturing images at a magnification of 25,000× and a defocus range between −3 to −6 μm.
Western Blotting
Samples containing ∼12 μg of protein from the Raw, 20K EVs and 120K EVs fractions were mixed with lithium dodecyl sulfate (LDS) 4× sample buffer (Invitrogen, cat# NP0007), supplemented with 5 mM dithiothreitol (DTT) and incubated at 80 °C for 10 min before loading onto NuPAGE 4–12% Bis-Tris 10-well SDS-PAGE gels (Invitrogen, cat# NP0321BOX) for electrophoretic separation. Postseparation, the proteins were transferred onto nitrocellulose membranes (Whatman Protran, Sigma–Aldrich). To assess the efficiency of blotting and for slicing the membranes into segments representing various molecular weight ranges, Ponceau S staining was applied. After rinsing off the Ponceau S stain, the membranes were blocked in a 5% BSA solution in PBS-T buffer (PBS with 0.1% Tween-20, pH 7.4). This was followed by an overnight incubation with primary antibodies, each diluted to 1:1000. The applied antibodies were from Cell Signaling Systems (CD54/ICAM-1 [E3Q9N] Rabbit mAb #67836; Flotillin-1 [D2 V7J] Rabbit mAb #18634; CD9 [D8O1A] Rabbit mAb #13174) and Santa Cruz Biotechnology Inc. (ACTB [C4] Mouse mAb sc-47778). Subsequently, the membranes were washed three times with PBS-T before incubation with HRP-conjugated secondary antibodies for 1 h (goat antirabbit IgG, Jackson Immunoresearch; #111–036–045, and goat antimouse IgG, Jackson Immunoresearch; #115–036–062), each diluted 1:5000 in PBS-T with 5% w/v skim milk. The final steps involved a 2 min treatment with Novex ECL chemiluminescent substrate and capturing the results by exposing high-performance chemiluminescence films (Amersham Hyperfilm). These films were then developed using a Kodak Medical X-ray processor (Carestream Health) and scanned with a CanoScan 8800 scanner (Canon).
Data Analysis
The raw files encompassing mass spectrometry data recorded by DIA, were processed using Spectronaut v. 17.1 (Biognosys AG, Schlieren, Switzerland). We processed DDA raw files by using MaxQuant v. 1.6.17.0. For DDA and DIA analyses without a spectral library, both strategies utilized standard settings and was searched against a FASTA file containing the human proteome (SwissProt, with 20,431 sequences with signal peptides removed, downloaded 17th October, 2018). Additionally, the FASTA file included sequences of Lys-C and trypsin used in the digestion. Here, for the DIA FASTA-file search, the directDIA method was employed with carbamidomethyl included as a fixed modification while N-terminal acetylation and methionine oxidation was included as variable modifications. A Q-value cutoff of 1% against mutated decoys was applied as false-discovery rate. Label-free quantification (LFQ) was based on the area under the curve for each targeted ion without automatized data normalization to better highlight sample variability.
Spectral libraries used for the Spectronaut searches were created using DIANN v1.8 by searching the same two FASTA files with default settings, except for enabling “Generate spectral library,” activating “Ox(M),” setting Precursor charges to “2–5 Da,” selecting Neural network classifier “Double pass mode,” and “Smart profiling” for library generation. The spectral library encompassed samples from most subexperiments, meaning that all fractions were represented in combinations with a variety of different input sample volumes; digest methods described, and peptide loadings.
The experiment comparing different input sample volumes was repeated with ∼3 months in-between to assess reproducibility of the sequential ultracentrifugation. The MS2 intensities from each experiment were Z-transformed separately before being combined for unsupervised clustering. This approach allowed us to evaluate the clustering patterns across different experiments independently.
Further, proteins were rank ordered and plotted against their log2(MS2-intensities) to visualize the dynamic range of the quantified proteins. Venn diagrams, created using unique SwissProt IDs, were used to display proteins detected in more than one fraction. Additionally, we calculated the overall hydrophobicity of samples using a cumulative GRAVY score based on all unique peptides quantified.29 Finally, prediction of the subcellular location of proteins were obtained using the DeepLoc algorithm.30
Application to Individual Patient Samples
To evaluate the optimized workflow, we included eight age-matched samples from eight ALL patients (four patients FCM-positive CNS leukemia and four patients with FCM-negative CNS leukemia). Here, we compared the urea-based in-solution digestion with the PAC-based on-bead digestion using DIA MS analysis of the samples in combination with the developed gas-phase spectral library. An input sample volume of 500 μL CSF was used (standard volume of biobanked aliquots). The Raw, 20K EV and 120K EV fractions were generated by the sequential ultracentrifugation described above. Finally, t-test-based Volcano plots were computed to tentatively compare protein abundances for FCM-positive vs FCM-negative patients.
We used the Perseus v.1.6.15.0 software and R v. 4.3.2 for the proteome analyses described.31
Data Availability
All raw-files with mass spectrometry data recorded were uploaded to MS data repository and is accessible with the identifier PXD052666
Results
Cases and Nontumor Controls: CSF Pool
Table 1 provides a detailed overview of the samples used to obtain the CSF pools and can be summarized as follows. CSF was obtained from 30 patients in both the ALL cohort and the nontumor control group. The age of ALL patients ranged from 0.1 to 17.1 years, with a median age of 5.3 years and an interquartile range of 3.4 to 8.2 years. The age of the nontumor control individuals varied more widely, from 0.0 to 35.2 years, with a median age of 15.3 years and an interquartile range of 3.0 to 26.0 years. The brain tumor group consisted of 18 patients, with a median age of 8.5 years at the time of CSF sampling. The age range was 0.4–17.7 years, and the interquartile range was 4.6–15.4 years. This subgroup included various tumor types: four cases of anaplastic ependymoma, four pilocytic astrocytomas, three medulloblastomas, two craniopharyngiomas, two diffuse intrinsic pontine gliomas, two gliomas, and one atypical teratoid rhabdoid tumor (AT/RT).
Sequential Ultracentrifugation
To enrich different EV fractions from the CSF, sequential ultracentrifugation was employed as described.21 The fractionation process is outlined in Figure 1A. To assess the reproducibility of the fractionation process, we repeated the entire procedure twice, with approximately three months between each run. Unsupervised hierarchical clustering analysis of Z-scored MS2 protein intensities and heatmap visualization showed distinct clustering patterns. The 20K and 120K EV fractions formed distinct clusters that remained consistent across experimental batches, demonstrating both the robustness and reproducibility of the EV fractionation. In contrast, the clustering of the Raw and EV-depleted fractions was primarily influenced by the volume used for EvoTip loading (40 vs 80 μL). However, the subclusters within these main clusters correlated with the individual fractions (Figure 2A).
Figure 1.

Experimental design and workflow (created with BioRender.com). (A) Pooling of CSF in subsets of patients with ALL, a brain tumor or nontumor control. These disease-specific subgroup-pools were pooled 1:1:1 prior to each experiment and sequential ultracentrifugation. (B) workflow for tryptic digest methods. (C) Data acquisition modes. (D) workflow for data analysis. Created using BioRender.com.
Figure 2.

(A) Heatmap of unsupervised clusters comprising all fractions. The experiment was repeated two times (“Initial” is the first experiment, while “Repeated” was the same experiment 3 months later). (B) Dynamic range and proteome depth stratified for sample input volume and each fraction. The intensity corresponds to median of MS2-intensity across the workflow triplicates. (C) Predicted subcellular location using the DeepLoc algorithm.30 (D) Cryo-transmission electron microscopy of 20K and 120K EVs. (E) Western blotting probed for common EV markers. In this experiment, equal sample masses were loaded. (F) Comparison of MS2-intensities between fractions with emphasis on EV markers. In this experiment, equal volumes were loaded. (G) Nanoparticle tracking analysis of 20K and 120K EVs stratified for patients with ALL or a brain tumor as well as nontumor controls. (H) Biomarker coverage of previous reported potential biomarkers, summarized in ref (10).
The 20K EV fraction appeared most essential for increasing the number of quantifiable proteins by reducing the dynamic range of the sample. A higher sample volume of CSF resulted in a higher quantification of unique proteins in the 20K EV fraction (+33% when increasing from 400 to 800 μL), making the 120K EV-fractionation less necessary with increasing starting volume. This was attributed to a higher 20K EV yield as a consequence of a higher input sample volume, leading to a more complex proteome (Figure 2B).
Mapping the in silico predicted subcellular location to all identified proteins highlighted those proteins differed in their location depending on the fraction analyzed (Figure 2C). In the Raw fraction, proteins from the “Extracellular domain” constituted 92% of the total MS2 intensity, while those from the “Cytoplasm” made up only 6%. In contrast, cytoplasmic proteins accounted for 27.4% of the MS2 intensity in the 20K EV fraction while the “Extracellular domain” constituted 65.4% of the total MS2 intensity. Similar, but less pronounced, trends were observed for the 120K EV fraction while the EV-depl. fraction resembled the Raw fraction.
Table 2 provides an overview of the expected time required to obtain each individual fraction.
Table 2. Overview of Recommended Steps and Expected Time Consumption.
| steps | research aim | description | time consumption |
|---|---|---|---|
| thaw sample in slushed ice | input CSF volume: 400 μL (optimal). | ∼1 h for 400 μL CSF | |
| 20K EV enrichment | proteome characterization of raw CSF and/or 20K EVs | add PBS to 1000 μL final volume | ∼1.5 h |
| centrifuge 20,000g for 30 min | |||
| wash with PBS (15 min) | |||
| transfer supernatant (until 20 μL) to a new tube; add PBS to 1000 μL and mix | |||
| centrifuge 20,000g for 30 min | |||
| discard supernatant–leave 20 μL incl pellet (10 min) | |||
| proceed to digestion if CSF biomarker exploration is the main research aim | |||
| 120K EV enrichment | proteomic characterization of 120K EVs | use supernatant collected after the first 20k centrifugation | ∼4–5 h |
| balance samples and place in rotor, 30 min | |||
| centrifuge 120,000g for 90 min | |||
| wash with PBS (15 min) as described above | |||
| balance samples and place in rotor, 30 min | |||
| centrifuge 120,000g for 90 min | |||
| discard supernatant–leave 20 μL incl pellet (10 min) | |||
| tryptic digestion | input volume: 20 μL pellet from raw CSF, 20K and/or 120K EV fractionation. | PAC-based on-bead protocol: 2 h | |
| sample limitation or low protein input, chose urea-based in-solution | urea-based in-solution: 1 h | ||
| if high protein input, chose PAC-based on-bead digest | both | ||
| +1 h Lys-C incubation + overnight incubation with trypsin | |||
Enrichment of EVs
Cryo-TEM, Western blot and mass spectrometry analyses all provided evidence for the enrichment of EVs in the 20K and the 120K fractions. CryoEM showed distinct morphological characteristics of EVs in the 20K EV and 120K EV fractions (Figure 2D). Western blot analysis (Figure 2E), probed for common EV markers like ANXA5, CD9, and FLOT-1, showed enrichment in the 20K and 120K fractions, particularly in contrast to the Raw or EV-depleted fractions. Finally, MS data further supported the enrichment of EV-associated proteins in both the 20K EV and 120K EV fractions over the Raw fraction (Figure 2F).
Nanoparticle tracking analysis suggested a size distribution difference between the two EV fractions, indicating that the 120K EVs are smaller than the 20K EVs (Figure 2G). This is expected, as lower centrifugal force primarily affects larger vesicles, while higher centrifugal force affects smaller vesicles. It was further suggested that variation in 120K EV morphology could be correlated to disease origin; specifically, brain tumor patients had considerably smaller EVs in the 120K EV fraction compared to ALL and nontumor controls (Figure 2G).
Biomarker Coverage
We recently published and summarized current potential CSF biomarkers based on published literature applying mass spectrometry-based proteomics for biomarker search in pediatric neuro-oncology.10 We applied this aggregated knowledge to highlight potential protein biomarkers on dynamic range plots including all proteins quantified in the current study for both the RAW fraction and the 20K fraction showing the increased accessibility to quantify potential biomarkers with our approach (Figure 2H). Supporting Table 1 presents a ranking of each fraction based on the MS2 intensity levels of the included biomarkers, indicating which fraction shows the highest intensity.
CSF Sample Volume
As indicated in Figure 2B, the 20K EV fraction substantially enhances the proteome depth by almost doubling the number of proteins covered. Fractionation beyond this step became less advantageous with increased sample volumes of CSF. Figure 3A presents the estimates from an adjusted linear regression model, where the outcome variable is the total number of proteins in each fraction. The model adjusts to loaded peptide mass (A280), CSF sample volume, and spectral library utilization. The results indicated that using 800 μL of CSF as sampling volume increased the identification of more unique proteins in all fractions than using 200 μL. In contrast, no statistically significant benefits in quantification of unique proteins were observed for the Raw, 120K EV, or EV-depl. fractions when increasing CSF sample volume from 400 to 800 μL. The 20K EV fraction, however, demonstrated further enhanced (and statistically significant) protein quantification with a higher sample volume (i.e., 800 vs 400 μL; P-value <0.001, Figure 3A).
Figure 3.

(A) The total number of unique proteins was analyzed using a linear regression model, with adjustments for the mass loaded onto the EvoTip and the CSF input volume. This model evaluated the impact of these variables on the number of unique proteins while accounting for their interrelationships. P-values for statistical significance are provided below each estimate to indicate the significance of the CSF input variable. (B) Reproducibility based in workflow triplicates and measured by the coefficient of variation across different sample input volumes. (C) Total number of proteins quantified with and without application of the gas-phase spectral library. Stratified for input sample volume and volume-based load. (D) Total number of proteins quantified within each fraction as a function of peptide mass loaded (measured on A280). (E) Total sample hydrophobicity (all peptides summed) measured as Gravy score and as a function of peptide mass loaded (A280 based).
The coefficient of variation indicated that protein quantification was consistent when using sample volumes of 400–800 μL. However, lowering the sample volume to 200 μL led to decreased reproducibility of protein quantification (Figure 3B).
Including a spectral library based on gas-phase fractionation for each fraction was highly beneficial and increased the number of quantifiable proteins in all fractions (Figure 3C), independent of the CSF sample volume. Generally, the benefits of the gas-phase spectral library were reduced at lower sample volumes. Using peptide amount-based loading, no benefit in quantification of proteins was observed when increasing the loaded peptide amount above 1000 ng (in accordance with product specification for the EvoTips) (Figure 3D). Regardless of CSF sample volume used in the 120K EV fraction, the quantification of unique proteins was similar when the peptide amount-based load ranged from 750 to 1000 ng (Figure 3D). As suggested, a higher CSF sample volume correlated with higher quantification in the 20K EV fraction (and Raw, but this observation was attributed to correspondingly higher quantity of EVs in Raw) (Figure 3D). Increasing the peptide load toward the level of the Evotip capacity correlated with lower overall GRAVY scores, suggesting displacement of hydrophilic peptides by more hydrophobic peptides as these peptides bind stronger to the C18 material in the EvoTips (Figure 3E).
Protein Digestion Methods
To identify the best protein extraction and digestion method for the CSF fractions, we used a fixed CSF sample volume of 400 μL CSF to compare three different digestion methodologies. From the resulting digests, we loaded 750 ng of peptide mixtures onto EvoTips and analyzed using DIA and a 22 min (60 SPD) gradient.
Utilizing the spectral library, the highest quantification of unique proteins across all fractions was achieved with the PAC-based digest (n = 2989) followed by Urea (n = 2558) and the Direct digest (n = 2133) (Figure 4A). There were 631, 207, and 142 unique proteins exclusively quantified in the PAC, Urea and Direct digest, respectively. Except for the 120K EVs fraction, the PAC digest consistently outperformed the two other methods across all fractions, especially when using the spectral library (Figure 4B). The 20K EV fraction considerably increased the dynamic range covered compared to the Raw fraction. Fractionating beyond the 20K EVs fraction, however, offered no substantial increase in the detection of unique proteins with any digestion method applied (Figure 4C). When comparing the protein quantification associated with the EV fractions of PAC and Urea digests, the PAC-20K EVs was superior and quantified 530 unique proteins while 145 were uniquely quantified using the Urea counterpart (Figure 4D). The significance of the 120K EVs fraction was minimal in the PAC digest, with only 2.7% of unique proteins identified in this fraction (Figure 4D,E). In contrast, 7.9% of the unique proteins in the Urea digest were identified in the 120K EV fraction.
Figure 4.

(A) Venn diagram of total numbers of proteins identified across all fractions and by each digest method. The identified proteins were quantified from an input sample volume of 400 μL and search via the gas-phase spectral library. (B) The dynamic range and proteome depth in each digest and protein extraction method in within the individual fractions, stratified for usage of the gas-phase spectral library. All proteins identified were included. (C) The same as B, but now including the addition of unique proteins identified within each fraction. I.e., the unique proteins were allocated to the fraction in which it was first observed. (D) A Venn diagram illustrating the overlap between 20K and 120K EVs identified using the PAC-based vs urea-based digest (using the gas-phase spectral library). (E) Unique proteins identified in the 120K and EV-depleted fraction on top of what have been identified in the Raw +20K EV fraction. Here, only the PAC-based and urea-based approach and search using the gas-phase spectral library. (F) Missed cleavages in each fraction while stratified for digest and protein extraction method as well as usage of the gas-phase spectral library. (G) Similarly stratified, here showing unique proteins and unique peptides identified in each fraction. (H) Total sample hydrophobicity based on all peptides summed. The hydrophobicity was measured as the Gravy score. Stratified for each method used for protein extraction and fraction.
The PAC digest resulted in fewer missed cleavages, and applying a spectral library further decreased the percentage of missed cleavages in all fractions (Figure 4F). The Urea-20K EVs fraction showed the highest rate of missed cleavages, leading to increased sample complexity due to combinations of possible partially overlapping peptides. Consequently, while the absolute peptide quantification in the Urea-20K EV fraction was high and similar to that of PAC-20K EV, it did not result in a correspondingly higher number of quantified unique proteins (Figure 4G). In general, the PAC digest resulted in more hydrophilic peptides whereas the Urea digest was associated with quantification of more hydrophobic peptides (Figure 4H).
MS Acquisition Modes
We compared DIA vs DDA using LC-MS/MS gradient lengths of 100SPD, 60SPD and 30SPD. Corresponding to each individual gradient length, we loaded 100, 250, and 500 ng of the same sample, respectively. DIA consistently outperformed DDA, and application of the gas-phase spectral library increased the identification of unique proteins and peptides considerably (Figure 5A,B). When using the spectral library, the quantification of unique proteins increased by a median of 76% (range: 38–105%), while the median increase of unique peptide quantification was 67% (range: 29–84%) across all fractions.
Figure 5.

(A) Unique proteins identified
in each fraction, here stratified
for gradient length (100, 60, 40 SPD) while comparing DDA vs DIA,
with and without the gas-phase spectral library using the sample pool.
(B) The same, but for unique peptides. (C) Number of unique proteins
identified in this study vs previous literature. (D) The efficiency
of quantifying the number of unique proteins, here measured as 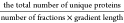 . (E) The overlap
between unique proteins
identified in this study and compared to previous studies on CSF from
healthy donors. (G) The same, but the CSF study was performed on EV-enriched
samples from pediatric patients with a brain tumor.
. (E) The overlap
between unique proteins
identified in this study and compared to previous studies on CSF from
healthy donors. (G) The same, but the CSF study was performed on EV-enriched
samples from pediatric patients with a brain tumor.
Comparison to Previous Studies
The total proteins identified
in our study using PAC-based (n = 2989) and urea-based
(n = 2558) digestion methods are comparable to that
of previous research, although these have quantified more than 3000
proteins in CSF (Figure 5C). Unlike previous studies requiring extensive fractionation and
MS time, our data was derived primarily from two fractions, Raw and
20K EVs, using a 60SPD (∼21 min gradient). Adjusting for efficiency,
i.e., comparing 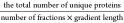 , our data demonstrates
an efficiency up
to 60-fold greater than previous studies (Figure 5D). This study identified proteins wherein
approximately 50% overlapped with those found in research on healthy
adult CSF, with 12.7% being unique to our study (Figure 5E). In comparison, about 75%
of the proteins identified here overlapped with those from another
study also enriching for EVs, leaving roughly 25% as unique to this
investigation (Figure 5F).
, our data demonstrates
an efficiency up
to 60-fold greater than previous studies (Figure 5D). This study identified proteins wherein
approximately 50% overlapped with those found in research on healthy
adult CSF, with 12.7% being unique to our study (Figure 5E). In comparison, about 75%
of the proteins identified here overlapped with those from another
study also enriching for EVs, leaving roughly 25% as unique to this
investigation (Figure 5F).
Application to Individual Samples
Finally, we compared the PAC vs Urea digestion protocols and applied them to eight individual ALL patient samples (n = 4 that were FCM-positive, and n = 4 that were FCM-negative patients). Four patients were included in both the PAC and Urea protocols. Limitations on available samples necessitated allocation of four unique patients to each separate protocol. The median age was 2.7 (range: 2.4–3.4) and 2.9 (range: 2.0–5.9) in the PAC vs Urea protocol, respectively.
Distinct clusters corresponding to the Raw, 20K EVs, and 120K EVs fractions were identified using both digestion methods, thus providing further evidence for the reproducibility of the EV fractionation (Figure 6A). When comparing the PAC protocol to the Urea protocol using the spectral library, 2004 and 2213 unique proteins were quantified, respectively (Figure 5B). However, the number of unique proteins quantified in the PAC-20K EV fraction was consistently lower than expected. This outcome was reproduced in three separate experiments, each time using different individual samples (due to sample limitation). In our optimization experiments, we used pooled CSF containing a wide range of patients linked to different medical conditions (ALL, brain tumors, and nontumor cases) and age groups (0 to 35 years). Therefore, this mixture demonstrated considerable diversity, unlike individual samples that naturally exhibited higher uniformity. In addition, the protein concentration in the 20K EV fraction is low, at just 4% of the Raw fraction based on a BCA assessment. This low concentration, combined with more uniform samples, may pose challenges for effective capture. Regardless, we increased the protein-to-bead ratio to 1:10 to mitigate issues related to a reduced bead volume (rather than the 1:2 protein-to-bead ratio used for the remaining fractions). Still, the critical step of rinsing the beads with 70% ethanol (EtOH) and 100% ACN (twice, to remove SDS), may inadvertently lead to the loss of some beads. Although this loss might be volumetrically small, it could significantly impact proteome depth in individual patient samples with a low protein concentration.
Figure 6.

Methodology applied to individual samples (in contrast to pooled samples). (A) A heatmap based on unsupervised clusters. Here showing that PAC-based on-bead digest performs instable compared to the urea-based in-solution digest. (B) Number of unique proteins identified across each fraction and within the PAC-based on-bead and urea-based in-solution digest. (C) Volcano plots comparing the log 2-transformed MS2-intensities obtained from analyzing four FCM-positive and four FCM-negative patients, here including Raw, 20K EVs and 120K EVs for both PAC-based on-bead and urea-based in-solution digest.
The Urea digest performed well and consistently by quantifying 2213 unique proteins. The 120K EVs fraction increased protein quantification by 12% (Figure 6B). Finally, volcano plots were generated by stratifying for the digest method and individual CSF fractions (Figure 6C). In addition to the lower protein identification observed with the PAC protocol, the results also suggested a skewed distribution toward FCM-negative patients in the 20K EV proteome.
Discussion
Implementing sequential ultracentrifugation in optimized workflows increases the proteome depth considerably. Fractions obtained by sequential ultracentrifugation consistently rendered distinct clusters by hierarchical clustering in the heatmap analysis, both for pooled and individual samples, suggesting good reproducibility. The increased number of proteins quantified was primarily achieved through the enrichment of EV-associated proteins, which reduced the dynamic range and allowed for a more detailed analysis of the CSF proteome. The 20K EV fraction alone led to approximately 30% increase in the quantification of unique proteins compared to the Raw fraction.8
The experiments in the present work aiming to optimize the workflow demonstrated that reducing CSF volumes below 400 μL resulted in fewer unique proteins quantified but increased the coefficient of variation, thereby adversely affecting the reproducibility. Increasing the CSF sample volume above 400 μL led to quantification of a higher number of unique proteins associated with 20K EVs fraction. The volume-dependency is likely because larger sample volumes yield a greater total number of EVs. Higher volumes of CSF contain more diverse and abundant EV populations, which translates into a broader range of detectable proteins. In contrast, smaller sample volumes are likely to experience higher relative losses during preparation due to nonspecific binding to surfaces and other inefficiencies. Fractionating beyond the 20K EV step increased the proteome depth further by approximately 10% in the urea-based in-solution digest, and was even less beneficial in the PAC-based on-bead digest. However, obtaining the 120K EV fraction is time-consuming and encompasses the rate limiting step of the fractionation protocol. The ultracentrifugation at 120,000g involves two steps, each requiring 90 min of run time. Due to the extreme forces generated at 120,000g, each sample tube must be carefully balanced and arranged to ensure that the difference in weight across the rotor does not exceed 10 μg. The hands-on time for obtaining the 120K EV fraction is between 4 to 5 h for 12 samples, not including the additional time required for the subsequent digestion step.
Future research focused on examining EV-associated diseases in CSF (or CNS) may still benefit from isolating the 120K EV fraction as well, whereas studies that focus on CSF identifying biomarkers may find it less beneficial. Compared to 20K EVs, the 120K EVs are smaller in size and express a different protein profile, which, as suggested by nanoparticle tracking analysis presented herein, may be related to the disease origin. Although the 120K fractionation did not significantly improve the number of quantified unique proteins, it may enable a more qualitative and detailed analysis of EVs enriched from CSF.
Further, obtaining the 120K EV fractions directly, without the intermediate step of the 20K EV fractionation, could provide valuable insights for refining enrichment strategies in future studies. However, this approach was not considered in the present study, as we prioritized maximizing the proteome depth while maintaining time efficiency.
Finally, the optimal load for LC-MS/MS analysis depends on the research aim. Increasing the peptide load may lead to the quantification of more unique proteins but may result in the disproportionate identification of hydrophobic proteins that could bias the data analysis. In the current work, loading 750 ng in combination with analysis in 60SPD DIA mode was most optimal. The inclusion of our gas-phase spectral library considerably increased the number of quantified proteins.
Previous studies on CSF have identified approximately 3000 proteins.32−36 However, these approaches have applied extensive fractionation and required substantial sample material and mass spectrometry time. In a previous study using Orbitrap-based LC-MS/MS analysis of CSF, 3081 unique proteins from a cohort of 21 neurological healthy individuals were identified.33 However, this result was obtained via mixed mode fractionation of 66 samples (obtaining 37 + 46 GeLC fractions) and high-abundant protein depletion from raw CSF prior to the mass spectrometry analysis of each fraction (using a 70 min gradient). Another study focused on analyzing a CSF pool mixed from 14 adults receiving “non-neurological operations” using high-pH reverse-phase liquid chromatography fractionation (60 fractions) of the digested sample prior to LC-MS/MS. This strategy resulted in quantitation of 3256 proteins including 2513 high confidence identifications.32 Similarly, extensive mass spectrometry time is required to obtain these numbers. Finally, in the most recent study, fractionation was also applied and subsequently analyzed using a 120 min gradient on a timsTOF instrument. This approach resulted in identification of 3174 unique proteins.34
Although the numbers of total unique proteins were
lower in our
study (the maximum was 2989 and 2558 unique proteins in PAC and urea-based
in-solution, respectively, and when including all fractions), the
overall efficiency was markedly superior when measured as 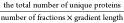 . Using this
measurement, our methodology
demonstrated up to a 60-fold increase in protein identification efficiency
compared to previous studies. The technological advancements in the
proteomics field, including faster and more sensitive instruments,
have naturally contributed to the increased efficiency compared to
studies published in 2015. However, in addition, the enrichment of
EVs through sequential ultracentrifugation, particularly the combination
of Raw and 20K EV fractionation, has significantly enabled to this
efficacy in the present study. Since there was no significant increase
in the quantification of unique proteins when proceeding with the
120K EVs and EV-depleted fractions, the efficiency consequently decreased
due to the considerable time required to obtain the 120K EV fraction.
Therefore, we recommend pursuing the 120K EVs only if they are specifically
relevant to the research question.
. Using this
measurement, our methodology
demonstrated up to a 60-fold increase in protein identification efficiency
compared to previous studies. The technological advancements in the
proteomics field, including faster and more sensitive instruments,
have naturally contributed to the increased efficiency compared to
studies published in 2015. However, in addition, the enrichment of
EVs through sequential ultracentrifugation, particularly the combination
of Raw and 20K EV fractionation, has significantly enabled to this
efficacy in the present study. Since there was no significant increase
in the quantification of unique proteins when proceeding with the
120K EVs and EV-depleted fractions, the efficiency consequently decreased
due to the considerable time required to obtain the 120K EV fraction.
Therefore, we recommend pursuing the 120K EVs only if they are specifically
relevant to the research question.
In comparison to our results, previous LC-MS/MS-based proteomic investigations of pediatric CSF have reported identification of fewer unique proteins. Regardless, all candidate biomarkers from these individual studies were also identified in this present paper by using the methods presented.10
A recent study also employed EV enrichment strategies via sequential ultracentrifugation to quantify the CSF proteome in pediatric solid brain tumors, thus paralleling the methods used in our present study (their settings yielded 22K EVs and 100K EVs). This resulted in the identification of 3072 unique proteins, but required considerable MS time (gradient lengths of 150 min for the Raw and 100 min for the EV fractions).37 Still, these results support that EV-enriched CSF can mitigate the limitation of a high dynamic range, thereby enabling deep proteome analysis
The pilot study aimed to evaluate the feasibility of applying the optimized protocol to individual samples from eight age-matched patients, reflecting true patient disease diversity, rather than just evaluating the protocol based on a homogeneous CSF pool. Thus, the goal was not to obtain or explore biological insights from these diseases but to demonstrate the potential applications of our optimized workflow. In this pilot study, we justified using samples from Day 15, where a treatment effect on CSF blasts is expected, rather than samples from Day 0. The priority was to avoid exhausting our unique Day 0 samples. The results indicated that the urea-based digest yielded more consistent and reliable outcomes for individual samples, contrasting with the pooled sample experiments where the PAC-digest appeared to perform better. Hence, the PAC-digest method shows its greatest advantage when dealing with higher protein concentrations; however, the urea-based approach offers greater stability and reproducibility making it particularly useful in scenarios where sample quantity is limited.
Finally, a major strength of our study was the availability of CSF from a diverse group of pediatric patients and noncancer children for pooling. This enabled us to create a homogeneous CSF pool that ensured material consistency across our experiments while reflecting various clinical situations. However, we were unable to include all relevant diagnoses in the CSF pool. For instance, although we would have preferred to include patients with T-ALL, this was not feasible due to sample limitations.
Recommendations
For the identification of cancer-associated biomarkers in CSF, sequential ultracentrifugation is advisible. While this study utilized pediatric CSF samples, the protocol is universally applicable in terms of attenuating the dynamic range. The PAC-based method exhibited enhanced effectiveness, but only at certain protein concentrations. BCA assay measurements indicated that a Raw fraction protein concentration of about 1 μg/μL is necessary for PAC-based on-bead digestion efficacy to exceed that of urea-based in-solution digestion. This protein input concentration approximates a protein concentration of 0.04 μg/μL in the 20K EV fraction. This, however, was not possible to validate and is suggestive. For samples with volume constraints or low input concentrations, the urea-based digestion is recommended. It is recommended that the CSF input volumes should not fall below 400 μL, given that lower volumes can negatively affect both the quantification of EV-associated proteins and the reproducibility, as reflected in the coefficient of variation. Fractionating beyond 20K EVs is time-consuming and does not increase the number of unique proteins, and is thus only advisible if the 120K EV proteome is specifically targeted. These recommendations support and adhere to “Recommendations for reproducibility of cerebrospinal fluid extracellular vesicle studies” published by the “International Society for Extracellular Vesicles Cerebrospinal Fluid Task Force”.38
Conclusions
Enriching CSF for EVs mitigates the challenges of a high dynamic range and substantially enhances the proteome depth. The PAC-based on-bead digest is optimal for high protein concentration inputs, whereas the urea-based in-solution digest is recommended for low protein concentrations or with limited samples. Additionally, using a gas-phase spectral library further increases proteome depth. The experiment-specific libraries generated in this present study are available via the PRIDE repository (for 40, 60, and 100 SPD).
Acknowledgments
Work at The Novo Nordisk Foundation Center for Protein Research (CPR) is funded in part by a generous donation from the Novo Nordisk Foundation (NNF14CC0001). This work is also a part of the Danish nationwide research program Childhood Oncology Network Targeting Research, Organisation & Life expectancy (CONTROL) and supported by the Danish Cancer Society (R-257-A14720) and the Danish Childhood Cancer Foundation (2019-5934 and 2020-5769). Establishing the biobank has been supported by Børne Hjernecancer Fonden (Children Brain cancer foundation, grant of 17/2 2022). Christian Mirian was funded by The Research Fund of Rigshospitalet, Copenhagen University Hospital, Grant Number: E-22093-09. This work was further supported by EPIC-XS, project number 823839, funded by the Horizon 2020 programme of the European Union and supported by the European Research Council ERC Synergy Grant 810057-HighResCells. We acknowledge the Core Facility for Integrated Microscopy, Faculty of Health and Medical Sciences, University of Copenhagen for help with characterizing the isolated EVs by cryo-electron microscopy
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jproteome.4c00471.
Table showing fractions categorized into highest to lowest MS2 intensity of previously reported biomarkers (PDF)
Author Contributions
◆ K.S. and J.V.O. share last authorship.
The authors declare no competing financial interest.
Supplementary Material
References
- Lun M. P.; Monuki E. S.; Lehtinen M. K. Development and functions of the choroid plexus–cerebrospinal fluid system. Nat. Rev. Neurosci. 2015, 16 (8), 445–457. 10.1038/nrn3921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vetter P.; Schibler M.; Herrmann J. L.; Boutolleau D. Diagnostic challenges of central nervous system infection: extensive multiplex panels versus stepwise guided approach. Clin. Microbiol. Infect. 2020, 26, 706–712. 10.1016/j.cmi.2019.12.013. [DOI] [PubMed] [Google Scholar]
- Lleó A.; Cavedo E.; Parnetti L.; Vanderstichele H.; Herukka S. K.; Andreasen N.; et al. Cerebrospinal fluid biomarkers in trials for Alzheimer and Parkinson diseases. Nat. Rev. Neurol. 2015, 11 (1), 41–55. 10.1038/nrneurol.2014.232. [DOI] [PubMed] [Google Scholar]
- Thilak S.; Brown P.; Whitehouse T.; Gautam N.; Lawrence E.; Ahmed Z.; Veenith T. Diagnosis and management of subarachnoid haemorrhage. Nat. Commun. 2024, 15 (1), 1850 10.1038/s41467-024-46015-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weston C. L.; Glantz M. J.; Connor J. R. Detection of cancer cells in the cerebrospinal fluid: Current methods and future directions. Fluids Barriers CNS 2011, 8, 14 10.1186/2045-8118-8-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SIOPE Embryonal Tumour group Clinical Practice Recommendations (Medulloblastoma) n.d.
- Thastrup M.; Duguid A.; Mirian C.; Schmiegelow K.; Halsey C. Central nervous system involvement in childhood acute lymphoblastic leukemia: challenges and solutions. Leukemia 2022, 36, 2751–2768. 10.1038/s41375-022-01714-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pui C.-H.; Howard S. C. Current management and challenges of malignant disease in the CNS in paediatric leukaemia. Lancet Oncol. 2008, 9, 257–268. 10.1016/S1470-2045(08)70070-6. [DOI] [PubMed] [Google Scholar]
- Hunger S. P.; Mullighan C. G. Acute Lymphoblastic Leukemia in Children. N. Engl. J. Med. 2015, 373, 1541–1552. 10.1056/NEJMra1400972. [DOI] [PubMed] [Google Scholar]
- Mirian C.; Thastrup M.; Mathiasen R.; Schmiegelow K.; Olsen J. V.; Østergaard O. Mass spectrometry-based proteomics of cerebrospinal fluid in pediatric central nervous system malignancies: a systematic review with meta-analysis of individual patient data. Fluids Barriers CNS 2024, 21, 14 10.1186/s12987-024-00515-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kardell O.; von Toerne C.; Merl-Pham J.; König A. C.; Blindert M.; Barth T. K.; et al. Multicenter Collaborative Study to Optimize Mass Spectrometry Workflows of Clinical Specimens. J. Proteome Res. 2024, 23, 117–129. 10.1021/acs.jproteome.3c00473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schraw J. M.; Woodhouse J. P.; Bernhardt M. B.; Taylor O. A.; Horton T. M.; Scheurer M. E.; et al. Comparison of the blood, bone marrow, and cerebrospinal fluid metabolomes in children with b-cell acute lymphoblastic leukemia. Sci. Rep. 2021, 11, 19613 10.1038/s41598-021-99147-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mo F.; Ma X.; Liu X.; Zhou R.; Zhao Y.; Zhou H. Altered CSF Proteomic Profiling of Paediatric Acute Lymphocytic Leukemia Patients with CNS Infiltration. J. Oncol. 2019, 2019, 3283629 10.1155/2019/3283629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo L.; Ren H.; Zeng H.; Gong Y.; Ma X. Proteomic analysis of cerebrospinal fluid in pediatric acute lymphoblastic leukemia patients: a pilot study. OncoTargets Ther. 2019, 12, 3859–3868. 10.2147/OTT.S193616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Priola G. M.; Foster M. W.; Deal A. M.; Richardson B. M.; Thompson J. W.; Blatt J. Cerebrospinal fluid proteomics in children during induction for acute lymphoblastic leukemia: A pilot study. Pediatr. Blood Cancer 2015, 62, 1190–1194. 10.1002/pbc.25420. [DOI] [PubMed] [Google Scholar]
- Bruschi M.; Petretto A.; Cama A.; Pavanello M.; Bartolucci M.; Morana G.; et al. Potential biomarkers of childhood brain tumor identified by proteomics of cerebrospinal fluid from extraventricular drainage (EVD). Sci. Rep. 2021, 11, 1818 10.1038/s41598-020-80647-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reichl B.; Niederstaetter L.; Boegl T.; Neuditschko B.; Bileck A.; Gojo J.; et al. Determination of a Tumor-Promoting Microenvironment in Recurrent Medulloblastoma: A Multi-Omics Study of Cerebrospinal Fluid. Cancers 2020, 12, 1350 10.3390/cancers12061350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spreafico F.; Bongarzone I.; Pizzamiglio S.; Magni R.; Taverna E.; De Bortoli M.; et al. Proteomic analysis of cerebrospinal fluid from children with central nervous system tumors identifies candidate proteins relating to tumor metastatic spread. Oncotarget 2017, 8, 46177–46190. 10.18632/oncotarget.17579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jankovska E.; Svitek M.; Holada K.; Petrak J. Affinity depletion versus relative protein enrichment: a side-by-side comparison of two major strategies for increasing human cerebrospinal fluid proteome coverage. Clin Proteomics 2019, 16, 9 10.1186/s12014-019-9229-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waury K.; de Wit R.; Verberk I. M. W.; Teunissen C. E.; Abeln S. Deciphering Protein Secretion from the Brain to Cerebrospinal Fluid for Biomarker Discovery. J. Proteome Res. 2023, 22, 3068–3080. 10.1021/acs.jproteome.3c00366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kverneland A. H.; Østergaard O.; Emdal K. B.; Svane I. M.; Olsen J. V. Differential ultracentrifugation enables deep plasma proteomics through enrichment of extracellular vesicles. Proteomics 2023, 23, 2200039 10.1002/pmic.202200039. [DOI] [PubMed] [Google Scholar]
- Thastrup M.; Marquart H. V.; Schmiegelow K. Flow Cytometric Detection of Malignant Blasts in Cerebrospinal Fluid: A Biomarker of Central Nervous System Involvement in Childhood Acute Lymphoblastic Leukemia. Biomolecules 2022, 12, 813 10.3390/biom12060813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thastrup M.; Marquart H. V.; Levinsen M.; Grell K.; Abrahamsson J.; Albertsen B. K.; et al. Flow cytometric detection of leukemic blasts in cerebrospinal fluid predicts risk of relapse in childhood acute lymphoblastic leukemia: a Nordic Society of Pediatric Hematology and Oncology study. Leukemia 2020, 34, 336–346. 10.1038/s41375-019-0570-1. [DOI] [PubMed] [Google Scholar]
- Batth T. S.; Tollenaere M. A. X.; Rüther P.; Gonzalez-Franquesa A.; Prabhakar B. S.; Bekker-Jensen S.; et al. Protein Aggregation Capture on Microparticles Enables Multipurpose Proteomics Sample Preparation. Mol. Cell. Proteomics 2019, 18, 1027–1035. 10.1074/MCP.TIR118.001270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bache N.; Geyer P. E.; Bekker-Jensen D. B.; Hoerning O.; Falkenby L.; Treit P. V.; et al. A Novel LC System Embeds Analytes in Pre-formed Gradients for Rapid, Ultra-robust Proteomics. Mol. Cell. Proteomics 2018, 17, 2284–2296. 10.1074/mcp.TIR118.000853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bekker-Jensen D. B.; Martínez-Val A.; Steigerwald S.; Rüther P.; Fort K. L.; Arrey T. N.; et al. A Compact Quadrupole-Orbitrap Mass Spectrometer with FAIMS Interface Improves Proteome Coverage in Short LC Gradients. Mol. Cell. Proteomics 2020, 19, 716–729. 10.1074/mcp.TIR119.001906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olsen J. V.; Macek B.; Lange O.; Makarov A.; Horning S.; Mann M. Higher-energy C-trap dissociation for peptide modification analysis. Nat. Methods 2007, 4, 709–712. 10.1038/nmeth1060. [DOI] [PubMed] [Google Scholar]
- Pino L. K.; Just S. C.; MacCoss M. J.; Searle B. C. Acquiring and Analyzing Data Independent Acquisition Proteomics Experiments without Spectrum Libraries. Mol. Cell. Proteomics 2020, 19, 1088–1103. 10.1074/mcp.P119.001913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kyte J.; Doolittle R. F. A simple method for displaying the hydropathic character of a protein. J. Mol. Biol. 1982, 157, 105–132. 10.1016/0022-2836(82)90515-0. [DOI] [PubMed] [Google Scholar]
- Armenteros J. J. A.; Sønderby C. K.; Sønderby S. K.; Nielsen H.; Winther O. DeepLoc: prediction of protein subcellular localization using deep learning. Bioinformatics 2017, 33, 3387–3395. 10.1093/bioinformatics/btx431. [DOI] [PubMed] [Google Scholar]
- Tyanova S.; Temu T.; Sinitcyn P.; Carlson A.; Hein M. Y.; Geiger T.; et al. The Perseus computational platform for comprehensive analysis of (prote)omics data. Nat. Methods 2016, 13 (9), 731–740. 10.1038/nmeth.3901. [DOI] [PubMed] [Google Scholar]
- Zhang Y.; Guo Z.; Zou L.; Yang Y.; Zhang L.; Ji N.; et al. A comprehensive map and functional annotation of the normal human cerebrospinal fluid proteome. J. Proteomics 2015, 119, 90–99. 10.1016/j.jprot.2015.01.017. [DOI] [PubMed] [Google Scholar]
- Guldbrandsen A.; Vethe H.; Farag Y.; Oveland E.; Garberg H.; Berle M.; et al. In-depth characterization of the cerebrospinal fluid (CSF) proteome displayed through the CSF proteome resource (CSF-PR). Mol. Cell. Proteomics 2014, 13, 3152–3163. 10.1074/mcp.M114.038554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macron C.; Lavigne R.; Galindo A. N.; Affolter M.; Pineau C.; Dayon L. Exploration of human cerebrospinal fluid: A large proteome dataset revealed by trapped ion mobility time-of-flight mass spectrometry. Data Brief 2020, 31, 105704 10.1016/J.DIB.2020.105704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smalley I.; Law V.; Law V.; Wyatt C.; Evernden B.; Fang B.; et al. Proteomic Analysis of CSF from Patients with Leptomeningeal Melanoma Metastases Identifies Signatures Associated with Disease Progression and Therapeutic Resistance. Clin. Cancer Res. 2020, 26, 2163–2175. 10.1158/1078-0432.CCR-19-2840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barkovits K.; Linden A.; Galozzi S.; Schilde L.; Pacharra S.; Mollenhauer B.; et al. Characterization of Cerebrospinal Fluid via Data-Independent Acquisition Mass Spectrometry. J. Proteome Res. 2018, 17, 3418–3430. 10.1021/acs.jproteome.8b00308. [DOI] [PubMed] [Google Scholar]
- Spinelli S.; Kajana X.; Garbarino A.; Bartolucci M.; Petretto A.; Pavanello M.; et al. Proteomic Profiling of Cerebrospinal Fluid and Its Extracellular Vesicles from Extraventricular Drainage in Pediatric Pilocytic Astrocytoma, towards Precision Oncology. Cancers 2024, 16, 1223 10.3390/cancers16061223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandau U. S.; Magaña S. M.; Costa J.; Nolan J. P.; Ikezu T.; Vella L. J.; et al. Recommendations for reproducibility of cerebrospinal fluid extracellular vesicle studies. J. Extracell. Vesicles 2024, 13, 12397 10.1002/jev2.12397. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All raw-files with mass spectrometry data recorded were uploaded to MS data repository and is accessible with the identifier PXD052666