

Abstract
Objective:
Elevated serum CGA is associated with intrinsic or treatment-related neuroendocrine differentiation (NED) in men with mCRPC. Fluctuations in serum CGA during treatment of mCRPC have had conflicting results. We analyzed the impact of (i) rising serum CGA and (ii) baseline CGA/PSA ratio during treatment to identify associations with AA therapy.
Methods:
Between June 2013 and August 2015, 92 men with mCRPC were enrolled in a prospective trial with uniform serum CGA processing performed before initiating abiraterone acetate/prednisone (AA/P) and serially after 12 weeks of AA/P treatments. Serum CGA was measured using a homogenous automated immunofluorescent assay. Patients receiving proton pump inhibitors or with abnormal renal function were excluded due to possible false elevations of serum CGA (n=21 excluded), therefore 71 patients were analyzed. All patients underwent a composite response assessment at 12-weeks. Kaplan Meier estimates and Cox Regression models were used to calculate the association with time-to-treatment failure analyses and overall survival.
Results:
An increase in chromogranin was associated with a lower risk of treatment failure (HR: 0.52, p= 0.0181). The median CGA/PSA ratio was 7.8 (2.6 -16.0) and an elevated pretreatment CGA/PSA ratio above the median was associated with a lower risk of treatment failure (HR: 0.54 p value=0.0185). An increase in CGA was not found to be associated with OS (HR: 0.71, 95% CI: 0.42–1.21, p = 0.207). An elevated baseline CGA/PSA ratio was not associated with OS (HR: 0.62, 95% CI: 0.37–1.03, p = 0.062). An increase in PSA after 12 weeks of treatment was associated with an increased risk of treatment failure (HR: 4.14, CI: 2.21–7.73, p = < 0.0001) and worse OS (HR: 2.93, CI: 1.57–4.45, p = < 0.0001).
Conclusions:
We show that an increasing chromogranin on AA/P and an elevated baseline CGA/PSA in patients with mCRPC were associated with a favorable response to AA/P with no changes in survival. There may be limited clinical utility in serum CGA testing to evaluate for lethal NED as AA/P did not induce lethal NED in this cohort. This highlights that not all patients with an increasing CGA have a worse OS.
Keywords: Castrate resistant, Prostate cancer, Chromogranin, Abiraterone acetate
Introduction
Androgen deprivation therapy (ADT) is the cornerstone treatment for men diagnosed with metastatic prostate cancer. After prolonged suppression of androgen receptor (AR) signaling during ADT, prostate cancer cells inevitably develop resistance to ADT and metastatic castrate-resistant prostate cancer (mCRPC) emerges. In mCRPC, neuroendocrine differentiation (NED) is a major factor associated with resistance to ADT1. Preclinical studies demonstrate that under the selective pressure of androgen targeted therapies, transdifferentiation into neuroendocrine prostate cancer (NEPC) occurs 2–4. Proposed mechanisms for transdifferentiation into NEPC involve overexpression of Aurora kinase A (AURKA ) and MYCN, downregulation of RE1-silencing transcription factor (REST), and loss of RB1.
Chromogranin-A (CGA), a 49 kDa protein produced in neuroendocrine cells, is a potential blood biomarker of NED. Neuroendocrine cells do not typically secrete PSA5 and the balance between PSA and CGA may be a surrogate for treatment emergent NEPC, therefore the ratio of CGA/PSA can be a useful novel biomarker for predicting neuroendocrine differentiation. Multiple studies identified that elevated CGA was associated adversely with progression free survival and overall survival 6–14. Fluctuations in serum CGA during mCRPC therapy are less well known with conflicting results 12,15–17.
The association of serial chromogranin with treatment failure has not been previously evaluated. Using a prospective cohort, we demonstrated that elevated baseline serum CGA was negatively associated with OS in men with mCRPC7. Here we analyzed the impact of (i) rising serum CGA and (ii) baseline CGA/PSA ratio during treatment to identify associations with AA/P therapy.
Materials and Methods
Patient selection:
In a multi-site prospective clinical and biospecimen collection study, 92 men with mCRPC were enrolled between June 2013 and August 2015 (NCT#01953640). Patients received only ADT as systemic therapy before enrollment. Eligibility criteria included men who had a histologic diagnosis of metastatic adenocarcinoma of the prostate and progressed on ADT [defined as at least a 20% increase in the sum of the longest diameters of measurable lesions, the appearance of new measurable lesions in lymph nodes (≥2.0 cm), viscera or soft tissue (≥1.0 cm), appearance of 2 or more new lesions on bone scan while on ADT, two consecutive increases in PSA levels documented over a previous reference value obtained at least one week apart]. Only men with sub-castrate levels of total testosterone (defined as ≤50 ng/dl) were included. Patients with high volume disease were defined as visceral metastases or greater than 4 bony metastases, 1 outside the axial skeleton.
Blood samples were collected for serum PSA and CGA after written informed consent was provided. After the first samples were collected, all patients were started on abiraterone acetate and prednisone (AA/P) and after 12 weeks of treatment with AA/P, a second set of blood samples was collected. Patients who received PPIs or with impaired renal and hepatic function were excluded, as artificial elevations in chromogranin-A levels have been reported previously with these conditions18.
Specimen methods:
After consent was obtained from the cohort, 6.0 mL blood was collected in BD SST™ vacutainers and centrifuged at 3000 r.p.m. for 10 min to generate serum. The serum was then separated into multiple aliquots and stored at −80°C. To maintain serum CGA stability, a protease inhibitor cocktail (10 mL PBS (Invitrogen No. 14190300);1 tablet complete of mini, EDTA-free protease inhibitor (Roche No. 11 836 170 001); Sodium Vanadate Na3VO4 and PMSF (Sigma No. P7626–5G)) was added after initial centrifugation dissolved in 5 μL/mL Na3VO4 with10 μL/mL PMSF and 10 mL PBS. This was then added to tick solution preparation of which 50 μL was added to each serum specimen for all assays.
Assay methods:
Serum chromogranin A (CGA) was measured in a homogeneous automated immunofluorescent assay. This assay is based on a variant of Forster resonance energy transfer called time-resolved amplified cryptate emission (TRACE). The TRACE assay used a chromogranin reference range of ≤ 93 ng/mL. CGA was sandwiched between the 2 antibodies labeled with europium cryptate (TRACE donor) and Alexa Fluor 647 (TRACE acceptor), bringing them into proximity. The antigen-antibody complex was excited with a nitrogen laser at 337 nm, with some fluorescent energy emitted at 620 nm and the rest emitted as fluorescence at 647 nm. A ratio of the energy emitted at 647 nm to that emitted at 620 nm is calculated for each sample. Signal intensity is proportional to the number of antigen-antibody complexes formed, and therefore to antigen concentration.
The serum PSA assay used a sandwich electrochemiluminescence immunoassay that employed a biotinylated monoclonal PSA-specific antibody and a monoclonal PSA-specific antibody labeled with ruthenium, forming a sandwich complex. Streptavidin-coated microparticles were added and the mixture was aspirated into the measuring cell where the microparticles are magnetically captured onto the surface of the electrode and the unbound substances were removed with ProCell. The application of voltage to the electrode induced the chemiluminescent emission, which was measured against a calibration curve to determine the amount of PSA in the specimens. The reference range for PSA in this assay was ≤ 7.2 ng/mL.
Statistical methods:
A change in CGA was defined as an absolute increase or decrease from the baseline CGA value. The median CGA/PSA was used as a cutpoint to evaluate for a change in the CGA/PSA ratio. The predetermined primary endpoints of this analysis were the association of serial serum chromogranin and CGA/PSA ratio with treatment failure (TTF), defined as the time from treatment initiation to AA/P discontinuation for any reason, and the coprimary endpoint was OS defined as the time from study enrolment to death or last follow-up. Cox proportional hazard regression analysis was performed on clinical parameters, change in CGA and CGA/PSA ratio for associations with OS and TTF. Kaplan–Meier method was used to compare an increase in CGA versus no increase in CGA to estimate the OS and TTF, with 95% confidence intervals using BlueSky Statistics (Chicago, IL, USA).
Results:
Patient characteristics
92 men were enrolled with CRPC, with 21 excluded due to PPI use or GFR <30. The median age was 71 years, and 35 (49%) patients had a Gleason score >=8. High volume disease was found in 39 patients. The median pretreatment CGA and PSA were 83 ng/ml (interquartile range (IQ): 53 – 127ng/ml) and 14.4 ng/ml (IQ: 6.4 – 40.2 ng/ml) respectively (Table 1). In univariate analysis, age, Gleason score, and volume of metastatic disease had no associations with OS.
Table: 1:
Clinical and laboratory characteristics of the patient cohort
| Overall | Overall (N=71) |
|---|---|
|
| |
| Age | |
| Median (IQ range) | 72.0 (65.5, 78.0) |
|
| |
| Baseline CGA | |
| Median (IQ range) | 83.0 (53.0, 127.0) |
|
| |
| CGA at 12 weeks post AA/P | |
| N-Miss | 4 |
| Median (IQ range) | 98.0 (58.0, 148.0) |
|
| |
| Change in CGA | |
| N-Miss | 4 |
| Decrease | 26/67 (38.8%) |
| Increase | 41/67 (61.2%) |
|
| |
| Baseline PSA | |
| Median (IQ range) | 14.45 (6.35, 40.18) |
|
| |
| PSA at 12 weeks post AA/P | |
| N-Miss | 1 |
| Median (IQ range) | 6.15 (0.89, 24.66) |
| Change in PSA | |
| N-Miss | 1 |
| Decrease | 54/70 (77.1%) |
| Increase | 16/70 (22.9%) |
|
| |
| Baseline CGA/PSA above or below the mean | |
| Below | 35/71 (49.3%) |
| Above | 36/71 (50.7%) |
|
| |
| Volume of metastatic disease at enrollment | |
| Low | 32/71 (45.1%) |
| High | 39/71 (54.9%) |
|
| |
| Gleason score ≥ 8 vs < 8 | |
| < 8 | 36/71 (50.7%) |
| ≥ 8 | 35/71 (49.3%) |
|
| |
| Second line treatment | |
| N-Miss | 15 |
| Docetaxel | 26/56 (46.4%) |
| Enzalutamide | 22/56 (39.3%) |
| Radium- 223 | 3/56 (5.4%) |
| Other | 5/56 (8.9%) |
Changes in Chromogranin A and PSA levels during treatment
In the subset of 67 men with an evaluable post-treatment CGA, the median CGA after 12 weeks of AA/P rose by 15 ng/mL to 98.0 ng/mL (58.0 -148.0 ng/mL). We observed a CGA rise in 41/67 (61.2%) patients (Figure 1). In this cohort, the median PSA after 12 weeks AA/P decreased by 8.35ng/mL to 6.15ng/mL (0.89 – 24.66ng/mL) and we observed a PSA rise in 16/70 (22.9%) patients (Figure 1). There was no demonstrated correlation between serum CGA levels and PSA (pearson= 0.642). The median baseline CGA/PSA ratio was 7.8. Thirty-five patients had a baseline CGA/PSA ratio >7.8 and of the 67 patients who had an available post treatment CGA and PSA, 50 (74.6%) had an increase in the CGA/PSA ratio.
Figure 1:
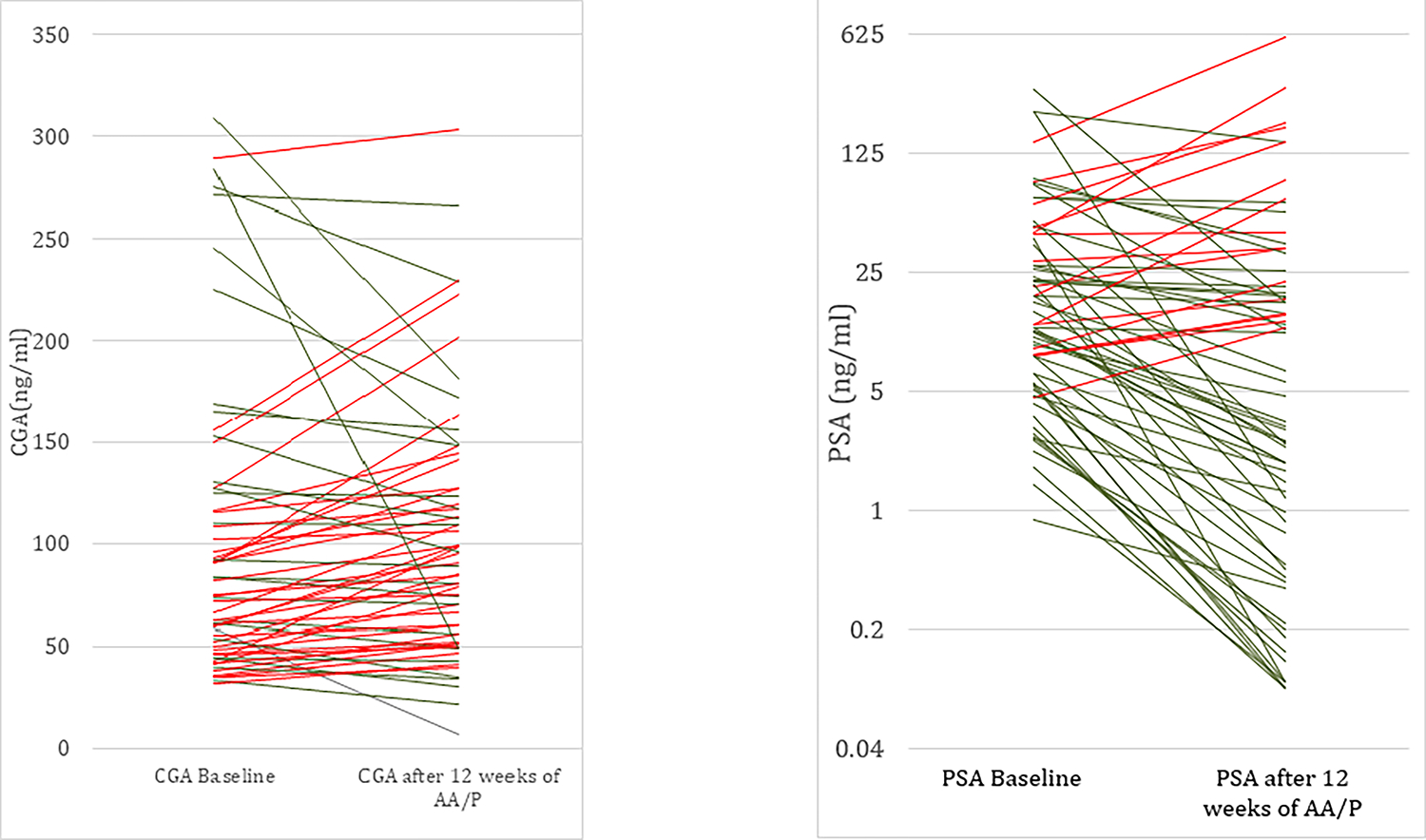
Parallel plots representation of CGA and PSA before and after 12 weeks of treatment with AA/P. (Red line: increase in CGA or PSA, Green line: decrease in CGA or PSA).
Serial chromogranin measurement and association with time to treatment failure
The median TTF was 10 months (IQ: 8 – 14 months) with 58/67 (87%) patients discontinuing treatment. The most common cause of treatment discontinuation was disease progression 55/67 (82%). A rise in CGA after 12 weeks of AA/P was associated with a lower risk of TTF (n=67; HR: 0.52, 95% CI: 0.30–0.89, p = 0.018). An elevated baseline CGA/PSA ratio was also associated with a lower risk of treatment failure (HR: 0.51, 95% CI: 0.30–0.86, p = 0.011) (Figure 2). An increase in PSA was associated with an increased risk of treatment failure (HR: 4.14, CI: 2.21–7.73, p = < 0.0001) (Table 2).
Figure 2:
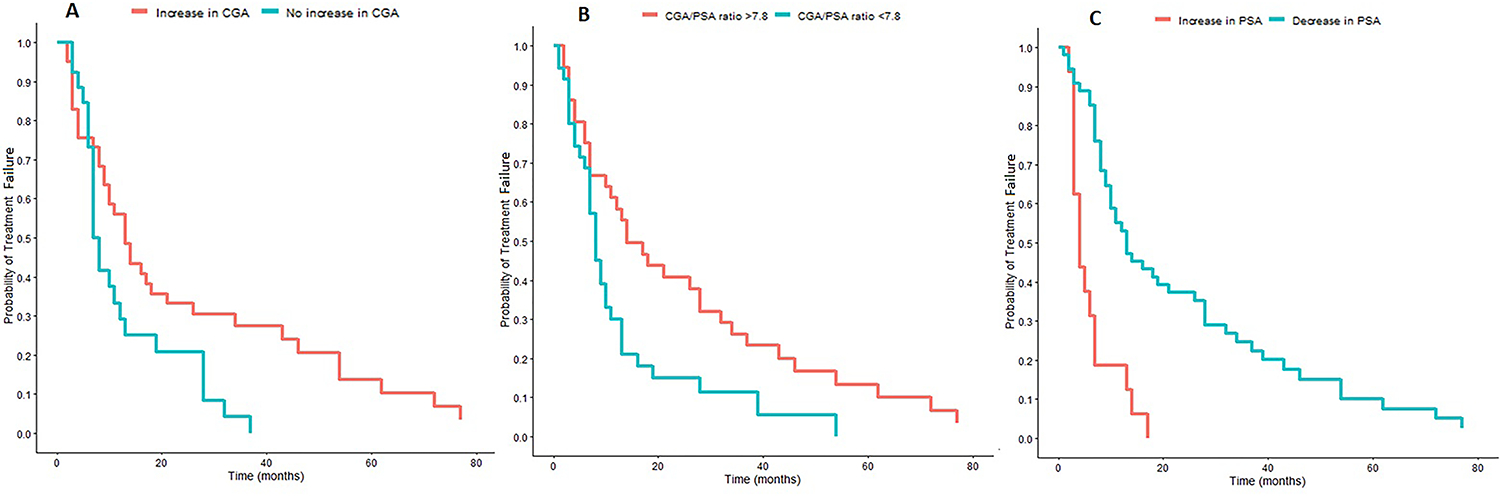
Kaplan–Meier curves demonstrating (A) change in CGA, (B) baseline CGA/PSA ratio above or below the median, and (C) change in PSA and association with TTF.
Table 2:
Cox regression univariate analysis for associations of clinical and laboratory parameters with time to treatment failure
| Cohort | Term | HR | P value | CI lower | CI upper |
|---|---|---|---|---|---|
| Study Cohort | Age | 1.01 | 0.55 | 0.98 | 1.04 |
| Increase in CGA | 0.52 | 0.0181 | 0.3 | 0.89 | |
| Increase in PSA | 4.14 | < 0.0001 | 2.21 | 7.73 | |
| Baseline GA/PSA ratio >7.8 | 0.51 | 0.011 | 0.3 | 0.86 | |
| High volume of metastatic disease | 1.1 | 0.710 | 0.67 | 1.79 | |
| Gleason score ≥ 8 | 1.28 | 0.332 | 0.78 | 2.09 |
Serial chromogranin measurement and association with overall survival
At a median follow-up of 6.4 years, 36/71 (51%) patients had died. An increase in post-treatment CGA was not found to be associated with OS (n=67; HR: 0.71, 95% CI: 0.42–1.21, p = 0.207). An elevated baseline CGA/PSA ratio was also not associated with OS (HR: 0.62, 95% CI: 0.37–1.03, p = 0.062). An increase in PSA was associated with a worse OS (HR: 2.93, CI: 1.57–4.45, p = < 0.0001) (Table 3).
Table 3:
Cox regression univariate analysis for associations of clinical and laboratory parameters with overall survival
| Cohort | Term | HR | P value | CI lower | CI upper |
|---|---|---|---|---|---|
| Study Cohort | Age | 1.03 | 0.064 | 1 | 1.07 |
| Increase in CGA | 0.71 | 0.208 | 0.42 | 1.21 | |
| Increase in PSA | 2.93 | 0.001 | 1.57 | 5.45 | |
| Baseline GA/PSA ratio >7.8 | 0.61 | 0.065 | 0.37 | 1.03 | |
| High volume of metastatic disease | 1.28 | 0.346 | 0.61 | 1.68 | |
| Gleason score ≥ 8 | 1.03 | 0.064 | 1 | 1.07 |
Discussion
Treatment emergent neuroendocrine differentiation in the setting of mCRPC is associated with a poor prognosis 1. A decrease in AR regulated protein expression and an increase in neural and neuroendocrine pathways is one mechanism of resistance 4. Based on previous clinical studies, it is thought that neuroendocrine serum marker levels may fluctuate during therapy and could therefore predict acquired resistance to novel therapies due to NED 4,12,14,15. In addition, elevation of serum CGA has shown concordant results with detection of NED in tumor tissue19,20 As CGA is produced in neuroendocrine cells, we hypothesized that a rising CGA may indicate early treatment emergent neuroendocrine transdifferentiation and is associated with poorer response to AA/P.
Our results indicate the inverse relationship. We demonstrate that rising CGA levels after 12 weeks of treatment with AA/P was associated with a lower risk of treatment failure. Interestingly a CGA/PSA ratio above the median was also associated with a lower risk of treatment failure. We believe that this decreased risk of treatment failure and rise in chromogranin is due to AA/P exerting selective pressure on tumor cells towards neuroendocrine differentiation while treating the predominant androgen-sensitive cells. This is due to the biological heterogeneity in the treatment of prostate adenocarcinoma, as characteristic molecular alterations seen in NEPC such as induction of neuroendocrine and neural programs and loss of androgen-regulated protein expression can also be seen in AR-driven CRPC4. It is also important to note that neuroendocrine differentiation is only one of the possible mechanisms of resistance to AA/P with AR splice variants, CYP17A1 upregulation, alternative ligand synthesis, or DNA repair alterations being other possible causes21,22.
We subsequently evaluated the change in chromogranin on OS in this cohort. Our data shows that an increase in chromogranin early into AA/P was not associated with a difference in OS. This was also seen when we evaluated the baseline CGA/PSA ratio. These findings are in contrast to 3 previous studies that showed increased serum CGA or in combination with NSE were associated with adverse PFS or OS12,14,15. However, the clinical heterogeneity between these studies makes comparison difficult. Fan et al used a LDN ELISA Kit for serum CGA, several patients were pre-treated with docetaxel and their Kaplan Myer estimates showed prolonged survival in patients with increasing CGA, with the opposite seen in univariate and multivariate analysis. Szarvas et al used KRYPTOR assay similar to this study, however patients were pre-treated with various agents before starting enzalutamide (ENZ)/AA/P. We attempted to validate their findings which showed a decreased OS with a 20% increase and a 50% increase in CGA, but we could not detect any correlations between these marker level changes and OS (Supplementary figure 3). Dong et al reported an adverse PSA-PFS and radiographic PFS with increasing CGA after 6 months of treatment with AA/P, however AA/P and the duration of AA/P treatment were not independent factors influencing the expression of NED. This again highlights that the influence of AA/P on NED of different patients might be diverse, as there was no significant difference of NED markers at baseline and after the failure of AA/P treatment in their study12. This data was also consistent with Von Harengberg et al who found the treatment of AA/P and the duration of AA/P treatment in 16 chemotherapy naive patients with CRPC were not independent factors influencing the expression of NED17.
Our clinical findings likely reflect the biological plasticity of mCRPC and neuroendocrine differentiation as a resistance mechanism. Although most patients had an increase in CGA, an increase in PSA after 12 weeks of AA/P was associated with a worse OS. This increase in CGA during treatment with AA/P was also demonstrated by Fan et al in which 52.5% of patients had an increase in serum CGA after 3 months of treatment with AA/P15. However, in this study an increase in CGA trended to a worse OS. The neuroendocrine or neuroendocrine-like tumor cells in prostate adenocarcinoma can sometimes have a gain or loss in expression of neuroendocrine markers with even reappearance of epithelial morphology owing to the diverse nature of these cells under androgen deprivation23–25. Neuroendocrine tumor cell heterogeneity in prostate cancer was also highlighted by Grobholz et al in which neuroendocrine cell growth patterns and clusters determined prostate cancer cell proliferation and PSA relapse-free survival 26.
This study has several strengths compared to previously published literature. This was the first independent cohort to evaluate serial chromogranin measurement and its clinical effect on treatment failure. This was done following the REMARK criteria for biomarker studies 27. In addition, compared to previous reports we also excluded patients on PPI therapy and with low GFR. This study does have limitations, mainly the small sample size of the prospective cohort which can affect the OS results. We also acknowledge that 12 weeks may not capture all patients with NED as NEPC can develop over an extended period of time. The next phase of this study will be to validate these findings in a larger prospective cohort.
Conclusion
Increasing serum CGA and an elevated CGA/PSA ratio are associated with a favourable response to AA/P with no difference in overall survival, however an increase in PSA after 12 weeks of AA/P is associated with an adverse OS. Based on our findings, there is limited clinical utility to pursue serum CGA testing to evaluate for lethal NED, as AA/P did not induce lethal NED in this cohort. This demonstrates that not all patients with increasing serum CGA have a worse OS. Further studies in large prospective cohorts with histological evaluation are needed to assess if rising serum CGA during therapy reflects neuroendocrine differentiation and may indicate a sign of treatment effect targeting the androgen susceptible cells.
Supplementary Material
Acknowledgements
This study was supported by National Institutes of Health grants U19 GM61388 (The Pharmacogenomics Research Network), R01 CA196648, R01 GM28157, Prostate Cancer Foundation, Department of Defense, and Private and philanthropy funding sources: i) Mayo Clinic Center for Individualized Medicine; ii) A.T. Suharya and Ghan D.H, Gail and Joseph Gassner; iii) Mayo Clinic Schulze Cancer for Novel Therapeutics in Cancer Research.
Footnotes
Conflict of interest
The authors declare no potential conflicts of interest.
References
- 1.Terry S, Beltran H. The Many Faces of Neuroendocrine Differentiation in Prostate Cancer Progression. Frontiers in Oncology. 2014;4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lin D, Wyatt AW, Xue H, et al. High Fidelity Patient-Derived Xenografts for Accelerating Prostate Cancer Discovery and Drug Development. Cancer Research. 2014;74(4):1272–1283. [DOI] [PubMed] [Google Scholar]
- 3.Dang Q, Li L, Xie H, et al. Anti-androgen enzalutamide enhances prostate cancer neuroendocrine (NE) differentiation via altering the infiltrated mast cells → androgen receptor (AR) → miRNA32 signals. Molecular Oncology. 2015;9(7):1241–1251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Beltran H, Tomlins S, Aparicio A, et al. Aggressive Variants of Castration-Resistant Prostate Cancer. Clinical Cancer Research. 2014;20(11):2846–2850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gkolfinopoulos S, Tsapakidis K, Papadimitriou K, Papamichael D, Kountourakis P. Chromogranin A as a valid marker in oncology: Clinical application or false hopes? World J Methodol. 2017;7(1):9–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cabrespine A, Guy L, Gachon F, Curé H, Chollet P, Bay J-O. Circulating Chromogranin A and Hormone Refractory Prostate Cancer Chemotherapy. Journal of Urology. 2006;175(4):1347–1352. [DOI] [PubMed] [Google Scholar]
- 7.Giridhar KV, Sanhueza C, Hillman DW, et al. Serum chromogranin-A-based prognosis in metastatic castration-resistant prostate cancer. Prostate Cancer and Prostatic Diseases. 2018;21(3):431–437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Conteduca V, Burgio SL, Menna C, et al. Chromogranin A is a potential prognostic marker in prostate cancer patients treated with enzalutamide. The Prostate. 2014;74(16):1691–1696. [DOI] [PubMed] [Google Scholar]
- 9.Burgio SL, Conteduca V, Menna C, et al. Chromogranin A predicts outcome in prostate cancer patients treated with abiraterone. Endocrine-Related Cancer. 2014;21(3):487–493. [DOI] [PubMed] [Google Scholar]
- 10.Fan L, Wang Y, Chi C, et al. Chromogranin A and neurone-specific enolase variations during the first 3 months of abiraterone therapy predict outcomes in patients with metastatic castration-resistant prostate cancer. BJU International. 2017;120(2):226–232. [DOI] [PubMed] [Google Scholar]
- 11.Heck MM, Thaler MA, Schmid SC, et al. Chromogranin A and neurone-specific enolase serum levels as predictors of treatment outcome in patients with metastatic castration-resistant prostate cancer undergoing abiraterone therapy. BJU International. 2017;119(1):30–37. [DOI] [PubMed] [Google Scholar]
- 12.Dong B, Fan L, Wang Y, et al. Influence of abiraterone acetate on neuroendocrine differentiation in chemotherapy-naive metastatic castration-resistant prostate cancer. The Prostate. 2017;77(13):1373–1380. [DOI] [PubMed] [Google Scholar]
- 13.Conteduca V, Scarpi E, Salvi S, et al. Plasma androgen receptor and serum chromogranin A in advanced prostate cancer. Scientific Reports. 2018;8(1):15442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Szarvas T, Csizmarik A, Fazekas T, et al. Comprehensive analysis of serum chromogranin A and neuron-specific enolase levels in localized and castration-resistant prostate cancer. BJU International. 2021;127(1):44–55. [DOI] [PubMed] [Google Scholar]
- 15.Fan L, Wang Y, Chi C, et al. Chromogranin A and neurone-specific enolase variations during the first 3 months of abiraterone therapy predict outcomes in patients with metastatic castration-resistant prostate cancer. BJU Int. 2017;120(2):226–232. [DOI] [PubMed] [Google Scholar]
- 16.Szarvas T, Csizmarik A, Fazekas T, et al. Comprehensive analysis of serum chromogranin A and neuron-specific enolase levels in localized and castration-resistant prostate cancer. BJU Int. 2021;127(1):44–55. [DOI] [PubMed] [Google Scholar]
- 17.von Hardenberg J, Schwartz M, Werner T, et al. Influence of abiraterone acetate on circulating neuromediators in chemotherapy-naïve castration-resistant prostate cancer. The Prostate. 2016;76(7):613–619. [DOI] [PubMed] [Google Scholar]
- 18.Korse CM, Muller M, Taal BG. Discontinuation of proton pump inhibitors during assessment of chromogranin A levels in patients with neuroendocrine tumours. British Journal of Cancer. 2011;105(8):1173–1175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Berruti A, Mosca A, Porpiglia F, et al. Chromogranin A Expression in Patients With Hormone Naïve Prostate Cancer Predicts the Development of Hormone Refractory Disease. Journal of Urology. 2007;178(3):838–843. [DOI] [PubMed] [Google Scholar]
- 20.Angelsen A, Syversen U, Stridsberg M, Haugen OA, Mjølnerød OK, Waldum HL. Use of neuroendocrine serum markers in the follow-up of patients with cancer of the prostate. The Prostate. 1997;31(2):110–117. [DOI] [PubMed] [Google Scholar]
- 21.Annala M, Vandekerkhove G, Khalaf D, et al. Circulating Tumor DNA Genomics Correlate with Resistance to Abiraterone and Enzalutamide in Prostate Cancer. Cancer Discovery. 2018;8(4):444–457. [DOI] [PubMed] [Google Scholar]
- 22.Nakazawa M, Paller C, Kyprianou N. Mechanisms of Therapeutic Resistance in Prostate Cancer. Curr Oncol Rep. 2017;19(2):13–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Terry S, Maillé P, Baaddi H, et al. Cross modulation between the androgen receptor axis and protocadherin-PC in mediating neuroendocrine transdifferentiation and therapeutic resistance of prostate cancer. Neoplasia. 2013;15(7):761–772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wafa LA, Palmer J, Fazli L, et al. Comprehensive expression analysis of l-dopa decarboxylase and established neuroendocrine markers in neoadjuvant hormone-treated versus varying Gleason grade prostate tumors. Human Pathology. 2007;38(1):161–170. [DOI] [PubMed] [Google Scholar]
- 25.Wright ME, Tsai M-J, Aebersold R. Androgen Receptor Represses the Neuroendocrine Transdifferentiation Process in Prostate Cancer Cells. Molecular Endocrinology. 2003;17(9):1726–1737. [DOI] [PubMed] [Google Scholar]
- 26.Grobholz R, Griebe M, Sauer CG, Michel MS, Trojan L, Bleyl U. Influence of neuroendocrine tumor cells on proliferation in prostatic carcinoma. Human Pathology. 2005;36(5):562–570. [DOI] [PubMed] [Google Scholar]
- 27.McShane LM, Altman DG, Sauerbrei W, Taube SE, Gion M, Clark GM. REporting recommendations for tumour MARKer prognostic studies (REMARK). Eur J Cancer. 2005;41(12):1690–1696. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.