

ABSTRACT
Gene expression systems that transcend species barriers are needed for cross-species analysis of gene function. In particular, expression systems that can be utilized in both model and pathogenic bacteria underpin comparative functional approaches that inform conserved and variable features of bacterial physiology. In this study, we develop replicative and integrative vectors alongside a novel, IPTG-inducible promoter that can be used in the model bacterium Escherichia coli K-12 as well as strains of the antibiotic-resistant pathogen, Acinetobacter baumannii. We generate modular vectors that transfer by conjugation at high efficiency and either replicate or integrate into the genome, depending on design. Embedded in these vectors, we also developed a synthetic, IPTG-inducible promoter, PabstBR, that induces to a high level but is less leaky than the commonly used trc promoter. We show that PabstBR is titratable at both the population and single-cell levels, regardless of species, highlighting the utility of our expression systems for cross-species functional studies. Finally, as a proof of principle, we use our integrating vector to develop a reporter for the E. coli envelope stress σ factor, RpoE, and deploy the reporter in E. coli and A. baumannii, finding that A. baumannii does not recognize RpoE-dependent promoters unless RpoE is heterologously expressed. We envision that these vector and promoter tools will be valuable for the community of researchers who study the fundamental biology of E. coli and A. baumannii.
IMPORTANCE
Acinetobacter baumannii is a multidrug-resistant, hospital-acquired pathogen with the ability to cause severe infections. Understanding the unique biology of this non-model bacterium may lead to the discovery of new weaknesses that can be targeted to treat antibiotic-resistant infections. In this study, we provide expression tools that can be used to study the gene function in A. baumannii, including in drug-resistant clinical isolates. These tools are also compatible with the model bacterium, Escherichia coli, enabling cross-species comparisons of gene function. We anticipate that the use of these tools by the scientific community will accelerate our understanding of Acinetobacter biology.
KEYWORDS: synthetic biology, gene expression, cloning, shuttle vector, Tn7 vector
INTRODUCTION
Historically, research in bacterial genetics focused on specific model organisms, such as Escherichia coli K-12, due to a lack of techniques, tools, reagents, genome sequences, and general knowledge of non-model bacteria (1, 2). As a result, much of our current understanding about the basic physiology of Gram-negative bacteria comes from E. coli (3, 4). Although most core cellular processes are likely conserved, gene function and regulation can vary subtly or even dramatically across species boundaries (4, 5). Such deviation is obvious in pathogens such as Acinetobacter baumannii, which has adopted many traits that are distinct from E. coli K-12—most notably extreme antibiotic resistance (6–8). With advances in DNA sequencing and synthesis as well as tools that democratize genetic analysis across species (e.g., CRISPR approaches (9)), there now exists an enormous opportunity to shrink the knowledge and technique gaps between model bacteria and clinically relevant pathogens. One simple approach to bridge the gap would be to develop systems capable of assessing gene function in both model and pathogenic bacteria, such that the function of any gene could be readily compared in different strain or species backgrounds. Indeed, many genetic tools have been developed that function in both E. coli and A. baumannii (9–14).
In this study, we focus on genetic tools applicable to the antibiotic-resistant pathogen, A. baumannii. A. baumannii is considered an “urgent threat” by the Centers for Disease Control and Prevention due to its ability to resist nearly all available antibiotic treatments (15). Although some promising new anti-Acinetobacter compounds that target lipooligosaccharide (LOS) transport (macrocyclic peptide antibiotics) or the LOS flippase, MsbA (cerastecins) have recently been discovered (16–18), more work is needed in this area as Acinetobacter is adept at acquiring and developing new resistance mechanisms (19–21). A. baumannii is poorly studied compared to E. coli K-12 and even other Gram-negative pathogens such as Pseudomonas aeruginosa; however, understanding the distinct physiology of A. baumannii is critical to developing new treatments (22, 23). For instance, lipid A, an essential component of the outer membrane in most Gram-negatives and a binding site for the antibiotic colistin (24), is not essential for viability in many A. baumannii strains, including clinical isolates (25). Furthermore, regulation of stress pathways that could play roles in antibiotic resistance, tolerance, or persistence is distinct in A. baumannii compared to other γ-proteobacteria, as A. baumannii lacks conserved transcription factors such as the stationary-phase sigma (σ) factor, RpoS (26, 27).
Although vectors that are capable of replicating in or integrating into E. coli and A. baumannii are currently available (10–13), such vectors could still be improved upon. Replicative shuttle vectors typically combine the high-copy, ColE1 origin of replication in E. coli with the pWH1266 (10) origin for A. baumannii or utilize the pRSF1010 (11–13) origin, which is functional in both species. The pWH1266 and pRSF1010 origins are compatible in A. baumannii, enabling expression from two replicative vectors in the same cell (11). Integrative vectors based on the site-specific transposon Tn7 insert DNA cargo into the genome downstream of the glmS gene and have been used extensively in E. coli (28), A. baumannii (29–31), and many other species (32, 33). However, many of these vectors were not designed to contain easily swappable modules (e.g., different antibiotic markers) outside of standard multiple cloning sites (MCSs), although pMMB67-derived vectors (pRSF1010 origin) are available with additional markers (13). Existing vectors typically employ inducible promoters that are either native to or designed for use in E. coli (34, 35). These include E. coli native promoters such as Plac and ParaBAD that can be induced with IPTG or arabinose, respectively (10, 36), or semi-synthetic promoters such as Ptac and Ptrc, which are IPTG-inducible (11). Unfortunately, characteristics of these promoters pose challenges for precise control of expression. For instance, ParaBAD expression cannot be titrated with sub-saturating concentrations of its inducer, arabinose, due to “all or nothing” effects that result in a fraction of cells inducing at high levels, while others show minimal activity (37–39). Ptac and Ptrc are sufficiently leaky such that genes placed under their control often complement deletion phenotypes in the absence of the inducer (34, 40, 41), and full induction often results in overexpression toxicity (42). It is unknown whether Ptac and Ptrc are titratable in A. baumannii. A titratable promoter with less leakiness and a lower maximal level of expression would be ideal for physiological expression and gene function studies in A. baumannii.
In this work, we generate useful reagents for gene function studies in A. baumannii and E. coli. We create modular vectors that replicate or integrate in both species and carry the novel promoter PabstBR, which can be induced and titrated with IPTG. In a proof-of-principle experiment, we combine all three reagents to probe the activity of the E. coli envelope stress σ factor, RpoE, in both species.
RESULTS AND DISCUSSION
Modular replicative and integrating vectors for E. coli and A. baumannii
We sought to construct a modular set of replicative and integrative vectors that could be used to examine gene function in A. baumannii and E. coli. Our shuttle vector (Fig. 1A) replicates in E. coli using the medium-copy origin, p15A (20–30 copies per cell (43)) and in A. baumannii using the low-copy origin pWH1266 (~nine copies per cell (44)). Our integrating vector (Fig. 1A) inserts into the genomes of E. coli and A. baumannii downstream of glmS using the Tn7 transposase (provided on a separate plasmid (9, 32)). Both vectors have an antibiotic module flanked by XhoI sites, which provide a facile way of removing the existing resistance marker, while simultaneously linearizing the plasmid as a substrate for Gibson assembly (45). In this study, we have provided hygromycin, apramycin, and kanamycin versions of both replicative and integrative vectors. We note that hygromycin and apramycin are attractive resistance markers for studying multidrug-resistant pathogens, given that neither antibiotic is used against A. baumannii clinically (29, 46). FRT sites in the integrative vector allow for optional FLP recombinase-mediated excision of the antibiotic marker (47, 48). The cloning module, or multiple cloning site (MCS), has several restriction sites for cloning genes of interest (Fig. 1B and C). Although other sites can be used, we recommend cloning into NcoI because it contains a translation start codon (ATG) in alignment with a strong upstream ribosome binding site (RBS) taken from the classic expression vector pTrc99a (49). The promoter module exists between AatII and NcoI sites for the replicating vector and SpeI and NcoI sites for the integrating vector. We provide these vectors with a novel, IPTG-inducible promoter (PabstBR, described below), but other promoters and RBSs of interest can be readily swapped into the module. Additionally, both the replicative and integrative vectors can be used in the same strain as multiple markers are available and only one vector replicates, ruling out compatibility issues.
Fig 1.

Modular replicative and integrative expression vectors. (A) Circular plasmid map and features of the replicative shuttle vector containing both E. coli and A. baumannii origins of replication (top) and the Tn7 expression vector containing a transposon that will integrate into the chromosomal attTn7 site (bottom). Available antibiotic resistance cassettes (AbR) are listed. Maps are adapted from SnapGene (GSL Biotech). (B and C) Linear maps showing the modular promoter region and multiple cloning sites (MCS) for the replicative plasmid and Tn7 vector. The NcoI site provides an ATG start codon optimally proximal to a strong ribosome binding site (RBS).
We next determined the efficiency of transfer for both vectors into E. coli and A. baumannii. Both vectors contain oriT sites, enabling transfer by conjugation from E. coli cells that are auxotrophic for diaminopimelic acid (DAP-) to DAP+ recipient bacteria, followed by antibiotic selection to recover only vector-containing recipients. Additionally, both vectors can be transferred by electroporation into competent recipient cells, if desired. To quantify the efficiency of transfer by conjugation, we mated DAP- E. coli donor cells (E. coli K-12 WM6026) with model strains of E. coli K-12 (BW25113) and A. baumannii (ATCC 17978). We found that both vectors were transferred at efficiencies consistent with use in downstream experiments ranging in scale from individual genes to large libraries (Fig. S1a and b). Transfers of both the replicative and integrative vectors were highly efficient in E. coli (>10−1 efficiencies for both vectors) and A. baumannii (>10−2 and 10−4 efficiencies for replicative and integrative vectors, respectively). Importantly, our observed transfer efficiencies were on par with those needed for library construction for genome-scale experiments (9). We note that we observed instances of unintended integration of the Tn7 vector backbone in both E. coli and A. baumannii (i.e., co-integrates (50)). The presence of such co-integrates in recipient colonies can be tested by screening for the ampR/bla gene (which confers carbenicillin resistance) present in the vector backbone. We patched 40 transconjugants for each organism, and while the frequency of integration with the vector backbone was relatively low (≤3/40 for each), we recommend testing transconjugants to verify the insertion accuracy (Fig. S1c). To facilitate this process in drug-resistant A. baumannii strains, we have also provided Tn7 vectors with a hygromycin or apramycin resistance marker in the vector backbone (Table S2). Taken together, we have created modular replicative and integrative vectors for E. coli and A. baumannii, which can be transferred at efficiencies that are useful for a variety of applications.
A tightly regulated, IPTG-inducible promoter for E. coli and A. baumannii
We sought to develop an IPTG-inducible promoter with low leakiness and high expression for A. baumannii. We previously found that a broadly utilized synthetic promoter in E. coli, PLlacO-1, was unstable when used to express a toxic protein in A. baumannii (dCas9) (31). When we selected for mutants with stable expression of dCas9, we found that lacO repeats in the promoter had collapsed, creating a new IPTG-regulated promoter (Fig. 2A, Acinetobacter Suppressor of Toxicity or Pabst). We hypothesized that this promoter was weaker due to its success at repressing toxicity. To measure promoter activity in A. baumannii, we cloned Pabst upstream of a gene encoding superfolder green fluorescent protein (sfgfp) in our replicative vector (Fig. 2B). Our measurements confirmed that Pabst expression was very weak, with less than twofold increase in expression at saturating levels of the inducer. This weak activity is likely due to the divergence between the Pabst −35 element (TTATAA) and the consensus σ70 -35 (TTGACA), especially at the −33 position (A versus G, respectively).
Fig 2.

PabstBR promoter construction and expression. (A) Promoter sequences showing the homologous recombination event in lacO repeat regions (red) of the PLlacO-1 sequence that produces Pabst, which contains a −35-like region (yellow). Site-directed mutagenesis reverts the −35 region back to consensus (orange) to create PabstBR. (B) Dot plots showing sfGFP fluorescence from replicative vectors containing sfgfp under Pabst, PabstBR, or Ptrc promoters in A. baumannii ATCC 17978 with no IPTG (left) or 1 mM IPTG (right). Values were normalized to empty vector controls, and sample means are represented by a solid horizontal line (n = 3 biological replicates). Asterisks and ns indicate significant and nonsignificant sample differences, respectively (Welch’s t-tests; P-values < 0.05).
To generate a new promoter with higher activity but without repeating lacO elements, we used site-directed mutagenesis to replace the Pabst −35 sequence with a consensus −35 (Fig. 2A). We found that the new promoter, PabstBR (Acinetobacter suppressor of toxicity with better regulation), showed significantly higher induction than Pabst (~150-fold; Welch’s t-test, P = 0.003) in A. baumannii (Fig. 2B). PabstBR also showed ~threefold reduced leakiness compared to Ptrc, a popular IPTG-inducible promoter used in both E. coli (34) and A. baumannii (11), although induction at saturating levels of IPTG was somewhat lower (~threefold) than Ptrc. With reduced leakiness and a more physiologically appropriate expression range, PabstBR has advantages for complementation and expression with reduced toxicity (40, 51).
PabstBR expression is titratable at the population and single-cell levels
Investigators frequently titrate promoter activity to determine expression–phenotype relationships and avoid toxic overexpression. To determine if PabstBR expression is titratable at the population level, we induced the expression of PabstBR-sfgfp at varying concentrations of IPTG from both our replicative and integrative vectors in E. coli K-12 BW25113 and A. baumannii ATCC 17978 (Fig. 3A and B). We found that PabstBR was titratable in all tested contexts. Plasmid-borne PabstBR showed similar patterns of IPTG induction in both E. coli and A. baumannii and had ~tenfold higher level of maximal expression compared to an integrated copy. Unexpectedly, Tn7-integrated PabstBR showed a higher apparent level of expression in A. baumannii compared to E. coli at nearly every concentration of IPTG, including saturating concentrations (Fig. 3B). In addition to ATCC 17978, the A. baumannii field uses strains ATCC 19606 and AB5075 as antibiotic-susceptible and -resistant models, respectively. To test PabstBR titratability in those strain backgrounds, we again expressed PabstBR-sfgfp at varying IPTG concentrations (Fig. S2). As expected, we found that PabstBR was titratable at the population level.
Fig 3.

Titration of PabstBR expression at the population level. Titration of the expression from (A) the replicative plasmid or (B) the Tn7 transposon. Plots shown are normalized sfGFP levels expressed from PabstBR across IPTG concentrations for E. coli BW25113 and A. baumannii ATCC 17978. Error bars represent standard deviation (n = 3 biological replicates for replicative vector; n = 6 biological replicates for the Tn7 transposon).
Inducible promoters can erroneously appear to be titratable at the population level due to varying subpopulations of fully induced cells, as is seen in systems with active transport and feedback of inducer molecules (e.g., arabinose and ParaBAD (37)). To rule out this possibility, we measured induction of PabstBR-sfgfp at varying concentrations of IPTG in single cells using flow cytometry (Fig. 4A and B; Fig. S3) and microscopy (Fig. S4). We measured PabstBR expression from replicative vectors as we reasoned that variations in plasmid copy number would be more likely to have a subpopulation effect. We found that PabstBR was fully titratable at the single-cell level in E. coli K-12 BW25113 and A. baumannii ATCC 17978. Distributions of sfGFP fluorescence were unimodal at all IPTG concentrations in both species, consistent with relatively uniform induction of PabstBR at the single-cell level. Although increasing concentrations of IPTG fully shifted the sfGFP distributions in A. baumannii, the distributions were wider than those seen in E. coli for unknown reasons (Fig. 4B). One possibility to explain increased expression variation in A. baumannii is simply that the pWH1266 origin has intrinsically greater plasmid copy number variation than p15A, although testing plasmid copy number at the single-cell level is fraught with challenges (52). We conclude that PabstBR is titratable at the single-cell level, enabling gene function studies with precise levels of expression.
Fig 4.

Titration of PabstBR expression at the single-cell level. Titration of sfgfp expression from the replicative expression vector, under control of PabstBR, in (A) E. coli BW25113 or (B) A. baumannii ATCC 17978. Ridgeline plots depict overlapping density plots of sfGFP fluorescence for cells induced at different IPTG concentrations, measured by flow cytometry (~70,000 cells per sample). EV are empty vector (no GFP) control samples in 1 mM IPTG. Asterisks indicate significant sample differences (Welch’s t-tests; P-values < 0.05).
Modular vectors and PabstBR enable gene regulation studies in E. coli and A. baumannii
As a proof of principle to demonstrate the utility of our PabstBR vector set in studying the gene function, we investigated RpoE-dependent promoter activity in E. coli and A. baumannii. RpoE, also known as σE, is an extracytoplasmic function (ECF) σ factor that regulates the envelope stress response in E. coli and related γ-proteobacteria (53–56). Species as distant from E. coli as Pseudomonas aeruginosa have a functional ortholog (AlgU, 66% identity) that recognizes the same DNA sequence as RpoE (57); however, a BLAST search of the A. baumannii genome recovered no hits for RpoE. To determine if A. baumannii recognizes RpoE-dependent promoters, we cloned the autoregulated rpoE promoter (PrpoE) from E. coli into our integration vector upstream of a gene encoding monomeric red fluorescent protein (mrfp) as a reporter. We integrated this construct into both E. coli and A. baumannii and found that PrpoE was only active in E. coli (Fig. 5A and B). To determine if the promoter could be recognized in A. baumannii in the presence of RpoE, we cloned the rpoE gene into our replicating vector under the control of PabstBR. We found that the expression of rpoE in A. baumannii was sufficient to increase transcription from PrpoE (Fig. 5A). This suggested that A. baumannii lacks an activity identical to that E. coli RpoE and that no other factors in A. baumannii can recognize PrpoE. As expected, we also found that overexpression of rpoE in E. coli resulted in increased PrpoE activity (Fig. 5B). However, it remained a formal possibility that a divergent version of RpoE exists in A. baumannii with slightly different promoter recognition preferences. To rule out this possibility, we cloned three additional RpoE-dependent promoters from E. coli (PmicA, PrybB, and PyicJ) and tested their activity in A. baumannii using our reporter assay. Consistent with a lack of RpoE activity in A. baumannii, these promoters were inactive, unless E. coli rpoE was heterologously expressed (Fig. S5). These results demonstrate the ability to utilize our integrative and replicative expression systems together, in the same strain, to better understand the biology and gene function in both E. coli and A. baumannii.
Fig 5.
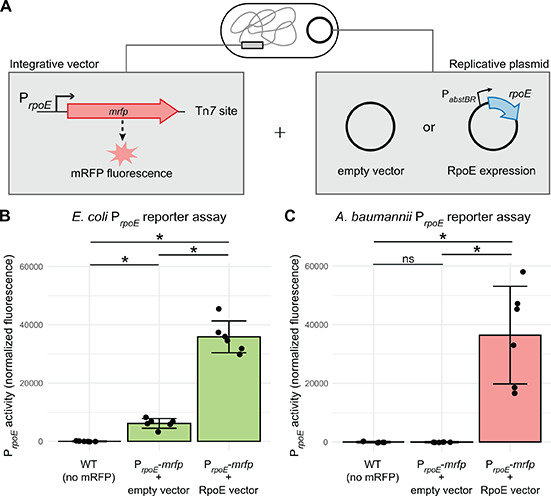
Modular integrative and replicative vectors facilitate a functional reporter assay. (A) Graphical depiction of reporter assay experiments. Strains contain an mRFP reporter under control of the E. coli–native rpoE promoter (PrpoE) in the attTn7 site (constructed using the Tn7 vector) and either a PabstBR-rpoE overexpression vector or empty vector control (replicative plasmid). (B and C) Bar graphs of mRFP fluorescence from PrpoE with and without the expression of RpoE in trans from the replicative plasmid in E. coli or A. baumannii. As RpoE is native to E. coli, the E. coli strains also carry a copy of the rpoE gene on the chromosome. Fluorescence is normalized to no mRFP controls, and individual data points and standard deviation are displayed (n = 6 biological replicates). Asterisks and ns indicate significant and nonsignificant sample differences, respectively (Welch’s t-tests; P-values < 0.05).
Conclusion
In this study, we have provided modular vectors that replicate and integrate into E. coli and A. baumannii and a titratable, IPTG-inducible promoter, PabstBR. We envision that our vectors will be valuable for complementation studies, particularly for comparing the function of genes in E. coli to those found in A. baumannii. We predict that our tools will allow for precise tuning of gene expression to achieve physiological or somewhat higher levels of expression, while avoiding toxicity from extreme high-level overexpression. As such, our vectors could also be used for expressing gene fusions with fluorescent proteins for localization studies. The high integration efficiencies make library-scale experiments possible, as we have previously shown for our Tn7-based CRISPR interference (CRISPRi) studies (9). Given the host ranges of our vector components, we expect our vectors to be broadly useful for gene function studies in Acinetobacter species not tested here, including multidrug-resistant isolates.
The tools we created in this study are not without limitations. Both the replicative and integrative systems described here use IPTG-inducible promoters that cannot be induced independently if both systems are employed in the same cell. Furthermore, PabstBR was induced to a lower level in AB5075 compared to 17978 and 19606. Although AB5075 is a resistant isolate and 17978/19606 are sensitive, we cannot say for certain if lower induction is a general feature of resistant strains or something peculiar to the AB5075 background because only three strain backgrounds were tested. As is the case with other Tn7-based integration systems (28), insertion of our integrative vectors downstream of glmS precludes the introduction of additional Tn7 insertions due to target site immunity. This could also be seen as a benefit when delivering a single genetic payload to the cell is critical (e.g., introduction of CRISPRi guide RNAs (9)).
MATERIALS AND METHODS
Strains and growth conditions
Strains are listed in Table S1. Escherichia coli and Acinetobacter baumannii were grown in Lennox lysogeny broth (LB) at 37°C shaking in a flask at 250 rpm, in a culture tube on a roller drum at max speed, in a 96-well plate shaking at 900 rpm, or in a plate reader with shaking (Tecan Infinite Mplex or Tecan Sunrise). The culture medium was solidified with 1.5% agar for growth on plates. Antibiotics were added when necessary: 100 µg/mL ampicillin (amp), 30 µg/mL kanamycin (kan), 50 µg/mL apramycin (apr), and 150 µg/mL hygromycin (hyg) for E. coli and 150 µg/mL carbenicillin (carb), 60 µg/mL kanamycin (kan), 100 µg/mL apramycin (apr), and 150 µg/mL hygromycin (hyg) for A. baumannii. Diaminopimelic acid (DAP) was added at 300 µM to support the growth of E. coli dap- donor strains. IPTG (isopropyl b-D-1-thiogalactopyranoside) was added at varying concentrations from 0 to 1 mM, as indicated in the figures or figure legends. Strains were preserved in 15% glycerol at −80°C. Plasmids were propagated in E. coli strain BW25141 attTn7::acrIIA4 (sJMP3053) or in E. coli strain DH10B (sJMP1) for DNA extraction and analysis or in E. coli strain WM6026 attTn7::acrIIA4 (sJMP3257) for conjugation.
General molecular biology techniques
A complete list of plasmids and oligonucleotides is given in Tables S2 and S3. Oligonucleotides were synthesized by Integrated DNA Technologies (Coralville, IA). Plasmid DNA was purified using the GeneJet Plasmid Miniprep kit (Thermo) or the Purelink HiPure Plasmid Midiprep kit (Invitrogen K210005). PCR was performed according to the manufacturer’s directions using Q5, OneTaq, or Phusion DNA Polymerases (NEB). DNA was digested with restriction enzymes from NEB. PCR products were purified with the DNA Spin and Concentrate kit (Zymo Research) following the manufacturer’s instructions or gel-purified from the kit (Zymo Research). Plasmids were assembled using the NEBuilder HiFi DNA assembly kit (NEB). DNA was quantified on a NanoDrop Lite or Qubit. Plasmids and recombinant strains were sequenced via Sanger sequencing by Functional Biosciences or Oxford Nanopore sequencing by Plasmidsaurus.
Construction of replicative expression vectors
Details for construction of expression vectors are listed under “Construction/notes” for corresponding vectors (Table S2). Briefly, base replicative expression plasmid construction was performed using HiFi assembly with (i) p15A origin of replication and oriT from pJMP3262, (ii) pWH1266 origin of replication from pJMP3347, (iii) pTrc99a plasmid base including lacI and MCS from pJMP3067, and (iv) kanR marker from pJMP3341 to create plasmid pJMP3649. To swap the promoters, pJMP3649 was cut with AatII and NcoI enzymes and HiFi-assembled with gblocks containing the desired promoters to create plasmids pJMP3651 (Pabst, kanR) and pJMP3653 (PabstBR, kanR). To swap the resistance markers, pJMP3653 was cut with the XhoI enzyme and HiFi-assembled with gblocks containing the desired resistance markers to create plasmids pJMP3664 (PabstBR, aprR) and pJMP3665 (PabstBR, hygR). To test the expression of genes from these vectors, the kanR versions of the vectors with Ptrc, Pabst, and PabstBR (pJMP3649, pJMP3651, and pJMP3653, respectively) were cut with NcoI and BamHI enzymes and HiFi-assembled with the sfgfp gene amplified from pJMP2748 to create plasmids pJMP3650, pJMP3652, and pJMP3654.
Construction of PabstBR
Site-directed mutagenesis of the Pabst promoter was performed by single-primer high-fidelity Phusion PCR using pJMP3407 and oJMP2167. The PCR product was treated with DpnI, electroporated into sJMP3053, and selected on kan to make plasmid pJMP4481 containing the PabstBR promoter. The mutation was verified by whole-plasmid sequencing with Plasmidsaurus.
Conjugative-based transfer of expression vectors
Replicative vector
Donor Dap- E. coli mating strain containing the desired replicative expression vector and recipient strain (A. baumannii or E. coli) were both scraped off an agar plate into LB at OD600 of ~3. Strains were mixed at equal ratios, placed on a 0.45-µm filter on an LB plate, and incubated upright at 37°C for ~3 hours. Filters were vortexed in LB media to remove cells and plated onto LB plates supplemented with appropriate antibiotics.
Tn7 integrating vector
Conjugation was performed similarly to above, except with the addition of a donor Dap- E. coli strain carrying a Tn7 transposase plasmid (tri-parental mating) for E. coli, A. baumannii ATCC 17978, and AB5075 strains. For A. baumannii ATCC 19606, quad-parental mating was performed, using an additional Dap- donor E. coli strain (sJMP4061) harboring a helper plasmid that contains additional mating machinery to improve the efficiency. Tn7 matings were performed for ~4 hours before plating on LB plates supplemented with the appropriate antibiotic.
Tenfold serial dilutions were spotted (10 µL) on LB and LB with antibiotics. Transfer efficiencies were calculated as transformants or transconjugants (colony-forming units or CFUs on selective plates) divided by total number of cells (CFUs on LB only).
Promoter activity assays
Promoter activities were assayed using the sfGFP expression vectors. Promoter-sfgfp or empty vector strains were grown to saturation in LB supplemented with appropriate antibiotic and IPTG inducer, washed several times with 1 x PBS to remove all media, and GFP fluorescence and OD600 were measured in a Tecan Infinite Mplex plate reader. Values were normalized to OD600 readings and were background-subtracted using empty vector cells.
Flow cytometry
Cells containing either a PabstBR-sfgfp vector or empty vector control were grown in LB supplemented with kan and varying concentrations of IPTG to either saturation or the mid-log growth phase in tubes. Cells were fixed with formaldehyde (5% final), washed, and resuspended in 1 x PBS. GFP fluorescence was measured by flow cytometry on an LSR Fortessa instrument (BD Biosciences) at ~100,000 events/sample. Data were analyzed in FlowJo (FlowJo, LLC) using singlet gates and dead cell or debris exclusion gates, as previously described (58). Gates were set using no IPTG controls and applied to the remaining samples.
Microscopy
Cells containing either a PabstBR-sfgfp vector or empty vector control were grown in LB supplemented with kan and varying concentrations of IPTG to the mid-log growth phase in tubes. Cells were fixed with formaldehyde (5% final), washed, and resuspended in 1 x PBS. Ten microliters of each sample was spotted on a glass slide for microscopy. Bacteria were imaged with a Nikon Ti-E inverted microscope with an Orca Fusion BT digital CMOS camera (Hamamatsu) using NIS-Elements. Fluorescence images were collected using the Prior Lumen 200 metal halide light source and a FITC-specific filter set. Fluorescence was calculated as mean intensity per cell using Fiji (59).
ACKNOWLEDGMENTS
We thank Colin Manoil for providing AB5075 WT strain and Quanjiang Ji for pSGAb-km (Addgene plasmid no. 121999). We also thank the UWCCC Flow Cytometry lab for equipment access and assistance (NIH Special BD LSR Fortessa Project 1S100OD018202-01).
This work was supported by the National Institutes of Health under award numbers K22AI137122 and 1R35GM150487-01. J.S.T. was funded by an NSF GRFP and the SciMed Graduate Research Scholars program.
Contributor Information
Jason M. Peters, Email: jason.peters@wisc.edu.
Philip N. Rather, Emory University School of Medicine, Atlanta, Georgia, USA
DATA AVAILABILITY
Plasmids and their sequences are available from Addgene under accession numbers 222348-222357. R code for data analysis and graphs can be found at https://github.com/jasonpeterslab/Aba-Eco-vectors-2024. Data available on request.
SUPPLEMENTAL MATERIAL
The following material is available online at https://doi.org/10.1128/spectrum.01306-24.
Fig. S1 to S5.
Tables S1 to S3.
ASM does not own the copyrights to Supplemental Material that may be linked to, or accessed through, an article. The authors have granted ASM a non-exclusive, world-wide license to publish the Supplemental Material files. Please contact the corresponding author directly for reuse.
REFERENCES
- 1. Blount ZD. 2015. The unexhausted potential of E. coli. Elife 4:e05826. doi: 10.7554/eLife.05826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Nora LC, Westmann CA, Guazzaroni ME, Siddaiah C, Gupta VK, Silva-Rocha R. 2019. Recent advances in plasmid-based tools for establishing novel microbial chassis. Biotechnol Adv 37:107433. doi: 10.1016/j.biotechadv.2019.107433 [DOI] [PubMed] [Google Scholar]
- 3. Ruiz N, Silhavy TJ. 2022. How Escherichia coli became the flagship bacterium of molecular biology. J Bacteriol 204:e0023022. doi: 10.1128/jb.00230-22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Hanage WP. 2016. Not so simple after all: bacteria, their population genetics, and recombination. Cold Spring Harb Perspect Biol 8:a018069. doi: 10.1101/cshperspect.a018069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Ramon C, Stelling J. 2023. Functional comparison of metabolic networks across species. Nat Commun 14:1699. doi: 10.1038/s41467-023-37429-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Peleg AY, Seifert H, Paterson DL. 2008. Acinetobacter baumannii: emergence of a successful pathogen. Clin Microbiol Rev 21:538–582. doi: 10.1128/CMR.00058-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Nowak J, Zander E, Stefanik D, Higgins PG, Roca I, Vila J, McConnell MJ, Cisneros JM, Seifert H, MagicBullet Working Group WP4 . 2017. High incidence of pandrug-resistant Acinetobacter baumannii isolates collected from patients with ventilator-associated pneumonia in Greece, Italy and Spain as part of the MagicBullet clinical trial. J Antimicrob Chemother 72:3277–3282. doi: 10.1093/jac/dkx322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Wong D, Nielsen TB, Bonomo RA, Pantapalangkoor P, Luna B, Spellberg B. 2017. Clinical and pathophysiological overview of acinetobacter infections: a century of challenges. Clin Microbiol Rev 30:409–447. doi: 10.1128/CMR.00058-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Peters JM, Koo BM, Patino R, Heussler GE, Hearne CC, Qu J, Inclan YF, Hawkins JS, Lu CHS, Silvis MR, Harden MM, Osadnik H, Peters JE, Engel JN, Dutton RJ, Grossman AD, Gross CA, Rosenberg OS. 2019. Enabling genetic analysis of diverse bacteria with Mobile-CRISPRi. Nat Microbiol 4:244–250. doi: 10.1038/s41564-018-0327-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Lucidi M, Runci F, Rampioni G, Frangipani E, Leoni L, Visca P. 2018. New shuttle vectors for gene cloning and expression in multidrug-resistant Acinetobacter species. Antimicrob Agents Chemother 62:e02480-17. doi: 10.1128/AAC.02480-17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Jie J, Chu X, Li D, Luo Z. 2021. A set of shuttle plasmids for gene expression in Acinetobacter baumannii. PLoS One 16:e0246918. doi: 10.1371/journal.pone.0246918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Fürste JP, Pansegrau W, Frank R, Blöcker H, Scholz P, Bagdasarian M, Lanka E. 1986. Molecular cloning of the plasmid RP4 primase region in a multi-host-range tacP expression vector. Gene 48:119–131. doi: 10.1016/0378-1119(86)90358-6 [DOI] [PubMed] [Google Scholar]
- 13. Tucker AT, Nowicki EM, Boll JM, Knauf GA, Burdis NC, Trent MS, Davies BW. 2014. Defining gene-phenotype relationships in Acinetobacter baumannii through one-step chromosomal gene inactivation. MBio 5:e01313-14. doi: 10.1128/mBio.01313-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Banta AB, Myers KS, Ward RD, Cuellar RA, Place M, Freeh CC, Bacon EE, Peters JM. 2024. A targeted genome-scale overexpression platform for proteobacteria. bioRxiv. doi: 10.1101/2024.03.01.582922 [DOI]
- 15. Anonymous . 2019. Antibiotic resistance threats in the United States, 2019
- 16. Zampaloni C, Mattei P, Bleicher K, Winther L, Thäte C, Bucher C, Adam J-M, Alanine A, Amrein KE, Baidin V, et al. 2024. A novel antibiotic class targeting the lipopolysaccharide transporter. Nature New Biol 625:566–571. doi: 10.1038/s41586-023-06873-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Falcone M, Tiseo G, Leonildi A, Della Sala L, Vecchione A, Barnini S, Farcomeni A, Menichetti F. 2022. Cefiderocol-compared to colistin-based regimens for the treatment of severe infections caused by carbapenem-resistant Acinetobacter baumannii. Antimicrob Agents Chemother 66:e0214221. doi: 10.1128/aac.02142-21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Wang H, Ishchenko A, Skudlarek J, Shen P, Dzhekieva L, Painter RE, Chen Y-T, Bukhtiyarova M, Leithead A, Tracy R, et al. 2024. Cerastecins inhibit membrane lipooligosaccharide transport in drug-resistant Acinetobacter baumannii. Nat Microbiol 9:1244–1255. doi: 10.1038/s41564-024-01667-0 [DOI] [PubMed] [Google Scholar]
- 19. Héritier C, Poirel L, Lambert T, Nordmann P. 2005. Contribution of acquired carbapenem-hydrolyzing oxacillinases to carbapenem resistance in Acinetobacter baumannii. Antimicrob Agents Chemother 49:3198–3202. doi: 10.1128/AAC.49.8.3198-3202.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Lupo A, Haenni M, Madec JY. 2018. Antimicrobial resistance in Acinetobacter spp. and Pseudomonas spp. Microbiol Spectr 6. doi: 10.1128/microbiolspec.ARBA-0007-2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Valentine SC, Contreras D, Tan S, Real LJ, Chu S, Xu HH. 2008. Phenotypic and molecular characterization of Acinetobacter baumannii clinical isolates from nosocomial outbreaks in Los Angeles County, California. J Clin Microbiol 46:2499–2507. doi: 10.1128/JCM.00367-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Zhao J, Zhu Y, Han J, Lin YW, Aichem M, Wang J, Chen K, Velkov T, Schreiber F, Li J. 2020. Genome-scale metabolic modeling reveals metabolic alterations of multidrug-resistant Acinetobacter baumannii in a murine bloodstream infection model. Microorganisms 8:1793. doi: 10.3390/microorganisms8111793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Moffatt JH, Harper M, Harrison P, Hale JDF, Vinogradov E, Seemann T, Henry R, Crane B, St Michael F, Cox AD, Adler B, Nation RL, Li J, Boyce JD. 2010. Colistin resistance in Acinetobacter baumannii is mediated by complete loss of lipopolysaccharide production. Antimicrob Agents Chemother 54:4971–4977. doi: 10.1128/AAC.00834-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Moran AP. 2009. Microbial glycobiology: structures, relevance and applications. Elsevier. [Google Scholar]
- 25. Boll JM, Crofts AA, Peters K, Cattoir V, Vollmer W, Davies BW, Trent MS. 2016. A penicillin-binding protein inhibits selection of colistin-resistant, lipooligosaccharide-deficient Acinetobacter baumannii. Proc Natl Acad Sci U S A 113:E6228–E6237. doi: 10.1073/pnas.1611594113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Robinson A, Brzoska AJ, Turner KM, Withers R, Harry EJ, Lewis PJ, Dixon NE. 2010. Essential biological processes of an emerging pathogen: DNA replication, transcription, and cell division in Acinetobacter spp. Microbiol Mol Biol Rev 74:273–297. doi: 10.1128/MMBR.00048-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Casella LG, Weiss A, Pérez-Rueda E, Antonio Ibarra J, Shaw LN. 2017. Towards the complete proteinaceous regulome of Acinetobacter baumannii. Microb Genom 3:mgen000107. doi: 10.1099/mgen.0.000107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. McKenzie GJ, Craig NL. 2006. Fast, easy and efficient: site-specific insertion of transgenes into enterobacterial chromosomes using Tn7 without need for selection of the insertion event. BMC Microbiol 6:39. doi: 10.1186/1471-2180-6-39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Sykes EME, Deo S, Kumar A. 2020. Recent advances in genetic tools for Acinetobacter baumannii. Front Genet 11:601380. doi: 10.3389/fgene.2020.601380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Pérez-Varela M, Tierney ARP, Kim JS, Vázquez-Torres A, Rather P. 2020. Characterization of RelA in Acinetobacter baumannii. J Bacteriol 202:e00045-20. doi: 10.1128/JB.00045-20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Ward RD, Tran JS, Banta AB, Bacon EE, Rose WE, Peters JM. 2024. Essential gene knockdowns reveal genetic vulnerabilities and antibiotic sensitivities in Acinetobacter baumannii MBio 15:e0205123. doi: 10.1128/mbio.02051-23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Choi KH, Gaynor JB, White KG, Lopez C, Bosio CM, Karkhoff-Schweizer RR, Schweizer HP. 2005. A Tn7-based broad-range bacterial cloning and expression system. Nat Methods 2:443–448. doi: 10.1038/nmeth765 [DOI] [PubMed] [Google Scholar]
- 33. Banta AB, Enright AL, Siletti C, Peters JM. 2020. A high-efficacy CRISPR interference system for gene function discoveryin Zymomonas mobilis. Appl Environ Microbiol 86:e01621-20. doi: 10.1128/AEM.01621-20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Terpe K. 2006. Overview of bacterial expression systems for heterologous protein production: from molecular and biochemical fundamentals to commercial systems. Appl Microbiol Biotechnol 72:211–222. doi: 10.1007/s00253-006-0465-8 [DOI] [PubMed] [Google Scholar]
- 35. Lozano Terol G, Gallego-Jara J, Sola Martínez RA, Martínez Vivancos A, Cánovas Díaz M, de Diego Puente T. 2021. Impact of the expression system on recombinant protein production in Escherichia coli BL21. Front Microbiol 12:682001. doi: 10.3389/fmicb.2021.682001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Müller-Hill B, Crapo L, Gilbert W. 1968. Mutants that make more lac repressor. Proc Natl Acad Sci U S A 59:1259–1264. doi: 10.1073/pnas.59.4.1259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Siegele DA, Hu JC. 1997. Gene expression from plasmids containing the araBAD promoter at subsaturating inducer concentrations represents mixed populations. Proc Natl Acad Sci U S A 94:8168–8172. doi: 10.1073/pnas.94.15.8168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Mäkelä J, Kandhavelu M, Oliveira SMD, Chandraseelan JG, Lloyd-Price J, Peltonen J, Yli-Harja O, Ribeiro AS. 2013. In vivo single-molecule kinetics of activation and subsequent activity of the arabinose promoter. Nucleic Acids Res 41:6544–6552. doi: 10.1093/nar/gkt350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Megerle JA, Fritz G, Gerland U, Jung K, Rädler JO. 2008. Timing and dynamics of single cell gene expression in the arabinose utilization system. Biophys J 95:2103–2115. doi: 10.1529/biophysj.107.127191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Chaperon DN. 2006. Construction and complementation of in-frame deletions of the essential Escherichia coli thymidylate kinase gene. Appl Environ Microbiol 72:1288–1294. doi: 10.1128/AEM.72.2.1288-1294.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Mutsuda M, Michel KP, Zhang X, Montgomery BL, Golden SS. 2003. Biochemical properties of CikA, an unusual phytochrome-like histidine protein kinase that resets the circadian clock in Synechococcus elongatus PCC 7942. J Biol Chem 278:19102–19110. doi: 10.1074/jbc.M213255200 [DOI] [PubMed] [Google Scholar]
- 42. Hannig G, Makrides SC. 1998. Strategies for optimizing heterologous protein expression in Escherichia coli. Trends Biotechnol 16:54–60. doi: 10.1016/s0167-7799(97)01155-4 [DOI] [PubMed] [Google Scholar]
- 43. Lutz R, Bujard H. 1997. Independent and tight regulation of transcriptional units in Escherichia coli via the LacR/O, the TetR/O and AraC/I1-I2 regulatory elements. Nucleic Acids Res 25:1203–1210. doi: 10.1093/nar/25.6.1203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Anderson SE, Chin CY, Weiss DS, Rather PN. 2020. Copy number of an integron-encoded antibiotic resistance locus regulates a virulence and opacity switch in Acinetobacter baumannii AB5075. MBio 11:e02338-20. doi: 10.1128/mBio.02338-20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Gibson DG, Young L, Chuang R-Y, Venter JC, Hutchison CA III, Smith HO. 2009. Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat Methods 6:343–345. doi: 10.1038/nmeth.1318 [DOI] [PubMed] [Google Scholar]
- 46. Drew RH. 2022. Aminoglycosides. Wolters Kluwe, UpToDate. [Google Scholar]
- 47. Hoang TT, Karkhoff-Schweizer RR, Kutchma AJ, Schweizer HP. 1998. A broad-host-range Flp-FRT recombination system for site-specific excision of chromosomally-located DNA sequences: application for isolation of unmarked Pseudomonas aeruginosa mutants. Gene 212:77–86. doi: 10.1016/s0378-1119(98)00130-9 [DOI] [PubMed] [Google Scholar]
- 48. Choi KH, Schweizer HP. 2006. mini-Tn7 insertion in bacteria with single attTn7 sites: example Pseudomonas aeruginosa. Nat Protoc 1:153–161. doi: 10.1038/nprot.2006.24 [DOI] [PubMed] [Google Scholar]
- 49. Amann E, Ochs B, Abel KJ. 1988. Tightly regulated tac promoter vectors useful for the expression of unfused and fused proteins in Escherichia coli. Gene 69:301–315. doi: 10.1016/0378-1119(88)90440-4 [DOI] [PubMed] [Google Scholar]
- 50. May EW, Craig NL. 1996. Switching from cut-and-paste to replicative Tn7 transposition. Science 272:401–404. doi: 10.1126/science.272.5260.401 [DOI] [PubMed] [Google Scholar]
- 51. Wen X, Zhang Y, Cheng H, An J, Guo Y, Wang L, Wang M. 2021. A CRISPR/dCas9-assisted system to clone toxic genes in Escherichia coli. Biochim Biophys Acta 1865:129994. doi: 10.1016/j.bbagen.2021.129994 [DOI] [PubMed] [Google Scholar]
- 52. Tal S, Paulsson J. 2012. Evaluating quantitative methods for measuring plasmid copy numbers in single cells. Plasmid 67:167–173. doi: 10.1016/j.plasmid.2012.01.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Rhodius VA, Suh WC, Nonaka G, West J, Gross CA. 2006. Conserved and variable functions of the σE stress response in related genomes. PLoS Biol 4:e2. doi: 10.1371/journal.pbio.0040002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Lima S, Guo MS, Chaba R, Gross CA, Sauer RT. 2013. Dual molecular signals mediate the bacterial response to outer-membrane stress. Science 340:837–841. doi: 10.1126/science.1235358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Firoved AM, Boucher JC, Deretic V. 2002. Global genomic analysis of AlgU (σE)-dependent promoters (sigmulon) in Pseudomonas aeruginosa and implications for inflammatory processes in cystic fibrosis. J Bacteriol 184:1057–1064. doi: 10.1128/jb.184.4.1057-1064.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Mathur J, Davis BM, Waldor MK. 2007. Antimicrobial peptides activate the Vibrio cholerae σE regulon through an OmpU-dependent signalling pathway. Mol Microbiol 63:848–858. doi: 10.1111/j.1365-2958.2006.05544.x [DOI] [PubMed] [Google Scholar]
- 57. Schulz S, Eckweiler D, Bielecka A, Nicolai T, Franke R, Dötsch A, Hornischer K, Bruchmann S, Düvel J, Häussler S. 2015. Elucidation of sigma factor-associated networks in Pseudomonas aeruginosa reveals a modular architecture with limited and function-specific crosstalk. PLoS Pathog 11:e1004744. doi: 10.1371/journal.ppat.1004744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Staats J, Divekar A, McCoy JP, Maecker HT. 2019. Guidelines for gating flow cytometry data for immunological assays, p 81–104. In McCoy JJP (ed), Immunophenotyping: methods and protocols. Springer New York, New York, NY. [DOI] [PubMed] [Google Scholar]
- 59. Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, Tinevez JY, White DJ, Hartenstein V, Eliceiri K, Tomancak P, Cardona A. 2012. Fiji: an open-source platform for biological-image analysis. Nat Methods 9:676–682. doi: 10.1038/nmeth.2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1 to S5.
Tables S1 to S3.
Data Availability Statement
Plasmids and their sequences are available from Addgene under accession numbers 222348-222357. R code for data analysis and graphs can be found at https://github.com/jasonpeterslab/Aba-Eco-vectors-2024. Data available on request.