

Summary
Rapeseed (Brassica napus) silique is the major carbohydrate source for seed development, and the final silique length has attracted great attention from breeders. However, no studies had focused on the dynamic character of silique elongation length (SEL). Here, the dynamic SEL investigation in a natural population including 588 lines over two years indicate that dynamic SEL during 0–20 days after flowering was the most essential stage associated with seed number per silique (SPS) and thousand seed weight (TSW). Then, nine loci were identified to be associated with SEL based on GWAS analysis, among which five SNPs (over 50%) distributed on the A02 chromosome within 6.08 to 6.48 Mb. Subsequently, we screened 5078 differentially expressed genes between two extreme materials. An unknown protein, BnaA02.SE, was identified combining with GWAS and RNA‐Seq analysis. Subcellular localization and expression profiles analysis demonstrated that BnaA02.SE is a chloroplast‐ and nucleus‐localized protein mainly expressed in pericarps and leaves. Furthermore, transgenic verification and dynamic cytological observation reveal that overexpressed BnaA02.SE can promote silique elongation by regulating JA and IAA contents, affecting cell proliferation and expansion, respectively, and finally enhance seed yield by influencing SPS and TSW. Haplotype analysis reveal that the homologs of BnaA02.SE may also be involved in silique elongation regulation. Our findings provided comprehensive insights into a newly SEL trait, and cloned the first gene (BnaA02.SE) controlling silique elongation in B. napus. The identified BnaA02.SE and its homologs can offer a valuable target for improving B. napus yield.
Keywords: Brassica napus, silique elongation, GWAS, transcriptome analysis, BnaA02.SE, functional research
Introduction
Rapeseed (Brassica napus), an allopolyploid species hybridized from Brassica rapa and Brassica oleracea (Chalhoub et al., 2014), is an essential oilseed crop with strongly high economic value worldwide. Therefore, improving B. napus yield has become an important research goal for breeders, and the seed production of B. napus is mainly determined on three component traits: silique number per plant (SPP), seed number per silique (SPS) and thousand seed weight (TSW). And B. napus silique, which are not only sink organs for accumulating photosynthetic products from leaves and stems but also perform mainly photosynthesis source organs that can supply nutrients and energy for seed development (Bennett et al., 2011; King et al., 1997; Wang et al., 2023), were highly associated with the mainly yield‐determining factors (Chen et al., 2007; Zhang et al., 2011). Thus, the B. napus silique has been considered as a favourable organ for yield enhancement in breeding programmes.
Silique can be divided into two mainly parts in B. napus, the seeds and pericarps. The pericarps provide a protection for seed from abiotic and biotic stresses, and more importantly, act as irreplaceable photosynthetic organs in continuously supplying nutrients for seed development. In B. napus, the pericarps become the major photosynthetic organs when leaf declined rapidly after flowering (Bennett et al., 2011). The net CO2 fixation rate in pericarps was as high as 35% in B. napus compared to that in leaves (King et al., 1998), which is significantly higher than that in pericarps of other species like chickpea (<5%) (Furbank et al., 2004). Additionally, more than 48% of the total nitrogen were found in mature seeds and pericarps in B. napus (Rossato et al., 2001). Therefore, the substance storage in seeds is determined by the availability of assimilates directly or indirectly provided by pericarps. Due to the close correlation between the pericarps and silique characteristics, such as the length of the pericarps is equal to the silique length (SL), and the width of the pericarps is also same as silique width (SW). So up to date, abundant researches focus on the SL or its related traits in B. napus (Dong et al., 2018; Fu et al., 2015; Li et al., 2014; Song et al., 2020; Wang et al., 2021; Yang et al., 2017; Zhang et al., 2011). However, most studies only focus on the final SL during the mature stage (Liu et al., 2015; Shi et al., 2019; Yang et al., 2012). Obviously, the mature pericarps will lose its photosynthetic capacity. Additionally, Wand et al. reported that silique complete their morphogenesis before 18 DAF (not mature stage) based on the dynamic SL investigation among the six SL extreme materials (Wang et al., 2021). Therefore, a better dynamic investigations of the pericarps (or silique elongation), rather than just studying its final morphology during maturity with weak photosynthesis, should be of critical essential for the enhancement of seed yield.
Many studies have illustrated that almost all the yield‐related traits are quantitative traits controlled by polygenes in B. napus (Lu et al., 2016, 2017; Pal et al., 2021; Zhang et al., 2022, 2023). Genome‐wide association study (GWAS) have been considered effective tools for screening candidate genes with complex quantitative traits in B. napus, such as plant height (Li et al., 2016), flowering time (Li et al., 2015), branching morphogenesis (He et al., 2017; Sun et al., 2016), oil content (Xiao et al., 2019; Zhao et al., 2022), harvest index (Lu et al., 2016; Luo et al., 2015) and seed weight (Dong et al., 2018; Pal et al., 2021). In recent years, GWASs have also been applied to screen candidate genes associated with SL. Such as 742 SNPs significantly associated with SL were identified by using GWAS analysis based on a natural population in B. napus (Dong et al., 2018). A PAV‐based genome‐wide association study (PAV‐GWAS) identified causal structural variations with SL in a nested association mapping population with ZS11 (Song et al., 2020). Wang et al. revealed several QTLs related to SL by combining GWAS and transcriptomic analysis (Wang et al., 2021). However, because of the complex genome of B. napus, only four known functional genes, named BnaA.ARF18, BnaA09.CYP78A9, BnaC7.ROT3 and Bna.A05DAD1, has been successfully cloned and proved to be key regulators in controlling final SL in B. napus (Liu et al., 2015, 2021; Shi et al., 2019; Zhou et al., 2022), while no genes (especially unknown proteins) related to the dynamic trait of silique elongation have been cloned, resulting in the genetic determinism of such important traits not being elucidated. So it is necessary to screen key candidate genes in regulating the dynamic traits of silique elongation through GWAS or transcription analysis.
In this study, we identified five overlapping and stable significant SNPs located on chromosome A02 across two natural environments by GWAS analysis with the dynamic trait of SEL. Meanwhile, combined with GWAS and transcriptome analysis between two extreme materials with fast and slow elongation of silique, we identified an unknown protein (BnaA02.SE), which homologous to an uncharacterized conserved protein (DUF2358, At1G65230) located on chloroplast. Transgenic functional verification, dynamic cytological observation and hormone property testing indicate that BnaA02.SE‐overexpressing lines can promote silique elongation by influencing JA and IAA metabolism to promote cell proliferation and expansion in the pericarp. Haplotype analysis further reveal that the homologs of BnaA02.SE (BnaC02.SE) may also play essential roles in determining SEL. Our study provides new insight into the molecular mechanism of the dynamic characteristics with silique elongation and the cloning of the first unknown gene can further enrich the gene resources for high‐yield breeding in B. napus.
Results
New awareness and SNP identification by GWAS with the dynamic character of silique elongation
The whole period of silique development is divided into three stages, 0‐10, 10‐20 and 20–30 days after flowering (DAF), named the A, B and C stages, respectively. And SL at 10, 20 and 30 DAF was measured and named SLA, SLB and SLC. Then, the dynamic characters of SEL were calculated with the formula of SLA, SLB‐SLA and SLC‐SLB, named as SELA, SELB and SELC. Based on the statistical analysis of SL traits (Table S1), we found that SL ranged from 3.49 to 10.88 cm in 2019CQ and 3.98 to 12.42 cm in 2021CQ, with coefficients of variation of 21.74% and 20.99%, respectively. Correspondingly, in 2019CQ, the dynamic SEL ranged from 3.39 ± 0.83, 2.92 ± 1.37 and 0.44 ± 0.54 cm during the A, B and C stages. And 3.42 ± 0.65, 2.58 ± 1.25 and 0.79 ± 0.65 cm SEL were observed in 2021CQ (Table S2). The dynamic SL among the 588 lines show that silique elongate fast during A and B stages, whereas hardy SL elongate in stage C, especially in 2019 environments (Figure 1a). Additionally, the dynamic SEL characteristics show that over 50% of the SL was completed during stage A (56% ± 13% in 2019CQ and 54% ± 11% in 2021CQ), and stage B exhibited a shorter SEL than that in stage A (44% ± 13% in 2019CQ and 40% ± 15% in 2021CQ). All of those resulting in hardly SEL occurred in stage C (7% ± 6% in 2019CQ and 12% ± 9% in 2021CQ) (Figure 1b). Suggesting that the early stage (A and B stages before 20 DAF) is the most essential period for silique development.
Figure 1.
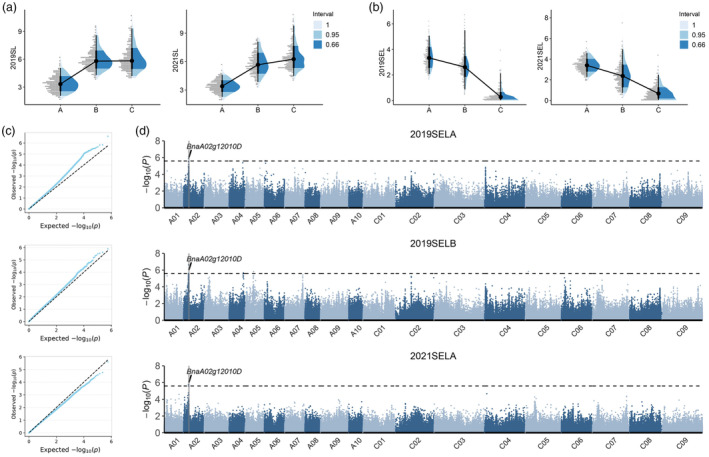
SNP identification of the newly dynamic characters of silique elongation with GWAS analysis. (a, b) Dynamic statistical analysis with the SL and corresponding silique elongation length among 588 materials over two different years, Letter of A, B and C represent the three stages with 0–10, 10–20 and 20–30 days after flowering, respectively; (c, d) Quantile‐quantile plots and manhattan plots resulting from the GWAS results for silique elongation length, the black dashed line represents the Bonferroni‐adjusted significance threshold.
The phenotypic statistics show that all SL and dynamic SEL traits exhibit continuous variation and approximated a normal distribution (Figure 1a,b), indicating that SL and SELs were both quantitative traits. Correlation analysis results show that both SELA and SELB exhibit significant positive correlations with the final SL in both the 2019 and 2021 environments: SELA showed a positive correlation trend with SPS and TSW, whereas only SELA and SPS reached extremely significant differences. Additionally, SELB showed significant differences from the TSW trait (Table S3). The results show that dynamic SEL and final SL possess higher relevance, and SEL is also a yield‐related trait highly associated with SPS and TSW. Thus, GWAS with SELA and SELB were conducted using the P + K module (Price et al., 2006). The quantile‐quantile (QQ) plots results show that this model is highly reliable for use in significant SNP identification (Figure 1c). Nine significant SNPs (P < 2.59 × 10−6) were finally identified, which were unevenly distributed across 5 different chromosomes, with A02 and A06 in the 2019SELA environment and A02, A04, A05 and C05 in 2019SELB, whereas only two SNPs mapped on chromosome A02 in 2021SELA (Figure 1d). Individual SNPs explained 3.73%–5.06% of the phenotypic variation (R 2). Detailed information on the nine significant SNPs is listed in Table S4. In total, only one SNP was distributed on chromosomes A04, A05, A06 and C05, and the remaining five SNPs (over 50%) were distributed on chromosome A02, indicating a high possibility of some key genes located on A02 in regulating the dynamic development of siliques.
Comparative analysis of two B. napus lines using RNA‐Seq and Identification of key BnaA02.SE
Two extreme materials with significant differences in the SEL trait were selected for transcriptome analysis. Material Y5 reached the final SL faster than Y8, whereas Y8 and Y5 had similar final SL (To eliminate interference from genes that control SL) (Figure 2a,b). Y5 elongates 3.3 and 2.5 cm during stages A and B, but Y8 elongates only 2.5 and 1.8 cm, respectively (Figure 2c). Considering that 10 and 20 DAF were the mainly essential stage for silique development (Figure 1a,b), and in order to unify the periods of GWAS and transcriptome analysis, pericarp with 10 DAF (10Sp) and 20 DAF (20Sp), including four independent samples and a total of 12 libraries (three biological repetitions per sample), was extracted for transcriptome analysis. A total of 3015 and 4152 differentially expressed genes (DEGs) were identified between Y8 and Y5 in 10SP and 20SP, respectively. We then selected the common trend DEGs between 10SP and 20SP (indicated in Figure 2d except red circle), and 3117 upregulated and 1961 downregulated DEGs were finally identified (indicated in Figure 2d with green box). All the DEGs and their FPKMs are shown in Table S5. Furthermore, Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis showed that most DEGs were enriched in plant hormone signal transduction and carbon metabolism pathways (Figure 2e), which were highly consistent with the previous research (Wang et al., 2021). Additionally, qRT‐PCR results were highly consistent with the RNA‐Seq data (Figure S1), which can fully prove the accuracy and reliability of the RNA‐Seq.
Figure 2.
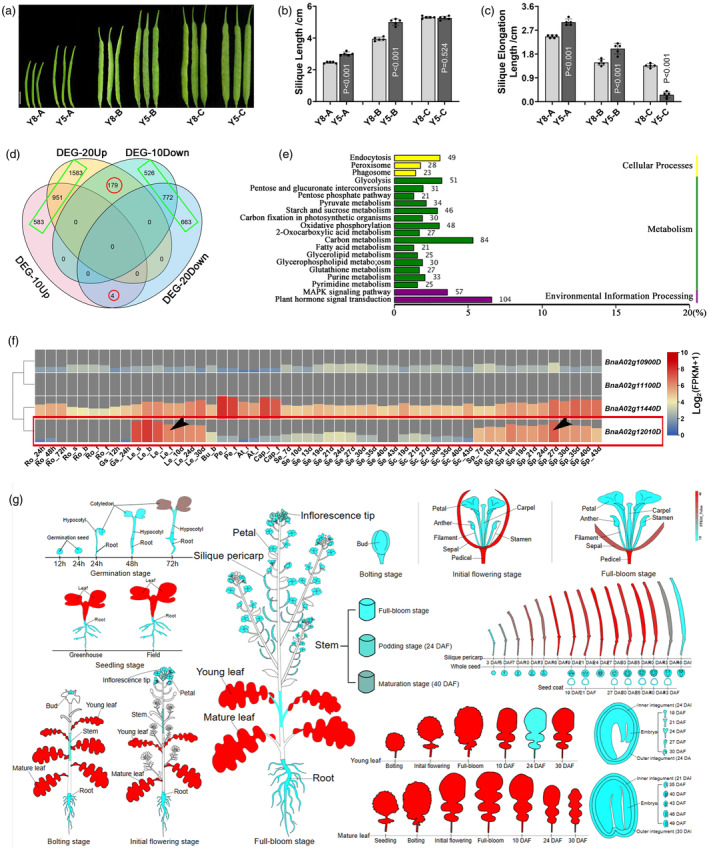
Transcriptomic analysis with dynamic silique elongation length and the identification of BnaA02.SE. (a) Dynamic silique phenotypes between the two extreme materials, scale bare = 2 cm; (b, c) Statistical analysis with traits of SL and corresponding silique elongation length, the value of n = 5 was used for statistical analysis; (d) Venn plot of DEGs with up‐/downregulated between 10SP and 20SP; (e) KEGG pathway analysis among the DEGs; (f) Heatmap of the four common DEGs in the ZS11 accession, Ro: root; Gs: germinated seeds; Le: leaf; Bu: bud; Pe: petal; At: anther; Cap: capillament; Se: seed; Sc: seed coat; Sp: silique pericarp; The abbreviations of b, i and f behind the horizontal line refer to bolting time, initial flowering stage and full flowering stage, respectively. The abbreviations 12, 24, 48 and 72 h indicate hours after seed germination; The numbers 3, 5, 7, 10 d etc., represent the days after flowering; (g) Expression pattern of BnaA02g12010D during the whole period of ZS11.
To select key candidate gene for further functional research, genes associated with dynamic SEL were determined by GWAS analysis with 350 kb flanking regions on either side of the significant SNPs, and a total of 559 and 119 genes were identified in the 2019 and 2021 environments, respectively (Table S6). Furthermore, combined with the RNA‐Seq analysis, four candidate genes were differentially expressed between extreme materials (Figure S2). Then, transcriptome data in accession ZS11 with 53 tissues (Chao et al., 2020) were downloaded to clarify tissue‐specific expression patterns (Table S7). The heatmap shows that BnaA02g10900D and BnaA02g11100D hardly expressed among the 53 organs, whereas BnaA02g11440D was expressed in almost all test tissues. Only BnaA02g12010D exhibited significant spatiotemporal expression specificity (indicated in Figure 2f with black arrow). Moreover, the expression pattern throughout the entire plant growth period also proved that BnaA02g12010D expressed higher in leaves and pericarps than that in roots, floral organs and seed tissues (https://brassica.biodb.org/) (Figure 2g). Thus, we finally chose the unknown protein BnaA02g12010D (named as BnaA02.SE), which was identified by RNA‐Seq and GWAS analysis in both 2019SELA, 2019SELB and 2021SELA within 6.08 Mb to 6.48 Mb on Chromosome A02 (Figure S3), for functional study.
Expression pattern and subcellular localization of BnaA02.SE
To analyse the sequence variations of BnaA02.SE between the extreme lines, we cloned the BnaA02.SE and no sequence variation was found between material Y8 and Y5 (Figure S4). Indicating that the different phenotypic characteristics with DEL are not caused by the sequence diverse of BnaA02.SE, but may be due to the different expression level of BnaA02.SE. Then in order to verify the expression pattern of BnaA02.SE, qRT‐PCR was performed among multiple tissues of ZS11 and the results show that BnaA02.SE was mainly expressed in leaves and pericarps, with a relatively low expression level in buds and flowers, and almost no expression in seeds (Figure 3a), which was completely consistent with the online data (https://brassica.biodb.org/). To further investigate the expression profiles of BnaA02.SE in detail, we analysed its transcriptional level among multiple silique‐related extreme materials. The results reveal that BnaA02.SE expressed higher in material Y5 (fast SEL) than Y8 (slow SEL) in both 10Sp and 20Sp (Figure 3b). Similarly, BnaA02.SE exhibit higher transcriptional level in long silique material (Y3) than that in short (Y6) (Figure 3c). These results indicate that BnaA02.SE has a pericarp‐specific expression pattern, and it may be a key regulator in controlling SL by influencing silique elongation.
Figure 3.
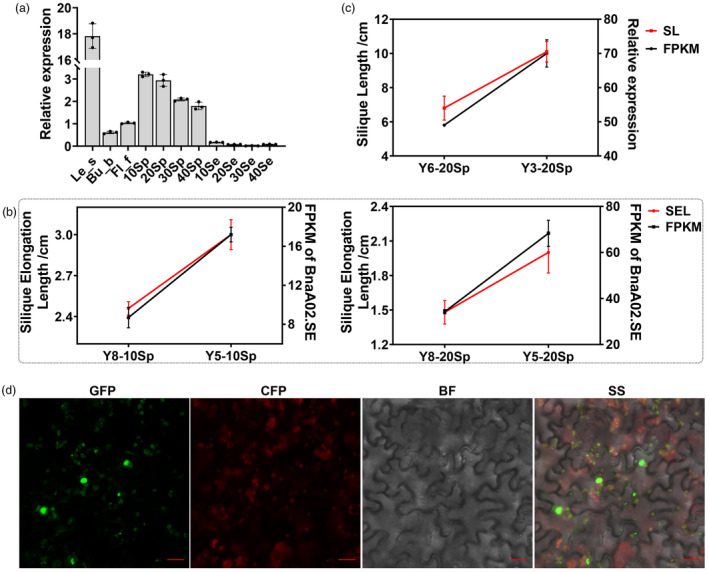
Expression profiles of BnaA02.SE and subcellular localization of BnaA02.SE protein. (a) qRT‐PCR verification of BnaA02.SE in various tissues in ZS11; (b, c) Joint analysis between transcriptional level of BnaA02.SE and the dynamic silique elongation length as well as SL traits; (d) Subcellular localization of BnaA02.SE protein in tobacco (Nicotiana benthamiana), BF, bright field; CFP, chloroplast protein observed using the red fluorescence channel; GFP, green fluorescent protein‐tagged target protein observed using the green fluorescence channel; SS, superimposed signal image; Scale bar = 20 μm.
To determine the subcellular localization of BnaA02.SE in plant cell, we tagged the coding sequence of BnaA02.SE with green fluorescent protein (GFP) and expressed the construction with the control of the 35S:pEarleyGate101‐GFP. The results show that the BnaA02.SE‐GFP fusion protein was mainly located to the nucleus and chloroplast (Figure 3d), which was consist with its transcriptional profiles that highly expressed in the two mainly photosynthetic organs (leaves and silique pericarps).
Enhancing seed yield by overexpressing BnaA02.SE in B. napus
To ascertain whether BnaA02.SE own functions in regulating silique development, a fragment of BnaA02.SE with 867 bp was amplified and cloned (Figure S5a). Three homozygous positive lines were generated based on qRT‐PCR verification, in which BnaA02.SE expressed 7.1, 6.8 and 9 times higher compared with control ZS11 (Figure S5b). OE‐lines exhibit little vigorous growth than that in control in both the seeding and flowering stages (Figure 4a,b). During the harvest stage, the plant height in ZS11 was only 170.2 cm, whereas that in the three OE lines was 185.2, 183.2 and 187 cm, respectively, and the difference reached an extremely significant level (Figure 4c,d). Moreover, the main inflorescence length in the three OE lines exhibited a 9.8% increase compared with that in ZS11 (Figure 4e), which resulted in a significant difference from the trait of silique numbers of the main inflorescence (Figure 4f). However, there was no difference in branching number between OE lines and ZS11, which both fluctuated around 8 (Figure 4g). The trait of silique numbers per plant in ZS11 was 394 on average, whereas the three OE lines exhibited 452, 459.2 and 471.2, respectively, but the difference did not reach significance (Figure 4h). Finally, OE lines can harvest 34.9, 35.2 and 35.6 g seed yield, higher than that in the control with 29.5 g, indicating that overexpressed BnaA02.SE can achieve a 19.2% increase in seed yield per plant (Figure 4i, Table S8).
Figure 4.
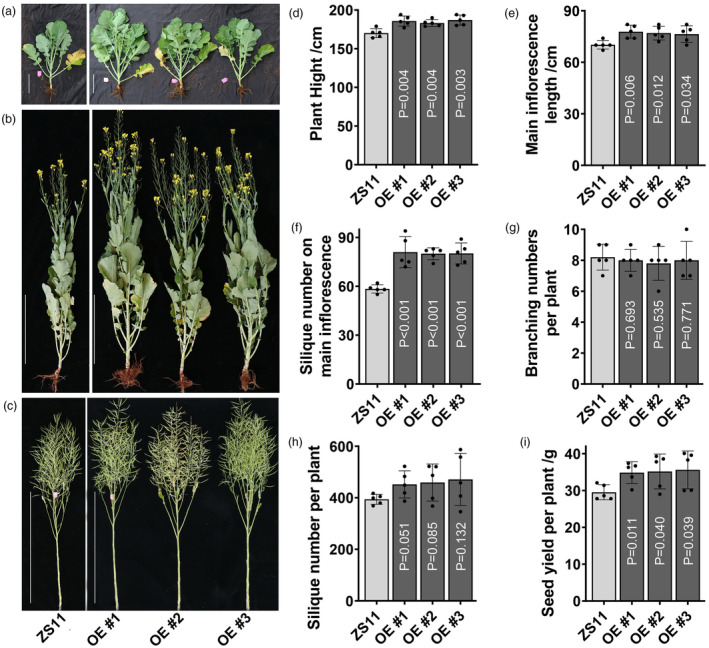
Phenotypic characteristics and statistical analysis of the plant characteristics between transgenic and control plants. (a–c) Phenotypic characteristics of the plants with seeding, flowering and harvest time, scale bare = 20, 50 and 100 cm, respectively; (d–i) Statistical data of the traits of plant height, main inflorescence length, silique number of main inflorescence, branching numbers, SPP and seed yield per plant, the value of n = 5 was used for statistical analysis with all those investigated characteristics.
BnaA02.SE regulates SPS and TSW traits by affecting silique elongation
Siliques in OE lines elongate 7.5 cm in average during 0‐10 DAF stage, reach significant difference compare with ZS11 with 5.5 cm; During 10–20 DAF, 2.2 and 3.7 cm of silique elongation length (SEL) occurred in ZS11 and OE lines, respectively; In contrast, silique of ZS11 show longer elongation length than OE line after 20DAF (Figure 5a,b). Correspondingly, the difference in silique elongation led to a 20% longer SL in the OE lines (11.4 cm on average) than in ZS11 (9.4 cm) (Figure 5c). Moreover, a 40% increase in silique volume (SV) occurred in the OE lines compared with the control (Figure 5d). Finally, all these changes with silique elongation resulted in differences in seed‐related traits (Figure 5e). Silique with OE lines had 27, 26.2 and 26.5 SPS, respectively, whereas ZS11 contained only 24.4 (Figure 5f). Additionally, the OE lines showed 15% higher TSW than the control (Figure 5g). All the phenotypic data are shown in Table S9. These results indicate that overexpression of BnaA02.SE can enhance two factors (SPS and TSW) influencing B. napus yield by regulating silique elongation.
Figure 5.
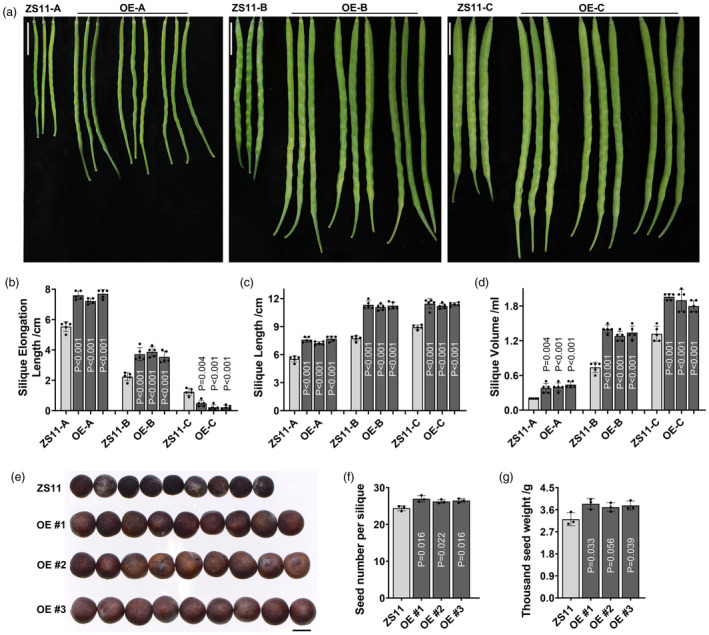
Phenotypic characteristics and statistical analysis of the silique characters among the transgenic plants. (a) Phenotypic traits with dynamic SL between control ZS11 and overexpression BnaA02.SE lines with three replications, OE‐A, OE‐B and OE‐C represent three different stages with 10, 20 and 30 days after flowering, scale bare = 2 cm; (b–d) Statistical analysis with characters of silique elongation length, SL and silique volume, the value of n = 5 was used for statistical analysis of related characters; (e) Phenotypic characters with seed, scale bare = 2 mm; (f, g) Statistical analysis with SPS and TSW, the value of n = 3 was used for statistical analysis.
BnaA02.SE affects cell proliferation and expansion by altering JA and IAA contents
To elucidate the mechanism of overexpressed BnaA02.SE in elongating siliques, we compared the DEGs in silique with 10 DAF between OE lines and ZS11. A total of 2585 DEGs were identified (Figure S6a), and the KEGG results showed that most genes (10.19%) were enriched in the plant hormone signal transduction pathway (ko04075) (indicated with red font in Figure S6b). Moreover, Gene Ontology (GO) enrichment analysis showed that DEGs were highly associated with the regulation of the jasmonic acid mediated signalling pathway in biological processes (GO2000022) (indicated with red font in Figure S6c). And the transcriptome analysis show that genes belongs to JAZ and ARF subfamily expressed significantly higher and lower than that in control, respectively (Figure 6b,c). All the DEGs and their FPKM_values are shown in Table S10. Correspondingly, the LC/MS results show a lower JA content in the OE lines (20.6 ng/g) than that in the control (37.8 ng/g) (Figure 6d). The OE lines possessed a 6.13 ng/g IAA content, which was significantly higher than the 2.97 ng/g content in ZS11 (Figure 6e). Dynamic cytological observation show that the OE lines have 102.4 and 28.2 cell numbers at 10 DAF and 20 DAF, respectively, whereas ZS11 only contains 87.4 and 20.6 cells, respectively (Figure 6f,g). Correspondingly, the cell size in ZS11 (1779.2 and 4326 μm2 in 10 and 20 DAF) was significantly larger than that in the OE lines (994 and 3120 μm2). However, at 30 DAF, the OE had a larger cell size (7742 μm2) than ZS11 (4406 μm2) (Figure 6f,h). Therefore, we speculate that BnaA02.SE may affect silique elongation by promoting cell proliferation and expansion through influencing JA and IAA content (Figure 6).
Figure 6.
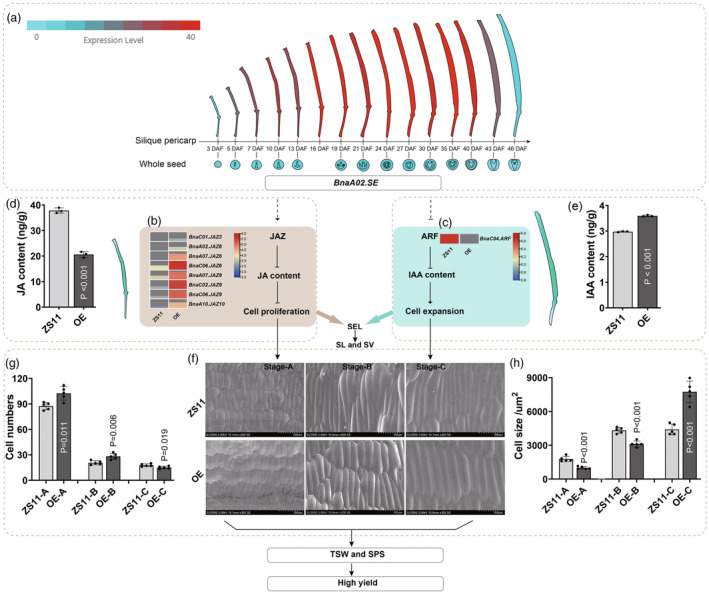
Proposed module by which BnaA02.SE overexpression to promote cell proliferation and expansion. (a) The pericarp‐specific expression level of BnaA02.SE; (b, c) Expression level of genes related to JA and IAA mediated signalling pathway; (d, e) JA and IAA content with n = 3 for statistical analysis; (f) Scanning electron microscopy observation, Stage a–c represent 10, 20 and 30 DAF, respectively; (g, h) Statistic analysis of cell number and size in pericarp at 10, 20 and 30 DAF, the value of n = 5 was used for the data statistic.
Candidate gene analysis of BnaA02.SE and haplotype analysis
Since BnaA02.SE is an unknown protein without any reference, thereby BLAST analysis was performed using BnaA02.SE sequence as the query, the results show that there are only one (At1G65230), one (Bra022487), one (Bol04532) and two (BnaA02g12010D and BnaC02g16480D) homologs in A. thaliana, B. rape, B. oleracea and B. napus, respectively (Figure S7). It is worth noting that At1G65230, an unknown protein located on chloroplast, is expressed mainly in leaves and pericarps (Figure S8), which results are completely consistent with BnaA02.SE (Figures 2g and 3d). Moreover, sequence alignment reveals high similarity between BnaA02.SE and BnaC02g16480D (named as BnaC02.SE), which both possess five exons (Figure 7a,c), and the similarity with the protein sequence between them was as high as 93.1%. Additionally, the transcriptional data from BrassicaEDB (https://brassica.biodb.org/) demonstrated that BnaC02.SE exhibit almost identical expression patterns with BnaA02.SE (Figure S9). Suggesting the highly conservative of BnaA02.SE and BnaC02.SE compared to At1G65230.
Figure 7.
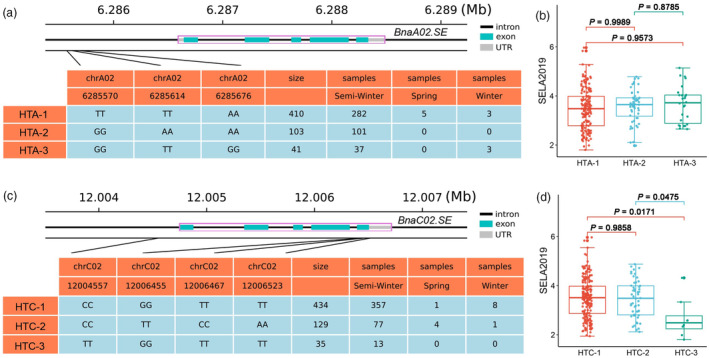
Haplotype analysis of BnaA02.SE and its homologs in natural germplasms. (a) Haplotype frequency of BnaA02.SE; (b) Silique elongation length of BnaA02.SE haplotype; (c) Haplotype frequency of BnaC02.SE; (d) Silique elongation length of BnaC02.SE haplotype.
We thereby conducted a haplotype analysis of these two homologs in natural germplasm. Unfortunately, none SNP were identified among the five exons in BnaA02.SE. We also calculate the mean phenotypic values of SEL among the three BnaA02.SE haplotypes (designed as HTA‐1 to HTA‐3) (Figure 7a), however, all the three haplotypes show no significant differences in SEL over two years (Figures 7b and S10a, Table S11). For BnaC02.SE, two of the four SNPs were identified in the fifth exon of BnaC02.SE, and three haplotypes divided into HTC‐1 to HTC‐3 were obtained (Figure 7c). Compared with that of HTC‐3, the HTC‐1 and HTC‐2 were both significantly higher in 2019SELA, while no significant differences were found in 2019SELB and 2021SELA (Figures 7d and S10b, Table S11). Indicating that the SNPs upstream BnaA02.SE are not associated with SEL, whereas haplotype HTC‐3 has a negative effect on SEL for BnaC02.SE.
Discussion
The newly dynamic trait of silique elongation was a quantitative characteristic highly associated with TSW and SPS
Brassica napus yield is dependents on three mainly components, i.e. SPP, SPS and TSW. Evidently, each of them is highly affected by the development of silique, which is the major photosynthetic organs when leaf declined rapidly after flowering (Bennett et al., 2011). Currently, researches associated with silique development have only focused on the final SL or its related traits, including the final breadth, thickness, and volume (Wang et al., 2016; Zhao et al., 2021) during the mature stage with weak photosynthesis, but pay little attention to the dynamic traits with SEL during the whole period of silique growth. Thus in this work, a detailed phenotypic statistics with the dynamic SEL trait among 588 materials were calculated (Table S1). The results show that faster elongation of the silique lead to longer silique and larger area of silique pericarp during the early stage of silique development. Since chloroplast is rich in mesocarp of silique pericarp (Sessions and Zambryski, 1995) and endows silique pericarps with photosynthetic capacity (Bennett et al., 2011). Therefore, the higher photosynthetic ability with faster elongation silique can provide more assimilates and nutrients to fuel seeds forming higher TSW. On the other hand, longer siliques have a larger space to hold more embryos and thereby conduct more SPS (Jiao et al., 2021). Moreover, the dynamic SEL characteristics show a near‐normal distribution curve, taken together, our data suggested that the newly SEL is a quantitative trait highly associated with TSW and SPS.
Identification of BnaA02.SE associated with the dynamic SEL trait
During the past decades, abundant SNPs associated with final SL were identified through GWAS, QTL or transcriptome analysis (Dong et al., 2018; Fu et al., 2015; Song et al., 2020; Wang et al., 2016, 2021; Yang et al., 2012, 2017; Zhang et al., 2011). Among them, only a few major QTLs own large effects, while most are minor QTLs with small effects. Up to date, a total of three QTLs controlling SL have been validated by transgenic analysis, among them, two of which are major QTLs located on A09 chromosomes (Liu et al., 2015; Shi et al., 2019). However, because of the complex genome, genes in B. napus possess multiple copies (Liu et al., 2018; Yang et al., 2023; Zhang et al., 2020, 2023) and undergo severe functional differentiation (Froschel et al., 2019; Han et al., 2020; Zheng et al., 2020). All these factors result to a difficulty in cloning key genes underlying minor QTLs in allopolyploid B. napus. To the best of our knowledge, these phenomenon not only occurred in B. napus, but only a small number of minor QTLs have been cloned in rice (Chan et al., 2020; Li et al., 2020). Whereas, one minor QTL (cqSL‐C7) explained 8%–14% of phenotypic variations with a small effect was successfully cloned and proved to be key factors in regulating SL in B. napus (Zhou et al., 2022). Indicating that some key candidate genes underlying minor QTLs with small effect can also possess essential roles in regulating SL in B. napus. However, none reports currently focus on the identification of genes related to dynamic SEL trait, even no QTLs related to SEL with neither large nor small effect were successfully cloned in B. napus. Thus, we performed GWAS analysis with dynamic SEL based on 588 natural populations, which provided a high‐resolution genomic variation map and was used to screen multiple quantitative traits in our previous research (Lu et al., 2019). Finally, nine SNPs were identified (Figure 1d), and eight of them overlapped with other silique‐related traits in previous studies (Dong et al., 2018; Lu et al., 2016, 2017; Luo et al., 2017; Zhao et al., 2016) (Table S4), supporting the high reliability of the SNPs detected in our study. Furthermore, combined with the analysis of GWAS and transcriptome, only one common candidate DEG (named as BnaA02.SE) own the pericarp‐specific expression patterns (Figure 2f,g), which was identified in the 2019SELA, 2019SELB and 2021SELA environments within 6.08 Mb to 6.48 Mb on Chromosome A02 with small effect (Figure S3). Additionally, BnaA02.SE was mainly located on nucleus and chloroplast, which may play an essential role in the photosynthesis of leaves and pericarps. Therefore, we choose BnaA02.SE for further functional research. This approach was also choose for identification of BnaFAX1‐1, a candidate gene contributing to biological yield and seed oil content by functional verification in our previous study (Xiao et al., 2021).
BnaA02.SE regulates silique elongation by affecting cell proliferation and expansion in B. napus
Based on the characterization of previously cloned candidate genes regulating SL, it is widely accepted that diverse hormones play essential roles in controlling silique development in B. napus. Such as auxins were proved to be predominant plant hormones that control SL (Liu et al., 2015; Shi et al., 2019), and BnaC7.ROT3 may regulate SL via influencing brassinosteroid (BR) biosynthesis (Zhou et al., 2022). Moreover, Liu et al. reported that B. napus SL is highly mediated through crosstalk between jasmonate acid (JA) and auxin (IAA) signalling (Liu et al., 2021). In this study, transcriptome analysis between BnaA02.SE OE‐lines and ZS11 also showed enrichment of the most significant pathway in hormone transduction (Figure S6b). Moreover, a set of genes (BnaJAZ3s, BnaJAZ6s, BnaJAZ9s and BnaJAZ10s), which has been confirmed to be negative regulators within the JA mediated pathway belonging to jasmonate‐zim‐domain protein family (Ghorbel et al., 2021; Liu et al., 2021), expressed higher in BnaA02.SE OE‐lines, resulting in lower JA content than control. Similarly, the latest reports also proved that the accumulation of transcriptional suppressor FvJAZ can relieve the inhibition of the target transcription factor FvMYC2, then promote the expression of downstream genes manipulated by FvMYC2, especially FvLOX3, and subsequently affect JA synthesis (Wang et al., 2024). This results was coincide with the speculation of JAZ inhibits the synthesis of JA in this study. On the other hand, BnaA09.ARF18 was reported to controlling SL by affecting cell elongation (Liu et al., 2015). Coincidently, a homologous ARF located on chromosome C04 was found to be significantly downregulated in BnaA02.SE overexpression lines, leading to a higher IAA content than that of the control (Figure 6).
Furthermore, BnaA05.DAD1, BnaC07.ROT3 and BnaA09.ARF18 were reported to controlling SL by regulating cell size (Liu et al., 2015; Zhou et al., 2022). In contrast, OE‐lines with BnaA02.SE had a small cell size during 10‐20 DAF, which exhibited significantly more cell numbers, and the cell size of the OE‐lines became larger than ZS11 at 30 DAF. Considering that JAs are one of the predominant plant hormones in regulating cell numbers (Chen et al., 2011), while IAA can increase cell volume through promoting cell expansion (Wang et al., 2001), we therefore propose a module of the mechanism by which BnaA02.SE regulate silique elongation in B. napus: The transgenic expression of BnaA02.SE leading to expression changes of a set of genes belonging to JA mediated pathway and then reduced the JA content, thereby promoting cell proliferation during the early stage of silique development. BnaA02.SE was constitutive overexpression during the later stage due to the use of 35S promoter, and IAA content was increased significantly by inhibiting the expression level of ARF homologous, which thereby promoting cell expansion. Both cell proliferation and expansion can promote silique elongation, and the longer siliques increase the photosynthetic area and enhance the amount of carbohydrates available for seed development in silique. The overexpression of BnaA02.SE thus increases the SPS and TSW, contributing to a slight increase with seed yield in final.
Future prospects and the availability of BnaA02.SE and its homologous
In soybean, pod length is significantly associated with seed size (Bravo et al., 1980). Similarly, the differences with SL can alter the photosynthetic area of silique pericarp and finally influence the total carbohydrate gross and seed weight in B. napus (Li et al., 2018). Additionally, the changing of SL can also take effect on the silique space to hold more embryos to produce more seed numbers (Bennett et al., 2011). Our results also showed that a slight increase in seed yield with BnaA02.SE OE‐lines were mainly caused by the enhancement of seed size, SPS and TSW (Figure 5). Although the control ZS11 is a wildly planted cultivar known for its long silique and high yield characteristics in China (Zhou et al., 2022), overexpressed BnaA02.SE can also promote silique elongation and improve seed yield in ZS11. Therefore, we speculate that overexpressing BnaA02.SE has a great potential to increase seed yield by influencing silique development in B. napus. However, haplotype analysis reveal some limitation on the application of BnaA02.SE for fast elongation of silique in B. napus breeding to some extent (Figure 7a,b). But interestingly, its homologs BnaC02.SE may own the similar functions in regulating SEL, which the haplotype HTC‐1 and HTC‐2 in BnaC02.SE had significantly longer SEL than that with HTC‐3 (Figure 7c,d). Though BnaA02.SE and its homologs BnaC02.SE own minor effect on SEL, the coordination of these two homologs may be helpful to effectively influence the B. napus SEL. Coincidently, Zhou et al. also found this similar phenomenon in long silique breeding (Zhou et al., 2022): no significant differences among the six haplotypes in the causal BnaC7.ROT3 underlying cqSL‐C7 locus, whereas some haplotypes identified in its homologs (BnaA3.ROT3 and BnaA1.ROT3) has a stable and slight effect on SL. Considering that serious functional redundancy among homologs occurred in allopolyploid B. napus (Han et al., 2020; Zheng et al., 2020), the pyramiding of homologs with minor loci (such as BnaC7.ROT3 and BnaA02.SE in this study) may be of great significance for high yield breeding in allotetraploid B. napus.
In conclusion, we revealed an unknown protein BnaA02.SE, which were identified from both RNA‐Seq and GWAS analysis with minor effect in regulating SEL in B. napus. And functional analysis show that BnaA02.SE controls the dynamic elongation of silique by influencing cell proliferation and expansion, and then take effect on final SL. This study provides new insights to the regulatory mechanism of final SL forming in B. napus. Haplotypes analysis elucidated that functional redundancy may exhibit between BnaA02.SE and its homologs BnaC02.SE. Finally, the functions of BnaC02.SE need to be further verified by transgenic characterization and the deep insights into the synergistic regulatory mechanism of silique development between BnaA02.SE and BnaC02.SE need to be explored in future.
Materials and methods
Plant material and trait evaluation
All plants used for GWAS analysis in our study comprised 588 accessions, including 7 from Australia, 13 from North America, 102 from Europe and 466 from Asia. According to their growth habit, all the 588 accessions were divided into 3 ecotypes, including spring (86 accessions), semi‐winter (428 accessions) and winter (74 accessions) (Figure S11). All the accessions were preserved at Southwest University, and planted with normal growing season in experimental farm with Southwest University in Beibei, Chongqing, China (29°45′N, 106°22′E, 238.6 m). All the field experiment followed a randomized complete‐block design with two biological replications. Each accession was cultivated in two rows with 10–12 plants per row, with 20 cm plant spacing and 40 cm row spacing. All the accessions were planted in the same filed over two years with 2019 and 2021. Field managements were conducted based on the standard agricultural practices. During the initial flowering stage, five individual plants were selected to mark the flowering period. Then, the SLs were measured and the dynamic trait of SEL was calculated.
Genome‐wide association analysis
Genome‐wide association study analysis of the dynamic SEL trait was performed based on the phenotypic data. All the parameters of the GWAS analysis, including the population structure, relative kinship and SNP genotyping were described in our previous study (Lu et al., 2019). The GWAS analysis was cultured by TASSEL5.2 software using P + K models (Bradbury et al., 2007). The QQ and Manhattan plots were generated by the R package.
Transcriptome analysis
Samples of pericarp were collected with 10 DAF and 20 DAF (10Sp and 20Sp) from 5 independent plants, and total RNA was extracted respectively. The sequencing library preparation and reaction were performed by Novogene Technology Corporation (Beijing, China). Gene expression levels were calculated with FPKM, and DEGs were obtained based on the Cuffdiff, with the parameters of |log2 fold change | > 2.0 and FDR (false discovery rate) < 0.05 (Xiao et al., 2019). And all the raw data can be downloaded from the NCBI SRA database with the accession no. PRJNA1096371.
Haplotype analysis
The genotypic data among the 588 accessions with nature population were collected from our previous study (Lu et al., 2019). Phenotypic data used for haplotype analysis are listed in Table S1. The missing genotype was imputed by Beagle 5 (Browning et al., 2018). SnpEff 5.0 was selected for annotating the effects of nonsynonymous variations (Cingolani et al., 2012). Online analysis tools with Hastat and Haplot (https://github.com/swu1019lab) were used for gene haplotype analysis and visualization, respectively.
Gene expression analyses
Total RNA was extracted with RNeasy Extraction Kit (Invitrogen, Carlsbad, CA) and cDNA was produced through Reverse Transcription Kit (TaKaRa Biotechnology, Dalian, China). Then qRT‐PCR was performed by BIO‐RAD system in triplicates. All the relative expression level of each gene was calculated using the 2−ΔΔCt method. The results were visualized with GraphPad Prism5.0 software. And all the primers used in this study were shown in Table S12.
Vector constructs and transgenic plant obtaining
Tissues of leaves, flowers, seeds and pericarps from ZS11 were mixed as the samples to clone candidate BnaA02.SE full coding DNA sequence. Then, the PCR products were ligated into the pEarleyGate101 vector with attR1 and attR2 sets to produce 35S:pEarleyGate101‐BnaA02.SE. The 35S:pEarleyGate101‐BnaA02.SE was further introduced into Agrobacterium strain GV3101 for transformation of the ZS11 with the simplified floral dip method (Ali et al., 2022). And qRT‐PCR was conducted to screen the positive transgenic plants.
Subcellular localization
The cDNA sequence without the termination codon of the BnaA02.SE was connected to pEarleyGate101 vector for constructing a GFP fusion plasmid. Agrobacterium GV3101 were transformed with the plasmid BnaA02.SE‐GFP, and the 35S:pEarleyGate101 was selected as control. The transient expression experiment in tobacco (Nicotiana benthamiana) was performed to analyse the subcellular localization of BnaA02.SE, which was determined as previously described (Zhou et al., 2022). And the fluorescence signals were imaged by a laser scanning confocal microscopy (Carl Zeiss, LSM780).
Phenotypic investigation of the transgenic plants
All the transgenic B. napus plants were grown restricted within an isolated experimental station in Southwest University. Three replicates of BnaA02.SE overexpression lines named as OE #1, #2 and #3 were selected for phenotypic investigation. Each replicate was planted ten individual plants and together with the control. During the mature period, five individual plants were harvested from each transgenic line. The plant height, main inflorescence length, and silique number of inflorescence and per plant were measured, and the seed yield per plant was weighted after natural air drying. Additionally, 30 siliques from the main inflorescence were collected, and all the seed numbers (SN) were counted by a tablet machine, and SPS was calculated as SN/30. The seed weight of 30 siliques (SW) was investigated by weighing scales, and TSW was calculated with SW/SN * 1000. For the trait of SL, we measured five SLs of the main inflorescence at 10, 20 and 30 DAF. SV was estimated based on the volume change following submersion of siliques using a cylinder containing water.
Cytological observation with scanning electron microscopy (SEM)
Scanning electron microscopy observation was performed to investigate the cellular morphology using a HITACHI SEM SU3500 (Hitachi, Tokyo, Japan). The parameters were as follows: temperature of −25 °C, voltage of 5000 V, and magnification with 300 times (Lima et al., 2019). Three biological replies were designed, and at least five siliques were collected from each OE lines and ZS11. ImageJ software (National Institute of Health) was chosen to calculate the cell number and size (Giaretta et al., 2018).
Quantification of JA and IAA in B. napus
Fresh samples with 10Sp from OE lines and ZS11 were collected. Hormone extraction and qualitative testing were performed according to the methods described by Zeng et al. (Zeng et al., 2019).
Statistical analysis
All statistical data were analysed by IBM SPSS statistics 20 (IBM, New York) software. GraphPad Prism 9.5 software (GraphPad Software, California) was used to visualize the charts. One‐way analysis of variance (ANOVA) was used to analyse the significance of the differences.
Conflict of interest
All the authors declare no conflict of interest.
Author contributions
L.Y.Z., B.Y. and C.Z. collected the phenotypic data in field; X.D.L. and B.Y. completed the GWAS and RNA‐Seq analysis; S.C. and S.R.X. completed the transgenic functional analysis; T.T.S., Z.Y.Y. and X.Z.K. completed data analysis; C.M.Q., K.L. and J.N.L. reviewed the manuscript; L.Y.Z. wrote this paper.
Funding information
This work was funded by National Key Research and Development Program of China (2023YFF1000700); National Natural Science Foundation of China (32201741 and 32360492); Scientific Innovation of 2030 Project (2022ZD0400802); Special fund for youth team of the Southwest Universities (SWU‐XJPY202306); Chongqing Modern Agricultural Industry Technology System (COMAITS202304).
Supporting information
Figure S1 qRT‐PCR verification of the RNA‐Seq data.
Figure S2 Venn plot of candidate genes identified by RNA‐Seq and GWAS over two different environments.
Figure S3 Comparison of the GWAS results in A02 chromosome in 2019SELA, 2019SELB and 2021SELB environments.
Figure S4 Sequence alignment of BnaA02.SE between the two extreme materials and ZS11 accession.
Figure S5 Sequence amplification of BnaA02.SE (a) and transgenic plants screening with qRT‐PCR (b).
Figure S6 Heatmap (a), KEGG (b) and GO (c) analysis among the 2585 DEGs between transgenic and control lines.
Figure S7 Identification of the homologs of BnaA02.SE from B. napus, B. oleracea, B. rapa and A. thaliana.
Figure S8 Expression pattern of homologs At1G65230 throughout the entire reproductive period.
Figure S9 Expression pattern of homologs BnaC02.SE during the whole period of ZS11.
Figure S10 Haplotype frequency of BnaA02.SE (a) and BnaC0.SE (b) in 2019SELB and 2021SELA.
Figure S11 The 588 accessions collected worldwide used for GWAS analysis.
Table S1 Phenotypic data of the SL and SEL characters among the 588 materials in two environments.
Table S2 Phenotypic statistics of different traits in natural population with two years.
Table S3 Correlation analysis between SEL and silique‐related traits.
Table S4 Markers with significant SNPs associated with silique elongation and candidate genes within confidence interval.
Table S5 FPKM values of DEGs screed by RNA‐Seq analysis.
Table S6 Candidate genes screened by GWAS.
Table S7 FPKM value of the four common DEGs among 53 tissues in ZS11.
Table S8 Phenotypic data between the transgenic and control plants.
Table S9 Phenotypic data with the silique of the transgenic and control plants.
Table S10 Transcriptome data between transgenic and control plants.
Table S11 Haplotype analysis of BnaA02.SE and its homologs.
Table S12 All the primers used in this study.
Contributor Information
Cunmin Qu, Email: drqucunmin@swu.edu.cn.
Kun Lu, Email: drlukun@swu.edu.cn.
Jiana Li, Email: ljn1950@swu.edu.cn.
Data availability statement
The data that supports the findings of this study are available in the supplementary material of this article.
References
- Ali, I. , Sher, H. , Ali, A. , Hussain, S. and Ullah, Z. (2022) Simplified floral dip transformation method of Arabidopsis thaliana . J. Microbiol. Methods, 197, 106492. [DOI] [PubMed] [Google Scholar]
- Bennett, E.J. , Roberts, J.A. and Wagstaff, C. (2011) The role of the pod in seed development: strategies for manipulating yield. New Phytol. 190(4), 838–853. [DOI] [PubMed] [Google Scholar]
- Bradbury, P.J. , Zhang, Z. , Kroon, D.E. , Casstevens, T.M. , Ramdoss, Y. and Buckler, E.S. (2007) TASSEL: software for association mapping of complex traits in diverse samples. Bioinformatics, 23(19), 2633–2635. [DOI] [PubMed] [Google Scholar]
- Bravo, J.A. , Fehr, W.R. and Rodriguez, d.C.S. (1980) Use of pod width for indirect selection of seed weight in soybeans. Crop Sci. 20(4), 507–510. [Google Scholar]
- Browning, B.L. , Zhou, Y. and Browning, S.R. (2018) A one‐penny imputed genome from next‐generation reference panels. Am. J. Hum. Genet. 103(3), 338–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chalhoub, B. , Denoeud, F. , Liu, S. , Parkin, I.A. , Tang, H. , Wang, X. , Chiquet, J. et al. (2014) Plant genetics. Early allopolyploid evolution in the post‐neolithic Brassica napus oilseed genome. Science, 345(6199), 950–953. [DOI] [PubMed] [Google Scholar]
- Chan, A.N. , Wang, L.L. , Zhu, Y.J. , Fan, Y.Y. , Zhuang, J.Y. and Zhang, Z.H. (2020) Identification through fine mapping and verification using CRISPR/Cas9‐targeted mutagenesis for a minor QTL controlling grain weight in rice. Theor. Appl. Genet. 134, 327–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chao, H. , Li, T. , Luo, C. , Huang, H. , Ruan, Y. , Li, X. , Niu, Y. et al. (2020) BrassicaEDB: a gene expression database for Brassica crops. Int. J. Mol. Sci. 21(16), 5831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, Q. , Sun, J. , Zhai, Q. , Zhou, W. , Qi, L. , Xu, L. , Wang, B. et al. (2011) The basic helix‐loop‐helix transcription factor MYC2 directly represses PLETHORA expression during jasmonate‐mediated modulation of the root stem cell niche in Arabidopsis. Plant Cell, 23(9), 3335–3352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, W. , Zhang, Y. , Liu, X. , Chen, B. , Tu, J. and Tingdong, F. (2007) Detection of QTL for six yield‐related traits in oilseed rape (Brassica napus) using DH and immortalized F(2) populations. Theor. Appl. Genet. 115(6), 849–858. [DOI] [PubMed] [Google Scholar]
- Cingolani, P. , Platts, A. , Wang le, L. , Coon, M. , Nguyen, T. , Wang, L. , Land, S.J. et al. (2012) A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso‐2; iso‐3. Fly (Austin), 6(2), 80–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong, H.L. , Tan, C. , Li, Y. , He, Y. , Wei, S. , Cui, Y. , Chen, Y. et al. (2018) Genome‐wide association study reveals both overlapping and independent genetic loci to control seed weight and silique length in Brassica napus. Front. Plant Sci. 9, 921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Froschel, C. , Iven, T. , Walper, E. , Bachmann, V. , Weiste, C. and Dröge‐Laser, W. (2019) A gain‐of‐function screen reveals redundant ERF transcription factors providing opportunities for resistance breeding toward the vascular fungal pathogen Verticillium longisporum . Mol. Plant‐Microbe Interact. 32(9), 1095–1109. [DOI] [PubMed] [Google Scholar]
- Fu, Y. , Wei, D. , Dong, H. , He, Y. , Cui, Y. , Mei, J. , Wan, H. et al. (2015) Comparative quantitative trait loci for silique length and seed weight in Brassica napus . Sci. Rep. 5, 14407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furbank, R.T. , White, R. , Palta, J.A. and Turner, N.C. (2004) Internal recycling of respiratory CO2 in pods of chickpea (Cicer arietinum L.): the role of pod wall, seed coat, and embryo. J. Exp. Bot. 55(403), 1687–1696. [DOI] [PubMed] [Google Scholar]
- Ghorbel, M. , Brini, F. , Sharma, A. and Landi, M. (2021) Role of jasmonic acid in plants: the molecular point of view. Plant Cell Rep. 40(8), 1471–1494. [DOI] [PubMed] [Google Scholar]
- Giaretta, E. , Mordenti, A.L. , Canestrari, G. , Brogna, N. , Palmonari, A. and Formigoni, A. (2018) Assessment of muscle Longissimus thoracis et lumborum marbling by image analysis and relationships between meat quality parameters. PLoS One, 13(8), e0202535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han, S. , Khan, M.H.U. , Yang, Y. , Zhu, K. , Li, H. , Zhu, M. , Amoo, O. et al. (2020) Identification and comprehensive analysis of the CLV3/ESR‐related (CLE) gene family in Brassica napus L. Plant Biol. (Stuttg). 22(4), 709–721. [DOI] [PubMed] [Google Scholar]
- He, Y.J. , Wu, D. , Wei, D. , Fu, Y. , Cui, Y. , Dong, H. , Tan, C. et al. (2017) GWAS, QTL mapping and gene expression analyses in Brassica napus reveal genetic control of branching morphogenesis. Sci. Rep. 7, 15971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiao, Y. , Zhang, K. , Cai, G. , Yu, K. , Amoo, O. , Han, S. , Zhao, X. et al. (2021) Fine mapping and candidate gene analysis of a major locus controlling ovule abortion and seed number per silique in Brassica napus L. Theor. Appl. Genet. 134(8), 2517–2530. [DOI] [PubMed] [Google Scholar]
- King, S.P. , Badger, M.R. and Furbank, R.T. (1998) CO2 refixation characteristics of developing canola seeds and silique wall. Funct. Plant Biol. 25(3), 377–386.. [Google Scholar]
- King, S.P. , Lunn, J.E. and Furbank, R.T. (1997) Carbohydrate content and enzyme metabolism in developing canola siliques. Plant Physiol. 114(1), 153–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, F. , Chen, B. , Xu, K. , Gao, G. , Yan, G. , Qiao, J. , Li, J. et al. (2016) A genome‐wide association study of plant height and primary branch number in rapeseed (Brassica napus). Plant Sci. 242, 169–177. [DOI] [PubMed] [Google Scholar]
- Li, L. , Long, Y. , Zhang, L. , Dalton‐Morgan, J. , Batley, J. , Yu, L. , Meng, J. et al. (2015) Genome wide analysis of flowering time trait in multiple environments via high‐throughput genotyping technique in Brassica napus L. PLoS One, 10(3), e0119425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, N. , Shi, J. , Wang, X. , Liu, G. and Wang, H. (2014) A combined linkage and regional association mapping validation and fine mapping of two major pleiotropic QTLs for seed weight and silique length in rapeseed (Brassica napus L.). BMC Plant Biol. 14, 114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, N. , Song, D. , Peng, W. , Zhan, J. , Shi, J. , Wang, X. , Liu, G. et al. (2018) Maternal control of seed weight in rapeseed (Brassica napus L.): the causal link between the size of pod (mother, source) and seed (offspring, sink). Plant Biotechnol. J. 17, 736–749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, Q. , Lu, L. , Liu, H. , Bai, X. , Zhou, X. , Wu, B. , Yuan, M. et al. (2020) A minor QTL, SG3, encoding an R2R3‐MYB protein, negatively controls grain length in rice. Theor. Appl. Genet. 133(8), 2387–2399. [DOI] [PubMed] [Google Scholar]
- Lima, D.M. , Linhares, T.S. , Lima, S.N.L. , Carvalho, E.M. , Loguercio, A.D. , Bauer, J. and Carvalho, C.N. (2019) Effect of sonic application of self‐adhesive resin cements on push‐out bond strength of glass fiber posts to root dentin. Materials (Basel), 12(12), 1930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, B. , Seong, K. , Pang, S. , Song, J. , Gao, H. , Wang, C. , Zhai, J. et al. (2021) Functional specificity, diversity, and redundancy of Arabidopsis JAZ family repressors in jasmonate and COI1‐regulated growth, development, and defense. New Phytol. 231(4), 1525–1545. [DOI] [PubMed] [Google Scholar]
- Liu, J. , Hua, W. , Hu, Z. , Yang, H. , Zhang, L. , Li, R. , Deng, L. et al. (2015) Natural variation in ARF18 gene simultaneously affects seed weight and silique length in polyploid rapeseed. Proc. Natl. Acad. Sci. USA, 112(37), E5123–E5132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, M. , Chang, W. , Yu, M. , Fan, Y. , Shang, G. , Xu, Y. , Niu, Y. et al. (2021) Overexpression of DEFECTIVE IN ANTHER DEHISCENCE 1 increases rapeseed silique length through crosstalk between JA and auxin signaling. Ind. Crop. Prod. 168(1), 113576. [Google Scholar]
- Liu, P. , Zhang, C. , Ma, J.Q. , Zhang, L.Y. , Yang, B. , Tang, X.Y. , Huang, L. et al. (2018) Genome‐wide identification and expression profiling of cytokinin oxidase/dehydrogenase (CKX) genes reveal likely roles in pod development and stress responses in oilseed rape (Brassica napus L.). Genes, 9(3), 168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu, K. , Peng, L. , Zhang, C. , Lu, J. , Yang, B. , Xiao, Z. , Liang, Y. et al. (2017) Genome‐wide association and transcriptome analyses reveal candidate genes underlying yield‐determining traits in Brassica napus . Front. Plant Sci. 8, 206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu, K. , Wei, L. , Li, X. , Wang, Y. , Wu, J. , Liu, M. , Zhang, C. et al. (2019) Whole‐genome resequencing reveals Brassica napus origin and genetic loci involved in its improvement. Nat. Commun. 10(1), 1154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu, K. , Xiao, Z. , Jian, H. , Peng, L. , Qu, C. , Fu, M. , He, B. et al. (2016) A combination of genome‐wide association and transcriptome analysis reveals candidate genes controlling harvest index‐related traits in Brassica napus . Sci. Rep. 6, 36452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo, X. , Ma, C. , Yue, Y. , Hu, K. , Li, Y. , Duan, Z. , Wu, M. et al. (2015) Unravelling the complex trait of harvest index in rapeseed (Brassica napus L.) with association mapping. BMC Genomics, 16(1), 379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo, Z. , Wang, M. , Long, Y. , Huang, Y. , Shi, L. , Zhang, C. , Liu, X. et al. (2017) Incorporating pleiotropic quantitative trait loci in dissection of complex traits: seed yield in rapeseed as an example. Theor. Appl. Genet. 130(8), 1569–1585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pal, L. , Sandhu, S.K. , Bhatia, D. and Sethi, S. (2021) Genome‐wide association study for candidate genes controlling seed yield and its components in rapeseed (Brassica napus subsp. napus). Physiol. Mol. Biol. Plants, 27(9), 1933–1951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price, A.L. , Patterson, N.J. , Plenge, R.M. , Weinblatt, M.E. , Shadick, N.A. and Reich, D. (2006) Principal components analysis corrects for stratification in genome‐wide association studies. Nat. Genet. 38(8), 904–909. [DOI] [PubMed] [Google Scholar]
- Rossato, L. , Laine, P. and Ourry, A. (2001) Nitrogen storage and remobilization in Brassica napus L. during the growth cycle: nitrogen fluxes within the plant and changes in soluble protein patterns. J. Exp. Bot. 52(361), 1655–1663. [PubMed] [Google Scholar]
- Sessions, R.A. and Zambryski, P.C. (1995) Arabidopsis gynoecium structure in the wild and in ettin mutants. Development, 121(5), 1519–1532. [DOI] [PubMed] [Google Scholar]
- Shi, L.L. , Song, J. , Guo, C. , Wang, B. , Guan, Z. , Yang, P. , Chen, X. et al. (2019) A CACTA‐like transposable element in the upstream region of BnaA9.CYP78A9 acts as an enhancer to increase silique length and seed weight in rapeseed. Plant J. 98(3), 524–539. [DOI] [PubMed] [Google Scholar]
- Song, J.M. , Guan, Z. , Hu, J. , Guo, C. , Yang, Z. , Wang, S. , Liu, D. et al. (2020) Eight high‐quality genomes reveal pan‐genome architecture and ecotype differentiation of Brassica napus . Nat. Plants, 6(1), 34–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun, C. , Wang, B. , Wang, X. , Hu, K. , Li, K. , Li, Z. , Li, S. et al. (2016) Genome‐wide association study dissecting the genetic architecture underlying the branch angle trait in rapeseed (Brassica napus L.). Sci. Rep. 6, 33673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, C. , Yang, J. , Chen, W. , Zhao, X. and Wang, Z. (2023) Contribution of the leaf and silique photosynthesis to the seeds yield and quality of oilseed rape (Brassica napus L.) in reproductive stage. Sci. Rep. 13(1), 4721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, J. , Fan, Y. , Mao, L. , Qu, C. , Lu, K. , Li, J. and Liu, L. (2021) Genome‐wide association study and transcriptome analysis dissect the genetic control of silique length in Brassica napus L. Biotechnol. Biofuels, 14(1), 214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, W. , Ouyang, J. , Li, Y. , Zhai, C. , He, B. , Si, H. , Chen, K. et al. (2024) A signaling cascade mediating fruit trait development via phosphorylation‐modulated nuclear accumulation of JAZ repressor. J. Integr. Plant Biol, 1–20 (Early Access). [DOI] [PubMed] [Google Scholar]
- Wang, X.D. , Chen, L. , Wang, A. , Wang, H. , Tian, J. , Zhao, X. , Chao, H. et al. (2016) Quantitative trait loci analysis and genome‐wide comparison for silique related traits in Brassica napus . BMC Plant Biol. 16, 71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, Z. , Cao, W. , Dai, T. and Zhou, Q. (2001) Effects of exogenous hormones on floret development and grain setin wheat. Plant Growth Regul. 35(3), 225–231. [Google Scholar]
- Xiao, Z. , Zhang, C. , Tang, F. , Yang, B. , Zhang, L. , Liu, J. , Huo, Q. et al. (2019) Identification of candidate genes controlling oil content by combination of genome‐wide association and transcriptome analysis in the oilseed crop Brassica napus . Biotechnol. Biofuels, 12, 216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao, Z.C. , Tang, F. , Zhang, L. , Li, S. , Wang, S. , Huo, Q. , Yang, B. et al. (2021) The Brassica napus fatty acid exporter FAX1‐1 contributes to biological yield, seed oil content, and oil quality. Biotechnol. Biofuels, 14(1), 190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, B. , Zhang, L. , Xiang, S. , Chen, H. , Qu, C. , Lu, K. and Li, J. (2023) Identification of Trehalose‐6‐phosphate synthase (TPS) genes associated with both source‐/sink‐related yield traits and drought response in rapeseed (Brassica napus L.). Plants (Basel), 12(5), 981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, P. , Shu, C. , Chen, L. , Xu, J. , Wu, J. and Liu, K. (2012) Identification of a major QTL for silique length and seed weight in oilseed rape (Brassica napus L.). Theor. Appl. Genet. 125(2), 285–296. [DOI] [PubMed] [Google Scholar]
- Yang, Y. , Shen, Y. , Li, S. , Ge, X. and Li, Z. (2017) High density linkage map construction and QTL detection for three silique‐related traits in orychophragmus violaceus derived Brassica napus population. Front. Plant Sci. 8, 1512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng, J. , Zhang, M. , Hou, L. , Bai, W. , Yan, X. , Hou, N. , Wang, H. et al. (2019) Cytokinin inhibits cotton fiber initiation by disrupting PIN3a‐mediated asymmetric accumulation of auxin in the ovule epidermis. J. Exp. Bot. 70(12), 3139–3151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, C. , Chang, W. , Li, X. , Yang, B. , Zhang, L. , Xiao, Z. , Li, J. et al. (2022) Transcriptome and small RNA sequencing reveal the mechanisms regulating harvest index in Brassica napus . Front. Plant Sci. 13, 855486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, L. , Yang, B. , Zhang, C. , Chen, H. , Xu, J. , Qu, C. , Lu, K. et al. (2023) Genome‐wide identification and posttranscriptional regulation analyses elucidate roles of key argonautes and their miRNA triggers in regulating complex yield traits in rapeseed. Int. J. Mol. Sci. 24(3), 2543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, L. , Yang, G. , Liu, P. , Hong, D. , Li, S. and He, Q. (2011) Genetic and correlation analysis of silique‐traits in Brassica napus L. by quantitative trait locus mapping. Theor. Appl. Genet. 122(1), 21–31. [DOI] [PubMed] [Google Scholar]
- Zhang, L. , Zhang, C. , Yang, B. , Xiao, Z. , Ma, J. , Liu, J. , Jian, H. et al. (2020) Genome‐wide identification and expression profiling of monosaccharide transporter genes associated with high harvest index values in rapeseed (Brassica napus L.). Genes (Basel), 11(6), 653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao, C. , Xie, M. , Liang, L. , Yang, L. , Han, H. , Qin, X. , Zhao, J. et al. (2022) Genome‐wide association analysis combined with quantitative trait loci mapping and dynamic transcriptome unveil the genetic control of seed oil content in Brassica napus L. Front. Plant Sci. 13, 929197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao, W. , Wang, X. , Wang, H. , Tian, J. , Li, B. , Chen, L. , Chao, H. et al. (2016) Genome‐wide identification of QTL for seed yield and yield‐related traits and construction of a high‐density consensus map for QTL comparison in Brassica napus . Front. Plant Sci. 7, 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao, X. , Yu, K. , Pang, C. , Wu, X. , Shi, R. , Sun, C. , Zhang, W. et al. (2021) QTL analysis of five silique‐related traits in Brassica napus L. across multiple environments. Front. Plant Sci. 12, 766271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng, M. , Zhang, L. , Tang, M. , Liu, J. , Liu, H. , Yang, H. , Fan, S. et al. (2020) Knockout of two BnaMAX1 homologs by CRISPR/Cas9‐targeted mutagenesis improves plant architecture and increases yield in rapeseed (Brassica napus L.). Plant Biotechnol. J. 18(3), 644–654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou, X. , Zhang, H. , Wang, P. , Liu, Y. , Zhang, X. , Song, Y. , Wang, Z. et al. (2022) BnaC7.ROT3, the causal gene of cqSL‐C7, mediates silique length by affecting cell elongation in Brassica napus . J. Exp. Bot. 73(1), 154–167. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 qRT‐PCR verification of the RNA‐Seq data.
Figure S2 Venn plot of candidate genes identified by RNA‐Seq and GWAS over two different environments.
Figure S3 Comparison of the GWAS results in A02 chromosome in 2019SELA, 2019SELB and 2021SELB environments.
Figure S4 Sequence alignment of BnaA02.SE between the two extreme materials and ZS11 accession.
Figure S5 Sequence amplification of BnaA02.SE (a) and transgenic plants screening with qRT‐PCR (b).
Figure S6 Heatmap (a), KEGG (b) and GO (c) analysis among the 2585 DEGs between transgenic and control lines.
Figure S7 Identification of the homologs of BnaA02.SE from B. napus, B. oleracea, B. rapa and A. thaliana.
Figure S8 Expression pattern of homologs At1G65230 throughout the entire reproductive period.
Figure S9 Expression pattern of homologs BnaC02.SE during the whole period of ZS11.
Figure S10 Haplotype frequency of BnaA02.SE (a) and BnaC0.SE (b) in 2019SELB and 2021SELA.
Figure S11 The 588 accessions collected worldwide used for GWAS analysis.
Table S1 Phenotypic data of the SL and SEL characters among the 588 materials in two environments.
Table S2 Phenotypic statistics of different traits in natural population with two years.
Table S3 Correlation analysis between SEL and silique‐related traits.
Table S4 Markers with significant SNPs associated with silique elongation and candidate genes within confidence interval.
Table S5 FPKM values of DEGs screed by RNA‐Seq analysis.
Table S6 Candidate genes screened by GWAS.
Table S7 FPKM value of the four common DEGs among 53 tissues in ZS11.
Table S8 Phenotypic data between the transgenic and control plants.
Table S9 Phenotypic data with the silique of the transgenic and control plants.
Table S10 Transcriptome data between transgenic and control plants.
Table S11 Haplotype analysis of BnaA02.SE and its homologs.
Table S12 All the primers used in this study.
Data Availability Statement
The data that supports the findings of this study are available in the supplementary material of this article.