

Abstract
The proceedings from the 30th August 2023 (Day 2) of the workshop “Physiologically Based Biopharmaceutics Models (PBBM) Best Practices for Drug Product Quality: Regulatory and Industry Perspectives” are provided herein. Day 2 covered PBBM case studies from six regulatory authorities which provided considerations for model verification, validation, and application based on the context of use (COU) of the model. PBBM case studies to define critical material attribute (CMA) specification settings, such as active pharmaceutical ingredient (API) particle size distributions (PSDs) were shared. PBBM case studies to define critical quality attributes (CQAs) such as the dissolution specification setting or to define the bioequivalence safe space were also discussed. Examples of PBBM using the credibility assessment framework, COU and model risk assessment, as well as scientific learnings from PBBM case studies are provided. Breakout session discussions highlighted current trends and barriers to application of PBBMs including: (a) PBBM credibility assessment framework and level of validation, (b) use of disposition parameters in PBBM and points to consider when iv data are not available, (c) conducting virtual bioequivalence trials and dealing with variability, (d) model acceptance criteria, and (e) application of PBBMs for establishing safe space and failure edges.
Keywords: Bioequivalence, bioequivalence safe space, context of use, dissolution, drug product quality, IVIVC, IVIVR, model credibility assessment framework, modeling, PBBM
1. Introduction
Physiologically Based Biopharmaceutics Modeling (PBBM) is an evolving tool which can be used throughout drug product development, regulatory approval, and life cycle management.1,2 PBBM focuses on providing a mechanistic understanding of the interaction between drug product quality attributes and physiology and then quantifying that interaction with regard to influence on in vivo drug performance. Effective utilization of PBBM requires a consistent approach to model development, verification, validation, and application.1,2
The workshop entitled “Physiologically Based Biopharmaceutics Modeling (PBBM) Best Practices for Drug Product Quality: Regulatory and Industry Perspectives”,3 sponsored by FDA in collaboration with M-CERSI (University of Maryland Center of Excellence in Regulatory Science and Innovation) was held August 29th–31st 2023 at the Universities at Shady Grove, Rockville.1,3,4 This Perspective reports on Day 2 of the workshop, which focused on considerations for PBBM verification, validation, and application. The agenda and presentations are available online at: “Physiologically Based Biopharmaceutics Models (PBBM) Best Practices for Drug Product Quality: Regulatory and Industry Perspectives”.3
The morning session included presentations from regulatory agencies on the analysis of three representative PBBM case studies, followed by a panel discussion centered around best practices for model validation. The afternoon session consisted of four parallel breakout (BO) sessions and covered the following topics:
Model development: data inputs, disposition, and absorption parameters, dealing with sparse data.
Model verification, model validation, and acceptance criteria for PBBM including the potential use of model risk-based analyses described in the credibility assessment framework and model COU.5−8
-
Model application:
Virtual bioequivalence (VBE) trials vs single representative modeling, dealing with within- and between-subject variability and parameter uncertainty.
Establishing bioequivalence safe space and failure edges, use of nonbioequivalent (BE) batches, and alternative in vitro in vivo correlations or relationships (IVIVC/R).
2. PBBM Case Studies
Three PBBM case studies provided by industry representatives prior to the workshop were presented and discussed by scientists from various health authorities. The model’s question of interest, COU,5−8 and discussions are summarized below and the presentations are available online.3 Following the health authority presentations, the fevipiprant PBBM case study9 was presented. Moreover, additional PBBM studies, such as GSK3640254, Drug C, and elagolix,3 were discussed throughout Day 2 by industry representatives and have been compiled in Table 1.
Table 1. Examples Case Studies of Possible Applicability of Risk-Informed Credibility Assessment Framework in PBBM with COU to Support Drug Product Quality (Modified from Min Li.3).
| Case, Question of Interest | COU | Decision Consequence | Model Influence | Model Riska | Regulatory Impact | Clinical data used for validation | Comments |
|---|---|---|---|---|---|---|---|
| Case study 4: Can the acceptance criteria for the drug substance particle size distribution (D10, D50, D90) of “EMD Compound A” be widened based on a PBBM approach?3 | PBBM developed using particle size distribution from nonmicronized, micronized, and fine-micronized drug substance. Validation with clinical data for different formulations and doses. Wider, yet clinically relevant drug substance acceptance criteria (as D10, D50, and D90) proposed based on the PBBM analysis. | Low (1) | Medium (2) | Low (2) | Low (2) | Oral solution; SAD PK (micronized and nonmicronized API in capsules); relBA and BE of various tablet formulations; no non-BE batch. All PK data in HV. | Model built using iv microdosing data. Lysosomal trapping of drug in enterocytes assumed, and model was adopted accordingly (fit fu,ent). Low CL drug with no significant first pass metabolism. Compartmental PK model used to model postabsorptive drug disposition. |
| Case study 5: Is the OOS batch based on the QC dissolution method, bioequivalent to the reference product?3 | PBBM developed using PBDT and validated using clinical data with different formulations. VBE trials to demonstrate that the OOS batch is BE to the reference batch eliminating the requirement for a clinical study. PBBM submitted as part of data package to justify QC dissolution spec widening | Low (1) | Medium (2) | Low (2) | Medium/High (eliminate clinical study) | Oral solution, BE and non-BE batches | PBBM built using PBDT data as input, not QC dissolution data |
| Case study 6: Is X% of polymorphic impurity allowed in the drug product?3 | PBBM developed with PBDT as input. PBBM used to simulate VBE trials which are used to define a certain % where there would be no influence on the extent of absorption and plasma PK, eliminating the requirement for a clinical study. PBBM submitted as part of data package to justify the % of polymorphic impurity | Low (1) | Medium–High (3–4) | Medium (3) | Medium/High (eliminate clinical study) | Parallel and crossover design, fasted state, different formulations, effect of process and scaling, PSD | PopPK used to parametrize disposition parameters. No iv data available |
| Case study, Drug Cb.12 How do you conduct extrapolation of bioequivalence study results obtained in male subjects to both genders? | Model was utilized to demonstrate that BE study results obtained in male subjects can be extrapolated to females | Medium (2) | Medium (2) | Low (2) | Low (2) | Pilot and pivotal batches (model differentiated between BE and non-BE batches) | Model was built wherein appropriate inputs for dissolution, enzymes and transporters are included. |
| Case Study GSK3640254.3 How do you support a biopredictive dissolution method? | Important for internal decision making, risk assessment. Changes in disintegration time (DT) due to process changes but within the clinically relevant dissolution safe space. Model demonstrated no clinically relevant changes in DT | Medium (2) | Medium (2) | Low (2) | Low (2), GSK3640254 development was terminated | Human relBA data (capsule to tablet). TIM1 data of reference and stretched batch | QC method was not biorelevant, but it was biopredictive. It was assessed using DLM scalar in ADAM model. A non-BE batch could not be produced within the formulation design space. |
| Case Study GSK3640254b,3 How do support clinically relevant dissolution specification | Important for internal decision making, risk assessment | Medium (2) | Medium (2) | Low (2) | Low (2), GSK3640254 development was terminated | Human relBA data (capsule to tablet). TIM1 data of reference and “stretched” batch | Clinically relevant model informed dissolution safe space, defined based on PK/PD relationship, was wider that then changes observed with QC method between reference and “stretched” batches. |
| Case Study Fevipiprantb:39 How do you to establish dissolution bioequivalence safe space? | PBBM to define BE safe space with QC method for 450 mg dose. Dissolution profiles are used as an input to the PBBM, the PBBM is then used to predict Cmax and AUC. | Medium (2) | Medium (2) | Low (2) | Medium (2), widening of dissolution specification; Fevipiprant development was discontinued | The PBBM performance was demonstrated for various oral dosage forms (150–450 mg), including the non-BE batches in fasted HVs. To define the safe space at 450 mg, simulations were performed using theoretical, virtual dissolution profiles | A specification of Q= 80% dissolved after 60 min for an IR oral solid dosage form reflected the boundaries of the safe space. |
| Case study 11. Elagolixb Justifying widening of dissolution specification without a non-BE lot. (US FDA Product Quality Review Available29) | PBPK model based on DDI was verified/validated using in vitro dissolution and in vivo data from pivotal and commercial materials, which are BE to the reference. This model predicted similar exposures from lots with slower dissolution profiles (75% slower dissolution rate led to 14% difference in exposure which was within 80–125% of reference,27,30 which resulted in widening of dissolution specifications and approved specifications.29 | Medium (2) | Medium (2) | Low (2) | Medium (2), widening of dissolution specification | Reference, Phase III and commercial lots were evaluated. Both Phase III and commercials lots were found BE to the reference in two separate PK studies. Slower-releasing lots were not tested in vivo but were evaluated with the PBBM/PBPK model. The dissolution safe space was extrapolated to slower dissolving lots using PBPK modeling. | |
| Q1. Can PBPK model reasonably describe elagolix PK after input of dissolution data? | Agency response to Q1 (R1). Yes. With the incorporation of in vitro dissolution profiles, the ratio of predicted Cmax and AUC by PBPK model to respective clinical observations were within 0.80–1.25. | ||||||
| Q2. Can PBPK models provide a reasonable prediction of the impact of slow dissolution on in vivo exposure ? | R2. The slower dissolution would not significantly affect the in vivo exposure of elagolix. See reference for further details. | ||||||
| Q3. Can modeling support a clinically relevant dissolution acceptance criterion? | R3. Yes, the PBPK model supported a clinically relevant dissolution acceptance criterion. |
2.1. Regulatory Presentation Case 4
Luiza Borges (ANVISA) presented on the regulatory discussions of case study 4 (from Merck Healthcare KGaA).3
Question of Interest
Can the acceptance criteria for the drug substance particle size distribution (D10, D50, and D90) of “EMD Compound A” be widened based on a PBBM approach?
Context of Use
During registration of “EMD Compound A”, the acceptance criteria for the drug substance particle size were set based on statistical evaluation of representative batches. To elucidate whether a PBBM approach is acceptable to widen drug substance particle size specifications and thus support setting active pharmaceutical ingredient (API) acceptance criteria, PBBM was utilized to build a relationship between the API particle size and the drug’s absorption and pharmacokinetics (PK). For this purpose, the PBBM was developed using particle size distribution from non-micronized, micronized, and fine-micronized drug substances and subsequently validated with clinical data for different formulations and doses. Wider, yet clinically relevant drug substance acceptance criteria (as D10, D50, and D90) were then proposed based on the PBBM analysis.
Discussion
“EMD Compound A” is a Biopharmaceutics Classification System (BCS) IV hydrochloride salt drug substance commercialized as an immediate release (IR) tablet with a micronized drug to increase exposure. Microdose intravenous (iv) and oral solution data were used to parameterize a 2-compartment disposition model and percentages of presystemic hepatic and intestinal metabolism were set, further supported by mass balance and drug–drug interaction (DDI) studies. In vitro solubility was measured in aqueous buffers with 100 mM sodium chloride added to account for decreased drug solubility due to the chloride common ion effect as well as biorelevant fasted and fed state simulated intestinal fluids (FaSSIF and FeSSIF). To match the measured in vitro solubility in the pH range between 1.0 and 6.8, the pKa value was fitted, resulting in a substantially different value from the one measured in vitro using the shake flask method. Other input parameter values were supported by parameter sensitivity analysis (PSA), including the effective human jejunal permeability (Peff) estimated based on Caco-2 studies, precipitation time (GastroPlus default value), and fraction unbound in enterocytes (fitted based on PK data from the oral solution). Lysosomal trapping was proposed as a plausible mechanism for the observed late tmax for EMD Compound A. For model validation and application, various solid oral formulations were included; however, without their in vitro dissolution data, alternate approaches, API PSD and Johnson dissolution model, were applied. It is recognized that dissolution kinetics of the primary API particles are not usually representative of the drug product dissolution, considering the effects of excipients and manufacturing processes (e.g., CMAs and CPPs) on formulations. To link API PSD to dissolution, it is critical for a robust PBBM to be enabled by an understanding of formulation properties and key controlling factors in the dissolution process. For model validation, varied data on doses, formulations (capsule and tablets, non-micronized, micronized, and fine micronized API) and prandial state (fed and fasted) from five clinical studies were applied with single representative simulations. The absence of a non-bioequivalent (non-BE) result at the clinically relevant dose might impact the strength of the validation, but the major limitation seems to be the lack of population simulations with the data set available. Model application assessed the impact of variations of API PSD at the clinically relevant dose level, keeping a constant D10, D50, and D90 ratio based on results of the reference batch. The impact of different API PSD ratios and VBE comparison to the reference PK data set were not investigated. The conclusion was that this PBBM, as presented, is not sufficient to support setting of API PSD specification, and possible approaches for model refinement were suggested to improve the robustness of the PBBM.3
2.2. Regulatory Presentation Case 5
Mary Malamatari (MHRA) presented on the regulatory discussions of case study 5 (Janssen).3
Question of Interest
Is the out-of-specification (OOS) batch, based on the quality control (QC) dissolution method, bioequivalent to the reference product?
Context of Use
For this drug product, the QC method is not considered physiologically relevant. Thus, the OOS batch was first tested in a physiologically based dissolution test (PBDT) mimicking the fed state. A PBBM was developed using PBDT profiles as input and validated with clinical data for different formulations including a non-bioequivalent batch. The PBBM was then used to predict the plasma concentration time profile of the OOS batches and simulate VBE trials. The purpose of the VBE trials was to demonstrate that the OOS batch is bioequivalent to the reference batch and therefore eliminate the need for a clinical study. The PBBM was submitted as part of a data package to health authorities to justify a widening of QC dissolution specifications.
Discussion
Drug A is a weak base with low solubility and high permeability (BCS class II compound). The drug product is an IR solid oral dosage form that should be administered with food. During stability testing, two batches of the drug product exhibited OOS results for the QC dissolution testing.
The aim of this study was to investigate the impact on drug exposure of failing the QC dissolution specification. A mechanistic absorption model was developed in GastroPlus using compound-specific parameters (i.e., LogD, pKa, solubility, and Peff). PBDT was established as a two-phase dissolution approach using biorelevant media mimicking the fed state (acetate buffer pH 4.9 followed by FeSSIF pH 5). The z-factor10 (an option to simulate the dissolution/release from solid oral dosage forms) fitted to the PBDT profiles was used to integrate dissolution data into the model. The distribution, metabolism, and excretion of the drug were simulated using a compartmental model.
The PBBM was validated by simulating the results of a relative bioavailability (relBA) study. The model was able to differentiate between BE and non-BE drug formulations/batches. VBE trials were performed by comparing the OOS batches with a reference batch, which were predicted to be BE for both Cmax and AUC0–72h under fed conditions. In conclusion, despite some concerns to be addressed, the PBBM approach was deemed satisfactory overall and no impact on drug exposure is expected for these two stability batches for which OOS dissolution results were obtained using the QC method during stability testing.
2.3. Regulatory Presentation Case 6
Shinichi Kijima (PMDA) presented on the regulatory discussions of case study 6 (Janssen).3
Question of Interest
Is X% of polymorphic impurity allowed in the drug product?
Context of Use
A PBBM was developed using PBDT profiles as input and validated with clinical data for different drug product critical quality attributes including a non-bioequivalent batch. PBBM was then used to predict the plasma concentration time profiles of the observed PK data from changes in formulation and processing and to simulate VBE trials. The purpose of the VBE trials was to simulate the influence of different percentages of polymorphic impurity in the drug product on the extent of absorption and plasma PK. VBE trials were then used to define the safe space for polymorphic impurity content, eliminating the need to conduct a clinical study. The PBBM was submitted as part of a data package to health authorities to justify the % of polymorphic impurity.
Discussion
A certain amount of polymorphic impurity in the final drug product can potentially influence the absorption and plasma PK and can be a CBA. Case study 6 (Drug X) involved a BCS II drug substance formulated as an IR oral dosage form. A mechanistic absorption model was developed in GastroPlus using compound-specific parameters (i.e., LogD, pKa, solubility, Peff). PBDT was established as a two-phase dissolution approach using biorelevant media mimicking the fasted and fed state. The Z-factor fitted to the PBDT profiles was used to integrate dissolution into the model. The distribution, metabolism, and excretion of the drug were estimated from a PopPK model. The PBBM was developed to model whether the presence of X% of a polymorphic impurity with slower dissolution in the drug product would have an impact on the systemic exposure or clinical performance of the drug. The validated PBBM, which can predict the in vivo relevance of changes in formulation and process parameters, was applied to assess the impact of polymorphic impurity in the drug product on in vivo exposure. PK predictions for different % of polymorphic impurity (0–100%) were conducted using VBE trials (n = 10 trials, n = 20 virtual subjects, healthy volunteers (HV), per trial) and an X% threshold was determined for which there would be no impact on the extent of absorption and PK. The PBBM showed generally good predictive performance and was therefore deemed acceptable for the presented purpose. However, it was noted that the acceptable threshold value (“X%”) was dependent on the evaluation of the VBE trials. For example, a more conservative value of “X” may need to be assigned if the chosen variability setting is assessed as uncertain.
2.4. PBBM Industry Case Studies
Industry case studies are summarized in Table 1. PBBM was used to aid in setting a clinically relevant dissolution specification (CRDS) for fevipiprant, which is a low molecular weight BCS class IV drug substance.9 This case study included clinical PK data for two doses with BE and clinically observed non-BE data. PBBM allowed the successful definition of the BE safe space for the QC dissolution method.3,9 Human (iv) microdosing data were used to describe disposition parameters.11 A second case study described the use of PBBM for safe space analysis for molnupiravir capsules.3 Another case study described informally in Day 2 discussions was the use of PBBM to justify extrapolation of BE study results observed for an IR product in male subjects to female subjects (see Supporting Information, Drug C).12
2.5. Panel Discussion
Following the PBBM case study presentations, the panel discussion brought together the following representatives from multiple health authorities: Rebecca Moody (FDA), Luiza Borges (ANVISA), Maria Malamatari (MHRA), Flora Musuamba Tshinanu (Belgium FAMHP), Shereeni Veerasingham (Health Canada), Shinichi Kijima (PMDA), and Paul Seo (FDA). The moderators were Tycho Heimbach (Merck & Co., Inc. Rahway, NJ, USA) and Claire Mackie (Janssen).
Key topics encompassed the workflow of PBBM, the intricacies of fitting the Z-factor model to the in vitro dissolution data, the limitations related to micelle-containing media, the connection between PBDT and QC release media, the necessity of non-BE batches, and the complementary role of animal PK data in model validation. This section provides a comprehensive overview of the discussion.
Q1: What Can Sponsors Do to Overcome Nonavailability of iv Data to Validate a PBBM?
There was acknowledgment that human iv data are not always available; however, it is considered the gold standard in building an understanding of the drug disposition. Other data sets that could be very useful to support model parameter selection include oral solution data, single oral ascending dose (SAD), multiple ascending dose (MAD) outputs, metabolism DDI studies, pH-mediated DDI studies, mass balance studies, and PopPK data. A consensus among workshop participants was that the PBBM community (industry and regulators) is continuously learning and evolving. The credibility assessment framework,5−8,13 a risk-based analysis built around the intended use of the model (case-by-case evaluation of when and where data are necessary, decision consequence) could be a way to move the field forward. There was a proposal to compile a PBBM template outlining the principles and methodologies, including the credibility assessment, which was endorsed by nearly 100 attendees in a workshop survey.1
Q2: How Are Model Risk and Regulatory Impact Considered for Setting Validation Criteria?
The use of the model credibility assessment framework and the COU assessments as proposed by Kuemmel et al., the FDA, Rusten et al., Musuamba et al. and the European regulatory network (EMA and EU NCAs), was encouraged for PBBM applications.5−8 This framework, shown in Figure 1, has already been implemented in PBPK DDI and specific population modeling, as well as in medical device modeling. The American Society of Mechanical Engineers (ASME) guidelines, for example, could be leveraged, as they emphasize gathering all available information to assess risk, address the central question of interest, and evaluate any potential risks to the patient as well as the regulatory impact.13
Figure 1.
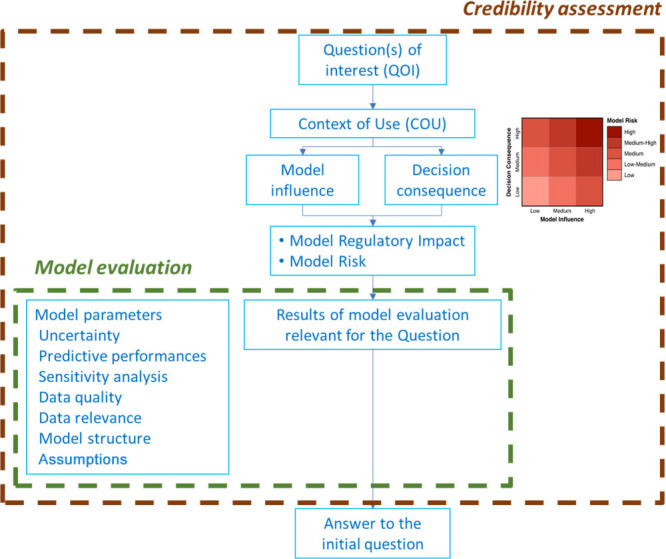
Possible scheme for the model credibility assessment. This schematic includes concepts on model risk, the model risk grid,13 along with model influence and decision consequence.5−8,13
PBBMs may have lower acceptance rates due to validation concerns. Therefore, a clearly defined context of use, model influence, decision consequence, model risk, and regulatory impact together with cross-discipline reviews within regulatory authorities could increase the impact of PBBMs in regulatory filings, while improving review efficiency and driving acceptance of PBBMs.
Q3: Is There a Minimum Number of Data Sets Recommended for Model Validation?
Participants stressed that the minimum number of data sets recommended for model validation is contingent upon several critical factors, including model risk, regulatory impact, and the extent of uncertainty inherent in the specific modeling COU. Hence, the approach to data set requirements should be tailored to the unique characteristics and demands of each modeling scenario, ensuring that validation efforts align with the associated risks and regulatory impact.
Q4: How Essential Is a Non-BE Batch for Model Validation?
The necessity of non-BE batches or batches with nonacceptable bioavailability for model validation was discussed.14 Participants understood that formulating a non-BE batch can present significant practical challenges and may be infeasible. The introduction of a non-BE batch should be guided by meaningful changes in, for example, formulation (CFVs) or API particle size (CMAs) or slight adjustments in process parameters (CPPs). Depending on the intended purpose, it could be acceptable to validate PBBMs using batches manufactured within the identified specification range. For example, BE batches that exhibit differences between in vitro and in vivo release profiles may help to define the edge of dissolution failure and support a dissolution safe space, which could improve confidence in the model.
Q5: What Can Be Done to Promote and Encourage PBBM and Model-Informed Drug Development (MIDD) in Global Drug Development?
Workshop participants engaged in a constructive dialogue about the role of regulatory agencies in advancing the adoption of PBBM and MIDD across the global drug development landscape. Regulatory representatives confirmed their commitment to promoting a well-justified and adequately implemented PBBM in regulatory decision-making processes. They highlighted the significance of workshops and similar platforms as valuable opportunities for mutual learning and knowledge exchange, benefiting both regulators and industry stakeholders.
To facilitate the integration of PBBM and MIDD, the following key recommendations emerged from the discussion:
Proactive Engagement: Regulators encouraged companies to initiate meetings as soon as possible when considering submitting a PBBM to agencies. Regulators strongly encouraged transparent and clear communication throughout the drug development process. Early discussions on modeling strategies and acceptance criteria may help streamline the regulatory expectations.
Case Studies/Submissions: Case study evaluation has been very useful to gain experience. Regulators encouraged industry to submit more applications to continue the growth for both industry and agencies. More submissions help agencies fine-tune what they are looking for. In the field of biopharmaceutics, agencies are educating themselves on the different modeling platforms and providing people with an opportunity to review. Companies are likely using many more PBBMs for internal decision making without filing them. However, sharing them could help regulators. Case studies can also help in building collaborations with academia.
Agency Feedback on PBBM: Participants stated that feedback with more context may be helpful. Certainly, if questions would arise on the model, providing context for the question would be very useful. The credibility framework can also be applied (Figure 1).5−8,13
Publication of Guidance(s): Guidances can serve as confirmation for what agencies would like to see and accept. These documents would serve as valuable resources for agencies to frame what they would like to see and to guide the industry in incorporating PBBM effectively.
Bridging Gaps: Recognizing the need for alignment between clinical pharmacology teams and CMC teams, regulators emphasized the importance of bridging this gap to enhance collaboration and streamline drug development processes both within companies and as reviewers.
Credibility Assessment Framework:(5−8,13) Regulators underlined the potential advantages to adopting a credibility assessment framework in that it may provide a structured approach to model intended use, model development, model validation, and regulatory decision-making. Clear explanation of inputs and justification within the PBBM report is essential for the regulators to follow the model build, validation, and intended use and ensure transparency in thinking. This can help reduce the scientific burden for regulators reviewing the models. In effect the pharmaceutical industry is the driving force and needs to put regulators in a comfortable position to support the models.
Scientific Advisory and Presubmission Meetings: MHRA suggested the use of scientific advisory meetings, while Health Canada suggested presubmission meetings as effective avenues for aligning regulatory expectations. EMA would also encourage scientific advice, qualification advice and opinions to engage with the agency. PMDA has seen less submissions and would like to have further discussions with Japanese stakeholders. In addition, participants discussed the postsubmission phase, highlighting the importance of connecting with the designated project manager responsible for query responses specifically for generic drug developers. This collaborative approach aims to address modeling-related queries effectively, ensuring a smoother regulatory evaluation process.
These collective insights serve as a promising foundation for efforts toward harmonization and advancement of PBBM and MIDD practices in global drug development.
Q6: Can a Single Model Be Used for Multiple Purposes, and Can Animal Data Be Utilized for Model Validation?
As to whether a single model can be used for multiple purposes, regulatory agencies’ experts expressed their willingness to evaluate models for multiple purposes if well justified but emphasized the importance of presenting the specific questions and objectives up front. If a model is found to be suitable and robust for addressing multiple inquiries, it may receive regulatory acceptance. It was also recognized that a single model might be suitable for one purpose but not necessarily for another. Therefore, the feasibility of using a single model for multiple purposes remains contingent upon the specific context and goals, with decisions made on a case-by-case basis. The key criterion is whether the model can effectively address all relevant questions and objectives. It should be noted that the concept of using a single model for multiple purposes is consistent with the newer model master file (MMF) framework concept, where MMFs are viewed as portable, reusable, generalizable, and sharable models that have received regulatory acceptance and have undergone full V&V.15 As for utilization of animal data, there was flexibility in considering the acceptability of using animal data for model validation or parameter estimation, and the acceptability determination would be made on a case-by-case basis. While no specific recommendations were made for a particular animal model or species, the rat absorption has been reported to be predictive of human absorption16,17 and the dog model has been used in human formulation selection and optimization to overcome pH-mediated DDIs.18
Q7: Where Does the Flexibility in Acceptance Criteria Lie?
The workshop participants reached a consensus on the importance of establishing predefined acceptance criteria and possibly discussing them with regulators ahead of the model development and validation process. These criteria provide clear benchmarks against which models can be assessed and validated. In particular, discussions revolved around validation requirements given a drug’s efficacy, safety, and therapeutic window. It was acknowledged that the determination of the acceptance criteria should be guided by the specific context and goals of the modeling exercise. It should be noted that the IQ consortium provided detailed suggestions for proposed acceptance criteria, as part of docket feedback to the FDA PBPK for oral biopharmaceutics applications draft guidance.14
A recurring theme throughout the discussion was the emphasis on the totality of the evidence. Participants recognized that while predefined acceptance criteria provide essential guidelines, the final evaluation and decision-making process should consider all available evidence. This holistic approach ensures that the model’s performance is assessed comprehensively, considering various data sources, model application, COU and model risk. The flexibility in acceptance criteria is balanced by the overarching principle of evaluating models based on the totality of evidence, thereby promoting rigorous and scientifically sound decision-making in drug development.
3. Breakout Sessions
The participants included scientists from the pharmaceutical industry, modeling and simulation software companies, academia, and regulatory agencies. The overview of Day 2 presentations and BO sessions are presented in Table 2.
Table 2. PBBM Breakout Sessions.

3.1. BO Session F: Considerations for Model Development: Data Inputs, Disposition, and Absorption Parameters, Dealing with Sparse Data
Session F speakers were Tycho Heimbach (Merck & Co., Inc. Rahway, NJ, USA), David Turner (Certara), and Rebecca Moody (FDA). Moderators were Lanyan (Lucy) Fang (FDA) and Cordula Stillhart (Roche) with Philip Bransford (Vertex Pharmaceuticals) and Xiaojun Ren (Novartis) as scribes.
Presentations:
-
(1)
Establishing the safe space via physiologically based biopharmaceutics modeling. Case study: fevipiprant/QAW039 (Tycho Heimbach).3,9
-
(2)
Approaches for obtaining disposition parameters for PBPK/PBBM (David Turner)
-
(3)
Considerations for model development—data inputs, disposition, and absorption parameters, dealing with sparse data (Rebecca Moody)
A decision tree for obtaining disposition parameters in PBBM was discussed (Figure 2). The decision tree aimed to establish considerations for how to construct the disposition model with iv data, with only oral data, or with only preclinical data (by using various scaling methods). The participants emphasized that the decision tree should not be overprescriptive for the oral solution approach and should describe it as a high bioavailability drug product (DP).
Figure 2.

Presented decision tree for human iv data generation used in PBBM.20−23
When only oral PK data of a low bioavailability compound are available for constructing a PBBM, more elaboration was requested. The participants acknowledged that this scenario poses additional challenges and complexities. Specific considerations and strategies for modeling the PK of low bioavailability compounds may include the established preclinical scaling methods.
The preference for bottom-up approaches over top-down approaches in modeling was highlighted. The participants emphasized the importance of incorporating physiological and mechanistic understanding into the models as well as utilizing relevant in vitro data to inform the model development process.
The participants expressed the need for illustrative examples to aid in understanding the practical implementation of PBBM without human iv data. These examples would ideally showcase successful PBBM development and applications, such as DDI assessments and justifying dissolution specifications, even in the absence of iv data.
It was acknowledged that there is no stringent regulatory requirement to routinely generate iv data for oral products and that obtaining approval for iv studies can often be challenging. The decision to conduct iv studies is driven by factors such as the clinical use of the iv formulation, the need to understand DDI, or the need for an enabled formulation, particularly for drugs with low bioavailability. If a BCS-based biowaiver can be applied, iv studies are often not conducted.
3.2. BO Session G: Considerations for Model Validation, Model Acceptance/Verification Criteria in PBBM in View of Available Clinical Data, and Model Risks (Model Risk and Impact)
The Session G speaker was Min Li (FDA). Moderators were Shereeni Veerasingham (Health Canada) and Nikunjkumar Patel (Certara Inc.) with David Sperry (Lilly), and Hansong Chen (FDA) as scribes.
Session G discussed considerations for model verification, model validation, and acceptance criteria for PBBM using risk-based analysis principles as described in the credibility assessment framework.5−8 It is important to note that other interpretations of model qualification, verification, and validation exist in scientific literature.24
The discussion focused on streamlining and mapping of the terminologies used in the PBBM area, as harmonization of terminologies used for modeling methods is currently in discussion for the upcoming ICH M15 guideline: General Principles for Model-Informed Drug Development. Although different interpretations of model validation and verification exist in scientific literature for PBPK/PBBM, the overarching aim of the ICH M15 guidance is to arrive at a harmonized set of terminologies to support clear and coherent communication, interpretation, and scientific/regulatory review of the PBBM submissions. Verification is considered to be the “process of determining that a computational model accurately represents the underlying mathematical model and its solution from the perspective of the intended uses of modeling and simulation”.5,25 Model validation can be considered a process of determining the degree to which the model or simulation is an accurate representation of the real world.5 Musuamba et al.6 further simplified verification as “solving the equation right” meaning activities related to numerical code and confirmation of the accuracy of mathematical computation of the software platform and validation as “solving the right equations” meaning the activities related to assessment of the predictive performance of the model in comparison to observed in vivo data.6 Examples for the possible applicability of the risk-informed Credibility Assessment Framework for PBBM have been compiled in Table 1.3
Model Verification
Model verification focuses on the correctness of the mathematical model structure and, in general terms, encompasses the question “Does the model make sense?” and entails software quality control. While the applicant is responsible for the content of a regulatory application, it was considered that the technical verification of modeling software relates to software quality control, and therefore, the modeling platform providers (in the case of a commercial software package) would play a primary role in model verification. It was mentioned that many model equations for commercial modeling and simulation software are already verified, for example, based on scientific publications in peer-reviewed journals. Installation of modeling platforms should undergo qualification to establish confidence in the ability of the installed platform in a particular computational environment to solve the modeling equation and obtain the expected outcome. This may be termed software installation qualification. The choice of modeling should be determined by the model developer and applicant. In general, regulators may provide suggestions during presubmission stages, but the choice of a particular modeling platform over another does not have to be justified.
Model Validation
In general, data sets used for model validation vary based on the question of interest, the modeling approach and associated risk, as well as the regulatory impact.5−8 Validation data sets should confirm the predictive ability of the model for the question of interest. Industry participants questioned how regulatory agencies weighted the relative value of validation data sets. Participants noted that crossover design studies would be preferred to parallel design studies as they provide standardization of the study subjects which facilitates the identification of formulation related differences. Data sets from single dose studies may be preferred to those from multiple dose studies when VBE trials are used as multiple dosing may result in changes in the range as well as a shift in the PK parameter confidence intervals.26 A practice for extended retention of batches used in clinical trials was proposed to facilitate alternative future use with consideration of batch expiry dates when these are used.
In cases in which there are subsequent submissions of models for a drug product, it is critical that the question of interest and COU for the model at each submission are considered. When an applicant submits the same model for a second application, the question of interest could be different from that for the initial model. While the technical part of the model may be the same, some parts of the model may need to be updated or additional validation may be needed to demonstrate that the model is able to reliably address a different question of interest. For example, the initial PBBM might have been developed to define appropriate drug product dissolution acceptance criteria, but the PBBM might be utilized later to support a biowaiver request for a postapproval formulation change. Additional validation of the model would be necessary to support the biowaiver request, given its higher regulatory impact and risk. For a new submission, it is helpful to reference the submission history of the initial model. Using PBBM version numbers and subsequent model adaptations would facilitate referencing, provide continuity to the initial model, and aid appropriate regulatory review.
Participants discussed the recommendation in the FDA draft guidance, The Use of Physiologically Based Pharmacokinetic Analyses—Biopharmaceutics Applications for Oral Drug Product Development, Manufacturing Changes, and Controls,14 for using batches that exhibit unacceptable bioavailability (non-BE batch) for model validation, in addition to those that exhibit acceptable bioavailability. A non-BE batch differs from the target one in formulation or a critical bioavailability attribute and, ideally, exhibits a dissimilar dissolution profile (similarity factor (f2) < 50). The FDA draft guidance recommends this approach to increase confidence in the model. Batches that only exhibit “acceptable” performance may not be adequate for validation, especially when a model is employed to propose a wide dissolution safe space. To gain confidence in the predictive ability of the model over a wider dissolution range, validation using a data set from a non-BE batch that is not bioequivalent to the target one would be very useful.14 This can allow a dissolution edge of failure for bioequivalence to be determined within the validated range.
Industry representatives noted that the intentional development of a non-BE batch is usually not an objective during the clinical development program. Furthermore, the formulation of a non-BE batch might present practical challenges, and therefore validation using a non-BE batch might not be feasible for most drug products. Having clinical PK data for a non-BE batch is generally an unintentional outcome during drug development rather than by design. Regulators acknowledged this challenge and suggested that a pilot batch formulated during the early development stage that shows that non-BE could be used for the model validation. If a non-BE batch is not practical, then the dissolution safe space based on bioequivalent batches may be able to accommodate the change in manufacturing, formulation, and life-cycle needs for the drug product. There are also examples of a PBBM safe space approach without a non-BE batch to support widening of dissolution specifications (Case Study 11, Table 1).27,28 Overall, while a non-BE batch is not required for PBBM validation, it is valuable for improving confidence in the model when available.
There can be uncertainty in the translation of an experimentally obtained value. For example, drug permeability can be predicted using different in vitro and in silico tools, and there could be uncertainty in the translation of the experimental values to effective permeability (Peff) values. Similarly, it can be challenging to predict in vivo precipitation based on the results of in vitro experiments. Participants emphasized the need for critical evaluation of links between assumptions made in a model. For example, precipitation time for a drug should be considered in the context of drug solubility given the link between the two in terms for the impact on the model. Overall, the key model assumptions should be justified scientifically. Participants discussed what type of data can be used to justify the assumptions. It was concluded that any available data can be employed to verify the assumptions, including animal data and in vitro data. Sometimes, PSA can be used to support an assumption. Participants also suggested that multiparameter analyses could be used to visualize trends and provide insights. In addition, an understanding of whether a process or a parameter has a significant impact on drug absorption could be inferred by an evaluation of dose–exposure relationships.
As default values in PBBM platforms might not represent all the physiological parameters that are important for the COU, scaling factors are occasionally necessary but should be justified. The discussion focused on guidance for changing scaling factors and, in particular, if there are any situations where changing scaling factors would not be acceptable. Empirical scaling factors are scientifically justified and are more readily accepted than theoretical scaling factors. There was agreement that, in principle, any value could be used if adequately scientifically justified or at a minimum rationalized. Participants emphasized that the changed scaling factors should be consistently applied to the model.
Considerations for Appropriate Model Acceptance Criteria
For BE studies, there are established BE acceptance criteria. However, there are no pre-established acceptance criteria available for PBBM. A risk-based approach is used to assess PBBM by considering the regulatory impact and risk of the model and the overall evidence, including available clinical data. The credibility assessment framework can be used to determine, standardize, and communicate the model risk.5−8 The model developer and applicant would use credibility items, such as the model influence and decision consequence, in evaluating the model risk (Figure 1). This, in conjunction with model regulatory impact, would inform evaluation practices and facilitate consistent decision making.
Model validation targets and criteria should be specified a priori. PBBM validation comprises comparisons of the predicted and observed in vivo drug concentration versus time profiles as well as PK parameter estimates, e.g., maximum concentration (Cmax), time to maximum concentration (Tmax), and area under the concentration versus time curve (AUC). Statistical analysis of PK parameters may be presented as percent prediction error (%PE), fold error (FE), average fold error (AFE), absolute average fold error (AAFE), average absolute prediction error (AAPE%), or another suitable approach to quantitate the prediction error. While there is no consensus on appropriate model validation criteria, participants suggested that it would depend, in part, on the COU. Criteria for VBE trials include the geometric means of the PK parameters as well as the associated confidence intervals, and considerations of BSV and WSV are important. Mispredictions and failure to meet acceptance criteria for validation data sets should be reviewed critically with consideration of the relevance of the data set (which is part of AC) for the question of interest and COU. Detailed IQ consortium docket feedback on PBBM acceptance criteria have been provided online as part of the PBPK for Oral Biopharmaceutics Applications draft guidance.14
3.3. BO Session H: Considerations for the Model Application: VBE Trials vs Single Representative Modeling, Dealing with Within- and Between-Subjects Variability and Parameter Uncertainty
Session H speakers were Amin Rostami-Hodjegan (Certara and The University of Manchester) and Viera Lukacova (Simulations Plus). Moderators were Duxin Sun (University of Michigan) and Jean-Flaubert Nguefack (Sanofi) with Tessa Carducci (Merck & Co., Inc. Rahway, NJ, USA) and Manuela Grimstein (FDA) as scribes.
Presentation: Dealing with Within- and Between-Subjects Variability (WSV and BSV) and Parameter Uncertainty (Amin Rostami-Hodjegan)
The key question debated was: How should we consider variability in a VBE study?
One of the reasons for considering variability is that one cannot assume that the reference and test products will have the same variability in vivo as demonstrated by Bego et al.31 The variability in physiology is the same for each formulation; however, the interaction of the physiology with each formulation will likely be different. This gives rise to the concept of formulation dependent WSV, i.e., different sensitivities of a formulation or drug to different components of the GI tract. Physiological WSV determines the observed WSV in PK which can be modeled in PBBM, as shown schematically in Figure 3.31
Figure 3.

Propagation of WSV in the physiological attributes of the GI tract through the interaction with attributes of the API and the formulation can be modeled with PBBM/PBPK. Figure is from Bego et al., https://creativecommons.org/licenses/by/4.0/.31
The “right way” to account for WSV when conducting a VBE study was also discussed. A suggested approach is to propagate the known variability in physiology a priori via PBPK analysis through the inclusion of % CV in relevant gastrointestinal properties. The values or measurements for all possible sources of variability are unknown. Therefore, there is a call out to academia and industry to sponsor more studies in which the relevant data can be collected to further explore the variability in physiological parameters. A better understanding will then permit the relevant ranges to be included in a PBBM analysis. Together with WSV, apparent BSV also requires consideration as single sampling from each individual is actually a hybrid measurement of BSV and WSV.31
To examine the likelihood of passing the BE criteria and incorporating BSV the right number of subjects (N) to include in a VBE study was discussed. The consensus is that the N should not go beyond the N that would be used in a clinical BE study for that drug product.
Combining these elements, an example of a VBE study for budesonide (local PD effect on the GI tract) was discussed.32 The workflow for the PBBM analyses should include considerations about varying dissolution profiles, local vs systemic PK, and disease effect (VBE in HVs and in the target population of Crohn’s disease). The following should be considered when incorporating WSV in VBE in PBBM.31,32 (1) WSV of PK is not independent of formulation; (2) WSV in physiology propagates to PK and manifests itself as WSV in PK; (3) WSV of PK can be simulated using WSV of physiology via population based PBPK; (4) The same WSV of GI tract physiology can lead to a varying WSV in PK for different formulations; (5) WSV for physiology of the GI tract is typically not measured, although advances have been made,33 but can be estimated; (6) BE based on systemic exposure is not equal to BE based on the local gut concentration–time profile.
Presentation: Considerations for Model Application: VBE Trials Vs Single Representative Modeling (Viera Lukacova, SimulationsPlus)
The presentation touched on the PBPK model application for VBE trials, starting with the use of individual subject data versus the average PK profiles in development and validation of a baseline PBPK model. Advantages and limitations of both were briefly discussed. The critical aspect is to understand the data and confirm that we know what “average” means. This will ensure that we account for the right absorption, distribution, metabolism, and elimination processes and that we justify the observed PK profiles. The topic of uncertainty versus variability was covered.34 Uncertainty is the “unknown” lack of precision. To address uncertainty, it is advised to evaluate parameter(s) via conducting PSA analyses. For BSV, the GastroPlus software incorporates population variability in a virtual population by differences in physiology between each subject. The algorithm implemented in the software to generate virtual subject populations incorporates reported BSV and accounts for known covariates between physiological parameters. For parameters where the distribution is unknown or covariates are not established, a log-normal distribution is used (e.g., formulation variability and intersubject variability). For including WSV in VBE crossover trials, the following points should be considered:
-
(1)
Does the virtual trial reproduce the observed clinical PK data? Compare predicted vs observed mean PK profile using the 90% confidence interval of the mean and the probability at 90%
-
(2)
Verify variability settings, additional variability will be added to parameters with known within subject variability. Variability is typically closer to the mean with a higher number of simulations
-
(3)
How many subjects should be used in a VBE study? Conduct simulations of the reference formulation against itself (including WSV) with different subject N on X number of trials, until all pass BE criteria. Find the minimum N needed for BE to meet the 90% confidence interval criteria.
Discussion
This breakout session took into consideration the following questions:
Q1. Intra- versus Intersubject Variability: Which One Is Larger?
Clinical BE studies should be powered to address BSV and WSV; therefore, it is preferable that a VBE study should match the clinical setting and estimate what the variability will be based on the variability from the available PK parameters, e.g., Cmax BSV. It is desirable to include physiological variability in PBBM, if known.
If we have a new formulation A, with corresponding BSV and WSV, when designing the clinical BE study, it will be powered by assuming that the test formulation will have the same variability as the formulation A. In this case, consider formulation dependent WSV. We cannot assume that reference and test formulations in the clinic will have the same variability. Data from Meyer et al.35 has shown that “test” vs “test” were more variable than the comparison of “reference” vs “reference”. One formulation could show more sensitivity to certain physiological parameters than the other formulation. This led to the question as to whether there are ways to evaluate the risk of differences in variability between reference vs test in vitro? Unfortunately, the answer is no, we cannot determine this difference in variability based on in vitro dissolution between the two formulations unless the in vitro data are modeled and analyzed to include all parameters that make up the observed dissolution profile and other attributes (for instance, influence of pH, common salt effect, viscosity, shear stress, bile salts). These can be integrated with the GI tract attributes and lead to a different WSV of the PK profile for the test vs reference product. However, in vitro set ups commonly do not contain sufficient details to translate them via fully mechanistic models (as opposed to empirical scalers) to the in vivo behavior. To conclude, in most cases, BSV is larger than WSV.
Q2. How Do You Consider Intra- Or Intersubject Variability in the Model?
If the test has higher variability than the reference, then it was suggested to increase WSV in the virtual model. Creating a safe space for model variability can be considered.
Q3. Parameter Optimization Considering These Variabilities
When assessing individual data versus average PK profiles, it is important modelers understand the data and know what the “average” represents. One needs to account for the right ADME process, and physiological parameters must be taken into account to justify the observed PK profile. Furthermore, when accounting for the right parameters in the model, fitting may be possible, e.g., optimizing permeability after accounting for lag time. If parameters are fitted to describe the observed variability, they should remain within physiologically realistic boundaries, and the assumptions should be clearly noted.
System parameters should not be different for virtual populations in different simulations just to cover the observed data. These parameters should remain as a robust set that can answer many different drugs and formulations; hence, they cannot be modified with every simulation for a new drug/formulation. The only exception for considering different systems parameters relates to cases where the target population itself is different than the typical population (e.g., cancer patients, elderly, pediatrics, different ethnic groups, different disease populations).
Q4. What is the Correct Sample Size for the VBE? What Main Factors Drive the VBE Sample Size Estimation?
Using a very high number of subjects, N, in a VBE study would create a very small confidence interval. VBE should not be powered with a very high N aiming to pass BE. For a clinical BE study powered to address the BSV and WSV, the corresponding VBE should match the clinical design. For the clinical study as well as the VBE, the aim would be to achieve 80% (e.g., 8 out of 10 trials) or higher of the virtual trials passing BE. With respect to sensitivity on the variability of the new formulations, one could consider creating a safe space to demonstrate that within the chosen formulation variables no impact on the PK is expected and VBE would be achieved. Using the Monte Carlo simulation approach of 1000 trials and checking the pass/fail ratio together with the N of VBE trials aiming to achieve an 80% pass, the results will show the expected differences in Cmax and AUC.
Q5. If a VBE Is Comparing Dosage Strengths (e.g., 120 mg versus 2 × 60 mg) and the Baseline Model for 2 × 60 mg Was Built Using the Johnson Dissolution Model with PSD Data, What Is the Best Way to Bridge the Formulations if We Do Not Have a Biopredictive Dissolution Method Mimicking the Drug Dissolution Process In Vivo?
If there is clinical BE established for the higher strength of the same formulation, a biowaiver for the lower strength may be requested, demonstrating equivalence through the dissolution profiles and an in vitro bridge. Alternatively, one can consider using the model if it can be demonstrated that the API PSD is driving the dissolution.
Q6. How Many Clinical (relBA/BE Trials) Are Required to Validate the Use of PBBM/VBE Trials?
Agencies present suggested that there was no fixed number. Some health authority scientists stated that N = 3 could be enough, with one clinical BE study powered to understand variability and two relBA studies. Others stated that 7–10 clinical studies may be needed. Each PBBM would be considered based on the question and COU, totality of data, and anticipated regulatory impact.5 They would also consider clinical factors, other clinical pharmacology data, and a risk-based approach.
Q7. If There Was a Non-BE batch, Should the Model Be Able to Reproduce This in the Validation Step?
The model should be able to reproduce a non-BE outcome. If the model is robust and reproduces all available clinical data, the non-BE batch may not be needed. The use of an independent study arm, e.g., an acid reducing agent study arm (with lower exposure) might be useful as a surrogate for a nonacceptable BE batch.
Q8. With a Biopredictive Dissolution Method Mimicking the Drug Dissolution Process In Vivo, Is It Mandatory to Compare the Simulated Cp-Time Profile of the Reference Strength Using Both the Johnson Model and the Biopredictive Dissolution Data even if f2 Value Is >50 in the Biopredictive Dissolution?
Biorelevant indicates that the dissolution media contain endogenous surfactants which mimic the in vivo GI fluid make up. Biopredictive indicates that the dissolution method (e.g., QC or other method) can pick up clinically relevant changes in systemic PK following oral administration.14 A PBBM can be built and verified by, e.g., inputting the data, i.e., Johnson model and z-factor.36 Comparison with the clinical data will demonstrate which dissolution data modeling method(s) is acceptable, well describes the input functions, and is biopredictive.
Q9. What Is Currently Known about the Variability of Drug Solubility in Human Intestinal Fluids (HIFs)?
Drug solubility in HIF as well as composition of HIF aspirates has been reported to be variable.37−39 To understand solubility variability Abuhassan et al. proposed solubility measurement in eight different FaSSIF media, around the FaSSIFv1 composition, varying total bile salt, phospholipid, total free fatty acid, cholesterol and pH.37,40 A 9-point design of experiment (DOE, 8 varied FaSSIF compositional values and FaSSIFv1) could be applied to investigate drug solubility in vitro and provide statistical solubility limits.37,40,41 As a multidimensional approach covered greater than 90 percent of the variability in fasted intestinal fluid composition from a study with 20 human volunteers,37,40,41 the upper and lower fasted intestinal solubility limits from this DoE and distributions could be applied to the biopharmaceutics classification system and could be useful as PBBM model inputs.
3.4. BO Session I: Considerations for Model Application: Establishing Safe Space and Failure Edges, Non-BE Batches, and Alternative IVIVR/C
The Session I speakers were Konstantinos Stamatopoulos (GSK), Xavier Pepin (Simulations Plus), and Siri Kalyan Chirumamilla (Certara). Moderators were Haritha Mandula (FDA) and Rob Ju (Abbvie), with Michael Wang (Merck & Co., Inc. Rahway, NJ, USA) and Joan Zhao (FDA) as scribes.
Presentation: Development and Application of PBBM to Define Dissolution Safe Space for a BCS IV Zwitterionic Lipophilic, IR Drug Product (Konstantinos Stamatopoulos, GSK)
A strategy was described to integrate in vitro solubility and permeability data into a PBBM to predict the food effect observed in clinical studies for a BCS IV zwitterionic drug substance (GSK3640254, GSK254).42 The model was developed and verified using clinical data of an immediate release (IR) tablet (10–320 mg) obtained in HVs under fasting and fed conditions. The solubility of GSK254 was a function of its ionization state, media composition, and pH, whereas its permeability, determined using MDCK cell lines, was enhanced by the presence of mixed micelles. In vitro data alongside PBBM suggested that the positive food effect observed in the clinical studies was attributed to micelle-mediated enhanced solubility and permeability. The biorelevant media, containing oleic acid and cholesterol in levels representing fasted or fed states, enabled the model to appropriately capture the magnitude of the food effect. The model accurately predicted the results of the food effect with predictions being within a 2-fold error and 70% being within 1.25-fold.
The PBBM was applied to define a clinically relevant dissolution safe space using criteria relevant to the target efficacious concentration of the drug 24 h postdose (C24h). In particular, 20–24% changes in Cmax were not clinically relevant with respect to the efficacy of GSK254 determined by C24h. The PBBM suggested that ≥70% of the drug should be dissolved within 1 h to ensure no impact on the efficacy of the drug.
This work showed that the predictive power of PBBM is improved when the understanding of the food effect goes beyond the typical approach (e.g., simple use of the typical FaSSIF and FeSSIF media) as well as when high quality in vitro data is utilized. Furthermore, although the integration of all of the available in vitro data might not be always possible or the model might not include all of the underlying mechanisms, the in vitro data should be used (if possible) to inform the right assumptions and adjustments to the model to describe the in vivo data. For instance, if TIM-1 data43,44 was lacking, then the potential explanation about the differences between the two meals observed in the exposure of GSK254 could be based on variability in the gastric emptying rather than differences in food composition (e.g., fatty acids). A PSA might show this impact; however, that would lead to wrong conclusions. Thus, the need for exploring how the composition might alter the absorption and hence the exposure of this class of drug is emphasized as a potential future work.
The developed model strategy can be effectively adopted to increase the confidence of using PBBMs to predict the food effect of BCS class IV drugs.
Presentation: Safe Spaces (Xavier Pepin, SimulationsPlus)
Safe spaces can be defined in different ways (Figure 4) for critical bioavailability attributes (CBAs). Using VBE or real BE studies, a safe space is defined by the boundaries of products which are bioequivalent to one another (upper panel).14 For example, the upper edge of the safe space can be a rapidly dissolving tablet or an oral solution, while the lower edge could be a slower dissolving tablet. Using exposure-response, a safe space can be defined by the products that show similar clinical efficacy and are equally safe to the patients (lower panel). Depending on the size of the therapeutic interval and the shape of the pharmacokinetic/pharmacodymanic (PK–PD) and pharmacokinetic/toxicodymanic (PK–TD) relationships, the size of the safe space based on efficacy and safety could be larger than that based only on bioequivalence.
Figure 4.
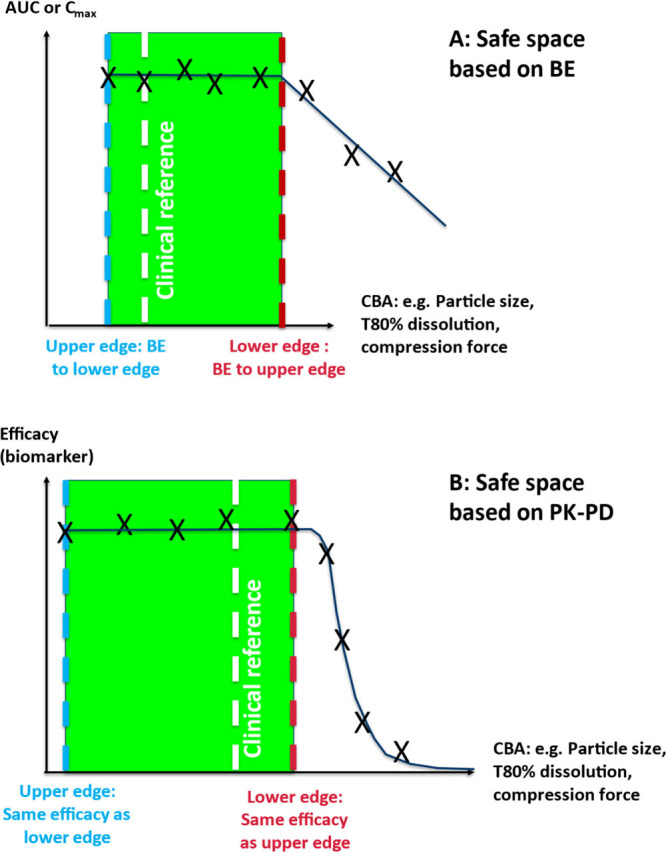
Definition of safe spaces based on bioequivalence (top) or PK–PD (lower panel).
To illustrate this concept of safe spaces, three brief case studies were presented as shown below:
Case study 1: Zolpidem hemitartrate (BCS class I): Safe space for dissolution based on VBE for 5 mg IR products
Case study 2: Acyclovir (BCS class III–IV): Safe space based on extrapolation for 800 mg reference and generic products
Case study 3: Acalabrutinib maleate (BCS class II): Safe space for dissolution based on PBBM+PK/PD
Case Study 1
A reference and test product comprising 5 mg of zolpidem hemitartrate were tested in pH 6.8, 50 rpm (salt solubility of 2.2 mg/mL). The dissolution profiles for these two batches were not comparable using the f2 criterion. A PBPK model was developed in GastroPlus based on literature data for zolpidem iv PK and immediate and modified release tablets, together with mechanistic clearance in the liver and the gut mediated by CYP3A4.45−51 The dissolution of drug products was mechanistically integrated using the P-PSD approach52,53 with two bins (Figure 5).
Figure 5.

2-bin P-PSDs derived for 5 mg of zolpidem hemitartrate REF and TEST products using 900 mL of dissolution medium in USP 2, 50 rpm, phosphate buffer pH 6.8.
A series of 10 VBE studies were then conducted with N = 25 subjects each (default) using the reference 5 mg tablet administered twice in a crossover fashion and applying default physiologic WSV. A feature by which the volumes, transit times, pH, and bile salt concentration are varied slightly within subjects during a crossover trial was included. Results from this series of VBE trials showed that 50% of the trials were unsuccessful in demonstrating BE between the REF product and itself. Upon data analysis, it was found that the WSV for zolpidem is 30% and it was decided to increase the number of subjects to 32 in order to power the crossover VBE adequately.54 With this new number of subjects, the 10 crossover VBE studies of REF vs REF passed. In addition, the 10 crossover VBE studies of TEST vs REF enabled the prediction of bioequivalence between the two batches of products despite differences in dissolution profiles.
Case Study 255
Acyclovir IR tablets are BCS class III–IV products. Acyclovir is a weak base (pKa 2.27) in the physiological region. Its Peff is estimated at 0.3 × 10–4 cm/s, and its solubility is 2.33 mg/mL at pH 5.8 with a maximal oral dose of 800 mg. The drug distribution and elimination are subject to the synthesis of OCT1 transporters, which show polymorphism across different populations. Dissolution studies conducted with 400 mg of Zovirax (reference) tablets and two generic products A and B in USP2, 50 rpm, 900 mL of simulated intestinal fluid showed noncomparable dissolution profiles according to f2 criterion. There was no major difference in excipients in the three formulations, and no excipient was included that could alter GI physiology or drug permeability. Despite differences in dissolution profiles, the two generic products were bioequivalent to the reference listed drug. A PBPK model was developed with GastroPlus based on iv and oral PK profiles from the literature. SIF dissolution data was used as input in the PBBM with a direct input of dissolution.55 The BE observed between these batches of products was compared through a series of VBE studies. To define the edge of the safe space, virtual dissolution profiles were generated by stretching the time scale for dissolution based on dissolution data measured on the reference product; i.e., the percent dissolved was kept constant, but the time for each data point was stretched by a fixed factor. A time stretch of 7-fold was needed to achieve a reduction of Cmax and AUC GMR to 0.9, an arbitrary threshold where the authors assumed that BE could still be demonstrated, thus defining the edge of the safe space in terms of dissolution.
Case Study 356
100 mg acalabrutinib free-base capsules are associated with label restrictions for patients undergoing ARA treatment since acalabrutinib solubility is insufficient at pH 6 to allow full drug dissolution in the stomach.57 With 20–40% hematological cancer patients estimated to take ARAs,58 the development of a formulation which could avoid ARA restriction was needed. Acalabrutinib maleate shows a higher surface solubility compared to the free base, leading to faster and complete dissolution in all tested media. Using the NDA dissolution method (QC dissolution method previously approved for the free base drug product) for 100 mg acalabrutinib maleate tablets (AMT), P-PSD can be fitted to clinical and virtual batches of drug products to provide a mechanistic model to integrate in a PBBM. To justify the dissolution specifications for 100 mg AMT, a PBBM previously developed for the free base52,57 was adapted to the salt formulation by changing the drug solubility vs pH profile. The model was validated based on the results of study ACE-HV-115 comparing an oral solution to three tablets of drug products comprising drug substances of increasing particle size.59 In the fasted state, the results showed that all investigational medicinal products were bioequivalent. In addition, the impact of food and PPI pretreatment could be adequately predicted by the model.56 Two virtual batches (VB) of 100 mg of AMT, i.e., passing at Q80% = 20 min (VBA) and passing at Q80% = 30 min (VBB) with the NDA dissolution method, were generated. PBBM predictions for VBA and VBB and the clinical batches in the fasted state showed that all of these batches were anticipated to be bioequivalent. In stomach conditions following PPI treatment, the relative exposure of batches compared to the clinical reference under acidic stomach conditions showed that VBB and VBA would fail BE criteria. VBA/ref AUC GMR would be around 0.8 if VBA was administered with a PPI. Using PBBM predicted exposure for these batches under PPI treatment and historical data showing the evolution of BTK occupancy by acalabrutinib as a function of Cmax or AUC, i.e., coupling the PBBM data with a PK–PD model, the efficacy of batches VBA and VBB could still be demonstrated since they were expected to lead to drug exposure which would engage the pharmacological target at levels comparable to those reached during phase 3 efficacy studies. It was concluded that a dissolution specification of 20–30 min was acceptable, since batches dissolving faster than this limit would lead to safe and effective exposure to the drug.
Take Home Messages
Depending on the BCS class of the drug substance and the physicochemical properties of the drug product, safe spaces can be defined using PBBM, either by univariate changes of a dissolution parameter such as the Z-factor or by generating virtual dissolution profiles and integrating the dissolution in the PBBM using the same method as that used for model validation. The use of a mechanistic dissolution model for IR products offers the advantages of predicting different prandial states and the effect of an ARA. It is important when running VBE studies to apply WSV. When combined with mechanistic models for dissolution, WSV leads to different in vivo dissolution for each dosing occasion. This could lead to failing VBE studies, as was shown for zolpidem when the size of the VBE is not adapted to the WSV. Overall, combining mechanistic IR dissolution models and VBE represents powerful tools to determine the edge of failure or allow bridging of formulations even if the dissolution data indicate a lack of similarity. With BCS class III–IV drugs, the dissolution safe space could be large, but care should be taken to evaluate any changes of formulation that may impact the GI physiology or the drug permeability independently from dissolution.
Case Study: Bioequivalence Safe Space in Healthy Volunteers vs Patients (Siri Kalyan Chirumamilla, Certara UK Limited, Simcyp Division)
Safe spaces for clinically relevant attributes were generated using PBBM with generally healthy volunteer physiological parameters. While the safe space generated using healthy volunteer physiology can be extended/relevant for patient populations for most drug products, it might not be the case for a few drug products.
BMS had developed a salt form to overcome the poor performance of a BCS class II basic drug free form when dosed with the ARA famotidine and a preclinical dog PK study showed higher AUC for the salt form compared to the free form in the presence of the ARA.60 Simcyp Simulator ADAM Model61 was used to develop a model for free form by using the human PK data without an ARA, then verified by predicting human PK with an ARA. The free form model was extended to predict salt form PK by using the Ksp Salt model, Mechanistic surface pH, and Two Solid States Model of the Simcyp Simulator. The developed model predicted a higher fraction absorbed for the salt form compared to free form in the presence of an ARA, similar to what was observed in the dog PK study.18
Though salts are used to improve solubility and dissolution, they tend to disproportionate during manufacture or storage to partly convert into free form. The Simcyp VBE module and two solid state models are used to generate safe space for % salt disproportionation in the dosage form. The safe space was generated using HV physiology and in patient physiology (elevated gastric pH mimicking dosage with ARA famotidine). The model predicted a wider space for HVs but a much narrower space using patient physiology. While the presence of 30% of the free form in the salt dosage form failed VBE in patients, it passed in HVs.
Though salt forms improve the dissolution of the drug, the presence of the free form in the dosage form did not alter the PK in HVs and gave wider safe space for salt disproportionation due to the precipitation of the dissolved salt form as free form inside the gastrointestinal tract. However, at elevated pH, the salt form exhibits favorable surface pH compared to the free form, driving dissolution and supersaturation as well as leading to a higher fraction being absorbed. Consequently, in patients dosed with an ARA, the PK is more sensitive to the extent of disproportionation (% salt form), which results in a narrow safe space.
Therefore, patient populations, for example, cancer patients, patients on co-medications, and Japanese elderly (achlorhydria), could have elevated gastric pH, which might influence the safe space as shown for the salt form in this case study. When in vitro dissolution is used as direct input into the PBBM, the in vivo dissolution will not be sensitive to the changing physiology between subjects and within subjects.62 Therefore, the generated safe space using this approach might not be accurate or relevant for the target population. Mechanistic models that consider both physiological parameters and drug form (salt/free form) can incorporate the effect of patient characteristics on the generated safe space which are relevant for the target drug product users.
In this context, another case example was shared. Elagolix has good aqueous solubility and low to moderate permeability and exhibits rapid absorption after oral administration with no meaningful effect of food on its pharmacokinetics.27 The original PBPK model for elagolix was developed to address metabolic and transporter DDI questions in Phase II of drug development.29,63 The model was refined to include in vitro dissolution as model input and validated with available clinical PK data including food effect study PK. The model was then employed to compare the slow dissolution of a commercial batch to a clinical batch and conclude that 30% slower release would still be bioequivalent to the reference commercial batch, thus allowing widening of the dissolution specification. A non-bioequivalence batch was not used during model validation.27,29 Subsequently the model was further refined to include a mechanistic dissolution model to differentiate tablet versus capsule formulations to support a VBE assessment between the fixed dose combination product and the elagolix tablet formulation.30 This case study demonstrates the advantage of developing PBPK models early in drug development and refinement as drug development progresses to answer different questions throughout the product life cycle.
Q1. Which Biopharmaceutic Properties of the Drug Substance Are Critical to and Should Be Considered to Establish a Model for Safe Space? That Is, Are There Any “Red Flags” In a Drug’s Biopharmaceutics Properties (e.g., Active Transporter, Regional Absorption, Precipitation) That Could Affect Model Development for the Bioequivalence Safe Space? In Such Case, if PBBM/IVIVCR Cannot Be Developed, How Would Alternative Models, Such As Nonlinear IVIVC (or Others), Be Used? How Would Noncompendial In Vitro Methods That Assess These Red Flag Properties Be Used?
Various scenarios were discussed which were (a) efflux which may change with dissolution change releasing a different percentage of drug, (b) solid form change during product manufacture which may impact dissolution, (c) solid form changes in the GI, (d) drugs with high logP, and (e) uncertainty of data generated with in vitro permeability studies.
Q2. What Tools (e.g., Models, Data) Are Available to Detect/Avoid an Undesired Outcome from BA Studies Designed to Establish Safe Space Where PBBM Cannot Be Developed (Due to Properties of the API and/or Drug Product) and Formulations Tested Are Non-BE to the Reference?
The participants shared the following procedures: (a) biorelevant dissolution models, e.g., 2-stage dissolution or TIM or other in vitro models prior to BA or BE studies, (b) preclinical models and deconvoluted data can be useful but need to be carefully evaluated to better predict clinical outcome, (c) fit for purpose PBBMs empower dissolution testing in very early stages to support PK BA/BE prediction, (d) ideally, include non-BE batches to increase confidence in safe space or IVIVC models. However, non-BE batches may be impractical.
Q3. In Principle, for IR Formulations of BCS III/IV Drug Substances, Dissolution Is Not Likely to Affect in Vivo Performance if the Release Rate Is Not Too Slow and the Probability of Achieving BE of Tested Formulations Is High. How Can Models (or Other Tools) Be Used to Establish the Threshold of Drug Release Rate to Establish the Safe Space and “Failure Edge” for Low-Permeability Drug Molecules?
The participants stated that it is possible to establish a wide safe space with a properly established PBBM as systemic exposure would be permeation rate controlled64 for BCS III/IV compounds. Thus, there would be opportunities for the extension of a BCS-based biowaiver.
Q4. Related to Question Q3, for BCS I/II Drug Substances, Dissolution Likely Affects in Vivo Performance and Non-BE Results from Tested Formulations Are More Likely than for BCS III/IV Molecules. A Safe Space Can Still Be Developed if a Model Is Available. However, If a Model Is Not Possible, We Run the Risk of Conducting BA Studies in a Clear Safe Space. One Mitigation Option Is to Perform Iterations of BA Studies to Find a Formulation That Is Right on the Failure Edge of BE. Nevertheless, if This Occurs, More than One BA Study Is Likely Required, And the Outcome Remains Uncertain. Under This Circumstance, What Alternative Models (Tools) Can Be Used to Help Establish Safe Space? If PBBM Is Not Successful, IVIVC and/or IVIVR Might Be the Options to Try
Participants discussed that performing interpolation between BE and non-BE lots is not recommended for an IVIVR. In contrast, IVIVC and PBBM can allow interpolation to define the edge of the safe space.
Q5. Larger Formulation Variations, Which Likely Lead to Non-BE Results, Are Often Required to Establish a Safe Space. However, A Key Criterion of Testing Formulation Variations Is These Variations Do Not Alter the Drug Release Mechanism or Else It May Result in Unexpected in Vivo Performance and/or Compromise the Development of a Model. What Data Are Needed to Support the Maintenance of the Release Mechanism?
Release mechanism may be affected for non-BE formulation changes such as different excipient release or different image sizes etc. Participants suggested that the images/picture/visualization technique (computed tomography and magnetic resonance) might be provided to show if such larger formulation variations would change the release mechanism. Also, multivariable dissolution studies can help with extending to other variations that were tested in the safe space studies.
Q6. Depending on the Properties of API, Formulations/Processes, and In Vitro Methods, It Is Possible That a Safe Space Is Narrower than +10%. What Mitigation Strategies (e.g., Developing a New In Vitro Method) Should Be Considered if This Occurs? Better Yet, How Can Modeling and Simulation Be Used to Predict This Undesired Outcome before Conducting the BA Studies?
Health authorities usually grant a ±10% window for in vitro drug dissolution method acceptance criteria. If a wider window is needed based on the data, further optimizing the dissolution method might be considered or an acceptable modeling and simulation approach may be used to support a wider window.
4. Conclusions and Future Directions
Day 2 continued to build on lessons from Day 1 in that the audience learned about specific feedback on three PBBM case studies from the regulatory perspective,3 including discussion on whether a PBBM could be considered as acceptable for the intended use, together with some direction for future submissions. The morning panel discussion with regulatory colleagues focused predominantly on model validation and application. Key takeaways were as follows: (1) For a specific question of interest and COU, PBBM influence and decision consequence define the model risk which will drive the level of data needed to evaluate the credibility of the model.5−8 (2) The introduction of a non-BE batch to help in model robustness should be guided by meaningful changes and be feasible to produce. (3) Cross discipline reviews among regulators will facilitate the credibility assessment of models. (4) Facilitating the future of integration of PBBM and MIDD in global drug development will include proactive engagement and continuous education from the regulatory perspective and bridging gaps between clinical pharmacology and CMC for industry. The afternoon discussions, as with Day 1, aimed to develop best practices, decision trees, and approaches for model development, model validation, and model application which would be beneficial for future PBBM submissions. Key topics in the breakout sessions covered how to establish disposition parameters, considerations for model validation criteria in light of available clinical data and model application (model risk vs decision consequence), dealing with WSV, BSV, and parameter uncertainty which needs to be considered from both the physiology and formulation perspectives, and establishing safe spaces and failure edges for dissolution specification development. It is suggested that in future PBBM submissions, the sponsor clarify the question of interest and the COU of the PBBM together with the model risk and for regulators to give detailed feedback to industry on the submissions, thereby setting clear expectations and increasing reliable PBBM development, which will ultimately benefit drug product quality for patients.
Acknowledgments
The meeting organizers are extremely grateful to Keiasia Robinson and Dana Hammell (University of Maryland) and the FDA Office of Regulatory Science and Innovation (ORSI) for significantly contributing to the success of the workshop. The authors would like to acknowledge the collaborative effort of regulators who participated in the review of PBBM case studies and the open and constructive way they shared their views on the scientific approach and data submitted. In parallel, the authors would like to thank the IQ consortium company members for taking the time to develop PBBMs around relevant biopharmaceutical questions, prepare the submissions, and make this exercise possible. The authors would like to thank Simulations Plus and Certara for sharing their advice and training when requested to use their respective modeling platforms. This research was supported by the Food and Drug Administration (FDA) of the U.S. Department of Health and Human Services (HHS) as part of a financial assistance award 5U01FD005946 totaling $5,000 with 100% funded by FDA/HHS. The collaboration work was supported by the IQ consortium.
Glossary
List of Abbreviations
- AAPE
Absolute Average Prediction Error
- AAFE
Average Fold Error
- AC
Acceptance Criteria
- ANVISA
Brazilian Health Regulatory Agency
- ANDA
Abbreviated New Drug Application
- API
Active Pharmaceutical Ingredient
- ARA
Acid Reducing Agent
- AUC
Area Under the Curve
- BA
Bioavailability
- BCS
Biopharmaceutical Classification System
- BE
Bioequivalent/Bioequivalence
- BO
Breakout Session
- BSV
Between Subject Variability
- Caco-2
Human Colon Adenocarcinoma Cell Line
- CBA
Critical Bioavailability Attribute(s)
- CFV
Critical Formulation Variable
- CMA
Critical Material Attribute
- Cmax
Maximum Plasma Concentration
- CMC
Chemistry and Manufacturing Controls
- COU
Context of Use
- CPP
Critical Process Parameter
- CQA
Critical Quality Attribute
- CRDS
Clinically Relevant Dissolution Specification
- DDI
Drug–Drug Interaction
- DP
Drug Product
- DT
Disintegration Time
- EMA
European Medicines Agency
- eCTD
Electronic Common Technical Document
- EU
European Union
- FDA
U.S. Food and Drug Administration
- FaSSIF
Fasted State Simulated Intestinal Fluid
- FAMHP
Belgian Federal Agency for Medicines and Health Products
- FE
Fold Error
- FeSSIF
Fed State Simulated Intestinal Fluid
- HV
Healthy Volunteers
- GI
Gastrointestinal
- IND
Investigational New Drug
- IR
Immediate Release
- IQ
International Consortium for Innovation & Quality in Pharmaceutical Development
- iv
Intravenous
- IVIVC
In vitro–in vivo correlation
- IVIVR
In vitro–in vivo relationship
- MAA
Marketing Authorization Application
- MAD
Multiple Ascending Dose
- MCERSI
University of Maryland Center of Excellence in Regulatory Science and Innovation
- MDCK
Madin–Darby canine kidney
- MHRA
Medicine and Healthcare products Regulatory Agency, UK
- MIDD
Model Informed Drug Development
- MR
Modified Release
- MS
Microsoft
- N
Number of Subjects
- NCE
New Chemical Entity
- NDA
New Drug Application
- NME
New Molecular Entity
- NMT
Not More Than
- non-BE
Non-bioequivalent/bioequivalence
- OC
Organizing Committee
- OGD
Office of Generic Drugs
- OOS
Out Of Specification
- Peff
Effective Permeability
- PBDT
Physiologically Based Dissolution Testing
- PBBM
Physiologically Based Biopharmaceutics Model (Modeling)
- PBPK
Physiologically Based Pharmacokinetics
- PD
Pharmacodynamics
- %PE
Percent Prediction Error
- PK
Pharmacokinetics
- PMDA
Pharmaceuticals and Medical Devices Agency, Japan
- PopPK
Population Pharmacokinetics
- PPI
Proton Pump Inhibitor
- P-PSD
Product Particle Size Distribution
- PSA
Parameter Sensitivity Analysis
- PSD
Particle Size Distribution
- QbD
Quality by Design
- QC
Quality Control
- QOI
Question of Interest
- relBA
Relative Bioavailability
- relBE
Relative Bioequivalence
- SAD
Single Ascending Dose
- SIF
Simulated Intestinal Fluid
- SUPAC
Scale Up and Post Approval Changes
- Tox
Toxicology
- VBE
Virtual Bioequivalence
- V&V
Verification and Validation
- WSV
Within Subject Variability
- US/USA
United States of America
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.molpharmaceut.4c00758.
Case study for Drug C wherein the modeling approach was utilized to justify the absence of impact of gender on bioequivalence study results; the outcome from modeling exercise indicated that the bioequivalence study results obtained in male subjects can be extrapolated and is ascribed to both genders (PDF)
This article reflects the views of the individual authors and should not be construed to represent the views, policies, or nomenclature of their organizations, including ANVISA, FAMHP, EMA, FDA, Health Canada, MHRA, Norwegian Medical Products Agency, Swedish Medical Products Agency, and the PMDA.
The authors declare the following competing financial interest(s): Tycho Heimbach, David Turner, Cordula Stillhart, Philip Bransford, Xiaojun Ren, Nikunj Patel, David Sperry, Amin Rostami-Hodjegan, Viera Lukacova, Jean-Flaubert Nguefack, Tessa Carducci, Xavier Pepin, Masoud Jamei, Konstantinos Stamatopoulos, Maitri Sanghavi, Christer Tannergren, Tzuchi Rob Ju, Christian Wagner, Michael Wang, Gregory Rullo, Amitava Mitra, James Polli, Sivacharan Kollipara, Claire Mackie are employees of their respective companies and have ownership, options, and/or interests in their respective stock.
Special Issue
Published as part of Molecular Pharmaceuticsspecial issue “2023 PBBM Workshop for Drug Product Quality”.
Supplementary Material
References
- Mackie C.; Arora S.; Seo P.; Moody R.; Rege B.; Pepin X.; Heimbach T.; Tannergren C.; Mitra A.; Suarez-Sharp S.; et al. Physiologically Based Biopharmaceutics Modeling (PBBM): Best Practices for Drug Product Quality, Regulatory and Industry Perspectives: 2023 Workshop Summary Report. Mol. Pharmaceutics 2024, 21, 2065. 10.1021/acs.molpharmaceut.4c00202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu F.; Shah H.; Li M.; Duan P.; Zhao P.; Suarez S.; Raines K.; Zhao Y.; Wang M.; Lin H. P.; et al. Biopharmaceutics Applications of Physiologically Based Pharmacokinetic Absorption Modeling and Simulation in Regulatory Submissions to the U.S. Food and Drug Administration for New Drugs. AAPS J. 2021, 23 (2), 31. 10.1208/s12248-021-00564-2. [DOI] [PubMed] [Google Scholar]
- Physiologically Based Biopharmaceutics Modeling (PBBM) Best Practices for Drug Product Quality Workshop. University of Maryland Center of Excellence in Regulatory Science and Innovation (M-CERSI), 2023. https://cersi.umd.edu/physiologically-based-biopharmaceutics-modeling-pbbm-best-practices-drug-product-quality (accessed February 24th, 2024).
- Pepin X.; Arora S.; Borges L.; Cano-Vega M.; Carducci T.; Chatterjee P.; Chen G.; Cristofoletti R.; Dallmann A.; Delvadia P.; et al. Parameterization of Physiologically Based Biopharmaceutics Models: Workshop Summary Report. Mol. Pharmaceutics 2024, 21, 3697. 10.1021/acs.molpharmaceut.4c00526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuemmel C.; Yang Y.; Zhang X.; Florian J.; Zhu H.; Tegenge M.; Huang S. M.; Wang Y.; Morrison T.; Zineh I. Consideration of a Credibility Assessment Framework in Model-Informed Drug Development: Potential Application to Physiologically-Based Pharmacokinetic Modeling and Simulation. CPT Pharmacometrics Syst. Pharmacol 2020, 9 (1), 21–28. 10.1002/psp4.12479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Musuamba F. T.; Skottheim Rusten I.; Lesage R.; Russo G.; Bursi R.; Emili L.; Wangorsch G.; Manolis E.; Karlsson K. E.; Kulesza A.; et al. Scientific and regulatory evaluation of mechanistic in silico drug and disease models in drug development: Building model credibility. CPT Pharmacometrics Syst. Pharmacol 2021, 10 (8), 804–825. 10.1002/psp4.12669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pappalardo F.; Russo G.; Tshinanu F. M.; Viceconti M. In silico clinical trials: concepts and early adoptions. Brief Bioinform 2019, 20 (5), 1699–1708. 10.1093/bib/bby043. [DOI] [PubMed] [Google Scholar]
- Skottheim Rusten I.; Musuamba F. T. Scientific and regulatory evaluation of empirical pharmacometric models: An application of the risk informed credibility assessment framework. CPT Pharmacometrics Syst. Pharmacol 2021, 10 (11), 1281–1296. 10.1002/psp4.12708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kourentas A.; Gajewska M.; Lin W.; Dhareshwar S. S.; Steib-Lauer C.; Kulkarni S.; Hirsch S.; Heimbach T.; Mueller-Zsigmondy M. Establishing the Safe Space via Physiologically Based Biopharmaceutics Modeling. Case Study: Fevipiprant/QAW039. AAPS J. 2023, 25 (1), 25. 10.1208/s12248-023-00787-5. [DOI] [PubMed] [Google Scholar]
- Takano R.; Sugano K.; Higashida A.; Hayashi Y.; Machida M.; Aso Y.; Yamashita S. Oral Absorption of Poorly Water-Soluble Drugs: Computer Simulation of Fraction Absorbed in Humans from a Miniscale Dissolution Test. Pharm. Res. 2006, 23 (6), 1144–1156. 10.1007/s11095-006-0162-4. [DOI] [PubMed] [Google Scholar]
- Rowland M. Microdosing: a critical assessment of human data. J. Pharm. Sci. 2012, 101 (11), 4067–4074. 10.1002/jps.23290. [DOI] [PubMed] [Google Scholar]
- Boddu R.; Kollipara S.; Vijaywargi G.; Ahmed T. Power of integrating PBPK with PBBM (PBPK-BM): a single model predicting food effect, gender impact, drug-drug interactions and bioequivalence in fasting & fed conditions. Xenobiotica 2023, 53 (4), 260–278. 10.1080/00498254.2023.2238048. [DOI] [PubMed] [Google Scholar]
- U.S. FDA Assessing the Credibility of Computational Modeling and Simulation in Medical Device Submissions, Guidance for Industry and Food and Drug Administration Staff. 2023. https://www.fda.gov/regulatory-information/search-fda-guidance-documents/assessing-credibility-computational-modeling-and-simulation-medical-device-submissions (accessed 2024 May 13th).
- U.S. FDA The Use of Physiologically Based Pharmacokinetic Analyses — Biopharmaceutics Applications for Oral Drug Product Development, Manufacturing Changes, and Controls. 2020. https://www.fda.gov/regulatory-information/search-fda-guidance-documents/use-physiologically-based-pharmacokinetic-analyses-biopharmaceutics-applications-oral-drug-product.
- Fang L.; Gong Y.; Hooker A. C.; Lukacova V.; Rostami-Hodjegan A.; Sale M.; Grosser S.; Jereb R.; Savic R.; Peck C.; Zhao L. The Role of Model Master Files for Sharing, Acceptance, and Communication with FDA. AAPS J. 2024, 26 (2), 28. 10.1208/s12248-024-00897-8. [DOI] [PubMed] [Google Scholar]
- Chiou W. L.; Barve A. Linear correlation of the fraction of oral dose absorbed of 64 drugs between humans and rats. Pharm. Res. 1998, 15 (11), 1792–1795. 10.1023/A:1011981317451. [DOI] [PubMed] [Google Scholar]
- Peternel L.; Kristan K.; Petruševska M.; Rižner T. L.; Legen I. Suitability of isolated rat jejunum model for demonstration of complete absorption in humans for BCS-based biowaiver request. J. Pharm. Sci. 2012, 101 (4), 1436–1449. 10.1002/jps.23027. [DOI] [PubMed] [Google Scholar]
- Chirumamilla S. K.; Banala V. T.; Jamei M.; Turner D. B. Mechanistic PBPK Modelling to Predict the Advantage of the Salt Form of a Drug When Dosed with Acid Reducing Agents. Pharmaceutics 2021, 13 (8), 1169. 10.3390/pharmaceutics13081169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ezuruike U.; Zhang M.; Pansari A.; De Sousa Mendes M.; Pan X.; Neuhoff S.; Gardner I. Guide to development of compound files for PBPK modeling in the Simcyp population-based simulator. CPT: Pharmacometrics & Systems Pharmacology 2022, 11 (7), 805–821. 10.1002/psp4.12791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao J.; Ma F.; Yu J.; Bruyn T. D.; Ning M.; Bowman C.; Chen Y. Shared learning from a physiologically based pharmacokinetic modeling strategy for human pharmacokinetics prediction through retrospective analysis of Genentech compounds. Biopharmaceutics & Drug Disposition 2023, 44 (4), 315–334. 10.1002/bdd.2359. [DOI] [PubMed] [Google Scholar]
- Ring B. J.; Chien J. Y.; Adkison K. K.; Jones H. M.; Rowland M.; Jones R. D.; Yates J. W.; Ku M. S.; Gibson C. R.; He H.; et al. PhRMA CPCDC initiative on predictive models of human pharmacokinetics, part 3: comparative assessement of prediction methods of human clearance. J. Pharm. Sci. 2011, 100 (10), 4090–4110. 10.1002/jps.22552. [DOI] [PubMed] [Google Scholar]
- Jones R. D.; Jones H. M.; Rowland M.; Gibson C. R.; Yates J. W. T.; Chien J. Y.; Ring B. J.; Adkison K. K.; Ku M. S.; He H.; et al. PhRMA CPCDC initiative on predictive models of human pharmacokinetics, part 2: Comparative assessment of prediction methods of human volume of distribution. J. Pharm. Sci. 2011, 100 (10), 4074–4089. 10.1002/jps.22553. [DOI] [PubMed] [Google Scholar]
- Frechen S.; Rostami-Hodjegan A. Quality Assurance of PBPK Modeling Platforms and Guidance on Building, Evaluating, Verifying and Applying PBPK Models Prudently under the Umbrella of Qualification: Why, When, What, How and By Whom?. Pharm. Res. 2022, 39 (8), 1733–1748. 10.1007/s11095-022-03250-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Assessing Credibility of Computational Modeling through Verification and Validation: Application to Medical Devices; VV40-2018; ASME, New York, NY, 2018. [Google Scholar]
- El-Tahtawy A. A.; Jackson A. J.; Ludden T. M. Comparison of Single and Multiple Dose Pharmacokinetics Using Clinical Bioequivalence Data and Monte Carlo Simulations. Pharm. Res. 1994, 11 (9), 1330–1336. 10.1023/A:1018906931100. [DOI] [PubMed] [Google Scholar]
- Mukherjee D.; Chiney M. S.; Shao X.; Ju T. R.; Shebley M.; Marroum P. Physiologically based pharmacokinetic modeling and simulations to inform dissolution specifications and clinical relevance of release rates on elagolix exposure. Biopharm Drug Dispos 2022, 43 (3), 98–107. 10.1002/bdd.2315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- U.S. FDA CDER NDA 216196 Integrated Review. 2022. https://www.accessdata.fda.gov/drugsatfda_docs/nda/2022/216196Orig1s000IntegratedR.pdf (accessed 2024 May 5th).
- U.S. FDA NDA 210450, ORILISSA (elagolix) Tablets. 2018. https://www.accessdata.fda.gov/drugsatfda_docs/nda/2018/210450Orig1s000ChemR.pdf (accessed 2024 May 5th).
- Mukherjee D.; Chen M. J.; Shao X.; Ju T. R.; Shebley M.; Marroum P. Virtual Bioequivalence Assessment of Elagolix Formulations Using Physiologically Based Pharmacokinetic Modeling. AAPS J. 2023, 25 (3), 30. 10.1208/s12248-023-00794-6. [DOI] [PubMed] [Google Scholar]
- Bego M.; Patel N.; Cristofoletti R.; Rostami-Hodjegan A. Proof of Concept in Assignment of Within-Subject Variability During Virtual Bioequivalence Studies: Propagation of Intra-Subject Variation in Gastrointestinal Physiology Using Physiologically Based Pharmacokinetic Modeling. AAPS J. 2022, 24 (1), 21. 10.1208/s12248-021-00672-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han C.; Sun T.; Chirumamilla S. K.; Bois F. Y.; Xu M.; Rostami-Hodjegan A. Understanding Discordance between In Vitro Dissolution, Local Gut and Systemic Bioequivalence of Budesonide in Healthy and Crohn’s Disease Patients through PBPK Modeling. Pharmaceutics 2023, 15 (9), 2237. 10.3390/pharmaceutics15092237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thwaites P. A.; Yao C. K.; Halmos E. P.; Muir J. G.; Burgell R. E.; Berean K. J.; Kalantar-zadeh K.; Gibson P. R. Review article: Current status and future directions of ingestible electronic devices in gastroenterology. Alimentary Pharmacology & Therapeutics 2024, 59 (4), 459–474. 10.1111/apt.17844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller N. A.; Reddy M. B.; Heikkinen A. T.; Lukacova V.; Parrott N. Physiologically Based Pharmacokinetic Modelling for First-In-Human Predictions: An Updated Model Building Strategy Illustrated with Challenging Industry Case Studies. Clin Pharmacokinet 2019, 58 (6), 727–746. 10.1007/s40262-019-00741-9. [DOI] [PubMed] [Google Scholar]
- Meyer M. C.; Straughn A. B.; Jarvi E. J.; Patrick K. S.; Pelsor F. R.; Williams R. L.; Patnaik R.; Chen M. L.; Shah V. P. Bioequivalence of methylphenidate immediate-release tablets using a replicated study design to characterize intrasubject variability. Pharm. Res. 2000, 17 (4), 381–384. 10.1023/A:1007560500301. [DOI] [PubMed] [Google Scholar]
- Kollipara S.; Bhattiprolu A. K.; Boddu R.; Ahmed T.; Chachad S. Best Practices for Integration of Dissolution Data into Physiologically Based Biopharmaceutics Models (PBBM): A Biopharmaceutics Modeling Scientist Perspective. AAPS PharmSciTech 2023, 24 (2), 59. 10.1208/s12249-023-02521-y. [DOI] [PubMed] [Google Scholar]
- Abuhassan Q.; Khadra I.; Pyper K.; Augustijns P.; Brouwers J.; Halbert G. W. Fasted intestinal solubility limits and distributions applied to the biopharmaceutics and developability classification systems. Eur. J. Pharm. Biopharm. 2022, 170, 160–169. 10.1016/j.ejpb.2021.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de la Cruz-Moreno M. P.; Montejo C.; Aguilar-Ros A.; Dewe W.; Beck B.; Stappaerts J.; Tack J.; Augustijns P. Exploring drug solubility in fasted human intestinal fluid aspirates: Impact of inter-individual variability, sampling site and dilution. Int. J. Pharm. 2017, 528 (1), 471–484. 10.1016/j.ijpharm.2017.05.072. [DOI] [PubMed] [Google Scholar]
- Riethorst D.; Mols R.; Duchateau G.; Tack J.; Brouwers J.; Augustijns P. Characterization of Human Duodenal Fluids in Fasted and Fed State Conditions. J. Pharm. Sci. 2016, 105 (2), 673–681. 10.1002/jps.24603. [DOI] [PubMed] [Google Scholar]
- Abuhassan Q.; Khadra I.; Pyper K.; Halbert G. W. Small scale in vitro method to determine a bioequivalent equilibrium solubility range for fasted human intestinal fluid. Eur. J. Pharm. Biopharm 2021, 168, 90–96. 10.1016/j.ejpb.2021.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pyper K.; Brouwers J.; Augustijns P.; Khadra I.; Dunn C.; Wilson C. G.; Halbert G. W. Multidimensional analysis of human intestinal fluid composition. Eur. J. Pharm. Biopharm. 2020, 153, 226–240. 10.1016/j.ejpb.2020.06.011. [DOI] [PubMed] [Google Scholar]
- Stamatopoulos K.; Ferrini P.; Nguyen D.; Zhang Y.; Butler J. M.; Hall J.; Mistry N. Integrating In Vitro Biopharmaceutics into Physiologically Based Biopharmaceutic Model (PBBM) to Predict Food Effect of BCS IV Zwitterionic Drug (GSK3640254). Pharmaceutics 2023, 15, 521. 10.3390/pharmaceutics15020521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hens B.; Sarcevica I.; Tomaszewska I.; McAllister M. Digitalizing the TIM-1 Model Using Computational Approaches–Part Two: Digital TIM-1 Model in GastroPlus. Mol. Pharmaceutics 2023, 20 (11), 5429–5439. 10.1021/acs.molpharmaceut.3c00423. [DOI] [PubMed] [Google Scholar]
- Blanquet S.; Zeijdner E.; Beyssac E.; Meunier J.-P.; Denis S.; Havenaar R.; Alric M. A Dynamic Artificial Gastrointestinal System for Studying the Behavior of Orally Administered Drug Dosage Forms Under Various Physiological Conditions. Pharm. Res. 2004, 21 (4), 585–591. 10.1023/B:PHAM.0000022404.70478.4b. [DOI] [PubMed] [Google Scholar]
- Andreas C. J.; Pepin X.; Markopoulos C.; Vertzoni M.; Reppas C.; Dressman J. Mechanistic investigation of the negative food effect of modified release zolpidem. European journal of pharmaceutical sciences 2017, 102, 284–298. 10.1016/j.ejps.2017.03.011. [DOI] [PubMed] [Google Scholar]
- Durand A.; Thénot J. P.; Bianchetti G.; Morselli P. L. Comparative Pharmacokinetic Profile of Two Imidazopyridine Drugs: Zolpidem and Alpidem. Drug Metabolism Reviews 1992, 24 (2), 239–266. 10.3109/03602539208996294. [DOI] [PubMed] [Google Scholar]
- Gertz M.; Harrison A.; Houston J. B.; Galetin A. Prediction of human intestinal first-pass metabolism of 25 CYP3A substrates from in vitro clearance and permeability data. Drug Metab. Dispos. 2010, 38 (7), 1147–1158. 10.1124/dmd.110.032649. [DOI] [PubMed] [Google Scholar]
- Pichard L.; Gillet G.; Bonfils C.; Domergue J.; Thénot J. P.; Maurel P. Oxidative metabolism of zolpidem by human liver cytochrome P450S. Drug metabolism and disposition: the biological fate of chemicals 1995, 23 (11), 1253–1262. [PubMed] [Google Scholar]
- Salva P.; Costa J. Clinical pharmacokinetics and pharmacodynamics of zolpidem. Therapeutic implications. Clin Pharmacokinet 1995, 29 (3), 142–153. 10.2165/00003088-199529030-00002. [DOI] [PubMed] [Google Scholar]
- Von Moltke L. L.; Greenblatt D. J.; Granda B. W.; Duan S. X.; Grassi J. M.; Venkatakrishnan K.; Harmatz J. S.; Shader R. I. Zolpidem metabolism in vitro: responsible cytochromes, chemical inhibitors, and in vivo correlations. Br. J. Clin. Pharmacol. 1999, 48 (1), 89–97. 10.1046/j.1365-2125.1999.00953.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon S.; Jeong S.; Jung E.; Kim K. S.; Jeon I.; Lee Y.; Cho J.-Y.; Oh W.-Y.; Chung J.-Y. Effect of CYP3A4 metabolism on sex differences in the pharmacokinetics and pharmacodynamics of zolpidem. Sci. Rep. 2021, 11 (1), 19150 10.1038/s41598-021-98689-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pepin X. J. H.; Sanderson N. J.; Blanazs A.; Grover S.; Ingallinera T. G.; Mann J. C. Bridging in vitro dissolution and in vivo exposure for acalabrutinib. Part I. Mechanistic modelling of drug product dissolution to derive a P-PSD for PBPK model input. Eur. J. Pharm. Biopharm. 2019, 142, 421–434. 10.1016/j.ejpb.2019.07.014. [DOI] [PubMed] [Google Scholar]
- Pepin X.; Goetschy M.; Abrahmsén-Alami S. Mechanistic models for USP2 dissolution apparatus, including fluid hydrodynamics and sedimentation. J. Pharm. Sci. 2022, 111, 185. 10.1016/j.xphs.2021.10.006. [DOI] [PubMed] [Google Scholar]
- Davit B.; Chen M.-L.; Conner D.; Haidar S.; Kim S.; Lee C.; Lionberger R.; Makhlouf F.; Nwakama P.; Patel D.; et al. Implementation of a Reference-Scaled Average Bioequivalence Approach for Highly Variable Generic Drug Products by the US Food and Drug Administration. AAPS J. 2012, 14 (4), 915–924. 10.1208/s12248-012-9406-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- García M. A.; Bolger M. B.; Suarez-Sharp S.; Langguth P. Predicting Pharmacokinetics of Multisource Acyclovir Oral Products Through Physiologically Based Biopharmaceutics Modeling. J. Pharm. Sci. 2022, 111 (1), 262–273. 10.1016/j.xphs.2021.10.013. [DOI] [PubMed] [Google Scholar]
- Pepin X.; McAlpine V.; Moir A.; Mann J. Acalabrutinib Maleate Tablets: The Physiologically Based Biopharmaceutics Model behind the Drug Product Dissolution Specification. Mol. Pharm. 2023, 20, 2181. 10.1021/acs.molpharmaceut.3c00005. [DOI] [PubMed] [Google Scholar]
- Pepin X. J. H.; Moir A. J.; Mann J. C.; Sanderson N. J.; Barker R.; Meehan E.; Plumb A. P.; Bailey G. R.; Murphy D. S.; Krejsa C. M.; et al. Bridging in vitro dissolution and in vivo exposure for acalabrutinib. Part II. A mechanistic PBPK model for IR formulation comparison, proton pump inhibitor drug interactions, and administration with acidic juices. Eur. J. Pharm. Biopharm. 2019, 142, 435–448. 10.1016/j.ejpb.2019.07.011. [DOI] [PubMed] [Google Scholar]
- Smelick G. S.; Heffron T. P.; Chu L.; Dean B.; West D. A.; Duvall S. L.; Lum B. L.; Budha N.; Holden S. N.; Benet L. Z.; et al. Prevalence of acid-reducing agents (ARA) in cancer populations and ARA drug-drug interaction potential for molecular targeted agents in clinical development. Mol. Pharmaceutics 2013, 10 (11), 4055–4062. 10.1021/mp400403s. [DOI] [PubMed] [Google Scholar]
- Sharma S.; Pepin X.; Burri H.; Zheng L.; Kuptsova-Clarkson N.; de Jong A.; Yu T.; MacArthur H. L.; Majewski M.; Byrd J. C.; et al. Bioequivalence and Relative Bioavailability Studies to Assess a New Acalabrutinib Formulation That Enables Coadministration With Proton-Pump Inhibitors. Clinical Pharmacology in Drug Development 2022, 11, 1294. 10.1002/cpdd.1153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gesenberg C.; Mathias N. R.; Xu Y.; Crison J.; Savant I.; Saari A.; Good D. J.; Hemenway J. N.; Narang A. S.; Schartman R. R.; et al. Utilization of In Vitro, In Vivo and In Silico Tools to Evaluate the pH-Dependent Absorption of a BCS Class II Compound and Identify a pH-Effect Mitigating Strategy. Pharm. Res. 2019, 36 (12), 164. 10.1007/s11095-019-2698-0. [DOI] [PubMed] [Google Scholar]
- Jamei M.; Turner D.; Yang J.; Neuhoff S.; Polak S.; Rostami-Hodjegan A.; Tucker G. Population-based mechanistic prediction of oral drug absorption. AAPS J. 2009, 11 (2), 225–237. 10.1208/s12248-009-9099-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jamei M.; Abrahamsson B.; Brown J.; Bevernage J.; Bolger M. B.; Heimbach T.; Karlsson E.; Kotzagiorgis E.; Lindahl A.; McAllister M.; et al. Current status and future opportunities for incorporation of dissolution data in PBPK modeling for pharmaceutical development and regulatory applications: OrBiTo consortium commentary. Eur. J. Pharm. Biopharm. 2020, 155, 55–68. 10.1016/j.ejpb.2020.08.005. [DOI] [PubMed] [Google Scholar]
- Chiney M. S.; Ng J.; Gibbs J. P.; Shebley M. Quantitative Assessment of Elagolix Enzyme-Transporter Interplay and Drug-Drug Interactions Using Physiologically Based Pharmacokinetic Modeling. Clin Pharmacokinet 2020, 59 (5), 617–627. 10.1007/s40262-019-00833-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laisney M.; Heimbach T.; Mueller-Zsigmondy M.; Blumenstein L.; Costa R.; Ji Y. Physiologically Based Biopharmaceutics Modeling to Demonstrate Virtual Bioequivalence and Bioequivalence Safe-space for Ribociclib which has Permeation Rate-controlled Absorption. J. Pharm. Sci. 2022, 111 (1), 274–284. 10.1016/j.xphs.2021.10.017. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.